

Abstract
There are over 420 human Solute Carrier transporters (SLCs) from 65 families that are expressed ubiquitously in the body. The SLCs mediate the movement of ions, drugs, and metabolites across membranes, and their dysfunction has been associated with a variety of diseases, such as diabetes, cancer, and central nervous system disorders. Thus, the SLCs are emerging as important targets for therapeutic intervention. Recent technological advancements in experimental and computational biology allow better characterization of SLC pharmacology. Here we describe recent approaches to modulate SLC transporter function, with an emphasis on the use of computational approaches and computer-aided drug design to study nutrient transporters. Finally, we discuss future perspectives in the rational design of SLC drugs.
Keywords: solute carrier, protein structure prediction, computer-aided drug design, structure-based drug discovery, membrane transporter
SLC pharmacology
Transport of solutes by Solute Carrier (SLC) transporters is essential for many of life’s processes, such as importing nutrients into cells, generating electrical and chemical signals, regulating cell volume, as well as the delivery and excretion of drugs. In humans, there are over 420 SLCs that are grouped into 65 families based on their sequence, function, and number of transmembrane (TM) regions [1]. A large number of SLCs are implicated in a wide array of diseases, including central nervous system (CNS) and metabolic disorders, as well as diabetes and cancer [2]. For example, genetic variations in the norepinephrine transporter (NET, SLC6A2) are associated with attention-deficit hyperactivity disorder (ADHD) [3], and upregulation of the neutral amino acid transporter, alanine serine cysteine transporter 2 (ASCT2, SLC1A5) is linked to poor prognosis in non-small cell lung cancer (NSCLC) [4]. In addition, the majority of the SLCs are capable of being modulated by small molecules, such as their natural substrates (e.g., organic ions and cations, see Glossary), as well as prescription drugs and phospholipids, which can bind in substrate or allosteric binding sites [5]. Thus, SLCs are emerging as important, druggable targets.
However, compared to other protein families of similar size, such as GPCRs or protein kinases, only few SLCs have been targeted with tool compounds or clinically approved drugs [6, 7]. Furthermore, most drugs targeting the SLCs focus on a small subset of proteins, such as neurotransmitter transporters belonging to the SLC6 family, including the γ-aminobutyric acid transporter (GAT1, SLC6A1), norepinephrine transporter (NET, SLC6A2), dopamine transporter (DAT, SLC6A3), and serotonin transporter (SERT, SLC6A4) for the treatment of CNS diseases and disorders [6]. Interestingly, from 2015 to 2018, out of 172 novel drug approvals by the U.S. Food and Drug Administration (FDA), four drugs target three SLCs including: the vesicular monoamine transporter 2 (VMAT2, SLC18A2), the uric acid transporter (URAT1, SLC22A12) and the sodium-glucose transporter (SGLT2, SLC5A2) (Table 1)i,ii In addition, the Japanese Pharmaceuticals and Medical Devices agency (PMDA) has recently approved elobixibat, an inhibitor of the apical sodium bile acid transporter (ASBT, SLC10A2) for treating chronic idiopathic constipation [8] (Table 1). While the number of drugs targeting SLCs is still low, “drugging” previously unexplored SLCs suggests that there are other promising therapeutic SLC targets.
Table 1.
FDA and PMDA approval of SLC drugs from 2015 to 2018.
| aSLC | bOther name | cDrug | dYear | Indication | Mechanism of action | References / Resources |
|---|---|---|---|---|---|---|
| SLC10A2 | ASBT | Elobixibate (A3309) |
2018 | Chronic idiopathic constipation | Increases bile acid delivery to colon to accelerate and increase colonic secretion. | [8] |
| SLC5A2 | SGLT2 | SteglatroTM (ertugliflozin) |
2017 | Type II diabetes mellitus | Increases urinary glucose excretion via SGLT2 inhibition to reduce blood glucose levels. | iv |
| SLC18A2 | VMAT2 | Ingrezza™ (valbenazine) |
2017 | Tardive dyskinesia | Reversible inhibition of VMAT2 activity. | v |
| SLC18A2 | VMAT2 | Austedo™ (deutetrabenazine) |
2017 | Chorea associated with Huntington’s disease; Tardive dyskinesia | Mechanism of action for anti-chorea effects is currently unknown. | vi |
| SLC22A12 | URAT1 | Zurampic™ (lesinurad) |
2015 | Hyperuricemia associated with gout | Inhibition of URAT1 and organic anion transporter 4 (OAT4) (to a lesser extent), to increase uric acid excretion and reduce serum uric acid levels. | vii |
SLC marks the primary SLC target of the newly approved drug
Other name corresponds to alternative name of the SLC gene or protein
Drug is the brand name of the drug with the generic name in parenthesis
Year marks the approval year of the drug by the FDA
Elobixibat is a PMDA-approved drug. All others are FDA-approved.
Most of drug discovery efforts toward the human SLCs have focused on high throughput [9] and phenotypic screens [10], optimization of known inhibitors with medicinal chemistry and molecular design [11], as well as ligand-based computational approaches such as quantitative structure-activity relationship (QSAR) and pharmacophore modeling (reviewed in [7, 12]). One weakness of ligand-based approaches is their limited ability to guide the design of unique chemical scaffolds [7]. Computer-aided structure-based drug design offers the advantages of an in-depth understanding of the three-dimensional (3D) protein binding site and possible insight into different conformational states; and when combined with virtual screening of large compound libraries, it can lead to the discovery of novel chemical scaffolds [13].
SLC structure and mechanism
An important step toward increasing the number of tool compounds and eventual drugs targeting the SLCs, requires the characterization of SLC structures in different conformational states in complex with their ligands. Technical limitations, such as the lack of inadequate biological regents (e.g., antibodies) and poor protein expression by conventional techniques have hindered the development of pharmacological agents targeting SLCs [6]. Furthermore, only a small number of atomic-resolution structures of human SLCs have been determined [14], making it difficult to elucidate molecular interactions between small molecules and transporters, and to rationally design relevant chemical tools. Encouragingly, in recent years there has been a surge in the number of experimentally determined structures of SLCs and their homologs from other organisms [5]. For example, structures of important human SLCs, such as the electrogenic sodium bicarbonate cotransporter (NBCe1, SLC4A4) [15], as well as the amino acid transporters, ASCT2 [16] and large neutral amino acid transporter (LAT1, SLC7A5) [17] have been determined at near atomic-resolution using cryo-electron microscopy (cryo-EM). Although most cryo-EM SLC structures are at insufficient resolution for describing the molecular interactions useful for rational drug design, this is anticipated to change in the near future due to various advances, such as those made in detector technology and data processing [18].
