

DNA damage and type I interferons (IFNs) contribute to inflammatory responses after traumatic brain injury (TBI). TBI-induced activation of microglia and peripherally-derived inflammatory macrophages may lead to tissue damage and neurological deficits.
Keywords: interferon-β, neurodegeneration, neuroinflammation, neuroprotection, traumatic brain injury, type I interferons
Abstract
DNA damage and type I interferons (IFNs) contribute to inflammatory responses after traumatic brain injury (TBI). TBI-induced activation of microglia and peripherally-derived inflammatory macrophages may lead to tissue damage and neurological deficits. Here, we investigated the role of IFN-β in secondary injury after TBI using a controlled cortical impact model in adult male IFN-β-deficient (IFN-β−/−) mice and assessed post-traumatic neuroinflammatory responses, neuropathology, and long-term functional recovery. TBI increased expression of DNA sensors cyclic GMP-AMP synthase and stimulator of interferon genes in wild-type (WT) mice. IFN-β and other IFN-related and neuroinflammatory genes were also upregulated early and persistently after TBI. TBI increased expression of proinflammatory mediators in the cortex and hippocampus of WT mice, whereas levels were mitigated in IFN-β−/− mice. Moreover, long-term microglia activation, motor, and cognitive function impairments were decreased in IFN-β−/− TBI mice compared with their injured WT counterparts; improved neurological recovery was associated with reduced lesion volume and hippocampal neurodegeneration in IFN-β−/− mice. Continuous central administration of a neutralizing antibody to the IFN-α/β receptor (IFNAR) for 3 d, beginning 30 min post-injury, reversed early cognitive impairments in TBI mice and led to transient improvements in motor function. However, anti-IFNAR treatment did not improve long-term functional recovery or decrease TBI neuropathology at 28 d post-injury. In summary, TBI induces a robust neuroinflammatory response that is associated with increased expression of IFN-β and other IFN-related genes. Inhibition of IFN-β reduces post-traumatic neuroinflammation and neurodegeneration, resulting in improved neurological recovery. Thus, IFN-β may be a potential therapeutic target for TBI.
SIGNIFICANCE STATEMENT TBI frequently causes long-term neurological and psychiatric changes in head injury patients. TBI-induced secondary injury processes including persistent neuroinflammation evolve over time and can contribute to chronic neurological impairments. The present study demonstrates that TBI is followed by robust activation of type I IFN pathways, which have been implicated in microglial-associated neuroinflammation and chronic neurodegeneration. We examined the effects of genetic or pharmacological inhibition of IFN-β, a key component of type I IFN mechanisms to address its role in TBI pathophysiology. Inhibition of IFN-β signaling resulted in reduced neuroinflammation, attenuated neurobehavioral deficits, and limited tissue loss long after TBI. These preclinical findings suggest that IFN-β may be a potential therapeutic target for TBI.
Introduction
Traumatic brain injury (TBI) is the leading cause of morbidity and mortality in developed countries (Maas et al., 2017). TBI induces delayed, secondary molecular and cellular injury responses, including chronic neuroinflammation, which contribute to progressive tissue loss and neurological impairments (Loane et al., 2014; Pischiutta et al., 2018). Clinical studies have shown that TBI induces chronic neurodegeneration that is associated with cognitive impairments and late-onset dementias (Mortimer et al., 1985; Salib and Hillier, 1997; Plassman et al., 2000). In addition, experimental models of repeated mild or moderate-to-severe TBI result in persistent brain inflammation and neurodegeneration leading to long-term motor and cognitive function deficits (Pierce et al., 1998; Dixon et al., 1999; Aungst et al., 2014; Loane et al., 2014; Mouzon et al., 2014). Microglia, the primary innate immune cells in brain, are chronically activated for months to years following moderate-to-severe TBI in humans and in animal models (Ramlackhansingh et al., 2011; Johnson et al., 2013; Smith et al., 2013; Loane et al., 2014). Experimental evidence demonstrates that selected, delayed anti-inflammatory therapies reduce post-traumatic microglial activation and mitigate behavioral deficits and tissue loss after TBI (Byrnes et al., 2012; Piao et al., 2013).
Detection of nucleic acids by the innate immune system is essential for the host response during a viral infection. A number of immune sensors capable of recognizing cytosolic DNA have been identified, including the PHYIN family members AIM2, IFI16 (mouse homolog IFI204), and the enzyme cyclic GMP-AMP synthase (cGAS). Activation of cGAS leads to activation of stimulator of interferon genes (STING) and the induction of type I interferons (IFNs; Almine et al., 2017). Type I IFNs are the main regulators of the host anti-viral response; in the absence of IFN signaling, mice are more susceptible to viral infection (Müller et al., 1994; Pinto et al., 2014). Following CNS injury, damage-associated molecular patterns, such as cytosolic and mitochondrial DNA, are released from injured neurons and initiate innate immune signaling that activates glia and drives secondary neuroinflammation (Walko et al., 2014; Chin, 2019; Wang et al., 2019). Microglia and astrocytes express members of the PHYIN family, which are upregulated during neurodegenerative disease (Cox et al., 2015). Inhibition of STING signaling reduces neuroinflammation and neurodegeneration in an experimental model of prion disease (Nazmi et al., 2019). In addition, type I IFNs contribute to the inflammatory response during normal aging and in age-related neurodegenerative disorders (Baruch et al., 2014; Taylor et al., 2014; Roy et al., 2020). However, the role of type I IFNs within the CNS is model-dependent; whereas IFNs in neurodegenerative models appear to be detrimental (Taylor et al., 2014), IFN-β ameliorates neuroinflammatory responses in multiple sclerosis (MS; Prinz et al., 2008; Owens et al., 2014), possibly by reducing production of IL-12 (Byrnes et al., 2002), and is the first disease-modifying drug approved for relapsing-remitting MS (Paty and Li 1993; Jacobs et al., 2000).
Although several studies have implicated type I IFNs in models of age-related neurodegeneration, the role of type I IFNs following CNS injury is poorly understood. One study reported that inhibition of type I IFN signaling is protective during the acute phase after TBI (Karve et al., 2016). However, their role in development of chronic neurological deficits and progressive neurodegeneration following TBI has not been studied. Work in prion models indicate that neurodegeneration is IFN-dependent, with peripheral inflammation further increasing IFN-related gene expression and exacerbating disease progression (Field et al., 2010; Nazmi et al., 2019). Given this recent evidence, we examined the role of IFN-β in the development of neuroinflammation, chronic neurodegeneration, and neurological impairments in a well characterized rodent TBI model, using genetic and pharmacological approaches.
Materials and Methods
Animals.
Studies were performed using adult male (10- to 12-week-old) IFN-β-deficient (IFN-β−/−; provided by Stefanie Vogel, UMB), or age-matched C57BL/6J (WT) male mice (Jackson Laboratories). IFN-β−/− mice have a targeted mutation in the gene that encodes IFN-β (Deonarain et al., 2003). IFN-β−/− mice were originally obtained from Dr. Eleanor Fish (University of Toronto) and were backcrossed to ∼N = 10 C57BL/6J mice. Mice were housed in the Animal Care facility at the University of Maryland School of Medicine under a 12 h light/dark cycle, with ad libitum access to food and water. All surgical procedures were performed in accordance with protocols approved by the Institutional Animal Care and Use Committee at the University of Maryland School of Medicine.
Controlled cortical impact.
Our custom-designed controlled cortical impact (CCI) device consists of a microprocessor-controlled pneumatic impactor with a 3.5 mm diameter tip as described previously (Loane et al., 2009). Briefly, mice were anesthetized with isoflurane evaporated in a gas mixture containing 70% N2O and 30% O2 administered through a nose mask. Mice were placed on a heated pad and core body temperature was maintained at 37°C. The head was mounted in a stereotaxic frame, a 10 mm midline incision was made over the skull and the skin and fascia were reflected. A 5 mm craniotomy was made on the central aspect of the left parietal bone. The impounder tip of the injury device was then extended to its full stroke distance (44 mm), positioned to the surface of the exposed dura, and reset to impact the cortical surface. Moderate-level CCI was induced using an impactor velocity of 6 m/s and deformation depth of 2 mm. After injury, the incision was closed with interrupted 6-0 silk sutures, anesthesia was terminated, and the animal was placed into a heated cage to maintain normal core temperature for 45 min post-injury. Sham animals underwent the same procedure as CCI mice except for the impact.
Intracerebroventricular guide cannula implantation and osmotic pump infusion.
Before CCI, the right lateral ventricle of the mouse was stereotaxically perforated with a brain infusion kit 3 (ALZET, DURECT; coordinates: 0.7 mm posterior to the bregma, 1.5 mm lateral to the bregma, 2 mm deep). Immediately following CCI on the left parietal cortex, the infusion cannula was connected to an osmotic minipump (ALZET, pump model 1007D) that was implanted subcutaneously in the animal's back, just behind the scapula. Osmotic pumps were primed for ∼8 h before implantation and were either filled with 0.5 mg/ml αIFNAR neutralizing antibody (MAR1-5A3, Invitrogen) or an equal concentration isotype control mouse IgG1 (clone Mg1-45, BioLegend). Once implanted, the pumps continually infused αIFNAR or control IgG1 into the lateral ventricle for 3 d at a rate of 0.5 μl/h.
Study 1.
WT sham-injured or CCI mice (n = 5–6) were anesthetized (100 mg/kg sodium pentobarbital, i.p.) at 3 d post-injury (dpi). Mice were transcardially perfused with ice-cold 0.9% saline (100 ml). Ipsilateral cortical and hippocampal tissue were rapidly dissected and snap-frozen on liquid nitrogen for RNA or protein extraction. An additional group of WT sham-injured or CCI mice (n = 7) were anesthetized (100 mg/kg sodium pentobarbital, i.p.) at 60 dpi, ipsilateral cortical tissue was rapidly dissected and snap-frozen on liquid nitrogen for RNA extraction.
Study 2.
WT and IFN-β−/− sham-injured (n = 6) or CCI (n = 6) of mice were anesthetized (100 mg/kg sodium pentobarbital, i.p.) at 3 dpi and transcardially perfused with ice-cold 0.9% saline (100 ml). Ipsilateral cortical and hippocampal tissue were rapidly dissected and snap-frozen on liquid nitrogen for RNA and protein extraction.
Study 3.
WT and IFN-β−/− sham-injured or CCI (n = 8–15) mice were anesthetized (100 mg/kg sodium pentobarbital, i.p.). All animals underwent motor function testing (beam walk) on 1, 3, 7, 14, 21, and 28 dpi (Henry et al., 2019). Cognitive function was assessed using the Y-maze (8 dpi; Henry et al., 2019) and the novel object recognition test (NOR; 18 dpi; Piao et al., 2013). At 28 dpi animals were anesthetized (100 mg/kg sodium pentobarbital, i.p.) and transcardially perfused with ice-cold 0.9% saline (100 ml), followed by 300 ml of 4% paraformaldehyde. Brains were removed and postfixed in 4% paraformaldehyde overnight, and cryoprotected in 30% sucrose for histological analysis.
Study 4.
Antibody to the type I IFN receptor (αIFNAR; 0.5 mg/ml) or isotype control IgG1 was delivered intracerebroventricularly via osmotic pump infusion to WT CCI mice (n = 7–9/group). The dose of αIFNAR was chosen based on prior studies demonstrating neutralization of type I IFN in a mouse model of aging (Baruch et al., 2014). Sham WT mice (n = 6) were used as control. All animals underwent motor function testing (beam walk) on 1, 3, 7, 14, 21, and 28 dpi, and cognitive function was assessed using the Y-maze (8 dpi) and NOR test (18 dpi). At 28 dpi animals were anesthetized (100 mg/kg sodium pentobarbital, i.p.) and transcardially perfused with ice-cold 0.9% saline (100 ml), followed by 300 ml of 4% paraformaldehyde. Brains were removed and postfixed in 4% paraformaldehyde overnight, and cryoprotected in 30% sucrose for histological analysis.
Quantitative real-time PCR.
