

Abstract
The AMPA receptor (AMPAR) subunit GluA3 has been suggested to shape synaptic transmission and activity-dependent plasticity in endbulb-bushy cell synapses (endbulb synapses) in the anteroventral cochlear nucleus, yet the specific roles of GluA3 in the synaptic transmission at endbulb synapses remains unexplored. Here, we compared WT and GluA3 KO mice of both sexes and identified several important roles of GluA3 in the maturation of synaptic transmission and short-term plasticity in endbulb synapses. We show that GluA3 largely determines the ultrafast kinetics of endbulb synapses glutamatergic currents by promoting the insertion of postsynaptic AMPARs that contain fast desensitizing flop subunits. In addition, GluA3 is also required for the normal function, structure, and development of the presynaptic terminal which leads to altered short term-depression in GluA3 KO mice. The presence of GluA3 reduces and slows synaptic depression, which is achieved by lowering the probability of vesicle release, promoting efficient vesicle replenishment, and increasing the readily releasable pool of synaptic vesicles. Surprisingly, GluA3 also makes the speed of synaptic depression rate-invariant. We propose that the slower and rate-invariant speed of depression allows an initial response window that still contains presynaptic firing rate information before the synapse is depressed. Because this response window is rate-invariant, GluA3 extends the range of presynaptic firing rates over which rate information in bushy cells can be preserved. This novel role of GluA3 may be important to allowing the postsynaptic targets of spherical bushy cells in mice use rate information for encoding sound intensity and sound localization.
SIGNIFICANCE STATEMENT We report novel roles of the glutamate receptor subunit GluA3 in synaptic transmission in synapses between auditory nerve fibers and spherical bushy cells (BCs) in the cochlear nucleus. We show that GluA3 contributes to the generation of ultrafast glutamatergic currents at these synapses, which is important to preserve temporal information about the sound. Furthermore, we demonstrate that GluA3 contributes to the normal function and development of the presynaptic terminal, whose properties shape short-term plasticity. GluA3 slows and attenuates synaptic depression, and makes it less dependent on the presynaptic firing rates. This may help BCs to transfer information about the high rates of activity that occur at the synapse in vivo to postsynaptic targets that use rate information for sound localization.
Keywords: Endbulb, synaptic depression, plasticity, development, lateral superior olive, rate-invariance
Introduction
In the cochlear nucleus, the first central nucleus in the auditory system, EPSCs at synapses between auditory nerve terminals and bushy cells (endbulb synapses) are extremely fast which allows the preservation of the temporal features of sound. EPSCs in endbulb synapses are mediated by AMPA receptors (AMPARs) with exceptionally fast kinetics of channel gating (Raman and Trussell, 1992; Isaacson and Walmsley, 1996), which result from the predominance of GluA3 and GluA4 subunits in their flop isoforms (Geiger et al., 1995; Wang et al., 1998; Lawrence and Trussell, 2000; Gardner et al., 2001; Schmid et al., 2001; Sugden et al., 2002). Flop subunits greatly accelerate the decay of EPSCs (Trussell and Fischbach, 1989; Gardner et al., 2001; Pei et al., 2007; Cao and Oertel, 2010), because they have faster rates of desensitization than their flip counterparts (Sommer et al., 1990; Mosbacher et al., 1994). The developmental increase of flop subunits largely contributes to the developmental speeding of EPSCs in endbulb synapses (Trussell and Fischbach, 1989; Lawrence and Trussell, 2000).
Another prominent feature of synaptic transmission in endbulb synapses is short-term synaptic depression (Wu and Oertel, 1987; Oleskevich et al., 2004; Wang and Manis, 2008; Yang and Xu-Friedman, 2009, 2015; Cao and Oertel, 2010), a form of plasticity that is believed to improve encoding of temporal information by reducing responses to non-phase-locked inputs (Taschenberger and von Gersdorff, 2000; Brenowitz and Trussell, 2001) and to negatively impact the transmission of presynaptic firing rate, or rate information, by decreasing postsynaptic firing reliability (Wang and Manis, 2006; Yang and Xu-Friedman, 2009; Wang et al., 2010). Yet, endbulb synapses can accurately transmit rate information even at high firing rates, suggesting that synaptic depression must be circumvented in some way to allow rate information for a wide range of firing rates (MacLeod et al., 2007; Bagnall et al., 2008; Lindner et al., 2009; Yang et al., 2009; Turecek et al., 2017).