The recent structures have revealed that apart from their diverse functions, the SLC families cluster to evolutionarily unrelated structural classes or folds, where the most common folds are the major facilitator superfamily (MFS) and leucine transporter (LeuT) folds [19]. Notably, although the SLCs generally follow an ‘alternating access’ transport mechanism [20, 21], each structural class uses a distinct type of conformational changes to transport substrates (Figure 1). For example: (i) MFS proteins use a ‘rocker-switch’ mechanism (e.g. GLUT1, SLC2A1), in which the substrate binds to the extracellular facing binding site, triggering conformational changes to an occluded state, followed by an inward-facing state where the substrate is released [22]. (ii) Transporters with the LeuT fold use a ‘rocking bundle’ or ‘gated-pore’ mechanism (e.g. SERT, SLC6A4), in which a mobile bundle domain undergoes large hinge-like rearrangements to release the substrate to the intracellular side, while the scaffold domain remains static [23, 24]. (iii) Several structural families (e.g., SLC1 and SLC13)) utilize an ‘elevator’ mechanism, in which the scaffold domain remains static as the mobile domain moves up and down like its namesake, to transport the substrate across the membrane [25–27]. Notably, knowledge of the transport mechanism, including relevant conformational changes, is important for devising a strategy for the development of small molecule modulators.
Figure 1. Different SLC alternating access transport mechanisms.
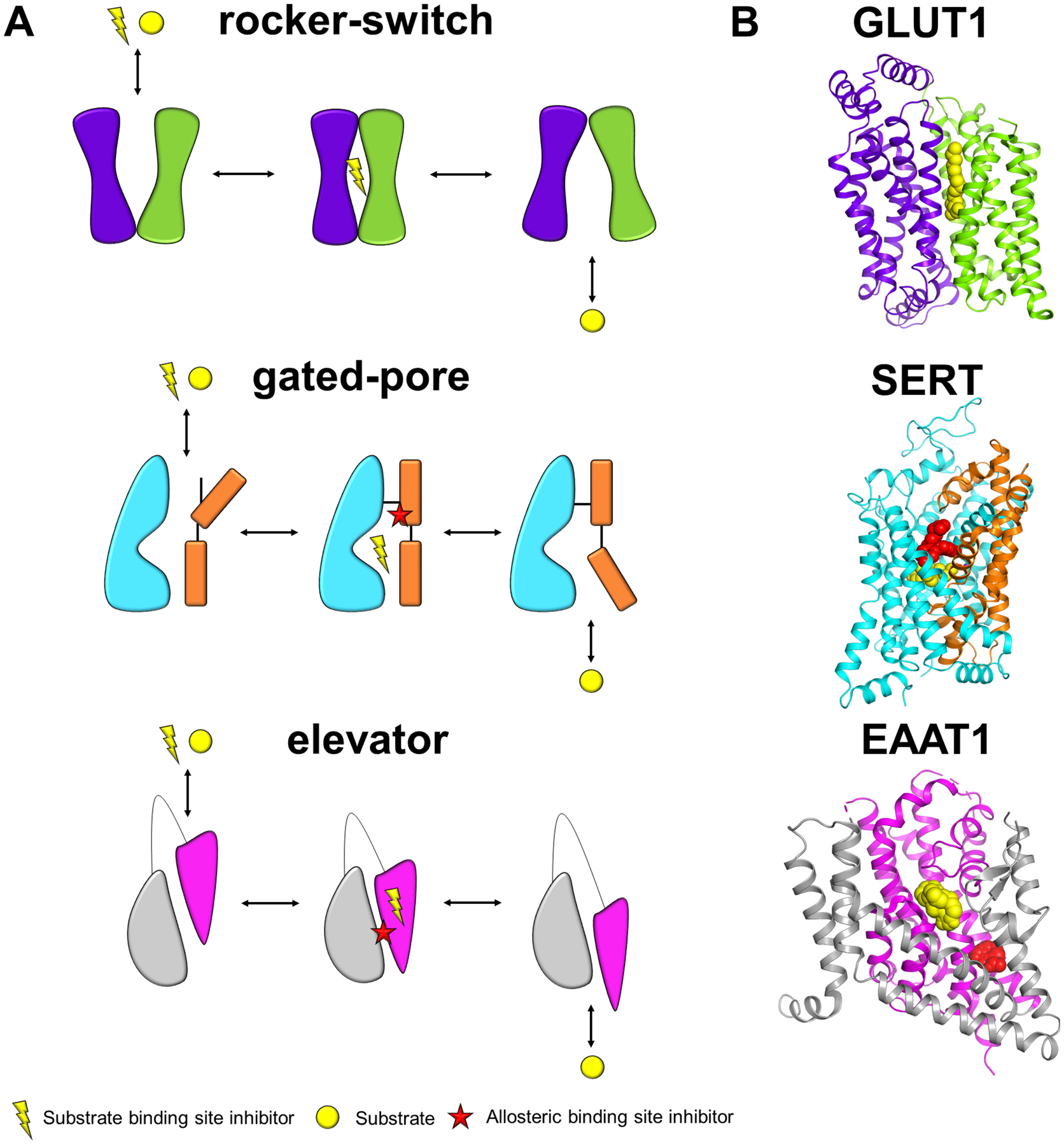
(A) For each transport mechanism, there is a representative schematic of an unbound outward-open conformation that is accessible to both inhibitor and substrate, an occluded inhibitor bound state, representative of when inhibitor binding blocks substrate transport and an inward open conformation, which is accessible to substrate from the intracellular side of the membrane. The colors denote the two different domains involved in the mechanism. The figure only shows one protomer even if more than one subunit is involved. The rocker-switch mechanism, the substrate binds to the extracellular facing binding site, triggering conformational changes to an occluded state, followed by an inward-facing state where the substrate is released. Rocking bundle or gated-pore mechanism has a mobile bundle domain (in orange) that undergoes large hinge-like rearrangements to release the substrate to the intracellular side, while the scaffold domain (in cyan) remains static. The elevator mechanism has a mobile domain (pink) that moves up and down, relative to a scaffold domain (gray), to transport the substrate across the membrane. (B) Representative structures of transporters using the transport mechanisms in (A) where the colors correspond to the respective domains as shown in (A), substrate binding site and allosteric site inhibitors are shown in yellow and red spheres, respectively. PDB IDs: GLUT1: 4PYP, SERT: 5I73, EAAT1: 5LLM.
