

Abstract
Alternative non-B form DNA structures, also called secondary structures, can form in certain DNA sequences under conditions that produce single-stranded DNA, such as during replication, transcription, and repair. Direct links between secondary structure formation, replication fork stalling, and genomic instability have been found for many repeated DNA sequences that cause disease when they expand. Common fragile sites (CFSs) are known to be AT-rich and break under replication stress, yet the molecular basis for their fragility is still being investigated. Over the past several years, new evidence has linked both the formation of secondary structures and transcription to fork stalling and fragility of CFSs. How these two events may synergize to cause fragility and the role of nuclease cleavage at secondary structures in rare and CFSs are discussed here. We also highlight evidence for a new hypothesis that secondary structures at CFSs not only initiate fragility but also inhibit healing, resulting in their characteristic appearance.
Keywords: Common fragile sites, rare fragile sites, DNA secondary structures, fork stalling, nuclease cleavage
I. Fragile sites: history and definition
In 1970, the term fragile site was used to refer to recurrent breaks found near the haptoglobin locus of human chromosome 16 (1). Since then, the term fragile site has been expanded to define loci that exhibit chromosome fragility as visible gaps and breaks on metaphase chromosomes or by physical or genetic assays of chromosome breakage. There are two main classes of fragile sites: rare fragile sites and common fragile sites (CFSs), which are defined by their frequency of expression (breakage) in the population. Rare fragile sites are present in less than 5% of individuals, are usually caused by expanded repetitive DNA elements, and are inherited in a Mendelian fashion (reviewed in (2, 3)). Common fragile sites are present in all individuals and are mainly visible after partial inhibition of DNA synthesis (reviewed in (4, 5)). CFS expression is induced by drugs that cause replication stress: aphidicolin, which inhibits DNA polymerase α, δ and ε elongation, hydroxyurea (HU), which depletes dNTP pools, or caffeine, which inhibits the kinase Ataxia Telangiectasia and Rad3 related (ATR), which detects stalled forks and activates the DNA damage checkpoint (3). Over 85 CFSs have been identified to date (2, 3).
II. Formation of secondary structures in DNA
Most DNA conforms to the canonical right-handed double helix structure known as B form DNA. Alternative, or secondary, DNA structures can form at DNA of certain sequence and composition. Secondary structures are preferentially formed during processes where DNA is unwound, such as replication, transcription, and repair, allowing the conversion of B-DNA to alternative DNA structures (Figure 1). Secondary structures are often formed within repetitive DNA sequences that can pair out of register after double-stranded DNA (dsDNA) is denatured, resulting in misalignment of the two strands. If the misalignment is not corrected, expansions or contractions in repeat length will result. A longer number of repeat units can give rise to a larger and more stable alternative DNA structure.
Figure 1. . Formation of DNA secondary structures during normal cellular processes.
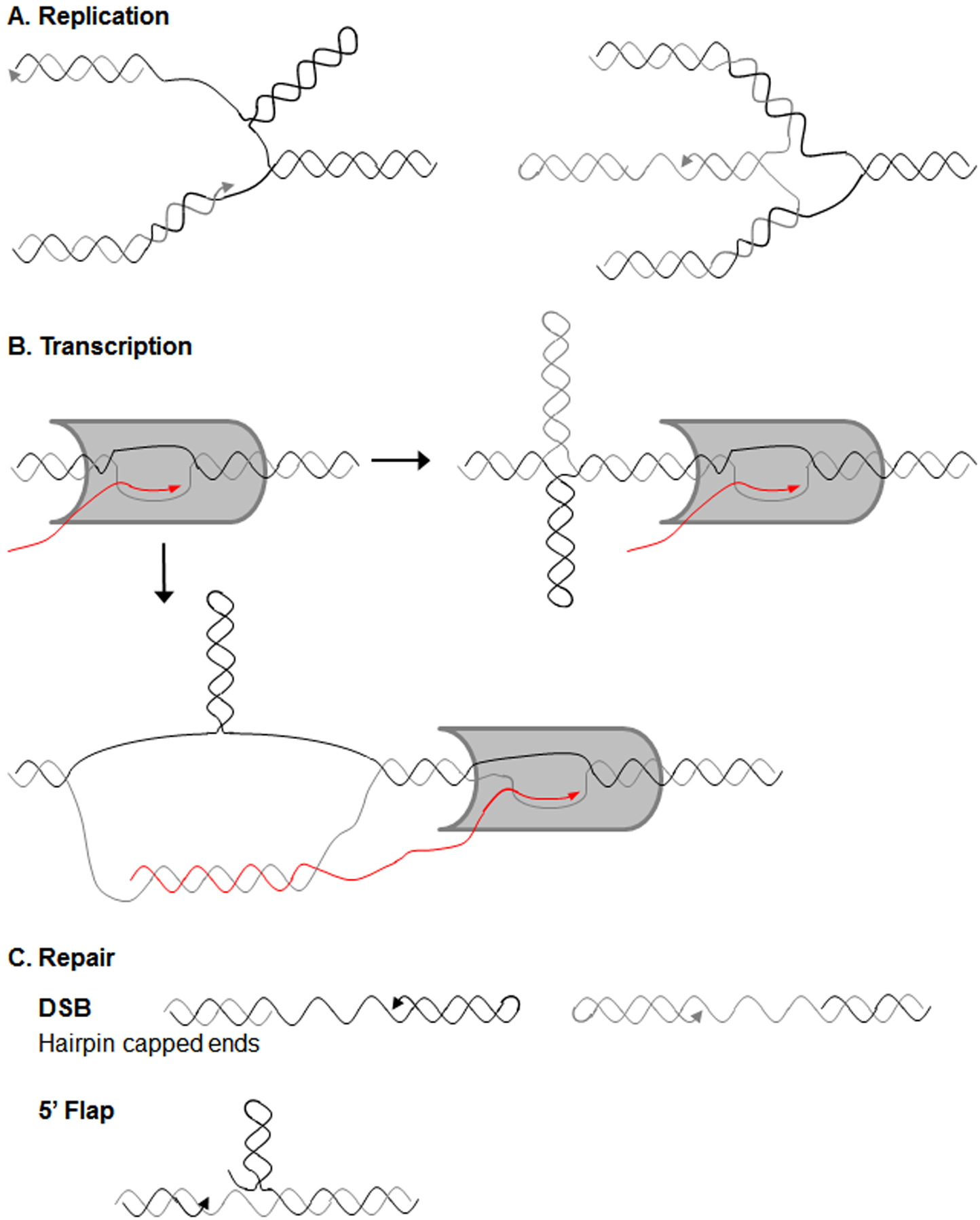
A) DNA secondary structures can form during replication, such as on the single-stranded lagging strand after DNA unwinding (left side) or on a ssDNA end created by fork reversal (right side). B) During transcription, RNA polymerase II (grey protein) passage results in increased negative supercoiling behind the polymerase, which can promote secondary structure formation, such as the cruciform depicted. Additionally, certain sequences form especially stable R-loops behind RNA polymerase II, which could allow secondary structure formation on the non-template strand. C) Secondary structures may also arise when DNA is single-stranded during repair. Hairpins formed on ssDNA ends exposed by resection can inhibit efficient DNA repair. Secondary structure formation on a displaced flap may render it unligatable, leading to nicks and DSBs (a 5’ flap is shown).
During replication, the DNA strands must be separated to expose the DNA template, which gives an opportunity for alternative DNA structures to form (Figure 1A). The lagging strand template is especially vulnerable to structure formation as it becomes transiently single-stranded. Indeed, studies of expandable CAG/CTG repeats show that when the more stable CTG hairpin structure is on the lagging strand template, this leads to bypass of the structure and deletions in the repetitive DNA, while CTG hairpins on the nascent lagging strand result in more expansions of the repetitive DNA (reviewed in (6)). Though formation of structures on the leading strand template has not been documented experimentally, they are potentially even more deleterious to replication fork progression. Another area of vulnerability is the 5’ end of the nascent lagging strand, which becomes single-stranded when displaced by polymerase synthesizing the incoming Okazaki fragment. If this flap forms a structure it can inhibit processing of the 5’ ends by FEN1 (S. cerevisiae Rad27), resulting in either ligation of extra sequence into the DNA backbone and a repeat expansion, or inhibition of ligation and a persistent nick, which can lead to fragility (reviewed in (6–8)). This same mechanism of structures interfering with flap ligation can occur during gap repair (Figure 1C) (reviewed in (6, 9)). Secondary structures could also form during fork reversal (Figure 1A), leading to either repeat length changes or fork breakage if they inhibit fork restart (reviewed in (10, 11)).
Transcription can play a role in DNA secondary structure formation and repeat length changes, as it involves DNA unwinding and local chromatin changes that favor structure formation (Figure 1B). It is notable that many expandable repeats are located within transcribed regions (reviewed in (6, 12)). The process of RNA polymerase II traveling along the DNA template generates positive supercoils ahead of the polymerase and excess negative supercoils behind the polymerase, which can allow for DNA secondary structure formation. Increased negative supercoiling stabilizes many different secondary structures in repetitive DNA, including hairpins, triplexes, slipped-strand DNA, Z-DNA, and cruciforms (reviewed in (13–15)). Increased negative supercoiling also results in deletions at secondary structure-forming GAA and CGG repeats in E. coli (16). RNA polymerase II transiently pauses when transcribing through repetitive DNA, possibly due to intrastrand structure formation (15, 17). Interestingly, convergent transcription through CAG repeats as well as other CG-rich repeats causes cellular stress and apoptosis (18–20); it is possible that convergent transcription (or convergent transcription and replication) could generate excessive positive supercoils that would inhibit further elongation (15, 21). Head-on collisions of replication and transcription machinery can also cause DNA breaks and genome instability (reviewed in (22, 23)). Transcription start sites are another potential hotspot for formation of single-stranded DNA and DNA structures, in fact this may be a physiological role of structure formation in genomes (24). Hairpin and stem-loop secondary structures are predicted to form 10 to 50 nucleotides upstream of RNA polymerase II promoter proximal pause sites (24). These structures are thought to initiate RNA polymerase II pausing either by directly binding proteins that mediate promoter-proximal pausing or by stabilizing the formation of RNA:DNA hybrids (24).
Another effect of transcription is the potential formation of R-loops, which are RNA:DNA hybrids that can form behind RNA polymerase by re-hybridization of the RNA transcript to its DNA template (Figure 1B). Certain DNA sequences, especially sequences that are G-rich on the non-template strand, form stable R-loops, which can stall both replication and transcription and lead to DNA damage and genome instability (reviewed in (25–27)). There is evidence for the formation of stable R-loops at many repetitive secondary structure-forming sequences, such as CAG, CGG, GAA, and G4C2 repeats (reviewed in (27–29)). The stabilization of one strand in an RNA:DNA hybrid can promote secondary structure formation on the exposed non-template DNA strand (15, 30, 31). Exposure of ssDNA by formation of R-loops or DNA structures can lead to breaks induced by cytosine deamination and reactive oxygen species (32, 33).
DNA repair also provides an opportunity for DNA secondary structure formation. When a double strand break (DSB) is formed, end resection leads to a single-stranded DNA end that is normally coated by RPA but can also fold back into a DNA structure (Figure 1C). The competition between RPA binding and structure formation can be influenced by the sequence and affinity of RPA binding (34, 35). Recent results from our lab indicate that DNA structure formation in this situation can significantly impair healing (36). Data from model systems has shown that DSB repair proteins such as Rad51/Rad52, Mre11 (human MRE11), Exo1, and Dnl4 (human LIG4) are important for preventing fragility at DNA structures (37). Gap repair provides yet another opportunity for secondary structure formation, both on the ssDNA gap itself, or on the 5’ flap if there is displacement synthesis. Formation of structures during gap repair is a primary means of disease-causing CAG repeat expansions (38, 39) (Figure 1C).
Though in general repair proteins are believed to mitigate the formation of DNA structures in order to allow repair or replication to proceed, some can play a role in stabilizing DNA structures. For CAG repeats, MutSβ stabilizes hairpin formation (40) and leads to increased replication fork stalling (41). Extensive research has implicated MutSβ in stabilizing hairpins during base excision repair, to facilitate CAG expansions and CGG expansions and contractions (reviewed in (6, 9)). In addition, the MutLα endonuclease, which is recruited by MutSβ, has been shown to cause breaks at GAA repeats (42). At CAG repeats R-loop-dependent structures stimulate MutLγ-mediated breaks and instability (33).