Total RNA was extracted from snap-frozen sham and TBI cortical and hippocampal tissue from WT and IFN-β−/− mice using an RNeasy isolation kit (Qiagen) with on-column DNase treatment (Qiagen). cDNA synthesis was performed using a Verso cDNA RT kit (ThermoFisher Scientific) according to the manufacturer's instructions. Quantitative real-time PCR was performed using TaqMan gene expression assays (IFN-β, Mm00439552_s1; IRF1, Mm01288580_m1; IRF3, Mm00516784_m1; IRF4, Mm00516431_m1; IRF5, Mm00496477_m1; IRF7, Mm00516793_g1; ISG15, Mm01705338_s1; MX1, Mm00487796_m1; IFI204, Mm00492602_m1; NOX2, Mm01287743_m1; TNF-α, Mm00443258_m1; IL-6, Mm00446190_m1; IL-1β, Mm01336189_m1; CCL5, Mm01302427_m1; CXCL10, Mm00445235_m1; CD11b (ITGAM), Mm00434455_m1; GFAP, Mm01253033_m1; YM1, Mm00657889_m1; Arg1, Mm00475988_m1; SOCS3, Mm00545913_s1; IL-10, Mm00439614_m1; TGF-β, Mm00441724_m1; and GAPDH, Mm99999915_g1; Applied Biosystems) on an ABI 7900 HT FAST Real Time PCR machine (Applied Biosystems). Samples were assayed in duplicate in one run (40 cycles), composed of three stages: 50°C for 2 min, 95°C for 10 s for each cycle (denaturation), and finally the transcription step at 60°C for 1 min. Gene expression was calculated relative to the endogenous control sample (GAPDH) to determine relative expression values, using the 2-ΔΔCt method (where Ct is the threshold cycle; Livak and Schmittgen, 2001).
Western blot analysis.
Proteins from ipsilateral cortical and hippocampal tissue were extracted using RIPA buffer, equalized, and loaded onto 5–20% gradient gels for SDS PAGE (Bio-Rad). Proteins were transferred onto nitrocellulose membranes, and then blocked for 1 h in 5% milk in 1 × TBS containing 0.05% Tween 20 (TBS-T) at room temperature. The membrane was incubated in rabbit anti-cGAS (1:1000; Cell Signaling Technology), rabbit anti-STING (1:500; Cell Signaling Technology), rabbit anti-STAT1 (1:1000; Cell Signaling Technology), or mouse anti-β-Actin (1:5000; Sigma-Aldrich) overnight at 4°C, then washed three times in TBS-T, and incubated in appropriate HRP-conjugated secondary antibodies (Jackson ImmunoResearch Laboratories) for 2 h at room temperature. Membranes were washed three times in TBS-T, and proteins were visualized using SuperSignal West Dura Extended Duration Substrate (ThermoFisher Scientific). Chemiluminescence was captured ChemiDoc XRS+ System (Bio-Rad), and protein bands were quantified by densitometric analysis using Bio-Rad Molecular Imaging Software. The data presented reflects the intensity of target protein band normalized to the intensity of the endogenous control for each sample (expressed in arbitrary units).
Immunofluorescence imaging.
Coronal brain sections from sham and CCI mice at ∼−1.70 mm from the bregma were selected, and standard immunostaining techniques were used (Henry et al., 2019). Briefly, 20 μm brain sections were washed three times with 1× PBS, blocked for 1 h in goat serum containing 0.4% Triton X-100, and incubated overnight at 4°C with a combination of primary antibodies, including mouse anti-gp91phox (NOX2, 1:1000; BD Biosciences) and rat anti-CD68 (1:1000; AbD Serotec). Sections were washed three times with 1× PBS and incubated with appropriate AlexaFluor-conjugated secondary antibodies (Life Technologies) for 2 h at room temperature. Sections were washed three times with 1× PBS, counterstained with 4′,6-diamidino-2-phenylindole (DAPI; 1 μg/ml; Sigma-Aldrich), and mounted with glass coverslips using hydromount solution (National Diagnostics). Images were acquired using a fluorescent Nikon Ti-E inverted microscope, at 10× (Plan Apo: 10× NA: 0.45) or 20× (Plan APO: 20× NA: 0.75) magnification. Exposure times were kept constant for all sections in each experiment. Analysis was performed using Nikon NIS-Elements software (AR 4.20.01) to identify nuclei count (DAPI), CD68+, and NOX2+ cells on the best focused image per region (automatically determined via software). For quantification, the total signal intensity of NOX2 within CD68+ regions was summed across each field and normalized to the nuclear count per field. The normalized sum intensity data from at least three fields was averaged for each animal.
Stereology
Lesion volume.
Sixty micrometer coronal sections from mice were stained with cresyl violet (FD NeuroTechnologies), dehydrated, and mounted for analysis. Lesion volume was quantified based on the Cavalieri method of unbiased stereology using Stereoinvestigator Software (MBF Biosciences) as described previously (Kumar et al., 2013). Briefly, lesion volume was quantified by outlining the missing tissue on the injured hemisphere using the Cavalieri estimator with a grid spacing of 0.1 mm. Every eighth section from a total of 96 sections was analyzed beginning from a random start point.
Neuronal cell loss.
Cresyl violet-stained 60 μm coronal sections were used to quantify neuronal densities in the dentate gyrus region of the hippocampus of both sham and CCI mice. The optical fractionator method of stereology was used as previously described (Kumar et al., 2016). Briefly, every fourth 60 μm section between −1.22 and −2.54 mm from bregma was analyzed beginning from a random start point. A total of five sections were analyzed. The optical dissector had a size of 50 × 50 μm in the x-axis and the y-axis, respectively, with a height of 10 μm and a guard zone of 4 μm from the top of the section. The sampled regions of the dentate gyrus was demarcated in the injured hemisphere and cresyl violet-positive cells were counted using Stereoinvestigator Software (MBF Biosciences). The volume of the dentate gyrus was measured using the Cavalieri estimator method with a grid spacing of 50 μm. The number of surviving neurons in each field was divided by the volume of the region of interest to obtain the cellular density expressed in counts/mm3.
Neurobehavioral testing
Beam walk.
Motor function recovery was assessed using a beam walk test as described previously (Loane et al., 2009). The beam walk tests fine motor coordination differences and consists of a narrow wooden beam (5 mm wide and 120 mm length), which is suspended 300 mm above a tabletop. Mice were placed on one end of the beam, and the number of foot faults of the right hindlimb recorded over 50 steps. Mice were trained on the beam walk for 3 d before CCI and tested through 28 dpi.
Y-maze spontaneous alternation.
The Y-maze test assesses spatial working memory and was performed as previously described (Kumar et al., 2016). Briefly, the Y-maze (Stoelting) consisted of three identical arms, each arm 35 cm long, 5 cm wide, and 10 cm high, at an angle of 120° with respect to the other arms. One arm was randomly selected as the “start” arm, and the mouse was placed within and allowed to explore the maze freely for 5 min. Arm entries (Arms A–C) were recorded by analyzing mouse activity using ANY-maze software (Stoelting). An arm entry was attributed when all four paws of the mouse entered the arm, and an alternation was designated when the mouse entered three different arms consecutively. The percentage of alternation was calculated as follows: total alternations × 100/(total arm entries − 2). If a mouse scored >50% alternations (the chance level for choosing the unfamiliar arm), this was indicative of spatial working memory.
Novel object recognition.
NOR was performed on 17–18 dpi to assess nonspatial hippocampal-mediated memory, as previously described (Piao et al., 2013). Mice were placed in an open field (22.5 × 22.5 cm) and two identical objects were placed near the left and right corners of the open field for training (familiar phase). Mice were allowed to freely explore until they spent a total of 20 s exploring the objects (exploration was recorded when the front paws or nose contacted the object). The time spent with each object was recorded using Any-Maze software (Stoelting). After 24 h, object recognition was tested by substituting a novel object for a familiar training object (the novel object location was counterbalanced across mice). Because mice inherently prefer to explore novel objects, a preference for the novel object [more time than chance (15 s) spent with the novel object] indicates intact memory for the familiar object.
Statistical analysis.
Mice that were ear tagged and housed five per cage were randomly removed one at a time from the cage and assigned to groups until sufficient numbers were reached for each group. Blinding was performed as follows: (1) individual who administered drugs was blinded to treatment group, and (2) behavioral and stereological analyses were performed by individuals blinded to injury or treatment groups. Quantitative data were expressed as mean ± SEM. Normality testing was performed and datasets passed normality (D'Agostino and Pearson omnibus normality test), and therefore parametric statistical analysis was performed. Statistical analysis was performed using a two-way ANOVA with Tukey post hoc tests, or one-way ANOVA followed by Tukey post hoc analysis to identify differences between groups. When comparisons were made between two conditions, an unpaired Student's t test was performed. Statistical analyses were performed using Prism v8 for Windows (GraphPad Software). Significance level was set as p < 0.05.
Results
TBI results in the activation of cGAS/STING pathway and the induction of type I IFN responses in the injured cortex and hippocampus
Ipsilateral cortical and hippocampal tissue were collected at 72 h after moderate-level CCI for expression analysis of IFN-β and IFN-stimulated genes (ISGs). Activation of cGAS and STING induces the production of type I IFNs (Almine et al., 2017; Cheng et al., 2018). Therefore, we evaluated the cortical expression of cGAS and STING by Western blot (Fig. 1A) and demonstrated that TBI increased protein expression of cGAS (t(8) = 4.395, p = 0.0023 vs sham, t test; 1B) and STING (t(8) = 6.202, p = 0.0003 vs sham; 1C). In addition, IFN-β mRNA expression was also increased in the cortex at 72 h post-injury (t(8) = 3.885, p = 0.006 vs sham; Fig. 1D). We next examined upregulation of genes required for downstream signaling pathways. STAT1 is a mediator of type I IFN-mediated responses (Ivashkiv and Donlin, 2014). STAT1 protein was increased in the cortex of TBI mice (t(8) = 3.442, p = 0.0088 vs sham; Fig. 1E). We also evaluated interferon regulatory factors (IRFs), transcription factors that induce type I IFNs (IRF3, IRF7) and propagate type I IFN responses (IRF1, IRF4, and IRF5; Honda et al., 2006; Günthner and Anders, 2013; Lazear et al., 2013). TBI increased mRNA expression of IRF3 (t(8) = 7.064, p = 0.0002 vs sham; Fig. 1F), IRF1 (t(8) = 5.047, p = 0.002 vs sham; 1G), IRF4 (t(8) = 3.165, p = 0.016 vs sham; 1H), IRF5 (t(8) = 6.935, p = 0.0002 vs sham; 1I), and IRF7 (t(8) = 2.417, p = 0.046 vs sham; 1J). Similar protein and mRNA expression patterns were observed in the ipsilateral hippocampus at 72 h post-injury (Fig. 1K–T).
Figure 1.

Type I IFN response in the injured brain following moderate-level controlled cortical impact. Cortical expression of cGAS and STING protein was assessed by Western immunoblotting (A, representative blot) in the ipsilateral cortex sham and TBI mice at 72 h post-injury. TBI increased cortical expression of cGAS (p = 0.0023; B) and STING (p = 0.023; C) protein compared with sham mice. TBI significantly increased the expression of IFN-β mRNA in the cortex of TBI mice (p = 0.006; D). The expression of STAT1 protein was significantly increased in the cortex of TBI mice compared with sham mice (p = 0.006; E; A, representative blot). mRNA expression of IRF family members was assessed, TBI significantly increased cortical IRF1 (p = 0.002; F), IRF3 (p = 0.0002; G), IRF4 (p = 0.016; H), IRF5 (p = 0.0002; I), and IRF7 (p = 0.046; J) mRNA expression. Expression of cGAS and STING protein was assessed by Western immunoblotting (K, representative blot) in the hippocampus of sham and TBI mice at 72 h post-injury. TBI increased expression of cGAS (t(8) = 3.645, p = 0.0065; L) and STING (t(8) = 2.956, p = 0.0182; M) protein compared with sham mice. IFN-β mRNA expression was significantly increased in the hippocampus of TBI mice (t(8) = 5.447, p = 0.0006; N). The expression of STAT1 protein was significantly increased in the hippocampus of TBI mice compared with sham mice (t(8) = 3.134, p = 0.014; O). mRNA expression of IRF family members was assessed, TBI significantly increased hippocampal IRF1 (t(8) = 9.671, p < 0.0001; P), IRF3 (t(8) = 3.523, p = 0.00078; Q), IRF4 (t(8) = 8.455, p < 0.0001; R), IRF5 (t(8) = 11.92, p < 0.0001; S), and IRF7 (t(8) = 3.921, p = 0.0044; T) mRNA expression. Data expressed as mean ± SEM (n = 5/group). *p < 0.05, **p < 0.01, ***p < 0.001, Student's t test.
IFN-β deficiency attenuates type I IFN signature in the brain following TBI
Having demonstrated a type I IFN signature in the cortex and hippocampus following TBI, we investigated the possible role of IFN-β on the inflammatory response after TBI, using mice with a targeted mutation in IFN-β (designated IFN-β−/−). At 72 h post-injury, TBI increased cGAS (F(1,19) = 43.71, p < 0.0001; two-way ANOVA; Fig. 2B) and STING (F(1,19) = 32.15; p < 0.0001; 2C) protein expression comparably in both WT and IFN-β−/− mice compared with sham operated counterparts. TBI also increased STAT1 protein expression in the cortex of WT mice (F(1,19) = 16.06, p = 0.0008; Fig. 2D), but levels were reduced in IFN-β−/− mice. There was a significant effect of genotype (F(1,19) = 8.866, p = 0.0077), and post hoc analysis demonstrated a significant reduction in STAT1 protein expression in IFN-β−/− TBI mice (p = 0.024, WT TBI vs IFN-β−/−TBI).
