

Abstract
Objective:
Calcification of atherosclerotic plaque is traditionally associated with increased cardiovascular event risk; however, recent studies have found increased calcium density to be associated with more stable disease. HMG-CoA reductase inhibitors or “statins” reduce cardiovascular events. Invasive clinical studies have found that statins alter both the lipid and calcium composition of plaque, but the molecular mechanisms of statin-mediated effects on plaque calcium composition remain unclear. We recently defined a macrophage Rac-IL-1β signaling axis to be a key mechanism in promoting atherosclerotic calcification and sought to define the impact of statin therapy on this pathway.
Approach and Results:
Here, we demonstrate that statin therapy is independently associated with elevated coronary calcification in a high-risk patient population and that statins disrupt the complex between Rac1 and its inhibitor, RhoGDI, leading to increased active (GTP-bound) Rac1 in primary monocytes/macrophages. Rac1 activation is prevented by rescue with the isoprenyl precursor geranylgeranyl diphosphate. Statin treated macrophages exhibit increased activation of NF-κB, increased IL-1β mRNA, and increased Rac1-dependent IL-1β protein secretion in response to inflammasome stimulation. Using an animal model of calcific atherosclerosis, inclusion of statin in the atherogenic diet led to a myeloid Rac1-dependent increase in atherosclerotic calcification, which was associated with increased serum IL-1β expression, increased plaque Rac1-activation, and increased plaque expression of the osteogenic markers, ALP and RUNX2.
Conclusion:
Statins are capable of increasing atherosclerotic calcification through disinhibition of a macrophage Rac1-IL-1β signaling axis.
Graphical Abstract
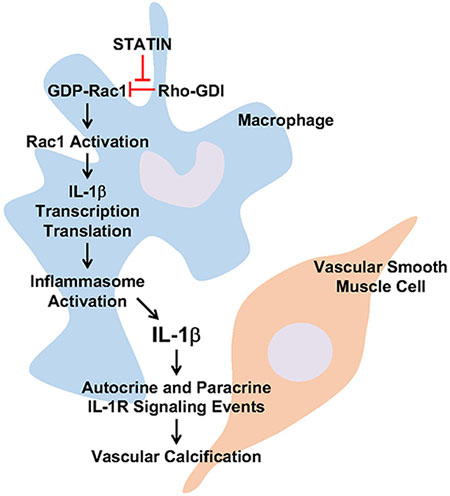
Introduction:
Ischemic heart disease caused by atherosclerosis is the leading cause of morbidity and mortality in the world1. Calcification of atherosclerotic plaques is predictive of total atherosclerotic burden and risk of cardiovascular and all-cause mortality2, 3. Rapid progression of vascular calcification is associated with a poor prognosis and increased risk of events4, 5. Yet, there has been growing evidence recently that certain types of densely calcified plaques can be associated with more stable disease3, 6–10, highlighting our limited understanding of the determinants of event risk with regard to atherosclerotic calcification.
For prevention of cardiovascular events, the current standard of care is pharmacotherapy with 3-hydroxy-3-methylglutaryl coenzymeA (HMG-CoA) reductase inhibitors or “statins”11–15. Statins are extremely successful at reducing cardiovascular events, and consequently, guidelines have moved toward more aggressive statin use16–20. Statins were developed as lipid lowering agents, but multiple studies have demonstrated that statins reduce the risk of myocardial infarction out of proportion to their lipid lowering effects16–19. These “pleiotropic effects” of statins have been attributed to the possibility that statins affect isoprenyl-based post-translational modification of protein, given isoprenyl groups are derived from precursor molecules within the cholesterol synthesis pathway downstream of mevalonate. The effects of statin therapy on atherosclerotic calcification have been controversial with initial studies suggesting statins reduce or slow atherosclerotic calcification and later studies indicating the opposite21–24. Moreover, recent studies have identified an association between statin therapy and increased plaque calcium composition relative to lipid composition, yet the mechanism that determines this association remains undefined22, 25, 26.
Racs are small GTPases and key signal transducers in inflammatory cells, influencing the expression of growth factors and cytokines27–31. Racs are post-translationally modified by an isoprenyl lipid group, specifically geranylgeranyl diphosphate (GGPP), which is thought to regulate subcellular localization32. Rac1 expression is ubiquitous, whereas Rac2 is primarily limited to cells of hematopoietic origin33. Rac1 and Rac2 share significant identity, except for a hypervariable region upstream of their isoprenylation site29. Though Racs often compensate for each other in terms of cellular function, Racs can also compete with each other for primary regulators, indicating that changes in gene expression of one Rac can simultaneously influence the activation state of the other34. Recently, our laboratory identified Rac2 as a major regulatory determinant of the degree of macrophage IL-1β expression by its regulation of Rac1 activity upstream of NF-κB and reactive oxygen species production35. Consequently, the Rac2 gene deletion model led to progressive inflammatory atherosclerotic calcification that was dependent on IL-1β signaling, and Rac2−/− macrophages demonstrated increases in Rac1 activation and Rac1-dependent IL-1β expression. The activity of Rac1 is regulated by three major enzymes: 1) guanine nucleotide exchange factors (GEFs), which activate Rac1 by promoting the exchange of GDP for GTP; 2) guanine nucleotide dissociation inhibitors (GDIs), which limit the access of Rac1 to GEFS; and 3) GTPase-activating proteins (GAPs), which lead to inactivation of Rac1 by accelerating its intrinsic GTPAse activity36.
Statins are known to inhibit mevalonate synthesis upstream of both cholesterol and geranylgeranyl diphosphate synthesis, and thus statins can inhibit the isoprenylation of Rac1 in addition to cholesterol synthesis37, 38. Current literature suggests that statin treatment disrupts Rac1 isoprenylation leading to reduced membrane association of Rac1 and consequently reduced membrane-dependent Rac1 functions38. However, the effects of statin treatment on Rac1 activation (GTP-binding) and downstream effector signaling are less clear and somewhat controversial. In fact, recently the genetic knockdown of the geranylgeranyl transferase enzyme that covalently attaches the isoprenyl group to Rac1 demonstrated constitutive GTP-binding of Rac1 and hyperactivation of Rac1-dependent pathways consequent to the loss of isoprenylation39.
In this manuscript, we sought to explore the relationship between statin use and coronary calcification in patients who have undergone noncontrast computed tomography followed by the quantification of Rac1 activity in peripheral blood monocytes of a subset of patients. To define a potential mechanistic relationship, we turned to preclinical models and assessed the effect of statins on macrophage Rac1 activity, Rac1-dependent IL-1β expression, and consequent atherosclerotic calcification in a mouse model of hypercholesterolemia. We hypothesize that, by inhibiting the isoprenylation of Rac1, statins may disrupt the regulation of Rac1 activity, leading to increased Rac1 activity, IL-1β expression, and consequent Rac1-dependent atherosclerotic calcification. Defining potential pleiotropic mechanisms whereby statins modulate plaque phenotype may lead to the development of novel therapeutic strategies for reducing cardiovascular event risk.
Materials and Methods:
The data that support the findings of this study are available from the corresponding author upon reasonable request. Because of the sensitive nature of some data collected for this study, requests to access the dataset from qualified researchers trained in human subject confidentiality protocols may be sent to the office of the Associate Chief of Staff for Research at Providence VA Medical Center at Research Services (151), 830 Chalkstone Avenue, Providence, RI 02908, or faxed to 401-457-3305; all data, analytical methods, and study materials are available upon request to researchers, who meet the Institutional Review Board criteria for access to VA confidential research data, for purposes of reproducing the results or replicating the study.
Materials
Anti-Rac1 mouse monoclonal antibody (clone 23A8) was purchased from Abcam (Cat#97732). Anti-activated Rac1 mouse monoclonal IgM antibody was purchased from New East Biosciences (Cat#26903). Anti-Actin, Alpha-smooth muscle-Cy3 antibody was purchased through Sigma Aldrich (Cat#C6198). PAK1 PBD agarose beads were purchased from Cell Biolabs (Cat#STA-411). Alexa Fluor-conjugated secondary antibodies, DAPI (4,6 diamidino-2-phenylindole), and RPMI 1640 culture medium were purchased from Fisher Scientific. Fluoroshield Mounting Medium with DAPI was purchased from Abcam (Cat#ab104139). Cholesterol and endotoxin-free BSA were purchased from Sigma-Aldrich. Primers used in quantitative real-time PCR were all purchased from IDT. Human Monocyte Isolation Kit and EasySep Buffer was purchased through StemCell (Cat#19359 and #20144, respectively). Sequences for murine primers were as follows: IL-1β, 5’-CCCTGCAGCTGGAGAGTGTGGA-3’ (sense) and 5’-CTGAGCGACCTGTCTTGGCCG-3’ (antisense); TNF-α, 5’-CACGTCGTAGCAAACCACCAA −3’(sense) and 5’-AGCAAATCGGCTGACGGTGT-3’ (antisense); GAPDH, 5’-AAGGCCGGGGCCCACTTGAA-3’ (sense) and 5’-GGACTGTGGTCATGAGCCCTTCCA-3’ (antisense); Rac1, 5’-CTACCCGCAGACAGACGTG-3’ (sense) and 5’-AGATCAAGCTTCGTCCCCAC-3’ (antisense); HPRT, 5’-GACCGGTCCCGTCATGCCGA-3’ (sense) and 5’-TGGCCTCCCATCTCCTTCATGACA-3’ (antisense).
Human Subjects Research
The patient studies comply with the Declaration of Helsinki and were approved by the Institutional Review Board of the Providence VA Medical Center. We evaluated a total of 465 US Veteran patients who had undergone LCSCT between December 1, 2013 and March 31, 2014.
Coronary Artery Calcium Scoring
Studies at the Providence VA Medical Center were performed using a 128-slice CT scanner (Siemans SOMATOM Definition AS) with a 128 mm x 0.6 mm collimation, using a helical acquisition protocol. Rotation time was 0.5 seconds, and pitch was 0.84. Studies were not ECG-gated. Scanning field of view was set to 380 mm. Matrix size was 512 x 512. Based on these parameters, the minimum area required to identify calcium was 0.55 mm2. The average tube voltage was 120 kV, and the average tube current was 40 mAmp. Image reconstruction slice thickness was between 1.0 and 1.25 mm.
Coronary artery calcium scoring (CACS) from lung cancer screening CT (LCSCT) were calculated using the Agatston method40. CACS was performed using previously described methods for other epidemiologic studies, and images were viewed and scored using a CarestreamVue PACS (Carestream Health) imaging workstation41–43. Briefly, the calcium scoring application in the CarestreamVue PACS displays axial image slices for the reader. The reader scrolls through axial slices and identifies coronary arteries with potential calcium to be scored. The reader indicates the appropriate coronary vessel to the program by circling a region of interest. Pixels of attenuation coefficient measurements above 130 HU and an area ≥0.55 mm2 within the region of interest are identified by the program as calcified are incorporated into the calcium score. All scans were scored by the preventive cardiology fellow (J.L.C) after being trained on 50 scans by the CCT board-certified cardiologist (A.R.M.). Readers were blinded to patient clinical data. Inter-observer agreement was quantified for total CACS values and the overall kappa was found to be very good at 0.907(0.859-0.956).
Covariates
The VA electronic medical record was searched for patient demographics and cardiovascular covariates including age, sex, race, and BMI. Medical history included smoking status, hypertension, hypertensive medication, cholesterol medication, diabetes, and fasting lipid profile. From the acquired data, the ASCVD risk scores were calculated.
Human CD14+CD16− Monocyte Isolation
Peripheral blood CD14+CD16− monocytes from a subset of patients either taking or not taking statin medication were isolated from 20 mL of blood using negative selection by the EasySep Human Monocyte Isolation Kit (StemCell Technologies, Cat#19359) per manufacturer protocols44. Briefly, the buffy coat from whole blood was extracted using 1500rpm centrifugation at room temperature and a subsequent Ficoll density gradient to separate out RBCs. Cells were pelleted and washed with EasySep Buffer (StemCell, Cat#20144). EasySep Human Monocyte Isolation Kit was used to further isolate monocytes and eliminate any remaining platelets or extraneous cell types via negative selection and magnet filtration. Purified monocytes were allowed to adhere to glass coverslips in complete RPMI for at least 2 hours. Adherent monocytes were washed with PBS, then fixed with 4% PFA and stored at 4°C for subsequent staining. Fixed monocytes were washed in PBS and incubated in 0.1% (V/V) Triton X in PBS for 10 minutes. After washing, they were incubated in 5% goat serum in 0.1% (V/V) Triton X-PBS for 1 hour. They were then washed and covered with primary antibody, Rac1-GTP (New East Biosciences, Cat#26903), overnight at 4°C. Primary antibody was washed with PBS and secondary antibody, Goat Anti-Mouse IgM AlexaFluor488 (Invitrogen, Cat#A21042), was added for 1 hour. Cells were washed and coverslips were plated to microscopy slides using Fluoroshield Mounting Medium with DAPI (Abcam, Cat#ab104139), and sealed with clear adhesive. Imaging was performed on the Nikon Eclipse 80i inverted microscope, and LUTs and exposure were standardized among all fields of view. Immunofluorescence was quantified using ImageJ45. Each individual data point represents an average CTCF of 4 fields of view with at least 20 cells per field of view.