III. Types of secondary structures and links to fork stalling and chromosome fragility
Examples abound of secondary structures formed from repetitive DNA in the genome, and there are many links to their ability to cause replication fork stalling, chromosome fragility, and genome instability. These links are less clear for CFSs, however there are parallels since CFS regions also contain repetitive and structure-forming sequences and are also regions with difficulty completing replication. The yeast genome exhibits an increase in recombination events at DNA sequences and subregions associated with replication fork stalling (43), illustrating the connection between replication fork stalling, fragility, and genome rearrangements. Below, we outline common repetitive DNA tracts and discuss evidence of the known links between replication fork stalling and chromosome fragility at these sites.
Mononucleotide runs are the simplest repetitive element and consist of repetition of the same base. Mononucleotide runs can result in slipped-strand DNA, where DNA strands mispair and anneal out-of-register, resulting in ssDNA loops interspersed within dsDNA. A or T mononucleotide runs can also serve as DNA unwinding elements (DUEs) due to the strong unwinding capability of AT-rich DNA (Figure 2). In vitro, polymerase δ holoenzyme accumulated at an (A)28 mononucleotide run from one subregion of CFS FRA16D, a (T)19 run from different subregion of FRA16D, and a (T)22 mononucleotide run from CFS FRA3B, especially after aphidocolin-induced replication stress (44). Mononucleotide runs, and especially A-tracts, had the strongest correlation with cancer deletion breakpoints out of all potential non-B-DNA forming sequences probed in 46,000 cancer genomes (45). Interestingly, early replicating fragile sites (ERFSs) are located close to origins (46) and new data shows that poly(dA:dT) tracts adjacent to origins are preferential sites of fork stalling and fork collapse under conditions of HU-induced replication stress in mouse B cells (47). DSB hotspots were also located by END-seq at poly(dA:dT) tracts within the mouse WWOX gene in a region corresponding to the CFS FRA16D AT-rich core in human cells and within the mouse FHIT gene which is the location of CFS FRA3B in humans; these data suggest that poly(dA:dT) tracts far from origins and within gene bodies are also fragile (47). The intergenic breaks at poly (dA:T) sites were present both when cells were exposed to a low dose of HU or no exogenous replication stress, emphasizing the role of sequence in their fragility. In contrast, a study that mapped DSBs by a different technique (BrITL) in conditions of ATR inhibition did not identify mononucleotide A:T runs, but rather break sites were enriched at structure-forming repeats in both mouse and human cells (covered below) (48).
Figure 2. DNA secondary structures and links to fragility.

DNA secondary structures are depicted on the left, corresponding sequences and locations in the human genome that have been linked to fragility listed in the middle, and references on the right. FS, Fragile Site.
Another interesting sequence is the ATTCT repeat at the ATXN10 locus which can expand to cause spinocerebellar ataxia type 10 (SCA10). ATTCT repeats are a DUE as detected by atomic force microscopy (49) and can function as a replication origin in lymphoblastoid cells (50). ATTCT tracts of 80 or 160 repeats were shown to cause fragility when placed on a yeast chromosome (51). The Debatisse lab found that two mammalian DNA replication origins exhibit higher levels of fragility than neighboring DNA upon aphidicolin treatment, providing a connection between replication origin sequences and fragility (52).
Regularly alternating purines and pyrimidines, especially GC and GT repeats, can result in left-handed Z-DNA (21, 53) (Figure 2). DNA sequences predicted to form a mixture of Z-DNA and triplexes reduced plasmid replication rates by up to 50 % compared to a control plasmid and stalled replication by 2D gel analysis in mammalian cells (54). Z-DNA induces DSBs in bacterial and mammalian cells (55) and a (CG)14 Z-DNA-forming repeat caused a 20x increase in mutation rate and nearby deletions in mammalian cells and mice (53). Sequences capable of forming Z-DNA have been found associated with breakage hotspots in the c-myc proto-oncogene in B-cell precursors from acute lymphoblastic leukemia patients and in lymphoid tumors (53).
G-quadruplex structures are four-stranded structures of Hoogsteen-bonded guanines (21, 56) (Figure 2). Interspersed guanine runs – optimally 4 repeats of at least 3 Gs in a row separated by a spacer of 1 to 7 nucleotides, can result in a G-quadruplex structure forming on one DNA strand, leaving the partner DNA strand single-stranded (21, 57). G-quadruplexes have been mapped to breakpoint regions of cancer-causing translocations in humans, implicating them in causing fragility (58–62). G-quadruplexes are involved in immunoglobulin class switching where they are stabilized by R-loops and promote AID-induced nicks (63). G-quadruplexes also can form within telomeric DNA, and telomeres show characteristics of fragile sites when under replication stress (64, 65). In fact an interstitial telomeric sequence appears as a CFS, with increased breakage in conditions where the telomere binding protein TRF1, which is needed for TTAGGG repeat-associated replication, is limiting (65). These data show that an aphidicolin-inducible CFS can be caused by a specific difficult-to-replicate DNA sequence. The helicases FANCJ, WRN, BLM/Sgs1, RTEL, and the PIF1 family helicases are especially important for faithful replication and preventing deletions of G-quadruplexes (57, 66–71).
DNA hairpin or stem-loop structures occur when one DNA strand base-pairs with itself rather than the strand of opposite polarity and are formed by many different types of sequences (Figure 2). Hairpins formed on opposite strands in an offset configuration are referred to as slipped-strand structures (Figure 2). Many expandable trinucleotide repeats (TNRs) that have been shown to be associated with chromosome fragility form hairpin structures, including CAG, CTG, CGG, and CCG sequences. In addition, many other types of repeats such as AT-rich repeats and inverted repeats (IRs), which are often found within CFS regions, form stem-loop structures.
Expansions of CGG TNRs cause fragile X syndrome and FRAXE mental retardation and are visible on human metaphase chromosomes as rare fragile sites (72). Expanded CGG repeats can adopt abnormal secondary structures including hairpin and G-quadruplex structures (6), though in an in vivo yeast replication system, fork stalling at a CGG repeat was associated with hairpin and not G-quadruplex formation (73). CGG repeat-induced fork stalling in mammalian cells correlates well with the threshold for expansions in humans (74). Further, expanded CGG tracts at the FMR1 locus cause late replication and problems with replication elongation (6). Expanded CGGs also cause chromosome fragility in a recombination-based yeast system (75). Fragility is exacerbated in the absence of the Srs2 helicase, which facilitates replication of CGG repeats via its helicase activity and ability to bind PCNA (73).
CAG/CTG repeat expansion is the cause of multiple heritable degenerative diseases including Huntington’s disease, myotonic dystrophy type 1, and several spinocerebellar ataxias (6, 38). Both the CAG strand and complementary CTG strand are able to form hairpins, which may make these structures especially likely to expand. CAG/CTG repeats cause fork stalling in both mammalian and yeast cells, and seem to be especially prone to fork reversal as visualized by EM in vitro and by 2D gels in vivo (76, 77). Expanded CAG/CTG repeats also cause fragility when inserted on a yeast chromosome (78–80). In primate cells, DSBs near CTG repeats result in deletions, with deletion length correlating with repetitive DNA length (81). Cells from myotonic dystrophy patients containing expanded CTG repeats have increased formation of micronuclei, a consequence of chromosome breakage (82). These studies all implicate hairpin structures as a cause for DNA breakage. Not surprisingly, cells have mechanisms for unwinding hairpins to prevent genome instability. In yeast, both Srs2 and Sgs1 helicases can unwind CAG and CTG hairpins in vitro, and both also protect against fragility of expanded CAG/CTG repeats (83, 84). The human RTEL helicase can unwind CAG and CTG hairpins in vitro, substitute for yeast Srs2 in protecting against fragility, and prevent CAG expansions in a human cell system (85). The Sgs1 homolog WRN was also found to unwind CAG/CTG repeat hairpins and prevent repeat contractions (86), and the FANCJ helicase suppresses microsatellite and CAG/CTG repeat instability (87, 88). Thus, multiple helicases likely cooperate to unwind hairpin structures and prevent repeat instability and chromosome fragility.
Expanded GAA repeats form triple helical structures called triplex or H-DNA (89–91) (Figure 2). Expanded GAA repeats cause replication fork stalling in S. cerevisiae in a length-dependent manner when present in greater than 20–40 copies on the lagging strand (92). GAA repeats are fragile sites on yeast chromosomes (42) and in human cells (93). Further, GAA and GAAA predicted triplex-forming repeats are enriched near translocation and deletion breakpoints in cancer genomes (45). A naturally occurring H-DNA forming sequence in the c-myc promoter maps to the Bcl-2 major breakpoint region and implicates secondary structures in common c-myc translocations that occur in lymphomas (94–97). Though expanded triplet repeats have not been associated with CFSs, the principles governing their fragility may be similar to structure-forming sequences within CFSs.
A cruciform structure is formed when two hairpins or stem-loops are located directly across from one another (Figure 2). Cruciform structures form within double-stranded DNA, thus they require conditions where the double helix can be easily unwound, such as AT rich sequences within negatively supercoiled DNA. For example, AT/TA dinucleotide repeats can participate in intrastrand base-pairing on both strands to form a cruciform. Cruciforms form at AT repeats on plasmids in bacteria and yeast when the stem length exceeds 22 bp (98–101). When an AT repeat within the Flex1 subregion of CFS FRA16D exceeded 46 bp (23 repeats) it caused replication fork stalling, which correlates well with the length requirements for cruciform formation (102). The Flex1 repeat induced breakage of a yeast chromosome (102), and recent results from our lab show that breakage increases with AT repeat length, proportional to fork stalling ability (36). These results directly link cruciform formation, fork stalling, and chromosome fragility. Polymorphic (AT)-rich DNA was enriched when compared to 20,000 cancer genome translocation breakpoints and deletion breakpoint data from the COSMIC database, and different patient translocation/deletion breakpoints were found at the same nucleotide position, supporting the capability of these sequences to form secondary structures and break in human cells (45).
Palindromic AT-rich repeats (PATRRs) can form large stem-loop secondary structures and are found at the breakpoints of known recurrent translocations, such as the t(11;22) translocation between PATRR11 and PATRR22, which causes Emanuel syndrome. Cruciform cleavage and resolution at PATRR11 and PATRR22 was proposed to be the basis of chromosomal translocations between these sequences (103). PATRR-mediated translocations occur de novo in sperm cells but not in somatic cells, implying that these translocations are not mediated by the process of replication (104, 105). Interestingly, PATRR11 resides within the CFS FRA11G sequence, which indicates that the region is also prone to fragility in somatic cells (106). Additionally, mild replication stress by siRNA against DNA polymerase α leads to deletions but not translocations at PATRR11 in cultured cell lines (107). These data suggest that translocations at PATRRs in germ cells and deletions in somatic cells occur through separate mechanisms. PATRRs 11 and 22 are both associated with non-recurrent translocations as well, and have been proposed to play a role in genome evolution (108).
Inverted repeats (IRs) are two sequences with complementary DNA on the same strand that are facing towards one another across a center of symmetry (Figure 2). Note that many dinucleotide repeats are also inverted repeats. IRs can form hairpins in ssDNA and certain IRs can form cruciforms in supercoiled dsDNA (109, 110). IRs form hairpins in vivo in S. cerevisiae when complementary sequences are as short as 7 bp (111). Due to their secondary structure, IRs stall replication forks in prokaryotes and eukaryotes, including mammalian cells (112, 113). Fork stalling decreased along with decreased percent homology and increased spacer length between the IRs, implying that the more stable a secondary structure, the more difficult it is for replication machinery to traverse (112). Similarly, the spacer distance between IRs inversely correlates with deletion and recombination rates in S. cerevisiae (114). IRs are sites of recombination and deletions both on yeast chromosomes (114, 115) and in human cells (113). Computational analysis of almost 20,000 translocation and 46,000 deletion breakpoints from cancer genomes revealed that small IRs of 7–30 bp in length were enriched within 200 bp of breakpoints. (45). Alu-repeats are roughly 300 bp in length and are the most common type of long IR in the human genome. Closely spaced Alu IRs form hairpins that stall forks and cause chromosome breakage (112, 116). Reduced levels of polymerases α and δ in S. cerevisiae show increased chromosome loss, recombination, and translocations at transposons that can form long IRs (117, 118).