Anatomical studies suggest that the GluA3 subunit plays a preponderant role in activity-dependent plasticity (Whiting et al., 2009; Wang et al., 2011; Clarkson et al., 2016) and normal auditory processing (García-Hernández et al., 2017). Upregulation of GluA3, but not GluA4, is believed to underlie hearing recovery after transient hearing loss (Clarkson et al., 2016; García-Hernández et al., 2017). However, the specific role of GluA3 in the synaptic transmission in endbulb synapses remains unexplored. Loss of GluA3 decreases the amplitude and increases the latency of wave II of the auditory brainstem responses (García-Hernández et al., 2017), suggesting that activity in the cochlear nucleus has been altered. Furthermore, loss of GluA3 produces ultrastructural changes in presynaptic and postsynaptic terminals of endbulb synapses (García-Hernández et al., 2017; Rubio et al., 2017). These changes will have consequences on synaptic efficacy and plasticity (Taschenberger et al., 2002; Cathala et al., 2005; Freche et al., 2011; Allam et al., 2015), a hypothesis that we address in this study by investigating synaptic transmission and short-term plasticity in endbulb synapses of GluA3 KO and WT mice.
Our results identify several important roles of GluA3 in synaptic transmission in endbulb synapses. GluA3 makes synaptic transmission ultrafast by promoting the insertion of flop subunits in the postsynaptic terminal. Furthermore, GluA3 is necessary for normal function and maturation of the presynaptic terminal, which ultimately shape short-term plasticity. GluA3 reduces and slows synaptic depression by lowering the probability of vesicle release, promoting efficient vesicle replenishment, and increasing the readily releasable pool of synaptic vesicles. Interestingly, we also found that GluA3 makes the speed of synaptic depression rate-invariant. We propose that the slower and rate-invariant speed of depression conferred by GluA3 allows the synapse to preserve rate information over a wide range of firing rates, despite strong synaptic depression. In this scenario, GluA3 is needed for accurate transfer of rate information from spherical BCs to postsynaptic targets that use rate information for encoding sound intensity and sound localization.
Materials and Methods
Mice.
Brain slices were obtained from CD57B6J WT mice (n = 21 animals) and GluA3 KO mice (n = 21 animals) of both sexes, aged from postnatal day (P) 17 to P21. GluA3 KO mice were kindly provided by Dr. Sakimura (García-Hernández et al., 2017). Genotyping of these mice was also confirmed post hoc. All experiments were performed in accordance with the guidelines of the University of Pittsburgh Animal Care and Use Committee.
Slice preparation and electrophysiological recordings.
Transverse slices (300 μm thick) containing the AVCN were cut with a vibratome (Leica) in aCSF containing the following (in mm): 125 NaCl, 2.5 KCl, 1 MgCl2, 2 CaCl2, 1.25 NaH2PO4, 25 NaHCO3, and 10 glucose, bubbled with 95% O2/5% CO2, pH 7.4, at room temperature. Slices were stored at 32°C for 1 h in a custom interface chamber before being transferred to the recording chamber for electrophysiology recordings (Weisz et al., 2016). During recordings, slices were continuously bathed in recirculating aCSF bubbled with 95% O2/5% CO2 at 30−33°C (inline heater, SH-27B; Warner Instruments). The aCSF used for recordings had the same composition as the aCSF used for cutting with the addition of 2 μm strychnine, 30 μm gabazine, and 50 μm APV to isolate AMPA receptor- mediated EPSCs. Cyclothiazide (50 μm) was added for desensitization experiments. No tetrodotoxin was added because sEPSCs at the endbulb reflect quantal release (Isaacson and Walmsley, 1996; Bellingham et al., 1998; Lu et al., 2007; Fig. 1).
