

Abstract
Background
Neonatal acute respiratory distress syndrome (ARDS) is a common clinical syndrome caused by lung immaturity and the abnormal synthesis of pulmonary surfactant in preterm newborns, and it has high morbidity and mortality rates. The present study investigated the roles of interleukin-37 (IL-37) in the pathogenesis of neonatal ARDS and the underlying biochemical mechanism.
Material/Methods
We used 6-day-old neonatal C57BL/6 mice to establish the ARDS model. Inflammatory cytokines levels were measured with enzyme-linked immunosorbent assay (ELISA) Kits. The pathological morphology of lung tissues was observed by hematoxylin-eosin (HE) staining. The expression levels of proteins were assessed by Western blotting and apoptotic cells were detected via TUNEL assay. Further, the expression of nucleotide-bound oligomerization domain (Nod)-like receptor P3 (NLRP3) was detected with immunohistochemistry and Western blotting.
Results
IL-37 attenuated lipopolysaccharide (LPS)-induced cell apoptosis and excessive inflammatory cytokines levels, including IL-1β, IL-8, TNF-α, and MCP-1, and ameliorated lung pathological manifestations in an LPS-induced neonatal ARDS model. Moreover, IL-37 suppressed the abnormal expression of proteins related to the CXCR4/SDF-1 chemokine axis and NLRP3 inflammasome pathway.
Conclusions
The present results suggest that IL-37 protect against LPS-induced lung injury through inhibition of inflammation and apoptosis in lung tissue in an LPS-induced neonatal ARDS model. Hence, IL-37 may be considered as a potential therapeutic agent for neonatal ARDS.
MeSH Keywords: Anti-Inflammatory Agents; Inflammasomes; Mice, 129 Strain; Respiratory Distress Syndrome, Newborn
Background
Critical illness syndrome caused by trauma or sepsis is characterized by a dysregulated immune response and can lead to organ dysfunction, increased morbidity, and eventual death [1–4]. Despite advances in therapeutic methods [5,6], organ dysfunction syndromes, including acute respiratory distress syndrome (ARDS), can be remarkably difficult to diagnose early, and they can complicate therapy status and accelerate disease progression in patients. Neonatal ARDS is a common clinical syndrome characterized by refractory respiratory failure with hypoxemia, and it has high morbidity and mortality rates. It is well established that neonatal ARDS is caused by excessive inflammation, lung immaturity, and abnormal synthesis of pulmonary surfactant in premature infants [7]. Recent studies have reported that excessive inflammation plays an important role in the pathogenesis of neonatal ARDS [8]. The pathogenesis of this neonatal condition is based on uncontrolled inflammatory response in lungs and impaired integrity of alveolar-capillary membranes, which leads to accumulation of high-protein edema fluid in the lung tissues [9,10]. However, current clinical immune-targeted or pharmacologic therapies have numerous limitations, which has stimulated interest in investigating the fundamental cellular mechanism related to the regulation of acute lung inflammation and injury in ARDS and developing novel and effective therapeutic strategies for neonatal ARDS [11,12].
Interleukin (IL)-37 belongs to the IL-1 family, but it is different from most IL-1 family members, which are pro-inflammatory factors, because of its notable anti-inflammation actions. Thus, IL-37 has emerged as a natural suppressor of innate immune response [13], whose function is reported to be caspase-1-dependent. Caspase-1 processing takes part in intracellular IL-37 maturation and mature IL-37 secretion [14]. As a fundamental inhibitor of innate immunity, IL-37 protects against pulmonary damage through inhibiting pro-inflammatory cytokines production in murine aspergillosis [15]. IL-37 was also demonstrated to suppress pertussis toxin-induced inflammatory reaction by Smad3 signaling in autoimmune myositis rats [16]. Thus, it appears that IL-37 has potential therapeutic effects on neonatal ARDS.
In the present study, we investigated the roles of IL-37 in the pathogenesis of neonatal ARDS and its underlying biochemical mechanism to help develop a clinical therapeutic strategy for neonatal ARDS.
