

Summary
Neurogenin3 (NEUROG3) is required for endocrine lineages formation of the pancreas and intestine. Patients with NEUROG3 mutations are born with congenital malabsorptive diarrhea due to complete loss of enteroendocrine cells, whereas endocrine pancreas development varies in an allele-specific manner. These findings suggest a context dependent requirement for NEUROG3 in pancreas versus intestine. We utilized human tissue differentiated from NEUROG3−/− pluripotent stem cells for functional analyses. Most disease-associated alleles had hypomorphic or null phenotype in both tissues, whereas the S171fsX68 mutation had reduced activity in the pancreas but largely null in the intestine. Biochemical studies revealed NEUROG3 variants have distinct molecular defects with altered protein stability, DNA binding and gene transcription. Moreover, NEUROG3 was highly unstable in the intestinal epithelium, explaining the enhanced sensitivity of intestinal defects relative to the pancreas. These studies emphasize that studies of human mutations in the endogenous tissue context may be required to assess structure-function relationships.
Graphical Abstract
Zhang et al. use a human pluripotent stem cell-based system to study the impact of patient-derived mutations in NEUROG3 on the development of pancreatic and intestinal endocrine cells. This system recapitulates patient phenotypes, identifies hypomophic alleles, and uncovers the mutations’ effects on dimerization, DNA binding, transcription, and protein stability.
Introduction
Endocrine cells in the pancreas and throughout the gastrointestinal tract secrete hormones that regulate many bodily functions including satiety, digestion, motility, nutrient absorption, and blood glycemia. The development of both pancreatic and intestinal endocrine cells is dependent on the basic helix-loop-helix (bHLH) transcription factor Neurogenin3 (NEUROG3). In mice, loss of Neurog3 results in the absence of pancreatic and intestinal endocrine cells, causing diabetes and a failure in nutrient absorption (Gradwohl et al., 2000; Jenny et al., 2002). Patients with homozygous or compound heterozygous NEUROG3 mutations are similarly born without any intestinal enteroendocrine cells (EECs), resulting in malabsorptive diarrhea (Hancili et al., 2017; Pauerstein et al., 2015; Pinney et al., 2011; Germán-Díaz et al., 2017; Rubio-Cabezas et al., 2011, 2014; Sayar et al., 2013; Ünlüsoy Aksu et al., 2016; Wang et al., 2006). Interestingly, however, some patients maintain sufficient glycemic control into adulthood indicating a functional endocrine pancreas, while others are diabetic at birth (neonatal diabetes) (Table S1).
The cause of variability in human phenotypes is not known; however, it is possible that each mutation has unique effects on the structure and function of NEUROG3. Alternatively, humans may not have an absolute dependence on NEUROG3 for pancreatic endocrine cell development (Rubio-Cabezas et al., 2014). This latter possibility was tested using CRISPR-mediated gene disruption of NEUROG3 in human pluripotent stem cells, which upon differentiation failed to generate any human pancreatic endocrine cell in vitro or following engraftment and growth of pancreatic progenitors in mice (McGrath et al., 2015). These data suggest that null alleles of NEUROG3 should result in a complete loss of both pancreatic and intestinal endocrine cells. Since some NEUROG3 mutations are likely to be hypomorphic, this can explain why some patients retain some pancreatic endocrine function. However, it remains unclear why hypomorphic NEUROG3 mutations result in a complete loss of intestinal EECs.
Neurog3 promotes endocrine cell specification through regulating target genes encoding transcription factors such as NeuroD1 (Huang et al., 2000), Nkx2–2 (Prado et al., 2004), Pax4 (Smith et al., 2003; Sosa-Pineda et al., 1997), Arx (Collombat et al., 2003), Rfx6 (Soyer et al., 2010), Nkx6–1 (Henseleit et al., 2005; Sander et al., 2000), among others. As with other bHLH family members Neurog3 can bind a core E-box motif, CANNTG as a heterodimer with E-proteins such as E47 (Jones, 2004; Longo et al., 2008), while some bHLH proteins have been proposed to act as homodimer independent of an E-protein partner (Lee et al., 2005). Once bound to DNA, Neurog3 acts as a transcriptional activator through recruitment of coactivators, such as p300/CBP and PCAF, to activate downstream targets (Breslin et al., 2007).
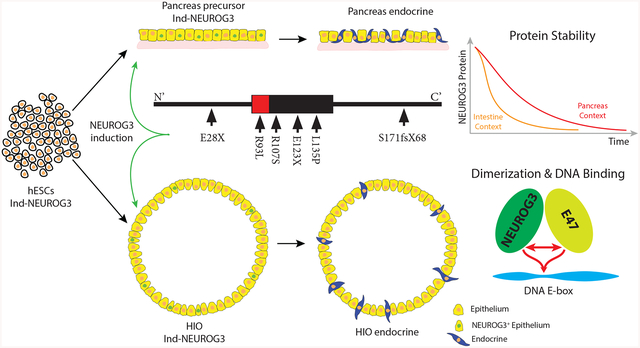
While none of the patient-derived NEUROG3 mutations have been studied in the context of human endocrine cell development, several have been studied in cancer cell lines and by over/misexpression in model organisms (Pauerstein et al., 2015; Pinney et al., 2011; Rubio-Cabezas et al., 2011; Wang et al., 2006). Given the context-dependent roles of NEUROG3, and possible species differences, we investigated the impact of NEUROG3 mutations during development of human pancreatic precursors (McGrath et al., 2015) and intestinal EECs (Spence et al., 2011) derived from human pluripotent stem cells (PSCs). NEUROG3−/− PSCs failed to form any pancreatic or intestinal endocrine cells, while endocrine specification was fully rescued by expression of physiologic levels of wild-type NEUROG3. To investigate the mechanism of patient mutations in NEUROG3 on the differentiation of pancreatic and intestinal endocrine cells, we expressed physiologic levels of NEUROG3 mutant proteins R93L, R107S, E123X, L135P, S171fsX68 and E28X in NEUROG3−/− hESCs and tested for their ability to rescue pancreatic and intestinal endocrine cell formation. R93L, R107S and S171fsX68 recapitulate the patient phenotype with development of some pancreatic endocrine cells, but not intestinal EECs. In contrast, E123X, L135P and E28X were devoid of functional activity in either context regardless of expression levels, consistent with the reported phenotypes in these patients. Biochemical analysis of each mutant protein revealed three types of molecular defects: reduced (R107S and E123X) and increased (S171fsX68) protein stability; diminished (R93L, R107S, S171fsX68) or abolished (E123X and L135P) DNA binding activity in vitro and by ChIP; and diminished (R107S) or abolished (E123X and L135P) E47 heterodimer formation. Moreover we identified that the half-life of NEUROG3 in intestinal EECs is half that of pancreatic cells, which could explain why mutations that reduce NEUROG3 activity all result in loss of EECs and an intestinal pathology.
Results
Generation of culture system to study the effects of NEUROG3 patient mutations on human pancreatic and intestinal endocrine cell development.
To map the effects of NEUROG3 mutations on human pancreatic and intestinal endocrine cells, we established a tetracycline NEUROG3-inducible system in NEUROG3-deficient (NEUROG3−/−) hESCs (Figures S1A–S1C) (McCracken et al., 2011; McGrath et al., 2015; Spence et al., 2011). We chose an inducible tetracycline strategy so that we could express tagged wild-type and mutant forms of NEUROG3 to resemble physiological level. Moreover, we controlled the onset of NEUROG3 expression to mirror the start of endogenous expression. We first confirmed that NEUROG3−/− hESCs were unable to give rise to pancreatic endocrine cells, as previously reported (Figures 1A and 1B) (McGrath et al., 2015). Similarly, human intestinal organoids (HIOs) derived from NEUROG3−/− hESCs did not develop EECs as measured by the pan-endocrine markers CHGA and Synaptophysin (Figures 1D and 1E).
Figure 1:

Expression of NEUROG3 rescues both pancreatic and intestinal endocrine cell formation in NEUROG3-null pancreatic precursors and HIOs. (A-C) Immunofluorescence analysis of CHGA, PDX1 and NKX2–2 of pancreatic endocrine derived from NEUROG3+/+ hESCs (A,A’), NEUROG3-null hESCs with a tetracycline inducible NEUROG3WT construct without doxycycline (0ng/ml) (B,B’) or with doxycycline (100ng/ml 8-hour) (C,C’).
(D-F) Immunofluorescence analysis of CHGA, SYP and ECAD in NEUROG3+/+ (D, D’), NEUROG3WT (0ng/ml) (E,E’), and NEUROG3WT (100ng/ml 8-hour) (F,F’) 35-day HIOs.
(G) Comparison of CHGA+ endocrine cell percentage in NEUROG3+/+ and NEUROG3WT (0 and 100ng/ml 8-hour) pancreatic precursors and HIOs.
(H) Immunofluorescence analysis of INS, GCG, and SST expression in NEUROG3+/+ or NERUOG3WT pancreatic precursors.
(I-J) Quantification of endogenous NEUROG3 expression per cell in pancreatic precursors derived from NEUROG3+/+ hESCs or tet-induced NEUROG3WT expression in NEUROG3-null hESCs. Quantification was based on immunofluorescence (I) and flow-cytometry (J).
(K) Quantification of induced NEUROG3WT protein levels in the epithelium of HIOs as compared to endogenous levels in HIOs treated with the MEK inhibitor PD0325901. For all experiments, the data is representative of a minimum of 3 separate experiments. A minimum of 18 organoids were assessed. Scale bar = 100 μm and 25 μm. Data are represented as mean ± SD. See also Figures S1 and S2.
We next determined if expression of wild-type NEUROG3 to physiological level was sufficient to rescue endocrine cell development in NEUROG3−/− pancreatic cultures. Endogenous NEUROG3 expression is first detected approximately 8–9 days after the start of differentiation (Figure S1D). We therefore induced NEUROG3 expression by adding doxycycline at day 7, 8, or 9 of differentiation, which promoted the formation of pancreatic endocrine cells (Figures S1E). Remarkably, induction of NEUROG3 expression at day 5 or 6 did not result in endocrine cell induction, demonstrating that pancreas precursors are not endocrine competent at this stage of development. We further determined that while an 8-hour pulse with 100ng/ml dox induced NEUROG3 protein in more cells than found in wild-type (NEUROG3+/+) cultures, their per cell levels were comparable as measured by quantitative immunofluorescence (Figures 1I, S1D, S1F and S1H) and flow cytometry (Figure 1J). Moreover, we found that the number of dox-induced pancreatic endocrine cells were comparable to those observed in NEUROG3+/+ hESC-derived pancreatic cultures (Figures 1C, 1G and 1H).
Intestinal enteroendocrine cells are initially observed in the embryonic intestine at E15.5 in mice (Jenny et al., 2002). In PSC-derived human intestinal organoids (HIOs), EECs were consistently observed by day 35 of differentiation. We therefore chose Day 28 as the time point to induce NEUROG3 expression (Figure S1B). Following an 8-hour pulse with 100ng/ml doxycycline, we observed expression of NEUROG3 in the epithelium (Figure S1G and S1J), albeit at low levels relative to the pancreatic cultures. However, this level of NEUROG3 expression was sufficient to induce EECs in numbers comparable to NEUROG3+/+ derived HIOs (Figures 1F and 1G). Due to the rarity of EECs in the intestine, detection of endogenous NEUROG3 in HIOs was not possible (S1J). However, we did detect a few cells expressing endogenous NEUROG3 in transplanted HIOs and in HIO cultures with enhanced EEC differentiation caused by adding mitogen-associated protein kinase (MAPK) pathway inhibitor PD0325901 (PD03) (Basak et al., 2017) (Figure S1G). A quantitative comparison of NEUROG3 protein levels from these endogenous cultures and our dox-induced system again revealed similar NEUROG3 protein levels (Figure 1K).