A recent genome-wide analysis of break sites in conditions of ATR inhibition alone or in combination with low-dose aphidocolin was accomplished using two different methods: RPA ChIP and a new high-resolution break detection method, Breaks Identified by TdT Labeling (BrITL) (48). In mouse embryonic fibroblasts, there was a strong enrichment for microsatellite repeats CAGAGG and CACAG (and derivatives), which were shown to form a structure and pause replication, as well as IRs and quasi-palindromes. In human breast cancer cells, structure-forming repeats were also highly enriched at break sites, but interestingly the repeat sequences were different from those identified in mouse cells. In human cells there was a significant bias for AT-rich repeats that form stable stem-loop structures, including perfect AT repeats and PATTRs, with a greater-than-expected overlap with CFSs. Other quasi- and perfect palindromes, including inverted Alu elements, were also identified. This study reinforces the link between DNA structures and fragility and indicates that the exact type of structure and base composition can differ among organisms. Interestingly, poly (dA:T) sequences were not enriched in this study (48) as they were in (47). This could be due to the break detection methods used, which had significant differences. Notably, the End-seq method starts with an end resection step followed by dA tailing and ligation of a hairpin adaptor, whereas BrITL labels ends directly by addition of a ddUTP-biotin conjugate. The latter may work better for recovering hairpin-capped ends, which can form when breaks occur within a stem-loop structure (see section VII).
IV. Secondary structures at CFSs: indirect and direct connections to fragility
CFSs are large regions compared to the defined DNA structures listed above, and the basis of their fragility is less well understood. CFSs tend to finish replication late in the cell cycle (3, 119, 120). Several lines of evidence suggest CFSs are enriched in their ability to form secondary structures, and that those structures play a role in CFS expression (121).
Computational analysis revealed that CFSs have high A/T content (>70%) and are enriched in interrupted runs of AT/TA dinucleotide repeats that are predicted to form stem-loop and hairpin structures by Mfold (122, 123). The programs TwistFlex and FlexStab were created to identify DNA regions with high flexibility between bases, and thus flexibility peaks in the program tend to be AT-rich and capable of forming secondary structures (122). TwistFlex identified the Flex1 and Flex5 subregions of FRA16D as likely to be forming secondary structures (124). Flex1 is the highest flexibility peak predicted in FRA16D by the FlexStab program. It is a roughly 300 bp subregion of FRA16D that contains a perfect AT dinucleotide repeat that is highly polymorphic in humans (125) and predicted to form a cruciform structure (102). The Freudenreich lab showed that as AT repeat length increased at Flex1 replication fork stalling increased, as measured by observing replication intermediates in yeast by 2D gels (102). We recently obtained direct evidence that increased numbers of AT repeats within the Flex1 region cause higher levels of chromosome fragility, in a manner consistent with cruciform formation (36). These results directly link AT repeat length, fork stalling, and fragility.
The Flex1 AT dinucleotide repeats within FRA16D coincides with deletion breakpoints in multiple cancer cell lines: AGS, HCT116, CO-116, KM12C (primary) and KM12SM (metastasis) cell lines all had Flex1-spanning deletions (124–126). An analysis of 3,131 cancer specimens in one study and 746 cancer cell lines in a separate study determined that WWOX (which contains FRA16D) was the third most common site of deletions in the entire human genome (126, 127). An intriguing possibility is that longer AT alleles at FRA16D Flex1 are associated with an increased likelihood of deletion in cells undergoing replication stress, such as cancer cells. Allelic imbalance and loss of heterozygosity (LOH) occurs preferentially at CFSs in both cancerous and pre-cancerous cells, and thus they may play a role in cancer development (128–130). In fact overexpression of oncogenes results in replication stress, termed oncogene-induced replication stress, which results in fragility, deletions, and rearrangements at CFSs ((131) reviewed in (120, 132, 133)). Interestingly, 40–80% of cancer cell lines with a deletion in FRA16D also contain a deletion in FRA3B and vice versa, which supports the idea of shared mechanisms of CFS fragility (125, 134, 135).
Consistent with the studies that showed that the Flex1 AT repeat stalls replication in yeast, DNA replication dynamics in human cells indicates that AT-rich sequences with the potential to form secondary structures block replication forks traversing CFSs. DNA combing of FRA16C revealed that replication forks are blocked under normal replication conditions in a lymphoblastoid cell line at long (>400 bp) AT-rich sequences (136). The correlation of block sites and AT flexibility peaks was statistically significant and occurred both without and with aphidicolin replication stress (136). Notably, FISH probing of CFSs FRA16C and FRA10E found that they colocalize to the same regions as rare fragile sites FRA16B and FRA10B, respectively, which contain expanded AT-rich repeats predicted to form stem-loop structures (123, 137, 138). FRA7E also contains a long interrupted AT/TA dinucleotide repeat (~300 bp) predicted to form multiple stem-loop structures (123). At FRA18C, a paternal aphidicolin-sensitive fragile site coincided with a chromosome truncation in the daughter, and the breakpoint was in a region that was highly enriched in AT-rich sequences (139). This case implicates in vivo fragility at AT/TA dinucleotide repeat sequences in genome rearrangements.
Single molecule analysis of replicated DNA (SMARD) through CFS FRA16D revealed that stalling occurred at multiple pause sites within the FRA16D AT-rich fragility core in FANCD2−/− lymphoblasts, instigating activation of dormant origins (140). The activation of the origins in the region adjacent to the fragility core generated the 3’ to 5’ replication forks required to complete replication and compensate for the 5’ to 3’ replication stalling observed in the fragility core. Similarly, SMARD through the FRA6E fragility core revealed replication fork pausing in FANCD2−/− lymphoblasts (140). A previous study of FRA6E using molecular combing analysis of primary human peripheral lymphocytes also showed a switch from bidirectional to unidirectional replication upon aphidicolin treatment (141). Thus when forks stall in conditions of replication stress, such as inhibition of polymerase by aphidicolin or the absence of the FANCD2 protein that facilitates replication of paused forks, this results in an altered replication program for the region.
The Wang lab used Mfold (142) to computationally predict the secondary structure forming capability of CFS DNA (143). The capability of 300 nt segments to form secondary structures were analyzed, as this is the size of an Okazaki initiation zone in mammalian cells and thus could reasonably be single-stranded during DNA replication. Windows of fragile versus non-fragile DNA on human chromosome 10 were grouped to look for clusters of sequences forming stable secondary structures with a more negative ΔG. CFSs FRA10G, FRA10D, and FRA10F were identified as enriched in low free-energy segments per section compared to non-fragile DNA. They confirmed the ability of these sequences to form hairpins or cruciforms by an in vitro assay, denaturing the DNA fragments predicted to form secondary structures and allowing them to re-duplex in various concentrations of NaCl. These data validate the use of Mfold to predict secondary structure formation by DNA sequence. FRA3B and FRA16D were also evaluated and each had eight and three regions, respectively, predicted to form stem loop structures in ssDNA. These regions correlated with known breakpoint and LOH regions in various types of cancers (143). Another genome-wide computational approach by the Eckert and Makova labs found that aphidicolin-induced CFSs have high DNA flexibility and are enriched in Alu repeats and mononucleotide microsatellites (144). Cytogenetically mapped aphildicolin-inducible CFSs co-located with evolutionarily conserved chromosomal breakpoints (144). Collectively, these data show a correlation between structure-forming capability, replication fork blockage, and fragility at CFSs.
V. Secondary structures and their relation to other theories for CFS expression
In contrast to the studies listed above, analysis of replication dynamics through a 1.5 Mb region of the FRA3B region by molecular combing led to the conclusion that there were no specific fork stalling regions, but rather that the late replication was due to a paucity of origins in the region (145). This is consistent with one of the findings from the analysis of FRA16C that there is a lack of back-up origins available to fire in the fragile region (136). The region of FRA3B analyzed spanned a roughly 1.5 Mb portion of the 4.5 Mb site, and did not include two long AT-rich sequences that would be predicted to cause fork stalling and impair fork speed due to an ability to form abnormal secondary DNA structures (136). One reason that replication is not completed in the core of FRA3B until G2 phase could be due to fork stalling at these secondary DNA structures at the boundaries of the fragile region. The long distance from an origin approaching from the other direction could exacerbate the situation, leading to incomplete replication. Indeed, it may take a combination of fork stalling at DNA structures and origin paucity to create a recurrent fragile site; since there are many origin-poor regions and many potential DNA structures in the human genome it is unlikely that just one of these elements would lead to a failure to finish replication. Nonetheless, it is clear that widely spaced origins can be an important contributing factor to creation of a fragile site.
Another element that influences CFS expression is transcriptional status of the gene, as transcription and transcription-replication collisions have been linked to CFS expression. CFSs tend to be in present in large genes between 300 kb and 2 Mb in length (3, 146–149). Some very long genes that contain CFSs take more than one cell cycle to transcribe (150). CFS expression also varies by cell type when tested in epithelial, erythroid, and fibroblast cell lines (148, 151, 152). As it is known that transcriptional profiles vary across cell types, transcription through CFSs was proposed to play a role in their fragility. Interestingly, FRA16D is fragile in multiple cell types, though not to the same extent, suggesting that its fragility is at least partly governed by something inherently fragile in its sequence or genomic location (148). The presence of a fork-stalling structure within a CFS region could synergize with the likelihood of occurrence of transcription-replication collisions. For example, if a structure-induced stall occurred at a co-directional replication fork within the gene body, this would increase the likelihood that the converging replication fork would approach, which would then be head-on with transcription and more likely to collide with RNA polymerase and cause a break (Figure 3).
Figure 3. Model for how secondary structures could promote downstream transcription-replication collisions at FRA16D and other CFSs.
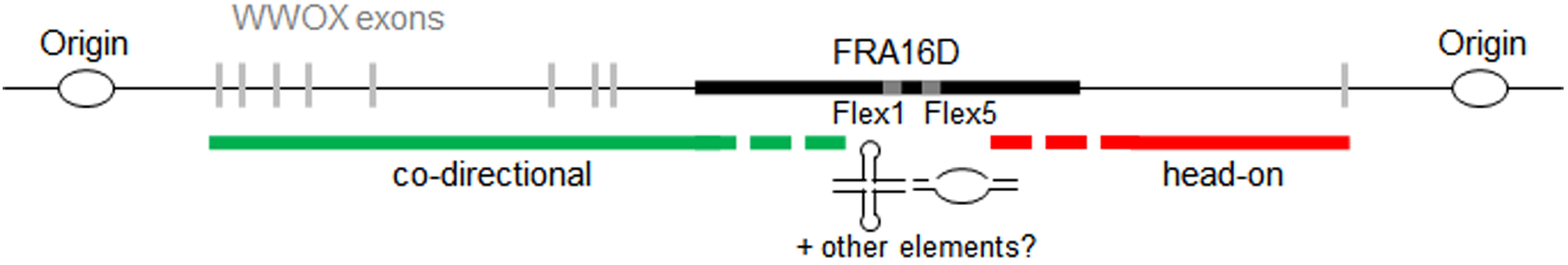
Secondary structures in CFSs can result in replication fork stalling, which could lead to replication of the downstream sequence from the converging fork. Since CFSs tend to be located in large genes that take a long time to transcribe, unscheduled transcription-replication collisions could occur, resulting in a region of unreplicated DNA between the structure-induced stall and the collision-induced stall.
R-loops can form co-transcriptionally and are stabilized by G-rich DNA and the formation of secondary structures on the opposite DNA strand (reviewed in (25)). There is evidence to support a role for R-loops in CFS fragility. Helmrich et al. found evidence for in vivo R-loops at FHIT/FRA3B which were reduced upon the addition of RNaseH (150). It is now known that R-loops are ubiquitous in the human genome, so their presence alone is not diagnostic of a problem that will lead to breaks (153). However, siRNA knockdown of RNaseH1 resulted in an increase in the number of breaks detected at CFSs FRA3B, FRA16D, and FRA7K, indicating that RNaseH1 functions to protect CFS regions by removing RNA:DNA hybrids (150). In addition, treatment with RNaseH1 reduced the number of fork stalls in the FRA16D and FRA6E regions in FANCD2−/− cells, implicating R-loops in causing fork stalling in this region (140). R-loops form stably at several structure-forming repetitive DNA sequences and there is evidence of an interplay between R-loops, DNA structure formation, and fragility (27, 30). R-loops are associated with both replication fork stalling (154) and DNA breaks. For example, at CAG repeats, R-loop-dependent cytosine deamination occurs, leading to base excision repair (BER) processing and APE1/Apn1-dependent breaks (33). Nucleotide excision repair (NER) nucleases XPF and XPG can also process R-loop DNA structures, as well as cruciform and triplex DNA structures, resulting in DSBs and genomic instability (27, 155, 156). Given the interplay between DNA structure and R-loop formation, it is possible that these two mechanisms are working together to cause fragility, or they could be independent events.