In addition, the newly determined SLC structures facilitate modeling the human SLC transporters with homology modeling, which relies on the structures of homolog proteins as templates [28], or integrative modeling, which uses restraints derived from experimental data (e.g., from cryo-EM or cross-linking data) [29]. Specifically, it is now possible to model the structures of many previously unmodelable SLC transporter targets, or SLCs with known structures in unknown conformations. For example, the crystal structure of a zebrafish homolog of the lysosomal sodium-coupled neutral amino acid transporter 9 (SLC38A9) has been recently determined [30]. SLC38A9 is associated with mTOR activation in cancer [31] and shares a sequence identity of 61.9% with the zebrafish homolog, making it useful to generate homology models suitable for rational design. The new structural information on SLCs, combined with superior computational power and the maturation of computational chemistry tools, such as ligand docking [13], next generation membrane protein Molecular Dynamics (MD) simulation approaches [32], free energy perturbation estimation [33], as well as advanced machine learning (ML) architectures (e.g., autoencoder) [34] is expected to expedite the characterization of human SLCs. Here, we outline recent studies characterizing the substrate specificities of biomedically important SLC nutrient transporters, and the discovery of small molecule modulators of these proteins using computer-aided drug design (CADD).
Structure-based ligand design for the nutrient transporter, ASCT2
The SLC1 family has seven members, including five excitatory amino acid transporters (EAATs) that transport glutamate (SLC1A1–3, SLC1A6, 7) and two neutral amino acid transporters ASCT1 (SLC1A4) and ASCT2 (SLC1A5). Over the past decade, structures of SLC1 members from human (i.e., EAAT1 and ASCT2) and their prokaryotic homologs GltPh (reviewed in [35]) and GltTk [36] have been determined. Particularly, much of what is known about the structure and dynamics of the human SLC1 members has been through the characterization of GltPh with a variety of biophysical approaches, such as Double Electron-Electron Resonance (DEER) [37], single-molecule Fluorescence Resonance Energy Transfer (smFRET) [38], and high-speed atomic force microscopy (AFM) [39]. Taken together, these studies describe a trimeric configuration and elevator transport mechanism (Figure 1A) conserved across organisms.
The SLC1 family includes several putative drug targets, such as EAAT2 for Alzheimer’s disease (AD) [40] and ASCT2 for cancer [4, 41, 42], however, there are no clinically approved drugs targeting this family. Specifically, ASCT2 is frequently upregulated in various cancer types, including triple negative breast cancer [41] and NSCLC [4], where this transporter is thought to play a key role in glutamine import, thereby fueling cancer cells [42]. Thus far, efforts to inhibit ASCT2 have largely focused on substrate like-inhibitors that likely bind the substrate binding site [43–47].
Over the past decade, ASCT2 has been modeled in the outward-open and outward-occluded conformations based its homologs’ structures [43–47]. The initial GltPh-based models were used to guide the design of novel ASCT2 inhibitors such as 1,2,3-dithiazoles [48], serine biphenyl-4-carboxylate [43], L-γ-Glutamyl-p-nitroanilide (GPNA) analogs (Nγ-glutamylanilides) [47] and 2-amino-4-bis(aryl-oxybenzyl)aminobutanoic acids [49]. Additionally, recent ASCT2 models based on similar structures revealed previously uncharacterized druggable pockets, pocket A (PA) and pocket B (PB) (Figure 2, left inset). PB is formed in ASCT2 by a substitution of an arginine residue in EAAT1 (R479) to a cysteine residue in ASCT2 (C467) (Figure 2, left inset). C467 was proposed by to be a key substrate specificity determinant in the SLC1 family [44, 50]. Further, virtual screening of large compound libraries from the ZINC databaseiii, combined with cellular uptake and electrophysiology assays, identified new ASCT2 ligands, including five substrate-like compounds and two ASCT2 inhibitors (e.g., γ−2-fluorobenzyl proline) [44]. This study highlighted the utility of virtual screening to capture unexplored chemical spaces of transporter ligands, which have been further optimized to obtain higher affinity inhibitors [51].
Figure 2. Substrate and allosteric binding sites of EAAT1 and ASCT2.
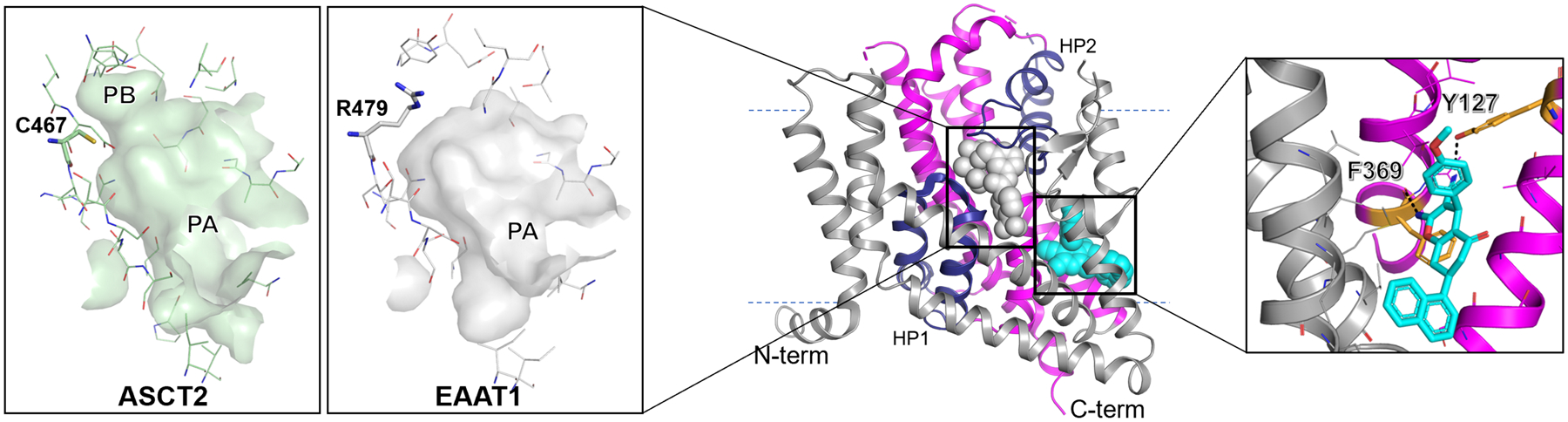
Here, gray represents the scaffold domain and; dark blue hairpins 1 and 2 (HP1 and 2) and pink comprise the transport domain. The substrate and allosteric binding sites of EAAT1 (PDB ID: 5MJU) are shown in gray and cyan spheres respectively. Here, dotted lines show the approximate location of the membrane. Inset left: Surface representations of the substrate binding site of the outward-open conformation for ASCT2 and EAAT1. Pockets A and B (PA and PB) are highlighted and residues impacting substrate specificity and binding site shape are shown as sticks with oxygen, nitrogen, and sulfur atoms shown in red, blue, and yellow. Inset right: The allosteric inhibitor UCPH101 bound to EAAT1. Key residues making polar contacts with UCPH101 are highlighted as orange sticks. Images were generated with PyMOL (https://pymol.org/2/).