Patient NEUROG3 mutations recapitulate pancreatic and intestinal phenotypes
Since expression of physiologic NEUROG3 levels rescued development of normal numbers of pancreatic and intestinal endocrine cells in a NEUROG3-null background, we used this system to define how six disease associated mutations affect NEUROG3 activity in their native context. R93L is located in the basic region predicted for DNA interaction, while R107S, E123X and L135P are located in the helix-loop-helix domain, which in NEUROD is essential for E47 heterodimerization (Longo et al., 2008) (Figure 2A). Two additional mutations, E28X and S171fsX68, map outside of the bHLH domain. We determined how each NEUROG3 mutation functionally differed by examining their ability to rescue development of pancreatic endocrine cells in a NEUROG3−/− background. NEUROG3R93L, NEUROG3R107S, and NEUROG3S171fsX68 were competent to induce endocrine cell specification, albeit at significantly reduced numbers relative to NEUROG3WT, suggesting these mutations are hypomorphic (Figures 2B–2D, 2G, 2P and 2R). Consistent with this idea, we found that while the direct target genes NEUROD1, NKX2–2, and PAX4 were robustly activated by NEUROG3WT, each were weakly activated by the NEUROG3R93L, NEUROG3R107S and NEUROG3S171fsX68 proteins (Figure S3C). This phenotype is similar to that observed in patients harboring these mutations, where they are born with some endocrine pancreas function that diminishes over time. In contrast, NEUROG3E123X, NEUROG3L135P and NEUROG3E28X were unable to specify any endocrine cells in pancreatic precursors or induce downstream target genes, suggesting complete loss of function for these mutations (Figures 2E, 2F, 2H, 2P and 2R). These findings are mostly consistent with the reported data of patients harboring these mutations in both alleles, who are born with neonatal diabetes.
Figure 2:

Recapitulating NEUROG3 mutation phenotypes in hESC-derived pancreatic precursors and HIOs. (A) Schematic of NEUROG3, the NEUROG3-E47-DNA complex and the conserved basic helix-loop-helix region of NEUROG across the species. Mutations are indicated by the arrows.
(B-H) Immunofluorescence analysis of CHGA, PDX1 and NKX2–2 of pancreatic endocrine with 100ng 8-hour induction of NEUROG3 variants, 3 days post induction.
(I-O) Immunofluorescence analysis of CHGA, SYP and ECAD of 35-day-old HIOs with 100ng/ml 8-hour induction of NEUROG3 variants, 7 days post induction.
(P-Q) Quantification of CHGA+ endocrine cell percentage of pancreatic endocrine (P) and HIOs (Q) with 100ng/ml 8-hour induction of NEUROG3 variants.
(R-S) qPCR analysis of CHGA transcripts in pancreatic endocrine (R) and HIOs (S) with 100ng/ml 8-hour induction of NEUROG3 variants.
For all experiments, the data is representative of a minimum of 3 separate experiments. A minimum of 18 organoids were assessed. Scale bar = 100 μm. Data are represented as mean ± SD. *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001, and ****p ≤ 0.0001 for one-way ANOVA with Tukey HSD, Tuckey-Kramer (unequal sample size) or Dunnett T3 (unequal variance) post hoc test.
None of the mutations in NEUROG3 affected mRNA levels (Figure 3O), protein nuclear localization (Figure S3F) or antibody epitopes since the NEUROG3 antibody and an N’ terminal HA tag equally stained each variant (Figures S2A and S2B). Expression levels were also not likely affected by number of integrated viral copies (Figures S2C and S2D) or silencing since we determined that most cells carry one integrated virus (Charrier et al., 2011; Figliozzi et al., 2016) and there is no silencing as long as we maintain selection (Figure S2E). While doxycycline was shown to alter cell metabolism and proliferation in human cell lines and stem cells, the dose and incubation time used in our culture systems were much lower and shorter than what is reported to alter cell metabolism (Ahler et al., 2013; Chang et al., 2014), and we did not observe any effect of doxycycline on cell proliferation in either pancreas or HIOs (Figures S2F and S2G).
Figure 3:

Identification of hypomorphic and null NEUROG3 mutations. (A-F) Immunofluorescence analysis of CHGA, PDX1 and NKX2–2 of pancreatic endocrine with 100ng/ml 24-hour induction of NEUROG3 variants.
(G-H) Immunofluorescence analysis of CHGA, SYP and ECAD of 35-day-old HIOs with 100ng/ml 24-hour induction of NEUROG3 variants.
(M-N) Quantification of CHGA+ cell percentage of pancreatic endocrine (M) and HIOs (N) with 100ng/ml 8-hour and 24-hour induction of NEUROG3 variants. Duplicated data (100ng/ml 8-hour) in Figures 2P and 2Q for comparison.
(O-P) qPCR analysis of induced NEUROG3 (O) and CHGA (P) transcripts in pancreatic endocrine with 100, 300ng/ml 8-hour and 100ng/ml 24-hour induction of NEUROG3 variants.
(Q-R) qPCR analysis of induced NEUROG3 (Q) and CHGA (R) transcripts in HIOs with 100, 300ng/ml 8-hour and 100ng/ml 24-hour induction of NEUROG3 variants.
For all experiments, the data is representative of a minimum of 3 separate experiments. A minimum of 18 organoids were assessed. Scale bar = 100 μm. Data are represented as mean ± SD. *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001, and ****p ≤ 0.0001 for one-way ANOVA with Tukey HSD, Tuckey-Kramer (unequal sample size) or Dunnett T3 (unequal variance) post hoc test. See also Figures S3 and S4.
We next investigated the ability of NEUROG3 mutant proteins to promote EEC formation. Induced NEUROG3WT, NEUROG3R93L, NEUROG3R107S, NEUROG3E123X, NEUROG3L135P, NEUROG3S171fsX68 and NEUROG3E28X mRNA were expressed at comparable levels (Figures 3Q, S4A and S4B). All NEUROG3 mutant proteins were detectable with the exception of NEUROG3E28X (Figure S4A). However, EECs were detectable only in the epithelium of NEUROG3WT HIOs (Figures 2I). Similarly to what is observed in patients, there was a complete absence of endocrine formation when we expressed physiologic levels of NEUROG3R93L, NEUROG3R107S, NEUROG3E123X, NEUROG3L135P, NEUROG3S171fsX68 and NEUROG3E28X (Figures 2I–2O, 2Q and 2S). We also observed that none of the mutants robustly activated expression of NEUROD1, NKX2–2, and PAX4 compared to NEUROG3WT (Figure S4C). Of note, while the doxycycline-inducible system drives NEUROG3 expression in the mesenchyme, NEUROG3 does not alter mesenchyme identity as measured by RNA-seq (data not shown). Taken together, expressing NEUROG3 mutant proteins at physiological levels in vitro recapitulated the reported patient phenotypes in both pancreas and intestinal cultures.
Identification of NEUROG3 mutations with hypomorphic activity in the pancreas and intestine
Since patients with NEUROG3 mutations NEUROG3R93L, NEUROG3R107S, and NEUROG3S171fsX68 retain functional pancreatic endocrine cells, it suggests that these mutations are hypomorphic in the pancreas. If so, then increasing the levels of the mutant proteins should restore endocrine cell formation in the pancreas. To test this idea, we used this highly tractable system to increase mutant NEUROG3 level by adding more doxycycline (300ng/ml) or by extending the time of 100ng/ml treatment from 8 to 24 hours. In pancreatic progenitor cultures, we found that increasing NEUROG3R93L, NEUROG3R107S, or NEUROG3S171fsX68 mRNA levels or extending the time of dox treatment to 24 hours resulted in increased pancreatic endocrine cells (Figures 3A–3C, 3F, 3M, S3D and S3E) and enhanced induction of target gene expression (Figures 3P and S3C). We observed a more robust endocrine cell rescue by extending the duration rather than increasing the induction levels. In contrast, increasing the expression of NEUROG3E123X or NEUROG3L135P failed to induce either pancreatic endocrine cells or target genes (Figures 3D, 3E, 3M, 3P and S3C). These results are consistent with NEUROG3R93L, NEUROG3R107S, or NEUROG3S171fsX68 behaving as hypomorphic alleles and NEUROG3E123X or NEUROG3L135P behave as null alleles in the pancreas.
In the intestine, all NEUROG3 mutants failed to induce EECs when expressed at physiologic levels. It is possible that all mutations function as null alleles in the intestine, or that the reduced activity of hypomorphic mutant proteins was insufficient to promote EEC development. To discriminate between these two possibilities, we increased the levels of NEUROG3 proteins similarly to the experiment in pancreas cultures and found that NEUROG3R93L and NEUROG3R107S were able to rescue EECs whereas NEUROG3E28X, NEUROG3E123X, and NEUROG3L135P were not (Figures 3G–3I, 3N, 3R and S4C–S4E). Interestingly, while the NEUROG3S171fsX68 mutant was hypomorphic in the pancreas, it did not rescue EECs when over expressed in the intestine (Figures 3L, 3N, 3R and S4C–S4E). These findings suggest that the S171fsX68 mutation disrupts a protein function that is required in the intestine but not the pancreas. Taken together, these data show that NEUROG3R93L and NEUROG3R107S function as hypomorphic alleles, whereas the NEUROG3E28X, NEUROG3E123X, and NEUROG3L135P alleles are functional nulls in both the pancreas and intestine. Moreover, each of the mutations that reduces NEUROG3 activity results in total loss of EECs, whereas pancreatic endocrine cells are present yet reduced, which phenocopies patients harboring these hypomorphic alleles.
Effects of mutations on NEUROG3 stability in pancreatic endocrine cells
Two mutants, NEUROG3R107S and NEUROG3E123X showed reduced protein levels in the pancreas while NEUROG3E28X protein was not detectable (Figures S3A and S3B), suggesting that mutations may affect protein stability. To study how NEUROG3 mutations affect protein stability in pancreatic cells, NEUROG3 proteins were expressed in day 9 pancreatic precursors by treating with doxycycline for 24 hours, followed by doxycycline removal and the addition of the translation inhibitor cycloheximide (CHX) to prevent further protein synthesis. Protein lysates were harvested at various time points following dox removal, NEUROG3 protein levels were quantified by western blot, and protein degradation was blocked by proteasome inhibition (Figures 4A). Using first-order kinetics, we observed that the half-life of NEUROG3WT, NEUROG3R93L, and NEUROG3L135P were approximately 1 hour, whereas that of NEUROG3R107S was reduced by 50%. The NEUROG3E123X mutation results in a truncated, unstable protein with a 75% reduction in half-life (Figure 4B). We were not able to detect NEUROG3E28X protein by western blot (data not shown) or immunostaining (Figures S3A and S4A). Interestingly, the half-life of NEUROG3S171fsX68 was significantly increased to over 1.5 hours (Figures 4B and 4C). Despite the protein stability differences, mRNA levels were comparable across all conditions (Figure 4D). The reduced protein stability of NEUROG3R107S did not fully explain the impaired ability of this protein to generate endocrine cells since increasing NEUROG3R107S levels only partially rescued this phenotype (Figures S5A and S5B). These data suggest that the NEUROG3R107S protein has other functional deficiencies.
Figure 4:

NEUROG3 mutations alter protein stability and protein phosphorylation. (A) Western blot analysis of NEUROG3 and ACTIN in pancreatic precursors with 100ng/ml 24-hour induced NEUROG3 variants. Arrows indicate multiple bands representing different forms of NEUROG3.