Some very recent publications have connected transcription to replication origin usage. The Debatisse lab found that high transcription of large genes resulted in a shift in their replication pattern from late to mid-S phase, likely giving the cells more time to complete synthesis of the regions before M phase (Blin et al., 2018 BioRxiv.org https://doi.org/10.1101/286807). The Smith lab reported that replication stress by HU results in redistribution of replication termination relative to transcription, setting up a situation where replication and transcription are no longer co-directional, so that head-on collisions are more likely to occur (Chen et al., 2018 BioRxiv.org https://doi.org/10.1101/324079). In cells exposed to oncogene-induced replication stress, inappropriate activation of intergenic origins leads to transcription-replication conflicts and fork collapse (157). Thus, other types of replication stress, such as inhibition of DNA polymerase progression by DNA structures, could also lead to a re-programming of origin firing, resulting in collisions between replication and transcription (Figure 3). This would further impact the ability of genes containing DNA structure-induced stalls to finish replication. For example, FRA16D is within one of the last introns of the very large WWOX gene. A replication stall at the Flex1 AT repeat could lead to activation of a dormant origin towards the end of the gene or simply allow time for the fork from the next intergenic origin to enter the 3’ end of WWOX. This would lead to a head-on orientation of transcription and replication for the region downstream of the Flex1-induced stall (Figure 3). A replication-transcription collision in this area would result in a second stall, preventing replication of the intervening region and appearance of a metaphase gap. This model could explain why there is evidence for both structure-induced fork stalling and transcription-replication collisions as the cause of fragility at CFSs.
VI. Cleavage at secondary structures within CFSs is a cause of fragility
Breakage at secondary structures could occur due to exposure of associated ssDNA, which is labile and prone to environmental damage and strand breaks. Another potential mechanism is physical breakage of unreplicated sister chromatids that persist into mitosis (158). Alternatively, breakage at secondary structures can occur actively by targeted cleavage of DNA by nucleases (Figure 4). There are many nucleases that cleave stalled replication forks and recombination intermediates that can arise from broken forks (159, 160). This mechanism can be either replication-associated or not. For example, XPF-ERCC1 (S. cerevisiae Rad1-Rad10) cleaves both cruciform structures and H-DNA independently of replication, suggesting that structures are recognized as DNA damage by the cell and subsequent NER results in their cleavage (97, 113, 161).
Figure 4. Secondary structures at CFSs can impede replication, resulting in fork stalling and cleavage to cause fragility.
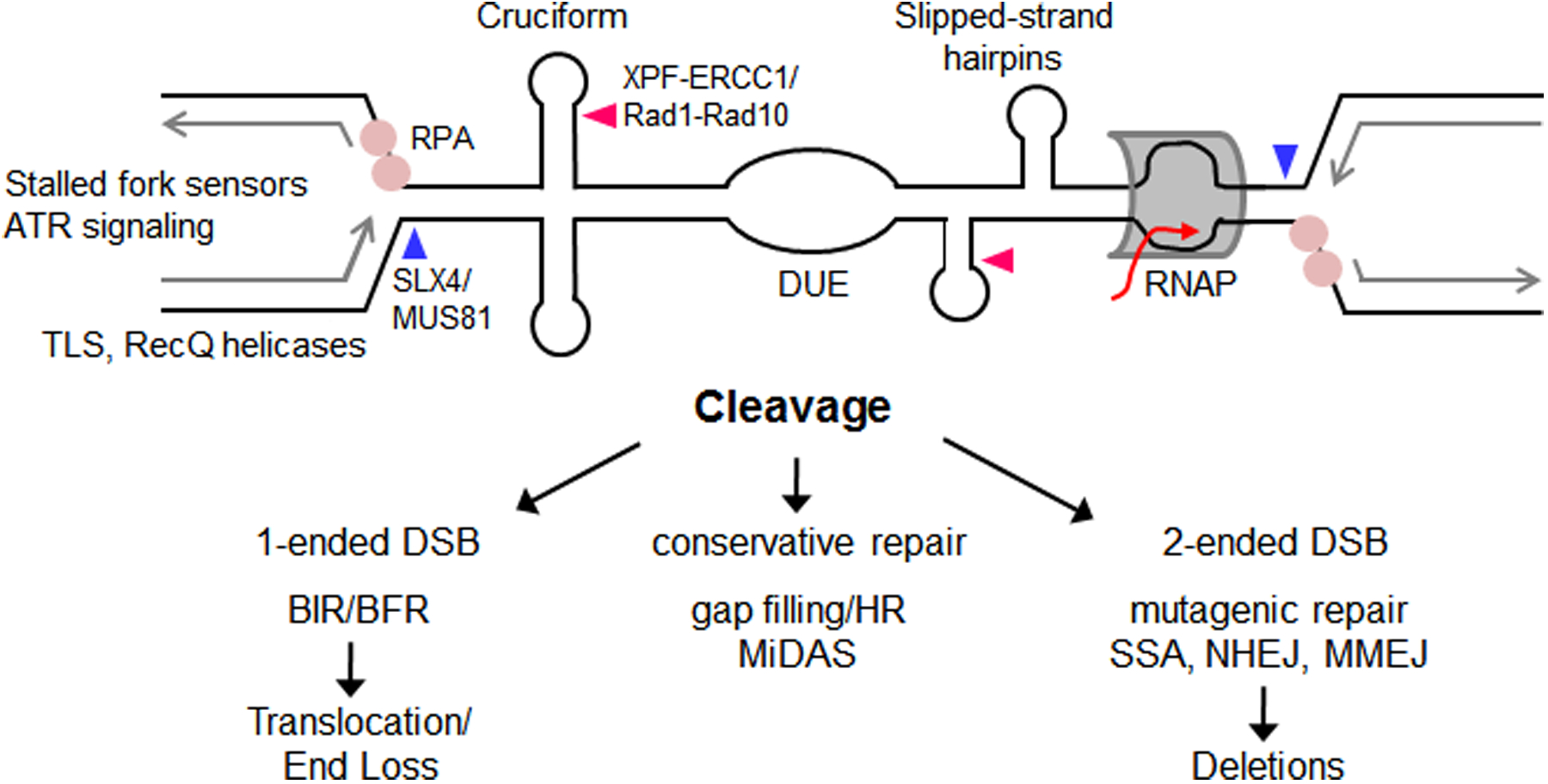
Multiple secondary structures at CFSs may progressively impede replication. Replication impairment can potentially be resolved by helicases such as WRN and RecQ1 (191, 192). TLS synthesis could also facilitate replication, or gap filling if structures are bypassed. If the replisome becomes uncoupled from the replicative helicase, ssDNA stretches can result in RPA binding, activation of stalled fork sensors, and ATR signaling. A stalled fork (top left side) can result in the approach of a converging fork from the opposite direction (top right side) that in turn could stall at other DNA structures or due to a transcription-replication collision, leading to an unreplicated area. Impaired replication can be resolved by structure-specific endonuclease (SSE) cleavage at secondary structures or at stalled and reversed forks caused by secondary structures. Cleavage of a stalled fork by MUS81-EME1/Mus81-Mms4 would result in a one-ended break that could engage in break-induced replication (BIR) using another chromosome as a template for repair (left pathway). Alternatively, broken fork repair (BFR) using the other sister chromatid as a template for repair or gap filling will result in conservative repair without loss of sequence (middle pathway). Cleavage of forks by MUS81/Mus81 at two stalled forks (blue arrows), or across the base of a 4-way junction (not shown), will result in a two-ended break, which can heal with fidelity (for example using homologous recombination (HR); middle pathway) or in a manner that results in deletions (for example by single strand annealing (SSA), non-homologous end joining (NHEJ), or microhomology-mediated end joining MMEJ; right pathway). Other SSEs (e.g. XPF-ERCC1/Rad1-Rad10 (pink arrows), or SLX1-SLX4/Slx1-Slx4 (not shown)) may also be involved in cleavage. Gap filling or HR-dependent mechanisms require DNA synthesis, which can manifest as MiDAS if it occurs in M phase. Human proteins are listed first, with S. cerevisiae homologs after the slash.
Work from the Hickson, Rosselli, and Debatisse labs showed that human complexes MUS81-EME1 (S. cerevisiae Mus81-Mms4) and ERCC1 (S. cerevisiae Rad1-Rad10) are required for CFS expression and sister chromatid separation (162, 163), pointing to a role for structure-specific endonucleases (SSEs) at CFSs. These SSEs are known to target both 3-way junctions that occur at stalled forks and recombination intermediates (160, 164). It was proposed that SSEs induce local fragility at CFSs to avoid global genomic fragility that could occur if the sister chromatids are mechanically separated while still unreplicated and attached to one another (163, 165). If CFSs do not undergo SSE-induced cleavage and sister chromatid separation, DNA bridges can persist during nuclear division, resulting in mechanical breakage of DNA, chromosome mis-segregation and 53BP1 body formation in G1 phase (162, 163, 166). Mammalian cells depleted for MUS81 by short hairpin RNA had increased FRA16D-containing micronuclei and decreased appearance of metaphase breaks and gaps (162, 163). Since these studies used whole chromosomes, they could not point to a specific DNA sequence or sequences that were targeted by SSEs. However, it is interesting to note that Mus81 resolves a cruciform formed on a plasmid in vivo in S. cerevisiae (101). New data from our lab show that Mus81 causes fragility at the Flex1 secondary structure forming subregion of FRA16D, but only when the AT repeat number is sufficient to cause cruciform formation and fork stalling. These results implicate SSEs directly in cleavage of secondary structures at CFSs or replication stall sites caused by the structure (36) (Figure 4). Depending on the location and number of cleavage events, several types of repair can be envisioned, some conservative (e.g. retaining the DNA sequence) and some resulting in deletions or chromosome rearrangements (Figure 4). If multiple secondary structures or stalled forks are cleaved, deletions of the intervening DNA can occur. It is possible that cancer cells gain a replicative advantage by accumulating deletions at CFSs since they are impediments to efficient replication fork progression. Multiple cleavage sites can result in multiple secondary structure-capped DNA ends that will need to be processed, for example by MRN-CtIP/MRX-Sae2 or XPF/Rad1, before healing at regions of homology (Figure 1C). In this case, the location and usage of regions of homology flanking the break will determine the extent of deletion.
CFSs have difficulty replicating, and the Hickson lab found that under replication stress, CFSs incorporate the DNA synthesis label 5-ethynyl-2’-deoxyuridine (EdU) during mitosis (167). This mechanism, Mitotic DNA Synthesis (MiDAS), is dependent on MUS81 activity and its recruitment by the SLX4 scaffold protein. MiDAS is a mechanism for cells to avoid the negative consequences of going through mitosis with unreplicated DNA, which would lead to DNA bridges connecting sister chromatids and subsequent DSBs. Stalled replication forks can be stabilized to prevent Exo1-mediated degradation of the DNA, allowing for rescue by a converging fork or by homologous recombination. In S. pombe, Rad51 and Rad52 prevent arrested replication forks from being degraded, and in their absence mitotic sister chromatid bridging is increased (168). We have found that S. cerevisiae SSEs Mus81, Rad1-Rad10, and Slx1-Slx4 are all important in causing fragility at the Flex1 structure-forming subregion of FRA16D (36). The involvement of all of these components suggests that they are functioning in a SSE super complex as has as has been shown for the human complex in vitro (169), and that one important target of SSEs at CFSs in human cells is forks stalled at DNA structures.
VII. Healing of breaks within DNA structures can lead to inefficient and mutagenic repair
After DSBs occur, they must be healed properly in order to prevent genomic rearrangements. If a DSB occurs during the G1 phase of the cell cycle, end joining pathways of healing predominate, sometimes resulting in small insertions or deletions (indels) (170). In situations where DSBs are formed in S or G2 phases of the cell cycle, they are first resected to create RPA-coated ssDNA stretches through the action of MRE11-RAD50-NBS1 (Mre11-Rad50-Xrs2 in S. cerevisiae) and CtIP (Sae2 in S. cerevisiae), followed by long range resection by Exo1 and Dna2 nucleases (the latter facilitated by a RecQ helicase such as Sgs1 in S. cerevisiae) (170, 171). RPA is then replaced with Rad51, to facilitate a homology search and strand invasion into an appropriate template, usually the sister chromatid (170, 171). If a sister chromatid template is not available, break-induced replication (BIR) can occur whereby the broken DNA invades a non-homologous chromosome. BIR can complete replication through the telomere, or be aborted and lead to further genome rearrangements by template switch or other alternate healing mechanisms (171–173). If a DSB occurs during replication, the broken DNA end can invade the sister chromatid in order to restart replication, also known as broken fork repair (BFR) (170, 174, 175). MiDAS at CFSs may represent a BFR- or BIR-like mechanism of responding to breaks at CFSs where missing information is copied from the sister chromatid or homolog using a Rad52-dependent mechanism (167, 176, 177) (Figure 4).