Animal Studies
The Providence VA Medical Center Institutional Animal Care and Use Committees approved all housing protocols and experimental animal procedures. ApoE−/−, Rac1fl/fl, and CSF1Rmcm mice were purchased from The Jackson Laboratory and have been previously described46–48. All animal studies adhered to the guidelines for experimental atherosclerosis studies as described in the recent AHA Scientific Statement49. All animal studies adhered to the guidelines as described in the ATVB Council Statement for considering “sex difference” as a biological variable50.
High Fat Diet
Male and female animals, 8-10 weeks old, were fed a high fat diet (HFD; 20% fat by weight with 1.25% cholesterol, ENVIGO TD.02028) or HFD+Statin (atorvastatin at dry weight concentration of 60 mg/kg of HFD chow, ENVIGO TD.150052) ad libitum for up to 20 weeks. A subset of ApoE−/−,Rac1fl/fl, and CSF1Rmcm mice underwent intraperitoneal injection of tamoxifen at 2 mg daily for 10 days in order to induce myeloid Rac1 gene deletion prior to HFD+Statin treatment (HFD+Statin+mR1KO).
Macrophage Cell Cultures
Bone marrow-derived macrophages (BMDMs) were obtained by in vitro differentiation of primary femur marrow cells with an established protocol that yields >97% CD11bhiF4/80hi cells30, 51. Briefly, femurs and tibiae from ApoE−/− and CSF1RmcmRac1fl/flApoE−/− mice were dissected, cleaned, disinfected in 70% ethanol, and washed with fully supplemented RPMI 1640 medium (10% FBS, 10 mM HEPES pH 7.4, 2 mM L-glutamine, 100 units/ml penicillin, 10 µg/ml streptomycin, and 50 μM 2-ME). Red blood cells were removed by ammonium-chloride-potassium lysis buffer (0.15 M NH4Cl, 1 mM KHCO3, 0.1 mM Na2EDTA, adjusted to pH 7.4) and subsequent centrifugation. Remaining bone marrow cells were then cultured at a density of 3.5 x 106 cells/10 cm petri dish in fully supplemented RPMI with 30% (v/v) L929 cell-conditioned medium. Cells were matured to phenotypic macrophages over 6-8 days. Nonadherent cells were washed away with PBS. Adherent cells were recovered with gentle pipetting in PBS with 1 mM EDTA. Flow cytometric analysis confirmed the recovered cells to be >97% CD11b+F4/80+30, 51.
Cholesterol Crystal Inflammasome Assay
Cholesterol crystals were generated as previously described52. Cholesterol (Sigma) was dissolved in 1-propanol at a concentration of 2 mg/ml. Crystals were precipitated overnight at room temperature by dilution to 40% 1-propanol with water. Crystals were pelleted by centrifugation and dried at 70°C. Pellets were re-suspended in 0.1% FBS in PBS at a concentration of 50 mg/ml. Average crystal size of 1-2 μm was confirmed by microscopy. BMDMs (+/− priming with low dose (10 ng/ml) LPS for 2 hours) were then either exposed to 1000 μg/ml cholesterol crystals or vehicle control for 24 hours. Culture supernatants were collected and IL-1β expression quantified by enzyme-linked immunosorbent assay (ELISA) per manufacturer protocols (BioLegend, Cat#432604 and 430904, respectively).
NF-κB Assay
BMDMs were transfected with NF-κB Firefly Luciferase Plasmid (Promega) and pRL-TK Renilla control plasmid (Promega), using the Amaxa Mouse Macrophage Nucleofector Kit (Lonza). 12 hours after transfection, BMDMs were primed with LPS (10 ng/mL) for 2 hours and then exposed to 1000 μg/ml cholesterol crystals for 12 hours. Luciferase activity in cell lysates was measured by Dual-Glo ®Luciferase Assay System (Promega).
Reactive Oxygen Species (ROS) Assay
BMDMs were primed with 10 ng/mL LPS for 2 hours at 37 °C and then cells were exposed to 1000 μg/ml cholesterol crystals for up to 3 hours at 37 °C. ROS detection was carried out using ROS-Glo™ H2O2 Assay (Promega) per manufacturer protocols.
Rac1 Activation Assay
Rac1 activity was measured by an affinity precipitation assay with Pak1 PBD agarose beads53. BMDMs were lysed in 500 μl lysis buffer (20 mM HEPES, pH 7.4, 150 mM NaCl, 1% Triton X-100, 4 mM EDTA, 4 mM EGTA, 10% glycerol, supplemented with protease inhibitor cocktail [Roche]). Ten percent of the whole cell lysate was set-aside for Rac1 loading control. To the remainder of the sample, 10 μg of Pak1 PBD beads were added, followed by a 45-minute incubation at 4°C. Beads were washed with lysis buffer and subsequently brought up in Laemmli sample buffer. Activated (GTP-bound) Rac1 was detected by immunoblot after affinity precipitation, and whole cell lysates were used as loading controls.
For active Rac1 immunofluorescence staining, human monocyctes or BMDMs were adhered, fixed and permeabilized, followed by primary staining with mouse anti-activated Rac1 (New East Biosciences) and secondary staining with Alexa Fluor 488 Goat Anti-Mouse IgM (Invitrogen). Corrected total cell fluorescence was quantified by ImageJ45. Each individual data point represents an average CTCF of 4 fields of view with at least 20 cells per field of view.
Proximity Ligation Assay
To demonstrate a complex between RhoGDI and Rac1 in adhered BMDMs, a proximity ligation assay kit was used as per manufacturer protocols54–56. Primary antibodies to RhoGDI and Rac1 were used, and secondary antibodies were conjugated to oligonucleotides for ligation and subsequent rolling circle amplification. Quantification of proximity ligation signal was carried out using ImageJ45. Each individual data point represents an average of 4 fields of view from 3 slides with at least 50 cells per field of view.
Serum Protein and Cholesterol Analysis
Yale Mouse Metabolic Phenotyping Center quantified serum total cholesterol, LDL, and triglyceride concentrations, utilizing the Roche COBAS Mira Plus automated chemistry analyzer. IL-1β and TNF-α were measured in serum samples using ELISA kits (Biolegend) according to manufacturer’s protocols.
RNA Isolation and qRT-PCR
RNA from BMDMS was isolated using RNeasy® Mini Kit (QIAGEN, Cat#74106). RNA was quantified by NanoDrop 2000 (Thermo Scientific) and then reverse transcribed by iScript (Bio-Rad, Cat#1708891) protocol for subsequent qRT-PCR of transcript expression. GAPDH and HPRT expression were used for normalization.
Histology and Microscopy
Animals were euthanized, and tissue was perfusion fixed with 4% paraformaldehyde. Aortas harvested and fixed for 12 hours with paraformaldehyde (4%, buffered neutral) overnight at 4°C. Aortas were then cleaned under dissection microscope. Traditional hematoxylin and eosin (H&E), Masson’s trichrome, and Alizarin red staining protocols were performed on 10-μm thick cross sections at the level of the aortic sinus, between 1400-1700 μm from the most inferior section of the valve tissue facing the left ventricular outflow tract. Quantifications of plaque area, plaque collagen area, and calcium area were carried out using ImageJ45. Each individual data point represents the average quantification from 3 aortic valve sinuses taken from the 6-8 adjacent sections per slide of on aortic sinus. Some ascending aortas (1-2 mm) at the level of the arch inferior to the brachiocephalic branch were fixed for 12 hours with paraformaldehyde (4%, buffered neutral) overnight at 4°C. Aortas were then cleaned under dissection microscope, placed in 30% Sucrose for up to 1 week, and embedded in Optimal Cutting Temperature (OCT) compound in pre-labeled Cryomold squares (Tissue-Tek®) followed by dry ice for 10-15 minutes. Tissue was sectioned at 10 μm thickness by cryostat. After sectioning, slides were dried at room temperature for 1-2 hours before immunostaining.
Sections (10 μm thickness) were immunostained Cy3-conjugated anti-α-smooth muscle actin (Sigma-Aldrich, Cat#C6198), anti-alkaline phosphatase (Abcam, Cat#Ab65834), anti-RUNX2 (Abcam, Cat#Ab76956), rat anti-mouse CD68 (BioLegend #137002), or mouse anti-activated Rac1 (New East Biosciences, Cat#26903). Unconjugated primary antibody staining was visualized with Goat Anti-Rat IgG H&L Cy5 (Abcam, Cat#Ab6565) and Goat Anti-Mouse IgG, IgM (H+L) AlexaFluor488 (Invitrogen, Cat#A10680). All sections were counterstained with DAPI. Bright field and immunofluorescence microscopy of sectioned tissue were carried out on a Nikon Eclipse 80i inverted microscope. Images were taken with Nikon Plan Apochromat 2X (numerical aperture 0.1), 4X (numerical aperture 0.2), 10x (numerical aperture 0.3), 20x (numerical aperture 0.75) objective lenses. Quantifications of percent area positive activated Rac1 staining, RUNX2 staining, or ALP staining per plaque were carried out using ImageJ45. Each individual data point represents the average quantification of each animal using the average area of images captured from 4-6 adjacent sections per slide.
Plaque Calcium and Lipid Quantification
One day prior to euthanasia, some animals underwent intraperitoneal injection with 2 nmol of IRDye 680RD BoneTag Optical Probe (LI-COR, Cat#926-09374). Animals were euthanized, and tissue perfusion fixed with 4% paraformaldehyde (buffered neutral). Thoracic aortas underwent microdissection and subsequently imaged ex vivo with the Odyssey Imaging System (LI-COR). Calcification was quantified with Image Studio Lite (LI-COR). Aortas were scanned using the following acquisition settings: 1) 700 nm intensity of 3; 2) resolution of 21 microns; and 3) focus offset of 0 mm. Image LUTS scale was set from a minimum of 50 to a maximum of 1500, with a k of 0.2. Calcification was quantified by outlining the entire aorta as the region of interest and determining the ratio of signal intensity to aorta area.
Plaque lipid burden was quantified in a subset of aortas by en face staining with Oil Red O (Sigma) as previously described57. Aortas were fixed overnight at 4°C in neutral buffered 4% paraformaldehyde in PBS, dissected luminal surface is exposed, and pinned to a silicon plate. Aortas were then washed in PBS and then covered in propylene glycol for 2 minutes at room temperature for dehydration. Aortas were covered by 0.5% Oil Red O in isopropanol for 2 hours at room temperature and then washed in 85% propylene glycol and then in PBS until background staining is cleared. Percent area positive for Oil Red O staining was quantified using Image J45.
Statistical analysis
All statistical data were analyzed with the use of Stata/SE (version 15.1; StataCorp LLC, College Station, TX), Prism 7 (GraphPad) or R software version 3.3.1 (R Core Team). Baseline demographics and clinical characteristics were compared between the cohort that was not taking a statin and cohort taking a statin. Continuous variables were tested for normality and equal variance using Shapiro-Wilk test for normality and one-way ANOVA to test for equal variance. Results are presented as mean (standard error) for continuous variables with normal distribution or equal variance and as median (interquartile range) for continuous variables without normal distribution, and as number (percentage) for categorical data. The t test was used to compare normally distributed continuous variables between 2 independent groups. The Mann-Whitney U test was used for continuous variables not normally distributed, and Chi-square test was used for categorical variables. Adjustment for cardiovascular covariates was carried out using Stata. A 2-sided P <0.05 was considered statistically significant.
Results:
Statin treatment is independently associated with increased coronary calcification in patients undergoing noncontrast lung cancer screening chest CT.
Table 1 defines the baseline demographics and clinical characteristics of the patient population. We evaluated a total of 465 patients who had undergone lung cancer screening computed tomography (LCSCT) between December 1, 2013 and March 31, 2014. All patients were US Veterans with a mean age of 67 (SE: 0.3)-years-old. The majority of the patients were Caucasian, male, and current smokers with an elevated cardiac risk as evidenced by a mean atherosclerotic cardiovascular disease (ASCVD) score of 21.6 (SE: 0.6)%. The mean cholesterol was 177.6 (SE: 2.0) mg/dL and the median body mass index (BMI) was 28.0 (IQI: 24.7, 31.9) kg/m2. Approximately 28% of the patients had a history of diabetes and 7% of patients had a family history of early CAD. Eighteen percent of patients had known CAD; approximately 5% had prior myocardial infarction (MI) and 5% had prior CABG. The median coronary artery calcification score (CACS) as determined LCSCT was 595 (IQI: 125, 1570).
Table I.