(B) Quantification of induced NEUROG3 variants half-life in pancreatic precursors using first order decay function.
(C) Induced NEUROG3 variants decay curve in pancreatic precursors over the 180-minute time course.
(D) qPCR analysis of induced NEUROG3 transcripts in pancreatic precursors with 100ng/ml 24-hour induction of NEUROG3 variants.
(E) Western blot analysis of NEUROG3 and ACTIN in pancreatic precursors with (+) or without (−) phosphatase λ-PP treatment.
(F) Western blot analysis of NEUROG3 and ACTIN in human intestinal enteroids with 100ng/ml 24-hour induction of NEUROG3WT. Arrows indicate multiple bands representing different forms of NEUROG3.
(G) Quantification of induced NEUROG3WT half-life and decay curve in pancreatic precursors and human intestinal enteroids over a 180-minute time course using first order decay function. A comparison of data of NEUROG3WT half-life in pancreatic precursors (from figure 4B) with protein half-life in intestinal enteroids.
For all experiments, the data is representative of a minimum of 3 separate experiments. Data are represented as mean ± SD. *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001, and ****p ≤ 0.0001 for two-tailed student’s t-test post F-test or one-way ANOVA with Tukey HSD or Dunnett T3 (unequal variance) post hoc test. See also Figures S5 and S6. Uncropped images for Western blots have been deposited on Mendeley Dataset https://data.mendeley.com/datasets/dj53dkzwp5/draft?a=3581f584-ea06-4cef-9991-6d6b6e6ab3c4
NEUROG3 protein is unstable in intestinal EECs
While endogenous NEUROG3 is readily detectible in the pancreas, it is difficult to detect in the epithelium of HIOs (Figure S1J). Even tetracycline-induced NEUROG3 does not accumulate to high levels in the epithelium of HIOs (Figure 1K), suggesting that NEUROG3 protein may have a short half-life in the intestine. The tetracycline system also drives inappropriate NEUROG3 expression in the mesenchyme of HIOs (Figure S1J). Since mesenchymal expression of NEUROG3 confounded efforts to study the half-life of NEUROG3 in the epithelium, we developed a doxycycline-inducible system in NEUROG3−/− human intestinal enteroids (HIOE) that lack mesenchyme. We expressed NEUROG3 protein in enteroids and performed a time course of protein degradation as described above (Figures 4F). Using first-order kinetics, we observed that the half-life of NEUROG3WT in intestinal epithelium was 30 minutes, whereas in the pancreas it was 67 minutes (Figures 4G). There was some level of variability regarding the starting levels of NEUROG3 protein from experiment to experiment. However the starting level of NEUROG3 protein did not affect the kinetics of protein degradation, as can be seen by the fact that protein half-life was remarkably consistent from experiment to experiment in both the pancreas (N=6) and intestine (N=5). Interestingly NEUROG3S171fsX68, the one mutant that showed increased stability in the pancreas was also more stable in the intestine (Figures S5C and S5D) suggesting that the C-terminal residues downstream of amino acid 171 mediate protein stability. Nonetheless, NEUROG3S171fsX68 is unable to induce formation of EECs (Figure 3 and Table 1) suggesting that the C-terminus harbors another functional domain that is required in the intestine but not the pancreas. The combined effects of a high turnover rate in the intestine and the reduced activity of NEUROG3 mutants could explain why all hypomorphic mutations cause a strong intestinal phenotype.
Table 1:
Summary of biochemical and functional comparison of NEUROG3 mutations to WT NEUROG3.
| NEUROG3 Mutation |
Protein Stability |
E47 dimerization |
DNA binding | Endocrine rescue | ||
|---|---|---|---|---|---|---|
| High affinity | Low affinity | Pancreas | HIO | |||
| WT | +++ | +++ | +++ | +++ | +++ | +++ |
| E28X | − | − | − | − | − | − |
| R93L | +++ | +++ | ++ | + | ++ | ++ |
| R107S | ++ | + | +++ | + | ++ | ++ |
| L135P | +++ | − | − | − | − | − |
| E123X | + | − | − | − | − | − |
| S171fsX68 | ++++ | + | + | ++ | ++ | − |
Phosphorylation of NEUROG3 has been reported to regulate protein stability and transcriptional activity (Azzarelli et al., 2017; Krentz et al., 2017; Sancho et al., 2014). Consistent with this idea, western blot analysis of NEUROG3WT protein isolated from pancreatic lysates showed several bands on a western blot (Figure 4A), and treatment of extracts with λ-phosphatase resulted in the collapse of the higher molecular weight bands into one lower band. The same analysis was performed on NEUROG3R93L, NEUROG3R107S and NEUROG3L135P where they all behaved like NEUROG3WT, indicating that all were similarly phosphorylated. In contrast, mutations that affect the C-terminus of NEUROG3, NEUROG3E123X and NEUROG3S171fsX68, only had a single band (Figures 4E). These findings are consistent with reports showing that a phosphorylation site, Serine 183 in the C-terminal region of NEUROG3 affects its function (Krentz et al., 2017). However, we found an engineered phospho-null (S/T>A) mutation in S183 of human NEUROG3 (NEUROG3S183A) did not affect phosphorylation, stability, or induction of pancreatic and intestinal endocrine cells relative to NEUROG3WT (Figures S6B–S6G). In another report, two Neurog2 C-terminal phosphorylation sites were shown to regulate motor neuron specification in mice (Ma et al., 2008). Mutating both of these conserved sites in NEUROG3 (NEUROG3S204A207A) had no effect on protein phosphorylation, activity, or half-life (Figures S6B–S6G). Another putative phosphorylation site, T120, which is conserved in the bHLH domains of NEUROG family members (Quan et al., 2016) was shown to regulate NEUROG protein stability in Xenopus laevis eggs (Vosper et al., 2007). However, phosphorylation and protein stability of NEUROG3T120A were both normal, although endocrine formation was reduced likely due to changes in DNA binding (Figures S6B–S6D). Together, our data suggest that human NEUROG3 is phosphorylated, however the previously reported S183 and S204/S207 phosphorylation sites of NEUROG3 do not have a functional regulatory role during human endocrine cell development in the pancreas and intestine (Table S2).
Effects of NEUROG3 mutations on DNA binding affinity and E47 dimerization
bHLH transcription factors can bind to DNA targets as heterodimers with E proteins like E47 or as homodimers. To determine whether NEUROG3 mutations affect NEUROG3-E47-DNA complex formation, we utilized electrophoretic mobility shift assay (EMSA) to test the binding activity of NEUROG3 to a high-affinity E-box sequence from the NKX2–2 promoter. As expected, the E47 protein formed homodimer/DNA complexes on the high-affinity NKX2–2 E-box probe and increasing amounts of NEUROG3WT protein with E47 resulted in the formation of an additional faster-migrating NEUROG3-E47 heterodimer/DNA complex (Figure 5A). To better discriminate between the E47/E47/DNA complex and the NEUROG3/E47/DNA heterodimer, we created a small C-terminal truncation in NEUROG3WT−177 that had very similar DNA binding characteristics as full length NEUROG3WT−214 but migrated faster and was therefore easier to distinguish in EMSAs. In addition, we found that the NEUROG3WT−177 protein forms homodimers on the high affinity NKX2–2 promoter, albeit less readily than as a heterodimer with E47 (Figure 5C).
Figure 5:

Defective E-box DNA binding affinity of NEUROG3 mutations in vitro and on chromatin. (A-B) EMSA analysis (A) and quantification (B) of purified NEUROG3 variants binding activity to high affinity E-box.
(C) EMSA analysis of purified NEUROG3 variants homodimer binding activity to high affinity E-box.
(D-E) ChIP-qPCR analysis of 100ng/ml 8-hour (C) and 100ng/ml 24-hour (D) induced NEUROG3 variants binding activity to the high affinity E-box in the NKX2–2 promoter in pancreatic precursors.
(F-G) EMSA analysis (F) and quantification (G) of purified NEUROG3 variants binding activity to low affinity E-box. Red arrow indicates NEUROG3R107S/E47/DNA complex; red arrow with dot line indicates loss of NEUROG3R93L/E47/DNA complex.
(H) ChIP-qPCR analysis of induced NEUROG3 variants binding activity to low affinity E-box in the NEUROG3 promoter in pancreatic precursors.
For all experiments, the data is representative of a minimum of 2 separate experiments. Data are represented as mean ± SD. *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001, and ****p ≤ 0.0001 for two-tailed student’s t-test or one-way ANOVA with Dunnett or Dunnett T3 (unequal variance) post hoc test. Uncropped images for EMSAs have been deposited on Mendeley Dataset https://data.mendeley.com/datasets/dj53dkzwp5/draft?a=3581f584-ea06-4cef-9991-6d6b6e6ab3c4
We then determined how NEUROG3 mutations affect complex formation on the high affinity E-box site. The ability of NEUROG3R107S to form NEUROG3/E47/DNA complex was normal, whereas NEUROG3R93L and NEUROG3S171fsX68 binding was slightly reduced (Figures 5A, 5B and S7B). Neither NEUROG3E123X nor NEUROG3L135P formed E47/DNA complexes (Figure S7A) consistent with their inability to promote endocrine cell formation. Interestingly, we observed a more dramatic effect on homodimer binding to the high affinity E-box site with mutations in the bHLH domain, as no binding was observed with NEUROG3R93L, NEUROG3R107S, NEUROG3E123X, NEUROG3L135P and NEUROG3T120A (Figures 5B, S6H and S7C), whereas NEUROG3S171fsX68 did form homodimers with reduced efficiency (Figures 5B and S7D). In contrast, NEUROG3S183A and NEUROG3S204207A efficiently formed homodimer as wild-type (Figure S6H).
Since gel shifts may not accurately reflect NEUROG3 binding to the site in a cellular context, we performed ChIP-qPCR targeting the same E-box from the NKX2–2 promoter in pancreatic endocrine precursors. Binding of NEUROG3R107S, NEUROG3R93L and NEUROG3S171fsX68 to the NKX2–2 locus were similarly reduced compared to NEUROG3WT (Figures 5D and 5E). To validate that the binding of NEUROG3 to this Ebox site in vitro correlates with transcriptional activation, we tested the ability of this site to regulate transcription of a luciferase reporter in human pancreatic precursors. Consistent with their inability to bind NKX2–2 E-box, NEUROG3E123X and NEUROG3L135P were unable to activate the luciferase reporter in pancreatic endocrine cells, and the NEUROG3R93L, NEUROG3R107S, and NEUROG3S171fsX68 mutations that showed decreased DNA binding activity in vitro also exhibited compromised transcriptional activation compared to NEUROG3WT (Figure S7E).
Binding site affinity is often a key regulatory mechanism whereby transcription factors target distinct genes under different circumstances. To determine if the mutations differentially alter the ability of NEUROG3 to bind high vs low affinity binding sites, we analyzed NEUROG3 binding to a low-affinity E-box from the NEUROG3 promoter (Smith et al., 2004). NEUROG3WT and NEUROG3S171fsX68 could bind as heterodimers with E47, but unlike on the high affinity site they both failed to form homodimers on the low affinity E-box (Figure S7F). Intriguingly, NEUROG3R93L and NEUROG3R107S showed a more significant reduction in E47 heterodimer formation on this low affinity site relative to the high affinity NKX2–2 probe (Figures 5F, 5G and S7B). The reduced NEUROG3R107S/E47 heterodimer forming efficiency suggested its compromised interaction with E47 (Figure 5F red arrow). Moreover, as expected, the NEUROG3E123X and NEUROG3L135P proteins that failed to bind the high affinity E-box sequence were also unable to bind a low-affinity E-box site (Figure S7A). We further confirmed the effects of different mutations on binding to this low affinity site in chromatin by ChIPqPCR of pancreatic endocrine precursors. NEUROG3R107S and NEUROG3R93L had reduced binding as compared to NEUROG3WT (Figures 5H). NEUROG3S171fsX68 had slightly reduced DNA binding in vitro and complete loss of binding to this site in cellular chromatin unless expressed at supraphysiologic levels (Figures 5E, 5F and S7B).