Figure 2.

Type I IFN response following TBI is reduced in IFN-β−/− mice. Cortical expression of cGAS and STING protein was assessed by Western immunoblotting (A, representative blot) in the ipsilateral cortex WT and IFN-β−/−sham and TBI mice at 72 h post-injury. TBI significantly increased cortical expression of cGAS (p < 0.0001; B) and STING (p < 0.0001; C) protein in WT and IFN-β−/− mice. The expression of STAT1 protein was significantly increased in the cortex of TBI mice compared with sham mice (p = 0.0004; D); this TBI effect was significantly reduced in IFN-β−/− mice (p = 0.015, WT TBI vs IFN-β−/− TBI). TBI significantly increased mRNA expression of IRF1 (p < 0.0001; E), IRF3 (p = 0.0478; F), IRF4 (p = 0.0208; G), IRF5 (p < 0.0001; H), and IRF7 (p < 0.0001; I) in WT and IFN-β−/− mice. The TBI-induced increase in IRF1 and IRF7 was significantly reduced in IFN-β−/− mice (IRF1: p = 0.0016, IRF7: p < 0.0001; WT TBI vs IFN-β−/− TBI). TBI significantly increased the expression of the viral response genes ISG15 (p = 0.0005; J), MX1 (p < 0.0001; K), and IFI204 (p < 0.0001; L) in WT and IFN-β−/− mice. This TBI effect was significantly reduced in IFN-β−/− mice, ISG15 (p < 0.0001, WT TBI vs IFN−/− TBI), MX1 (p < 0.0001, WT TBI vs I IFN-β−/− TBI), and IFI204 (p < 0.0001, WT TBI vs IFN-β−/− TBI). Expression of cGAS and STING protein was assessed by Western immunoblotting (M, representative blot) in ipsilateral hippocampal sham and TBI mice at 72 h post-injury. TBI increased expression of cGAS (p < 0.0001; N) and STING (p < 0.0001; O) protein compared with sham mice, this TBI effect was significantly reduced in IFN-β−/− mice (cGAS: p = 0.0007, WT TBI vs IFN-β−/− TBI; STING: p = 0.0004, WT TBI vs IFN-β−/− TBI). The expression of STAT1 protein was significantly increased in the hippocampus of TBI mice compared with sham mice (p = 0.0014; P), the expression of STAT1 was significantly attenuated in IFN-β−/− TBI mice compared with WT TBI mice (p = 0.0073). TBI significantly increased hippocampal IRF7 (p = 0.0015; Q), ISG15 (p = 0.0034; R), MX1 (p = 0.0001; S), and IFI204 (p = 0.0052; T) mRNA expression. The TBI effect on all of these genes was significantly reduced in IFN-β−/− mice, IRF7 (p = 0.0002, WT TBI vs IFN-β−/− TBI), ISG15 (p = 0.0006, WT TBI vs IFN-β−/− TBI), MX1 (p < 0.0001, WT TBI vs IFN-β−/− TBI), and IFI204 (p = 0.0078, WT TBI vs IFN-β−/− TBI). Data expressed as Mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001 vs sham (effect of TBI) and +p < 0.05, ++p < 0.01, +++p < 0.001 WT TBI vs IFN-β−/− TBI. Two-way ANOVA (n = 6/group).
We next assessed IRF expression patterns in WT and IFN-β−/− mice. TBI significantly increased mRNA expression of IRF1 (F(1,19) = 108.1, p < 0.0001; Fig. 2E), IRF3 (F(1,19) = 4.4, p = 0.0478; 2F), IRF4 (F(1,19) = 6.3, p = 0.0208; 2G), IRF5 (F(1,19) = 42.5, p < 0.0001; 2H), and IRF7 (F(1,19) = 73.45, p < 0.0001; 2I). Although the TBI-induced increase in the expression of IRF3, IRF4, and IRF5 was comparable in WT and IFN-β−/− mice, there was a significant effect of genotype on IRF1 (F(1,19) = 12.5, p = 0.0025) and IRF7 (F(1,19) = 72.82, p < 0.0001) mRNA expression, as well as a significant interaction between TBI and genotype (IRF1: F(1,19) = 8.274, p = 0.0097; IRF7: F(1,19) = 54.58, p < 0.0001). Post hoc analysis demonstrated that IRF1 and IRF7 mRNA expression was significantly decreased in IFN-β−/− TBI mice (IRF1: p = 0.0016, IRF7: p < 0.0001, WT TBI vs IFN-β−/− TBI). We then examined mRNA expression of genes associated with IFN-mediated antiviral responses, including ISG15, MX1, and IFI204. TBI increased mRNA expression of ISG15 (F(1,19) = 17.78, p = 0.0005; Fig. 2J), MX1 (F(1,19) = 102.1, p < 0.0001; 2K), and IFI204 (F(1,19) = 126.0, p < 0.0001; 2L) in the cortex of WT TBI mice, but not IFN-β TBI mice. There was a significant genotype effect on all genes (ISG15: F(1,19) = 32.07, p < 0.0001; MX1: F(1,19) = 93.98, p < 0.0001; IFI204: F(1,19) = 66.29, p < 0.0001), and an interaction between TBI and genotype for ISG15 (F(1,19) = 12.06, p = 0.0025), MX1 (F(1,19) = 79.23, p < 0.0001), and IFI204 (F(1,19) = 63.07, p < 0.0001). Post hoc analysis revealed that ISG15, MX1, IFI204 mRNA expression was significantly decreased in IFN-β−/− TBI mice (ISG15: p < 0.0001; MX1: p < 0.0001; IFI204: p < 0.0001; WT TBI vs IFN-β−/− TBI).
In the hippocampus, cGAS and STING protein expression were also increased in WT TBI mice at 72 h post-injury (cGAS: F(1,16) = 43.07, p < 0.0001; Fig. 2M; STING: F(1,16) = 35.66, p < 0.0001; 2N). In contrast to the cortex, however, there was a significant genotype effect (cGAS: F(1,16) = 12.52, p = 0.0027; STING: F(1,16) = 14.09, p = 0.0017) and a significant interaction between TBI and genotype (cGAS: F(1,16) = 13.01, p = 0.0024; STING: F(1,16) = 14,47 p = 0.0016). Hippocampal cGAS and STING protein expression was significantly decreased in IFN-β−/− TBI mice (cGAS: p = 0.0007, STING: p = 0.0004; WT TBI vs IFN-β−/− TBI). Similarly, TBI-induced STAT1 protein expression was attenuated in IFN-β−/− TBI mice (Fig. 2O). There were main effects of TBI (F(1,16) = 14.82, p = 0.0014), and genotype (F(1,16) = 12.95, p = 0.0024), but no significant interactions between them. Post hoc analysis demonstrated that hippocampal STAT1 protein expression was significantly reduced in IFNβ−/− TBI mice (p = 0.0073, WT TBI vs IFN-β−/− TBI). Furthermore, TBI increased hippocampal mRNA expression of IRF7 (F(1,16) = 14.66, p = 0.0015; Fig. 2P), ISG15 (F(1,16) = 11.77, p = 0.0034; 2R), MX1 (F(1,16) = 26.01, p = 0.0001; 2S), and IFI204 (F(1,16) = 10.48, p = 0.0052; 2T) in WT mice. There was a significant genotype effect on all genes (IRF7: F(1,16) = 17.05, p = 0.0008; ISG15: F(1,16) = 12.61, p = 0.0027; MX1: F(1,19) = 21.99, p = 0.0002; IFI204: F(1,16) = 7.187, p = 0.0164), and a significant interaction between TBI and genotype (IRF7: F(1,16) = 13.80, p = 0.0019; ISG15: F(1,16) = 13.69, p = 0.0019; MX1: F(1,16) = 21.18, p = 0.0003; IFI204: F(1,16) = 7.195, p = 0.0164). Post hoc analysis revealed that hippocampal IRF7, ISG15, MX1, and IFI204 mRNA expression was significantly reduced in IFNβ−/− TBI mice (IRF7: p = 0.0002; ISG15, p = 0.0006; MX1: p < 0.0001; IFI204: p = 0.0078; WT TBI vs IFN-β−/− TBI). Overall, these data demonstrate that IFN-β plays an important role in the development of the type I IFN gene signature in the cortex and hippocampus acutely after TBI.
IFN-β deficiency results in an altered inflammatory response following TBI
We next examined classical neuroinflammatory responses following TBI. Cortical expression of proinflammatory genes, TNF-α, NOX2, IL-6, IL-1β, CCL5, CXCL10, and markers of glial activation, CD11b (for microglia) and GFAP (for astrocytes), were assessed in WT and IFN-β−/− mice at 72 h post-injury. TBI induced a robust increase in the mRNA expression of TNF-α (F(1,19) = 72.63, p < 0.0001; Fig. 3A), NOX2 (F(1,19) = 48.53, p < 0.0001; 3B), IL-6 (F(1,19) = 14.01, p = 0.0014; 3C), IL-1β (F(1,19) = 36.13, p < 0.0001; 3D), CCL5 (F(1,19) = 266.7, p < 0.0001; 3E), CXCL10 (F(1,19) = 29.99, p < 0.0001; 3F), CD11b (F(1,19) = 159.3, p < 0.0001; 3G), and GFAP (F(1,19) = 286.2, p < 0.0001; ANOVA; 3H). There was a significant genotype effect on TNF-α (F(1,19) = 6.355, p = 0.0208), NOX2 (F(1,19) = 8.937, p = 0.0075), CCL5 (F(1,19) = 27.37, p < 0.0001), and CXCL10 (F(1,19) = 24.73, p < 0.0001), and a significant interaction of TBI and genotype for all genes (TNF-α: F(1,19) = 6.121, p = 0.0230; NOX2: F(1,19) = 9.189, p = 0.0069; CCL5: F(1,19) = 28.72, p < 0.0001; and CXCL10: F(1,19) = 23.36, p < 0.0001). Post hoc analysis demonstrated a significant reduction in TNF-α, NOX2, CCL5 and CXCL10 mRNA expression in IFN-β−/− TBI mice (TNF-α: p = 0.0131; NOX2: p = 0.0027; CCL5: p < 0.0001; CXCL10: p < 0.0001; WT TBI vs IFN-β−/− TBI). Interestingly, both CD11b mRNA and GFAP mRNA were upregulated in WT and IFN-β−/− TBI mice to a comparable extent (Fig. 3G,H). Similar effects of IFN-β−/− genotype on mRNA expression patterns of TNF-α, NOX2, CCL5 and CXCL10 were observed in the ipsilateral hippocampus at 72 h post-injury (Fig. 3I–L). Overall, our data suggest a role for IFN-β in the induction of classical proinflammatory response acutely after TBI.
Figure 3.

IFNβ deficiency reduces the proinflammatory response following TBI. Cortical expression of a number of proinflammatory genes was assessed in sham and TBI at 72 h post-injury. TBI significantly increased cortical mRNA expression of TNF-α (p < 0.0001; A), NOX2 (p < 0.0001; B), IL-6 (p = 0.0014; C), IL-1β (p < 0.0001; D), CCL5 (p < 0.0001; E), CXCL10 (p < 0.0001; F), CD11b (ITGAM) (p < 0.0001; G), and GFAP (p < 0.0001; H) in WT and IFN-β−/− mice. The TBI-induced increase in TNFα, NOX2, CCL5, and CXCL10 was significantly reduced in IFN-β−/− mice (TNF: p = 0.0131; NOX2: p = 0.0027; CCL5: p < 0.0001; CXCL10: p < 0.0001; WT TBI vs IFN-β−/− TBI). Hippocampal mRNA expression of TNF-α, NOX2, CCL5 and CXCL10 was measured in sham and TBI mice at 72 h post-injury. TBI significantly increased hippocampal mRNA expression of TNF-α (F(1,16) = 9.745, p = 0.066; I), NOX2 (F(1,16) = 11.33, p = 0.0039; J), CCL5 (F(1,19) = 15.08, p = 0.0013; K), and CXCL10 (F(1,19) = 22.82, p = 0.0002; L) in WT and IFN-β−/− mice. There was a genotype effect on these genes (NOX2: F(1,16) = 4.975, p = 0.0404; CCL5: F(1,16) = 5.200, p = 0.0366; CXCL10: F(1,16) = 20.79, p = 0.0003), and a significant interaction between TBI and genotype (NOX2: F(1,16) = 5.736, p = 0.0292; CCL5: F(1,16) = 5.839, p = 0.0280; CXCL10: F(1,16) = 18.94, p = 0.0005). The expression of all genes was significantly reduced in IFN-β−/− TBI mice compared with WT TBI mice (TNF-α: p = 0.0458; NOX2: p = 0.0224; CCL5: p = 0.0203; CXCL10: p < 0.0001; WT TBI vs IFN-β−/− TBI). Data expressed as mean ± SEM. **p < 0.01, ***p < 0.001 vs sham (effect of TBI) and +p < 0.05, ++p < 0.01, +++p < 0.001 WT TBI vs IFN-β−/− TBI. Two-way ANOVA (n = 6/group).