Baseline Demographics and Clinical Characteristics of Patients.
| All Patients (N= 465) | No Statin (n=182) | Statin (n= 283) | P Value | |
|---|---|---|---|---|
| Age, years, mean (SE) | 67 (0.3) | 65 (0.6) | 68 (0.4) | 0.0004 |
| BMI, median (IQI) | 28.0 (24.7, 31.9) | 26.8 (24.3, 30.3) | 28.8 (25.1, 33.0) | 0.0006 |
| Male, n (%) | 453 (97.2) | 173 (95.0) | 280 (98.9) | 0.01 |
| Caucasian, n (%) | 435 (93.3) | 159 (87.3) | 276 (97.5) | <0.0001 |
| African American, n (%) | 22 (4.7) | 17 (9.3) | 5 (1.76) | <0.0001 |
| Diabetes Mellitus, n (%) | 131 (28.1) | 26 (14.2) | 105 (37.1) | <0.0001 |
| Hypertension, n (%) | 281 (60.3) | 91 (50.0) | 190 (67.1) | <0.0001 |
| Hyperlipidemia, n (%) | 334 (71.6) | 77 (42.3) | 257 (90.8) | <0.0001 |
| Total Cholesterol, mean (SE) | 177.6 (2.0) | 187.6 (3.2) | 171.1 (2.5) | 0.0001 |
| HDL Cholesterol, median (IQI) | 42 (26, 50) | 43 (37, 55) | 41 (36, 48) | 0.0071 |
| Statin Use, n (%) | 283 (60.8) | --- | --- | --- |
| Current Smoker, n (%) | 221 (47.4) | 95 (52.1) | 126 (44.5) | 0.106 |
| Family History of Early CAD, n (%) | 33 (7.0) | 14 (7.6) | 19 (6.7) | 0.688 |
| CAD, n (%) | 83 (17.8) | 14 (7.6) | 69 (24.3) | <0.0001 |
| Prior MI, n (%) | 24 (5.1) | 4 (2.1) | 20 (7.0) | 0.021 |
| Prior CABG, n (%) | 25 (5.3) | 2 (1.0) | 23 (8.1) | 0.001 |
| ASCVD Risk Score, mean (SE) | 21.6 (0.6) | 18.4 (0.8) | 23.7 (0.8) | <0.0001 |
| CACS, median (IQI) | 595 (125, 1570) | 307 (54, 814) | 941 (276, 2104) | <0.0001 |
BMI = body mass index; CAD = coronary artery disease; MI = myocardial infarction; CABG = coronary artery bypass graft surgery; ASCVD = atherosclerotic cardiovascular disease; CACS = coronary artery calcium score.
Of the 465 patients, there were 283 (approximately 60%) patients on statin therapy compared to 182 patients who were not taking a statin. Patients on statin therapy had been taking statin for a median of 56 (IQI: 24, 96) months. When comparing those on statin therapy with those not on statin therapy, the two groups were comparable in the percent of patients that were current smokers and that had a family history of early CAD. Patients on a statin were older, Caucasian, and with a higher BMI and had higher rates of hypertension, diabetes, CAD, prior MI, and prior revascularization. Patients in the statin group also had higher ASCVD risk scores with a median score of 23.7 (SE: 0.8) % compared to 18.4 (SE: 0.8) % (P <0.0001).
Patients on statin therapy had significantly higher CACS values with a median of 941 (IQI 276, 2104) compared to 307 (IQI 54, 814) (P <0.0001) (Table I and Fig. 1A,B). Coronary artery calcification was significantly associated with statin treatment (β-coefficient = 855.4 ± 141.2, P <0.0001) and remained significant after adjustment for comorbidities such as age, BMI, DM, HTN, HLD, CAD and active smoking (β-coefficient = 610.1 ± 158.6, P <0.0001). For those without known CAD and after controlling for ASCVD score, statin use was also significantly associated with coronary artery calcium (β-coefficient = 351.3 ± 121.5, P <0.004).
Figure 1. Statin treatment is associated with increased coronary calcification and peripheral blood CD14+CD16− monocytes from patients on statin are associated with increased Rac1 activation.
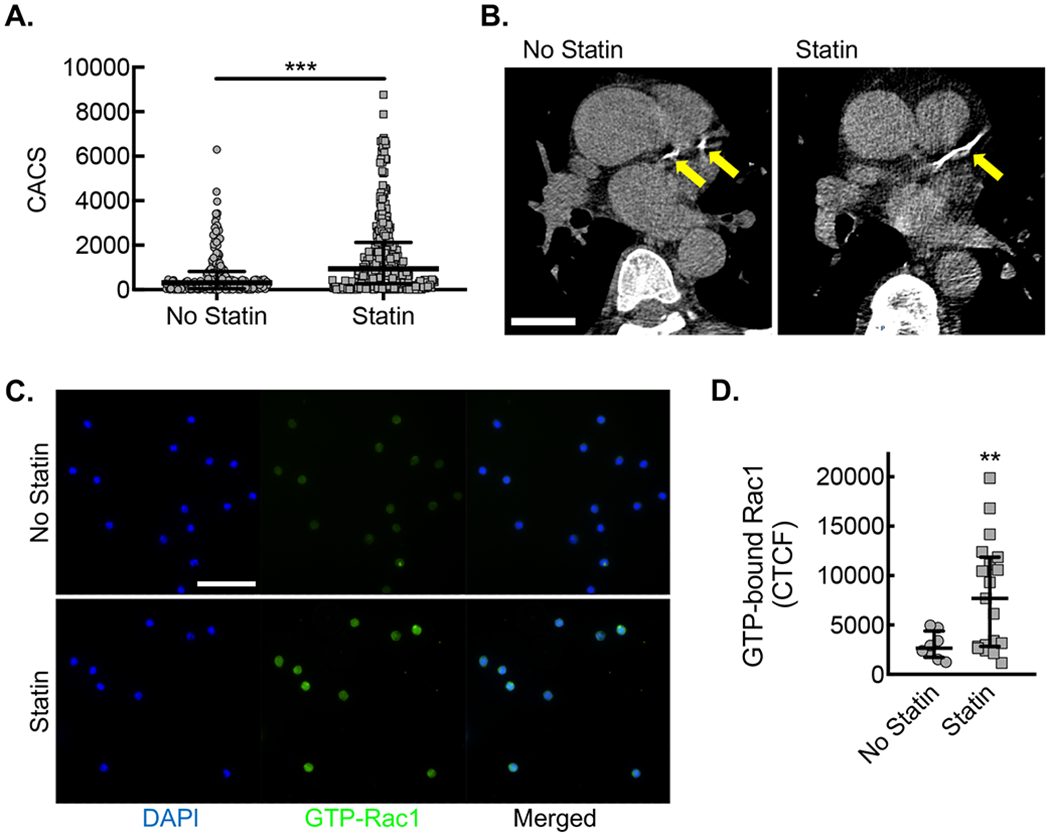
(A) CACS for each patient along with median and interquartile ranges for patients not taking statin (No statin, gray circles) vs. patients taking statin (Statin, gray squares) (***, P=0.0001 Mann-Whitney U; n=182 no statin and n=283 statin). (B) Example of differences in coronary calcium from patients of similar age not taking (No statin) or taking statin (Statin), using LCSCT axial images at the level of proximal left anterior descending coronary artery (LAD, yellow arrows) with total CACS of 283 and 412, respectively. Bar, 3 cm. (C) Immunofluorescent micrographs from CD14+ CD16− monocytes isolated from patients not taking statin (No statin, gray circles) vs. patients taking statin (Statin, gray squares) stained with antibody specific to activated Rac1 (green). Counterstaining with DAPI (blue). Bar, 50 μm. Quantification of Corrected Total Cell Fluorescence (CTCF) for activated Rac1 (**, P=0.029 Mann-Whitney U; No statin, n=8 patients; Statin, n=16 patients).
Peripheral blood CD14+CD16− monocytes from patients on statin are associated with increased Rac1 activation
To assess the impact of systemic statin treatment on monocyte Rac1 activity, we isolated peripheral blood monocytes from a subset of patients either taking or not taking statin. After isolation by negative selection, CD14+CD16− monocytes, known as an inflammatory subset of monocytes44, demonstrated increased Rac1 activation by immunofluorescence staining with an antibody specific to the GTP-bound (active) state of Rac1 (Fig. 1C,D).
Statin treatment of macrophages disrupted the isoprenyl-dependent complex between RhoGDI and Rac1.
To assess potential molecular mechanisms whereby statins may be permissive of or may promote Rac1 activation and consequently atherosclerotic calcification, and given our access to the relevant genetic strains, we moved to a mouse animal model for further in vitro and in vivo study. Rac1 is post-translationally modified by geranylgeranylation, which is known to direct Rac1’s subcellular localization32. Geranylgeranyl diphosphate (GGPP) is downstream of mevalonate in the cholesterol synthesis pathway, and thus GGPP synthesis can be inhibited by statins. In addition, the primary regulator of Rac1, Rho-specific guanine nucleotide dissociation inhibitor (RhoGDI), maintains Rac1 in the inactive (GDP-bound) state in the cell cytosol until Rac1 can be activated by a GEF36. Crystal structure analysis demonstrates that the principle binding interaction of Rho-GDI and Rac1 involves a lipophilic pocket that serves to hold the isoprenylation modification (Fig. 2A)58. We first sought to identify whether the complex between Rac1 and RhoGDI can be disrupted by statin treatment. Briefly, bone marrow-derived macrophages (BMDMs) from ApoE−/− mice were incubated with or without 10 μM atorvastatin, followed by proximity ligation assay to assess the molecular complex formation between Rac1 and RhoGDI. Proximity ligation assay demonstrated that the complex between Rac1 and RhoGDI in resting, vehicle control-treated BMDMs was inhibited after a 24-hour atorvastatin treatment (Fig. 2B,C). Moreover, loss of the Rac1-RhoGDI complex by statin treatment was rescued by isoprenyl precursors, farnesyl diphosphate (FPP) and GGPP, but the Rac1-RhoGDI complex was not rescued by the cholesterol synthesis precursor squalene. In summary, the association in macrophages between Rac1 and its inhibitor, RhoGDI, can be inhibited by atorvastatin, and this inhibition can be rescued by supplementation with isoprenyl precursors.
Figure 2. Statin treatment of primary bone derived marrow macrophages (BMDMs) disrupts the isoprenylation-dependent complex between Rho-GDI and Rac1.
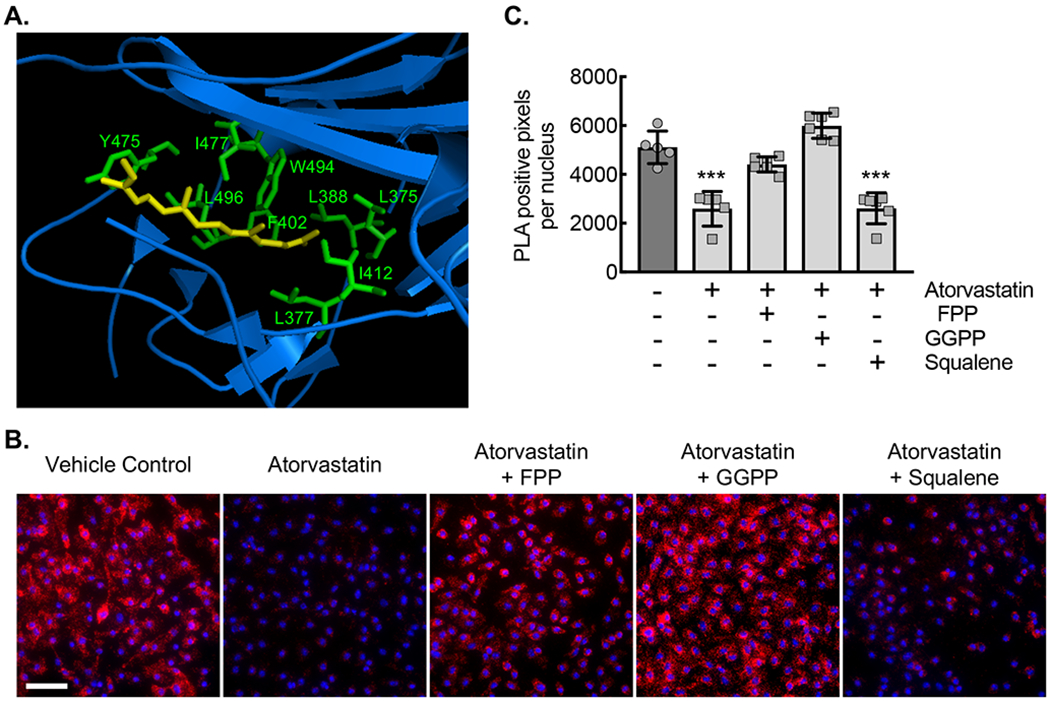
(A) Ribbon diagram modeling the interaction between GDP-bound (inactive) Rac1 and Rho-GDI. Yellow arrow denotes the isoprenyl group sitting in a hydrophobic pocket. Hydrophobic amino acids labeled in green. (B) Micrographs of ApoE−/− BMDMs incubated with vehicle control (DMSO) or with atorvastatin (10 μM) without or with either FPP (5 μM), GGPP (5 μM), or squalene (5 μM) for 24 hours, followed by proximity ligation assay to detect the Rho-GDI-Rac1 complex (red signal). Counterstaining with DAPI (blue). Bar, 50 μm. (C) Quantification of PLA signal from conditions in panel C (***, P<0.0001 ANOVA; n=6 biological replicates from 3 mice). Quantitative data are displayed as mean ± SD.
Macrophage treatment with statin led to increased Rac1 activation.