Several TLS polymerases are recruited to CFSs in order to prevent their expression (178–180). The Eckert lab investigated human polymerase δ holoenzyme transit throughout several subregions from CFSs that contain both poly(dA:dT) runs and sequences predicted to form hairpin structures. They investigated polymerase progression in vitro through a non-AT rich inverted repeat (IR) next to a A19 run from FRA16D, an (AT)25 repeat near an A22 run from FRA3B, and an interrupted (AT)24 repeat followed by a A28 run (Flex5) from FRA16D. They found that human TLS polymerases η and κ can switch with the replicative polymerase δ holoenzyme to alleviate pausing at IRs and mononucleotide runs (44). Thus, TLS polymerases may be important in replicating through mononucleotide runs or gaps formed due to bypass of secondary structures. TLS polymerases are also involved in repair of DSB breaks and could be especially important when repair through DNA structures must occur (181).
Mammalian CtIP and S. cerevisiae Sae2 are required to stimulate MRN/MRX processing of DSBs. Both the mammalian and yeast proteins are important in situations where broken DNA is blocked with structure-forming DNA such as hairpins, in order to cleave the blocked end and avoid deleterious consequences such as deletions and genomic rearrangements (159, 182, 183). We found that Sae2 is needed to heal after Flex1(AT)34-induced breaks in a yeast recombination-based system (36). Similarly CtIP is needed to heal after Flex1(AT)34-induced breaks in a mammalian cell mitotic recombination assay whereas it is not needed at “clean” I-SceI breaks that do not have secondary structure forming sequences (184). Sae2 is also needed for the repair of a TALEN-induced DSB at a hairpin-forming CTG sequence (185). Our data at Flex1 led us to propose a new theory for CFS fragility – that the regions are not only prone to breakage but also have difficulty healing after fragility due to neighboring secondary structures (36). Fragile sites have long been known to be associated with gene amplification through a breakage-fusion-bridge cycle (186, 187). Indeed, after DSB induction next to an IR, 5’ to 3’ end resection exposes DNA to form hairpin-capped DNA ends, resulting in amplification and genomic rearrangements (188–190). These results all provide evidence for a connection between secondary structures, fragility, and genomic instability.
VIII. Conclusions
Both common and rare fragile sites are associated with gaps and breaks on chromosomes that represent unreplicated regions prone to breakage. However, while rare fragile site fragility is generally caused by expanded repetitive DNA sequences that form secondary structures, the molecular basis of CFS fragility has been more elusive. Current evidence shows that CFSs are enriched in sequences with secondary structure forming capability, and the study of the Flex1 region of FRA16D supports a causative relationship between DNA structure formation, fork stalling, and fragility. This mechanism could synergize with additional issues at CFS regions, including late replication and co-occurrence of transcription and replication, increasing the chance of collisions. We propose that the characteristic expression of CFSs is a combination of a propensity for fork stalling, subsequent nuclease cleavage, and difficulty healing due to the presence of secondary structures at the broken ends. Thus, deletions of structure-forming, fork-stalling subregions of CFS sequences could give tumor cells a replicative advantage.
References
- 1.Magenis RE, Hecht F, and Lovrien EW (1970) Heritable fragile site on chromosome 16: probable localization of haptoglobin locus in man. Science 170, 85–87 [DOI] [PubMed] [Google Scholar]
- 2.Schwartz M, Zlotorynski E, and Kerem B (2006) The molecular basis of common and rare fragile sites. Cancer Lett 232, 13–26 [DOI] [PubMed] [Google Scholar]
- 3.Durkin SG, and Glover TW (2007) Chromosome fragile sites. Annu Rev Genet 41, 169–192 [DOI] [PubMed] [Google Scholar]
- 4.Techer H, Koundrioukoff S, Nicolas A, and Debatisse M (2017) The impact of replication stress on replication dynamics and DNA damage in vertebrate cells. Nat Rev Genet 18, 535–550 [DOI] [PubMed] [Google Scholar]
- 5.Irony-Tur Sinai M, and Kerem B (2018) DNA replication stress drives fragile site instability. Mutat Res 808, 56–61 [DOI] [PubMed] [Google Scholar]
- 6.Usdin K, House NC, and Freudenreich CH (2015) Repeat instability during DNA repair: Insights from model systems. Crit Rev Biochem Mol Biol 50, 142–167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Balakrishnan L, and Bambara RA (2013) Flap endonuclease 1. Annu Rev Biochem 82, 119–138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tsutakawa SE, Thompson MJ, Arvai AS, Neil AJ, Shaw SJ, Algasaier SI, Kim JC, Finger LD, Jardine E, Gotham VJB, Sarker AH, Her MZ, Rashid F, Hamdan SM, Mirkin SM, Grasby JA, and Tainer JA (2017) Phosphate steering by Flap Endonuclease 1 promotes 5’-flap specificity and incision to prevent genome instability. Nat Commun 8, 15855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Polyzos AA, and McMurray CT (2017) Close encounters: Moving along bumps, breaks, and bubbles on expanded trinucleotide tracts. DNA Repair (Amst) 56, 144–155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kim JC, and Mirkin SM (2013) The balancing act of DNA repeat expansions. Curr Opin Genet Dev 23, 280–288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Polleys EJ, House NCM, and Freudenreich CH (2017) Role of recombination and replication fork restart in repeat instability. DNA Repair (Amst) 56, 156–165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lin Y, and Wilson JH (2011) Transcription-induced DNA toxicity at trinucleotide repeats: double bubble is trouble. Cell Cycle 10, 611–618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Frank-Kamenetskii MD (1990) DNA Topology and Its Biological Effects, Cold Spring Harbor Laboratory Press, Plainview, NY [Google Scholar]
- 14.Sinden RR (1994) DNA structure and function, Academic Press, San Diego [Google Scholar]
- 15.Lin Y, Hubert L Jr., and Wilson JH (2009) Transcription destabilizes triplet repeats. Mol Carcinog 48, 350–361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Napierala M, Bacolla A, and Wells RD (2005) Increased negative superhelical density in vivo enhances the genetic instability of triplet repeat sequences. J Biol Chem 280, 37366–37376 [DOI] [PubMed] [Google Scholar]
- 17.Salinas-Rios V, Belotserkovskii BP, and Hanawalt PC (2011) DNA slip-outs cause RNA polymerase II arrest in vitro: potential implications for genetic instability. Nucleic Acids Res 39, 7444–7454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lin Y, Leng M, Wan M, and Wilson JH (2010) Convergent transcription through a long CAG tract destabilizes repeats and induces apoptosis. Mol Cell Biol 30, 4435–4451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Budworth H, and McMurray CT (2013) Bidirectional transcription of trinucleotide repeats: roles for excision repair. DNA Repair (Amst) 12, 672–684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lin WY, Lin Y, and Wilson JH (2014) Convergent transcription through microsatellite repeat tracts induces cell death. Mol Biol Rep 41, 5627–5634 [DOI] [PubMed] [Google Scholar]
- 21.Mirkin EV, and Mirkin SM (2007) Replication fork stalling at natural impediments. Microbiol Mol Biol Rev 71, 13–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hamperl S, and Cimprich KA (2016) Conflict Resolution in the Genome: How Transcription and Replication Make It Work. Cell 167, 1455–1467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Garcia-Muse T, and Aguilera A (2016) Transcription-replication conflicts: how they occur and how they are resolved. Nat Rev Mol Cell Biol 17, 553–563 [DOI] [PubMed] [Google Scholar]
- 24.Szlachta K, Thys RG, Atkin ND, Pierce LCT, Bekiranov S, and Wang YH (2018) Alternative DNA secondary structure formation affects RNA polymerase II promoter-proximal pausing in human. Genome Biol 19, 89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Santos-Pereira JM, and Aguilera A (2015) R loops: new modulators of genome dynamics and function. Nat Rev Genet 16, 583–597 [DOI] [PubMed] [Google Scholar]
- 26.Hamperl S, Bocek MJ, Saldivar JC, Swigut T, and Cimprich KA (2017) Transcription-Replication Conflict Orientation Modulates R-Loop Levels and Activates Distinct DNA Damage Responses. Cell 170, 774–786 e719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Freudenreich CH (2018) R-loops: targets for nuclease cleavage and repeat instability. Curr Genet 64, 789–794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Belotserkovskii BP, Mirkin SM, and Hanawalt PC (2013) DNA sequences that interfere with transcription: implications for genome function and stability. Chem Rev 113, 8620–8637 [DOI] [PubMed] [Google Scholar]
- 29.Salvi JS, and Mekhail K (2015) R-loops highlight the nucleus in ALS. Nucleus 6, 23–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Maizels N, and Gray LT (2013) The G4 genome. PLoS Genet 9, e1003468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Neil AJ, Liang MU, Khristich AN, Shah KA, and Mirkin SM (2018) RNA-DNA hybrids promote the expansion of Friedreich’s ataxia (GAA)n repeats via break-induced replication. Nucleic Acids Res 46, 3487–3497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pannunzio NR, and Lieber MR (2017) AID and Reactive Oxygen Species Can Induce DNA Breaks within Human Chromosomal Translocation Fragile Zones. Mol Cell 68, 901–912 e903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Su XA, and Freudenreich CH (2017) Cytosine deamination and base excision repair cause R-loop-induced CAG repeat fragility and instability in Saccharomyces cerevisiae. Proc Natl Acad Sci U S A 114, E8392–E8401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chen H, Lisby M, and Symington LS (2013) RPA coordinates DNA end resection and prevents formation of DNA hairpins. Mol Cell 50, 589–600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Deng SK, Yin Y, Petes TD, and Symington LS (2015) Mre11-Sae2 and RPA Collaborate to Prevent Palindromic Gene Amplification. Mol Cell 60, 500–508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kaushal S, Wollmuth CE, Barnes RP, Das K, Regan SB, Haouzi A, Lee SM, House NCM, Eckert KA, and Freudenreich CH (in revision) Sequence and nuclease requirements for breakage and healing of a structure-forming (AT)n sequence within fragile site FRA16D. [DOI] [PMC free article] [PubMed]
- 37.Sundararajan R, Gellon L, Zunder RM, and Freudenreich CH (2010) Double-strand break repair pathways protect against CAG/CTG repeat expansions, contractions and repeat-mediated chromosomal fragility in Saccharomyces cerevisiae. Genetics 184, 65–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.McMurray CT (2010) Mechanisms of trinucleotide repeat instability during human development. Nat Rev Genet 11, 786–799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liu Y, and Wilson SH (2012) DNA base excision repair: a mechanism of trinucleotide repeat expansion. Trends Biochem Sci 37, 162–172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pearson CE, Ewel A, Acharya S, Fishel RA, and Sinden RR (1997) Human MSH2 binds to trinucleotide repeat DNA structures associated with neurodegenerative diseases. Hum Mol Genet 6, 1117–1123 [DOI] [PubMed] [Google Scholar]
- 41.Viterbo D, Michoud G, Mosbach V, Dujon B, and Richard GF (2016) Replication stalling and heteroduplex formation within CAG/CTG trinucleotide repeats by mismatch repair. DNA Repair (Amst) 42, 94–106 [DOI] [PubMed] [Google Scholar]