Material and Methods
Animals
Neonatal C57BL/6 mice (6 days old) (Oriental Bio Service, Inc., Nanjing) were maintained in standard cages with a 12-h light/dark cycle at 22±2°C with 55–65% relative humidity and free access to food and water. Animal care and experimental procedures were performed in accordance with the guidelines established by the Institutional Animal Care and Use Committees at Huashan North Hospital (IACUC issue no. 20180406; date: 20180407).
Mouse model of neonatal acute respiratory distress syndrome (ARDS) and drug administration
The neonatal ARDS mouse model was established as previously described, with minor revision [17]. Thirty neonatal C57BL/6 mice were randomly divided into a normal saline control group (n=10) and an LPS-induced group (n=20). Mice in the LPS-induced group were injected intraperitoneally with LPS (10 mg/kg) purchased from Sigma-Aldrich (St. Louis, MO). Mice in the normal saline control group were injected with an equal volume of sterile saline. Ten mice in the LPS-induced group were intraperitoneally injected with 1 μg of recombinant IL-37 (R&D Systems, Shanghai, China) per mouse [15] 2 h before LPS injection. The other 10 LPS-induced mice and the 10 control mice received the same weight-based volume of vehicle. All mice were sacrificed 72 h after LPS or saline treatment and blood samples were collected.
ELISA assay
Blood was collected through eyeball extraction after LPS injection at 12 h and 24 h, and serum was separated through centrifugation at 3500 rpm at 4°C for 15 min. A specific ELISA kit (Invitrogen, Carlsbad, CA, USA) was used to measure levels of inflammatory cytokines (IL-1β, IL-8, TNF-α, and MCP-1) according to the manufacturer’s protocols. The color intensity was assayed at 450 nm with reference wave length using a microplate reader (Bio-Rad, Hercules, CA, USA).
Hematoxylin-eosin (HE) staining
At 12 h and 24 h after LPS injection, lung tissues were fixed in 4% paraformaldehyde for 24 h at room temperature and then embedded in paraffin. Lung tissues were cut into 5-μm-thick sections. Then, the sections were dewaxed with xylene and dehydrated with gradient ethanol. Slides were stained with HE. Finally, the sections were transparentized with xylene and observed using an inverted fluorescence microscope (Nikon Eclipse Ti; Nikon Corporation, Tokyo, Japan) at 200× magnification to confirm the pathological changes of lung tissues.
Western blotting
Lung tissues were homogenized to obtain protein samples with RIPA lysis buffer containing PMSF after washing with cold PBS. Then, the protein (25 μg) was separated by SDS-PAGE and transferred to PVDF membranes. The membranes were incubated overnight with appropriated primary antibody at 4°C after being blocked with 5% bovine serum albumin (BSA). Rabbit anti-ICAM-1 antibodies (1: 1000), rabbit anti-CXCR4 antibodies (1: 1000), rabbit anti-SDF-1 antibodies (1: 1000), rabbit anti-Bcl2 antibodies (1: 1000), rabbit anti-bax antibodies (1: 1000), rabbit anti-cleaved caspase3 antibodies (1: 500), rabbit anti-NLRP3 antibodies (1: 1000), rabbit anti-ASC antibodies (1: 1000), rabbit anti-pro-IL-1β antibodies (1: 1000), rabbit anti-IL-1β antibodies (1: 1000), rabbit anti-pro-caspase1 antibodies (1: 1000), and rabbit anti-caspase1 antibodies (1: 1000) were purchased from Cell Signaling Technology (Danvers, USA). Subsequently, PVDF membranes were incubated with the corresponding secondary antibodies and visualized using a Tanon-5200 Chemiluminescence Imager (Tanon, Shanghai, China) with enhanced chemiluminescence (ECL) method.
TUNEL assay
Apoptotic cells were detected via TUNEL assay. The ApopTag In Situ Apoptosis Detection Kit (Merck Millipore, Darmstadt, Germany) was used to detect apoptotic cells. Fluorescein isothiocyanate (FITC) (green) and 4′,6-diamidino-2-phenylindole (DAPI) (blue) were used to stain the apoptotic cells and the nuclei, respectively. In blinded fashion, we counted the number of positive cells in micrographs at 200× magnification).