Subsequent ASCT2 models based on the human EAAT1 structure [27] (sequence identity of 46%) provided a refined conformation of TM8 and hairpin 2 (HP2), and overall a more accurate model, as measured by its ability to enrich known ligands compared to decoy molecules [52]. The EAAT1-based ASCT2 models were sufficiently accurate to guide the development of sulfonamide and sulfonic acid ester linkers that specifically target PA, providing a more quantitative structure activity relationship (SAR) [53, 54]. For example, the experimentally determined Ki correlated with estimated free energy of binding using ligand docking and molecular mechanics generalized Born and surface area (MM-GBSA) calculations [53].
The new cryo-EM structures of ASCT2 in inward-facing occluded (3.85 Å) and open (3.6 Å) conformations, provided additional mechanistic details of the structure-function of this protein [16, 54]. For example, the inward-facing structure confirmed the role of HP2 in gating, as well as the presence of druggable pockets seen in the EAAT1-based models [16, 52]. Interestingly, the EAAT1-based outward homology models of ASCT2 showed better ligand enrichment than that obtained with the ASCT2 inward occluded cryo-EM structure [53]. This suggests that a reliable homology model in a pharmacologically relevant conformation can be as powerful a tool for rational design as a near-atomic resolution structure.
Elucidating the dynamics of SLC1 transporters
Visualization of transporter dynamics can be useful to describe the different structural states of the transport cycle, predict the effect of mutations on transport, as well as identify druggable pockets on the protein and estimate interactions between SLC and ligands, including potential drugs. A common approach to model conformational changes in SLC transporters is through MD simulations [7]. Progress in efficient simulation approaches, increased computational power and improved force fields, coupled with the growing number of SLC transporter structures have enabled researchers to address key questions in transporter biology. For example, the full transport cycle of a prokaryotic homolog of the human sugar transporter (SWEET, SLC50A1), was accurately captured with all-atom MD simulations [55]. Furthermore, MD simulations of GltPh have contributed to our knowledge of the sodium binding sites of the SLC1 transporters [56–59]. Additional studies have revealed mechanisms of uncoupled anion conductance [60], gating by HP2 [61], and ion-substrate coupling [62], as well as mechanisms of substrate binding [63] and translocation [64, 65] that occur in the SLC1 family.
While technological advancements have improved the ability to simulate longer timescales and the confidence of MD simulations, there are significant limitations that remain. Specifically, some transporters such as LeuT, have a timescale of transport ranging from milliseconds to seconds, making all-atom MD simulations extremely computationally demanding and often unfeasible [66]. Although methods such as steered MD [67] and umbrella sampling [68] can simulate at longer timescales, these approaches are often limited by the availability of suitable resolution protein structures and forcefields [32]. It should also be noted that large conformational changes are difficult to approximate with MD simulations, at a resolution sufficient for drug design.
Allosteric control of SLC1 transporters
Allosteric modulation is an attractive way to target transporters for inhibiting or enhancing their function [69]. Allosteric inhibitors can bind protein-specific subpockets and may be particularly useful to selectively target homologs or other proteins that share a similar substrate binding site.
Due in part to the highly conserved substrate binding site among SLC1 members, most currently available inhibitors are nonselective, hindering their clinical relevance. The SLC1 members, however, are amenable to allosteric modulation [70]. For example, an allosteric activator that increases the clearance of glutamate from synapses can be a potential drug for EAAT2, a target for AD therapy [71]. Indeed, the EAAT2 specific activator parawixin1, was recently found to increase EAAT2 activity by binding an allosteric site predicted computationally [72]. Interestingly, parawaxin1, sourced from spider venom, was not designed to specifically activate EAAT2. Notably, computational methods such as molecular docking and free energy perturbations are limited in their ability to distinguish between inhibitors, substrates, and activators [73].
Conversely, the EAAT1-specific inhibitor (UCPH101), was crystallized in an allosteric site located in the interface between the mobile and static domains of this protein, highlighting residues such as F369 and Y127 which provide critical interactions with the transporter (Figure 2, right inset). The UCPH101 binding site is thought to be distinct from the EAAT2 parawixin1 binding site [72]. The structural and biochemical data indicated that the compound can access the allosteric site from the intracellular side and its binding does not preclude substrate binding. Since the EAAT1, structure was solved as a trimer, the compound UCPH101 also added further support of independent transport by each subunit [27]. Notably, the structure with bound ligand also provided a framework for developing allosteric inhibitors specific to individual family members.
Design of ligands for ADME transporters
Membrane transporters, including several SLC families, can play an important role in drug absorption, distribution, metabolism and excretion (ADME) (i.e., ‘ADME transporters’) [74]. Examples include, members of the SLC22 family of organic cation, anion, and zwitterion transporters, which are expressed in the kidney, liver, and blood-brain-barrier, to regulate drug pharmacokinetics (PK) [75]. The SLC22 family members transport a broad range of drugs and prodrugs, such as metformin [76] for the treatment of type II diabetes, and gemcitabine, a first-line chemotherapy for NSCLC [77]. Additionally, drug-drug interactions are often mediated by ADME transporters similarly to those occurring in metabolic enzymes, and genetic variation in ADME transporters can lead to differential drug response among patients (i.e., pharmacogenomics) [74]. Key questions in drug discovery of ADME transporters include: (i) Can we develop predictive models for distinguishing between SLC variants leading to differential drug absorption and dynamics among patients? (ii) Can we identify SLC small molecule ligands, including inhibitors, substrates, or activators that may cause drug-drug interactions? and (iii) Can we rationally design efficient transporter substrates with optimal PK properties or bioavailability, including drugs that can be handled by transporter variants? Multiple recent studies have combined a range of ligand-based and structure-based computational and experimental methods to discover ligands for various ADME SLC targets, such as the organic cation transporter (OCT1, SLC22A1) [78] and the human intestinal transporter, the organic anion transporting polypeptides (OATP2B1, SLC21A9) [79], with varying levels of success, as well as explain the effects of mutations on drug uptake and dynamics (reviewed in [7, 12]).
An important group of ADME transporters include members of the SLC15 transporters that mediate the uptake of a broad range of peptides and peptide-like drugs. Particularly, the proton-dependent di/tri-peptide transporter 1 (PepT1, SLC15A1) is primarily responsible for intestinal absorption of luminal di- and tri-peptides from dietary protein digestion and peptide-like drugs, such as the β-lactam antibiotics cefadroxil and antiviral prodrug valacyclovir [80].The three-dimensional structures of mammalian SLC15 members are unknown. Our current understanding of PepT1 structure and drug-transporter interactions has primarily relied on structures of various prokaryotic homologs [81]. Recently determined crystal structures of PepTSh from the bacterium Staphylococcus hominis, bound to two drugs (i.e., valacyclovir and 5-aminolevulinic acid) propose a pharmacophore model for PepT1-drug recognition [82]. For example, the amino acid scaffold and ester linker of valacyclovir makes critical contacts with the binding site of PepTSh, providing guidance for future optimization for drugs with better absorption [82] (Figure 3).
