

Abstract
Aims
Remodelling of the extracellular matrix (ECM) is a key mechanism involved in the development and progression of heart failure (HF) but also functional in associated pulmonary hypertension (PH). Our aim was to identify plasma ECM proteins associated to end‐stage HF and secondary PH in relation to haemodynamics, before and after heart transplantation (HT).
Methods and results
Twenty ECM plasma proteins were analysed with proximity extension assay in 20 controls and 26 HF patients pre‐HT and 1 year post‐HT. Right heart catherization haemodynamics were assessed in the patients during the preoperative evaluation and at the 1 year follow‐up post‐HT. Plasma levels of prolargin and matrix metalloproteinase‐2 (MMP‐2) were elevated (P < 0.0001) in HF patients compared with controls and decreased (P < 0.0001) post‐HT towards controls' levels. The decrease in prolargin post‐HT correlated with improved mean right atrial pressure (r s = 0.63; P = 0.00091), stroke volume index (r s = −0.73; P < 0.0001), cardiac index (r s = −0.64; P = 0.00057), left ventricular stroke work index (r s = −0.49; P = 0.015), and N‐terminal pro brain natriuretic peptide (r s = 0.7; P < 0.0001). The decrease in MMP‐2 post‐HT correlated with improved mean pulmonary artery pressure (r s = 0.58; P = 0.0025), mean right atrial pressure (r s = 0.56; P = 0.0046), pulmonary artery wedge pressure (r s = 0.48; P = 0.016), and N‐terminal pro brain natriuretic peptide (r s = 0.56; P = 0.0029).
Conclusions
The normalization pattern in HF patients of plasma prolargin and MMP‐2 post‐HT towards controls' levels and their associations with improved haemodynamics indicate that prolargin and MMP‐2 may reflect, in part, the aberrant ECM remodelling involved in the pathophysiology of HF and associated PH. Their potential clinical use as biomarkers or targets for future therapy in HF and related PH remains to be investigated.
Keywords: Biomarkers, Extracellular matrix, Heart failure, Heart transplantation, Haemodynamics, Pulmonary hypertension
1. Introduction
The development and progression of heart failure (HF) are operated by a complex interplay of several pathogenic mechanisms, including haemodynamic overload, inflammation, and extracellular matrix (ECM) remodelling.1 The ECM is a scaffold of proteins contributing to the architecture of intact ventricles. An imbalance in the formation and degradation of the ECM is involved in impaired left ventricular (LV) function. For instance, ventricular remodelling due to excessive ECM production in hypertrophy due to pressure overload is characterized by increased LV stiffness as well as impaired relaxation and contraction.1, 2 Accordingly, secondary pulmonary hypertension (PH) develops from a rise in left‐sided filling pressures due to LV dysfunction. In this setting, the PH is enhanced by mechanical components of venous congestion including increased stiffness of the left atrium due to interstitial fibrosis, reduced left atrial compliance, and impaired contractility.3 This may further induce endothelial dysfunction with a concomitant vasoconstriction and progress into vascular remodelling, in which the ECM is involved.3, 4, 5
Blood‐borne biomarkers, including but not limited to proteomics, exhibit an important clinical utility and have become the subject of intense inquiry.6, 7 In addition to their clinical and prognostic value, as well as having a potential pivotal role in clinical decision making, biomarkers may provide important information on the pathophysiology of HF and aid in identifying new diagnostic and therapeutic approaches.1, 6, 7 Given the critical role of the ECM in cardiovascular development and pathophysiology,8, 9, 10 identifying alterations of ECM proteins, which may reflect and/or determine the homeostasis of cardiac remodelling and pulmonary vascular development, are of great importance. Moreover, biomarkers constitute a crucial step towards developing individualized therapies and more importantly, new therapies in the field of HF, as urged for in the guidelines of the European Society of Cardiology (ESC)11 and the American College of Cardiology Foundation/American Heart Association.12
In a previous study, we characterized the haemodynamics of patients at rest and exercise, before and after heart transplantation (HT), and found that post‐operative resting haemodynamics improved and were maintained throughout the first year after HT.13 The aim of the present study was to additionally investigate the plasma levels of ECM proteins in relation to improved haemodynamics before and 1 year after HT, to identify target ECM proteins that may be linked to end‐stage HF and PH, resolved in response to HT.
2. Methods
2.1. Blood samples and population selection
The present study was based on blood samples collected between October 2011 and February 2017 and stored in the Lund Cardio Pulmonary Register cohort of Region Skåne's biobank, initiated by Göran Rådegran 2011. Venous blood has been collected from participants (≥18 years), comprising healthy controls and heart transplant recipients before HT and during the 1 year follow‐up after HT, stored at −80°C.
Twenty healthy controls and 29 patients with underlying HF with or without additional PH were included. Controls without a past medical history of myocardial infarction (MI), HF, diabetes mellitus, or atrial fibrillation were included. Patients with missing haemodynamic values or persisting PH at the 1 year follow‐up after HT were excluded (n = 3).
The study was performed in accordance with the Declaration of Helsinki and Istanbul and approved by the regional ethical board in Lund, Sweden (diary numbers 2010/114, 2010/442, 2011/368, 2011/777, 2014/92, and 2015/270). All participants provided informed written consent.
2.2. Plasma protein analysis and storage
Twenty ECM plasma proteins and N‐terminal pro brain natriuretic peptide (NT‐proBNP) were analysed with proximity extension assay using Proseek Multiplex immunoassay reagent kits (Cardiovascular disease II, III, and Oncology II panels) using fixed plasma volumes from participants (Olink proteomics, Uppsala, Sweden).14 From the three panels, ECM proteins were extracted and included as a category, comprising collagen 1 alpha‐1 (I) chain, cysteine‐rich angiogenic inducer 61, decorin, glypican‐1, integrin α‐V, integrin β‐2, integrin β‐5, matrix metalloproteinase (MMP)‐2, MMP‐3, MMP‐7, MMP‐9, MMP‐12, melusin, metalloproteinase inhibitor 4, perlecan, prolargin (proline/arginine‐rich end leucine‐rich repeat protein), syndecan‐1, thrombospondin‐2, vascular endothelial cadherin (VE‐cadherin), and WNT1‐inducible‐signalling pathway protein 1 (WISP‐1). Internal controls were added to each sample, and external controls were added as separate samples for protein level normalization and to adjust for inter‐plate variations, respectively. Levels of the circulating proteins are expressed in arbitrary units, which reflects a linear normalized protein expression scale.
Plasma protein storage was standardized according to the procedures at our laboratory at Skåne University Hospital in Lund, Sweden. To ensure validity of results, the levels of plasma proteins were compared when stored for less than a year, 1, 2, 3, 4, and 5 years in each group (controls, pre‐HT, and post‐HT). Also, each protein was compared in all analysed groups for consistency. No clear pattern due to storage time or variation in distribution could be concluded.
2.3. Assessment of haemodynamics, renal function, and demographic characteristics
Haemodynamics were measured in supine position before HT and at the routine follow‐up 1 year after HT with right heart catheterization (RHC), using a Swan Ganz catheter (Baxter Health Care Corp, Santa Ana, CA), inserted predominantly via the right internal jugular vein. PH due to left heart disease (PH‐LHD), diagnosed by experienced cardiologists, was defined according to current ESC/European Respiratory Society guidelines as a pulmonary artery wedge pressure (PAWP) >15 mmHg, at a mean pulmonary artery pressure (mPAP) ≥25 mmHg.4 Isolated post‐capillary PH was defined as a diastolic pulmonary pressure gradient (DPG) <7 mmHg and/or pulmonary vascular resistance (PVR) ≤3 WU (wood units), and combined post‐capillary and pre‐capillary PH was defined as DPG ≥7 mmHg and/or PVR >3 WU.4
During RHC, mean arterial pressure, mPAP, mean right atrial pressure (MRAP), PAWP, systolic pulmonary artery pressure (SPAP), diastolic pulmonary artery pressure (DPAP), arterial oxygen saturation (SaO2), and mixed venous oxygen saturation (SvO2) were measured. Cardiac output (CO) was measured with thermodilution.4 Unless a LV assist device was used as a bridge to HT, the haemodynamic data closest to HT were used if more than one RHC was performed.
Haemodynamic parameters were calculated using the following formulae: cardiac index (CI) = CO/body surface area; stroke volume index (SVI) = CI/heart rate; DPG = DPAP − PAWP; transpulmonary pressure gradient = mPAP − PAWP; PVR = transpulmonary pressure gradient/CO; pulmonary arterial compliance = stroke volume/ (SPAP − DPAP); LV stroke work index (LVSWI) = (mean arterial pressure − PAWP) × SVI; and right ventricular stroke work index = (mPAP − MRAP) × SVI.