- 42.Kim HM, Narayanan V, Mieczkowski PA, Petes TD, Krasilnikova MM, Mirkin SM, and Lobachev KS (2008) Chromosome fragility at GAA tracts in yeast depends on repeat orientation and requires mismatch repair. EMBO J 27, 2896–2906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Song W, Dominska M, Greenwell PW, and Petes TD (2014) Genome-wide high-resolution mapping of chromosome fragile sites in Saccharomyces cerevisiae. Proc Natl Acad Sci U S A 111, E2210–2218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Barnes RP, Hile SE, Lee MY, and Eckert KA (2017) DNA polymerases eta and kappa exchange with the polymerase delta holoenzyme to complete common fragile site synthesis. DNA Repair (Amst) 57, 1–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bacolla A, Tainer JA, Vasquez KM, and Cooper DN (2016) Translocation and deletion breakpoints in cancer genomes are associated with potential non-B DNA-forming sequences. Nucleic Acids Res 44, 5673–5688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Barlow JH, Faryabi RB, Callen E, Wong N, Malhowski A, Chen HT, Gutierrez-Cruz G, Sun HW, McKinnon P, Wright G, Casellas R, Robbiani DF, Staudt L, Fernandez-Capetillo O, and Nussenzweig A (2013) Identification of early replicating fragile sites that contribute to genome instability. Cell 152, 620–632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tubbs A, Sridharan S, van Wietmarschen N, Maman Y, Callen E, Stanlie A, Wu W, Wu X, Day A, Wong N, Yin M, Canela A, Fu H, Redon C, Pruitt SC, Jaszczyszyn Y, Aladjem MI, Aplan PD, Hyrien O, and Nussenzweig A (2018) Dual Roles of Poly(dA:dT) Tracts in Replication Initiation and Fork Collapse. Cell 174, 1127–1142 e1119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shastri N, Tsai YC, Hile S, Jordan D, Powell B, Chen J, Maloney D, Dose M, Lo Y, Anastassiadis T, Rivera O, Kim T, Shah S, Borole P, Asija K, Wang X, Smith KD, Finn D, Schug J, Casellas R, Yatsunyk LA, Eckert KA, and Brown EJ (2018) Genome-wide Identification of Structure-Forming Repeats as Principal Sites of Fork Collapse upon ATR Inhibition. Mol Cell 72, 222–238 e211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Potaman VN, Bissler JJ, Hashem VI, Oussatcheva EA, Lu L, Shlyakhtenko LS, Lyubchenko YL, Matsuura T, Ashizawa T, Leffak M, Benham CJ, and Sinden RR (2003) Unpaired structures in SCA10 (ATTCT)n.(AGAAT)n repeats. J Mol Biol 326, 1095–1111 [DOI] [PubMed] [Google Scholar]
- 50.Liu G, Bissler JJ, Sinden RR, and Leffak M (2007) Unstable spinocerebellar ataxia type 10 (ATTCT*(AGAAT) repeats are associated with aberrant replication at the ATX10 locus and replication origin-dependent expansion at an ectopic site in human cells. Mol Cell Biol 27, 7828–7838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cherng N, Shishkin AA, Schlager LI, Tuck RH, Sloan L, Matera R, Sarkar PS, Ashizawa T, Freudenreich CH, and Mirkin SM (2011) Expansions, contractions, and fragility of the spinocerebellar ataxia type 10 pentanucleotide repeat in yeast. Proc Natl Acad Sci U S A 108, 2843–2848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Toledo F, Coquelle A, Svetlova E, and Debatisse M (2000) Enhanced flexibility and aphidicolin-induced DNA breaks near mammalian replication origins: implications for replicon mapping and chromosome fragility. Nucleic Acids Res 28, 4805–4813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Vasquez KM, and Wang G (2013) The yin and yang of repair mechanisms in DNA structure-induced genetic instability. Mutat Res 743–744, 118–131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Brinton BT, Caddle MS, and Heintz NH (1991) Position and orientation-dependent effects of a eukaryotic Z-triplex DNA motif on episomal DNA replication in COS-7 cells. J Biol Chem 266, 5153–5161 [PubMed] [Google Scholar]
- 55.Wang G, Christensen LA, and Vasquez KM (2006) Z-DNA-forming sequences generate large-scale deletions in mammalian cells. Proc Natl Acad Sci U S A 103, 2677–2682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gilbert DE, and Feigon J (1999) Multistranded DNA structures. Curr Opin Struct Biol 9, 305–314 [DOI] [PubMed] [Google Scholar]
- 57.Bochman ML, Paeschke K, and Zakian VA (2012) DNA secondary structures: stability and function of G-quadruplex structures. Nat Rev Genet 13, 770–780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Rimokh R, Rouault JP, Wahbi K, Gadoux M, Lafage M, Archimbaud E, Charrin C, Gentilhomme O, Germain D, Samarut J, and et al. (1991) A chromosome 12 coding region is juxtaposed to the MYC protooncogene locus in a t(8;12)(q24;q22) translocation in a case of B-cell chronic lymphocytic leukemia. Genes Chromosomes Cancer 3, 24–36 [DOI] [PubMed] [Google Scholar]
- 59.Busch K, Keller T, Fuchs U, Yeh RF, Harbott J, Klose I, Wiemels J, Novosel A, Reiter A, and Borkhardt A (2007) Identification of two distinct MYC breakpoint clusters and their association with various IGH breakpoint regions in the t(8;14) translocations in sporadic Burkitt-lymphoma. Leukemia 21, 1739–1751 [DOI] [PubMed] [Google Scholar]
- 60.Nambiar M, Goldsmith G, Moorthy BT, Lieber MR, Joshi MV, Choudhary B, Hosur RV, and Raghavan SC (2011) Formation of a G-quadruplex at the BCL2 major breakpoint region of the t(14;18) translocation in follicular lymphoma. Nucleic Acids Res 39, 936–948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Katapadi VK, Nambiar M, and Raghavan SC (2012) Potential G-quadruplex formation at breakpoint regions of chromosomal translocations in cancer may explain their fragility. Genomics 100, 72–80 [DOI] [PubMed] [Google Scholar]
- 62.Nambiar M, Srivastava M, Gopalakrishnan V, Sankaran SK, and Raghavan SC (2013) G-quadruplex structures formed at the HOX11 breakpoint region contribute to its fragility during t(10;14) translocation in T-cell leukemia. Mol Cell Biol 33, 4266–4281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Duquette ML, Handa P, Vincent JA, Taylor AF, and Maizels N (2004) Intracellular transcription of G-rich DNAs induces formation of G-loops, novel structures containing G4 DNA. Genes Dev 18, 1618–1629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sfeir A, Kosiyatrakul ST, Hockemeyer D, MacRae SL, Karlseder J, Schildkraut CL, and de Lange T (2009) Mammalian telomeres resemble fragile sites and require TRF1 for efficient replication. Cell 138, 90–103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Bosco N, and de Lange T (2012) A TRF1-controlled common fragile site containing interstitial telomeric sequences. Chromosoma 121, 465–474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zimmermann M, Kibe T, Kabir S, and de Lange T (2014) TRF1 negotiates TTAGGG repeat-associated replication problems by recruiting the BLM helicase and the TPP1/POT1 repressor of ATR signaling. Genes Dev 28, 2477–2491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Drosopoulos WC, Kosiyatrakul ST, and Schildkraut CL (2015) BLM helicase facilitates telomere replication during leading strand synthesis of telomeres. J Cell Biol 210, 191–208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Vannier JB, Sarek G, and Boulton SJ (2014) RTEL1: functions of a disease-associated helicase. Trends Cell Biol 24, 416–425 [DOI] [PubMed] [Google Scholar]
- 69.Geronimo CL, and Zakian VA (2016) Getting it done at the ends: Pif1 family DNA helicases and telomeres. DNA Repair (Amst) 44, 151–158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Crabbe L, Verdun RE, Haggblom CI, and Karlseder J (2004) Defective telomere lagging strand synthesis in cells lacking WRN helicase activity. Science 306, 1951–1953 [DOI] [PubMed] [Google Scholar]
- 71.Kruisselbrink E, Guryev V, Brouwer K, Pontier DB, Cuppen E, and Tijsterman M (2008) Mutagenic capacity of endogenous G4 DNA underlies genome instability in FANCJ-defective C. elegans. Curr Biol 18, 900–905 [DOI] [PubMed] [Google Scholar]
- 72.Verkerk AJ, Pieretti M, Sutcliffe JS, Fu YH, Kuhl DP, Pizzuti A, Reiner O, Richards S, Victoria MF, Zhang FP, and et al. (1991) Identification of a gene (FMR-1) containing a CGG repeat coincident with a breakpoint cluster region exhibiting length variation in fragile X syndrome. Cell 65, 905–914 [DOI] [PubMed] [Google Scholar]
- 73.Anand RP, Shah KA, Niu H, Sung P, Mirkin SM, and Freudenreich CH (2012) Overcoming natural replication barriers: differential helicase requirements. Nucleic Acids Res 40, 1091–1105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Voineagu I, Surka CF, Shishkin AA, Krasilnikova MM, and Mirkin SM (2009) Replisome stalling and stabilization at CGG repeats, which are responsible for chromosomal fragility. Nat Struct Mol Biol 16, 226–228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Balakumaran BS, Freudenreich CH, and Zakian VA (2000) CGG/CCG repeats exhibit orientation-dependent instability and orientation-independent fragility in Saccharomyces cerevisiae. Hum Mol Genet 9, 93–100 [DOI] [PubMed] [Google Scholar]
- 76.Cleary JD, Nichol K, Wang YH, and Pearson CE (2002) Evidence of cis-acting factors in replication-mediated trinucleotide repeat instability in primate cells. Nat Genet 31, 37–46 [DOI] [PubMed] [Google Scholar]
- 77.Pearson CE, Wang YH, Griffith JD, and Sinden RR (1998) Structural analysis of slipped-strand DNA (S-DNA) formed in (CTG)n. (CAG)n repeats from the myotonic dystrophy locus. Nucleic Acids Res 26, 816–823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Freudenreich CH, Kantrow SM, and Zakian VA (1998) Expansion and length-dependent fragility of CTG repeats in yeast. Science 279, 853–856 [DOI] [PubMed] [Google Scholar]
- 79.Jankowski C, Nasar F, and Nag DK (2000) Meiotic instability of CAG repeat tracts occurs by double-strand break repair in yeast. Proc Natl Acad Sci U S A 97, 2134–2139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Callahan JL, Andrews KJ, Zakian VA, and Freudenreich CH (2003) Mutations in yeast replication proteins that increase CAG/CTG expansions also increase repeat fragility. Mol Cell Biol 23, 7849–7860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Marcadier JL, and Pearson CE (2003) Fidelity of primate cell repair of a double-strand break within a (CTG).(CAG) tract. Effect of slipped DNA structures. J Biol Chem 278, 33848–33856 [DOI] [PubMed] [Google Scholar]
- 82.Casella M, Lucarelli M, Simili M, Beffy P, Del Carratore R, Minichilli F, Chisari C, and Simi S (2003) Spontaneous chromosome loss and colcemid resistance in lymphocytes from patients with myotonic dystrophy type 1. Cytogenet Genome Res 100, 224–229 [DOI] [PubMed] [Google Scholar]
- 83.Dhar A, and Lahue RS (2008) Rapid unwinding of triplet repeat hairpins by Srs2 helicase of Saccharomyces cerevisiae. Nucleic Acids Res 36, 3366–3373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Kerrest A, Anand RP, Sundararajan R, Bermejo R, Liberi G, Dujon B, Freudenreich CH, and Richard GF (2009) SRS2 and SGS1 prevent chromosomal breaks and stabilize triplet repeats by restraining recombination. Nat Struct Mol Biol 16, 159–167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Frizzell A, Nguyen JH, Petalcorin MI, Turner KD, Boulton SJ, Freudenreich CH, and Lahue RS (2014) RTEL1 inhibits trinucleotide repeat expansions and fragility. Cell Rep 6, 827–835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Chan NL, Hou C, Zhang T, Yuan F, Machwe A, Huang J, Orren DK, Gu L, and Li GM (2012) The Werner syndrome protein promotes CAG/CTG repeat stability by resolving large (CAG)(n)/(CTG)(n) hairpins. J Biol Chem 287, 30151–30156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Matsuzaki K, Borel V, Adelman CA, Schindler D, and Boulton SJ (2015) FANCJ suppresses microsatellite instability and lymphomagenesis independent of the Fanconi anemia pathway. Genes Dev 29, 2532–2546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Barthelemy J, Hanenberg H, and Leffak M (2016) FANCJ is essential to maintain microsatellite structure genome-wide during replication stress. Nucleic Acids Res 44, 6803–6816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Gacy AM, Goellner GM, Spiro C, Chen X, Gupta G, Bradbury EM, Dyer RB, Mikesell MJ, Yao JZ, Johnson AJ, Richter A, Melancon SB, and McMurray CT (1998) GAA instability in Friedreich’s Ataxia shares a common, DNA-directed and intraallelic mechanism with other trinucleotide diseases. Mol Cell 1, 583–593 [DOI] [PubMed] [Google Scholar]
- 90.Sakamoto N, Chastain PD, Parniewski P, Ohshima K, Pandolfo M, Griffith JD, and Wells RD (1999) Sticky DNA: self-association properties of long GAA.TTC repeats in R.R.Y triplex structures from Friedreich’s ataxia. Mol Cell 3, 465–475 [DOI] [PubMed] [Google Scholar]