A decrease in NEUROG3 complex formation on DNA could be due to a decrease in DNA binding or to a decrease in protein-protein dimer formation with the E47 protein. To test for loss of protein-dimerization, we developed a competition assay using an E-box sequence from a Dll1 promoter (Dll1 E-box) that is bound by E47 but not by Neurogenin proteins (Castro et al., 2006). In this assay, we tested the ability of NEUORG3 proteins to bind E47 as measured by loss of E47 homodimer/DNA complexes (Figure 6A).
Figure 6:

Defective NEUROG3-E47 dimerization capability of NEUROG3 mutations. (A) Schematic model of relative NEUROG3-E47 heterodimerization capability test by competition loss of E47 homodimer binding onto Dll-1 E-box.
(B) E47 heterodimerization analysis with purified NEUROG3 variants.
(C) Quantification of E47 heterodimerization with purified NEUROG3 variants by loss of the signal of E47 homodimer binding onto Dll-1 E-box.
(D) Quantification of E47 heterodimerization with purified NEUROG3 variants by loss of the signal of E47 homodimer binding onto Dll-1 E-box.
For all experiments, the data is representative of a minimum of 3 separate experiments. Data are represented as mean ± SD. *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001, and ****p ≤ 0.0001 for two-tailed student’s t-test or one-way ANOVA with Dunnett or Dunnett T3 (unequal variance) post hoc test. See also Figure S7.
NEUROG3WT−214, NEUROG3WT−177 and NEUROG3R93L proteins bound to E47 protein with comparable affinity (Figure 6B). In contrast, NEUROG3E123X or NEUROG3L135P were unable to bind E47 (Figures 6C and S7F). It’s not surprising as the second helix structure is essential for heterodimerization which is either lost in E123X or disrupted by the proline in L135P, known to break helix structure. NEUROG3R107S was partially compromised in its ability to heterodimerize with E47, while NEUROG3S171fsX68 had only a modest reduction in E47 binding (Figures 6B and 6D). These data suggest that the R93L mutation directly impairs the ability of NEUROG3 protein to contact DNA but this protein can readily form heterodimers with the E47 protein off of DNA in a manner similar to the wild-type NEUROG3 protein. In contrast, the R107S mutation has decreased affinity for the E47 protein. These findings are consistent with the fact that R93L locates in the basic region of the bHLH domain that interacts with the DNA backbone whereas R107S lies in the HLH domain predicted to interact with the HLH domain of E47 (Ishii et al., 2012).
Discussion
NEUROG3 is essential for development of endocrine cells in the human pancreas and intestine. All human patients with homozygous or compound heterozygous mutations in NEUROG3 have congenital malabsorptive diarrhea due to loss of EECs in the intestine while the loss of pancreatic endocrine lineages varies depending on the mutation. The key conclusions of this study are that (1) hESC-derived human pancreas and intestinal organoids harboring NEUROG3 mutations largely recapitulated the phenotypes observed in human patients, (2) R93L and R107S are hypomorphic mutations in both pancreas and intestine, E123X and L135P are loss-of-function mutations in both tissues, and S171fsX68 preferentially affects development of intestinal over pancreatic endocrine cells, (3) All mutations affect NEUROG3 activity through different molecular mechanisms including protein stability, NEUROG3-E47 heterodimerization capability, and DNA binding affinity (Table 1), and (4) NEUROG3 is intrinsically unstable in the intestine, which may explain why intestinal endocrine cell development is more sensitive to NEUROG3 mutations. In addition, we obtained different results regarding the functional impact of various NEUROG3 phosphorylation sites compared to previous studies performed in cancer cell lines and animal models. Altogether, these studies suggest that functional studies of human proteins should be done in relevant cell contexts, such as the developing human pancreas or intestine.
There are several advantages in using this inducible system to study the function of NEUROG3. First, our ability to regulate NEUROG3 transcription allowed us to focus on the downstream roles of NEUROG3 without transcriptional feedback loops that would alter NEUROG3 levels (Kim et al., 2012; Lee et al., 2001; Lynn et al., 2007; Wang et al., 2008; Yang et al., 2011). Second, we were able to match the timing and expression levels of endogenous NEUROG3 and to confirm protein levels with epitope tagged NEUROG3. Although the pulse of dox induced NEUROG3 protein in more cells than in wild-type, the per cell levels as well as endocrine formation were comparable. Third, the system allowed us to further increase NEUROG3 expression levels to discriminate which mutations were hypomorphic versus null in each cellular context. Fourth, the ability to shut off expression was essential to measure the protein half-life of NEUROG3 in the pancreas and intestine. Lastly, this system also resulted in discoveries such as the fact that cellular context is essential for endocrine competency. Hence, we found that pancreatic progenitors are only competent to form endocrine cells in response to NEUROG3 after day 7 of differentiation, which suggests that the timing of NEUROG3 transcription is coordinated to coincide with endocrine competency. Endocrine competency was also observed in HIOs, where NEUROG3 was ectopically expressed in both mesenchyme and epithelium, yet only the intestinal epithelium was competent to respond and produce EECs.
We observed that R93L and R107S are hypomorphic mutations, which is consistent with published findings (Pauerstein et al., 2015; Wang et al., 2006). At the molecular level NEUROG3R93L had reduced DNA binding affinity and NEUROG3R107S had reduced stability. R107S also showed reduced NEUROG3-E47 heterodimerization, thus impairing the ability of NEUROG3 to bind DNA. E28X, E123X and L135P mutations appeared to be null as we were unable to identify any cellular or biochemical activity. We also did not observe any changes in nuclear localization of NEUROG3 variants.
By design, this system is not affected by other regulatory inputs that affect endogenous NEUROG3 levels such as transcriptional autoregulation (Ejarque et al., 2013) and alternative splicing (Osipovich et al., 2014). However, the ability to experimentally control levels of NEUORG3 allowed us to study post translational regulation of NEUROG3. In the pancreas, endogenous NEUROG3 protein levels are low and it has a half-life of approximately one hour. In the intestinal epithelium, NEUROG3 is highly unstable, which may explain why detecting endogenous protein is difficult. Experimentally increasing NEUROG3 transcripts had little impact on the levels of NEUROG3 protein, suggesting that endocrine progenitors have mechanisms to limit the absolute levels of protein in a cell. Interestingly, when we override the temporal regulation of NEUROG3 by maintaining expression with a 24-hour dox-induction of NEUROG3 we see a much more robust induction of endocrine cells. This suggests that it is important to maintain a minimal threshold of NEUROG3 for effective target gene activation and endocrine cell generation.
Our studies on the S171fsX68 frameshift mutation in the C-terminus region of NEUROG3 were particularly informative. This allele is hypomorphic in the pancreas but largely null in the intestine, suggesting that the C-terminus of NEUROG3 might regulate transcription in the intestine possibly via a transactivation domain (Krentz et al., 2017; Smith et al., 2003). NEUROG3S171fsX68 is also highly stable and is not phosphorylated relative to NEUROG3WT protein suggesting that the C-terminus of the protein might regulate phosphorylation and degradation. Previous studies have linked proliferation of pancreatic progenitor cells with CDK dependent Neurog3 phosphorylation and degradation (Azzarelli et al., 2017; Krentz et al., 2017). Using adult intestinal organoids it was shown that EEC differentiation can be stimulated by inducing cell cycle arrest (Basak et al., 2017), and we similarly observed increased endogenous NEUROG3 protein levels using the same culture conditions with reduced cell proliferation. The cumulative data suggest that NEUROG3 is phosphorylated in active cycling cells, but upon exit from the cell cycle, cells accumulate NEUROG3 protein, which may drive endocrine cell fate. While also observed NEUROG3 phosphorylation, none of the previously reported phosphorylation sites (S183, S204 or S207) appeared to alter phosphorylation, stability or activity during human endocrine cell development. This discrepancy could be due to species or cell type differences, suggesting that structurefunction analyses are best done in the endogenous cellular context in order to reach biologically accurate conclusions.
Two additional mutations have been reported, P39fsX38 and Q4× that are predicted to be null alleles of NEUORG3 (Hancili et al., 2017; Pauerstein et al., 2015). The patient with the Q4× mutation had no pancreatic endocrine function and was born with neonatal diabetes. However, the patient with the P39fsX38 was not diagnosed with diabetes until 3-years and had low but detectable serum C-peptide (Pauerstein et al., 2015). Another curious mutation is the NEUROG3L135P variant, where two patients were born with neonatal diabetes, yet a third patient was not diagnosed with diabetes until age 13 ((Rubio-Cabezas et al., 2011, 2014)). NEUROG3L135P had no activity by any developmental or biochemical assay, consistent with the neonatal diabetes observed in two patients with the L135P allele. Neurog3 function is required in mice for development of pancreatic endocrine cells as well as mature pancreatic endocrine cells in vivo from transplanted human pancreatic progenitors (McGrath et al., 2015). It is therefore unclear how some pancreatic endocrine cells might form in a subset of patients harboring null alleles of NEUROG3. One possibility is that humans have a population of pancreatic endocrine cells that are NEUROG3 independent during embryonic development — for example, insulin-expressing cells specified during first wave of endocrine cell development. It is also possible that the diverse genetic backgrounds in humans results in some patients having compensatory gene expression of other NEUROG or NEUROD proteins that rescues development of some beta cells. While we do not know the answer to this fascinating conundrum, we do know that new human experimental systems like this one will likely enable the discovery of NEUROG3-dependent and independent pathways controlling human endocrine cell development.
STAR Methods
Contact for Reagent and Resource Sharing
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, James Wells (james.wells@cchmc.org).
Experimental Model and Subject Details
Pluripotent stem cell culture
The parent human embryonic stem cell line WA01 (H1) was obtained from WiCell. The NEUROG3−/− hESC line was created previously using CRISPR/Cas9 to disrupt endogenous expression with a frame-shift INDEL (McGrath et al., 2015). hESCs were maintained in mTeSR (StemCell Technologies) on hESC-qualified Matrigel (BD Biosciences) coated plates. Cells were routinely passaged every four days with Dispase (Invitrogen).
Plasmid construction
The human NEUROG3 cDNA was acquired from the Harvard PlasmID database (Plasmid ID HsCD00345748). The NEUROG3 cDNA was subcloned using the pENTR/D-TOPO kit (Thermofisher Scientific). The HA tag and various NEUROG3 mutations were added by site-directed mutagenesis. The various constructs were Gateway cloned into pINDUCER20 (Meerbrey et al., 2011, Addgene #44012) using LR Clonase. (Primers listed in supplemental materials).
Lentivirus Constructs
Vectors were packaged into high-titer lentivirus by the CCHMC viral production core. The virus was added to the media of newly plated hESCs following normal dispase passaging. The media was replaced with mTeSR containing selective antibiotic (G418, 500μg/ml) after 24 hours. All transduced cell lines were maintained under selection.