Next, we sought to determine whether treatment of primary mouse ApoE−/− BMDMs with atorvastatin would lead to increased Rac1 activation analogous to the human monocytes from patients on statin therapy. Using an activated Rac1 affinity precipitation assay, lysates from BMDMs incubated with atorvastatin for 24 hours demonstrated significantly increased levels of GTP-bound (active) Rac1 in an atorvastatin dose-dependent manner. (Fig. 3A,B). Using the monoclonal antibody specific to activated-Rac1, we confirmed that relative immunofluorescence in ApoE−/− BMDMs treated with 10 μM atorvastatin was also increased. (Fig. 3C,D). The increase in GTP-bound Rac1 caused by atorvastatin treatment was prevented by isoprenyl precursor rescue, partially by FPP and completely by GGPP (Fig. 3E,F). The cholesterol synthesis precursor squalene had no effect on the atorvastatin-induced activation of Rac1.
Figure 3. Statin treatment of BMDMs led to an isoprenylation-dependent increase in Rac1 activation.
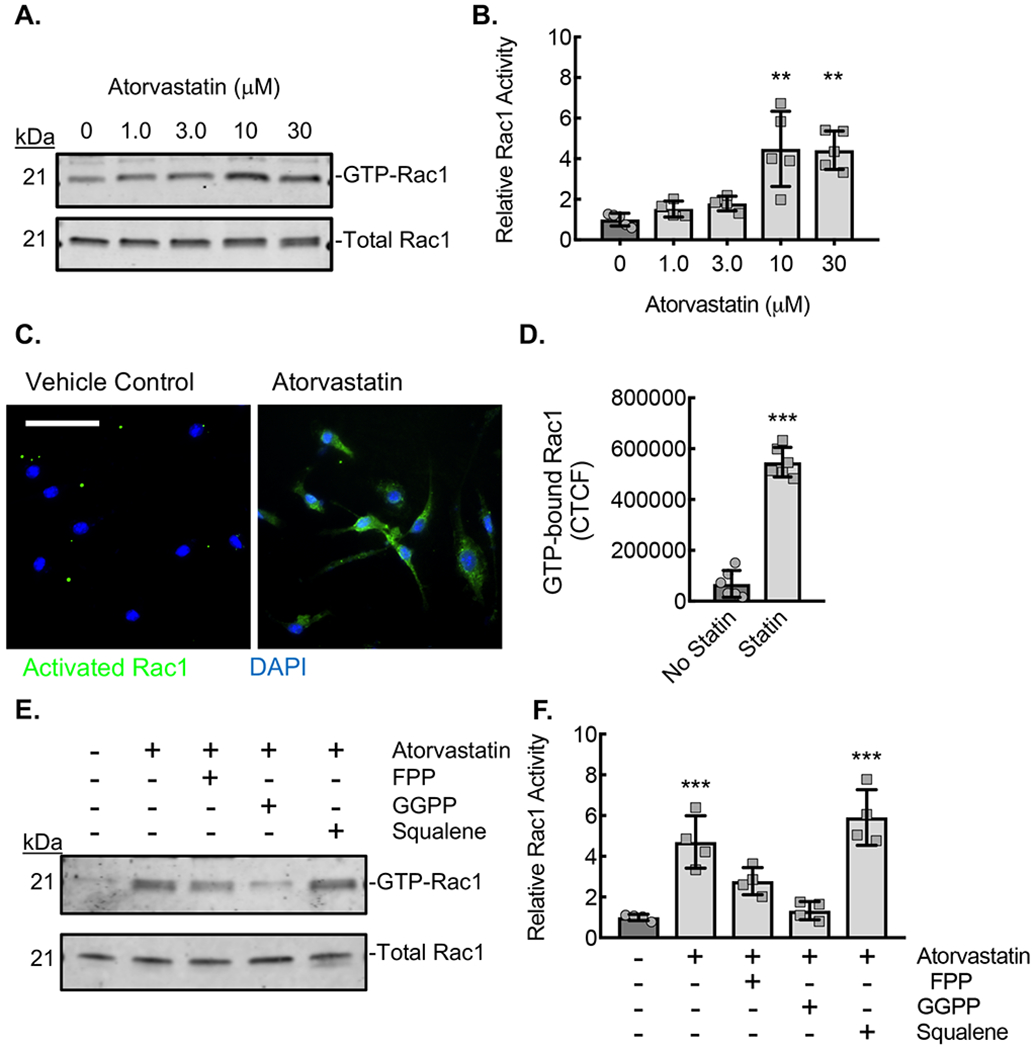
(A) Rac1 immunoblots from lysates of ApoE−/− BMDMs incubated with the indicated concentrations of atorvastatin for 24 hours, after which active (GTP-bound) Rac1 was affinity precipitated by PBD pulldown assay. (B) Densitometry quantification of the relative activated (GTP-bound) Rac1 to total Rac1 in lysates from panel A (**, P<0.0002; n=5 biological replicates from 5 mice; 3 male and 2 female). (C) Micrographs from BMDMs treated with atorvastatin (10 μM) and stained with antibody specific to activated (GTP-bound) Rac1 (green). Counterstaining with DAPI (blue). Bar, 50 μm. (D) Quantification of the Corrected Total Cell Fluorescence (CTCF) for active Rac1from conditions in panel C (***, P<0.0001 t-test; n=6 biological replicates from 3 mice; 2 male and 1 female). (E) Rac1 immunoblots from lysates of ApoE−/− BMDMs incubated with vehicle control (DMSO) or atorvastatin (10 μM) without or with either FPP (5 μM), GGPP (5 μM), or squalene (5 μM) for 24 hours, after which GTP-bound Rac1 was affinity precipitated by PBD pulldown. (F) Densitometry quantification of the relative active (GTP-bound) Rac1 to total Rac1 in lysates from panel E (**, P<0.0001; n=4 biological replicates from 4 mice; 2 male and 2 female). Quantitative data are displayed as mean ± SD.
Macrophage treatment with statin led to an increase in IL-1β expression that could be prevented by supplementation with isoprenyl precursors in a Rac1-dependent manner.
We previously demonstrated that increased Rac1 activation is associated with increased macrophage IL-1β expression in the setting of an inflammasome stimulation assay using a combination of LPS-priming and cholesterol crystal exposure35. Because statin treatment increased activation of Rac1, we sought to determine whether there were consequent increases in IL-1β production. We evaluated IL-1β expression from ApoE−/− BMDMs cultured under inflammasome stimulation conditions, involving LPS-priming with cholesterol crystal exposure, in the setting or absence of atorvastatin (Fig. 4A,B). As expected, we found BMDM IL-1β mRNA expression and mature IL-1β protein secretion into the culture media to be increased in a manner specific to the combination of low dose (10 ng/mL) LPS and cholesterol crystal exposure relative to either LPS or cholesterol crystal exposure alone. Moreover, treatment of the ApoE−/− BMDMs with 10 μM atorvastatin during the inflammasome stimulation led to significantly increased BMDM IL-1β mRNA expression and mature IL-1β protein secretion. In order to confirm the specificity of this statin-induced increase in IL-1β expression, we evaluated a second marker of general inflammation, TNF-α, and found that TNF-α expression was comparable between the vehicle control-treated and statin treated samples both at the mRNA and secreted protein levels (Fig. 4C,D). Finally, under conditions of the combination of low dose LPS and cholesterol crystal exposure, we demonstrated a statin concentration-dependent increase in mature IL-1β secretion into the culture media (Fig. 4E).
Figure 4. Statin treatment of BMDMs led to an Rac1 isoprenylation-dependent increase in IL-1β expression.
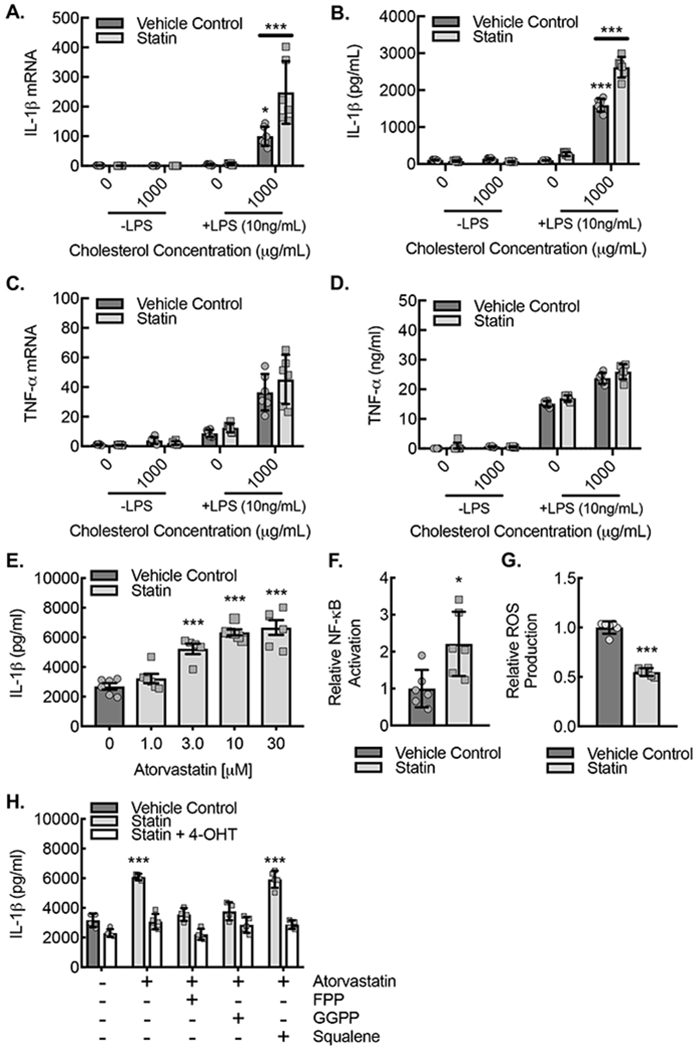
(A) ApoE−/− BMDMs were pretreated for 24 hours with or without atorvastatin (10 μM) and then primed with or without LPS (10 ng/ml) followed by exposure to cholesterol crystals (1000 μg/mL) or vehicle control for another 24 hours, and cell lysates underwent real-time PCR quantification of IL-1β mRNA (***, P<0.0001 ANOVA; n=6 biological replicates from 6 mice; 3 male and 3 female). (B) ApoE−/− BMDMs were treated as in (A) followed by by ELISA on culture supernatants for IL-1β (***, P<0.0001 ANOVA; n=6 biological replicates from 6 mice; 3 male and 3 female). (C) ApoE−/− BMDMs were treated as in (A) followed by real-time PCR quantification of TNF-α mRNA from cell lysates. (D) ApoE−/− BMDMs were treated as in (A) followed by ELISA on culture supernatants for TNF-α. (E) Resting or LPS-primed (10 ng/ml) ApoE−/− BMDMs were treated with or without 1000 μg/mL cholesterol crystals for 24 hours in the setting of indicated atorvastatin concentrations followed by ELISA on culture supernatants for IL-1β (**, P<0.0001 ANOVA; n=6 biological replicates from 6 mice; 3 male and 3 female). (F) Relative luciferase activity in lysates from BMDMs transfected with a NF-κB responsive luciferase construct and the treated with vehicle control (DMSO) or atorvastatin (10 μM) for 24 hours (*, P<0.05 t-test; n=6 biological replicates from 6 mice; 3 male and 3 female). (G) Relative luminescence as a measure of ROS production in BMDMs treated with vehicle control (DMSO) or atorvastatin (10 μM) for 24 hours (***, P<0.0001 t-test; n=6 biological replicates from 6 mice; 3 male and 3 female).
(I) BMDMs from ApoE−/−CSF1RmcmRac1fl/fl mice were treated with vehicle control or atorvastatin (10 μM) with or without either FPP (5 μM), GGPP (5 μM), or squalene (5 μM) supplementation for 24 hours with or without 4-hydroxytamoxifen (4-OHT) to induce Rac1 deletion and then were primed with or without LPS (10 ng/ml) and exposed to cholesterol crystals (1000 μg/mL) or vehicle control for 24 hours followed by ELISA on culture supernatants for IL-1β (**, P=0.0001; n=6 biological replicates from 3 mice; 2 male and 1 female). Quantitative data are displayed as mean ± SD.
NF-κB activation and reactive oxygen species (ROS) production are downstream effectors of Rac1 activity that promote priming and activation of the NLRP3 inflammasome protein complex that promotes IL-1β maturation28, 39, 59. Therefore, we sought to assess the effects of macrophage statin treatment on NF-κB activation and ROS production. Lysates from statin-treated BMDMs cultured in the setting of inflammasome stimulation with low-dose LPS priming coupled to cholesterol crystal exposure demonstrated increased NF-κB activation but reduced ROS production (Fig. 4G,H).