Previously, we demonstrated that a decrease in the proinflammatory response after TBI concurrently resulted in increased expression of proinflammatory and anti-inflammatory genes (Kumar et al., 2016; Barrett et al., 2017). Thus, we assessed the mRNA expression of genes associated with anti-inflammatory responses, such as Arg1, YM1, IL-10, SOCS3, and TGFβ, in the cortex at 72 h post-injury. TBI increased cortical expression of Arg1 (F(1,19) = 21.73, p = 0.0002; Fig. 4A), YM1 (F(1,19) = 56.16, p < 0.0001; 4B), IL-10 (F(1,19) = 60.13, p < 0.0001; 4C), SOCS3 (F(1,19) = 46.25, p < 0.0001; 4D), and TGFβ (F(1,19) = 131.7, p < 0.0001; 4E) in WT mice compared with sham mice. There was a genotype effect for YM1 (F(1,16) = 9.669, p = 0.0058), and IL-10 (F(1,16) = 24.55, p < 0.0001) only, and a significant interaction between TBI and genotype for YM1 (F(1,16) = 9.415, p = 0.0063) and IL-10 (F(1,16) = 23.63, p = 0.0001). YM1 and IL-10 mRNA expression was significantly reduced in the cortex of IFN-β−/− TBI mice (YM1, p = 0.0022; IL-10, p < 0.0001; WT TBI vs IFN-β−/− TBI). Similar effects of IFN-β−/− genotype on mRNA expression patterns of Arg1, YM1, SOCS3, and TGFβ were observed in the ipsilateral hippocampus at 72 h post-injury (Fig. 4F–I). IL-10 mRNA expression was undetectable in the hippocampus of all animals.
Figure 4.
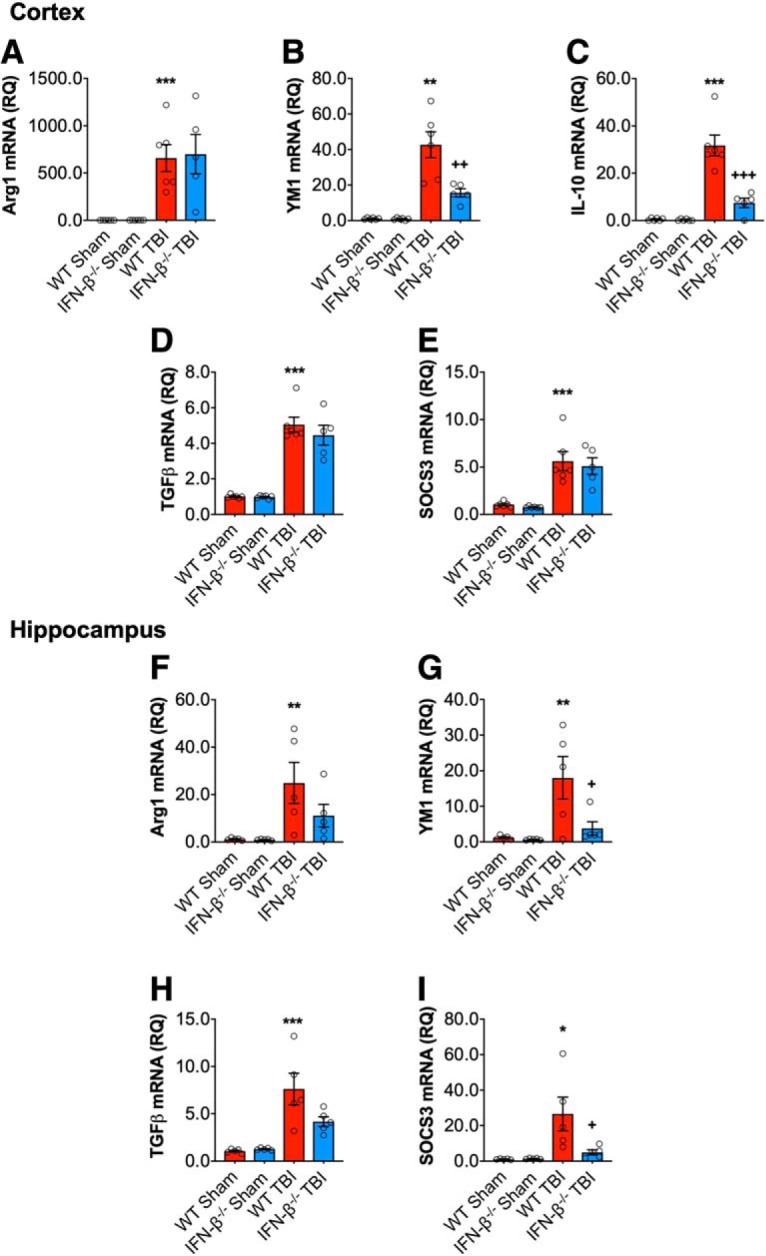
IFN-β deficiency alters the inflammatory response after TBI. Cortical mRNA expression of Arg1, YM1, IL-10, SOCS3, and TGFβ was assessed in WT and IFN-β−/− sham and TBI mice. TBI significantly increased cortical mRNA expression of Arg1 (p = 0.0002; A), YM1 (p < 0.0001; B), IL-10 (p < 0.0001; C), TGFβ (p < 0.0001; D), and SOCS3 (p < 0.0001; E) in WT and IFN-β−/− mice. The effect of TBI on YM1 (p = 0.0022, WT TBI vs IFN-β−/− TBI) and IL-10 (p < 0.0001, WT TBI vs IFN-β−/− TBI) was significantly reduced in IFN-β−/− TBI mice compared with WT TBI mice. Hippocampal mRNA expression of Arg1, YM1, SOCS3, and TGFβ was assessed in WT and IFN-β−/− Sham and TBI mice. TBI significantly increased hippocampal mRNA expression of Arg1 (F(1,16) = 11.70, p = 0.0035; F), YM1 (F(1,16) = 6.131, p = 0.0248; G), TGFβ (F(1,16) = 28.69, p < 0.0001; H), and SOCS3 (F(1,16) = 9.001, p = 0.0085; I) in WT and IFN-β−/− mice. There was a genotype effect on YM1 (F(1,16) = 4.737, p = 0.0449) and SOCS3 (F(1,16) = 4.907, p = 0.0416), as well as a significant interaction between TBI and genotype for YM1 (F(1,16) = 4.578, p = 0.0481) and SOCS3 (F(1,16) = 5.153, p = 0.0374). Post hoc analysis demonstrated decreased YM1 and SOCS3 expression in IFN-β−/− TBI mice (YM1: p = 0.0345; SOCS3: p = 0.0273; WT TBI vs IFN-β−/−TBI). Data expressed as mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001 versus sham (effect of TBI) and +p < 0.05, ++p < 0.01, +++p < 0.001 WT TBI versus IFN-β−/− TBI. Two-way ANOVA (n = 6/group).
IFN-β deficiency improves long-term motor and cognitive function recovery after TBI
To investigate the long-term consequences of IFN-β genetic deletion on TBI outcomes, we evaluated motor and cognitive recovery in sham and TBI WT and IFN-β−/− mice through 28 dpi. We performed a beam walk task to assess deficits in fine-motor coordination. TBI produced significant motor function impairments in WT TBI mice, with 50 ± 0 foot faults (FFs) at 1 dpi and persistent FFs through 28 dpi (47 ± 1 FF; p < 0.0001 vs WT sham; RM-ANOVA; Fig. 5A). In contrast, IFN-β−/− TBI mice exhibited improved motor performance with a reduced number of FFs at 7 (32 ± 2 FF; p < 0.0001 vs WT TBI), 14 (31 ± 2 FF; p < 0.0001 vs WT TBI), 21 (31 ± 3 FF; p < 0.0001 vs WT TBI), and 28 dpi (36 ± 3 FF; p < 0.0001 vs WT TBI).
Figure 5.
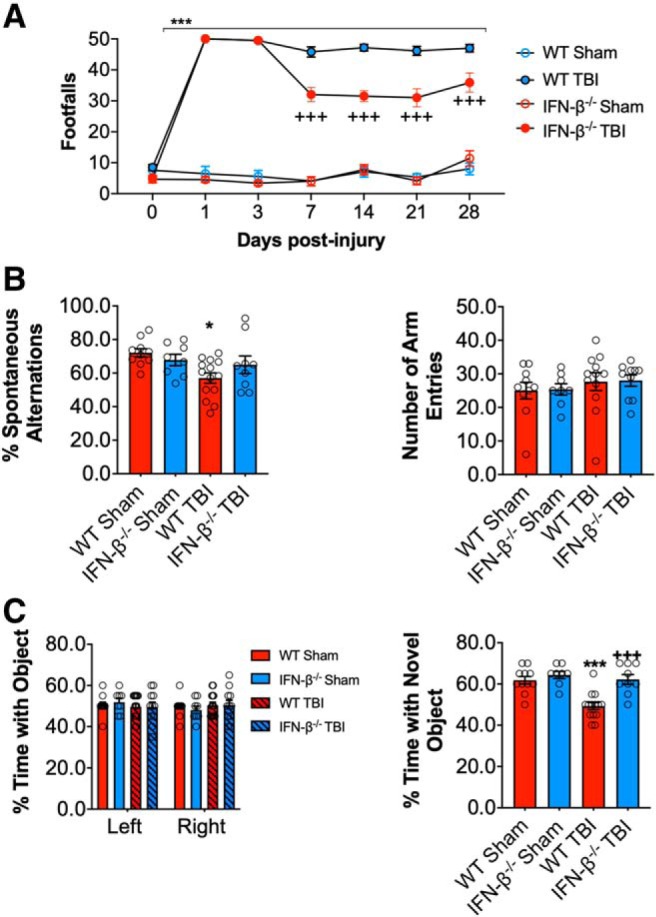
IFN-β deficiency improves motor and cognitive function recovery after TBI. Beam walk analysis of sham and TBI WT and IFN-β−/− mice. In WT mice TBI induced persistent deficits in fine motor coordination through 28 dpi (p < 0.0001, sham vs TBI; A). In contrast, IFN-β−/− TBI mice had significantly reduced fine motor coordination deficits at 14, 21, and 28 dpi (p < 0.0001, WT TBI vs IFN-β−/− TBI). TBI induces a significant decrease in the percentage spontaneous alternations in the Y-maze task in WT TBI mice compared with WT sham counterparts (p = 0.0134; B), at 8 dpi. In addition, there was no significant difference between groups in the number of entries in each arm of the Y-maze task. To assess nonspatial hippocampal-mediated memory, the NOR task was performed at 18 dpi. TBI mice exhibited a significant decrease in percentage time spent with the novel object compared with sham counterparts (p = 0.0008; C). However, the percentage time IFN-β−/− TBI mice spent with the novel object was comparable to sham animals and spent a significant more percentage of time with the novel object compared with WT TBI mice (p < 0.0001). Data expressed as mean ± SEM. *p < 0.05, ***p < 0.001 versus sham (effect of TBI) and +++p < 0.001 WT TBI versus IFN-β−/− TBI. A, Two-way repeated-measures ANOVA (n = 8–13/group) and (B, C) Two-way ANOVA (n = 8–13/group).
Hippocampal-dependent spatial working memory was also assessed using a Y-maze test at 8 dpi. TBI resulted in deficits in working memory in TBI mice (F(1,38) = 6.660, p = 0.0137; two-way ANOVA; Fig. 5B), but only WT TBI mice had significantly decreased spontaneous alternations (p = 0.0151 vs WT sham). Each group of mice had equivalent numbers of arm entries in the Y-maze test. To assess hippocampal-dependent nonspatial learning, we used a NOR test at 17 and 18 dpi. During the familiar phase of the test, there was no difference between groups with regard to time spent with either the right or left objects, indicating no side preference. Twenty-four hours later, mice were retested with a novel object. Compared with WT sham mice (61.8 ± 1.8%), WT TBI mice spent less time with the novel object (49.3 ± 1.7%; Fig. 5C). In contrast, both IFN-β−/− sham and TBI mice spent an increased amount of time with the novel object (62.2.0 ± 2.4% and 64.3 ± 1.8%, respectively; TBI effect: F(1,38) = 16.48, p = 0.0008; genotype effect: F(1,38) = 12.01, p = 0.0013; interaction: F(1,38) = 8.464, p = 0.0060; WT TBI vs IFN-β−/− TBI comparison, adjusted p < 0.0001; Fig. 5C).