Fig. 3. Structure of an ADME transporter homolog, PepTSh.
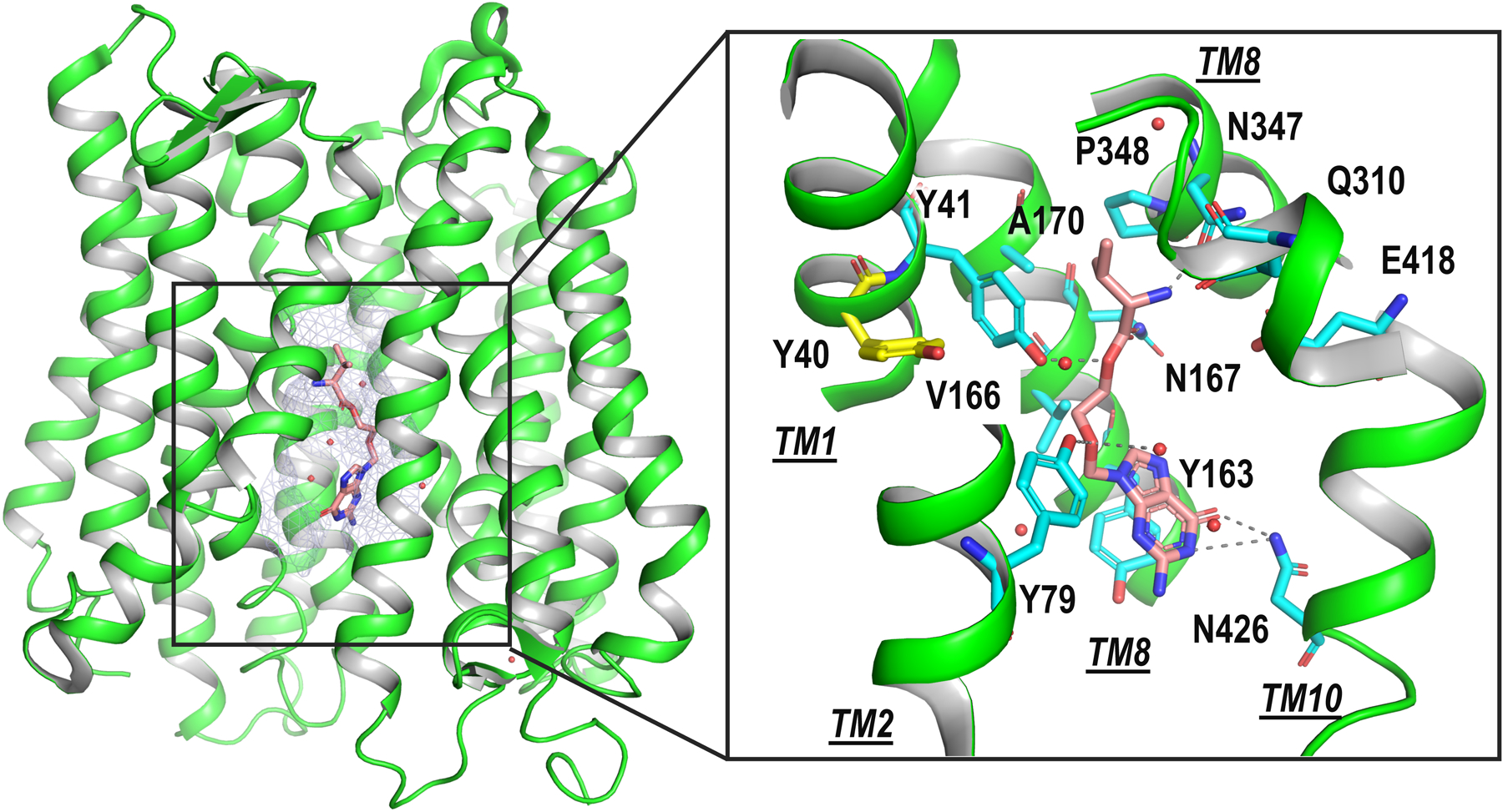
The crystal structure of PepTSh (green cartoon) in complex with the antiviral drug valacyclovir (peach sticks) is shown. The substrate binding site is shown in light blue mesh. Inset shows the magnified view of the interaction of valacycolovir with the binding site of PepSh. The sidechain atoms of key residues in PepTSh are illustrated with cyan sticks, with Y40, in yellow. Y40 is equivalent to F28 in the human PepT1. Rare mutation in this position in African Americans (F28Y) reduces substrate uptake by PepT1. Hydrogen bonds between binding site residues and valacycolvir are displayed as dashed gray lines. Images were generated with PyMOL (https://pymol.org/2/).
Samsudin et al. have recently used state-of-the-art computational techniques to describe the substrate specificity of PepT1 and its bacterial homologs [83]. Estimates of free energy of binding suggested that the properties of the N-terminus of the transported peptide ligands were more critical for transport than those of the C-terminus [83]. In parallel, Colas et al. have identified new substrates and inhibitors of PepT1 using a combined approach of homology modeling, virtual screening, and experimental testing with cellular uptake and electrophysiology assays, providing novel scaffolds for future tool compound development [84]. While both of these computer-guided studies attempted to estimate the free energy of binding of small molecules to PepT1, it is important to note that: (i) even if successful, computational prediction of affinity can usually determine only whether a small molecule likely to bind or not; and (ii) a compound’s affinity or potency (e.g., as measured with IC50) does not necessarily correlate with its ability as a substrate to get transported across the membrane [85].
Finally, PepT1 variants have been associated with reduced transport activity in specific populations [86]. For example, F28Y variation in PepT1 (Figure 3, inset) is a rare mutation in African Americans which reduces substrate uptake. Structures of PepT1 homologs and the homology model of PepT1 indicate that the mutation occurs near the substrate binding site (Figure 3, inset) which may have an effect on helix packing and binding site conformation, thereby hindering transport.
Concluding remarks and future outlook
The SLC transporters provide a wealth of underexplored therapeutic targets. Here, we first outlined the structure, function, and pharmacology of the human SLCs (Table 1, Figure 1). We then presented examples of how rational design was used to develop small molecule ligands targeting the substrate and allosteric sites of SLC1 transporters (Figure 2) and summarized key limitations of current computational approaches. Finally, we show how structures of SLC15 homologs can help explain intestinal drug absorption by the human PepT1 and the deleterious functional effect of a mutation in patients (Figure 3).
Many challenges remain in understanding SLC biology, as well as in pharmacological targeting of these proteins (see Outstanding Questions). Chemical tools for human SLCs are needed to characterize the structure and function of these biomedically important transporters, and to deorphanize SLCs with unknown function. Currently, rational design of SLC modulators is hampered by the limited understanding of transporter structure and dynamics. While CADD has proven useful for the development of tool compounds for SLCs, limitations remain. For example, at present, computational methods provide useful information about binding but not kinetics (e.g., residence time on target) [87]. Thus, they cannot distinguish between inhibitors that may bind tightly to the target, substrates that bind and unbind the target triggering transport, and activators that make the transporter more efficient [13].