Given that statin treatment can potentially influence the isoprenylation of multiple small GTPases, we sought to understand whether the statin-induced increases in IL-1β expression were dependent on Rac1, specifically. We crossed mice that express a tamoxifen inducible Cre recombinase in myeloid cells, CSF1Rmcm/+, with Rac1fl/fl mice to create mice with inducible macrophage deletion of Rac1 on an ApoE−/− background46, 47. BMDMs from ApoE−/−CSF1Rmcm/+Rac1fl/fl mice were treated with or without 4-hydroxytamoxifen (4-OH) and then stimulated with low-dose LPS plus cholesterol crystal exposure in the absence or presence of 10 μM atorvastatin along with isoprenyl and cholesterol synthesis precursors (Fig. 4H,I). In the vehicle control treated BMDMs, the statin-dependent increase in IL-1β production could be reduced toward baseline with isoprenyl precursors, FPP and GGPP, but not with cholesterol synthesis precursor, squalene, indicating the statin-induced increase in IL-1β secretion to be due to a loss of protein isoprenylation. Moreover, genetic deletion of Rac1 abrogated the statin induced increase in IL-1β secretion and any effects of the isoprenyl precursors at reducing IL-1β secretion, indicating that the statin induced increases in IL-1β secretion were in fact dependent on Rac1 and specifically on Rac1’s isoprenylation state.
Mice fed a HFD supplemented with atorvastatin revealed comparable cholesterol profile and lipid plaque burden.
We next sought to evaluate the effects of statin treatment on the ApoE−/−CSF1Rmcm/+Rac1fl/fl animal model of atherosclerosis. Vehicle control injected animals were fed a high fat diet (20% by fat with 1.25% cholesterol) supplemented without (HFD) or with atorvastatin (HFD+Statin) in the chow at a dose 60 mg/kg chow dry weight. A subgroup of animals on HFD+Statin also underwent a tamoxifen injections (2 mg intraperitoneally daily for 10 days) to induce myeloid Rac1 deletion prior to being fed in order to assess the impact of macrophage Rac1 expression on the statin-induced phenotype (HFD+Statin+mR1KO). All three groups (HFD, HFD+Statin, and HFD+Statin+mR1KO) were fed for 18 weeks. Animals on HFD+Statin demonstrated similar serum lipid panels, including total cholesterol, triglycerides, HDL, and LDL relative to animals on HFD (Fig. 5A–D). Animals on HFD+Statin+mR1KO demonstrated a modest increase in total cholesterol, triglycerides, and LDL. Aortas were harvested from the three groups of mice and demonstrated comparable levels of atherosclerotic lipid burden by en face Oil Red O staining (Fig. 5E,F). Moreover, traditional histological sectioning and staining at the level of the aortic sinus demonstrated comparable sinus plaque area and sinus plaque collagen area by hematoxylin and eosin and Masson’s trichrome staining in all three groups, HFD, HFD+Statin, and HFD+Statin+mR1KO, whereas statin treatment demonstrated a Rac1-dependent increase in sinus plaque calcium area by Alizarin Red staining (Fig. 5G–J).
Figure 5. ApoE−/−CSF1RmcmRac1fl/fl mice fed a HFD supplemented with atorvastatin revealed comparable cholesterol profile and plaque burden relative to those on HFD alone.
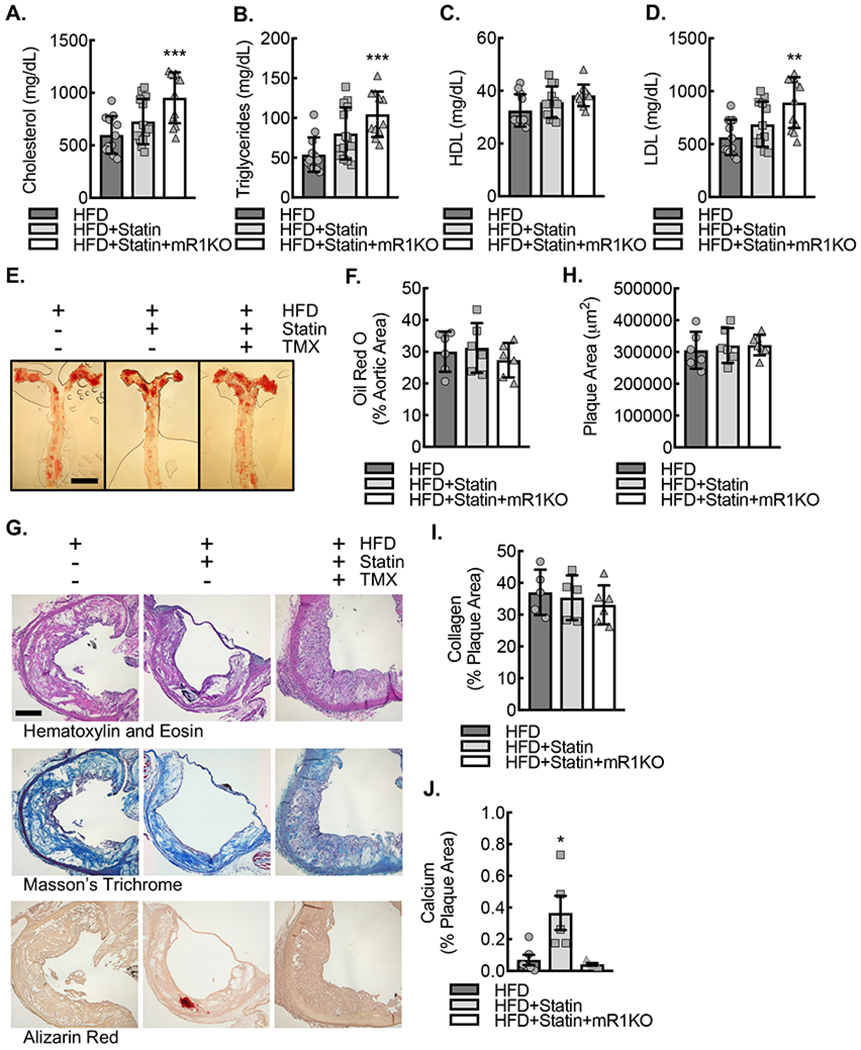
Serum total cholesterol (A), triglycerides (B), HDL (C), and LDL (D) concentrations after 18 weeks of HFD, HFD+Statin or HFD+Statin+mR1KO (**, P<0.002, ***, P<0.001 relative to HFD by ANOVA; n=12 mice; 6 male and 6 female). (E) En face Oil Red O lipid staining of aortic whole mounts after 18 weeks of HFD, HFD+Statin, or HFD+Statin+mR1KO, along with quantification (F) of percent aortic area stained positive for Oil Red O. Bar, 3mm. (G) Standard histology stained for hematoxylin and eosin, Masson’s trichrome, and Alizarin red of adjacent aortic sinus sections at the level of the aortic valve after 18 weeks of HFD, HFD+Statin, or HFD+Statin+mR1KO. Bar, 200 μm. (H) Quantification of average aortic plaque area by hematoxylin and eosin. (I) Quantification of percent plaque area staining positive for collagen by Masson’s trichrome. (J) Quantification of percent plaque area staining positive for Alizarin red. (*, P=0.012 by ANOVA; n=6 mice; 3 male and 3 female). Quantitative data are displayed as mean ± SD.
Mice fed a HFD supplemented with atorvastatin demonstrated myeloid Rac1-dependent elevations of global atherosclerotic calcification and of serum IL-1β protein.
Despite similarities of serum lipid panel and atherosclerotic lipid burden, there was histological evidence of increased atherosclerotic calcification in the mice on HFD+Statin, using Alizarin Red staining. We next assessed global levels of active atherosclerotic calcification in the aortas of the mice using a commercially available near-infrared conjugated bisphosphonate compound. Near-infrared conjugated bisphosphonate compounds can bind to hydroxyapatite deposited by osteoblast-like cells during the mineralization of calcium, allowing for ex vivo molecular imaging and quantification of the calcification process60, 61. Overall, the mice fed a HFD+Statin demonstrated increased aortic atherosclerotic calcification relative to animals on HFD, and myeloid Rac1 deletion abrogated the effect, supporting the statin-induced increase in atherosclerotic calcification to be Rac1-dependent (Fig. 6A,B). Moreover, the serum from statin-treated animals demonstrated Rac1-dependent increases in IL-1β protein levels (Fig. 6C). This statin-dependent increase in serum IL-1β appeared specific, whereas another general inflammatory cytokine, TNF-α, was comparable between HFD, HFD+Statin, and HFD+Statin+mR1KO treated animals (Fig. 6D).
Figure 6. ApoE−/−CSF1RmcmRac1fl/fl mice fed a HFD supplemented with atorvastatin demonstrate Rac1 dependent increases in both atherosclerotic calcification and systemic IL-1β expression.

(A) Ex vivo near-infrared calcium imaging from mice on normal chow (NC), HFD, HFD+Statin or HFD+Statin+mR1KO for 18 weeks along with quantification (B) of calcification signal relative to total aorta area (***, P<0.0001 relative to NC by ANOVA; ψ, P<0.0001 relative to all others by ANOVA; n= 12 mice; 6 male and 6 female). (C) Serum IL-1β concentrations by ELISA from ApoE−/−CSF1RmcmRac1fl/fl mice after 18 weeks of NC, HFD, HFD+Statin or HFD+Statin+mR1KO (***, P<0.0001 relative to NC by ANOVA; ϕ, P=0.005 relative to all others by ANOVA; n= 12 mice; 6 male and 6 female). (D) Serum TNF-α concentrations by ELISA from ApoE−/−CSF1RmcmRac1fl/fl mice after 18 weeks of NC, HFD, HFD+Statin or HFD+Statin+mR1KO (***, P<0.0001 relative to NC by ANOVA; n= 12 mice; 6 male and 6 female). Quantitative data are displayed as mean ± SD.
Of note, sex was analyzed as a biological variable for lipid panel, serum IL-1β, and near infrared calcification imaging using ANOVA, and we found no sex-dependent changes in our hands in this preclinical model, indicating that this type of Rac1-dependent increase in IL-1β and atherosclerotic calcification appears comparably relevant to both sexes (Supplemental Fig. I).
Statin treatment demonstrated increased plaque expression of activated (GTP-bound) Rac1 along with increased expression of osteogenic markers, ALP and RUNX2, in atherosclerotic lesions.
The inner aortic arch neointimal lesions of animals fed a HFD+Statin demonstrated increased expression of activated (GTP-bound) Rac1 which was abrogated by myeloid Rac1 deletion, consistent with our in vitro findings that statin induces activation of Rac1 in macrophages (Fig. 7A,B). Finally, neointimal plaque staining for the osteoblast markers, RUNX2 and alkaline phosphatase (ALP), were increased in the mice fed a HFD+Statin, and this increase was abrogated by myeloid Rac1 deletion, consistent with a myeloid Rac1-dependent expression of these markers (Fig. 7C–F).
Figure 7. ApoE−/−CSF1RmcmRac1fl/fl mice fed a HFD supplemented with atorvastatin demonstrate Rac1 dependent increases activated Rac1, ALP, and RUNX2 expression.