The creatinine‐based estimation of glomerular filtration rate (eGFR) of patients at the time of RHC was calculated using the revised Lund‐Malmö formula.15 Arteriovenous oxygen difference was calculated from SaO2, SvO2and haemoglobin. HF and immunosuppressive medications were based on the current consensus statement of ESC and The International Society of Heart and Lung Transplantation11, 16 (Table 1).
Table 1.
Demographic characteristics of the study population
| Variable | Controls (n = 20) | Pre‐HT (n = 26) | Post‐HT (n = 26) | |||
|---|---|---|---|---|---|---|
| n (%) | Median (IQR) | n (%) | Median (IQR) | n (%) | Median (IQR) | |
| Female, n (%) | 10 (50) | 5 (19.2) | ||||
| Age (years) | 20 (100) | 41 (27–51) | 26 (100) | 50 (45–61)† | 26 (100) | 52 (47–63) |
| BSA (m2) | 19 (95) | 1.9 (1.8–2.0) | 25 (96.2) | 2 (1.8–2.1) | 26 (100) | 2 (1.8–2.1) |
| Creatinine (μmol/L) | 25 (96.2) | 108 (90–123) | 26 (100) | 114 (97–142) | ||
| eGFR (mL/min/1.73 m2) | 25 (96.2) | 63 (55–71) | 26 (100) | 53 (43–72) | ||
| NT‐proBNP (AU) | 20 (100) | 1.1 (1.1–1.2) | 26 (100) | 24 (11–40)† | 26 (100) | 2 (1.4–5.8)† , ‡ |
| HFrEF (EF < 50%) | 24 (92.3) | |||||
| HFpEF (EF ≥ 50%) | 2 (7.7) | |||||
| PH‐LHD, n (%) | 19 (73.1) | |||||
| Ipc‐PH | 10 (52.6)a | |||||
| Cpc‐PH | 9 (47.4) | |||||
| Co‐morbidities | n (%) | n (%) | ||||
| Atrial fibrillation | 13 (50) | — | ||||
| Diabetes mellitus | 1 (3.8) | 9 (34.6) | ||||
| Hypertension | 5 (19.2) | 3 (11.5) | ||||
| Medications | n (%) | n (%) | ||||
| β‐blockers | 25 (96.2) | 9 (34.6) | ||||
| Angiotensin‐converting enzyme inhibitor | 11 (42.5) | — | ||||
| Angiotensin receptor blocker | 11 (42.5) | 10 (38.5) | ||||
| Mineralcorticoid receptor antagonist | 22 (84.6) | 3 (11.5) | ||||
| Furosemide | 24 (92.3) | 12 (46.2) | ||||
| Cordarone | 4 (15.4) | — | ||||
| Prednisolone | 1 (3.8) | 25 (96.2) | ||||
| Cyclosporine | — | 3 (11.5) | ||||
| Tacrolimus | — | 23 (88.5) | ||||
| Mycophenolate mofetil | — | 21 (80.8) | ||||
| Azathioprine | — | 5 (19.2) | ||||
| Sildenafil | — | 1 (3.8) | ||||
| Levosimendan | — | — | ||||
Statistical significance was considered P < 0.0003; false discovery rate (FDR) < 0.01. AU, arbitrary units; BSA, body surface area (BSA = weight0.425 × height0.725 × 0.007184)17; Cpc‐PH, combined post‐capillary and pre‐capillary PH; eGFR, estimation of glomerular filtration rate; HFpEF, heart failure with preserved ejection fraction; HFrEF, heart failure with reduced ejection fraction; HT, heart transplantation; Ipc‐PH, isolated post‐capillary PH; IQR, interquartile range; NT‐proBNP, N‐terminal pro brain natriuretic peptide; PH‐LHD, pulmonary hypertension due to left heart disease.
One patient had severe orthopnea during right heart catheterization when blood samples were collected, and pulmonary artery wedge pressure could not be measured. However, a second right heart catheterization as a part of the pre‐HT evaluation revealed that the patient had Ipc‐PH.
P < 0.0003; FDR < 0.01, vs. controls.
P < 0.0003; FDR < 0.01, vs. pre‐HT.
2.4. Study set‐up
To determine the relevance and relative importance of the various ECM plasma proteins, two criteria were applied: (i) a significant change in levels post‐HT vs. pre‐HT as well as in pre‐HT vs. the controls and (ii) post‐HT levels that develop towards the controls' plasma levels, displaying a pattern of normalization (Figure 1 A). These criteria excluded proteins whose alterations did not correspond to the development of NT‐proBNP levels and invasively measured haemodynamics. Next, to explore the presence of associations between the parameters' dynamics, correlations of changes between the proteins' levels and the improvement in haemodynamics were performed (Figure 1 B). ECM proteins were ranked according to the number of correlations present (Figure 1 C and D).
Figure 1.
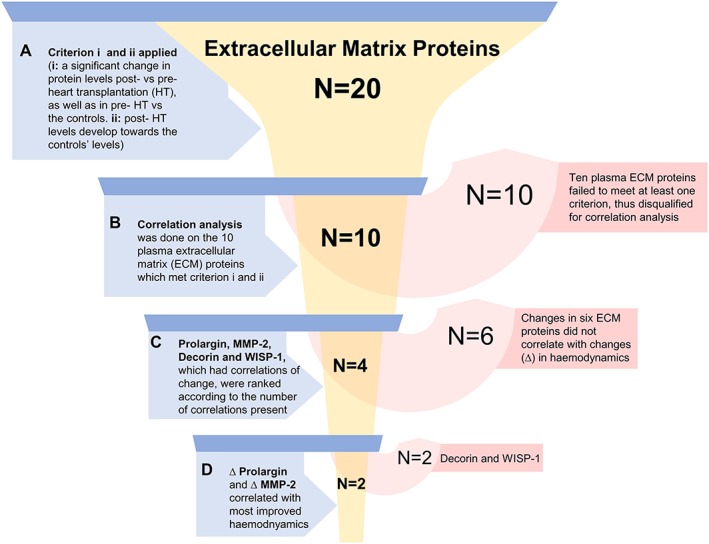
Overview of study set‐up. Inclusion steps (blue) and plasma protein exclusions (red) are shown. Statistical significance in (A) was P < 0.008; false discovery rate < 0.01. Statistical significance for correlations of change (Δ) in (B‐D) was P < 0.02; false discovery rate < 0.1. MMP‐2, matrix metalloproteinase‐2; WISP‐1, WNT1‐inducible‐signalling pathway protein 1.
2.5. Statistical analysis
Descriptive continuous variables are presented as median (interquartile range). Wilcoxon signed‐rank test and Mann–Whitney U test were used as appropriate. Tukey's fence was used to define outliers. Correlations were expressed by Spearman's rank coefficient (r s). To accommodate for mass significance, the two‐stage step‐up method of Benjamini, Krieger, and Yekutieli was used to calculate the false discovery rate (FDR) with a Q value of 1% for demographics, ECM plasma proteins, and haemodynamic parameters and 10% for correlations.18 P values less than the thresholds assessed by FDR were considered statistically significant. All analyses were performed using GraphPad Prism version 8.00 (GraphPad Software, La Jolla, California, USA, http://www.graphpad.com).
3. Results
3.1. Study population
Clinical and demographic characteristics of controls and heart transplant recipients are summarized in Table 1. The main underlying aetiology of the HF patients was dilated cardiomyopathy (65.4%), followed by ischaemic and hypertrophic cardiomyopathy or other (11.5%, respectively). NT‐proBNP was higher pre‐HT compared with controls and decreased post‐HT towards controls' levels (P < 0.001; FDR < 0.01). Plasma creatinine (P = 0.49) and creatinine‐based eGFR (P = 0.15) did not change pre‐HT vs. post‐HT (FDR < 0.01) (Table 1).
3.2. Extracellular matrix protein selection and qualification
The levels of 10 out of 20 ECM proteins were higher in severe HF patients before HT vs. controls and decreased following HT in the direction of the controls' levels, meeting the inclusion criteria [Figure 1 (A)]. The 10 qualified ECM proteins were decorin, MMP‐2, MMP‐9, MMP‐12, perlecan, prolargin, syndecan‐1, thrombospondin‐2, VE‐cadherin, and WISP‐1 (P < 0.008; FDR < 0.01) (Table 2). The ECM proteins that failed to meet either of the criteria were disqualified for correlation analysis (Figure 1 , Table 2).
Table 2.