Figure 1.

Spontaneous synaptic events in endbulb synapses differ between WT and GluA3 KO mice: In KO mice, sEPSCs have smaller amplitude, slower kinetics, and higher frequency. A, Representative traces showing sEPSCs (left) and their corresponding averaged events (right). WT, blue; KO, red. B–F, Cumulative distribution plots of average sEPSC amplitude (B), half-width (C), rise time (D), decay tau (E), and interevent intervals (F), for both genotypes (WT, n = 26 neurons from 19 animals; KO, n = 30 neurons from 21 animals; 200 events per neuron; Two sample Kolmogorov–Smirnov test: D = 0.26, for Amplitude; D = 0.18, for half-width; D = 0.11, for rise-time; D = 0.29, for decay tau; all ***p < 0.001).
Whole-cell patch-clamp recordings from BCs in the AVCN were performed using a MultiClamp 700B amplifier and a Digidata 1550 digitizer controlled by Clampex 10.4 software (Molecular Devices). BCs were identified by their characteristic firing pattern (few brief spikes) to prolonged depolarizing current injections (Oertel, 1983; Wu and Oertel, 1987; Cao et al., 2007; Lu et al., 2007). We recorded from BCs in the most anterior region of the VCN, a region that in mice is occupied mainly by small spherical BCs, as opposed to globular BCs that are most frequently found more posteriorly (Tolbert and Morest, 1982; Willard and Ryugo, 1983). Therefore, most of our recordings were likely made from small spherical BCs (Wright et al., 2014). Recordings were sampled at 50 kHz and filtered online at 10 kHz. Recording pipettes were pulled from 1.5 mm outer diameter borosilicate glass (Sutter Instruments) to tip resistances of 3–5 mΩ. The electrode solution used for all recordings contained the following (in mm): 140 K-gluconate, 4.5 MgCl2, 9 HEPES, 5 EGTA, 4 MG-ATP, 0.3 GTP, 4.4 creatine phosphate (Tris salt), and was adjusted to pH 7.3 with KOH. Neurons were voltage clamped at −70 mV and membrane voltages were adjusted online for a measured liquid junction potential of 14.2 mV. Series resistance (<10 MΩ) was compensated with a 60–80% correction and a 90% prediction (3 kHz bandwidth). The membrane resistance of BCs was calculated based on responses to +10 and −10 pA current pulses. BC membrane resistance was similar in both genotypes (WT, median = 82.4MΩ; KO, median = 91.0MΩ; Wilcoxon's rank-sum test W = 628, p = 0.5; data not shown).
Endbulb-elicited EPSCs were elicited by electrically stimulating the auditory nerve root with a minimal stimulation protocol (Fig. 2). Briefly, the stimulus level was increased until a threshold level was determined and an EPSC was elicited by a single endbulb (Fig. 2A). For each cell, the stimulus strength was adjusted to be slightly above the minimal strength that reliably elicited a response (Wang et al., 2010). Endbulb evoked EPSCs were distinguished by their large amplitude (>1 nA), all-or-none synaptic currents and brief time course (Isaacson and Walmsley, 1996; Wang et al., 2010; Fig. 2B). Stimulus pulses were generated by an ISO-Flex isolator (AMPI, Israel) and delivered through an extracellular saline-filled glass pipette (∼10 μm tip).
Figure 2.