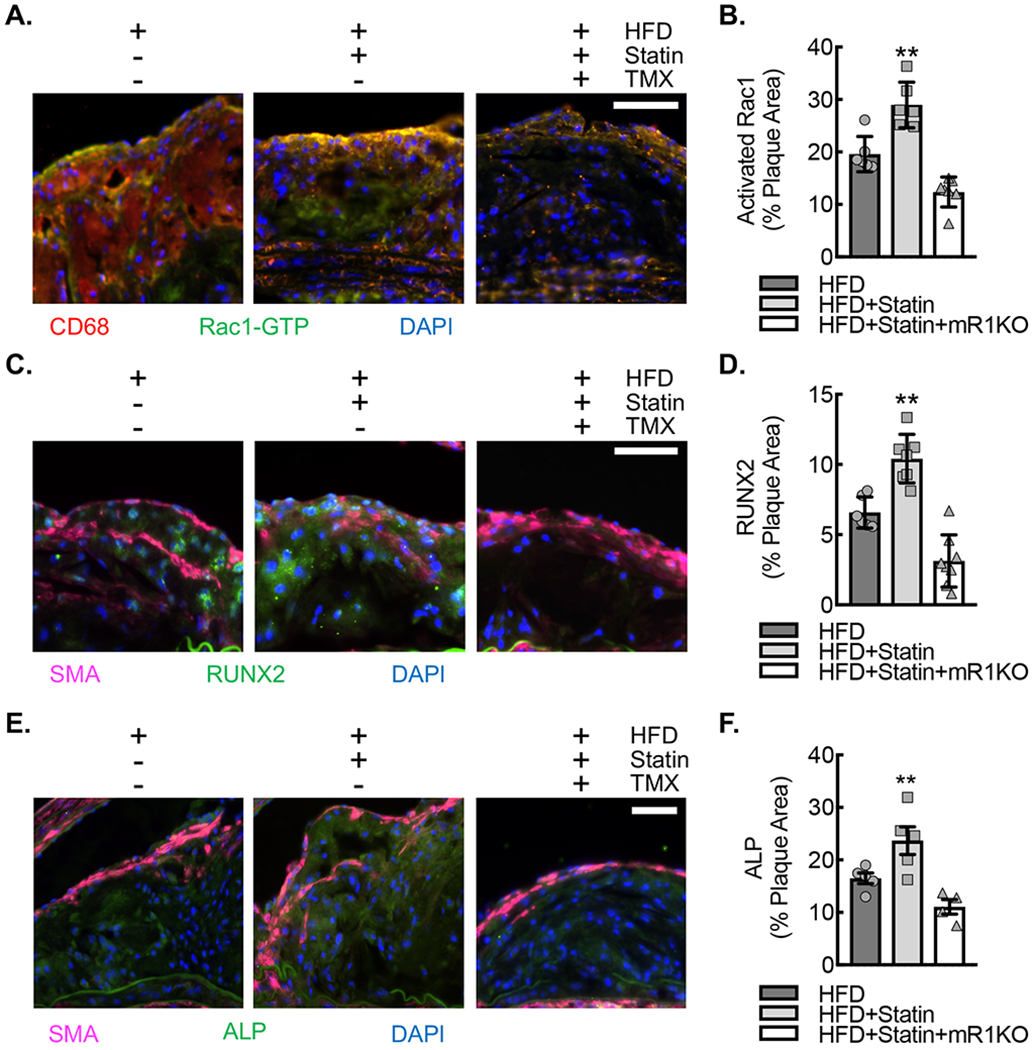
(A) Immunofluorescence micrographs of neointimal lesions in the ascending aortic arch lesser curvature of ApoE−/−CSF1RmcmRac1fl/fl mice fed HFD, HFD+Statin or HFD+Statin+mR1KO for 18 weeks that were stained for GTP-bound (activated) Rac1 (green) and CD68 (red). Nuclei counterstained with DAPI (blue). Bar, 50 μm. (B) Quantification of percent plaque area staining positive for activated Rac1 expression in aortic arch plaques from A (**, P<0.005 relative to others by ANOVA; n=6 animals; 3 male and 3 female). (C) Immunofluorescence micrographs of neointimal lesions derived as described in (A) that were stained for ALP (green) and SMA (magenta). Nuclei counterstained with DAPI (blue). Bar, 50 μm. (D) Quantification percent plaque area staining positive for ALP expression (*, P<0.05 relative to others by ANOVA; n=6 ; 3 male and 3 female). (E) Immunofluorescence micrographs of neointimal lesions derived as described in (A) that were stained for RUNX2 (green) and SMA (magenta). Nuclei counterstained with DAPI (blue). Bar, 50 μm. (F) Quantification percent plaque area staining positive for RUNX2 expression (**, P<0.005 relative to others by ANOVA; n=6; 3 male and 3 female). Quantitative data are displayed as mean ± SD.
Discussion:
Calcification of atherosclerotic plaque is common in human disease and has been identified as a marker of disease burden, and is associated with increased risk of cardiovascular events2, 3. However, recent data has demonstrated that the ultrastructure of calcium within individual plaques may predict vulnerability in some situations whereas stability in others, indicating there are likely additional factors that couple with calcification to alleviate or worsen risk3, 6–8, 62. Statins have had tremendous success as a family of therapeutic agents that can reduce cardiovascular events, but there is accumulating evidence that these pleiotropic effects are out of proportion to their lipid lowering effects, supporting the possibility that additional molecular mechanisms are at play in the stabilization of plaque16–20. Moreover, there has been recent controversy over whether statins can influence the calcium composition of atherosclerotic plaque with a growing pool of clinical data demonstrating that high-intensity statins may in fact increase atherosclerotic plaque calcification21–26. The potential molecular mechanisms of how statins may influence plaque calcification are largely unknown. In our database of patients who have undergone LCSCT, statin use was independently associated with increased coronary calcification when adjusted for other cardiovascular risk factors. Limitations of our clinical study include it being a single site, retrospective analysis of primarily male veterans, increasing risk of a selection bias. However, our findings are consistent with other clinical studies that demonstrate increased plaque calcification with statin use, supporting the importance of further study in this area21–26.
Recently, we identified Rac signaling to be a critical mediator of macrophage IL-1β expression and consequent IL-1β-mediated atherosclerotic calcification. To understand the potential molecular mechanisms whereby statins may influence atherosclerotic plaque calcification, we assessed the impact of statins on inflammatory monocyte Rac1 activity in patients followed by more mechanistic studies of macrophage Rac1 activity and Rac-mediated IL-1β expression in mice. Here we identified statin-induced inhibition of macrophage Rac1 isoprenylation to be a critical mediator of its complex formation with RhoGDI, resulting in increased Rac1 activation, increased activation of the Rac1 effector, NF-kB, and consequently increased IL-1β mRNA and mature protein production upon stimulation of the NLRP3 inflammasome. The data are supported by the ability of FPP to partially rescue and of GGPP to fully rescue the statin mediated effects on Rac1 activity and IL-1β expression, whereas squalene, which lies downstream of FPP in the cholesterol synthesis pathway, was unable to mitigate the statin mediated effects. Though statins are known to reduce the isoprenylation state of multiple Rac family members, conditional Rac1 knockdown, using an inducible myeloid specific promoter for Cre recombinase expression crossed with a loxP flanked Rac1, confirmed the statin-mediated effect on IL-1β expression to be specific to macrophage Rac1 isoprenylation.
Moreover, our in vivo studies using animals with inducible myeloid Rac1 deletion and HFD supplemented with atorvastatin further demonstrate statin use to be a determinant for the degree of atherosclerotic plaque calcification in a macrophage Rac1-dependent manner. We chose a dose of statin in the chow that appeared to impact the immune response but did not impact chronic lipid metabolism as cholesterol and plaque lipid burden were relatively similar between treatment groups. The estimated daily mouse ad libitum dosing of atorvastatin at 6 mg/kg was near the recommended high intensity statin dosing for humans (1.07 mg/kg), and consistent with other studies that suggest a higher dose is required to have a more significant impact on cholesterol and atherosclerosis burden in mice fed this type of high fat diet for over 20 weeks63. Importantly, this statin dose was associated with increased circulating IL-1β expression and increased plaque ALP and RUNX2 expression, consistent with our prior studies demonstrating that increased IL-1β expression appears to trigger an osteoblast-like differentiation of vascular smooth muscle cells leading to calcifying plaque35. Prior literature supports that a close association between inflammatory macrophages and calcifying vascular cells appears required to activate vascular smooth muscle cell to osteoblast-like phenotypic conversion, and a number of those studies involve macrophage-secreted factors like TNF-α, or in the case of our prior study, IL-1β35, 64–69. In our previous work, increased vascular smooth muscle cell osteogenic transcription factor expression, including RUNX2, SOX9, OSX, and MSX2, in response to IL-1β raised the question of possible directs mechanism of IL-1R signaling in the promotion of an osteogenic program in mesenchymal cells35. Further studies are required to address the molecular mechanisms that determine the IL-1β responsiveness along with consequent changes in gene programing in vascular smooth muscle cells in vivo. Here, we focused on mechanisms upstream of IL-1β that may be impacted by statin therapy, specifically Rac1-dependent IL-1β expression.
Our mechanistic study outlines a critical upstream signaling mechanism that may provide a fundamental understanding of the potential impact of statin therapy on immune responses within the plaque. Because statins are associated with reduced outcomes and plaque stabilization, the question naturally arises as to whether both Rac1-dependent IL-1β expression and atherosclerotic calcification are “good” or “bad”, forcing us to re-evaluate this binary perception of neointimal inflammation and calcification in the context of both the heterogeneous nature of plaques and the overall patient risk. In our prior studies of atherosclerotic calcification, we had found elevated levels of ROS associated with elevated Rac-IL-1β signaling, whereas in the setting of statin use, ROS production was reduced, consistent with other studies demonstrating a statin-mediated reduction of ROS70–72. It is possible that while some aspects of the immune response are elevated, others are blunted and the summation is a plaque with altered phenotypic features that favor reduced vulnerability to rupture.
Traditionally, plaque inflammation and calcification have been considered to be biologic processes associated with increased atherosclerotic plaque vulnerability and adverse cardiovascular events, but a number of recent studies have demonstrated that the relationship between statins, inflammation, and calcification may be much more complicated. The CANTOS trial recently demonstrated a significant reduction in secondary cardiovascular events with inhibition of the potent inflammatory cytokine, interleukin-1β (IL-1β), supporting the inflammation hypothesis of atherosclerosis73. However, it is important to note that the overall benefit to all-cause and cardiovascular mortality was restricted to a subset of patients who achieved reductions in the systemic inflammation marker, hsCRP74. Moreover, 93.4% of patients in the CANTOS trial were already on lipid lowering therapy, a majority of which were statins, indicating that the results of the CANTOS trial reflect, in part, the addition of systemic IL-1β inhibition to systemic statin therapy. In addition, there has been some recent evidence that certain types of densely calcified plaques are associated with more stable disease and that statin use may be associated with expansion of this densely calcified stable plaque phenotype3, 6–10, 22, 25, 26. Adding to the controversy, there is growing data in preclinical models that IL-1β antibody treatment (in absence of statin therapy) causes plaques with decreased lesion size but remodeling of the fibrous cap with increased macrophage content in a manner consistent with histological features of an increased vulnerable plaque phenotype75. This vulnerable histological phenotype has been supported by global and smooth muscle cell selective IL-1R gene knock down models as well75–77. Clearly, these conflicting studies around IL-1β indicate a need for more research dedicated to understanding the key cellular players and cellular signaling events involved in IL-1β-IL-1R signal-mediated atherosclerotic calcification both in the presence and absence of statin therapy, as overlapping pathways albeit with some likely variations analogous to the changes we identified with regard to ROS may be involved.
One major limitation to basic research in this area has been the lack of a standardized small animal model for plaque rupture that allows further dissection of potential molecular mechanisms determining plaque vulnerability. Thus, we are not able to comment on whether this statin-mediated mechanism of increased calcification in the setting of reduced ROS production can lead to plaque stabilization. Similar questions can be raised about inhibiting IL-1β and/or consequent IL-1R signaling in animal models leading to alterations in the histological features of plaque.
In summary, we find that statin use is independently associated with increased calcification in patients, and using an animal model of hypercholesterolemia, we present the first potential molecular mechanism whereby statins may promote calcification of atherosclerotic plaque through macrophage Rac1-dependent IL-1β expression. Modulation of Rac1-dependent IL-1β expression to influence plaque calcium composition and ultimately plaque phenotype may help pave the way for the development of a novel therapeutic strategy that can mitigate cardiovascular risk, as inhibitors of Rac1 and IL-1β already exist and some are already in use in clinical practice.
Supplementary Material
Highlights.
Statin therapy is associated with higher coronary artery calcium scores and increased macrophage Rac1 activity in patients.
Statins disrupt the complex between Rac1 and its primary inhibitor, RhoGDI, in macrophages, leading to Rac1 activation and Rac1-dependent IL-1β expression.
During experimental atherosclerosis, statin therapy leads to macrophage Rac1-dependent increases in IL-1β and atherosclerotic plaque calcification.
Acknowledgments:
Research reported in this publication was supported by a Research Project Grant NIH NHLBI R01HL139795 (A.R.M.) and an Institutional Development Award (IDeA) from NIH NIGMS P20GM103652 (A.R.M.) and. This work was also supported by Career Development Award Number 7IK2BX002527 from the United States Department of Veterans Affairs Biomedical Laboratory Research and Development Program (A.R.M.). GC is supported by R01HL128661, I01CX001892 and R01HL148727. The views expressed in this article are those of the authors and do not necessarily reflect the position or policy of the Department of Veterans Affairs or the United States government. This work was also supported in part by NIH NHLBI R25HL088992 (R.N. and J.N.).
Nonstandard Abbreviations and Acronyms:
- ALP
Alkaline phosphatase
- ASCVD
Atherosclerotic cardiovascular disease
- BMDM
Bone marrow derived macrophage
- BMI
Body mass index
- CABG
Coronary artery bypass graft surgery
- CACS
Coronary artery calcification score
- CAD
Coronary artery disease
- CD68
Cluster of Differentiation 68
- CT
Computed tomography
- DAPI
4′,6-diamidino-2-phenylindole
- DMSO
Dimethyl sulfoxide
- ELISA
Enzyme-linked immunosorbent assay
- FPP
Farnesyl diphosphate
- GDI
GDP-dissociation inhibitor
- GDP
Guanosine diphosphate
- GEF
Guanine nucleotide exchange factor
- GGPP
Geranylgeranyl diphosphate
- GTP
Guanosine diphosphate
- HFD
High fat diet (20%) supplemented by 1.25% cholesterol
- HMCOA
3-hydroxy-3-methylglutaryl coenzymeA
- IL-1β
Interleukin-1 beta
- IL-1R
Interleukin-1 Receptor
- LCSCT
Lung cancer screening CT
- LPS
Lipopolysaccharide
- MI
Myocardial infarction
- NF-κB
Nuclear factor kappa-light-chain-enhancer of activated B cells
- NLRP3
NACHT, LRR and PYD domains-containing protein 3
- PLA
Proximity ligation assay
- RhoGDI
Rho GDP-dissociation inhibitor
- ROS
Reactive oxygen species
- SMA
Smooth muscle actin
- SMC
Smooth muscle cell
- TNF-α
Tumor necrosis factor alpha
Footnotes
Conflicts of Interest: The authors have no disclosures to declare.