Characteristics of extracellular matrix plasma proteins and N‐terminal pro brain natriuretic peptide in the study population
| Plasma protein (AU) | Controls (n = 20) | Pre‐HT (n = 26) | Post‐HT (n = 26) | ∆ (Post‐HT − Pre‐HT) | P values | ||
|---|---|---|---|---|---|---|---|
| Median (IQR) | Median (IQR) | Median (IQR) | Median (IQR) | Post‐HT vs. Pre‐HT | C vs. Pre‐HT | C vs. Post‐HT | |
| COL1A1 | 4.5 (3.4–5.9) | 4.4 (3.7–5) | 5.1 (4.4–6.7) | 0.35 (−0.45 to 1.9) | 0.0559 | 0.869 | 0.0780 |
| CYR61 | 22 (17–27)† | 27 (22–32) | 18 (15–22) | −8.1 (−14 to −1.9)† | <0.0001* | 0.0575 | 0.0411 |
| Decorin | 20 (18–23) | 34 (27–38) | 26 (24–33) | −4.6 (−8.4 to −0.66) | 0.00104* | <0.0001* | <0.0001* |
| Glypican‐1 | 15 (12–17)† | 15 (13–17) | 15 (14–19) | 0.52 (−1.2 to 2.4)† | 0.381 | 0.743 | 0.230 |
| Integrin α‐V | 6.7 (6.1–7.4)† | 6.1 (5.3–7.4)† | 5.7 (4.9–6.2) | −0.59 (−1.4 to −0.12)† | 0.00115* | 0.300 | 0.000803* |
| Integrin β‐2 | 20 (17–22) | 15 (11–20)† | 18 (13–19) | 1.3 (−2.8 to 4.8) | 0.367 | 0.00501* | 0.0221 |
| Integrin β‐5 | 211 (132–236)† | 185 (145– 224) | 143 (129–176) | −29 (−72 to 0.53)† | 0.00130* | 0.833 | 0.0787 |
| Melusin | 31 (18–66) | 14 (6.2–26) | 15 (7.5–53) | 4.8 (−4.1 to 31) | 0.0493 | 0.00152* | 0.0819 |
| MMP‐2 | 7.7 (7.2–8.2) | 14 (12–16)† | 7.5 (6.2–8.9) | −6.4 (−9.4 to −4) | <0.0001* | <0.0001* | 0.576 |
| MMP‐3 | 89 (62–144) | 122 (83–224) | 338 (193–424) | 187 (101–269) | <0.0001* | 0.0519 | <0.0001* |
| MMP‐7 | 273 (222–372) | 797 (603–1043) | 986 (676–1174) | 85 (−73 to 212) | 0.0493 | <0.0001* | <0.0001* |
| MMP‐9 | 10 (7.5–14) | 17 (12–27) | 6.7 (4.6–9.6) | −9.3 (−20 to −4) | <0.0001* | 0.00196* | 0.00773* |
| MMP‐12 | 76 (54–101) | 121 (77–174) | 89 (64–144) | −30 (−62 to 1.6) | 0.00613* | 0.00432* | 0.213 |
| Perlecan | 66 (54–75) | 110 (82–133) | 97 (63–125) | −23 (−42 to −2.6) | 0.000935* | <0.0001* | 0.00108* |
| Prolargin | 64 (59–71) | 130 (105–139) | 84 (78–96) | −34 (−53 to −19) | <0.0001* | <0.0001* | <0.0001* |
| Syndecan‐1 | 56 (43–75)† | 122 (84–193)† | 75 (60–92) | −37 (−129 to −7.4)† | <0.0001* | <0.0001* | 0.0174 |
| Thrombospondin‐2 | 37 (34–39) | 53 (47–59)† | 41 (36–46) | −11 (−18 to −4.3) | <0.0001* | <0.0001* | 0.0298 |
| TIMP‐4 | 15 (13–17) | 19 (16–26) | 19 (14–23) | −3.6 (−9.7 to 5.3) | 0.136 | 0.00166* | 0.0353 |
| VE‐cadherin | 8.5 (6.7–10) | 11 (9.3–13)† | 7.8 (6.6–9.2) | −3.5 (−4.7 to −2) | <0.0001* | 0.00181* | 0.247 |
| WISP‐1 | 13 (11–15)† | 27 (20–38) | 19 (13–23) | −6.5 (−23 to −1.5)† | <0.0001* | <0.0001* | 0.00445* |
| NT‐proBNP | 1.1 (1.1–1.2) | 24 (11–40) | 2 (1.4–5.8) | −17 (−37 to −8.4) | <0.0001* | <0.0001* | <0.0001* |
AU, arbitrary units; C, controls; COL1A1, collagen alpha‐1 chain; CYR61, cysteine‐rich angiogenic inducer 61; HT, heart transplantation; IQR, interquartile range; MMP, matrix metalloproteinase; NT‐proBNP, N‐terminal pro brain natriuretic peptide; TIMP‐4, metalloproteinase inhibitor 4; VE‐cadherin, vascular endothelial cadherin; WISP‐1, WNT1‐inducible‐signalling pathway protein 1.
Indicates statistically significant values (P < 0.008; false discovery rate < 0.01).
Indicates n − 1.
3.3. Haemodynamics and correlations of change
Haemodynamic improvement was characterized by a decrease post‐HT in mPAP, PAWP, MRAP, and PVR, as well as an increase in pulmonary arterial compliance, CI, SVI, and LVSWI (Table 3). The Δ (delta; post‐HT − pre‐HT values) of the plasma proteins levels that met inclusion criteria correlated with the Δ of NT‐proBNP and nine haemodynamic parameters, which improved following HT (Table 4).
Table 3.
Haemodynamic characteristics of patients before and 1 year after heart transplantation
| Haemodynamic parameter | Pre‐HT (n = 26) | Post‐HT (n = 26) | Δ (Post‐HT − Pre‐HT) | P value | |||
|---|---|---|---|---|---|---|---|
| n | Median (IQR) | n | Median (IQR) | n | Median (IQR) | Post‐HT vs. Pre‐HT | |
| MAP (mmHg) | 25 | 82 (77–93) | 26 | 102 (91–108) | 25 | 15 (9–27) | <0.0001* |
| mPAP (mmHg) | 25 | 29 (24–38) | 26 | 14 (12–17) | 25 | −15 (−26 to −7.5) | <0.0001* |
| PAWP (mmHg) | 24 | 20 (18–25) | 26 | 7 (4–9.3) | 24 | −17 (−21 to −6.5) | <0.0001* |
| MRAP (mmHg) | 25 | 14 (7.5–18) | 25 | 3 (1–4) | 24 | −12 (−15 to −3.3) | <0.0001* |
| HR (beats/min) | 25 | 73 (69–82) | 26 | 82 (73–89) | 25 | 7 (−4 to 15) | 0.063 |
| CO (L/min) | 25 | 3.3 (2.6–4.1) | 26 | 5.5 (5–6.5) | 25 | 2.2 (1.2–2.9) | <0.0001* |
| CI (L/min/m2) | 25 | 1.8 (1.4–2.2) | 26 | 2.8 (2.6–3.2) | 25 | 1.1 (0.65–1.6) | <0.0001* |
| SV (mL/beat) | 25 | 48 (35–58) | 26 | 72 (66–78) | 25 | 23 (14–34) | <0.0001* |
| SVI (mL/beat/m2) | 25 | 25 (18–29) | 26 | 36 (33–40) | 25 | 12 (6.5–18) | <0.0001* |
| DPG (mmHg) | 24 | 1 (0–3.8) | 26 | 2 (−0.25 to 4) | 24 | 0 (−2 to 3.5) | 0.8 |
| TPG (mmHg) | 24 | 8.5 (6–12) | 26 | 8 (5–10) | 24 | −1.5 (−6 to 2) | 0.17 |
| PAC (mL/mmHg) | 25 | 2.2 (1.8–3.1) | 26 | 5.4 (4.1–6.6) | 25 | 3.2 (1.3–4) | 0.00029* |
| PVR (WU) | 24 | 2.4 (1.4–3.5) | 26 | 1.4 (0.89–1.9) | 24 | −1.3 (−1.9 to −0.036) | <0.0001* |
| PVRI (WU/m2) | 24 | 5.1 (2.9–6.9) | 26 | 2.8 (1.7–3.7) | 24 | −2.4 (−4 to −0.42) | <0.0001* |
| LVSWI (mmHg × mL/m2) | 24 | 1541 (1052–2007) | 26 | 3344 (3167–3810) | 24 | 1675 (1224–2532) | <0.0001* |
| RVSWI (mmHg × mL/m2) | 25 | 362 (294–615) | 25 | 429 (317–516) | 24 | 62 (−119 to 245) | 0.64 |
| a‐vO2 diff (mL O2/L) | 25 | 74 (63–81) | 23 | 42 (40–51) | 22 | −32 (−40 to −19) | <0.0001* |
| SaO2 (%) | 25 | 96 (94–97) | 23 | 97 (96–98) | 22 | 1.7 (−0.2 to 2.8) | 0.046 |
| SvO2 (%) | 25 | 52 (47–60) | 26 | 69 (66–72) | 25 | 17 (11–24) | <0.0001* |
One cardiac output (CO) value was calculated with indirect Fick before heart transplantation (HT). a‐vO2 diff, arteriovenous oxygen difference; CI, cardiac index; DPG, diastolic pulmonary pressure gradient; HR, heart rate; IQR, interquartile range; LVSWI, left ventricular stroke work index; MAP, mean arterial pressure; mPAP, mean pulmonary artery pressure; MRAP, mean right atrial pressure; PAC, pulmonary arterial compliance; PAWP, pulmonary artery wedge pressure; PVR, pulmonary vascular resistance; PVRI, pulmonary vascular resistance index; RVSWI, right ventricular stroke work index; SaO2, arterial oxygen saturation; SV, stroke volume; SVI, stroke volume index; SvO2, mixed venous oxygen saturation; TPG, transpulmonary pressure gradient; WU, wood units.