Differentiation of pancreatic precursors
Stem cells were dispersed with Accutase (StemCell Technologies), washed, collected, resuspended in mTeSR containing 10 M ROCK inhibitor (Y-27632, Tocris Bioscience), and plated at a concentration of 1×105 cells/cm2 on matrigel-coated, 24-well Nunclon plates (Delta treated). Differentiation was initiated when cells reached ~75% confluency, approximately 48 hours after plating. At the start of differentiation (day 0), cells were switched to RPMI 1640 supplemented with non-essential amino acids, 100ng/ml Activin A (Cell Guidance Systems) and 50ng/ml BMP-4 (R&D Systems). Day 1–2 media included 0.2% tetracycline-free FBS (Hyclone) and did not have BMP4. On day 3 the media was changed to RPMI 1640 containing 2% FBS, 50ng/ml FGF-7 (R&D Systems), and 50ng/ml Noggin (R&D Systems). On days 5 and 7 the media was switched to highglucose (HG) DMEM (Gibco) containing 50ng/ml Noggin, 2μM all-trans retinoic acid (Stemgent), and 1% (0.5×) B27 without vitamin A (Gibco). Finally, day 9–12 media was prepared using HG-DMEM supplemented with 1% B27 and 25ng/ml Noggin.
Differentiation and three-dimensional culture of intestinal organoids
Stem cells were handled as stated above, but only 0.5×105 cells were plated. Differentiation was initiated when cells reached ~40% confluency, approximately 48 hours after plating. Cells were treated with 100ng/ml Activin A for 3 consecutive days in RPMI 1640 (Invitrogen) with increasing concentrations of tetracycline-free FBS (0%, 0.2%, 2% on day 1, 2, 3, respectively). Definitive endoderm was then incubated for four days in DMEM-F12 containing 2% tetracycline-free FBS, 400ng ml−1 FGF4, and 3μM CHIR99021 (Stemgent). Spheroids were then collected and embedded in a 50μl bubble of Matrigel (BD Bioscience). The bubble was allowed to solidify for 30 min at 37C, and then overlaid with gut media (advanced DMEM/F12 (Invitrogen), L-glutamine, 10μM HEPES, 1× N2 supplement (R&D Systems), 1× B27 (Invitrogen), pen/strep, and 100ng ml−1 EGF (R&D Systems)). Gut media was replaced every four days.
Method Details
Immunofluorescent staining
Monolayers were fixed for 20 minutes at room temperature in 4% paraformaldehyde (PFA). Organoids were fixed for 30–120 minutes in 4% PFA at 4°C, cryopreserved overnight in 30% sucrose, frozen in OCT, then cryosectioned in 8–10 m increments. Prior to staining, monolayers and sections were blocked for 1 hour in 5% donkey serum and 0.1% Triton-X in PBS. Primary antibodies were diluted in PBS + 0.1% Tween and incubated with the samples overnight at 4°C. Samples were then washed 3× with PBS and incubated in secondary antibodies with DAPI in blocking buffer for 2 hours at room temperature. Samples were then washed 3× with PBS. Slides were then mounted using Fluormount-G (SouthernBiotech). All antibodies are listed in supplemental materials.
Confocal image acquisition and analysis
Images were captured using a Nikon A1R confocal microscope with PMT based detectors and motorized stage. The microscope has 405nm, 488nm, 561nm, and 640nm lasers with appropriate filters. All image analysis and quantification were performed using Nikon Elements. Figures were assembled using the Adobe Creative Suite.
Flow cytometry and cell analysis
Pancreatic monolayers on the indicated days were digested in to single cells with Accutase (Invitrogen) and transferred into FACS tubes with filter caps (40μm). The live cells were stained with LIVE/DEAD® Fixable Violet Dead Cell Stain Kit (Invitrogen) in staining buffer (2% FBS in BSA with 100μM DNAse and 10μM Y-27632) for 20 minutes on ice and washed 1× with PBS. Samples were fixed in 4% PFA for 30 minutes on ice, washed 3× with PBS, and incubated 1× with 0.1% Triton-X in PBS for 30 minutes at room temperature followed by a final wash with PBS. Samples were then blocked in 10% NDS in PBS for 30 minutes at room temperature. Primary antibodies were diluted in blocking buffer and incubated with the samples overnight at 4°C. Samples were then washed 3× with PBS and incubated in secondary antibodies in blocking buffer for 1 hour at room temperature. Samples were then washed 3× with PBS. Studies were performed using five laser flow cytometry LSR Fortessa II (BD) through sequential gating of LIVE/DEAD® Fixable Violet, ECAD, and NEUROG3. Data analysis and quantification were performed using BD FACSDiva.
Cell percentage transduction and MOI
Virus with each inducible NEUROG3 variant was added to the media of newly plated NEUROG3−/− hESCs in two different wells (1μl for each well) following normal Dispase passaging. The media was replaced with mTeSR with or without selective antibiotic (G418, 500μg/ml) after 24 hours, spare one well of NEUROG3−/− hESCs without viral transduction as negative control for G418 selection. On the day all cells appeared dead in the negative control well (~ 4 days), the cells in each well were collected and live cells were quantified by cell counting using Trypan Blue and a BIO-RAD automated cell counter. Percentage transduction (probability of the cells with at least one viral particle) was determined by selection over no selection ratio for each virus. Provided the fulfillment of premise of Poisson distribution (Bhukhai et al., 2018; Fehse et al., 2004; Figliozzi et al., 2016; Shabram and Aguilar-Cordova, 2000), MOI and the probability for any given number of viral particle in one cell for each virus were calculated through Poisson distribution (Figliozzi et al., 2016).
Human intestinal organoid derived enteroids and viral transduction
NEUROG3−/− HIO was transplanted under the kidney capsule of immunodeficient NSG mice for maturation as previous described (Watson et al., 2014). After approximately 10 weeks of in vivo growth, crypts were isolated from transplanted HIOs and plated in 3D as previous described (Mahe et al., 2015). NEUROG3−/− HIO derived enteroids (HIOE) were maintained in phenol red-free, growth factor-reduced Matrigel (Corning, Corning, NY) with basal growth medium (IntestiCult™ Organoid Growth Medium (Human) Component A and B (StemCell Technologies)). HIOE were routinely passaged every 7 days by mechanical dissociation using 25G needles. HIOE differentiation was performed 5 days following the passage by switching the basal growth medium to differentiation medium (IntestiCult™ Organoid Growth Medium (Human) Component A (StemCell Technologies) and gut media) and then collected for endocrine analysis after 5 days. All media was changed every 2–3 days. For lentiviral transduction, on the day of HIOE passaging, viruses carrying NEUROG3 variants were incubated with dissociated HIOE at 37°C for 30 minutes, resuspended in growth factor-reduced Matrigel (Corning, Corning, NY) and covered with basal growth medium. After 3 days, transduced HIOE were selected using G418 (250μg/ml).
ChIP-qPCR
On the indicated days, pancreatic differentiation with induced NEUROG3 variants were cross-linked in 1% formaldehyde in PBS for 12 minutes at room temperature and was stopped by replacing with 0.125M glycine. Nuclei were pelleted in lysis buffer (10mM Tris-HCl, 10mM NaCl, 0.2% NP-40). For chromatin fragmentation, cells were resuspended in Nuclear Lysis Buffer (20mM Tris-HCl pH=8, 0.1% SDS, 2mM EDTA) and sonicated in Diagenode Sonicator for 9 cycles of 20 seconds on, 60 seconds off at 4°C. The desired amount of fragmented chromatin was supplemented with Nuclear Lysis Dilution Buffer (20mM Tris-HCl pH=8, 0.1% SDS, 2mM EDTA, 150mM NaCl, 1% TritionX-100) and precleared with blocked Protein G magnetic beads (Thermo Fisher Scientific) with rotation at 4°C for 3 hours. 1% precleared samples were saved as input and each 25μg sample was incubated with 20μg Protein G magnetic beads preloaded with 5ug antibody overnight with rotation at 4°C. Beads were then washed sequentially at 4°C using: (1) Washing A (150 mM NaCl, 20 mM Tris-HCl (pH 8.0), 2mM EDTA, 0.1% SDS, 1% Triton-X, 0.1% sodium deoxycholate), (2) Washing B-1 (500 mM NaCl, 20 mM Tris-HCl (pH 8.0), 2mM EDTA, 0.1% SDS, 1% Triton-X, 0.1% sodium deoxycholate), (3) Washing B-1 (1M NaCl, 20 mM Tris-HCl (pH 8.0), 2mM EDTA, 0.1% SDS, 1% Triton-X, 0.1% sodium deoxycholate), (4) Washing B-1 (2M NaCl, 20 mM Tris-HCl (pH 8.0), 2mM EDTA, 0.1% SDS, 1% Triton-X, 0.1% sodium deoxycholate), (5) Washing C (50 mM Tris-HCl (pH 8.0), 2mM EDTA, 500Mm LiCl, 1% BP-40, 0.5% sodium deoxycholate) and final 2× wash with TE (10 mM Tris-HCl (pH 8.0), 10 mM EDTA). Protein-DNA complexes were eluted from the beads in Elution Buffer (50mM Tris-HCl pH=8, 10mM EDTA, 1% SDS) at 65° C for 30min. Cross-links were reversed at 65 °C overnight, and DNA was purified for real-time PCR with QIAquick PCR Purification Kit (Qiagen). Quantitative qPCR was performed using a QuantStudio 6 real-time PCR detection system (Thermo Fisher Scientific). Primers are listed in the supplemental materials.
RNA isolation and quantitative PCR
All RNA was column purified using a NucleoSpin RNA kit (Machery-Nagel) including an on-column DNAse digestion. cDNA was produced with the SuperScript VILO cDNA synthesis kit (Invitrogen) following the manufacturer’s instructions. 5ng of cDNA was amplified per reaction with QuantiTect SYBR Green (Qiagen) then amplified using a QuantStudio 6 real-time PCR detection system (Thermo Fisher Scientific). Primers are listed in the supplemental materials.
Western blotting, protein half-life and quantification
For protein half-life, HIOE and pancreatic precursors (differentiation Day 9) were treated with 100ng/ml Doxycycline for 24 hours to induce NEUROG3 expression. At time t=0, the medium with Doxycycline was withdrawn and 100μM cycloheximide (CHX) was applied to block further protein translation. At each indicated time point, harvested cells were immediately lysed in ice-cold RIPA buffer with protease inhibitors and stored for Western Blot. For phosphatase treatment, samples were first incubated with Lambda Protein Phosphotase for 1 hour at 30°C. All samples were mixed with 2× Laemmli buffer and boiled for 15 minutes at 100°C. Proteins were s eparated on Mini-PROTEAN® TGX™ Precast Gel (Bio-Rad) and transferred onto a PVDF membrane. Membranes were blocked in Blocking Buffer (TBS, Odyssey) for 1 hour followed by incubation with primary antibodies at 4°C overnight (diluted in Blo cking Buffer). Secondary antibodies were then applied at room temperature for 1 hour. Images were acquired and quantified on an Odyssey CLx Imaging System. NEUROG3 protein was quantified with the regions including all bands. Protein degradation half-lives were calculated using first-order rate kinetics in Excel. Specifically, data collected from different time points was plotted with exponential curve fitting (y-intercept=1) and half-life was calculated by ln2/|exponent of curve function|. Antibodies are listed in the supplemental materials.