IFN-β deficiency reduces microglial activation and neurodegeneration after TBI
To assess the effects of IFN-β deficiency on post-traumatic neuroinflammation we evaluated NOX2 expression in reactive microglia/macrophages (CD68+ cells) in the injured cortex at 28 dpi. TBI induced robust NOX2 expression in CD68+ microglia/macrophages in the cortex of WT and IFN-β−/− TBI mice (F(1,10) = 30.12, p = 0.0003; two-way ANOVA; Fig. 6A). There was a significant genotype effect (F(1,10) = 5.420, p = 0.0422), and post hoc analysis demonstrated that IFN-β deficiency was associated with reduced NOX2 expression in the injured cortex (p = 0.0207 IFN-β−/− TBI vs WT TBI).
Figure 6.
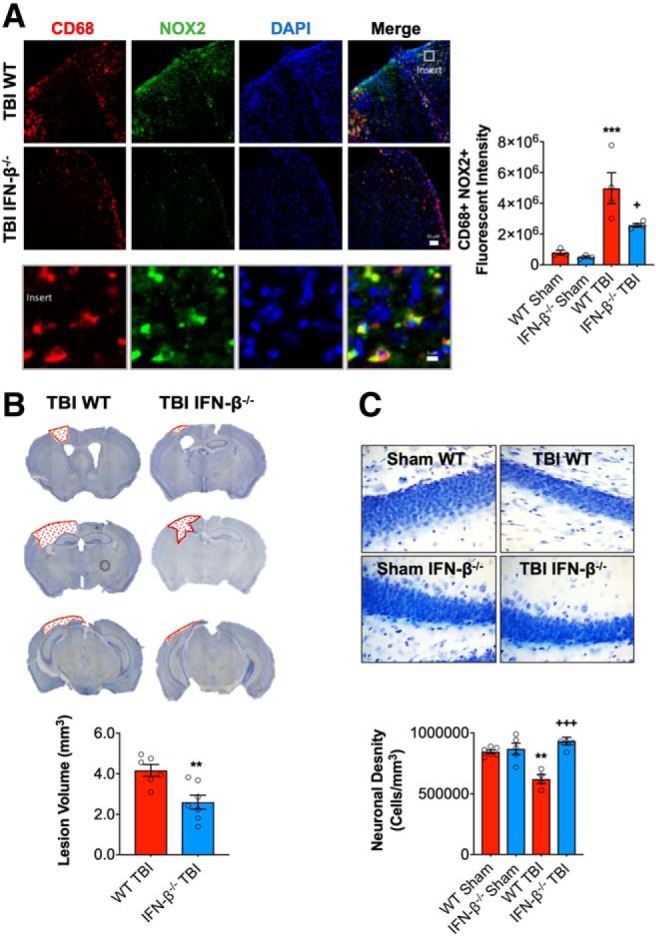
IFN-β deficiency reduces lesion volume and hippocampal neurodegeneration after TBI. Immunofluorescence analysis of NOX2 (green) and CD68 (red) demonstrates that injury-induced NOX2 expression in reactive microglia/macrophages was significantly decreased in IFN-β−/− TBI mice compared with WT TBI mice at 28 dpi (A). Scale bar, 50 μm. Representative images of cresyl violet stained coronal sections from WT and IFN-β−/− TBI mice at 28 dpi (B). Quantification of lesion volume in WT and IFN-β−/− TBI mice at 28 dpi. IFN-β−/− resulted in a significant reduction in TBI lesion volume (p = 0.003, B). Quantification of hippocampal neurodegeneration in WT and IFN-β−/− TBI mice at 28 dpi (C). In WT mice, TBI resulted in significant loss of hippocampal neurons compared with the WT sham group. In contrast, TBI in IFN-β−/− mice resulted in reduced neuronal loss, which was significantly different to the WT TBI group. Data expressed as mean ± SEM. ***p < 0.001 vs sham (effect of TBI) and +++p < 0.001 WT TBI vs IFN-β−/− **p = 0.0013 (Sham WT vs WT TBI), +p < 0.05 WT TBI vs IFN TBI. A, Two-way ANOVA (n = 4–5/group), B, Student's t test (n = 6–7/group), and C, Two-way ANOVA (n = 4–5/group).
To examine the effects of IFN-β deficiency on neurodegeneration, we quantified TBI-induced lesion volume and neuronal loss at 28 dpi. TBI-induced lesion volume was significantly reduced in IFN-β−/− TBI mice (2.6 ± 0.35 mm3) compared with WT TBI mice (4.17 ± 0.29 mm3; t(11) = 3.399, p = 0.003, t test; vs WT TBI; Fig. 6B). Furthermore, TBI resulted in hippocampal neurodegeneration with a significant loss of neurons in the dentate gyrus compared with sham WT mice (F(1,15) = 4.86, p = 0.0435; two-way ANOVA; Fig. 6C). There was a significant genotype effect (F(1,15) = 22.53, p = 0.0002) and an interaction of TBI and genotype (F(1,15) = 16.05, p = 0.0011), with neuronal density being equivalent to sham levels in the IFN-β−/− TBI mice. Notably, hippocampal neuronal loss was reduced in IFN-β−/− TBI mice (932,580 ± 32,045 cells/mm3) compared with WT TBI mice (621,054 ± 37,628 cells/mm3; p = 0.0001 vs WT TBI). Overall, these data demonstrate that IFN-β deficiency results in improvements in long-term motor and cognitive function recovery and reduced NOX2-associated microglial activation and related neurodegeneration after TBI.
Central administration of neutralizing αIFNAR improves acute phase neurological function, but fails to improve long-term neurological recovery or reduce neurodegeneration after TBI
We next investigated whether post-injury treatment of TBI mice with a neutralizing type I IFN receptor antibody (αIFNAR) could improve functional recovery. The absence of drug-based interventions that can specifically target IFN-β limited our ability to mimic the genetic knock-out. Because IFNAR is a shared IFN-α/β receptor, our treatment regime using αIFNAR inhibits more broad range type I IFN responses, with the advantage of potentially increasing therapeutic relevance. CCI mice received a central infusion of αIFNAR (12 μg/d) or an equal concentration of an isotype-matched control antibody (IgG1) via contralateral ICV administration using ALZET pumps, beginning at 30 min post-injury and continuing through 3 dpi. Motor and cognitive function recovery were assessed up to 28 dpi, and TBI neuropathology was also evaluated at 28 dpi.
Compared with sham mice, TBI resulted in fine-motor coordination deficits in IgG-treated TBI mice, with 50 ± 0 FFs at 1 dpi that persisted through 28 dpi (42 ± 2 FF; p < 0.0001 vs sham; RM-ANOVA; Fig. 7A). Administration of αIFNAR significantly improved motor function recovery at 14 dpi (34 ± 3 FF; p = 0.0431 vs TBI+IgG), although this improvement was not sustained. We also assessed hippocampal-dependent spatial working memory at 8 dpi using a Y-maze test. TBI induced cognitive deficits in the Y-maze test (F(2,18) = 5.723; p = 0.0119, ANOVA; Fig. 7B), such that IgG-treated TBI mice had reduced spontaneous alternations (p = 0.0245 vs sham; Fig. 7B). In contrast, αIFNAR-treated TBI mice exhibited increased spontaneous alternations (p = 0.0282 vs IgG-treated TBI mice; Fig. 7B), and performed to similar level as sham mice in this cognitive task. Each group of mice had approximately equal numbers of arm entries in the Y-maze test.
Figure 7.
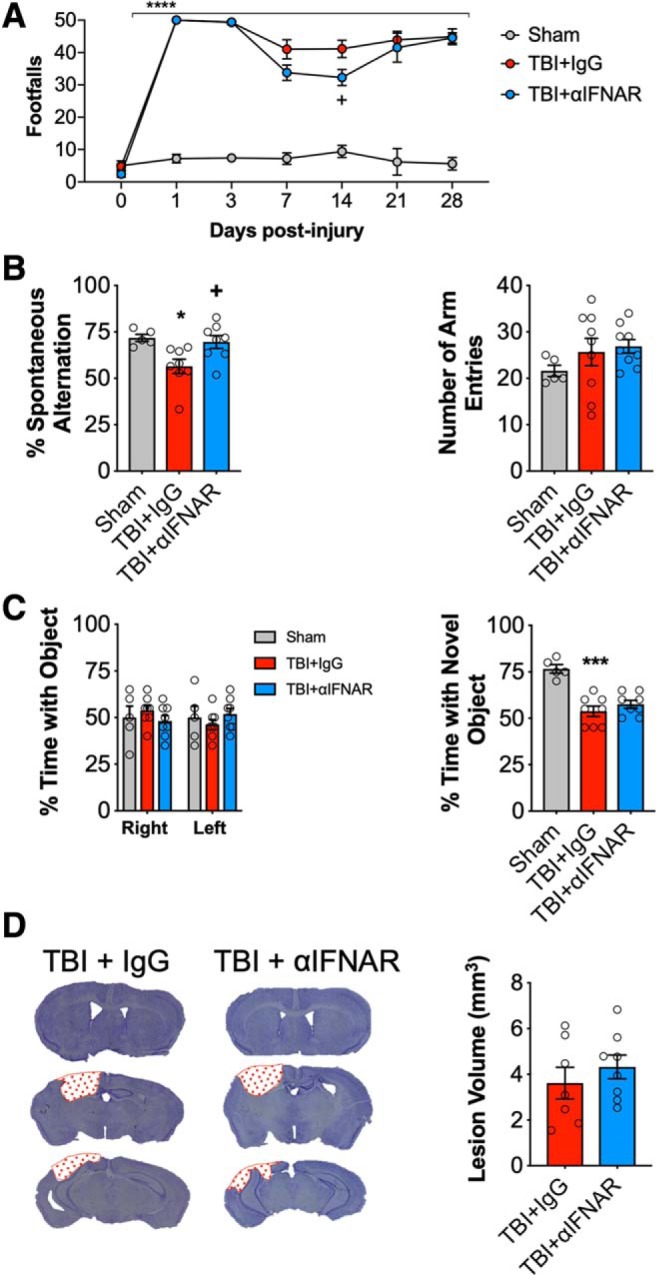
Early post-injury inhibition of type I IFN signaling provides transient improvements in neurological function following TBI that are lost at later time points. Beam walk analysis of sham, IgG-, and αIFNAR-treated TBI mice at 28 dpi. TBI induced persistent deficits in fine motor coordination in IgG- and αIFNAR-treated TBI mice through 28 dpi (p < 0.0001, vs sham; A). TBI induces a significant decrease in the percentage spontaneous alternations in the Y-maze task in IgG-treated TBI mice compared with sham counterparts (p = 0.0281, vs sham; B), at 8 dpi. No injury effect was observed in αIFNAR-treated TBI mice; in addition to this, αIFNAR-treated TBI mice exhibited a significant improvement in performance compared with IgG-treated TBI mice (p = 0.0331 vs IgG-treated TBI mice). No significant differences were observed between groups in the number of entries in each arm of the Y-maze task. To assess nonspatial hippocampal-mediated memory, the NOR task was performed at 18 dpi. TBI induced a significant decrease in percentage time IgG- and αIFNAR-treated TBI mice spent with the novel object compared with sham counterparts (TBI+IgG: p < 0.0001; TBI+αIFNAR: p = 0.0002, vs sham; C). There was no effect of treatment. Representative images for lesion volume in IgG-treated- and αIFNAR-treated TBI mice at 28 dpi (D). Stereological analysis revealed lesion volume to be similar in IgG- and αIFNAR-treated mice. Data expressed as mean ± SEM. *p < 0.05, ***p < 0.001, ****p < 0.0001 versus sham (effect of TBI), and +p < 0.05 TBI + IgG versus TBI+IFNAR. Two-way ANOVA (A; n = 5–9/group), one-way ANOVA (B, C; n = 5–9/group), and Student's t test (D; n = 8/9group).
We then assessed hippocampal-dependent nonspatial learning after TBI using NOR test at 17 and 18 dpi. During the familiar phase of the test, there was no difference between groups with regard to time spent with either the right or left objects, indicating no side preference. Twenty-four hours later, mice were retested with a novel object. Compared with WT sham mice (76.6 ± 2.3%; Fig. 7C), IgG-treated and αIFNAR-treated TBI mice spent less time with the novel object (53.8 ± 2.8% and 57.5 ± 2.1%, respectively; Fig. 7C). However, central administration of αIFNAR failed to improve working memory in this test (TBI effect, F(2,18) = 19.78, p < 0.0001 IgG-treated TBI mice vs sham; p = 0.002 αIFNAR-treated TBI mice vs sham). Finally, we quantified TBI-induced lesion volume in the ipsilateral cortex of IgG- and αIFNAR-treated TBI mice at 28 dpi. αIFNAR treatment failed to reduce TBI-induced lesion volume (3.6 ± 0.69 mm3) compared with IgG-treated TBI mice (4.1 ± 0.54 mm3; Fig. 7D).