Indicates statistically significant values (P < 0.0003; false discovery rate < 0.01).
Table 4.
Correlations of changes (Δ: delta) between plasma extracellular matrix proteins and haemodynamics as well as N‐terminal pro brain natriuretic peptide
| Variable (Δ) | Pulmonary parameters | Cardiac parameters | ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| mPAP (mmHg) | PVR (WU) | PAC (mL/mmHg) | MRAP (mmHg) | NT‐proBNP (AU) | PAWP (mmHg) | CI (L/min/m2) | SVI (mL/beat/m2) | LVSWI (mmHg × mL/m2) | ||||||||||
| n | r s (P value) | n | r s (P value) | n | r s (P value) | n | r s (P value) | n | r s (P value) | n | r s (P value) | n | r s (P value) | n | r s (P value) | n | r s (P value) | |
| Prolargin (AU) | 25 | 0.4 (0.049) | 24 | 0.37 (0.071) | 25 | −0.082 (0.7) | 24 | 0.63 (0.00091)* | 26 | 0.7 (<0.0001)* | 24 | 0.32 (0.13) | 25 | −0.64 (0.00057)* | 25 | −0.73 (<0.0001)* | 24 | −0.49 (0.015)* |
| MMP‐2 (AU) | 25 | 0.58 (0.0025)* | 24 | 0.46 (0.022) | 25 | 0.041 (0.85) | 24 | 0.56 (0.0046)* | 26 | 0.56 (0.0029)* | 24 | 0.48 (0.016)* | 25 | −0.19 (0.36) | 25 | −0.25 (0.23) | 24 | −0.31 (0.15) |
| Decorin (AU) | 25 | 0.43 (0.032) | 24 | 0.24 (0.26) | 25 | 0.0062 (0.98) | 24 | 0.53 (0.0081)* | 26 | 0.66 (0.0003)* | 24 | 0.34 (0.11) | 25 | −0.18 (0.38) | 25 | −0.36 (0.079) | 24 | −0.53 (0.008)* |
| WISP‐1 (AU) | 24 | 0.24 (0.27) | 23 | 0.21 (0.35) | 24 | −0.022 (0.92) | 23 | 0.67 (0.00049)* | 25 | 0.78 (<0.0001)* | 23 | 0.35 (0.097) | 24 | −0.19 (0.38) | 24 | −0.43 (0.034) | 23 | −0.42 (0.045) |
| VE‐cadherin (AU) | 25 | 0.09 (0.67) | 24 | 0.23 (0.29) | 25 | 0.18 (0.4) | 24 | 0.44 (0.031) | 26 | 0.55 (0.0033)* | 24 | 0.068 (0.75) | 25 | −0.14 (0.51) | 25 | −0.078 (0.71) | 24 | −0.31 (0.15) |
| Perlecan (AU) | 25 | 0.12 (0.56) | 24 | 0.17 (0.42) | 25 | 0.045 (0.83) | 24 | 0.32 (0.12) | 26 | 0.46 (0.019)* | 24 | 0.16 (0.44) | 25 | 0.2 (0.33) | 25 | 0.038 (0.86) | 24 | −0.21 (0.33) |
| Syndecan‐1 (AU) | 24 | 0.38 (0.069) | 23 | 0.23 (0.29) | 24 | −0.086 (0.69) | 23 | 0.4 (0.057) | 25 | 0.52 (0.0083)* | 23 | 0.28 (0.19) | 24 | −0.29 (0.17) | 24 | −0.28 (0.18) | 23 | −0.16 (0.46) |
| MMP‐9 (AU) | 25 | 0.25 (0.22) | 24 | 0.12 (0.57) | 25 | −0.16 (0.46) | 24 | 0.23 (0.28) | 26 | 0.17 (0.42) | 24 | 0.26 (0.21) | 25 | 0.0023 (0.99) | 25 | −0.13 (0.52) | 24 | −0.057 (0.79) |
| MMP‐12 (AU) | 25 | −0.19 (0.36) | 24 | 0.034 (0.88) | 25 | −0.068 (0.75) | 24 | −0.34 (0.11) | 26 | −0.31 (0.12) | 24 | −0.27 (0.21) | 25 | 0.43 (0.03) | 25 | 0.33 (0.11) | 24 | 0.067 (0.76) |
| Thrombospondin‐2 (AU) | 25 | 0.06 (0.78) | 24 | 0.14 (0.52) | 25 | −0.078 (0.71) | 24 | −0.16 (0.44) | 26 | 0.076 (0.71) | 24 | 0.11 (0.61) | 25 | −0.041 (0.85) | 25 | −0.084 (0.69) | 24 | −0.074 (0.73) |
r s indicates Spearman's rank correlation coefficient. AU, arbitrary units; CI, cardiac index; LVSWI, left ventricular stroke work index; MMP, matrix metalloproteinase; mPAP, mean pulmonary artery pressure; MRAP, mean right atrial pressure; NT‐proBNP, N‐terminal pro brain natriuretic peptide; PAC, pulmonary arterial compliance; PAWP, pulmonary artery wedge pressure; PVR, pulmonary vascular resistance; SVI, stroke volume index; VE‐cadherin, vascular endothelial cadherin; WISP‐1, WNT1‐inducible‐signalling pathway protein 1; WU, wood units.
Indicates statistically significant correlations (P < 0.02; false discovery rate < 0.1).
The decrease in plasma levels post‐HT of prolargin, MMP‐2, decorin, and WISP‐1 correlated with improvement in one or more haemodynamic parameters and NT‐proBNP following HT (Figures 2 A and 3 A, Table 4). ΔProlargin correlated with improved MRAP (r s = 0.63), SVI (r s = −0.73), CI (r s = −0.64), LVSWI (r s = 0.49), and NT‐proBNP (r s = 0.7) (Figure 2 ). ΔMMP‐2 correlated with improved mPAP (r s = 0.58), PAWP (r s = 0.48), MRAP (r s = 0.56), and NT‐proBNP (r s = 0.56) (Figure 3 ). ΔDecorin correlated with improved MRAP (r s = 0.53), LVSWI (r s = −0.53), and NT‐proBNP (r s = 0.66), whereas ΔWISP‐1 correlated with the Δ of MRAP (r s = 0.67) and NT‐proBNP (r s = 0.78) (Figure 3 ) (P < 0.02; FDR < 0.1) (Table 4). Notably, ∆PVR correlated with the decrease in MMP‐2 post‐HT, yet not significantly (r s = 0.46, P = 0.022). The Δ of MMP‐9 and MMP‐12, perlecan, syndecan‐1, thrombospondin‐2, and VE‐cadherin did not correlate with improved haemodynamics after HT (Table 4).
Figure 2.