REFERENCES
- 1.“The top 10 causes of death.” World Health Organization WHO Press, Geneva, 24 May 2018, https://www.who.int/en/news-room/fact-sheets/detail/the-top-10-causes-of-death [Google Scholar]
- 2.Budoff MJ, Mao S, Zalace CP, Bakhsheshi H, Oudiz RJ. Comparison of spiral and electron beam tomography in the evaluation of coronary calcification in asymptomatic persons. International journal of cardiology. 2001;77:181–188 [DOI] [PubMed] [Google Scholar]
- 3.Rennenberg RJ, Kessels AG, Schurgers LJ, van Engelshoven JM, de Leeuw PW, Kroon AA. Vascular calcifications as a marker of increased cardiovascular risk: A meta-analysis. Vascular health and risk management. 2009;5:185–197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Raggi P, Callister TQ, Shaw LJ. Progression of coronary artery calcium and risk of first myocardial infarction in patients receiving cholesterol-lowering therapy. Arteriosclerosis, thrombosis, and vascular biology. 2004;24:1272–1277 [DOI] [PubMed] [Google Scholar]
- 5.Raggi P, Cooil B, Shaw LJ, Aboulhson J, Takasu J, Budoff M, Callister TQ. Progression of coronary calcium on serial electron beam tomographic scanning is greater in patients with future myocardial infarction. The American journal of cardiology. 2003;92:827–829 [DOI] [PubMed] [Google Scholar]
- 6.Criqui MH, Denenberg JO, Ix JH, McClelland RL, Wassel CL, Rifkin DE, Carr JJ, Budoff MJ, Allison MA. Calcium density of coronary artery plaque and risk of incident cardiovascular events. JAMA : the journal of the American Medical Association. 2014;311:271–278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Motoyama S, Sarai M, Harigaya H, Anno H, Inoue K, Hara T, Naruse H, Ishii J, Hishida H, Wong ND, Virmani R, Kondo T, Ozaki Y, Narula J. Computed tomographic angiography characteristics of atherosclerotic plaques subsequently resulting in acute coronary syndrome. Journal of the American College of Cardiology. 2009;54:49–57 [DOI] [PubMed] [Google Scholar]
- 8.Puchner SB, Liu T, Mayrhofer T, Truong QA, Lee H, Fleg JL, Nagurney JT, Udelson JE, Hoffmann U, Ferencik M. High-risk plaque detected on coronary ct angiography predicts acute coronary syndromes independent of significant stenosis in acute chest pain: Results from the romicat-ii trial. Journal of the American College of Cardiology. 2014;64:684–692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Puri R, Nicholls SJ, Shao M, Kataoka Y, Uno K, Kapadia SR, Tuzcu EM, Nissen SE. Impact of statins on serial coronary calcification during atheroma progression and regression. Journal of the American College of Cardiology. 2015;65:1273–1282 [DOI] [PubMed] [Google Scholar]
- 10.Irkle A, Vesey AT, Lewis DY, Skepper JN, Bird JL, Dweck MR, Joshi FR, Gallagher FA, Warburton EA, Bennett MR, Brindle KM, Newby DE, Rudd JH, Davenport AP. Identifying active vascular microcalcification by (18)f-sodium fluoride positron emission tomography. Nat Commun. 2015;6:7495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Stone NJ, Robinson JG, Lichtenstein AH, Bairey Merz CN, Blum CB, Eckel RH, Goldberg AC, Gordon D, Levy D, Lloyd-Jones DM, McBride P, Schwartz JS, Shero ST, Smith SC Jr., Watson K, Wilson PW, Eddleman KM, Jarrett NM, LaBresh K, Nevo L, Wnek J, Anderson JL, Halperin JL, Albert NM, Bozkurt B, Brindis RG, Curtis LH, DeMets D, Hochman JS, Kovacs RJ, Ohman EM, Pressler SJ, Sellke FW, Shen WK, Smith SC Jr., Tomaselli GF, American College of Cardiology/American Heart Association Task Force on Practice G. 2013 acc/aha guideline on the treatment of blood cholesterol to reduce atherosclerotic cardiovascular risk in adults: A report of the american college of cardiology/american heart association task force on practice guidelines. Circulation. 2014;129:S1–45 [DOI] [PubMed] [Google Scholar]
- 12.Ridker PM, Danielson E, Fonseca FA, Genest J, Gotto AM Jr., Kastelein JJ, Koenig W, Libby P, Lorenzatti AJ, MacFadyen JG, Nordestgaard BG, Shepherd J, Willerson JT, Glynn RJ. Rosuvastatin to prevent vascular events in men and women with elevated c-reactive protein. The New England journal of medicine. 2008;359:2195–2207 [DOI] [PubMed] [Google Scholar]
- 13.Taylor F, Huffman MD, Macedo AF, Moore TH, Burke M, Davey Smith G, Ward K, Ebrahim S. Statins for the primary prevention of cardiovascular disease. The Cochrane database of systematic reviews. 2013;1:CD004816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Downs JR, Clearfield M, Weis S, Whitney E, Shapiro DR, Beere PA, Langendorfer A, Stein EA, Kruyer W, Gotto AM Jr. Primary prevention of acute coronary events with lovastatin in men and women with average cholesterol levels: Results of afcaps/texcaps. Air force/texas coronary atherosclerosis prevention study. JAMA : the journal of the American Medical Association. 1998;279:1615–1622 [DOI] [PubMed] [Google Scholar]
- 15.Shepherd J, Cobbe SM, Ford I, Isles CG, Lorimer AR, MacFarlane PW, McKillop JH, Packard CJ. Prevention of coronary heart disease with pravastatin in men with hypercholesterolemia. West of scotland coronary prevention study group. The New England journal of medicine. 1995;333:1301–1307 [DOI] [PubMed] [Google Scholar]
- 16.Heart Protection Study Collaborative G. Mrc/bhf heart protection study of cholesterol lowering with simvastatin in 20,536 high-risk individuals: A randomised placebo-controlled trial. Lancet. 2002;360:7–22 [DOI] [PubMed] [Google Scholar]
- 17.Ridker PM. Inflammatory biomarkers, statins, and the risk of stroke: Cracking a clinical conundrum. Circulation. 2002;105:2583–2585 [DOI] [PubMed] [Google Scholar]
- 18.Schwartz GG, Olsson AG, Ezekowitz MD, Ganz P, Oliver MF, Waters D, Zeiher A, Chaitman BR, Leslie S, Stern T, Myocardial Ischemia Reduction with Aggressive Cholesterol Lowering Study I. Effects of atorvastatin on early recurrent ischemic events in acute coronary syndromes: The miracl study: A randomized controlled trial. JAMA : the journal of the American Medical Association. 2001;285:1711–1718 [DOI] [PubMed] [Google Scholar]
- 19.White HD, Simes RJ, Anderson NE, Hankey GJ, Watson JD, Hunt D, Colquhoun DM, Glasziou P, MacMahon S, Kirby AC, West MJ, Tonkin AM. Pravastatin therapy and the risk of stroke. The New England journal of medicine. 2000;343:317–326 [DOI] [PubMed] [Google Scholar]
- 20.Stone NJ, Robinson JG, Lichtenstein AH, Bairey Merz CN, Blum CB, Eckel RH, Goldberg AC, Gordon D, Levy D, Lloyd-Jones DM, McBride P, Schwartz JS, Shero ST, Smith SC Jr., Watson K, Wilson PW, Eddleman KM, Jarrett NM, LaBresh K, Nevo L, Wnek J, Anderson JL, Halperin JL, Albert NM, Bozkurt B, Brindis RG, Curtis LH, DeMets D, Hochman JS, Kovacs RJ, Ohman EM, Pressler SJ, Sellke FW, Shen WK, Smith SC Jr., Tomaselli GF. 2013 acc/aha guideline on the treatment of blood cholesterol to reduce atherosclerotic cardiovascular risk in adults: A report of the american college of cardiology/american heart association task force on practice guidelines. Circulation. 2014;129:S1–45 [DOI] [PubMed] [Google Scholar]
- 21.Achenbach S, Ropers D, Pohle K, Leber A, Thilo C, Knez A, Menendez T, Maeffert R, Kusus M, Regenfus M, Bickel A, Haberl R, Steinbeck G, Moshage W, Daniel WG. Influence of lipid-lowering therapy on the progression of coronary artery calcification: A prospective evaluation. Circulation. 2002;106:1077–1082 [DOI] [PubMed] [Google Scholar]
- 22.Arad Y, Spadaro LA, Roth M, Newstein D, Guerci AD. Treatment of asymptomatic adults with elevated coronary calcium scores with atorvastatin, vitamin c, and vitamin e: The st. Francis heart study randomized clinical trial. Journal of the American College of Cardiology. 2005;46:166–172 [DOI] [PubMed] [Google Scholar]
- 23.Callister TQ, Raggi P, Cooil B, Lippolis NJ, Russo DJ. Effect of hmg-coa reductase inhibitors on coronary artery disease as assessed by electron-beam computed tomography. The New England journal of medicine. 1998;339:1972–1978 [DOI] [PubMed] [Google Scholar]
- 24.Hecht HS, Harman SM. Relation of aggressiveness of lipid-lowering treatment to changes in calcified plaque burden by electron beam tomography. The American journal of cardiology. 2003;92:334–336 [DOI] [PubMed] [Google Scholar]
- 25.Houslay ES, Cowell SJ, Prescott RJ, Reid J, Burton J, Northridge DB, Boon NA, Newby DE, Scottish Aortic S, Lipid Lowering Therapy IoRtI. Progressive coronary calcification despite intensive lipid-lowering treatment: A randomised controlled trial. Heart. 2006;92:1207–1212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Raggi P, Davidson M, Callister TQ, Welty FK, Bachmann GA, Hecht H, Rumberger JA. Aggressive versus moderate lipid-lowering therapy in hypercholesterolemic postmenopausal women: Beyond endorsed lipid lowering with ebt scanning (belles). Circulation. 2005;112:563–571 [DOI] [PubMed] [Google Scholar]
- 27.Heasman SJ, Ridley AJ. Mammalian rho gtpases: New insights into their functions from in vivo studies. Nature reviews. Molecular cell biology. 2008;9:690–701 [DOI] [PubMed] [Google Scholar]
- 28.Hordijk PL. Regulation of nadph oxidases: The role of rac proteins. Circulation research. 2006;98:453–462 [DOI] [PubMed] [Google Scholar]
- 29.Li B, Yu H, Zheng W, Voll R, Na S, Roberts AW, Williams DA, Davis RJ, Ghosh S, Flavell RA. Role of the guanosine triphosphatase rac2 in t helper 1 cell differentiation. Science. 2000;288:2219–2222 [DOI] [PubMed] [Google Scholar]
- 30.Morrison AR, Yarovinsky TO, Young BD, Moraes F, Ross TD, Ceneri N, Zhang J, Zhuang ZW, Sinusas AJ, Pardi R, Schwartz MA, Simons M, Bender JR. Chemokine-coupled beta2 integrin-induced macrophage rac2-myosin iia interaction regulates vegf-a mrna stability and arteriogenesis. The Journal of experimental medicine. 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Williams LM, Lali F, Willetts K, Balague C, Godessart N, Brennan F, Feldmann M, Foxwell BM. Rac mediates tnf-induced cytokine production via modulation of nf-kappab. Molecular immunology. 2008;45:2446–2454 [DOI] [PubMed] [Google Scholar]
- 32.Didsbury JR, Uhing RJ, Snyderman R. Isoprenylation of the low molecular mass gtp-binding proteins rac 1 and rac 2: Possible role in membrane localization. Biochemical and biophysical research communications. 1990;171:804–812 [DOI] [PubMed] [Google Scholar]
- 33.Filippi MD, Harris CE, Meller J, Gu Y, Zheng Y, Williams DA. Localization of rac2 via the c terminus and aspartic acid 150 specifies superoxide generation, actin polarity and chemotaxis in neutrophils. Nature immunology. 2004;5:744–751 [DOI] [PubMed] [Google Scholar]
- 34.Boulter E, Garcia-Mata R, Guilluy C, Dubash A, Rossi G, Brennwald PJ, Burridge K. Regulation of rho gtpase crosstalk, degradation and activity by rhogdi1. Nature cell biology. 2010;12:477–483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ceneri N, Zhao L, Young BD, Healy A, Coskun S, Vasavada H, Yarovinsky TO, Ike K, Pardi R, Qin L, Qin L, Tellides G, Hirschi K, Meadows J, Soufer R, Chun HJ, Sadeghi MM, Bender JR, Morrison AR. Rac2 modulates atherosclerotic calcification by regulating macrophage interleukin-1beta production. Arteriosclerosis, thrombosis, and vascular biology. 2017;37:328–340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Garcia-Mata R, Boulter E, Burridge K. The ‘invisible hand’: Regulation of rho gtpases by rhogdis. Nature reviews. Molecular cell biology. 2011;12:493–504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zeiser R, Maas K, Youssef S, Durr C, Steinman L, Negrin RS. Regulation of different inflammatory diseases by impacting the mevalonate pathway. Immunology. 2009;127:18–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rashid M, Tawara S, Fukumoto Y, Seto M, Yano K, Shimokawa H. Importance of rac1 signaling pathway inhibition in the pleiotropic effects of hmg-coa reductase inhibitors. Circulation journal : official journal of the Japanese Circulation Society. 2009;73:361–370 [DOI] [PubMed] [Google Scholar]