Type I IFN genes are chronically elevated in the injured cortex following TBI
The failure of short-term αIFNAR neutralization to reduce chronic neurological impairments and neurodegeneration after TBI indicates that IFN-related pathways are active for prolonged periods after TBI. Therefore, we set out to assess their expression during the chronic phase of TBI. Ipsilateral cortical tissue was collected at 60 dpi to quantify the expression of ISGs and proinflammatory genes. We determined that key ISGs and proinflammatory genes remain significantly upregulated in the chronically injured cortex. Although there was no effect of TBI on cGAS gene expression (Fig. 8A), we found that TBI increased mRNA expression of STING (t(12) = 4.593, p = 0.0006 vs sham; Fig. 8B), STAT1 (t(12) = 3.929, p = 0.0020 vs sham; 8C), IRF1 (t(12) = 3.197, p = 0.0077 vs sham; 8D), IRF7 (t(12) = 3.647, p = 0.046 vs sham; 8E), CXCL10 (t(12) = 3.220, p = 0.0074 vs sham; 8F), ISG15 (t(12) = 2.616, p = 0.0225 vs sham; 8G), and IFI204 (t(12) = 5.132, p = 0.0002 vs sham; 8H). Chronic TBI also increased mRNA expression of proinflammatory TNF-α (t(12) = 3.006, p = 0.0101 vs sham; Fig. 8I), NOX2 (t(12) = 4.591, p = 0.0005 vs sham; 8J), CCL5 (t(12) = 3.642, p = 0.003 vs sham; 8K), and glial activation markers CD11b (t(12) = 5.867, p < 0.0001 vs sham; 8L), CD68 (t(12) = 5.353, p = 0.0001 vs sham; 8M), and GFAP (t(12) = 5.381, p = 0.0001 vs sham; 8N).
Figure 8.
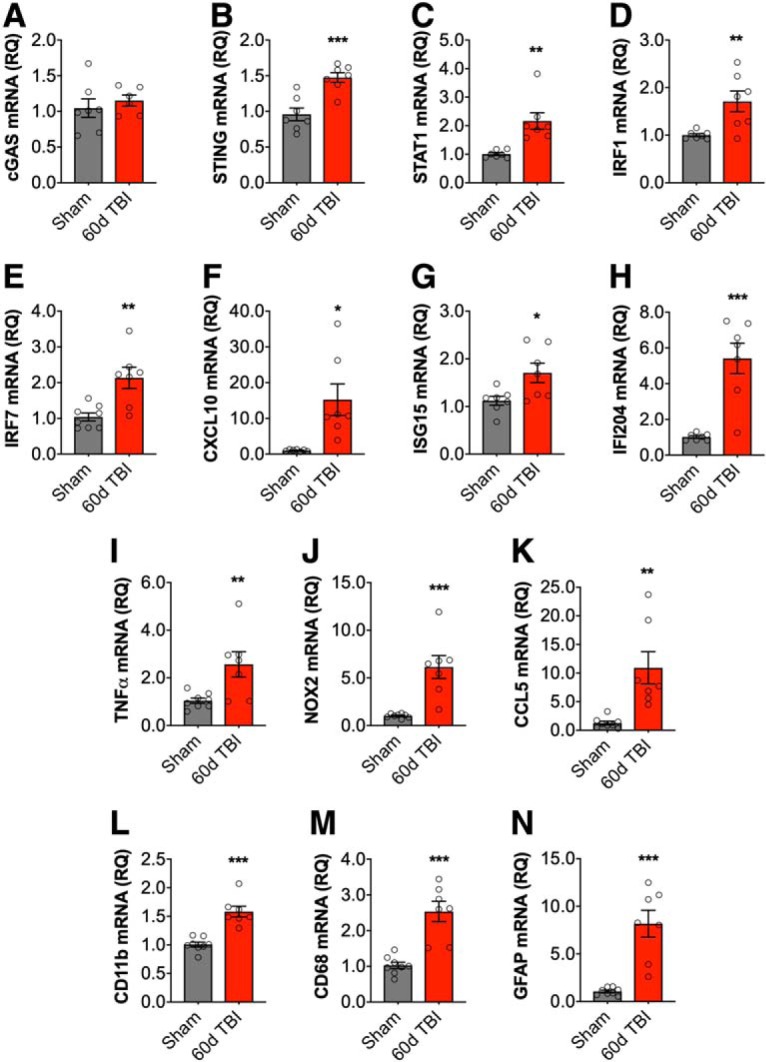
Type I IFN genes are chronically elevated in the injured cortex following TBI. Cortical mRNA expression of type I IFN-related genes was assessed at 60 dpi. Although there was no effect of TBI on cGAS gene expression (A), TBI significantly increased cortical STING (p = 0.0006; B), STAT1 (p = 0.0020; C), IRF1 (p = 0.0077; D), IRF7 (p = 0.046; E), CXCL10 (p = 0.0074; F), ISG15 (p = 0.0225; G), and IFI204 (p = 0.0002; H) mRNA expression. Chronic TBI also increased mRNA expression of TNF-α (p = 0.0101; I), NOX2 (p = 0.0005; J), CCL5 (p = 0.003; K), CD11b (p < 0.0001; L), CD68 (p = 0.0001; M), and GFAP (p = 0.0001; N). Data expressed as mean ± SEM (n = 7/group). *p < 0.05, **p < 0.01, ***p < 0.001, Student's t test.
Discussion
Prior preclinical studies have shown that IFNAR1 (IFN-α and -β receptor) knock-out mice have reduced lesion size and proinflammatory cytokine expression as well as increased levels of anti-inflammatory mediators following TBI (Karve et al., 2016). This indicates that type I IFNs contribute to secondary neuroinflammation after TBI and inhibiting these pathways will result in neuroprotection. However, studies based on IFNAR signaling cannot pinpoint the functional contribution of IFN-α and IFN-β during neuroprotection. Moreover, similar to its protective actions in MS (Scheu et al., 2019), exogenous administration of IFN-β following spinal cord injury improves functional recovery in mice, in part by reducing lipid peroxidation and inflammatory cytokines (Gok et al., 2007; Sandrow-Feinberg et al., 2010). This highlights the complex role of IFN-β during secondary injury and CNS repair. Here, our preclinical studies using IFN-β knock-out mice demonstrate that IFN-β drives secondary neuroinflammation because genetic deletion of IFN-β results in improved motor and cognitive function recovery following TBI and reduces chronic neurodegeneration. As such, this is the first study to implicate IFN-β in the development of neurological dysfunction and neurodegeneration following TBI.
DNA released from dying cells can act as an alarmin, resulting in the activation of the immune response (Almine et al., 2017). We demonstrate that cGAS and STING, critical components of the cytosolic DNA sensing machinery, are robustly upregulated in the injured cortex and hippocampus within 72 h of brain trauma. Others have shown that STING mRNA is induced immediately after TBI, and is associated with elevated expression of IFN-related and inflammatory genes (Abdullah et al., 2018). Activation of cGAS-STING pathway in our model was accompanied by a robust increase in IFN-β mRNA expression in the injured brain. Although a recent study demonstrates that neurons under ER stress upregulate IFN-β after TBI (Sen et al., 2020), it is also likely that other cells, including glia (Cox et al., 2015), produce IFN-β during secondary neuroinflammation. Importantly, it has also been reported that IFN-β mRNA is highly upregulated after 6 h of TBI in human postmortem tissue, and specifically in the injured hemisphere (Karve et al., 2016).
We also found that STAT1, a key mediator of type I IFN responses (Ivashkiv and Donlin, 2014), was also upregulated in the injured brain. IRFs are involved in the induction and amplification of type I IFN responses (Honda and Taniguchi, 2006). IRF3 is required for the production of IFN-β (Akira et al., 2006; Honda and Taniguchi, 2006; Hatesuer et al., 2017), whereas IRF1 and IRF5 contribute to the propagation of proinflammatory responses (Liu et al., 2003; Takaoka et al., 2005; Xie et al., 2016). Furthermore, IRF4 is involved in the induction of anti-inflammatory cytokines, IL-4 and IL-10 (Ahyi et al., 2009), whereas IRF7 amplifies type I IFN responses, such that deletion of IRF7 diminishes responses to inflammatory stimuli (Honda et al., 2005; Tanaka et al., 2015). We determined that IRF1, IRF3, IRF4, IRF5, and IRF7 mRNA expression were significantly upregulated within 72 h of brain trauma. Notably, TBI-induced STAT1 expression was significantly reduced in IFN-β−/− TBI mice, as were expression levels of IRF1 and IRF7 mRNA. The latter observation is in agreement with a previous report that IRF7 mRNA expression was decreased in IFNAR1 knock-out mice (Karve et al., 2016). Interestingly, lipopolysaccharide-induced STAT1, IRF1, and IRF7 changes have been shown to be reduced in IFN-β−/− macrophages compared with WT macrophages (Thomas et al., 2006; Sheikh et al., 2014). We also demonstrated that the TBI-related increase in viral response genes (ISG15, MX1, and IFI204) was significantly reduced in IFNβ−/− TBI mice.
Levels of proinflammatory cytokines and chemokines (TNF-α, NOX2, IL-6, CCL5, CXCL10) were reduced in the injured cortex of IFNβ−/− mice compared with their WT counterparts. These findings support prior reports that decreased type I IFN signaling is associated with decreased inflammatory gene expression during the acute phase after TBI (Karve et al., 2016; Abdullah et al., 2018). Inhibition of type I IFNs also reduce chronic neuroinflammation in models of AD (Minter et al., 2016), Parkinson's disease (Main et al., 2016), and prion disease (Nazmi et al., 2019). Although anti-inflammatory markers, Arg1, SOCS3, and TGFβ mRNA expression levels were similar in the cortex WT and IFNβ−/− mice, the expression levels of the IL-10 and YM1 were significantly decreased in IFN-β−/− TBI mice. IL-10 production in the CNS is dependent upon IFN-β activity (Lobo-Silva et al., 2017), and inhibition of type I IFN signaling in a model of prion disease reduces IL-10 expression levels (Nazmi et al., 2019). Furthermore, in a respiratory syncytial virus model, YM1 expression in peripheral macrophages also appears to be dependent upon IFN-β (Shirey et al., 2010). Overall, these findings suggest that IFN-β contributes to neuroinflammatory responses by promoting both proinflammatory and anti-inflammatory gene expression.
Proinflammatory gene expression was also significantly reduced in the hippocampus of IFN-β−/− TBI mice suggesting that IFN-β actively contributes to the propagation of post-traumatic inflammatory responses at distant sites to the primary lesion. Thus, targeted inhibition of IFN-β may limit the spread of damaging inflammatory cascades to other brain regions, thereby reducing functional loss (cognitive deficits). TBI-induced effects on STAT1 and IRF7 were also significantly decreased in the hippocampus of IFNβ−/− mice, along with a significant decrease in viral response genes (ISG15, MX1, and IFI204). Our data suggest that IFN-β upregulates IFI204, which may contribute to detrimental inflammatory processes and functional impairments in the hippocampus through a positive feedforward induction of type I IFNs (Storek et al., 2015; Almine et al., 2017). Although we demonstrated increased chronic expression of type I IFN related genes at 60 dpi, the current study was primarily focused on acute changes including the early transcriptional regulation of type I responses after TBI; future studies will examine chronic mechanisms involved in type I IFN activation, including protein level changes and cell-specific responses.
The long-term impact of selectively inhibiting type I IFNs on functional recovery and TBI neuropathology is unknown (Roselli et al., 2018). Here, we demonstrated that IFN-β−/− TBI mice had improved long-term motor and cognitive function recovery, which was accompanied by reduced microglial activation and neurodegeneration. We, and others, have shown that NOX2 is a key inflammatory mediator in post-traumatic neurodegeneration, as decreased NOX2 expression is associated with improved neurological function and attenuated neurodegeneration (Dohi et al., 2010; Zhang et al., 2012; Kumar et al., 2016; Ma et al., 2017). In the present study, we show that NOX2 expression in microglia was significantly reduced in IFN-β−/− mice compared with WT mice, suggesting that neuroprotection conferred by IFN-β−/− is mediated in part through reduced chronic microglial activation. IFNAR1 knock-out mice also have improvements in motor function recovery through 7 d post-TBI (Karve et al., 2016) due to loss of IFNAR signaling. Our neurobehavioral analysis through 28 dpi demonstrated sustained improvements in motor function in injured IFN-β−/− mice. Whereas previous work reported that inhibition of type I IFN signaling can improve cognitive function in normal aging (Baruch et al., 2014) and in AD models (Minter et al., 2016), our study is the first to demonstrate the long-term beneficial effects of IFN-β inhibition on cognitive function after TBI.