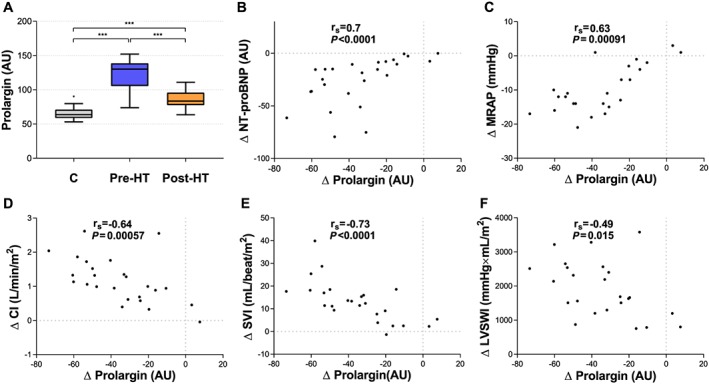
(A) Levels of plasma prolargin in controls (C), pre‐heart transplantation (HT), and post‐HT. (B–F) Correlations between changes in plasma prolargin and changes in haemodynamics as well as N‐terminal pro brain natriuretic peptide (NT‐proBNP). Statistical significance in (A) was considered P < 0.008; false discovery rate < 0.01. *** P < 0.0001. Statistical significance for correlations of change was considered P < 0.02; false discovery rate < 0.1. AU, arbitrary units; CI, cardiac index; LVSWI, left ventricular stroke work index; MRAP, mean right atrial pressure; SVI, stroke volume index.
Figure 3.
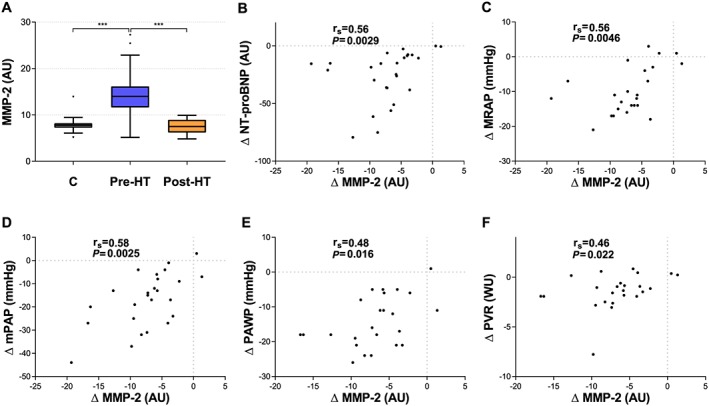
(A) Plasma levels of matrix metalloproteinase‐2 (MMP‐2) in controls (C), pre‐heart transplantation (HT), and post‐HT. (B–F) Correlations between changes in plasma MMP‐2 and changes in haemodynamics as well as N‐terminal pro brain natriuretic peptide (NT‐proBNP). In (F), a correlation of change was present with a P = 0.022. Statistical significance in (A) was considered P < 0.008; false discovery rate < 0.01. *** P < 0.0001. Statistical significance for correlations of change was considered P < 0.02; false discovery rate < 0.1. AU, arbitrary units; mPAP, mean pulmonary artery pressure; MRAP, mean right atrial pressure; PAWP, pulmonary artery wedge pressure; PVR, pulmonary vascular resistance.
4. Discussion
An important gap in the management of HF constitutes evidence for new therapies targeting ‘non‐myocytic compartments', involving anti‐fibrosis and ECM remodelling.11 Identifying the dynamics of plasma ECM proteins reflecting matrix remodelling, considered as one of the major classes contributing to the biomarker profile in HF patients,1 may establish a crucial step towards achieving new treatment strategies. By utilizing the unique reversibility potential of HT on HF and related PH, we found that plasma prolargin, MMP‐2, decorin, and WISP‐1 developed in accordance with recovered heart function. In specific, ∆prolargin and ∆MMP‐2 correlated with a wide range of improved haemodynamic parameters. Prolargin and MMP‐2 may therefore offer insight into new treatment strategies in HF and related PH, in addition to their potential pathophysiological significance.
Prolargin or proline/arginine‐rich end leucine‐rich repeat protein belongs to the family of small leucine‐rich repeat proteoglycans, expressed in collagen‐rich tissues.19 According to the open‐source database Human Protein Atlas (HPA) (proteinatals.org), prolargin RNA expression is abundant in a variety of tissues including cardiac muscle, lung tissue and smooth muscle. However, this data is based on non‐neoplastic and morphologically normal surgical material, where tissue remodeling,degradation and inflammation may still be present.30 Although highly unexplored, prolargin functions through direct inhibition of all complement pathways (classical, lectin, and alternative) by interfering in the formation of membrane attack complex. This has highlighted its potential function in limiting the inflammatory milieu of certain diseases, such as rheumatoid arthritis.20, 21 Intriguingly, accumulating evidence implicates complement activation as an important pathogenic mechanism in HF including cardiac remodelling. Activation of all complement pathways is systemically up‐regulated in congestive HF patients compared with healthy controls and negatively associated with improved LV ejection fraction.22 Herein, we report that plasma prolargin levels were elevated in severe HF before HT, potentially counteracting the complement activation in HF. Theoretically, this increase may exert a protective role against adverse cardiac remodelling resulting in impaired haemodynamics (Figure 4 ).23 This is supported by our findings as decreased levels of prolargin in response to HT correlated with improved CI, SVI, LVSWI, MRAP and decreased NT‐proBNP.
Figure 4.
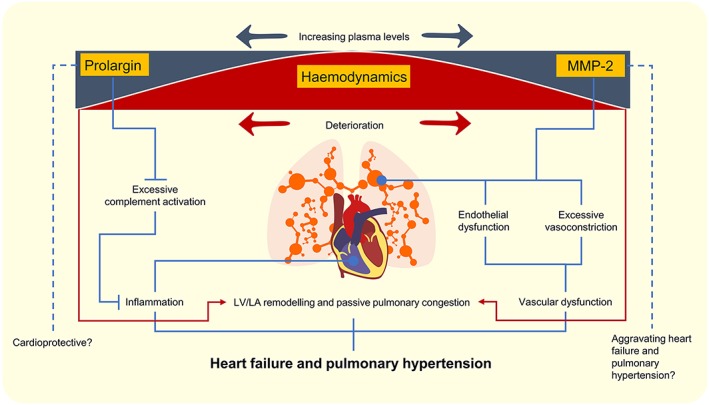
An illustration of plasma prolargin and matrix metalloproteinase‐2 (MMP‐2) levels in relation to haemodynamics, heart failure, and pulmonary hypertension. Increasing plasma levels of prolargin and MMP‐2 levels in relation to impaired haemodynamics may inhibit or mediate pathophysiological pathways functional in heart failure and pulmonary hypertension. Hypothetical conclusions are drawn in dotted lines. Red arrows and lines demonstrate deterioration of haemodynamics. LA, left atrial; LV, left ventricular.
Treatment with complement inhibition has previously been investigated in both animal models and patients. A systematic overview of early clinical trials using pexelizumab, an inhibitor of complement factor C5, on patients with ST‐elevation MI or coronary artery bypass graft demonstrated a mortality reduction.24 However, the subsequent APEX‐AMI trial failed to show any beneficial effects.25 It was later concluded that this inconsistency in the effect of pexelizumab may be ascribed to the use of inadequate concentrations, a late administration that precludes beneficial effects of complement inhibition following myocardial injury or other alternative mechanisms.26, 27 Thus, complement inhibition needs to be further investigated and may still be a plausible alternative treatment in HF. Given the important role of inflammation, where the complement system is involved, in cardiac remodelling and progression of HF,28, 29 it is reasonable to assume that treatment with prolargin may be a future alternative to explore in HF studies, as it may have additional or alternative mechanisms of complement inhibition compared with pexelizumab. However, whether increased prolargin is ascribed to HF and its circulating plasma levels are representative of the expression in cardiac tissue in advanced HF remain to be established.