In the near future, methods can be improved by using ML algorithms (reviewed in [88]). For example, recently, deep learning algorithms were used to aid in image processing and secondary structure assignment of cryo-EM maps [89], as well as to explore conformational space with MD simulations [90]. In addition, while ML approaches have been used for many years for drug prediction in QSAR, including for membrane transporters [7], the new deep learning architectures such as autoencoder can also be used to design drug-like chemicals de novo [34]. Notably, these emerging ML methodologies usually require large sets of active and inactive compounds in order to be optimally trained. However, for SLCs, often times this is simply not yet the case [6]. We expect that the advancements discussed in this review and renewed interest in SLCs in both academia and industry will lead to an expansion in tool compounds available for the further characterization of SLCs and the development of eventual SLC drugs.
Acknowledgements
We thank Da-Neng Wang (NYU) for valuable comments on the manuscript. We also thank Peter Man-Un Ung, Claire Colas, and Rayees Rahman (Mt. Sinai), and Christof Grewer (Binghamton University) and Jeff Holst (UNSW Sydney) for useful discussions. This work was supported in part by the National Institutes of Health grant R01 GM108911 to A. S. and grant T32 CA078207 to R.A.G.
GLOSSARY
- Activator
A molecule that enhances or increases the activity of an SLC by binding to the transporter in a substrate or allosteric site.
- Allosteric inhibitor
A molecule that binds at a different site than the substrate binding site and inhibits SLC transport.
- Alternating access
Conformational changes that cause the transporter to alternately expose its substrate-binding site to either side of the membrane.
- Druggable
Disease-related target that is amenable to small molecule binding and modulation.
- Molecular dynamics (MD) simulation
Application of Newton’s equations of motion to predict the positions of atoms in a biomolecular system as a function of time using a force field to specify the parameters in the system.
- Molecular mechanics generalized Born and surface area (MM-GBSA)
Molecular mechanics generalized Born and surface area, a popular method to estimate the free energy of the binding of small molecules to SLC transporters.
- Pharmacophore modeling.
Ligand or structure-based methods that use the spatial arrangement of physicochemical features from the ligand and structure, respectively, to develop quantitative models that are predictive of bioactivity.
- Quantitative structure-activity relationship (QSAR).
Structural descriptors of ligands, such as number of hydrogen bond acceptors and donors, size, charge, are used to develop a statistical model predicting biological activity.
- Substrate
A molecule that gets transported across the membrane by membrane transporter.
Footnotes
Publisher's Disclaimer: Disclaimer
Publisher's Disclaimer: Avner is co-founder of AIchemy, LLC.
References
- 1.Hediger Matthias A., C. B, Burrier Robert E., Bruford Elspeth A. (2013) Bioparadigms.
- 2.Lin L et al. (2015) SLC transporters as therapeutic targets: emerging opportunities. Nat Rev Drug Discov 14 (8), 543–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hahn MK and Blakely RD (2007) The functional impact of SLC6 transporter genetic variation. Annu Rev Pharmacol Toxicol 47, 401–441. [DOI] [PubMed] [Google Scholar]
- 4.Shimizu K et al. (2014) ASC amino-acid transporter 2 (ASCT2) as a novel prognostic marker in non-small cell lung cancer. Br J Cancer 110 (8), 2030–2039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schlessinger A et al. (2013) SLC Classification: An Update. Clin Pharmacol Ther 94 (1), 19–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cesar-Razquin A et al. (2015) A Call for Systematic Research on Solute Carriers. Cell 162 (3), 478–487. [DOI] [PubMed] [Google Scholar]
- 7.Schlessinger A et al. (2018) Molecular Modeling of Drug-Transporter Interactions-An International Transporter Consortium Perspective. Clin Pharmacol Ther 104 (5), 818–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chedid V et al. (2018) Elobixibat for the treatment of constipation. Expert Rev Gastroenterol Hepatol 12 (10), 951–960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hu G et al. (2013) New fluorescent substrate enables quantitative and high-throughput examination of vesicular monoamine transporter 2 (VMAT2). ACS Chem Biol 8 (9), 1947–1954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ulanovskaya OA et al. (2011) A pairwise chemical genetic screen identifies new inhibitors of glucose transport. Chem Biol 18 (2), 222–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nomura S et al. (2010) Discovery of canagliflozin, a novel C-glucoside with thiophene ring, as sodium-dependent glucose cotransporter 2 inhibitor for the treatment of type 2 diabetes mellitus. J Med Chem 53 (17), 6355–6360. [DOI] [PubMed] [Google Scholar]
- 12.Matsson P and Artursson P (2013) Computational prospecting for drug-transporter interactions. Clin Pharmacol Ther 94 (1), 30–32. [DOI] [PubMed] [Google Scholar]
- 13.Irwin JJ and Shoichet BK (2016) Docking Screens for Novel Ligands Conferring New Biology. J Med Chem 59 (9), 4103–4120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Colas C et al. (2016) SLC Transporters: Structure, Function, and Drug Discovery. Medchemcomm 7 (6), 1069–1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Huynh KW et al. (2018) CryoEM structure of the human SLC4A4 sodium-coupled acid-base transporter NBCe1. Nat Commun 9 (900), 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Garaeva AA et al. (2018) Cryo-EM structure of the human neutral amino acid transporter ASCT2. Nat Struct Mol Biol 25 (6), 515–521. [DOI] [PubMed] [Google Scholar]
- 17.Yan R et al. (2019) Structure of the human LAT1–4F2hc heteromeric amino acid transporter complex. Nature, 127–130. [DOI] [PubMed] [Google Scholar]
- 18.Ceska T et al. (2019) Cryo-EM in drug discovery. Biochem Soc Trans, 281–293. [DOI] [PubMed] [Google Scholar]
- 19.Schlessinger A et al. (2010) Comparison of human solute carriers. Protein Sci 19 (3), 412–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jardetzky O (1966) Simple allosteric model for membrane pumps. Nature 211 (5052), 969–970. [DOI] [PubMed] [Google Scholar]
- 21.Vidaver GA (1966) Inhibition of parallel flux and augmentation of counter flux shown by transport models not involving a mobile carrier. J Theor Biol 10 (2), 301–306. [DOI] [PubMed] [Google Scholar]
- 22.Law CJ et al. (2008) Ins and outs of major facilitator superfamily antiporters. Annu Rev Microbiol 62, 289–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Patlak CS (1957) Contributions to the theory of active transport: II. The gate type non-carrier mechanism and generalizations concerning tracer flow, efficiency, and measurement of energy expenditure. Bull. Math. Biol (19), 209–235. [Google Scholar]