Immunohistochemistry of NLRP3
Lung tissue sections were blocked with 1% normal fetal goat serum and then stained overnight with antibody against rabbit anti-NLRP3 polyclonal antibody (1: 200, Millipore, Billerica, MA, USA) at 4°C. Subsequently, the sections were incubated with alkaline biotin-conjugated secondary antibody for 2 h at 37°C. The immunoreactivity was visualized via the standard avidin–biotin complex reaction with diaminobenzidine (DAB) and observed under a conventional light microscope (Olympus PD70) at 200× magnification.
Statistical analysis
The group data are expressed as mean±standard error (SEM) and were analyzed with SPSS 17.0 software. The groups were compared with the 2-tailed unpaired t test or ANOVA, as appropriate, followed by Bonferroni’s multiple comparison post hoc test using GraphPad Prism 5.0. Differences at P<0.05 were considered to indicate statistically significant differences.
Results
IL-37 attenuated excessive release of inflammatory cytokines and lung pathological manifestations in the LPS-induced neonatal ARDS model
To detect the secretion levels of the inflammatory cytokines IL-1β, IL-8, TNF-α, and MCP-1, enzyme-linked immunosorbent assay (ELISA) kits were used at 12 and 48 h after LPS injection, showing that LPS injection increased the production of inflammatory cytokines. The levels of IL-1β, IL-8, TNF-α, and MCP-1 were significantly elevated and gradually increased with prolonged administration time in serum of mice in the LPS-induced group compared with the control group, showing the effect of IL-37 treatment (Figure 1A–1D).
Figure 1.

IL-37 attenuated excessive inflammatory cytokines release and lung pathological manifestations in an LPS-induced neonatal ARDS model. (A) Tumor necrosis factor-α (TNF-α), (B) interleukin-1β (IL-1β), (C) IL-8, and (D) monocyte chemotactic protein 1 (MCP-1) levels were detected with ELISA kits. (E) Representative images of hematoxylin and eosin (HE) staining lung tissues from different groups (×200 magnification). The data are expressed as means±SD from 3 independent experiments; *** P<0.001 vs. Control. ### P<0.001 vs. LPS.
HE staining was performed to observe the pathological changes of the lung tissues. As shown in Figure 1E, lung sections from LPS-induced neonatal mice confirmed alveolar congestion, hemorrhage, edema and inflammatory cell infiltration, which was more severe after LPS exposure for 24 h. In contrast, IL-37 treatment ameliorated the lung injury induced by inflammatory conditions.
IL-37 suppressed the CXCR4/SDF-1 chemokine axis in lung tissue of neonatal ARDS mice
The chemokine receptor4 (CXCR4)/stromal-derived factor-1 (SDF-1) chemokine axis plays key roles in inflammation of injured tissues. Western blotting was used to analyze the expression levels of the proteins CXCR4 and SDF-1 and intercellular adhesion molecule-1 (ICAM-1) in lung tissues. As shown in Figure 2A, 2B, the expression levels of CXCR4, SDF-1, and ICAM-1 were significantly increased in the LPS-induced group at 12 h and 24 h compared with normal mice. Moreover, mild increases were observed in protein expression at 24 h. However, IL-37 inhibited expression of CXCR4, SDF-1, and ICAM-1. Therefore, our results suggest that the CXCR4/SDF-1 chemokine axis is part of the mechanism underlying the inhibition effect of IL-37 on excessive inflammation.
Figure 2.