MMP‐2 belongs to the zinc‐dependent endopeptidase superfamily that participate in ECM degradation and remodelling.31According to HPA, both RNA and protein expression of MMP‐2 is found in cardiomyocytes, smooth muscle cells, lung tissue and other organs.30 MMP‐2 seems to be involved in the pathogenesis of hypertension, cardiac remodelling following myocardial injury and healing, as well as pressure overload.31, 32, 33, 34, 35 Myocardial expression of MMP‐2 has been shown to be increased in both experimental and clinical studies of HF and pressure overload.32, 33 In addition to previously observed elevated plasma MMP‐2 expression in congestive HF patients,34 congruent with our findings, we provide further evidence that plasma MMP‐2 decreases to the levels of healthy controls after HT. Interestingly, deletion of MMP‐2 in mice decreased myocardial remodelling in response to pressure overload32 but also attenuated early LV rupture and late cardiac remodelling following MI.35
Consistent with its detrimental effects on cardiac tissue, MMP‐2 cleaves the precursor big endothelin‐1 to form endothelin‐1 yielding vasoconstriction, suggesting MMP‐2 to be involved in the regulation of vascular tone and reactivity.31, 36 Interestingly, an increase in endothelin‐1 expression is a hallmark of endothelial dysfunction, implicated in the pathogenesis of PH‐LHD.37, 38 Moreover, a recent study revealed that angiotensin‐II‐induced endothelial dysfunction, and vascular injury and remodelling were dependent on the expression of MMP‐2 in the small mesenteric artery.31 Pulmonary vascular remodelling, including arterialization of pulmonary veins, but also intimal thickening and media hypertrophy which are characteristic of pulmonary arterial hypertension pathology, was shown in patients with PH associated with HF irrespective of ejection fraction.5, 39 Thus, vascular remodelling in end‐stage HF may be influenced by an elevation of MMP‐2 before HT, supported by ΔMMP‐2 correlation with ΔPVR. As decreased MMP‐2 was associated with improved PVR that if fixed preoperatively may impose a contraindication for HT,40 there is a potential for MMP‐2 to be a part of a multimarker panel as a biomarker to assess the reversibility potential of PVR. This is of importance as drug‐induced reversibility testing of PVR has the drawback of systemic hypotension.41 However, such investigation needs a larger population and an early post‐transplant follow‐up, as reduction in PVR is most abundant within the first month42 and is maintained throughout the first year after HT.13 Additionally, MMP‐2 may be a marker related to HF aggravation and passive pulmonary congestion, as indicated by the correlation of ΔMMP‐2 with ΔNT‐proBNP, ΔMRAP respectively ΔPAWP and ΔmPAP (Figure 4 ). Whether the cardiopulmonary expression of MMP‐2 is a main source of circulating MMP‐2 levels in end‐stage HF and/or PH‐LHD remains to be elucidated.
There is extensive evidence supporting the critical role of TGFβ in cardiac injury, repair, and remodelling. For instance, blockage of TGFβ in a rat model of cardiac pressure overload prevented myocardial fibrosis and reversed LV diastolic dysfunction.43 Decorin has anti‐fibrotic effects, through interaction with TGFβ and its downstream effector connective tissue growth factor (CTGF), decreasing collagen III production.19 Interestingly, smad3 signalling mediates TGFβ1‐induced CTGF expression and is essential for the pathogenesis of cardiac remodelling.44, 45 In the present study, given that decorin levels were high in severe HF patients compared with controls and decreased following HT, a decrease that was associated with improvement in MRAP, LVSWI, and NT‐proBNP, it is reasonable to hypothesize that higher levels of plasma decorin may have a cardioprotective role in HF by inhibiting the profibrotic activity of TGFβ and its effector CTGF, possibly in a smad3‐dependent manner, attenuating adverse cardiac remodelling.
Moreover, WISP‐1 is a member of the CCN family of growth factors, expressed by epithelium, heart, and lungs. WISP‐1 stimulates angiogenesis by regulating VEGF‐A in osteosarcoma,46 and WISP‐1 up‐regulation has been observed following MI. In addition to its proposed role in cardiac remodelling,47 we found that plasma WISP‐1 was elevated in end‐stage HF and that its decrease after HT was associated with reversal of pressure and volume overload. Although WISP‐1 expression is implicated in tissue repair, its relationship with different signalling pathways may mediate pulmonary fibrosis in mice and promote tumorigenesis.48 Whether increased expression of WISP‐1 acts as a friend or foe in end‐stage HF with or without related PH remains, however, uncertain. Notably, other ECM proteins including other MMPs and proteoglycans, showing a similar plasma expression pattern as prolargin, MMP‐2, decorin, and WISP‐1 that did not correlate with haemodynamics, may, however, also be collectively linked to the severity, progression, alleviation, or pathophysiology of HF with or without concomitant PH.
Although we consider the use of RHC and proximity extension assay as major strengths of our study, there are limitations worth addressing including the small and single centred nature of the study, as well as the somewhat younger controls. However, the number of the patients is in line with other similar studies, and controls are not required in pre‐post studies. Although we found no change in the patients' eGFR in response to HT, implying a minimal influence on plasma ECM protein expression, plasma levels may still be affected by patients' co‐morbidities, medications, or age. For instance, a large proportion of the preoperative population was prescribed both β‐blockers and angiotensin‐converting enzyme inhibitors, which are known to reduce adverse cardiac remodelling.49 In addition, the effect of immunosuppressive agents was not accounted for. To enable a reliable statistical adjustment for medications and other parameters, a larger sample size is required. However, as the present work is an initial hypothesis‐generating study, it is intriguing to map the plasma protein development including the potential influence of medications during HF and after HT.
To note, the present study does not address causality. To provide insight into the origin of expression and causality, future studies are needed to investigate whether plasma proteins are associated with altered tissue expression, specifically in the endothelium, heart and lung. Despite these limitations, our study offers new insights into important proteins that may be related to the pathophysiology of HF and related PH and may affect future treatment strategies.
In conclusion, the present study identifies the dynamics in HF, prior to and after HT, of plasma ECM proteins including prolargin, MMP‐2, decorin, and WISP‐1. Specifically, the normalization pattern in HF patients of plasma prolargin and MMP‐2 post‐HT towards controls' levels and their associations with improved haemodynamics indicate that MMP‐2 and prolargin may reflect, in part, the aberrant ECM remodelling functional in HF and related PH. Prolargin may potentially be cardioprotective in HF, as indicated by its established anti‐inflammatory effect through complement inhibition. Higher plasma MMP‐2 was associated with impaired haemodynamics and may thus be related to aggravation of HF and pulmonary vascular disease including passive pulmonary congestion. Thus, ECM‐related plasma proteins, particularly prolargin and MMP‐2, may be of specific interest as biomarker candidates for further research in the pathophysiology and future treatment of severe HF and related PH. Although causality and cardiovascular origin are yet to be proven, the present study identifies important dynamics of ECM proteins that may affect future management of HF and associated PH.
Conflict of interest
Mr Abdulla Ahmed and Mr Salaheldin Ahmed report no conflicts of interest. Mr Mattias Arvidsson reports an unrestricted research grant from The Swedish Society of Pulmonary Hypertension on behalf of Actelion Pharmaceuticals Sweden AB. Mr Bouzina reports an unrestricted research grant from The Swedish Society of Pulmonary Hypertension on behalf of GlaxoSmithKline. Dr Lundgren reports unrestricted research grants from The Swedish Society of Pulmonary Hypertension on behalf of Actelion Pharmaceuticals Sweden AB. Dr Rådegran reports unrestricted research grants from ALF and Actelion Pharmaceuticals Sweden AB, during the conduct of the study. Mr Abdulla Ahmed, Mr Salaheldin Ahmed, and Mr Mattias Arvidsson report no personal lecture fees. Mr Bouzina reports personal lecture fees from Actelion Pharmaceuticals Sweden AB outside the submitted work. Dr Lundgren reports personal lecture fees from Actelion Pharmaceuticals Sweden AB and GlaxoSmithKline outside the submitted work. Dr Rådegran reports personal lecture fees from Actelion Pharmaceuticals Sweden AB, GlaxoSmithKline, Bayer Health Care, and Nordic Infucare outside the submitted work. Dr Rådegran is, and has been primary, or co‐, investigator in clinical PAH trials for GlaxoSmithKline, Actelion Pharmaceuticals Sweden AB, Pfizer, Bayer, and United Therapeutics and in clinical heart transplantation immuno‐suppression trials for Novartis. The companies had no role in the data collection, analysis, and interpretation and had no right in disapproving of the manuscript.
Funding
The work was supported by unrestricted research grants from Avtal om Läkarutbildning och Forskning (ALF) and Actelion Pharmaceuticals Sweden AB. The funding organizations played no role in the collection, analysis, or interpretation of the data and had no right to restrict the publishing of the manuscript.
Acknowledgements
We acknowledge the support and assistance provided by the staff of the Haemodynamic Lab, The Section for Heart Failure and Valvular Disease, Skåne University Hospital, Lund, Sweden, and The Department of Clinical Sciences, Cardiology, Lund University, Lund, Sweden. We express our gratitude to Anneli Ahlqvist for the assistance, involving management of plasma samples and LCPR registration. Additionally, we acknowledge the biobank services and retrieval of blood samples from LCPR performed at Labmedicine Skåne, University and Regional Laboratories, Region Skåne, Sweden.
Ahmed, A. , Ahmed, S. , Arvidsson, M. , Bouzina, H. , Lundgren, J. , and Rådegran, G. (2020) Prolargin and matrix metalloproteinase‐2 in heart failure after heart transplantation and their association with haemodynamics. ESC Heart Failure, 7: 223–234. 10.1002/ehf2.12560.