- 24.West IC (1997) Ligand conduction and the gated-pore mechanism of transmembrane transport. Biochim Biophys Acta 1331 (3), 213–234. [DOI] [PubMed] [Google Scholar]
- 25.Mancusso R et al. (2012) Structure and mechanism of a bacterial sodium-dependent dicarboxylate transporter. Nature 491 (7425), 622–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Reyes N et al. (2009) Transport mechanism of a bacterial homologue of glutamate transporters. Nature 462 (7275), 880–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Canul-Tec JC et al. (2017) Structure and allosteric inhibition of excitatory amino acid transporter 1. Nature 544 (7651), 446–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Marti-Renom MA et al. (2000) Comparative protein structure modeling of genes and genomes. Annual review of biophysics and biomolecular structure 29, 291–325. [DOI] [PubMed] [Google Scholar]
- 29.Webb B et al. (2018) Integrative structure modeling with the Integrative Modeling Platform. Protein Sci 27 (1), 245–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lei HT et al. (2018) Crystal structure of arginine-bound lysosomal transporter SLC38A9 in the cytosol-open state. Nat Struct Mol Biol 25 (6), 522–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wyant GA et al. (2017) mTORC1 Activator SLC38A9 Is Required to Efflux Essential Amino Acids from Lysosomes and Use Protein as a Nutrient. Cell 171 (3), 642–654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vermaas JV et al. (2016) Microscopic Characterization of Membrane Transporter Function by In Silico Modeling and Simulation. Methods Enzymol 578, 373–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mobley DL and Gilson MK (2017) Predicting Binding Free Energies: Frontiers and Benchmarks. Annu Rev Biophys 46, 531–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gómez-Bombarelli R et al. (2018) Automatic Chemical Design Using a Data-Driven Continuous Representation of Molecules. ACS Cent Sci 4 (2), 268–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Drew D and Boudker O (2016) Shared Molecular Mechanisms of Membrane Transporters. Annu Rev Biochem 85, 543–572. [DOI] [PubMed] [Google Scholar]
- 36.Guskov A et al. (2016) Coupled binding mechanism of three sodium ions and aspartate in the glutamate transporter homologue Glt(Tk). Nat Commun 7, 1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hanelt I et al. (2013) Conformational heterogeneity of the aspartate transporter Glt(Ph). Nat Struct Mol Biol 20 (2), 210–214. [DOI] [PubMed] [Google Scholar]
- 38.Akyuz N et al. (2015) Transport domain unlocking sets the uptake rate of an aspartate transporter. Nature 518 (7537), 68–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ruan Y et al. (2017) Direct visualization of glutamate transporter elevator mechanism by high-speed AFM. Proc Natl Acad Sci U S A 114 (7), 1584–1588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lin CL et al. (2012) Glutamate transporter EAAT2: a new target for the treatment of neurodegenerative diseases. Future Med Chem 4 (13), 1689–1700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.van Geldermalsen M et al. (2016) ASCT2/SLC1A5 controls glutamine uptake and tumour growth in triple-negative basal-like breast cancer. Oncogene 35 (24), 3201–3208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Liu Y et al. (2018) The role of ASCT2 in cancer: A review. Eur J Pharmacol 837, 81–87. [DOI] [PubMed] [Google Scholar]
- 43.Albers T et al. (2012) Defining substrate and blocker activity of alanine-serine-cysteine transporter 2 (ASCT2) Ligands with Novel Serine Analogs. Mol Pharmacol 81 (3), 356–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Colas C et al. (2015) Ligand Discovery for the Alanine-Serine-Cysteine Transporter (ASCT2, SLC1A5) from Homology Modeling and Virtual Screening. PLoS Comput Biol 11 (10), 1–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Oppedisano F et al. (2010) Inactivation by Hg2+ and methylmercury of the glutamine/amino acid transporter (ASCT2) reconstituted in liposomes: Prediction of the involvement of a CXXC motif by homology modelling. Biochem Pharmacol 80 (8), 1266–1273. [DOI] [PubMed] [Google Scholar]
- 46.Pingitore P et al. (2013) Large scale production of the active human ASCT2 (SLC1A5) transporter in Pichia pastoris--functional and kinetic asymmetry revealed in proteoliposomes. Biochim Biophys Acta 1828 (9), 2238–2246. [DOI] [PubMed] [Google Scholar]
- 47.Schulte ML et al. (2015) 2-Substituted Ngamma-glutamylanilides as novel probes of ASCT2 with improved potency. Bioorg Med Chem Lett 25 (1), 113–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Oppedisano F et al. (2012) Inactivation of the glutamine/amino acid transporter ASCT2 by 1,2,3-dithiazoles: proteoliposomes as a tool to gain insights in the molecular mechanism of action and of antitumor activity. Toxicol Appl Pharmacol 265 (1), 93–102. [DOI] [PubMed] [Google Scholar]
- 49.Schulte ML et al. (2016) 2-Amino-4-bis(aryloxybenzyl)aminobutanoic acids: A novel scaffold for inhibition of ASCT2-mediated glutamine transport. Bioorg Med Chem Lett 26 (3), 1044–1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Scopelliti AJ et al. (2013) Molecular determinants for functional differences between alanine-serine-cysteine transporter 1 and other glutamate transporter family members. J Biol Chem 288 (12), 8250–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Singh K et al. (2017) Structure activity relationships of benzylproline-derived inhibitors of the glutamine transporter ASCT2. Bioorg Med Chem Lett 27 (3), 398–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Garibsingh RA et al. (2018) Homology Modeling Informs Ligand Discovery for the Glutamine Transporter ASCT2. Front Chem 6, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ndaru E et al. (2019) Novel alanine serine cysteine transporter 2 (ASCT2) inhibitors based on sulfonamide and sulfonic acid ester scaffolds. J Gen Physiol 151, 357–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Garaeva AA et al. (2019) A one-gate elevator mechanism for the human neutral amino acid transporter ASCT2. Nat Commun 10 (1), 3427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Latorraca NR et al. (2017) Mechanism of Substrate Translocation in an Alternating Access Transporter. Cell 169 (1), 96–107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Venkatesan S et al. (2015) Refinement of the Central Steps of Substrate Transport by the Aspartate Transporter GltPh: Elucidating the Role of the Na2 Sodium Binding Site. PLoS Comput Biol 11 (10), e1004551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Larsson HP et al. (2010) Evidence for a third sodium-binding site in glutamate transporters suggests an ion/substrate coupling model. Proc Natl Acad Sci U S A 107 (31), 13912–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bastug T et al. (2012) Position of the third Na+ site in the aspartate transporter GltPh and the human glutamate transporter, EAAT1. PLoS One 7 (3), e33058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Huang Z and Tajkhorshid E (2010) Identification of the third Na+ site and the sequence of extracellular binding events in the glutamate transporter. Biophys J 99 (5), 1416–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Machtens JP et al. (2015) Mechanisms of anion conduction by coupled glutamate transporters. Cell 160 (3), 542–53. [DOI] [PubMed] [Google Scholar]