- 91.Potaman VN, Oussatcheva EA, Lyubchenko YL, Shlyakhtenko LS, Bidichandani SI, Ashizawa T, and Sinden RR (2004) Length-dependent structure formation in Friedreich ataxia (GAA)n*(TTC)n repeats at neutral pH. Nucleic Acids Res 32, 1224–1231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Krasilnikova MM, and Mirkin SM (2004) Replication stalling at Friedreich’s ataxia (GAA)n repeats in vivo. Mol Cell Biol 24, 2286–2295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Kumari D, Hayward B, Nakamura AJ, Bonner WM, and Usdin K (2015) Evidence for chromosome fragility at the frataxin locus in Friedreich ataxia. Mutat Res 781, 14–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Raghavan SC, Swanson PC, Wu X, Hsieh CL, and Lieber MR (2004) A non-B-DNA structure at the Bcl-2 major breakpoint region is cleaved by the RAG complex. Nature 428, 88–93 [DOI] [PubMed] [Google Scholar]
- 95.Wang G, and Vasquez KM (2004) Naturally occurring H-DNA-forming sequences are mutagenic in mammalian cells. Proc Natl Acad Sci U S A 101, 13448–13453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Raghavan SC, Chastain P, Lee JS, Hegde BG, Houston S, Langen R, Hsieh CL, Haworth IS, and Lieber MR (2005) Evidence for a triplex DNA conformation at the bcl-2 major breakpoint region of the t(14;18) translocation. J Biol Chem 280, 22749–22760 [DOI] [PubMed] [Google Scholar]
- 97.Zhao J, Wang G, Del Mundo IM, McKinney JA, Lu X, Bacolla A, Boulware SB, Zhang C, Zhang H, Ren P, Freudenreich CH, and Vasquez KM (2018) Distinct Mechanisms of Nuclease-Directed DNA-Structure-Induced Genetic Instability in Cancer Genomes. Cell Rep 22, 1200–1210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.McClellan JA, Boublikova P, Palecek E, and Lilley DM (1990) Superhelical torsion in cellular DNA responds directly to environmental and genetic factors. Proc Natl Acad Sci U S A 87, 8373–8377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Bowater R, Aboul-ela F, and Lilley DM (1991) Large-scale stable opening of supercoiled DNA in response to temperature and supercoiling in (A + T)-rich regions that promote low-salt cruciform extrusion. Biochemistry 30, 11495–11506 [DOI] [PubMed] [Google Scholar]
- 100.Dayn A, Malkhosyan S, Duzhy D, Lyamichev V, Panchenko Y, and Mirkin S (1991) Formation of (dA-dT)n cruciforms in Escherichia coli cells under different environmental conditions. J Bacteriol 173, 2658–2664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Cote AG, and Lewis SM (2008) Mus81-dependent double-strand DNA breaks at in vivo-generated cruciform structures in S. cerevisiae. Mol Cell 31, 800–812 [DOI] [PubMed] [Google Scholar]
- 102.Zhang H, and Freudenreich CH (2007) An AT-rich sequence in human common fragile site FRA16D causes fork stalling and chromosome breakage in S. cerevisiae. Mol Cell 27, 367–379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Inagaki H, Kato T, Tsutsumi M, Ouchi Y, Ohye T, and Kurahashi H (2016) Palindrome-Mediated Translocations in Humans: A New Mechanistic Model for Gross Chromosomal Rearrangements. Front Genet 7, 125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Kurahashi H, and Emanuel BS (2001) Unexpectedly high rate of de novo constitutional t(11;22) translocations in sperm from normal males. Nat Genet 29, 139–140 [DOI] [PubMed] [Google Scholar]
- 105.Kato T, Kurahashi H, and Emanuel BS (2012) Chromosomal translocations and palindromic AT-rich repeats. Curr Opin Genet Dev 22, 221–228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Fechter A, Buettel I, Kuehnel E, Savelyeva L, and Schwab M (2007) Common fragile site FRA11G and rare fragile site FRA11B at 11q23.3 encompass distinct genomic regions. Genes Chromosomes Cancer 46, 98–106 [DOI] [PubMed] [Google Scholar]
- 107.Kurahashi H, Inagaki H, Kato T, Hosoba E, Kogo H, Ohye T, Tsutsumi M, Bolor H, Tong M, and Emanuel BS (2009) Impaired DNA replication prompts deletions within palindromic sequences, but does not induce translocations in human cells. Hum Mol Genet 18, 3397–3406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Babcock M, Yatsenko S, Stankiewicz P, Lupski JR, and Morrow BE (2007) AT-rich repeats associated with chromosome 22q11.2 rearrangement disorders shape human genome architecture on Yq12. Genome Res 17, 451–460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Lilley DM (1980) The inverted repeat as a recognizable structural feature in supercoiled DNA molecules. Proc Natl Acad Sci U S A 77, 6468–6472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Panayotatos N, and Wells RD (1981) Cruciform structures in supercoiled DNA. Nature 289, 466–470 [DOI] [PubMed] [Google Scholar]
- 111.Nag DK, and Petes TD (1991) Seven-base-pair inverted repeats in DNA form stable hairpins in vivo in Saccharomyces cerevisiae. Genetics 129, 669–673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Voineagu I, Narayanan V, Lobachev KS, and Mirkin SM (2008) Replication stalling at unstable inverted repeats: interplay between DNA hairpins and fork stabilizing proteins. Proc Natl Acad Sci U S A 105, 9936–9941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Lu S, Wang G, Bacolla A, Zhao J, Spitser S, and Vasquez KM (2015) Short Inverted Repeats Are Hotspots for Genetic Instability: Relevance to Cancer Genomes. Cell Rep [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Lobachev KS, Shor BM, Tran HT, Taylor W, Keen JD, Resnick MA, and Gordenin DA (1998) Factors affecting inverted repeat stimulation of recombination and deletion in Saccharomyces cerevisiae. Genetics 148, 1507–1524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Nasar F, Jankowski C, and Nag DK (2000) Long palindromic sequences induce double-strand breaks during meiosis in yeast. Mol Cell Biol 20, 3449–3458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Lobachev KS, Gordenin DA, and Resnick MA (2002) The Mre11 complex is required for repair of hairpin-capped double-strand breaks and prevention of chromosome rearrangements. Cell 108, 183–193 [DOI] [PubMed] [Google Scholar]
- 117.Lemoine FJ, Degtyareva NP, Lobachev K, and Petes TD (2005) Chromosomal translocations in yeast induced by low levels of DNA polymerase a model for chromosome fragile sites. Cell 120, 587–598 [DOI] [PubMed] [Google Scholar]
- 118.Lemoine FJ, Degtyareva NP, Kokoska RJ, and Petes TD (2008) Reduced levels of DNA polymerase delta induce chromosome fragile site instability in yeast. Mol Cell Biol 28, 5359–5368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Franchitto A (2013) Genome instability at common fragile sites: searching for the cause of their instability. Biomed Res Int 2013, 730714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Glover TW, Wilson TE, and Arlt MF (2017) Fragile sites in cancer: more than meets the eye. Nat Rev Cancer 17, 489–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Thys RG, Lehman CE, Pierce LC, and Wang YH (2015) DNA secondary structure at chromosomal fragile sites in human disease. Curr Genomics 16, 60–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Mishmar D, Rahat A, Scherer SW, Nyakatura G, Hinzmann B, Kohwi Y, Mandel-Gutfroind Y, Lee JR, Drescher B, Sas DE, Margalit H, Platzer M, Weiss A, Tsui LC, Rosenthal A, and Kerem B (1998) Molecular characterization of a common fragile site (FRA7H) on human chromosome 7 by the cloning of a simian virus 40 integration site. Proc Natl Acad Sci U S A 95, 8141–8146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Zlotorynski E, Rahat A, Skaug J, Ben-Porat N, Ozeri E, Hershberg R, Levi A, Scherer SW, Margalit H, and Kerem B (2003) Molecular basis for expression of common and rare fragile sites. Mol Cell Biol 23, 7143–7151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Ried K, Finnis M, Hobson L, Mangelsdorf M, Dayan S, Nancarrow JK, Woollatt E, Kremmidiotis G, Gardner A, Venter D, Baker E, and Richards RI (2000) Common chromosomal fragile site FRA16D sequence: identification of the FOR gene spanning FRA16D and homozygous deletions and translocation breakpoints in cancer cells. Hum Mol Genet 9, 1651–1663 [DOI] [PubMed] [Google Scholar]
- 125.Finnis M, Dayan S, Hobson L, Chenevix-Trench G, Friend K, Ried K, Venter D, Woollatt E, Baker E, and Richards RI (2005) Common chromosomal fragile site FRA16D mutation in cancer cells. Hum Mol Genet 14, 1341–1349 [DOI] [PubMed] [Google Scholar]
- 126.Bignell GR, Greenman CD, Davies H, Butler AP, Edkins S, Andrews JM, Buck G, Chen L, Beare D, Latimer C, Widaa S, Hinton J, Fahey C, Fu B, Swamy S, Dalgliesh GL, Teh BT, Deloukas P, Yang F, Campbell PJ, Futreal PA, and Stratton MR (2010) Signatures of mutation and selection in the cancer genome. Nature 463, 893–898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Beroukhim R, Mermel CH, Porter D, Wei G, Raychaudhuri S, Donovan J, Barretina J, Boehm JS, Dobson J, Urashima M, Mc Henry KT, Pinchback RM, Ligon AH, Cho YJ, Haery L, Greulich H, Reich M, Winckler W, Lawrence MS, Weir BA, Tanaka KE, Chiang DY, Bass AJ, Loo A, Hoffman C, Prensner J, Liefeld T, Gao Q, Yecies D, Signoretti S, Maher E, Kaye FJ, Sasaki H, Tepper JE, Fletcher JA, Tabernero J, Baselga J, Tsao MS, Demichelis F, Rubin MA, Janne PA, Daly MJ, Nucera C, Levine RL, Ebert BL, Gabriel S, Rustgi AK, Antonescu CR, Ladanyi M, Letai A, Garraway LA, Loda M, Beer DG, True LD, Okamoto A, Pomeroy SL, Singer S, Golub TR, Lander ES, Getz G, Sellers WR, and Meyerson M (2010) The landscape of somatic copy-number alteration across human cancers. Nature 463, 899–905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Bartkova J, Horejsi Z, Koed K, Kramer A, Tort F, Zieger K, Guldberg P, Sehested M, Nesland JM, Lukas C, Orntoft T, Lukas J, and Bartek J (2005) DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nature 434, 864–870 [DOI] [PubMed] [Google Scholar]
- 129.Gorgoulis VG, Vassiliou LV, Karakaidos P, Zacharatos P, Kotsinas A, Liloglou T, Venere M, Ditullio RA Jr., Kastrinakis NG, Levy B, Kletsas D, Yoneta A, Herlyn M, Kittas C, and Halazonetis TD (2005) Activation of the DNA damage checkpoint and genomic instability in human precancerous lesions. Nature 434, 907–913 [DOI] [PubMed] [Google Scholar]
- 130.Halazonetis TD, Gorgoulis VG, and Bartek J (2008) An oncogene-induced DNA damage model for cancer development. Science 319, 1352–1355 [DOI] [PubMed] [Google Scholar]
- 131.Miron K, Golan-Lev T, Dvir R, Ben-David E, and Kerem B (2015) Oncogenes create a unique landscape of fragile sites. Nat Commun 6, 7094. [DOI] [PubMed] [Google Scholar]
- 132.Macheret M, and Halazonetis TD (2015) DNA replication stress as a hallmark of cancer. Annu Rev Pathol 10, 425–448 [DOI] [PubMed] [Google Scholar]
- 133.Taylor EM, and Lindsay HD (2016) DNA replication stress and cancer: cause or cure? Future Oncol 12, 221–237 [DOI] [PubMed] [Google Scholar]
- 134.Mangelsdorf M, Ried K, Woollatt E, Dayan S, Eyre H, Finnis M, Hobson L, Nancarrow J, Venter D, Baker E, and Richards RI (2000) Chromosomal fragile site FRA16D and DNA instability in cancer. Cancer Res 60, 1683–1689 [PubMed] [Google Scholar]
- 135.Karras JR, Schrock MS, Batar B, and Huebner K (2016) Fragile Genes That Are Frequently Altered in Cancer: Players Not Passengers. Cytogenet Genome Res 150, 208–216 [DOI] [PubMed] [Google Scholar]
- 136.Ozeri-Galai E, Lebofsky R, Rahat A, Bester AC, Bensimon A, and Kerem B (2011) Failure of origin activation in response to fork stalling leads to chromosomal instability at fragile sites. Mol Cell 43, 122–131 [DOI] [PubMed] [Google Scholar]
- 137.Yu S, Mangelsdorf M, Hewett D, Hobson L, Baker E, Eyre HJ, Lapsys N, Le Paslier D, Doggett NA, Sutherland GR, and Richards RI (1997) Human chromosomal fragile site FRA16B is an amplified AT-rich minisatellite repeat. Cell 88, 367–374 [DOI] [PubMed] [Google Scholar]
- 138.Hewett DR, Handt O, Hobson L, Mangelsdorf M, Eyre HJ, Baker E, Sutherland GR, Schuffenhauer S, Mao JI, and Richards RI (1998) FRA10B structure reveals common elements in repeat expansion and chromosomal fragile site genesis. Mol Cell 1, 773–781 [DOI] [PubMed] [Google Scholar]