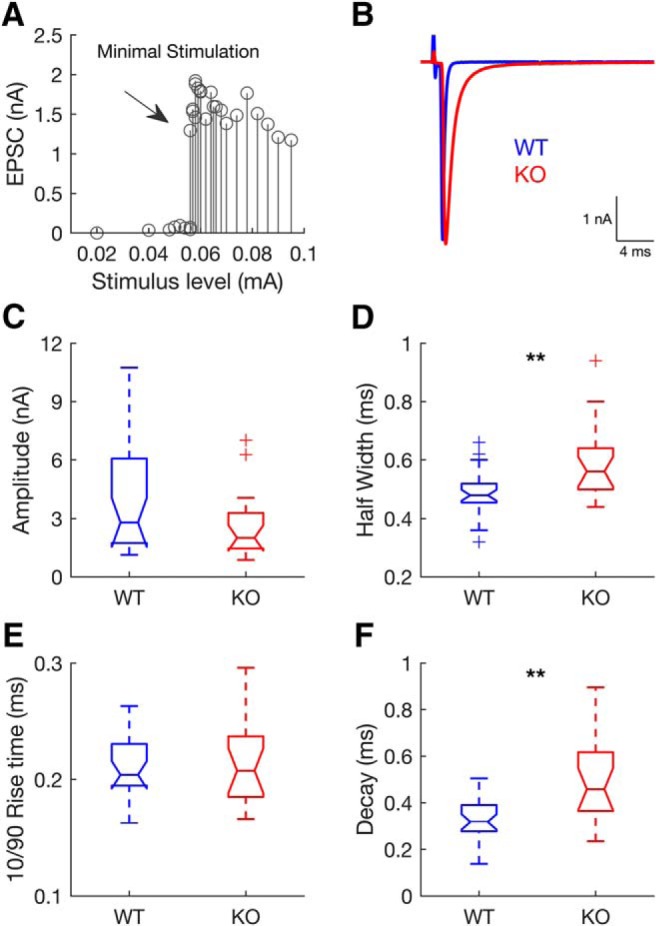
Evoked synaptic responses in endbulb synapses of WT and GluA3 KO mice. In the KO, AMPAR-mediated EPSCs have slower kinetics. A, Single endbulb-elicited EPSCs were elicited by increasing stimulus strength until an all-or-nothing EPSC was elicited (arrow). B, Example EPSCs from a WT (blue) and a KO (red) bushy cell. C–F, Distribution of EPSC amplitudes (C) and kinetics (D–F) displayed as notched boxplots (line in the middle: median; box: interquartile range; whiskers: full range excluding outliers; outliers: marked with + sign; notches: provide an approximate confidence interval for the population median) (WT, n = 29 from 21 animals; KO, n = 19 from 18 animals). Lack of GluA3 had no effect on EPSC amplitudes (C; Wilcoxon's rank-sum test: W = 779, p = 0.15) and rise times (E; Wilcoxon's rank-sum test: W = 490, p = 0.87; Notches between WT and KO boxplots overlap). Lack of GluA3 significantly increased EPSC half-widths (D; Wilcoxon's rank-sum test: W = 587, **p = 0.009) and decay time constants (F; Wilcoxon's rank-sum test: W = 558, **p = 0.001; notches between WT and KO boxplots do not overlap).
Electron microscopy.
Twelve mice of both sexes were used for ultrastructural morphometrical analysis of the synaptic vesicles size within endbulb terminals. Two mice per age (P20, P44 and P100) and genotype (WT and KO mice) were used. Mice were transcardially perfused with fixative, and brainstem sections with the cochlear nucleus were cut with a vibratome and processed for electron microscopy as previously described (García-Hernández et al., 2017). Ultrathin sections (80 nm) were cut with a Leica EM UC7 ultramicrotome (Leica Microsystems) and examined with a JEOL 1400 transmission electron microscope. Endbulb terminals on spherical bushy cells in the anteroventral cochlear nucleus were identified as previously described (García-Hernández et al., 2017). Images of endbulb synapses were digitally captured with an ORIUS SC200 CCD camera (Gatan). Images were assembled with Adobe Photoshop, modifying brightness and contrast if necessary.
Experimental design, data analysis, and statistics.
Electrophysiological traces were analyzed using Clampfit 10.4 (Molecular Devices) and MiniAnalysis (SynaptoSoft). Additional analyses of data, including statistics, were performed and figures were created using MATLAB (The MathWorks), except two-way repeated-measures ANOVAs, which were performed with SigmaPlot (Systat Software). Data were tested for normality using Shapiro–Wilk test, and for equal variances between groups using Brown–Forsythe test. Statistical tests were considered significant when p < 0.05.