References
- 1. Braunwald E. Heart failure. JACC Heart Fail 2013; 1: 1–20. [DOI] [PubMed] [Google Scholar]
- 2. Katz Arnold M, Zile MR. New molecular mechanism in diastolic heart failure. Circulation 2006; 113: 1922–1925. [DOI] [PubMed] [Google Scholar]
- 3. Vachiery JL, Adir Y, Barbera JA, Champion H, Coghlan JG, Cottin V, De Marco T, Galie N, Ghio S, Gibbs JS, Martinez F, Semigran M, Simonneau G, Wells A, Seeger W. Pulmonary hypertension due to left heart diseases. J. Am. Coll. Cardiol. 2013; 62: D100–D108. [DOI] [PubMed] [Google Scholar]
- 4. Galiè N, Humbert M, Vachiery J‐L, Gibbs S, Lang I, Torbicki A, Simonneau G, Peacock A, Vonk Noordegraaf A, Beghetti M, Ghofrani A, Gomez Sanchez MA, Hansmann G, Klepetko W, Lancellotti P, Matucci M, McDonagh T, Pierard LA, Trindade PT, Zompatori M, Hoeper M, Group ESCSD . 2015 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension: the Joint Task Force for the diagnosis and treatment of pulmonary hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS): Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC), International Society for Heart and Lung Transplantation (ISHLT). Eur Heart J 2016; 37: 67–119. [DOI] [PubMed] [Google Scholar]
- 5. Humbert M, Guignabert C, Bonnet S, Dorfmüller P, Klinger JR, Nicolls MR, Olschewski Andrea J, Pullamsetti SS, Schermuly RT, Stenmark Kurt R, Rabinovitch M. Pathology and pathobiology of pulmonary hypertension: state of the art and research perspectives. Eur Respir J 2018; 1801887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Braunwald E. Biomarkers in heart failure. N Engl J Med 2008; 358: 2148–2159. [DOI] [PubMed] [Google Scholar]
- 7. Ibrahim Nasrien E, Januzzi JL. Established and emerging roles of biomarkers in heart failure. Circ Res 2018; 123: 614–629. [DOI] [PubMed] [Google Scholar]
- 8. Lockhart M, Wirrig E, Phelps A, Wessels A. Extracellular matrix and heart development. Birth defects research. Part A, Clinical and molecular teratology 2011; 91: 535–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bloksgaard M, Lindsey M, Martinez‐Lemus LA. Extracellular matrix in cardiovascular pathophysiology. American Journal of Physiology‐Heart and Circulatory Physiology 2018; 315: H1687–H1690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Davis George E, Senger DR. Endothelial extracellular matrix. Circ Res 2005; 97: 1093–1107. [DOI] [PubMed] [Google Scholar]
- 11. Ponikowski P, Voors AA, Anker SD, Bueno H, Cleland JGF, Coats AJS, Falk V, González‐Juanatey JR, Harjola V‐P, Jankowska EA, Jessup M, Linde C, Nihoyannopoulos P, Parissis JT, Pieske B, Riley JP, Rosano GMC, Ruilope LM, Ruschitzka F, Rutten FH, van der Meer P, Group ESCSD . 2016 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure: the Task Force for the diagnosis and treatment of acute and chronic heart failure of the European Society of Cardiology (ESC) developed with the special contribution of the Heart Failure Association (HFA) of the ESC. Eur Heart J 2016; 37: 2129–2200. [DOI] [PubMed] [Google Scholar]
- 12. Yancy CW, Jessup M, Bozkurt B, Butler J, Casey DE, Drazner MH, Fonarow GC, Geraci SA, Horwich T, Januzzi JL, Johnson MR, Kasper EK, Levy WC, Masoudi FA, McBride PE, McMurray JJV, Mitchell JE, Peterson PN, Riegel B, Sam F, Stevenson LW, Tang WHW, Tsai EJ, Wilkoff BL. 2013 ACCF/AHA Guideline for the management of heart failure. J. Am. Coll. Cardiol. 2013; 62: e147. [DOI] [PubMed] [Google Scholar]
- 13. Lundgren J, Rådegran G. Hemodynamic characteristics including pulmonary hypertension at rest and during exercise before and after heart transplantation. J Am Heart Assoc 2015; 4: e001787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Assarsson E, Lundberg M, Holmquist G, Björkesten J, Bucht Thorsen S, Ekman D, Eriksson A, Rennel Dickens E, Ohlsson S, Edfeldt G, Andersson A‐C, Lindstedt P, Stenvang J, Gullberg M, Fredriksson S. Homogenous 96‐plex PEA immunoassay exhibiting high sensitivity, specificity, and excellent scalability. PLoS One 2014; 9: e95192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Nyman U, Grubb A, Larsson A, Hansson L‐O, Flodin M, Nordin G, Lindström V, Björk J. The revised Lund‐Malmö GFR estimating equation outperforms MDRD and CKD‐EPI across GFR, age and BMI intervals in a large Swedish population. Clin Chem Lab Med 2014; 52: 815. [DOI] [PubMed] [Google Scholar]
- 16. Costanzo MR, Dipchand A, Starling R, Anderson A, Chan M, Desai S, Fedson S, Fisher P, Gonzales‐Stawinski G, Martinelli L, McGiffin D, Parisi F, Smith J, Taylor D, Meiser B, Webber S, Baran D, Carboni M, Dengler T, Feldman D, Frigerio M, Kfoury A, Kim D, Kobashigawa J, Shullo M, Stehlik J, Teuteberg J, Uber P, Zuckermann A, Hunt S, Burch M, Bhat G, Canter C, Chinnock R, Crespo‐Leiro M, Delgado R, Dobbels F, Grady K, Kao W, Lamour J, Parry G, Patel J, Pini D, Pinney S, Towbin J, Wolfel G, Delgado D, Eisen H, Goldberg L, Hosenpud J, Johnson M, Keogh A, Lewis C, O'Connell J, Rogers J, Ross H, Russell S, Vanhaecke J. The International Society of Heart and Lung Transplantation Guidelines for the care of heart transplant recipients. J Heart Lung Transplant 2010; 29: 914–956. https://www.jhltonline.org/article/S1053-2498(10)00358-X/fulltext [DOI] [PubMed] [Google Scholar]
- 17. Du BD, Du BE. Clinical calorimetry: tenth paper a formula to estimate the approximate surface area if height and weight be known. Arch Intern Med 1916; XVII: 863–871. [Google Scholar]
- 18. Benjamini Y, Krieger AM, Yekutieli D. Adaptive linear step‐up procedures that control the false discovery rate. Biometrika 2006; 93: 491–507. [Google Scholar]
- 19. Hultgårdh‐Nilsson A, Borén J, Chakravarti S. The small leucine‐rich repeat proteoglycans in tissue repair and atherosclerosis. J Intern Med 2015; 278: 447–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Happonen KE, Fürst CM, Saxne T, Heinegård D, Blom AM. PRELP protein inhibits the formation of the complement membrane attack complex. J Biol Chem 2012; 287: 8092–8100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Birke MT, Lipo E, Adhi M, Birke K, Kumar‐Singh R. AAV‐mediated expression of human PRELP inhibits complement activation, choroidal neovascularization and deposition of membrane attack complex in mice. Gene Ther 2014; 21: 507–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Aukrust P, Gullestad L, Lappegård Knut T, Ueland T, Aass H, Wikeby L, Simonsen S, Frøland Stig S, Mollnes TE. Complement activation in patients with congestive heart failure. Circulation 2001; 104: 1494–1500. [DOI] [PubMed] [Google Scholar]
- 23. Zhang C, Li Y, Wang C, Wu Y, Cui W, Miwa T, Sato S, Li H, Song W‐C, Du J. Complement 5a receptor mediates angiotensin II‐induced cardiac inflammation and remodeling. Arterioscler Thromb Vasc Biol 2014; 34: 1240–1248. [DOI] [PubMed] [Google Scholar]
- 24. Mahaffey KW, Van de Werf F, Shernan SK, Granger CB, Verrier ED, Filloon TG, Todaro TG, Adams PX, Levy JH, Hasselblad V, Armstrong PW. Effect of pexelizumab on mortality in patients with acute myocardial infarction or undergoing coronary artery bypass surgery: a systematic overview. Am Heart J 2006; 152: 291–296. [DOI] [PubMed] [Google Scholar]