- 139.Debacker K, Winnepenninckx B, Ben-Porat N, FitzPatrick D, Van Luijk R, Scheers S, Kerem B, and Frank Kooy R (2007) FRA18C: a new aphidicolin-inducible fragile site on chromosome 18q22, possibly associated with in vivo chromosome breakage. J Med Genet 44, 347–352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Madireddy A, Kosiyatrakul ST, Boisvert RA, Herrera-Moyano E, Garcia-Rubio ML, Gerhardt J, Vuono EA, Owen N, Yan Z, Olson S, Aguilera A, Howlett NG, and Schildkraut CL (2016) FANCD2 Facilitates Replication through Common Fragile Sites. Mol Cell 64, 388–404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Palumbo E, Matricardi L, Tosoni E, Bensimon A, and Russo A (2010) Replication dynamics at common fragile site FRA6E. Chromosoma 119, 575–587 [DOI] [PubMed] [Google Scholar]
- 142.Zuker M (2003) Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res 31, 3406–3415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Dillon LW, Pierce LC, Ng MC, and Wang YH (2013) Role of DNA secondary structures in fragile site breakage along human chromosome 10. Hum Mol Genet 22, 1443–1456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Fungtammasan A, Walsh E, Chiaromonte F, Eckert KA, and Makova KD (2012) A genome-wide analysis of common fragile sites: what features determine chromosomal instability in the human genome? Genome Res 22, 993–1005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Letessier A, Millot GA, Koundrioukoff S, Lachages AM, Vogt N, Hansen RS, Malfoy B, Brison O, and Debatisse M (2011) Cell-type-specific replication initiation programs set fragility of the FRA3B fragile site. Nature 470, 120–123 [DOI] [PubMed] [Google Scholar]
- 146.Smith DI, Zhu Y, McAvoy S, and Kuhn R (2006) Common fragile sites, extremely large genes, neural development and cancer. Cancer Lett 232, 48–57 [DOI] [PubMed] [Google Scholar]
- 147.McAvoy S, Ganapathiraju SC, Ducharme-Smith AL, Pritchett JR, Kosari F, Perez DS, Zhu Y, James CD, and Smith DI (2007) Non-random inactivation of large common fragile site genes in different cancers. Cytogenet Genome Res 118, 260–269 [DOI] [PubMed] [Google Scholar]
- 148.Le Tallec B, Millot GA, Blin ME, Brison O, Dutrillaux B, and Debatisse M (2013) Common fragile site profiling in epithelial and erythroid cells reveals that most recurrent cancer deletions lie in fragile sites hosting large genes. Cell Rep 4, 420–428 [DOI] [PubMed] [Google Scholar]
- 149.Gao G, and Smith DI (2014) Very large common fragile site genes and their potential role in cancer development. Cell Mol Life Sci 71, 4601–4615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Helmrich A, Ballarino M, and Tora L (2011) Collisions between replication and transcription complexes cause common fragile site instability at the longest human genes. Mol Cell 44, 966–977 [DOI] [PubMed] [Google Scholar]
- 151.Le Tallec B, Dutrillaux B, Lachages AM, Millot GA, Brison O, and Debatisse M (2011) Molecular profiling of common fragile sites in human fibroblasts. Nat Struct Mol Biol 18, 1421–1423 [DOI] [PubMed] [Google Scholar]
- 152.Hosseini SA, Horton S, Saldivar JC, Miuma S, Stampfer MR, Heerema NA, and Huebner K (2013) Common chromosome fragile sites in human and murine epithelial cells and FHIT/FRA3B loss-induced global genome instability. Genes Chromosomes Cancer 52, 1017–1029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Sanz LA, Hartono SR, Lim YW, Steyaert S, Rajpurkar A, Ginno PA, Xu X, and Chedin F (2016) Prevalent, Dynamic, and Conserved R-Loop Structures Associate with Specific Epigenomic Signatures in Mammals. Mol Cell 63, 167–178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Gan W, Guan Z, Liu J, Gui T, Shen K, Manley JL, and Li X (2011) R-loop-mediated genomic instability is caused by impairment of replication fork progression. Genes Dev 25, 2041–2056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Sollier J, and Cimprich KA (2015) Breaking bad: R-loops and genome integrity. Trends Cell Biol 25, 514–522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Wang G, and Vasquez KM (2017) Effects of Replication and Transcription on DNA Structure-Related Genetic Instability. Genes (Basel) 8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Macheret M, and Halazonetis TD (2018) Intragenic origins due to short G1 phases underlie oncogene-induced DNA replication stress. Nature 555, 112–116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Ganem NJ, and Pellman D (2012) Linking abnormal mitosis to the acquisition of DNA damage. J Cell Biol 199, 871–881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Mimitou EP, and Symington LS (2009) DNA end resection: many nucleases make light work. DNA Repair (Amst) 8, 983–995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Rass U (2013) Resolving branched DNA intermediates with structure-specific nucleases during replication in eukaryotes. Chromosoma 122, 499–515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Zhang Y, Shishkin AA, Nishida Y, Marcinkowski-Desmond D, Saini N, Volkov KV, Mirkin SM, and Lobachev KS (2012) Genome-wide screen identifies pathways that govern GAA/TTC repeat fragility and expansions in dividing and nondividing yeast cells. Mol Cell 48, 254–265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Naim V, Wilhelm T, Debatisse M, and Rosselli F (2013) ERCC1 and MUS81-EME1 promote sister chromatid separation by processing late replication intermediates at common fragile sites during mitosis. Nat Cell Biol 15, 1008–1015 [DOI] [PubMed] [Google Scholar]
- 163.Ying S, Minocherhomji S, Chan KL, Palmai-Pallag T, Chu WK, Wass T, Mankouri HW, Liu Y, and Hickson ID (2013) MUS81 promotes common fragile site expression. Nat Cell Biol 15, 1001–1007 [DOI] [PubMed] [Google Scholar]
- 164.Wyatt HD, and West SC (2014) Holliday junction resolvases. Cold Spring Harb Perspect Biol 6, a023192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Minocherhomji S, and Hickson ID (2014) Structure-specific endonucleases: guardians of fragile site stability. Trends Cell Biol 24, 321–327 [DOI] [PubMed] [Google Scholar]
- 166.Lukas C, Savic V, Bekker-Jensen S, Doil C, Neumann B, Pedersen RS, Grofte M, Chan KL, Hickson ID, Bartek J, and Lukas J (2011) 53BP1 nuclear bodies form around DNA lesions generated by mitotic transmission of chromosomes under replication stress. Nat Cell Biol 13, 243–253 [DOI] [PubMed] [Google Scholar]
- 167.Minocherhomji S, Ying S, Bjerregaard VA, Bursomanno S, Aleliunaite A, Wu W, Mankouri HW, Shen H, Liu Y, and Hickson ID (2015) Replication stress activates DNA repair synthesis in mitosis. Nature 528, 286–290 [DOI] [PubMed] [Google Scholar]
- 168.Ait Saada A, Teixeira-Silva A, Iraqui I, Costes A, Hardy J, Paoletti G, Freon K, and Lambert SAE (2017) Unprotected Replication Forks Are Converted into Mitotic Sister Chromatid Bridges. Mol Cell 66, 398–410 e394 [DOI] [PubMed] [Google Scholar]
- 169.Wyatt HD, Laister RC, Martin SR, Arrowsmith CH, and West SC (2017) The SMX DNA Repair Tri-nuclease. Mol Cell 65, 848–860 e811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Symington LS, Rothstein R, and Lisby M (2014) Mechanisms and regulation of mitotic recombination in Saccharomyces cerevisiae. Genetics 198, 795–835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Haber JE (2016) A Life Investigating Pathways That Repair Broken Chromosomes. Annu Rev Genet 50, 1–28 [DOI] [PubMed] [Google Scholar]
- 172.Anand RP, Lovett ST, and Haber JE (2013) Break-induced DNA replication. Cold Spring Harb Perspect Biol 5, a010397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Malkova A, and Ira G (2013) Break-induced replication: functions and molecular mechanism. Curr Opin Genet Dev 23, 271–279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Mayle R, Campbell IM, Beck CR, Yu Y, Wilson M, Shaw CA, Bjergbaek L, Lupski JR, and Ira G (2015) DNA REPAIR. Mus81 and converging forks limit the mutagenicity of replication fork breakage. Science 349, 742–747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Hustedt N, and Durocher D (2016) The control of DNA repair by the cell cycle. Nat Cell Biol 19, 1–9 [DOI] [PubMed] [Google Scholar]
- 176.Bhowmick R, Minocherhomji S, and Hickson ID (2016) RAD52 Facilitates Mitotic DNA Synthesis Following Replication Stress. Mol Cell 64, 1117–1126 [DOI] [PubMed] [Google Scholar]
- 177.Sotiriou SK, Kamileri I, Lugli N, Evangelou K, Da-Re C, Huber F, Padayachy L, Tardy S, Nicati NL, Barriot S, Ochs F, Lukas C, Lukas J, Gorgoulis VG, Scapozza L, and Halazonetis TD (2016) Mammalian RAD52 Functions in Break-Induced Replication Repair of Collapsed DNA Replication Forks. Mol Cell 64, 1127–1134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Rey L, Sidorova JM, Puget N, Boudsocq F, Biard DS, Monnat RJ Jr., Cazaux C, and Hoffmann JS (2009) Human DNA polymerase eta is required for common fragile site stability during unperturbed DNA replication. Mol Cell Biol 29, 3344–3354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Bhat A, Andersen PL, Qin Z, and Xiao W (2013) Rev3, the catalytic subunit of Polzeta, is required for maintaining fragile site stability in human cells. Nucleic Acids Res 41, 2328–2339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Barnes R, and Eckert K (2017) Maintenance of Genome Integrity: How Mammalian Cells Orchestrate Genome Duplication by Coordinating Replicative and Specialized DNA Polymerases. Genes (Basel) 8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.McVey M, Khodaverdian VY, Meyer D, Cerqueira PG, and Heyer WD (2016) Eukaryotic DNA Polymerases in Homologous Recombination. Annu Rev Genet 50, 393–421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Makharashvili N, Tubbs AT, Yang SH, Wang H, Barton O, Zhou Y, Deshpande RA, Lee JH, Lobrich M, Sleckman BP, Wu X, and Paull TT (2014) Catalytic and noncatalytic roles of the CtIP endonuclease in double-strand break end resection. Mol Cell 54, 1022–1033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Cejka P (2015) DNA End Resection: Nucleases Team Up with the Right Partners to Initiate Homologous Recombination. J Biol Chem 290, 22931–22938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Wang H, Li Y, Truong LN, Shi LZ, Hwang PY, He J, Do J, Cho MJ, Li H, Negrete A, Shiloach J, Berns MW, Shen B, Chen L, and Wu X (2014) CtIP maintains stability at common fragile sites and inverted repeats by end resection-independent endonuclease activity. Mol Cell 54, 1012–1021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Mosbach V, Poggi L, Viterbo D, Charpentier M, and Richard GF (2018) TALEN-Induced Double-Strand Break Repair of CTG Trinucleotide Repeats. Cell Rep 22, 2146–2159 [DOI] [PubMed] [Google Scholar]
- 186.Coquelle A, Pipiras E, Toledo F, Buttin G, and Debatisse M (1997) Expression of fragile sites triggers intrachromosomal mammalian gene amplification and sets boundaries to early amplicons. Cell 89, 215–225 [DOI] [PubMed] [Google Scholar]
- 187.Haber JE, and Debatisse M (2006) Gene amplification: yeast takes a turn. Cell 125, 1237–1240 [DOI] [PubMed] [Google Scholar]
- 188.Butler DK, Yasuda LE, and Yao MC (1996) Induction of large DNA palindrome formation in yeast: implications for gene amplification and genome stability in eukaryotes. Cell 87, 1115–1122 [DOI] [PubMed] [Google Scholar]
- 189.Tanaka H, Tapscott SJ, Trask BJ, and Yao MC (2002) Short inverted repeats initiate gene amplification through the formation of a large DNA palindrome in mammalian cells. Proc Natl Acad Sci U S A 99, 8772–8777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Narayanan V, Mieczkowski PA, Kim HM, Petes TD, and Lobachev KS (2006) The pattern of gene amplification is determined by the chromosomal location of hairpin-capped breaks. Cell 125, 1283–1296 [DOI] [PubMed] [Google Scholar]
- 191.Pirzio LM, Pichierri P, Bignami M, and Franchitto A (2008) Werner syndrome helicase activity is essential in maintaining fragile site stability. J Cell Biol 180, 305–314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Lu X, Parvathaneni S, Hara T, Lal A, and Sharma S (2013) Replication stress induces specific enrichment of RECQ1 at common fragile sites FRA3B and FRA16D. Mol Cancer 12, 29. [DOI] [PMC free article] [PubMed] [Google Scholar]