Protein Purification and EMSA
Coding regions for all protein fragments used for EMSA were cloned in-frame with an N-terminal 6xHis-tag in the pET14b vector and expressed in BL21 cells. The mouse E47 protein containing the bHLH domain (amino acids 430 to 648) was purified under native conditions via Ni-chromatography as described previously (Uhl et al., 2010). A larger fragment of the E47 protein containing amino acids 271 to 648 was also constructed. The following NEUROG3 proteins were made: Wild-type NEUROG3, R107S, R93L, and L135P from amino acids 1–177; Wild-type full-length NEUROG3, E123X, and S171fsx68 contained all amino acids of the indicated variant. All NEUROG3 proteins included an N-terminal HA-tag. NEUROG3 proteins and the larger E47 protein were purified under denaturing conditions via Ni-chromatography as described previously (Witt et al., 2010). All experiments used the short E47 fragment (430 to 648) except the analysis of NEUROG3 phospho mutants (T120A and S204AS207A) which used the longer E47 (271 to 648). The following modification was made to the previous protocol, proteins were refolded while on Ni-beads during 3 washes in the native lysis buffer used for E47 purification. Expression of all proteins was confirmed via Coomassie staining, and protein concentrations were measured via Bradford Assays. EMSAs were performed as previously described using probes generated with 5’IREdye-700 labeled oligos from IDT (Uhl et al., 2016). The following modifications were made to the previously reported protocol: protein binding reactions were heated to 37°C for 40 minutes and allowed to cool to room temperature to promote homo-to heterodimer exchange between the E-proteins and NEUROG3 proteins. DNA probes were subsequently added and samples incubated at room temperature for an additional 10 minutes before loading onto the polyacrylamide gel. All EMSAs were imaged using a LICOR Clx scanner, and all quantification was performed using LICOR Image Studio software. Probes with a high affinity E-box site from the NKX2–2 promoter (sequence 5’-TTATTACCGCTGAACATATGGCCAATATTTTGACTTAGTGCGGGCGTGGCT-3’) and a low affinity site from the NEUROG3 Promoter (sequence 5’-CTTTGTCCGGAATCCAGCTGTGCCCTGCGGGGGAGTAGTGCGGGCGTGGCT-3’) were added to binding reactions to a final concentration of 0.034μM. For the experiments using high and low affinity probes, E47 was added in all indicated lanes to a concentration of 0.058 μM, and the indicated NEUROG3 protein was added in increasing concentrations from 0.019 μM to 1.2 μM. To assay for E-protein heterodimerization, a probe containing an E-box from the Dll1 gene that is specifically bound by other bHLH factors, including E-protein homodimers, but not by NEUROG3 complexes, was used (sequence 5’-AGAGAGCAGGTGCTGTAGTGCGGGCGTGGCT-3’) (Castro et al., 2006). E47 was added in all indicated lanes to a concentration of 0.058 μM, and the indicated NEUROG3 protein was added in increasing concentrations from 0.05 μM to 1.5 μM. Dimerization was measured by the ability of each NEUROG3 mutant to disrupt E-protein homodimer binding to the probe.
Luciferase reporter constructs and luciferase assay
NEUROG3 luciferase reporters were constructed by cloning multimerized E-box sequences from the NKX2–2 promoter regions upstream of a Tk promoter (Smith et al., 2004). Here the TK promoter was cloned upstream of Luciferase in the pGL4.10 Luciferase vector (Promega) between BglII and HindIII restriction sites, and 6 copies of the NKX2–2 high affinity E-box (5’-CTGAACATATGGCCAA-3’) were placed upstream of the Tk promoter. Two copies of double stranded oligonucleotides containing 3 repeats of the 16bp sequence containing the NKX2–2 E-box were cloned into the Tk-Luciferase vector between NheI and XhoI restriction sites (5’-CTAGaacatatggccaactgaacatatggccaactgaacatatggccaaGCTAGCGTAC-3’; 5’-TCGAGTACGCTAGCttggccatatgttcagttggccatatgttcagttggccatatgtt-3’). 0.9ng/μl of either the 6× NKX2–2 E-box-TK-luciferase reporter or a Tk-Luciferase (No E-box) reporter and 0.1ng/μl of a constitutively expressed Renilla Luciferase were transfected into 9-day pancreatic monolayer by Lipofectamine 3000 (Thermofisher Scientific) followed by NEUROG3 variants induction. Expression of NEUROG3 WT, R93L, R107S, E123X, L135P, or S171fsX68 was induced by adding either 100ng/ml or 300ng/ml dox. After 24 hour induction, the Promega Dual Luciferase Assay system was used according to manufacturer’s protocols to measure Luciferase activity in each condition. To control for transfection efficiency all Firefly luciferase measurements were normalized to Renilla levels and samples were normalized to the Tk-Luciferase (No E-box) reporter. All experimental conditions were tested in triplicate and representative data is shown in Figure S7E.
Quantification and Statistical Analysis
For all experiments, “N” represents the number of replicates performed from different differentiation or induction experiments. F-test and unpaired Student’s t-test of two samples was performed by Microsoft Excel. One-way ANOVA followed by Dunnett, Tukey HSD, Tuckey-Kramer (unequal sample size) or Dunnett T3 (unequal variance) post hoc test for multiple comparisons test of more than two samples was performed by SPSS 24 software, where *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001, and ****p ≤ 0.0001.
Data and Software Availability
Complete gels for western blots and EMSAs have been deposited on Mendeley Dataset https://data.mendeley.com/datasets/dj53dkzwp5/draft?a=3581f584-ea06-4cef-9991-6d6b6e6ab3c4.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Actin | NeoMarkers | Cat#ms-1295-p1 |
| Chromogranin A | Immunostar | Cat #20086 |
| E-Cadherin | R&D | Cat #AF648 |
| E-Cadherin | BD Transduction Lab | Cat #610182 |
| Glucagon | Zymed | Cat #18–0064 |
| Glucagon | Cell Marque | Cat #259R-15 |
| HA | Santa Cruz | Cat #sc-805 |
| HA | Roche | Cat #3F10 |
| Insulin | Dako | Cat #A0564 |
| Neurogenin 3 | R&D | Cat #AF3444 |
| Nkx2–2 | DSHB | Cat #74.5A5 |
| Pdx1 | Abcam | Cat #ab47383–100 |
| Somatostatin | Santa Cruz | Cat #sc-7819 |
| Synaptophysin | Synaptic Systems | Cat #101004 |
| Bacterial and Virus Strains | ||
| One Shot™ Stbl3™ Chemically Competent E. coli | Invitrogen | Cat #C737303 |
| One Shot™ TOP10 Chemically Competent E. coli | Invitrogen | Cat #C404003 |
| Lentiviral vectors pseudotyped with vesicular stomatitis virus (VSV) G-protein | CCHMC Viral Vector Core | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| recombinant human FGF4 | R&D Systems | Cat#235-F4 |
| recombinant human EGF | R&D Systems | Cat#236-EG-01M |
| recombinant human Noggin | R&D Systems | Cat#6057-NG |
| recombinant human BMP4 | R&D Systems | Cat#314-BP-050 |
| recombinant human FGF7 | R&D Systems | Cat#251-KG-050 |
| Activin A | Cell Guidance Systems | Cat#GFH6 |
| CHIR99021 | Stemgent | Cat#04–0004 |
| Y-27632 dihydrochloride | Tocris | Cat#1254 |
| Doxycycline | Sigma-Aldrich | Cat#D3447 |
| hESC-qualified Matrigel | BD Biosciences | Cat#354277 |
| mTeSR1 media | Stem Cell Technologies | Cat#5850 |
| RPMI 1640 Medium | Thermo Fisher Scientfic | Cat#11-875-093 |
| DMEM, High Glucose | Thermo Fisher Scientfic | Cat#11-965-092 |
| Advanced DMEM/F12 | Thermo Fisher Scientfic | Cat#12-634-010 |
| Growth Factor Reduced (GFR) Basement Membrane Matrix | Corning | Cat#356231 |
| Matrigel | BD Biosciences | Cat#354234 |
| Defined fetal bovine serum (dFBS) | Hyclone | Cat#SH30070.02 |
| Normal donkey serum | Jackson Immunoresearch Labs | Cat#017-000-121 |
| Odyssey® Blocking Buffer (TBS) | LI-COR | Cat#927–50000 |
| L-glutamine (100×) | Thermo Fisher Scientific | Cat#25030–081 |
| Pen/Strep (100×) | Thermo Fisher Scientific | Cat#15140–122 |
| 50× B27 supplement w/o Vitamin A | Thermo Fisher Scientific | Cat#12587–010 |
| Non-essential Amino Acids (100×) | Thermo Fisher Scientific | Cat#11140050 |
| HEPES Buffer | Thermo Fisher Scientific | Cat#15630080 |
| N2 Supplement | Thermo Fisher Scientific | Cat#17502–048 |
| Dispase | Thermo Fisher Scientific | Cat#17105–041 |
| Acutase | Thermo Fisher Scientific | Cat#A11105–01 |
| IntestiCult™ Organoid Growth Medium (Human) | Stem Cell Technologies | Cat#06010 |
| G-418 disulfate salt | Sigma-Aldrich | Cat#A1720 |
| Fluoromount-G® | SouthernBiotech | Cat#0100–01 |
| PD0325901 | Stem Cell Technologies | Cat#72182 |
| MG-132 | Selleckchem | Cat#S2619 |
| Cycloheximide | Sigma-Aldrich | Cat#C1988–1G |
| Critical Commercial Assays | ||
| Quantitect SYBR Green | Qiagen | Cat#204145 |
| QIAquick PCR Purification Kit | Qiagen | Cat#28104 |
| 8–16% Mini-PROTEAN® TGX™ Precast Protein Gels | Bio-Rad | Cat#4561104 |
| Nucleospin RNA | Macherey-Nagel | Cat#740955 |
| Lipofectamine™ 3000 Transfection Reagent | Invitrogen | Cat#L3000001 |
| SuperScript VILO cDNA synthesis kit | Invitrogen | Cat#11754250 |
| Dual-Luciferase® Reporter Assay System | Promega | Cat#E1910 |
| Lambda Protein Phosphatase | New England Biolabs | Cat#P0753S |
| LIVE/DEAD™ Fixable Violet Dead Cell Stain Kit | Invitrogen | Cat#L34963 |
| pENTR™/D-TOPO™ Cloning Kit | Invitrogen | Cat#K240020 |
| Gateway™ LR Clonase™ II Enzyme mix | Invitrogen | Cat#11791020 |
| Deposited Data | ||
| Western Blots and EMSA | This paper; Mendeley Data | https://data.mendeley.com/datasets/dj53dkzwp5/draft?a=3581f584-ea06-4cef-9991-6d6b6e6ab3c4 |
| Experimental Models: Cell Lines | ||
| Human: NEUROG3−/− H1 ES
background cells (passage 56) |
McGrath et al., 2015 | N/A |
| Human: H1 ES cells (passage 42): | CCHMC Pluripotent Stem cell core/WiCell Research Institute | NIH hESC-10–0043 |
| Oligonucleotides | ||
| Probes with high affinity E-box site from
NKX2–2 promoter: TTATTACCGCTGAACATATGGCCAATATTTTGACTTAGTGCGGGCGTGGCT |
IDT DNA | N/A |
| Probes with low affinity E-box site from
NEUROG3 promoter: CTTTGTCCGGAATCCAGCTGTGCCCTGCGGGGGAGTAGTGCGGGCGTGGCT |