To evaluate the translational potential of our genetic studies, we treated TBI mice with a type I IFN neutralizing antibody, αIFNAR. Central administration of αIFNAR initiated at 30 min post-injury and continued for 72 h improved neurological recovery during the subacute phase. Others have shown that αIFNAR treatment reduces the production of IFN-α and IFN-β, as well as decreases the production of IL-1β and IL-6 acutely after TBI (Karve et al., 2016). In our study, we show that αIFNAR treatment leads to improved motor and cognitive function recovery after TBI; however, beneficial effects were not sustained beyond 14 dpi, and treatment did not reduce lesion volume at 28 dpi. One possible explanation for the non-sustained improvements in functional recovery was that the duration of αIFNAR treatment was too short (0–3 dpi), allowing IFN-mediated signaling to be restored. Interestingly, RNA sequencing and pathway analyses indicate that immune responses associated with IFN-α and IFN-β signaling are chronically upregulated in isolated microglia up to 90 d post-TBI (Makinde et al., 2020). This suggests that type I IFN-related pathways are active for prolonged periods after TBI, and chronic administration of inhibitors will be required to improve long-term neurological recovery. Indeed, our data demonstrate that type I IFNs and microglial activation genes remain chronically elevated up to 60 dpi. It is also possible that central infusion of αIFNAR failed to reduce type I IFN-mediated changes in peripheral immune cells. In fact, previous data suggest that ablation of IFNAR signaling on hematopoietic cells is critical for neuroprotection following TBI (Karve et al., 2016). The use of a constitutive IFN-β knock-out mouse model is likely to have impacted peripheral immune responses, and this may have contributed to sustained neuroprotection in our genetic studies. Additional studies will be needed to investigate the functional role of IFN-β, and related type I IFNs, in peripheral immune cells following TBI.
In summary, TBI induces a robust neuroinflammatory response that is associated with increased expression of IFN-β and other IFN-related genes. Inhibition of IFN-β reduces post-traumatic neuroinflammation and neurodegeneration resulting in long-term motor and cognitive function recovery. These preclinical studies suggest that therapeutic inhibition of type I IFNs, and IFN-β in particular, may limit persistent neuroinflammation and the development of motor and cognitive function deficits following moderate-to-severe TBI.
Footnotes
This work was supported by National Institutes of Health Grants R01NS082308 (D.J.L.), R01NS037313 (A.I.F.), R01NS096002 (B.A.S.), R01NS110756 (D.J.L./A.I.F./B.A.S.), AI125215 (S.N.V.), U.S. Veterans Affairs Grant 1I01 RX001993 (B.A.S.), and Science Foundation Ireland Grant 17/FRL/4860 (D.J.L.).
The authors declare no competing financial interests.
References
- Abdullah A, Zhang M, Frugier T, Bedoui S, Taylor JM, Crack PJ (2018) STING-mediated type-I interferons contribute to the neuroinflammatory process and detrimental effects following traumatic brain injury. J Neuroinflammation 15:323. 10.1186/s12974-018-1354-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahyi AN, Chang HC, Dent AL, Nutt SL, Kaplan MH (2009) IFN regulatory factor 4 regulates the expression of a subset of Th2 cytokines. J Immunol 183:1598–1606. 10.4049/jimmunol.0803302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akira S, Uematsu S, Takeuchi O (2006) Pathogen recognition and innate immunity. Cell 124:783–801. 10.1016/j.cell.2006.02.015 [DOI] [PubMed] [Google Scholar]
- Almine JF, O'Hare CA, Dunphy G, Haga IR, Naik RJ, Atrih A, Connolly DJ, Taylor J, Kelsall IR, Bowie AG, Beard PM, Unterholzner L (2017) IFI16 and cGAS cooperate in the activation of STING during DNA sensing in human keratinocytes. Nat Commun 8:14392. 10.1038/ncomms14392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aungst SL, Kabadi SV, Thompson SM, Stoica BA, Faden AI (2014) Repeated mild traumatic brain injury causes chronic neuroinflammation, changes in hippocampal synaptic plasticity, and associated cognitive deficits. J Cereb Blood Flow Metab 34:1223–1232. 10.1038/jcbfm.2014.75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrett JP, Henry RJ, Villapol S, Stoica BA, Kumar A, Burns MP, Faden AI, Loane DJ (2017) NOX2 deficiency alters macrophage phenotype through an IL-10/STAT3 dependent mechanism: implications for traumatic brain injury. J Neuroinflammation 14:65. 10.1186/s12974-017-0843-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baruch K, Deczkowska A, David E, Castellano JM, Miller O, Kertser A, Berkutzki T, Barnett-Itzhaki Z, Bezalel D, Wyss-Coray T, Amit I, Schwartz M (2014) Aging: aging-induced type I interferon response at the choroid plexus negatively affects brain function. Science 346:89–93. 10.1126/science.1252945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byrnes AA, McArthur JC, Karp CL (2002) Interferon-beta therapy for multiple sclerosis induces reciprocal changes in interleukin-12 and interleukin-10 production. Ann Neurol 51:165–174. 10.1002/ana.10084 [DOI] [PubMed] [Google Scholar]
- Byrnes KR, Loane DJ, Stoica BA, Zhang J, Faden AI (2012) Delayed mGluR5 activation limits neuroinflammation and neurodegeneration after traumatic brain injury. J Neuroinflammation 9:43. 10.1186/1742-2094-9-43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng WY, He XB, Jia HJ, Chen GH, Jin QW, Long ZL, Jing ZZ (2018) The cGas-sting signaling pathway is required for the innate immune response against ectromelia virus. Front Immunol 9:1297. 10.3389/fimmu.2018.01297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chin AC. (2019) Neuroinflammation and the cGAS-STING pathway. J Neurophysiol 121:1087–1091. 10.1152/jn.00848.2018 [DOI] [PubMed] [Google Scholar]
- Cox DJ, Field RH, Williams DG, Baran M, Bowie AG, Cunningham C, Dunne A (2015) DNA sensors are expressed in astrocytes and microglia in vitro and are upregulated during gliosis in neurodegenerative disease. Glia 63:812–825. 10.1002/glia.22786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deonarain R, Verma A, Porter AC, Gewert DR, Platanias LC, Fish EN (2003) Critical roles for IFN-beta in lymphoid development, myelopoiesis, and tumor development: links to tumor necrosis factor alpha. Proc Natl Acad Sci U S A 100:13453–13458. 10.1073/pnas.2230460100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon CE, Kochanek PM, Yan HQ, Schiding JK, Griffith RG, Baum E, Marion DW, DeKosky ST (1999) One-year study of spatial memory performance, brain morphology, and cholinergic markers after moderate controlled cortical impact in rats. J Neurotrauma 16:109–122. 10.1089/neu.1999.16.109 [DOI] [PubMed] [Google Scholar]
- Dohi K, Ohtaki H, Nakamachi T, Yofu S, Satoh K, Miyamoto K, Song D, Tsunawaki S, Shioda S, Aruga T (2010) Gp91phox (NOX2) in classically activated microglia exacerbates traumatic brain injury. J Neuroinflammation 7:41. 10.1186/1742-2094-7-41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Field R, Campion S, Warren C, Murray C, Cunningham C (2010) Systemic challenge with the TLR3 agonist poly I: c induces amplified IFNα/β and IL-1β responses in the diseased brain and exacerbates chronic neurodegeneration. Brain Behav Immun 24:996–1007. 10.1016/j.bbi.2010.04.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gok B, Okutan O, Beskonakli E, Palaoglu S, Erdamar H, Sargon MF (2007) Effect of immunomodulation with human interferon-beta on early functional recovery from experimental spinal cord injury. Spine 32:873–880. 10.1097/01.brs.0000259841.40358.8f [DOI] [PubMed] [Google Scholar]
- Günthner R, Anders HJ (2013) Interferon-regulatory factors determine macrophage phenotype polarization. Mediators Inflamm 2013:731023. 10.1155/2013/731023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatesuer B, Hoang HT, Riese P, Trittel S, Gerhauser I, Elbahesh H, Geffers R, Wilk E, Schughart K (2017) Deletion of Irf3 and Irf7 genes in mice results in altered interferon pathway activation and granulocyte-dominated inflammatory responses to influenza A infection. J Innate Immun 9:145–161. 10.1159/000450705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henry RJ, Doran SJ, Barrett JP, Meadows VE, Sabirzhanov B, Stoica BA, Loane DJ, Faden AI (2019) Inhibition of miR-155 limits neuroinflammation and improves functional recovery after experimental traumatic brain injury in Mice. Neurotherapeutics 16:216–230. 10.1007/s13311-018-0665-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honda K, Taniguchi T (2006) IRFs: master regulators of signalling by toll-like receptors and cytosolic pattern-recognition receptors. Nat Rev Immunol 6:644–658. 10.1038/nri1900 [DOI] [PubMed] [Google Scholar]
- Honda K, Yanai H, Negishi H, Asagiri M, Sato M, Mizutani T, Shimada N, Ohba Y, Takaoka A, Yoshida N, Taniguchi T (2005) IRF-7 is the master regulator of type-I interferon-dependent immune responses. Nature 434:772–777. 10.1038/nature03464 [DOI] [PubMed] [Google Scholar]
- Honda K, Takaoka A, Taniguchi T (2006) Type I interferon [corrected] gene induction by the interferon regulatory factor family of transcription factors. Immunity 25:349–360. 10.1016/j.immuni.2006.08.009 [DOI] [PubMed] [Google Scholar]
- Ivashkiv LB, Donlin LT (2014) Regulation of type I interferon responses. Nat Rev Immunol 14:36–49. 10.1038/nri3581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobs LD, Beck RW, Simon JH, Kinkel RP, Brownscheidle CM, Murray TJ, Simonian NA, Slasor PJ, Sandrock AW (2000) Intramuscular interferon beta-1a therapy initiated during a first demyelinating event in multiple sclerosis. CHAMPS study Group. N Engl J Med 343:898–904. 10.1056/NEJM200009283431301 [DOI] [PubMed] [Google Scholar]
- Johnson VE, Stewart JE, Begbie FD, Trojanowski JQ, Smith DH, Stewart W (2013) Inflammation and white matter degeneration persist for years after a single traumatic brain injury. Brain 136:28–42. 10.1093/brain/aws322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karve IP, Zhang M, Habgood M, Frugier T, Brody KM, Sashindranath M, Ek CJ, Chappaz S, Kile BT, Wright D, Wang H, Johnston L, Daglas M, Ates RC, Medcalf RL, Taylor JM, Crack PJ (2016) Ablation of type-1 IFN signaling in hematopoietic cells confers protection following traumatic brain injury. eNeuro 3:ENEURO.0128–15.2016. 10.1523/ENEURO.0128-15.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar A, Stoica BA, Sabirzhanov B, Burns MP, Faden AI, Loane DJ (2013) Traumatic brain injury in aged animals increases lesion size and chronically alters microglial/macrophage classical and alternative activation states. Neurobiol Aging 34:1397–1411. 10.1016/j.neurobiolaging.2012.11.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar A, Barrett JP, Alvarez-Croda DM, Stoica BA, Faden AI, Loane DJ (2016) NOX2 drives M1-like microglial/macrophage activation and neurodegeneration following experimental traumatic brain injury. Brain Behav Immun 58:291–309. 10.1016/j.bbi.2016.07.158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazear HM, Lancaster A, Wilkins C, Suthar MS, Huang A, Vick SC, Clepper L, Thackray L, Brassil MM, Virgin HW, Nikolich-Zugich J, Moses AV, Gale M Jr, Früh K, Diamond MS (2013) IRF-3, IRF-5, and IRF-7 coordinately regulate the type I IFN response in myeloid dendritic cells downstream of MAVS signaling. PLoS Pathog 9:e1003118. 10.1371/journal.ppat.1003118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Cao S, Herman LM, Ma X (2003) Differential regulation of interleukin (IL)-12 p35 and p40 gene expression and interferon (IFN)-gamma-primed IL-12 production by IFN regulatory factor 1. J Exp Med 198:1265–1276. 10.1084/jem.20030026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2(−δδC(T)) method. Methods 25:402–408. 10.1006/meth.2001.1262 [DOI] [PubMed] [Google Scholar]