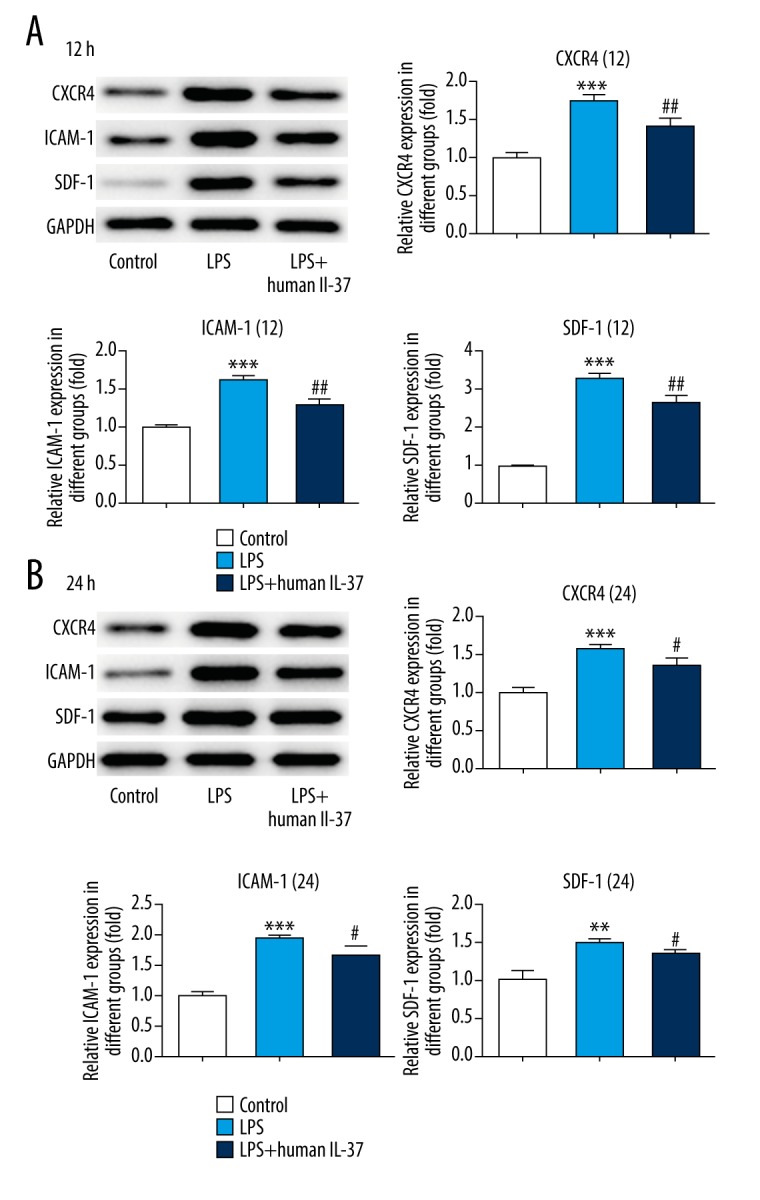
IL-37 suppressed inactivation of CXCR4/SDF-1 chemokine axis. (A) The expression levels of CXCR4, ICAM-1, and SDF-1 were measured by Western blotting at 12 h. (B) The expression levels of CXCR4, ICAM-1, and SDF-1 were measured by Western blotting at 24 h. The data are expressed as means±SD from 3 independent experiments; ** P<0.01, *** P<0.001 vs. Control. # P<0.05, ## P<0.01 vs. LPS.
IL-37 decreased the cell apoptosis rate induced by LPS
TUNEL assay was performed to detect cell apoptosis, indicating significant apoptosis in lung tissue induced by LPS compared to controls, while the number of apoptotic cells was markedly decreased after IL-37 treatment. The results show that lung tissues had a greater increase in the number of apoptotic cells 24 h after LPS injection (Figure 3A). The proteins related to cell apoptosis were analyzed by Western blotting, showing that the expression levels of Bax and cleaved caspase3 were enhanced in lung tissues of LPS-induced mice, while the expression level of Bcl-2 was suppressed. Interestingly, IL-37 corrected the abnormal expression of these proteins, indicating that IL-37 protected against LPS-induced lung injury (Figure 3B).
Figure 3.