To quantify the differences in spontaneous activity between WT and KO, spontaneous events (sEPSCs; 200 events per neuron) were merged into two overall distributions for WT versus KO for each parameter measured (amplitude, half-width, rise time, decay time, and interevent interval) and their distributions were compared using the two-sample Kolmogorov–Smirnov test ('kstest2′ function in MATLAB; WT, n = 26 neurons; GluA3 KO, n = 30 neurons; Fig. 1). To visualize the differences between their distributions, their empirical cumulative distribution functions were plotted (overlaying the distributions of both genotypes, for each parameter measured; the Kolmogorov–Smirnov test statistic [D] is the maximum difference between these functions; Fig. 1B–F). The decay time constants of sEPSCs were obtained by fitting the decay phase (10–90%) of the events with single exponential decay functions.
To compare evoked synaptic activity between WT and KO, pairwise statistical comparisons for endbulb elicited EPSC parameters (amplitude, half-width, rise time, and decay time) between genotypes were performed using two-sided Wilcoxon's rank-sum test (WT, n = 29 neurons; KO, n = 19 neurons; Fig. 2). Data are presented as notched boxplots (Fig. 2C–F). The tops and bottoms of each box are the 25th and 75th percentiles of the samples, respectively, and their distances are the interquartile ranges. The sample median is represented by the line in the middle of each box. Data points beyond the whisker length are marked as outliers (values that are >1.5 times the interquartile range away from the top or bottom of the box; displayed with a + sign). Notches display the variability of the median between samples (nonoverlapping notches between boxes indicate that their medians are significantly different at 5% significance level).
To evaluate whether the effect of cyclothiazide (CTZ) on the EPSC kinetics differed between WT and KO two-way repeated-measures ANOVAs (factors: genotype, drug, genotype × drug; one-factor repetition; repeated factor: drug) were performed. A significant interaction indicated that the impact of CTZ was different between WT and KO, i.e., there was a differential effect of CTZ between genotypes, and this was followed by post hoc comparisons (Holm–Sidak method; comparisons versus control group; control group for genotype: WT; control group for drug: CTR; n = 7 neurons, per genotype, per condition; Fig. 3). Because there are only two levels of repeated measures in the ANOVAs performed here the assumption of sphericity is met. This is true for all the repeated-measures ANOVAs performed in the present work.
Figure 3.
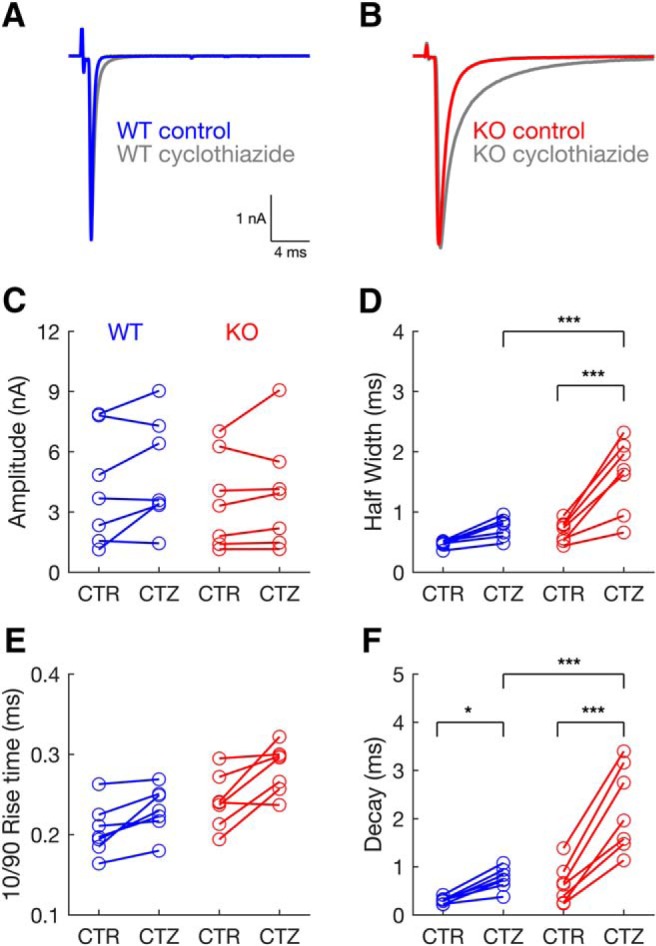