IDT DNA | N/A |
| Dll1 E-box bound by E-protein homodimers
but not NEUROG3 complexes: AGAGAGCAGGTGCTGTAGTGCGGGCGTGGCT |
Castro et al., 2006 | N/A |
| 5’ HA-F_Neurog3 ACCTGCGCCGGTAGAAAGGATGTACCCATACGATGTTCCAGATTACG |
IDT DNA | N/A |
| 5’ HA-R_Neurog3 GCGCACCCGAGGGTTGAGGCGTAGCGTAATCTGGAACATCGTATGGG |
IDT DNA | N/A |
| E123X (367G-T)-F CAAGATCTAGACGCTGCGCTTCGCCCACAACTAC |
IDT DNA | N/A |
| E123X (367G-T)-R CAGCGTCTAGATCTTGGTGAGCTTCGCGTCGTC |
IDT DNA | N/A |
| E28X (82G-T)-F GAGCCTCGGAAGACTAAGTGACCTGCCCC |
IDT DNA | N/A |
| E28X (82G-T)-R GGGGCAGGTCACTTAGTCTTCCGAGGCTC |
IDT DNA | N/A |
| L135P (404T-C)-F ACTACATCTGGGCGCCGACTCAAACGCTGCG |
IDT DNA | N/A |
| L135P (404T-C)-R CGCAGCGTTTGAGTCGGCGCCCAGATGTAGT |
IDT DNA | N/A |
| Neurog3 cDNA TOPO-F CACCTGCGCCGGTAGAAAG |
IDT DNA | N/A |
| Neurog3 cDNA TOPO-R ACAGACAGGTCCTTTCACAG |
IDT DNA | N/A |
| R107S (319C-A)-F ACTGGACGCCCTGAGCGGTGTCCTGCC |
IDT DNA | N/A |
| R107S (319C-A)-R GGCAGGACACCGCTCAGGGCGTCCAGT |
IDT DNA | N/A |
| R93L (278G-T)-F GCGAGCTCAATCGAATGCACAACCTCAACTCGGC |
IDT DNA | N/A |
| R93L (278G-T)-R TCGATTGAGCTCGCGGTCGTTGGCCTTCTTTCG |
IDT DNA | N/A |
| S171fsX68 (510G-GG)-F TGGGGGGTCCCTCTACTCCCCAGTCTCCCAGGC |
IDT DNA | N/A |
| S171fsX68 (510G-GG)-R GAGGGACCCCCCAGTCCCCGGGGGAACCG |
IDT DNA | N/A |
| S183A Phosphor_null-F GCCTGGCTCCCGCCGCGTCG |
IDT DNA | N/A |
| S183A Phosphor_null-R CGGGAGCCAGGCTGCCAGCCTGG |
IDT DNA | N/A |
| S204A Phosphor_null-F TCCGCCTGCTTGGCCCCAGGCAGTCTGGCTTTCT |
IDT DNA | N/A |
| S204A Phosphor_null-R AGACTGCCTGGGGCCAAGCAGGCGGAAAAGGTG |
IDT DNA | N/A |
| S207A Phosphor_null-F TTGGCCCCAGGCGCTCTGGCTTTCTCAGATTTTCTGTGAAAG |
IDT DNA | N/A |
| S207A Phosphor_ null-R GAGAAAGCCAGAGCGCCTGGGGCCAAGCAGG |
IDT DNA | N/A |
| T120A Phosphor_null-F GCTCGCCAAGATCGAGACGCTGC |
IDT DNA | N/A |
| T120A Phosphor_null-R TCTTGGCGAGCTTCGCGTCGTCTGG |
IDT DNA | N/A |
| Chga-qRT-PCR_F TGTGTCGGAGATGACCTCAA |
IDT DNA | N/A |
| Chga-qRT-PCR_R GTCCTGGCTCTTCTGCTCTG |
IDT DNA | N/A |
| Ind-Neurog3-qRT-PCR_F AGGTCACTTCGTCTTCCGAG |
IDT DNA | N/A |
| Ind-Neurog3-qRT-PCR_R ACAGACAGGTCCTTTCACAG |
IDT DNA | N/A |
| Neurod1-qRT-PCR_F ATCAGCCCACTCTCGCTGTA |
IDT DNA | N/A |
| Neurod1-qRT-PCR_R GCCCCAGGGTTATGAGACTAT |
IDT DNA | N/A |
| Nkx2–2-qRT-PCR_F GGAGCTTGAGTCCTGAGGG |
IDT DNA | N/A |
| Nkx2–2-qRT-PCR_R TCTACGACAGCAGCGACAAC |
IDT DNA | N/A |
| PAX4-qRT-PCR_F TGTGCAGAGATGATTCCTGG |
IDT DNA | N/A |
| PAX4-qRT-PCR_R GAGGGTCTGGTTTTCCAACA |
IDT DNA | N/A |
| Negative control-ChIP-qPCR _F ATGGTTGCCACTGGGGATCT |
IDT DNA | N/A |
| Negative control-ChIP-qPCR _R TGCCAAAGCCTAGGGGAAGA |
IDT DNA | N/A |
| Nkx2–2 promoter-ChIP-qPCR_F TGTTTTCCTTGCCGGACTGA |
IDT DNA | N/A |
| Nkx2–2 promoter-ChIP-qPCR _R CCTACCATGTGGCAGCTGAA |
IDT DNA | N/A |
| Neurog3 promoter-ChIP-qPCR_F CGAGAGAGCAAACAGAGCGG |
IDT DNA | N/A |
| Neurog3 promoter-ChIP-qPCR _R GCCACCGGCCAATCAGC |
IDT DNA | N/A |
| Recombinant DNA | ||
| pINDUCER20 | Meerbrey et al., 2011 | Addgene # 44012 |
| Software and Algorithms | ||
| Imaris | Bitplane | N/A |
| NIS Elements | Nikon | N/A |
| BD FACSDiva | BD Biosciences | N/A |
| PRISMv7 graphing software | GraphPad Software | N/A |
| SPSS Statistics 24 | IBM | N/A |
Highlights:
Human PSC-derived pancreas and intestinal endocrine cells to model NEUROG3 mutations
NEUROG3 mutations affect protein stability, dimerization and DNA binding
NEUROG3 protein is less stable in intestinal epithelium than in the pancreas
Reduced protein stability in intestine enhances the sensitive to NEUROG3 mutations
ACKNOWLEDGEMENTS
We would like to thank the members of the Wells laboratories for reagents and feedback. We also thank Chris Mayhew and Amy Pitstick from the Pluripotent Stem Cell Facility, Matthew Kofron and the Confocal Imaging Core. This work was supported by NIH grants R01NS044080, R01DK092456, U19 AI116491, P01 HD093363–01, UG3 DK119982 and the Digestive Disease Research Center (P30 DK078392).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
None of the authors have potential conflicts of interests to declare related to this work.
References:
- Ahler E, Sullivan WJ, Cass A, Braas D, York AG, Bensinger SJ, Graeber TG, and Christofk HR (2013). Doxycycline Alters Metabolism and Proliferation of Human Cell Lines. PLoS One 8, e64561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azzarelli R, Hurley C, Sznurkowska MK, Rulands S, Hardwick L, Gamper I, Ali F, McCracken L, Hindley C, McDuff F, et al. (2017). Multi-site Neurogenin3 Phosphorylation Controls Pancreatic Endocrine Differentiation. Dev. Cell 41, 274–286.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basak O, Beumer J, Wiebrands K, Seno H, Van Oudenaarden A, and Clevers H (2017). Induced Quiescence of Lgr5+ Stem Cells in Intestinal Organoids Enables Differentiation of Hormone-Producing Enteroendocrine Cells. Cell Stem Cell 20, 177–190.e4. [DOI] [PubMed] [Google Scholar]
- Bhukhai K, de Dreuzy E, Giorgi M, Colomb C, Negre O, Denaro M, Gillet-Legrand B, Cheuzeville J, Paulard A, Trebeden-Negre H, et al. (2018). Ex Vivo Selection of Transduced Hematopoietic Stem Cells for Gene Therapy of β-Hemoglobinopathies. Mol. Ther 26, 480–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breslin MB, Wang HW, Pierce A, Aucoin R, and Lan MS (2007). Neurogenin 3 recruits CBP co-activator to facilitate histone H3/H4 acetylation in the target gene INSM1. FEBS Lett. 581, 949–954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castro DS, Skowronska-Krawczyk D, Armant O, Donaldson IJ, Parras C, Hunt C, Critchley JA, Nguyen L, Gossler A, Göttgens B, et al. (2006). Proneural bHLH and Brn proteins coregulate a neurogenic program through cooperative binding to a conserved DNA motif. Dev. Cell 11, 831–844. [DOI] [PubMed] [Google Scholar]
- Chang MY, Rhee YH, Yi SH, Lee SJ, Kim RK, Kim H, Park CH, and Lee SH (2014). Doxycycline enhances survival and self-renewal of human pluripotent stem cells. Stem Cell Reports 3, 353–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charrier S, Ferrand M, Zerbato M, Précigout G, Viornery A, Bucher-Laurent S, Benkhelifa-Ziyyat S, Merten OW, Perea J, and Galy A (2011). Quantification of lentiviral vector copy numbers in individual hematopoietic colony-forming cells shows vector dose-dependent effects on the frequency and level of transduction. Gene Ther. 18, 479–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collombat P, Mansouri A, Hecksher-Sorensen J, Serup P, Krull J, Gradwohl G, and Gruss P (2003). Opposing actions of Arx and Pax4 in endocrine pancreas development. Genes Dev. 17, 2591–2603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ejarque M, Cervantes S, Pujadas G, Tutusaus A, Sanchez L, and Gasa R (2013). Neurogenin3 cooperates with Foxa2 to autoactivate its own expression. J. Biol. Chem 288, 11705–11717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fehse B, Kustikova OS, Bubenheim M, and Baum C (2004). Pois(s)on - It’s a question of dose. Gene Ther. 11, 879–881. [DOI] [PubMed] [Google Scholar]
- Figliozzi RW, Chen F, Chi A, and Hsia SCV (2016). Using the inverse Poisson distribution to calculate multiplicity of infection and viral replication by a high-throughput fluorescent imaging system. Virol. Sin 31, 180–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Germán-Díaz M, Rodriguez-Gil Y, Cruz-Rojo J, Charbit-Henrion F, Cerf-Bensussan N, Manzanares JM-L, and Moreno-Villares JM (2017). A new case of congenital malabsorptive diarrhea and diabetes secondary to mutant Neurogenin-3. Pediatrics 140. [DOI] [PubMed] [Google Scholar]
- Gradwohl G, Dierich a, LeMeur M, and Guillemot F (2000). Neurogenin3 Is Required for the Development of the Four Endocrine Cell Lineages of the Pancreas. Proc. Natl. Acad. Sci. U. S. A 97, 1607–1611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hancili S, Bonnefond A, Philippe J, Vaillant E, De Graeve F, Sand O, Busiah K, Robert JJ, Polak M, Froguel P, et al. (2018a). A novel NEUROG3 mutation in neonatal diabetes associated with a neuro-intestinal syndrome. Pediatr. Diabetes 19, 381–387. [DOI] [PubMed] [Google Scholar]
- Hancili S, Bonnefond A, Philippe J, Vaillant E, De Graeve F, Sand O, Busiah K, Robert JJ, Polak M, Froguel P, et al. (2018b). A novel NEUROG3 mutation in neonatal diabetes associated with a neuro-intestinal syndrome. Pediatr. Diabetes 19, 381–387. [DOI] [PubMed] [Google Scholar]
- Henseleit KD, Nelson SB, Kuhlbrodt K, Hennings JC, Ericson J, and Sander M (2005). NKX6 transcription factor activity is required for alpha-and beta-cell development in the pancreas. Development 132, 3139–3149. [DOI] [PubMed] [Google Scholar]
- Huang H-P, Liu M, El-Hodiri HM, Chu K, Jamrich M, and Tsai M-J (2000). Regulation of the Pancreatic Islet-Specific Gene BETA2 (neuroD) by Neurogenin 3. Mol. Cell. Biol 20, 3292–3307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishii R, Isogaya K, Seto A, Koinuma D, Watanabe Y, Arisaka F, Yaguchi S, Ikushima H, Dohmae N, Miyazono K, et al. (2012). Structure of a dominant-negative helix-loop-helix transcriptional regulator suggests mechanisms of autoinhibition. EMBO J. 31, 2541–2552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenny M, Uhl C, Roche C, Duluc I, Guillermin V, Guillemot F, Jensen J, Kedinger M, and Gradwohl G (2002). Neurogenin3 is differentially required for endocrine cell fate specification in the intestinal and gastric epithelium. EMBO J. 21, 6338–6347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones S (2004). An overview of the basic helix-loop-helix proteins. Genome Biol. 5, 226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim YS, Kang HS, Takeda Y, Hom L, Song HY, Jensen J, and Jetten AM (2012). Glis3 regulates neurogenin 3 expression in pancreatic β-cells and interacts with its activator, Hnf6. Mol. Cells 34, 193–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krentz NAJ, Van Hoof, D., Li Z, Nian C, German MS, Lynn FC, Krentz NAJ, Van Hoof, D., Li Z, Watanabe A, et al. (2017). Phosphorylation of NEUROG3 Links Endocrine Differentiation to the Cell Cycle in Pancreatic Progenitors. Dev. Cell 41, 129–142.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JC, Smith SB, Watada H, Lin J, Scheel D, Wang J, Mirmira RG, and German MS (2001). Regulation of the pancreatic pro-endocrine gene neurogenin3. Diabetes 50, 928–936. [DOI] [PubMed] [Google Scholar]