IL-37 decreased cell apoptosis rate. (A) Cell apoptosis was analyzed by TUNEL staining (×200 magnification). (B) The expression levels of Bax, Bcl-2, cleaved caspase-3, and caspase-3 were measured by Western blotting. The data are expressed as means±SD from 3 independent experiments; *** P<0.001 vs. Control. # P<0.05, ### P<0.001 vs. LPS.
IL-37 inactivated the NLRP3 inflammasome pathway in the LPS-induced neonatal ARDS model
Western blotting and immunohistochemistry were performed to detect the expression of proteins related to NLRP3 signaling. The results showed that the expression levels of NLRP3, ASC, pro-IL-1β, IL-1β, pro-caspase1, and caspase1 were significantly upregulated, and these levels were higher after 24-h LPS exposure. IL-37 treatment suppressed the expressions of NLRP3, ASC, IL-1β, and caspase1, while IL-37 had no effect on the increased expression levels of pro-IL-1β and pro-caspase1 induced by LPS, suggesting the inhibition effect of IL-37 on LPS-induced activation of NLRP3 signaling (Figure 4A, 4B). Moreover, immunohistochemistry results further confirmed that the expression level of NLRP3 was upregulated in lung tissues of LPS-induced mice, which were reversed by IL-37 treatment (Figure 4C).
Figure 4.

IL-37 inactivated NLRP3 inflammasome pathway in LPS-induced neonatal ARDS model induced by LPS. (A) The expression levels of NLRP3, ASC, IL-1β, pro-IL-1β, pro-caspase-1, and caspase-1 were measured by Western blotting at 12 h. (B) The expression levels of NLRP3, ASC, IL-1β, pro-IL-1β, pro-caspase-1, and caspase-1 were measured by Western blotting at 24 h. (C) NLRP3 expression level was analyzed by immunohistochemistry (×200 magnification). The data are expressed as means±SD from 3 independent experiments; ** P<0.01, *** P<0.001 vs. Control. # P<0.05, ### P<0.001 vs. LPS.
Discussion
Interleukin (IL)-37 is a member of the IL-1 family. However, IL-37 is different from most IL-1 family members due to its biological properties of downregulating inflammation. In recent years, numerous studies have reported that IL-37 binds to IL-18 receptor and IL-18 binding protein to forms a complex that inhibits IL-18 activity [18]. Sharma et al. demonstrated that nuclear translocation of IL-37 is related to caspase-1 processing and reduced pro-inflammatory cytokines production induced by LPS [19]. Pro-inflammatory cytokines, especially tumor necrosis factor-α (TNF-α), have been identified as key mediators of pathogenesis of neonatal acute respiratory distress syndrome (ARDS) [20–22]. In the present study, 6-day-old neonatal C57BL/6 mice were used to establish the ARDS model induced by LPS to assess the anti-inflammatory effect of IL-37 in neonatal ARDS, showing that IL-37 significantly attenuated pathological changes of the lung tissues of neonatal ARDS mice, and reduced LPS-induced inflammatory reactions and apoptosis, indicating that IL-37 may be an effective therapeutic agent for neonatal ARDS treatment.
ARDS is a serious disease lacking effective treatment and is characterized by inflammatory pulmonary edema. Davino-Chiovatto et al. reported that lung inflammation was successfully induced by LPS in a mouse model of ARDS [23], consistent with our results. IL-37 significantly reduced the elevated levels of the inflammatory cytokines IL-1β, IL-8, TNF-α, and MCP-1, and ameliorated lung injury. In addition, the chemokine receptor4 (CXCR4)/stromal-derived factor-1 (SDF-1) chemokine axis, which is involved in inflammation of injured tissues, plays a role in the effect of IL-37 on release of inflammatory cytokines. Previous studies have demonstrated that the SDF-1/CXCR4 axis is involved in the progression and disease severity of idiopathic inflammatory bowel diseases [24]. Shi et al. also suggested that the SDF-1/CXCR4 chemokine axis plays an important role in the process of early acute lung injury [25]. Consistent with our results, blocking the SDF-1/CXCR4 axis ameliorated pulmonary inflammation in lung damage [25]. IL-37 inhibited the abnormal SDF-1 and CXCR4 overexpression induced by inflammation, indicating that IL-37 may have a therapeutic effect on lung injury induced by inflammatory conditions. Furthermore, IL-37 treatment attenuated the cell apoptosis rate and expression levels of proteins related to apoptosis of lung tissues in LPS-induced mice. As demonstrated by Pibao et al., miR-150 attenuates LPS-induced acute lung injury through alleviating A549 cell apoptosis and release of inflammatory cytokines by targeting AKT3 [26]. Based on the evidence presented above, we hypothesized that IL-37 exerts a suppressive effect on the excessive inflammatory response and cell apoptosis in lung tissue in neonatal ARDS.