Blocking AMPAR desensitization with cyclothiazide (CTZ) had a stronger effect on EPSCs in GluA3 KO than in WT mice. A, Example EPSC from a WT bushy cell in control (blue) and in the presence of CTZ (gray). B, Example EPSC from a KO bushy cell in control (red) and in the presence of CTZ (gray). C–F, EPSC amplitude (C), half-width (D), rise time (E), and decay time (F) values in control (CTR) and in the presence of CTZ, in WT (blue panels on the left; n = 7) and KO (red panels on the right; n = 7). C, CTZ had no effect on EPSC amplitudes [2-way repeated-measures ANOVA: F(1,12) = 0.335, p = 0.573, genotype; F(1,12) = 4.738, p = 0.050, drug; F(1,12) = 0.640, p = 0.439, interaction]. D, CTZ prolonged EPSCs half-widths more strongly in KO mice than in WT mice [2-way repeated-measures ANOVA/Holm–Sidak method: F(1,12) = 23.295, p = <0.001, genotype × drug interaction; CTR vs CTZ, within genotype: KO, t = 7.110, p < 0.001; WT, t = 1.985, p = 0.071; WT vs KO, within CTZ: t = 4.964, p < 0.001]. E, CTZ had a similar effect on EPSC rise times in either genotype [2-way repeated-measures ANOVA: F(1,12) = 8.379, p = 0.01, genotype; F(1,12) = 21.836, p < 0.001, drug; F(1,12) = 1.097, p = 0.32, interaction]. F, CTZ prolonged more strongly EPSC decay times in KO than in WT mice [2-way repeated-measures ANOVA/Holm–Sidak method: F(1,12) = 13.135, p = 0.003, genotype × drug interaction; CTR vs CTZ, within genotype: KO, t = 9.613, p < 0.001; WT, t = 2.787, p = 0.016; WT vs KO, within CTZ: t = 5.389, p < 0.001]. *p < 0.05, ***p < 0.001; lower square brackets, CTR vs CTZ, within genotype; upper square brackets, WT versus KO, within CTZ.
To study short-term depression, trains of stimuli were delivered consisting of 10 repetitions of 20 pulses at different frequencies (10, 50, 100, and 300 Hz), every 2 s (WT, n = 18, 23, 29, and 13 neurons; KO, n = 6, 6, 17, and 14 neurons; for each frequency, respectively; Fig. 4). For each neuron, EPSC amplitudes evoked for each pulse were averaged over the 10 repetitions. EPSC amplitudes were then normalized to the amplitude of the first EPSC in the train. The normalized amplitudes over time (stimulus number) were fitted with a single exponential with two parameters (y = aebx + c), where b is the decay constant and c is the steady-state using the “fit” function in MATLAB. The fit function also computes the coefficient of determination (adjusted r2) of the whole fit and the 95% confidence intervals (CI) for the fitted parameters, which we used to determine the significance levels for comparisons of the decay and steady state between WT and KO (Figs. 4, 5, 6). The same experimental design and analysis was repeated in some neurons recorded under the effect of CTZ to control for desensitization (WT, n = 7 and 6 neurons; KO, n = 7 and 5 neurons; at 100 Hz and 300 Hz, respectively; Figs. 5, 6).
Figure 4.