- 61.Zomot E and Bahar I (2013) Intracellular gating in an inward-facing state of aspartate transporter Glt(Ph) is regulated by the movements of the helical hairpin HP2. J Biol Chem 288 (12), 8231–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Huang Z and Tajkhorshid E (2008) Dynamics of the extracellular gate and ion-substrate coupling in the glutamate transporter. Biophys J 95 (5), 2292–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Shrivastava IH et al. (2008) Time-resolved mechanism of extracellular gate opening and substrate binding in a glutamate transporter. J Biol Chem 283 (42), 28680–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.DeChancie J et al. (2011) The mechanism of substrate release by the aspartate transporter GltPh: insights from simulations. Mol Biosyst 7 (3), 832–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Stolzenberg S et al. (2012) Structural intermediates in a model of the substrate translocation path of the bacterial glutamate transporter homologue GltPh. J Phys Chem B 116 (18), 5372–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Cheng MH and Bahar I (2014) Complete mapping of substrate translocation highlights the role of LeuT N-terminal segment in regulating transport cycle. PLoS Comput Biol 10 (10), e1003879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Do PC et al. (2018) Steered Molecular Dynamics Simulation in Rational Drug Design. J Chem Inf Model 58 (8), 1473–1482. [DOI] [PubMed] [Google Scholar]
- 68.Bowman JD et al. (2019) Mechanism of Cardiac Troponin C Calcium Sensitivity Modulation by Small Molecules Illuminated by Umbrella Sampling Simulations. J Chem Inf Model 59 (6), 2964–2972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wodak SJ et al. (2019) Allostery in Its Many Disguises: From Theory to Applications. Structure 27 (4), 566–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Rives ML et al. (2017) Potentiating SLC transporter activity: Emerging drug discovery opportunities. Biochem Pharmacol 135, 1–11. [DOI] [PubMed] [Google Scholar]
- 71.Masliah E et al. (1996) Deficient glutamate transport is associated with neurodegeneration in Alzheimer’s disease. Ann Neurol 40 (5), 759–66. [DOI] [PubMed] [Google Scholar]
- 72.Kortagere S et al. (2018) Identification of Novel Allosteric Modulators of Glutamate Transporter EAAT2. ACS Chem Neurosci 9 (3), 522–534. [DOI] [PubMed] [Google Scholar]
- 73.Chen YC (2015) Beware of docking! Trends Pharmacol Sci 36 (2), 78–95. [DOI] [PubMed] [Google Scholar]
- 74.Giacomini KM et al. (2010) Membrane transporters in drug development. Nature Reviews Drug Discovery 9 (3), 215–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Koepsell H (2013) The SLC22 family with transporters of organic cations, anions and zwitterions. Mol Aspects Med 34 (2–3), 413–435. [DOI] [PubMed] [Google Scholar]
- 76.Pakkir Maideen NM et al. (2017) Drug Interactions of Metformin Involving Drug Transporter Proteins. Adv Pharm Bull 7 (4), 501–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Wang G et al. (2017) Combination of l-Carnitine with Lipophilic Linkage-Donating Gemcitabine Derivatives as Intestinal Novel Organic Cation Transporter 2-Targeting Oral Prodrugs. J Med Chem 60 (6), 2552–2561. [DOI] [PubMed] [Google Scholar]
- 78.Chen EC et al. (2017) Discovery of Competitive and Noncompetitive Ligands of the Organic Cation Transporter 1 (OCT1; SLC22A1). J Med Chem 60 (7), 2685–2696. [DOI] [PubMed] [Google Scholar]
- 79.Khuri N et al. (2017) Computational Discovery and Experimental Validation of Inhibitors of the Human Intestinal Transporter OATP2B1. J Chem Inf Model 57 (6), 1402–1413. [DOI] [PubMed] [Google Scholar]
- 80.Smith DE et al. (2013) Proton-coupled oligopeptide transporter family SLC15: physiological, pharmacological and pathological implications. Mol Aspects Med 34 (2–3), 323–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Newstead S (2017) Recent advances in understanding proton coupled peptide transport via the POT family. Curr Opin Struct Biol 45, 17–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Minhas GS and Newstead S (2019) Structural basis for prodrug recognition by the SLC15 family of proton-coupled peptide transporters. Proc Natl Acad Sci U S A 116 (3), 804–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Samsudin F et al. (2016) Accurate Prediction of Ligand Affinities for a Proton-Dependent Oligopeptide Transporter. Cell Chem Biol 23 (2), 299–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Colas C et al. (2017) Chemical Modulation of the Human Oligopeptide Transporter 1, hPepT1. Mol Pharm 14 (12), 4685–4693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Colas C et al. (2016) Computing Substrate Selectivity in a Peptide Transporter. Cell Chem Biol 23 (2), 211–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Rubio-Aliaga I and Daniel H (2008) Peptide transporters and their roles in physiological processes and drug disposition. Xenobiotica 38 (7–8), 1022–1042. [DOI] [PubMed] [Google Scholar]
- 87.Wang Y et al. (2015) In silico ADME/T modelling for rational drug design. Q Rev Biophys 48 (4), 488–515. [DOI] [PubMed] [Google Scholar]
- 88.Chan HCS et al. (2019) Advancing Drug Discovery via Artificial Intelligence. Trends Pharmacol Sci 40 (8), 592–604. [DOI] [PubMed] [Google Scholar]
- 89.Maddhuri Venkata Subramaniya SR et al. (2019) Protein secondary structure detection in intermediate-resolution cryo-EM maps using deep learning. Nat Methods. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Degiacomi MT (2019) Coupling Molecular Dynamics and Deep Learning to Mine Protein Conformational Space. Structure 27 (6), 1034–1040.e3. [DOI] [PubMed] [Google Scholar]
Resources
- i). https://www.fda.gov/Drugs/DevelopmentApprovalProcess/DrugInnovation.
- ii). https://www.centerwatch.com/drug-information/fda-approved-drugs.
- iii). http://zinc.docking.org/
- iv). https://www.centerwatch.com/drug-information/fda-approved-drugs/drug/100244/steglatro-ertugliflozin.
- v). https://www.centerwatch.com/drug-information/fda-approved-drugs/drug/100198/ingrezza-valbenazine.
- vi). https://www.centerwatch.com/drug-information/fda-approved-drugs/drug/100197/austedo-deutetrabenazine-
- vii). https://www.centerwatch.com/drug-information/fda-approved-drugs/drug/100127/zurampic-lesinurad.