Numerous studies have reported that the nucleotide-bound oligomerization domain (Nod)-like receptors (NLRs) play important roles in innate immune responses. NLRs are responsible for maturation and secretion of pro-inflammatory cytokines such as IL-1β and IL-18 [27]. The NLRP3 inflammasome, which is the best-defined NLR, can be activated in response to cellular stresses during cellular or tissue injury [28]. The inflammasome complex is composed of 3 proteins: the cytosolic sensor (NLRs or ALRs), the apoptosis-associated speck-like protein containing caspase-recruitment domain (ASC), and the effector molecule pro-caspase-1 [29]. The inflammasome adaptor ASC acts as a bridge between the sensor and pro-caspase-1 via molecular interactions. Activation of the NLRP3 inflammasome increases the inflammatory cytokines IL-18 and IL-1β, thereby aggravating excessive inflammation [30]. Inflammatory responses can be attenuated by inhibiting the activation of NLRP3 inflammasomes by elimination of damaged mitochondria [31]. In the present study, IL-37 remarkably decreased the expression of inflammasomes complex components and triggered the inactivation of NLRP3 inflammasomes signaling. These results further confirmed that NLRP3 inflammasomes signaling may also be involved in the mechanism underlying the inhibition effect of IL-37 on excessive inflammatory response induced by LPS challenge in lung tissue of neonatal AARDS mice.
Our study indicated that IL-37 inhibited excessive inflammatory response and cell apoptosis in lung tissue of neonatal AARDS mice and exerted a suppressive effect on the CXCR4/SDF-1 chemokine axis and NLRP3 inflammasome pathway ARDS. Our findings indicate that inactivation of the CXCR4/SDF-1 chemokine axis and NLRP3 inflammasome pathway may be involved in the mechanism underlying the therapeutic effect of IL-37 on neonatal AARDS. Our results provide novel insights and suggest possible therapeutic approaches for neonatal AARDS. The mechanism underlying the effect of IL-37 on neonatal ARDS warrants investigation in future studies.
Conclusions
We found that IL-37 ameliorated lung pathological manifestations in LPS-induced neonatal ARDS mice. IL-37 attenuated LPS-induced excessive inflammatory cytokines production and cell apoptosis rate. Furthermore, IL-37 inhibited abnormal activation of the CXCR4/SDF-1 chemokine axis and NLRP3 inflammasomes activation in lung tissue in an LPS-induced neonatal ARDS model. Thus, IL-37 has promise as a clinically therapeutic agent in protecting against pulmonary injury resulting from chronic inflammation.
Footnotes
Source of support: Departmental sources
Conflict of interest
None.