- Lee SK, Lee B, Ruiz EC, and Pfaff SL (2005). Olig2 and Ngn2 function in opposition to modulate gene expression in motor neuron progenitor cells. Genes Dev. 19, 282–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longo A, Guanga GP, and Rose RB (2008). Crystal structure of E47-NeuroD1/Beta2 bHLH domain-DNA complex: Heterodimer selectivity and DNA recognition. Biochemistry 47, 218–229. [DOI] [PubMed] [Google Scholar]
- Lynn FC, Smith SB, Wilson ME, Yang KY, Nekrep N, and German MS (2007). Sox9 coordinates a transcriptional network in pancreatic progenitor cells. Proc. Natl. Acad. Sci. U. S. A 104, 10500–10505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma YC, Song MR, Park JP, Henry Ho HY, Hu L, Kurtev MV, Zieg J, Ma Q, Pfaff SL, and Greenberg ME (2008). Regulation of Motor Neuron Specification by Phosphorylation of Neurogenin 2. Neuron 58, 65–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahe MM, Sundaram N, Watson CL, Shroyer NF, and Helmrath MA (2015). Establishment of human epithelial enteroids and colonoids from whole tissue and biopsy. J. Vis. Exp [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCracken KW, Howell JC, Wells JM, and Spence JR (2011). Generating human intestinal tissue from pluripotent stem cells in vitro. Nat. Protoc 6, 1920–1928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrath PS, Watson CL, Ingram C, Helmrath MA, and Wells JM (2015). The Basic Helix-Loop-Helix Transcription Factor NEUROG3 Is Required for Development of the Human Endocrine Pancreas. Diabetes 64, 2497–2505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meerbrey KL, Hu G, Kessler JD, Roarty K, Li MZ, Fang JE, Herschkowitz JI, Burrows AE, Ciccia A, Sun T, et al. (2011). The pINDUCER lentiviral toolkit for inducible RNA interference in vitro and in vivo. Proc. Natl. Acad. Sci. U. S. A 108, 3665–3670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osipovich AB, Long Q, Manduchi E, Gangula R, Hipkens SB, Schneider J, Okubo T, Stoeckert CJ, Takada S, and Magnuson M. a (2014). Insm1 promotes endocrine cell differentiation by modulating the expression of a network of genes that includes Neurog3 and Ripply3. Development 141, 2939–2949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pauerstein PT, Sugiyama T, Stanley SE, McLean GW, Wang J, Martín MG, and Kim SK (2015). Dissecting human gene functions regulating islet development with targeted gene transduction. Diabetes 64, 3037–3049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinney SE, Oliver-Krasinski J, Ernst L, Hughes N, Patel P, Stoffers DA, Russo P, and De León DD (2011). Neonatal diabetes and congenital malabsorptive diarrhea attributable to a novel mutation in the human neurogenin-3 gene coding sequence. In Journal of Clinical Endocrinology and Metabolism, pp. 1960–1965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prado CL, Pugh-Bernard AE, Elghazi L, Sosa-Pineda B, and Sussel L (2004). Ghrelin cells replace insulin-producing beta cells in two mouse models of pancreas development. Proc. Natl. Acad. Sci. U. S. A 101, 2924–2929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quan XJ, Yuan L, Tiberi L, Claeys A, De Geest N, Yan J, Van Der Kant R, Xie WR, Klisch TJ, Shymkowitz J, et al. (2016). Post-translational Control of the Temporal Dynamics of Transcription Factor Activity Regulates Neurogenesis. Cell 164, 460–475. [DOI] [PubMed] [Google Scholar]
- Rubio-Cabezas O, Jensen JN, Hodgson MI, Codner E, Ellard S, Serup P, and Hattersley AT (2011). Permanent neonatal diabetes and enteric anendocrinosis associated with biallelic mutations in NEUROG3. Diabetes 60, 1349–1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubio-Cabezas O, Codner E, Flanagan SE, Gómez JL, Ellard S, and Hattersley AT (2014). Neurogenin 3 is important but not essential for pancreatic islet development in humans. Diabetologia 57, 2421–2424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubio-Cabezas O, Gómez JL, Gleisner A, Hattersley AT, and Codner E (2016). Hypogonadotropic hypogonadism and short stature in patients with diabetes due to neurogenin 3 deficiency. J. Clin. Endocrinol. Metab 101, 3555–3558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rukstalis JM, and Habener JF (2009). Neurogenin3: a master regulator of pancreatic islet differentiation and regeneration. Islets 1, 177–184. [DOI] [PubMed] [Google Scholar]
- Sancho R, Gruber R, Gu G, and Behrens A (2014). Loss of Fbw7 reprograms adult pancreatic ductal cells into α, δ, and β cells. Cell Stem Cell 15, 139–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sander M, Sussel L, Conners J, Scheel D, Kalamaras J, Dela Cruz F, Schwitzgebel V, Hayes-Jordan A, and German M (2000). Homeobox gene Nkx6.1 lies downstream of Nkx2.2 in the major pathway of beta-cell formation in the pancreas. Development 127, 5533–5540. [DOI] [PubMed] [Google Scholar]
- Sayar E, Yilmaz A, Islek A, Elpek GO, Flanagan SE, and Artan R (2013). Chromogranin-A Staining Reveals Enteric Anendocrinosis in Unexplained Congenital Diarrhea. J. Pediatr. Gastroenterol. Nutr 57, e21. [DOI] [PubMed] [Google Scholar]
- Schwitzgebel VM, Scheel DW, Conners JR, Kalamaras J, Lee JE, Anderson DJ, Sussel L, Johnson JD, and German MS (2000). Expression of neurogenin3 reveals an islet cell precursor population in the pancreas. Development 127, 3533–3542. [DOI] [PubMed] [Google Scholar]
- Shabram P, and Aguilar-Cordova E (2000). Multiplicity of Infection/Multiplicity of Confusion. Mol. Ther 2, 420–421. [DOI] [PubMed] [Google Scholar]
- Smith SB, Gasa R, Watada H, Wang J, Griffen SC, and German MS (2003). Neurogenin3 and hepatic nuclear factor 1 cooperate in activating pancreatic expression of Pax4. J. Biol. Chem 278, 38254–38259. [DOI] [PubMed] [Google Scholar]
- Smith SB, Watada H, and German MS (2004). Neurogenin3 Activates the Islet Differentiation Program while Repressing Its Own Expression. Mol. Endocrinol 18, 142–149. [DOI] [PubMed] [Google Scholar]
- Sosa-Pineda B, Chowdhury K, Torres M, Oliver G, and Gruss P (1997). The Pax4 gene is essential for differentiation of insulin-producing beta cells in the mammalian pancreas. Nature 386, 399–402. [DOI] [PubMed] [Google Scholar]
- Soyer J, Flasse L, Raffelsberger W, Beucher A, Orvain C, Peers B, Ravassard P, Vermot J, Voz ML, Mellitzer G, et al. (2010). Rfx6 is an Ngn3-dependent winged helix transcription factor required for pancreatic islet cell development. Development 137, 203–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spence JR, Mayhew CN, Rankin SA, Kuhar MF, Vallance JE, Tolle K, Hoskins EE, Kalinichenko VV, Wells SI, Zorn AM, et al. (2011). Directed differentiation of human pluripotent stem cells into intestinal tissue in vitro. Nature 470, 105–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran R, Cho J, Ph D, Tsai M, Ph D, Treem WR, Hill ID, Vargas JH, Gershman G, Farmer DG, et al. (2007). Mutant Neurogenin-3 in Congenital Malabsorptive Diarrhea. N. Engl. J. Med 356, 1781–1782.17460236 [Google Scholar]
- Uhl JD, Cook TA, and Gebelein B (2010). Comparing anterior and posterior Hox complex formation reveals guidelines for predicting cis-regulatory elements. Dev. Biol 343, 154–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uhl JD, Zandvakili A, and Gebelein B (2016). A Hox Transcription Factor Collective Binds a Highly Conserved Distal-less cis-Regulatory Module to Generate Robust Transcriptional Outcomes. PLoS Genet. 12, 1–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ünlüsoy Aksu A, Eğritaş Gürkan Ö, Sarı S, Demirtaş Z, Türkyýlmaz C, Poyraz A, and Dalgýç B (2016). Mutant neurogenin-3 in a Turkish boy with congenital malabsorptive diarrhea. Pediatr. Int 58, 379–382. [DOI] [PubMed] [Google Scholar]
- Vosper JMD, Fiore-Heriche CS, Horan I, Wilson K, Wise H, and Philpott A (2007). Regulation of neurogenin stability by ubiquitin-mediated proteolysis. Biochem. J 407, 277–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Cortina G, Wu SV, Tran R, Cho J-H, Tsai M-J, Bailey TJ, Jamrich M, Ament ME, Treem WR, et al. (2006). Mutant Neurogenin-3 in Congenital Malabsorptive Diarrhea. N. Engl. J. Med 355, 270–280. [DOI] [PubMed] [Google Scholar]
- Wang S, Hecksher-Sorensen J, Xu Y, Zhao A, Dor Y, Rosenberg L, Serup P, and Gu G (2008). Myt1 and Ngn3 form a feed-forward expression loop to promote endocrine islet cell differentiation. Dev. Biol 317, 531–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson CL, Mahe MM, Múnera J, Howell JC, Sundaram N, Poling HM, Schweitzer JI, Vallance JE, Mayhew CN, Sun Y, et al. (2014). An in vivo model of human small intestine using pluripotent stem cells. Nat. Med 20, 1310–1314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witt LM, Gutzwiller LM, Gresser AL, Li-Kroeger D, Cook TA, and Gebelein B (2010). Atonal, senseless, and abdominal-A regulate rhomboid enhancer activity in abdominal sensory organ precursors. Dev. Biol 344, 1060–1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Chang BHJ, Yechoor V, Chen W, Li L, Tsai MJ, and Chan L (2011). The Krüppel-like zinc finger protein GLIS3 transactivates neurogenin 3 for proper fetal pancreatic islet differentiation in mice. Diabetologia 54, 2595–2605. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Complete gels for western blots and EMSAs have been deposited on Mendeley Dataset https://data.mendeley.com/datasets/dj53dkzwp5/draft?a=3581f584-ea06-4cef-9991-6d6b6e6ab3c4.