- 39.Khan OM, Ibrahim MX, Jonsson IM, Karlsson C, Liu M, Sjogren AK, Olofsson FJ, Brisslert M, Andersson S, Ohlsson C, Hulten LM, Bokarewa M, Bergo MO. Geranylgeranyltransferase type i (ggtase-i) deficiency hyperactivates macrophages and induces erosive arthritis in mice. The Journal of clinical investigation. 2011;121:628–639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Agatston AS, Janowitz WR, Hildner FJ, Zusmer NR, Viamonte M Jr., Detrano R. Quantification of coronary artery calcium using ultrafast computed tomography. Journal of the American College of Cardiology. 1990;15:827–832 [DOI] [PubMed] [Google Scholar]
- 41.Carr JJ, Nelson JC, Wong ND, McNitt-Gray M, Arad Y, Jacobs DR Jr., Sidney S, Bild DE, Williams OD, Detrano RC. Calcified coronary artery plaque measurement with cardiac ct in population-based studies: Standardized protocol of multi-ethnic study of atherosclerosis (mesa) and coronary artery risk development in young adults (cardia) study. Radiology. 2005;234:35–43 [DOI] [PubMed] [Google Scholar]
- 42.Sung JH, Yeboah J, Lee JE, Smith CL, Terry JG, Sims M, Samdarshi T, Musani S, Fox E, Ge Y, Wilson JG, Taylor HA, Carr JJ. Diagnostic value of coronary artery calcium score for cardiovascular disease in african americans: The jackson heart study. Br J Med Med Res. 2016;11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bailey G, Healy A, Young BD, Sharma E, Meadows J, Chun HJ, Wu WC, Choudhary G, Morrison AR. Relative predictive value of lung cancer screening ct versus myocardial perfusion attenuation correction ct in the evaluation of coronary calcium. PloS one. 2017;12:e0175678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Marzaioli V, Hurtado-Nedelec M, Pintard C, Tlili A, Marie JC, Monteiro RC, Gougerot-Pocidalo MA, Dang PM, El-Benna J. Nox5 and p22phox are 2 novel regulators of human monocytic differentiation into dendritic cells. Blood. 2017;130:1734–1745 [DOI] [PubMed] [Google Scholar]
- 45.Schneider CA, Rasband WS, Eliceiri KW. Nih image to imagej: 25 years of image analysis. Nature methods. 2012;9:671–675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Glogauer M, Marchal CC, Zhu F, Worku A, Clausen BE, Foerster I, Marks P, Downey GP, Dinauer M, Kwiatkowski DJ. Rac1 deletion in mouse neutrophils has selective effects on neutrophil functions. J Immunol. 2003;170:5652–5657 [DOI] [PubMed] [Google Scholar]
- 47.Qian BZ, Li J, Zhang H, Kitamura T, Zhang J, Campion LR, Kaiser EA, Snyder LA, Pollard JW. Ccl2 recruits inflammatory monocytes to facilitate breast-tumour metastasis. Nature. 2011;475:222–225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Piedrahita JA, Zhang SH, Hagaman JR, Oliver PM, Maeda N. Generation of mice carrying a mutant apolipoprotein e gene inactivated by gene targeting in embryonic stem cells. Proc Natl Acad Sci U S A. 1992;89:4471–4475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Daugherty A, Tall AR, Daemen M, Falk E, Fisher EA, Garcia-Cardena G, Lusis AJ, Owens AP 3rd, Rosenfeld ME, Virmani R, American Heart Association Council on Arteriosclerosis T, Vascular B, Council on Basic Cardiovascular S. Recommendation on design, execution, and reporting of animal atherosclerosis studies: A scientific statement from the american heart association. Arteriosclerosis, thrombosis, and vascular biology. 2017;37:e131–e157 [DOI] [PubMed] [Google Scholar]
- 50.Robinet P, Milewicz DM, Cassis LA, Leeper NJ, Lu HS, Smith JD. Consideration of sex differences in design and reporting of experimental arterial pathology studies-statement from atvb council. Arteriosclerosis, thrombosis, and vascular biology. 2018;38:292–303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Davies JQ, Gordon S. Isolation and culture of murine macrophages. Methods Mol Biol. 2005;290:91–103 [DOI] [PubMed] [Google Scholar]
- 52.Duewell P, Kono H, Rayner KJ, Sirois CM, Vladimer G, Bauernfeind FG, Abela GS, Franchi L, Nunez G, Schnurr M, Espevik T, Lien E, Fitzgerald KA, Rock KL, Moore KJ, Wright SD, Hornung V, Latz E. Nlrp3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature. 2010;464:1357–1361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.del Pozo MA, Price LS, Alderson NB, Ren XD, Schwartz MA. Adhesion to the extracellular matrix regulates the coupling of the small gtpase rac to its effector pak. The EMBO journal. 2000;19:2008–2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Soderberg O, Gullberg M, Jarvius M, Ridderstrale K, Leuchowius KJ, Jarvius J, Wester K, Hydbring P, Bahram F, Larsson LG, Landegren U. Direct observation of individual endogenous protein complexes in situ by proximity ligation. Nature methods. 2006;3:995–1000 [DOI] [PubMed] [Google Scholar]
- 55.Fredriksson S, Gullberg M, Jarvius J, Olsson C, Pietras K, Gustafsdottir SM, Ostman A, Landegren U. Protein detection using proximity-dependent DNA ligation assays. Nature biotechnology. 2002;20:473–477 [DOI] [PubMed] [Google Scholar]
- 56.Fredriksson S, Dixon W, Ji H, Koong AC, Mindrinos M, Davis RW. Multiplexed protein detection by proximity ligation for cancer biomarker validation. Nature methods. 2007;4:327–329 [DOI] [PubMed] [Google Scholar]
- 57.Rubin EM, Krauss RM, Spangler EA, Verstuyft JG, Clift SM. Inhibition of early atherogenesis in transgenic mice by human apolipoprotein ai. Nature. 1991;353:265–267 [DOI] [PubMed] [Google Scholar]
- 58.Grizot S, Faure J, Fieschi F, Vignais PV, Dagher MC, Pebay-Peyroula E. Crystal structure of the rac1-rhogdi complex involved in nadph oxidase activation. Biochemistry. 2001;40:10007–10013 [DOI] [PubMed] [Google Scholar]
- 59.Arbibe L, Mira JP, Teusch N, Kline L, Guha M, Mackman N, Godowski PJ, Ulevitch RJ, Knaus UG. Toll-like receptor 2-mediated nf-kappa b activation requires a rac1-dependent pathway. Nature immunology. 2000;1:533–540 [DOI] [PubMed] [Google Scholar]
- 60.Zaheer A, Lenkinski RE, Mahmood A, Jones AG, Cantley LC, Frangioni JV. In vivo near-infrared fluorescence imaging of osteoblastic activity. Nature biotechnology. 2001;19:1148–1154 [DOI] [PubMed] [Google Scholar]
- 61.Zaheer A, Murshed M, De Grand AM, Morgan TG, Karsenty G, Frangioni JV. Optical imaging of hydroxyapatite in the calcified vasculature of transgenic animals. Arteriosclerosis, thrombosis, and vascular biology. 2006;26:1132–1136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Joshi NV, Vesey AT, Williams MC, Shah AS, Calvert PA, Craighead FH, Yeoh SE, Wallace W, Salter D, Fletcher AM, van Beek EJ, Flapan AD, Uren NG, Behan MW, Cruden NL, Mills NL, Fox KA, Rudd JH, Dweck MR, Newby DE. 18f-fluoride positron emission tomography for identification of ruptured and high-risk coronary atherosclerotic plaques: A prospective clinical trial. Lancet. 2014;383:705–713 [DOI] [PubMed] [Google Scholar]
- 63.Zadelaar S, Kleemann R, Verschuren L, de Vries-Van der Weij J, van der Hoorn J, Princen HM, Kooistra T. Mouse models for atherosclerosis and pharmaceutical modifiers. Arteriosclerosis, thrombosis, and vascular biology. 2007;27:1706–1721 [DOI] [PubMed] [Google Scholar]
- 64.Aikawa E, Nahrendorf M, Figueiredo JL, Swirski FK, Shtatland T, Kohler RH, Jaffer FA, Aikawa M, Weissleder R. Osteogenesis associates with inflammation in early-stage atherosclerosis evaluated by molecular imaging in vivo. Circulation. 2007;116:2841–2850 [DOI] [PubMed] [Google Scholar]
- 65.Radcliff K, Tang TB, Lim J, Zhang Z, Abedin M, Demer LL, Tintut Y. Insulin-like growth factor-i regulates proliferation and osteoblastic differentiation of calcifying vascular cells via extracellular signal-regulated protein kinase and phosphatidylinositol 3-kinase pathways. Circulation research. 2005;96:398–400 [DOI] [PubMed] [Google Scholar]
- 66.Tintut Y, Patel J, Territo M, Saini T, Parhami F, Demer LL. Monocyte/macrophage regulation of vascular calcification in vitro. Circulation. 2002;105:650–655 [DOI] [PubMed] [Google Scholar]
- 67.Al-Aly Z, Shao JS, Lai CF, Huang E, Cai J, Behrmann A, Cheng SL, Towler DA. Aortic msx2-wnt calcification cascade is regulated by tnf-alpha-dependent signals in diabetic ldlr−/− mice. Arteriosclerosis, thrombosis, and vascular biology. 2007;27:2589–2596 [DOI] [PubMed] [Google Scholar]
- 68.Sage AP, Tintut Y, Demer LL. Regulatory mechanisms in vascular calcification. Nature reviews . Cardiology. 2010;7:528–536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Isoda K, Matsuki T, Kondo H, Iwakura Y, Ohsuzu F. Deficiency of interleukin-1 receptor antagonist induces aortic valve disease in balb/c mice. Arteriosclerosis, thrombosis, and vascular biology. 2010;30:708–715 [DOI] [PubMed] [Google Scholar]
- 70.Sumi D, Hayashi T, Thakur NK, Jayachandran M, Asai Y, Kano H, Matsui H, Iguchi A. A hmg-coa reductase inhibitor possesses a potent anti-atherosclerotic effect other than serum lipid lowering effects--the relevance of endothelial nitric oxide synthase and superoxide anion scavenging action. Atherosclerosis. 2001;155:347–357 [DOI] [PubMed] [Google Scholar]
- 71.Wagner AH, Kohler T, Ruckschloss U, Just I, Hecker M. Improvement of nitric oxide-dependent vasodilatation by hmg-coa reductase inhibitors through attenuation of endothelial superoxide anion formation. Arteriosclerosis, thrombosis, and vascular biology. 2000;20:61–69 [DOI] [PubMed] [Google Scholar]
- 72.Wassmann S, Laufs U, Muller K, Konkol C, Ahlbory K, Baumer AT, Linz W, Bohm M, Nickenig G. Cellular antioxidant effects of atorvastatin in vitro and in vivo. Arteriosclerosis, thrombosis, and vascular biology. 2002;22:300–305 [DOI] [PubMed] [Google Scholar]
- 73.Ridker PM, Everett BM, Thuren T, MacFadyen JG, Chang WH, Ballantyne C, Fonseca F, Nicolau J, Koenig W, Anker SD, Kastelein JJP, Cornel JH, Pais P, Pella D, Genest J, Cifkova R, Lorenzatti A, Forster T, Kobalava Z, Vida-Simiti L, Flather M, Shimokawa H, Ogawa H, Dellborg M, Rossi PRF, Troquay RPT, Libby P, Glynn RJ, Group CT. Antiinflammatory therapy with canakinumab for atherosclerotic disease. The New England journal of medicine. 2017 [DOI] [PubMed] [Google Scholar]
- 74.Ridker PM, MacFadyen JG, Everett BM, Libby P, Thuren T, Glynn RJ, Group CT. Relationship of c-reactive protein reduction to cardiovascular event reduction following treatment with canakinumab: A secondary analysis from the cantos randomised controlled trial. Lancet. 2018;391:319–328 [DOI] [PubMed] [Google Scholar]
- 75.Gomez D, Baylis RA, Durgin BG, Newman AAC, Alencar GF, Mahan S, St Hilaire C, Muller W, Waisman A, Francis SE, Pinteaux E, Randolph GJ, Gram H, Owens GK. Interleukin-1beta has atheroprotective effects in advanced atherosclerotic lesions of mice. Nature medicine. 2018;24:1418–1429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Alexander MR, Moehle CW, Johnson JL, Yang Z, Lee JK, Jackson CL, Owens GK. Genetic inactivation of il-1 signaling enhances atherosclerotic plaque instability and reduces outward vessel remodeling in advanced atherosclerosis in mice. The Journal of clinical investigation. 2012;122:70–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Rader DJ. Il-1 and atherosclerosis: A murine twist to an evolving human story. The Journal of clinical investigation. 2012;122:27–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.