- 25. Investigators TAA. Pexelizumab for acute ST‐elevation myocardial infarction in patients undergoing primary percutaneous coronary intervention: a randomized controlled trial. JAMA 2007; 297: 43–51. [DOI] [PubMed] [Google Scholar]
- 26. Martel C, Granger CB, Ghitescu M, Stebbins A, Fortier A, Armstrong PW, Bonnefoy A, Theroux P. Pexelizumab fails to inhibit assembly of the terminal complement complex in patients with ST‐elevation myocardial infarction undergoing primary percutaneous coronary intervention. Insight from a substudy of the Assessment of Pexelizumab in Acute Myocardial Infarction (APEX‐AMI) trial. Am Heart J 2012; 164: 43–51. [DOI] [PubMed] [Google Scholar]
- 27. Pischke SE, Gustavsen A, Orrem HL, Egge KH, Courivaud F, Fontenelle H, Despont A, Bongoni AK, Rieben R, Tønnessen TI, Nunn MA, Scott H, Skulstad H, Barratt‐Due A, Mollnes TE. Complement factor 5 blockade reduces porcine myocardial infarction size and improves immediate cardiac function. Basic Res Cardiol 2017; 112: 20–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Van Linthout S, Tschöpe C. Inflammation—cause or consequence of heart failure or both? Curr Heart Fail Rep 2017; 14: 251–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Timmers L, Pasterkamp G, de Hoog VC, Arslan F, Appelman Y, de Kleijn DP. The innate immune response in reperfused myocardium. Cardiovasc Res 2012; 94: 276–283. [DOI] [PubMed] [Google Scholar]
- 30. Thul PJ, Åkesson L, Wiking M, Mahdessian D, Geladaki A, Ait Blal H, Alm T, Asplund A, Björk L, Breckels LM, Bäckström A, Danielsson F, Fagerberg L, Fall J, Gatto L, Gnann C, Hober S, Hjelmare M, Johansson F, Lee S, Lindskog C, Mulder J, Mulvey CM, Nilsson P, Oksvold P, Rockberg J, Schutten R, Schwenk JM, Sivertsson Å, Sjöstedt E, Skogs M, Stadler C, Sullivan DP, Tegel H, Winsnes C, Zhang C, Zwahlen M, Mardinoglu A, Pontén F, von Feilitzen K, Lilley KS, Uhlén M, Lundberg E. A subcellular map of the human proteome. Science 2017; 356: eaal3321. [DOI] [PubMed] [Google Scholar]
- 31. Barhoumi T, Fraulob‐Aquino JC, Mian MOR, Ouerd S, Idris‐Khodja N, Huo K‐G, Rehman A, Caillon A, Dancose‐Giambattisto B, Ebrahimian T, Lehoux S, Paradis P, Schiffrin EL. Matrix metalloproteinase‐2 knockout prevents angiotensin II‐induced vascular injury. Cardiovasc Res 2017; 113: 1753–1762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Matsusaka H, Ide T, Matsushima S, Ikeuchi M, Kubota T, Sunagawa K, Kinugawa S, Tsutsui H. Targeted deletion of matrix metalloproteinase 2 ameliorates myocardial remodeling in mice with chronic pressure overload. Hypertension 2006; 47: 711–717. [DOI] [PubMed] [Google Scholar]
- 33. Polyakova V, Hein S, Kostin S, Ziegelhoeffer T, Schaper J. Matrix metalloproteinases and their tissue inhibitors in pressure‐overloaded human myocardium during heart failure progression. J Am Coll Cardiol 2004; 44: 1609–1618. [DOI] [PubMed] [Google Scholar]
- 34. Yamazaki T, Lee J‐D, Shimizu H, Uzui H, Ueda T. Circulating matrix metalloproteinase‐2 is elevated in patients with congestive heart failure. Eur J Heart Fail 2004; 6: 41–45. [DOI] [PubMed] [Google Scholar]
- 35. Hayashidani S, Tsutsui H, Ikeuchi M, Shiomi T, Matsusaka H, Kubota T, Imanaka‐Yoshida K, Itoh T, Takeshita A. Targeted deletion of MMP‐2 attenuates early LV rupture and late remodeling after experimental myocardial infarction. Am J Physiol Heart Circ Physiol 2003; 285: H1229–H1235. [DOI] [PubMed] [Google Scholar]
- 36. Fernandez‐Patron C, Radomski MW, Davidge ST. Vascular matrix metalloproteinase‐2 cleaves big endothelin‐1 yielding a novel vasoconstrictor. Circ Res 1999; 85: 906–911. [DOI] [PubMed] [Google Scholar]
- 37. Berthelot E, Bailly MT, Hatimi SE, Robard I, Rezgui H, Bouchachi A, Montani D, Sitbon O, Chemla D, Assayag P. Pulmonary hypertension due to left heart disease. Arch Cardiovasc Dis 2017; 110: 420–431. [DOI] [PubMed] [Google Scholar]
- 38. Ranchoux B, Harvey LD, Ayon RJ, Babicheva A, Bonnet S, Chan SY, Yuan JXJ. Perez VdJ. Endothelial dysfunction in pulmonary arterial hypertension: an evolving landscape (2017 Grover Conference Series). Pulm Circ 2017; 8: 2045893217752912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Fayyaz Ahmed U, Edwards William D, Maleszewski Joseph J, Konik Ewa A, DuBrock HM, Borlaug Barry A, Frantz Robert P, Jenkins Sarah M, Redfield MM. Global pulmonary vascular remodeling in pulmonary hypertension associated with heart failure and preserved or reduced ejection fraction. Circulation 2018; 137: 1796–1810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Mehra MR, Canter CE, Hannan MM, Semigran MJ, Uber PA, Baran DA, Danziger‐Isakov L, Kirklin JK, Kirk R, Kushwaha SS, Lund LH, Potena L, Ross HJ, Taylor DO, Verschuuren EAM, Zuckermann A. The 2016 International Society for Heart Lung Transplantation listing criteria for heart transplantation: a 10‐year update. J Heart Lung Transplant 2016; 35: 1–23. [DOI] [PubMed] [Google Scholar]
- 41. Pasero D, Rana NK, Bonato R, Ribezzo M, Ivaldi F, Ricci D, Grosso Marra W, Checco L, Lupo M, Boffini M, Rinaldi M. Inhaled nitric oxide versus sodium nitroprusside for preoperative evaluation of pulmonary hypertension in heart transplant candidates. Transplant Proc 2013; 45: 2746–2749. [DOI] [PubMed] [Google Scholar]
- 42. Vachiéry J‐L, Tedford RJ, Rosenkranz S, Palazzini M, Lang I, Guazzi M, Coghlan G, Chazova I, De Marco T. Pulmonary hypertension due to left heart disease. Eur Respir J 2018; 1801897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kuwahara F, Kai H, Tokuda K, Kai M, Takeshita A, Egashira K, Imaizumi T. Transforming growth factor‐β function blocking prevents myocardial fibrosis and diastolic dysfunction in pressure‐overloaded rats. Circulation 2002; 106: 130–135. [DOI] [PubMed] [Google Scholar]
- 44. Cheng J‐C, Chang H‐M, Leung PCK. Connective tissue growth factor mediates TGF‐β1‐induced low‐grade serous ovarian tumor cell apoptosis. Oncotarget 2017; 8: 85224–85233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Bujak M, Ren G, Kweon HJ, Dobaczewski M, Reddy A, Taffet G, Wang XF, Frangogiannis NG. Essential role of Smad3 in infarct healing and in the pathogenesis of cardiac remodeling. Circulation 2007; 116: 2127–2138. [DOI] [PubMed] [Google Scholar]
- 46. Tsai H‐C, Tzeng H‐E, Huang C‐Y, Huang Y‐L, Tsai C‐H, Wang S‐W, Wang P‐C, Chang A‐C, Fong Y‐C, Tang C‐H. WISP‐1 positively regulates angiogenesis by controlling VEGF‐A expression in human osteosarcoma. Cell Death Dis 2017; 8: e2750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Colston JT, de la Rosa SD, Koehler M, Gonzales K, Mestril R, Freeman GL, Bailey SR, Chandrasekar B. Wnt‐induced secreted protein‐1 is a prohypertrophic and profibrotic growth factor. Am J Physiol Heart Circ Physiol 2007; 293: H1839–H1846. [DOI] [PubMed] [Google Scholar]
- 48. Maiese K. Wisp1: Clinical insights for a proliferative and restorative member of the CCN family. Curr Neurovasc Res 2014; 11: 378–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Cohn JN, Ferrari R, Sharpe N. Cardiac remodeling—concepts and clinical implications: a consensus paper from an international forum on cardiac remodeling. J Am Coll Cardiol 2000; 35: 569–582. [DOI] [PubMed] [Google Scholar]