- Loane DJ, Pocivavsek A, Moussa CE, Thompson R, Matsuoka Y, Faden AI, Rebeck GW, Burns MP (2009) Amyloid precursor protein secretases as therapeutic targets for traumatic brain injury. Nat Med 15:377–379. 10.1038/nm.1940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loane DJ, Kumar A, Stoica BA, Cabatbat R, Faden AI (2014) Progressive neurodegeneration after experimental brain trauma: association with chronic microglial activation. J Neuropathol Exp Neurol 73:14–29. 10.1097/NEN.0000000000000021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lobo-Silva D, Carriche GM, Castro AG, Roque S, Saraiva M (2017) Interferon-beta regulates the production of IL-10 by toll-like receptor-activated microglia. Glia 65:1439–1451. 10.1002/glia.23172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma MW, Wang J, Dhandapani KM, Brann DW (2017) NADPH oxidase 2 regulates NLRP3 inflammasome activation in the brain after traumatic brain injury. Oxid Med Cell Longev 2017:6057609. 10.1155/2017/6057609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maas AIR, Menon DK, Adelson PD, Andelic N, Bell MJ, Belli A, Bragge P, Brazinova A, Buki A, Chesnut RM, Citerio G, Coburn M, Cooper DJ, Crowder AT, Czeiter E, Czosnyka M, Diaz-Arrastia R, Dreier JP, Duhaime AC, Ercole A, et al. (2017) Traumatic brain injury: integrated approaches to improve prevention, clinical care, and research. Lancet Neurol 16:987–1048. 10.1016/S1474-4422(17)30371-X [DOI] [PubMed] [Google Scholar]
- Main BS, Zhang M, Brody KM, Ayton S, Frugier T, Steer D, Finkelstein D, Crack PJ, Taylor JM (2016) Type-1 interferons contribute to the neuroinflammatory response and disease progression of the MPTP mouse model of Parkinson's disease. Glia 64:1590–1604. 10.1002/glia.23028 [DOI] [PubMed] [Google Scholar]
- Makinde HM, Just TB, Gadhvi GT, Winter DR, Schwulst SJ (2020) Microglia adopt longitudinal transcriptional changes after traumatic brain injury. J Surg Res 246:113–122. 10.1016/j.jss.2019.08.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minter MR, Moore Z, Zhang M, Brody KM, Jones NC, Shultz SR, Taylor JM, Crack PJ (2016) Deletion of the type-1 interferon receptor in APPSWE/PS1DeltaE9 mice preserves cognitive function and alters glial phenotype. Acta Neuropathol Commun 4:72. 10.1186/s40478-016-0341-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mortimer JA, French LR, Hutton JT, Schuman LM (1985) Head injury as a risk factor for Alzheimer's disease. Neurology 35:264–267. 10.1212/WNL.35.2.264 [DOI] [PubMed] [Google Scholar]
- Mouzon BC, Bachmeier C, Ferro A, Ojo JO, Crynen G, Acker CM, Davies P, Mullan M, Stewart W, Crawford F (2014) Chronic neuropathological and neurobehavioral changes in a repetitive mild traumatic brain injury model. Ann Neurol 75:241–254. 10.1002/ana.24064 [DOI] [PubMed] [Google Scholar]
- Müller U, Steinhoff U, Reis LF, Hemmi S, Pavlovic J, Zinkernagel RM, Aguet M (1994) Functional role of type I and type II interferons in antiviral defense. Science 264:1918–1921. 10.1126/science.8009221 [DOI] [PubMed] [Google Scholar]
- Nazmi A, Field RH, Griffin EW, Haugh O, Hennessy E, Cox D, Reis R, Tortorelli L, Murray CL, Lopez-Rodriguez AB, Jin L, Lavelle EC, Dunne A, Cunningham C (2019) Chronic neurodegeneration induces type I interferon synthesis via STING, shaping microglial phenotype and accelerating disease progression. Glia 67:1254–1276. 10.1002/glia.23592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owens T, Khorooshi R, Wlodarczyk A, Asgari N (2014) Interferons in the central nervous system: a few instruments play many tunes. Glia 62:339–355. 10.1002/glia.22608 [DOI] [PubMed] [Google Scholar]
- Paty DW, Li DK (1993) Interferon beta-1b is effective in relapsing-remitting multiple sclerosis: II. MRI analysis results of a multicenter, randomized, double-blind, placebo-controlled trial. UBC MS/MRI study group and the IFNB multiple sclerosis study Group. Neurology 43:662–667. 10.1212/WNL.43.4.662 [DOI] [PubMed] [Google Scholar]
- Piao CS, Stoica BA, Wu J, Sabirzhanov B, Zhao Z, Cabatbat R, Loane DJ, Faden AI (2013) Late exercise reduces neuroinflammation and cognitive dysfunction after traumatic brain injury. Neurobiol Dis 54:252–263. 10.1016/j.nbd.2012.12.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierce JE, Smith DH, Trojanowski JQ, McIntosh TK (1998) Enduring cognitive, neurobehavioral and histopathological changes persist for up to one year following severe experimental brain injury in rats. Neuroscience 87:359–369. 10.1016/S0306-4522(98)00142-0 [DOI] [PubMed] [Google Scholar]
- Pinto AK, Ramos HJ, Wu X, Aggarwal S, Shrestha B, Gorman M, Kim KY, Suthar MS, Atkinson JP, Gale M Jr, Diamond MS (2014) Deficient IFN signaling by myeloid cells leads to MAVS-dependent virus-induced sepsis. PLoS Pathog 10:e1004086. 10.1371/journal.ppat.1004086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pischiutta F, Micotti E, Hay JR, Marongiu I, Sammali E, Tolomeo D, Vegliante G, Stocchetti N, Forloni G, De Simoni MG, Stewart W, Zanier ER (2018) Single severe traumatic brain injury produces progressive pathology with ongoing contralateral white matter damage one year after injury. Exp Neurol 300:167–178. 10.1016/j.expneurol.2017.11.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plassman BL, Havlik RJ, Steffens DC, Helms MJ, Newman TN, Drosdick D, Phillips C, Gau BA, Welsh-Bohmer KA, Burke JR, Guralnik JM, Breitner JC (2000) Documented head injury in early adulthood and risk of Alzheimer's disease and other dementias. Neurology 55:1158–1166. 10.1212/WNL.55.8.1158 [DOI] [PubMed] [Google Scholar]
- Prinz M, Schmidt H, Mildner A, Knobeloch KP, Hanisch UK, Raasch J, Merkler D, Detje C, Gutcher I, Mages J, Lang R, Martin R, Gold R, Becher B, Brück W, Kalinke U (2008) Distinct and nonredundant in vivo functions of IFNAR on myeloid cells limit autoimmunity in the central nervous system. Immunity 28:675–686. 10.1016/j.immuni.2008.03.011 [DOI] [PubMed] [Google Scholar]
- Ramlackhansingh AF, Brooks DJ, Greenwood RJ, Bose SK, Turkheimer FE, Kinnunen KM, Gentleman S, Heckemann RA, Gunanayagam K, Gelosa G, Sharp DJ (2011) Inflammation after trauma: microglial activation and traumatic brain injury. Ann Neurol 70:374–383. 10.1002/ana.22455 [DOI] [PubMed] [Google Scholar]
- Roselli F, Chandrasekar A, Morganti-Kossmann MC (2018) Interferons in traumatic brain and spinal cord injury: current evidence for translational application. Front Neurol 9:458. 10.3389/fneur.2018.00458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy ER, Wang B, Wan YW, Chiu GS, Cole AL, Yin Z, Propson NE, Xu Y, Jankowsky JL, Liu Z, Lee VM, Trojanowski JQ, Ginsberg SD, Butovsky O, Zheng H, Cao W (2020) Type I interferon response drives neuroinflammation and synapse loss in Alzheimer disease. J Clin Invest. Advance online publication. Retrieved January 9, 2020. doi: 10.1172/JCI133737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salib E, Hillier V (1997) Head injury and the risk of Alzheimer's disease: a case control study. Int J Geriatr Psychiatry 12:363–368. 10.1002/(SICI)1099-1166(199703)12:3<363::AID-GPS515>3.0.CO;2-F [DOI] [PubMed] [Google Scholar]
- Sandrow-Feinberg HR, Zhukareva V, Santi L, Miller K, Shumsky JS, Baker DP, Houle JD (2010) PEGylated interferon-beta modulates the acute inflammatory response and recovery when combined with forced exercise following cervical spinal contusion injury. Exp Neurol 223:439–451. 10.1016/j.expneurol.2010.01.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheu S, Ali S, Mann-Nuttel R, Richter L, Arolt V, Dannlowski U, Kuhlmann T, Klotz L, Alferink J (2019) Interferon beta-mediated protective functions of microglia in central nervous system autoimmunity. Int J Mol Sci 20:190. 10.3390/ijms20010190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sen T, Saha P, Gupta R, Foley LM, Jiang T, Abakumova OS, Hitchens TK, Sen N (2020) Aberrant ER stress induced neuronal-IFNβ elicits white matter injury due to microglial activation and T-cell infiltration after TBI. J Neurosci 40:424–446. 10.1523/JNEUROSCI.0718-19.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheikh F, Dickensheets H, Gamero AM, Vogel SN, Donnelly RP (2014) An essential role for IFN-beta in the induction of IFN-stimulated gene expression by LPS in macrophages. J Leukoc Biol 96:591–600. 10.1189/jlb.2A0414-191R [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirey KA, Pletneva LM, Puche AC, Keegan AD, Prince GA, Blanco JC, Vogel SN (2010) Control of RSV-induced lung injury by alternatively activated macrophages is IL-4Rα-, TLR4-, and IFN-β-dependent. Mucosal Immunol 3:291–300. 10.1038/mi.2010.6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith C, Gentleman SM, Leclercq PD, Murray LS, Griffin WS, Graham DI, Nicoll JA (2013) The neuroinflammatory response in humans after traumatic brain injury. Neuropathol Appl Neurobiol 39:654–666. 10.1111/nan.12008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storek KM, Gertsvolf NA, Ohlson MB, Monack DM (2015) cGAS and Ifi204 cooperate to produce type I IFNs in response to Francisella infection. J Immunol 194:3236–3245. 10.4049/jimmunol.1402764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takaoka A, Yanai H, Kondo S, Duncan G, Negishi H, Mizutani T, Kano S, Honda K, Ohba Y, Mak TW, Taniguchi T (2005) Integral role of IRF-5 in the gene induction programme activated by toll-like receptors. Nature 434:243–249. 10.1038/nature03308 [DOI] [PubMed] [Google Scholar]
- Tanaka T, Murakami K, Bando Y, Yoshida S (2015) Interferon regulatory factor 7 participates in the M1-like microglial polarization switch. Glia 63:595–610. 10.1002/glia.22770 [DOI] [PubMed] [Google Scholar]
- Taylor JM, Minter MR, Newman AG, Zhang M, Adlard PA, Crack PJ (2014) Type-1 interferon signaling mediates neuro-inflammatory events in models of Alzheimer's disease. Neurobiol Aging 35:1012–1023. 10.1016/j.neurobiolaging.2013.10.089 [DOI] [PubMed] [Google Scholar]
- Thomas KE, Galligan CL, Newman RD, Fish EN, Vogel SN (2006) Contribution of interferon-beta to the murine macrophage response to the toll-like receptor 4 agonist, lipopolysaccharide. J Biol Chem 281:31119–31130. 10.1074/jbc.M604958200 [DOI] [PubMed] [Google Scholar]
- Walko TD 3rd, Bola RA, Hong JD, Au AK, Bell MJ, Kochanek PM, Clark RS, Aneja RK (2014) Cerebrospinal fluid mitochondrial DNA: a novel DAMP in pediatric traumatic brain injury. Shock 41:499–503. 10.1097/SHK.0000000000000160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang HC, Lin YT, Hsu SY, Tsai NW, Lai YR, Su BY, Kung CT, Lu CH (2019) Serial plasma DNA levels as predictors of outcome in patients with acute traumatic cervical spinal cord injury. J Transl Med 17:329. 10.1186/s12967-019-2084-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie C, Liu C, Wu B, Lin Y, Ma T, Xiong H, Wang Q, Li Z, Ma C, Tu Z (2016) Effects of IRF1 and IFN-beta interaction on the M1 polarization of macrophages and its antitumor function. Int J Mol Med 38:148–160. 10.3892/ijmm.2016.2583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang QG, Laird MD, Han D, Nguyen K, Scott E, Dong Y, Dhandapani KM, Brann DW (2012) Critical role of NADPH oxidase in neuronal oxidative damage and microglia activation following traumatic brain injury. PLoS One 7:e34504. 10.1371/journal.pone.0034504 [DOI] [PMC free article] [PubMed] [Google Scholar]