References
- 1.Herridge MS, Tansey CM, Matte A, et al. Functional disability 5 years after acute respiratory distress syndrome. N Engl J Med. 2011;364:1293–304. doi: 10.1056/NEJMoa1011802. [DOI] [PubMed] [Google Scholar]
- 2.Reilly JP, Anderson BJ, Mangalmurti NS, et al. The ABO histo-blood group and AKI in critically ill patients with trauma or sepsis. Clin J Am Soc Nephrol. 2015;10:1911–20. doi: 10.2215/CJN.12201214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rubenfeld GD, Caldwell E, Peabody E, et al. Incidence and outcomes of acute lung injury. N Engl J Med. 2005;353:1685–93. doi: 10.1056/NEJMoa050333. [DOI] [PubMed] [Google Scholar]
- 4.Shah CV, Localio AR, Lanken PN, et al. The impact of development of acute lung injury on hospital mortality in critically ill trauma patients. Crit Care Med. 2008;36:2309–15. doi: 10.1097/CCM.0b013e318180dc74. [DOI] [PubMed] [Google Scholar]
- 5.Brower RG, Matthay MA, Morris A, et al. Ventilation with lower tidal volumes as compared with traditional tidal volumes for acute lung injury and the acute respiratory distress syndrome. N Engl J Med. 2000;342:1301–8. doi: 10.1056/NEJM200005043421801. [DOI] [PubMed] [Google Scholar]
- 6.Guerin C, Reignier J, Richard JC, et al. Prone positioning in severe acute respiratory distress syndrome. N Engl J Med. 2013;368:2159–68. doi: 10.1056/NEJMoa1214103. [DOI] [PubMed] [Google Scholar]
- 7.De Bisschop B, Derriks F, Cools F. Early predictors for INtubation-SURfactant-Extubation failure in preterm infants with neonatal respiratory distress syndrome: A systematic review. Neonatology. :2019. doi: 10.1159/000501654. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 8.Li L, Dong L, Zhao D, et al. Classical dendritic cells regulate acute lung inflammation and injury in mice with lipopolysaccharideinduced acute respiratory distress syndrome. Int J Mol Med. 2019;44:617–29. doi: 10.3892/ijmm.2019.4208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.De Luca D, van Kaam AH, Tingay DG, et al. The Montreux definition of neonatal AARDS: biological and clinical background behind the description of a new entity. Lancet Respir Med. 2017;5:657–66. doi: 10.1016/S2213-2600(17)30214-X. [DOI] [PubMed] [Google Scholar]
- 10.Liu ZQ, Feng J, Shi LL, et al. Influences of miR-155/NF-kappaB signaling pathway on inflammatory factors in AARDS in neonatal pigs. Eur Rev Med Pharmacol Sci. 2019;23:7042–48. doi: 10.26355/eurrev_201908_18746. [DOI] [PubMed] [Google Scholar]
- 11.Ranieri VM, Rubenfeld GD, Thompson BT, et al. Acute respiratory distress syndrome: the Berlin Definition. JAMA. 2012;307:2526–33. doi: 10.1001/jama.2012.5669. [DOI] [PubMed] [Google Scholar]
- 12.Reilly JP, Christie JD, Meyer NJ. Fifty years of research in AARDS. Genomic contributions and opportunities. Am J Respir Crit Care Med. 2017;196:1113–21. doi: 10.1164/rccm.201702-0405CP. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nold MF, Nold-Petry CA, Zepp JA, et al. IL-37 is a fundamental inhibitor of innate immunity. Nat Immunol. 2010;11:1014–22. doi: 10.1038/ni.1944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liu L, Xue Y, Zhu Y, et al. Interleukin 37 limits monosodium urate crystal-induced innate immune responses in human and murine models of gout. Arthritis Res Ther. 2016;18:268. doi: 10.1186/s13075-016-1167-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Moretti S, Bozza S, Oikonomou V, et al. IL-37 inhibits inflammasome activation and disease severity in murine aspergillosis. PLoS Pathog. 2014;10:e1004462. doi: 10.1371/journal.ppat.1004462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yan P, Zhang Y, Wang C, et al. Interleukin-37 (IL-37) suppresses pertussis toxin-induced inflammatory myopathy in a rat model. Med Sci Monit. 2018;24:9187–95. doi: 10.12659/MSM.910904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ying L, Alvira CM. Developmental differences in focal adhesion kinase expression modulate pulmonary endothelial barrier function in response to inflammation. Am J Physiol Lung Cell Mol Physiol. 2018;315(1):L66–77. doi: 10.1152/ajplung.00363.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bufler P, Azam T, Gamboni-Robertson F, et al. A complex of the IL-1 homologue IL-1F7b and IL-18-binding protein reduces IL-18 activity. Proc Natl Acad Sci USA. 2002;99:13723–28. doi: 10.1073/pnas.212519099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sharma S, Kulk N, Nold MF, et al. The IL-1 family member 7b translocates to the nucleus and down-regulates proinflammatory cytokines. J Immunol. 2008;180:5477–82. doi: 10.4049/jimmunol.180.8.5477. [DOI] [PubMed] [Google Scholar]
- 20.Kambas K, Markiewski MM, Pneumatikos IA, et al. C5a and TNF-alpha up-regulate the expression of tissue factor in intra-alveolar neutrophils of patients with the acute respiratory distress syndrome. J Immunol. 2008;180:7368–75. doi: 10.4049/jimmunol.180.11.7368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Meduri GU, Kohler G, Headley S, et al. Inflammatory cytokines in the BAL of patients with AARDS. Persistent elevation over time predicts poor outcome. Chest. 1995;108:1303–14. doi: 10.1378/chest.108.5.1303. [DOI] [PubMed] [Google Scholar]
- 22.Yu LL, Zhu M, Huang Y, et al. Metformin relieves acute respiratory distress syndrome by reducing miR-138 expression. Eur Rev Med Pharmacol Sci. 2018;22:5355–63. doi: 10.26355/eurrev_201808_15737. [DOI] [PubMed] [Google Scholar]
- 23.Davino-Chiovatto JE, Oliveira-Junior MC, MacKenzie B, et al. Montelukast, leukotriene inhibitor, reduces LPS-Induced acute lung inflammation and human neutrophil activation. Arch Bronconeumol. 2019;55(11):573–80. doi: 10.1016/j.arbres.2019.05.003. [DOI] [PubMed] [Google Scholar]
- 24.Mrowicki J, Przybylowska-Sygut K, Dziki L, et al. The role of polymorphisms of genes CXCL12/CXCR4 and MIF in the risk development IBD the Polish population. Mol Biol Rep. 2014;41:4639–52. doi: 10.1007/s11033-014-3335-y. [DOI] [PubMed] [Google Scholar]
- 25.Shi H, Lu R, Wang S, et al. Effects of SDF-1/CXCR4 on acute lung injury induced by cardiopulmonary bypass. Inflammation. 2017;40:937–45. doi: 10.1007/s10753-017-0538-0. [DOI] [PubMed] [Google Scholar]
- 26.Li P, Yao Y, Ma Y, Chen Y. MiR-150 attenuates LPS-induced acute lung injury via targeting AKT3. Int Immunopharmacol. 2019;75 doi: 10.1016/j.intimp.2019.105794. 105794. [DOI] [PubMed] [Google Scholar]
- 27.Shaw MH, Reimer T, Kim YG, Nunez G. NOD-like receptors (NLRs): Bona fide intracellular microbial sensors. Curr Opin Immunol. 2008;20:377–82. doi: 10.1016/j.coi.2008.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lee S, Suh GY, Ryter SW, Choi AM. Regulation and function of the nucleotide binding domain leucine-rich repeat-containing receptor, pyrin domain-containing-3 inflammasome in lung disease. Am J Respir Cell Mol Biol. 2016;54:151–60. doi: 10.1165/rcmb.2015-0231TR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Atianand MK, Rathinam VA, Fitzgerald KA. SnapShot: Inflammasomes. Cell. 2013;153:272–272.e271. doi: 10.1016/j.cell.2013.03.009. [DOI] [PubMed] [Google Scholar]
- 30.Lupfer C, Thomas PG, Anand PK, et al. Receptor interacting protein kinase 2-mediated mitophagy regulates inflammasome activation during virus infection. Nat Immunol. 2013;14:480–88. doi: 10.1038/ni.2563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhong Z, Umemura A, Sanchez-Lopez E, et al. NF-kappaB restricts inflammasome activation via elimination of damaged mitochondria. Cell. 2016;164:896–910. doi: 10.1016/j.cell.2015.12.057. [DOI] [PMC free article] [PubMed] [Google Scholar]