Dynamics of short-term synaptic depression induced by train stimulation. In GluA3 KO mice, synaptic depression is both stronger and faster than in WT mice. A, Example traces recorded in the WT (blue) and KO (red) in response to stimulus trains with increasing frequency. B, Normalized EPSC amplitudes of the population vs stimulus number, for the different stimulation frequencies (WT: n = 18, 23, 29, and 22 neurons; KO: n = 6, 6, 17, and 14 neurons; for 10, 50, 100, and 300 Hz, respectively). Amplitude values are jittered in the x-axis (“jitterAmount” = 0.4 units in MATLAB). C, Decay curves and their respective confidence intervals (outer lines) derived from exponential fits to the data in B. Depression of EPSC amplitudes followed a single exponential ([y = aebx + c], where b is the decay, and c is the steady state). Gray dashed lines indicate the steady-state plateau reached during synaptic depression. Compared with WT mice, in KO mice synaptic depression was stronger and faster as evidenced by its lower steady state (gray dashed lines; all frequencies) and its faster decay (at 100 Hz and 300 Hz). * Statistical significance derived from the 95% confidence intervals calculated by the “fit” function in MATLAB.
Figure 5.

Stronger depression of the GluA3 KO synapse persists after blocking AMPAR desensitization with cyclothiazide (CTZ). A, Example traces from a WT neuron in response to 100 Hz frequency stimulation in control (blue) and in the presence of CTZ (cyan). B, Same as A but from a KO neuron in control (red) and in the presence of CTZ (orange). C, Plot on the left, normalized EPSC amplitudes of the population vs stimulus number at 100 Hz stimulation frequency, in control and in the presence of CTZ (same color code as in A and B; n = 7 neurons per genotype, per condition). Amplitude values are jittered in the x-axis (“jitterAmount” = 0.4 units in MATLAB). Right: decay curves and their respective confidence intervals (outer lines) derived from single exponential fits to the data plotted on the left. Dashed lines indicate the steady-state plateau reached during synaptic depression in the presence of CTZ for WT (cyan) and KO (orange) neurons. D, Same as in C but for 300 Hz stimulation frequency (WT: n = 6 neurons; KO: n = 5 neurons; per condition). For both stimulation frequencies, the stronger and faster depression in KO mice persisted during desensitization block. * Statistical significance between WT (cyan) and KO (orange) in the presence of CTZ, derived from the 95% confidence intervals calculated by the “fit” function in MATLAB.
Figure 6.

GluA3 is required for slower and rate-invariant speed of synaptic depression. A, Decay constants and their respective 95% confidence intervals (bars) obtained by the single exponential fits ([y = aebx + c], where b is the decay constant) to the train data at 10, 50, 100, and 300 Hz, for WT (blue) and KO (red) mice, same data as Figure 4 shows. B, Same as in A but for responses in the presence of CTZ at 100 and 300 Hz, for WT (cyan) and KO (yellow) mice, same data as Figure 5 shows. While in WT mice the speed of depression is similar between stimulation frequencies (rate-invariant), in KO mice the speed of depression increases with the increase in stimulation frequency (rate-dependent), both in control conditions (A) and in the presence of CTZ (B).
To determine the size of the readily releasable pool (RRP) and the initial probability of vesicle release (Pr) we used two different methods: the train method (Schneggenburger et al., 1999; Thanawala and Regehr, 2013; Fig. 7) and the EQ method (Fig. 7; Elmqvist and Quastel, 1965). For both methods, the normalized EPSC amplitudes for the 300 Hz trains were summed throughout the train stimulus to give a cumulative normalized EPSC amplitude curve, for each neuron (WT, n = 22 neurons; KO, n = 15 neurons; Fig. 7A,D). For the train method, a regression line was fitted to the final 5 points of the cumulative normalized EPSC and back-extrapolated to the y-axis (Fig. 7A). The y-intercept corresponds to RRPtrain (arrows in Fig. 7A), and Prtrain equals EPSC0 divided by RRPtrain (Schneggenburger et al., 1999; Thanawala and Regehr, 2013; Lu and Trussell, 2016; Fig. 7A–C). For the EQ method, first used in a mammalian CNS synapse by Taschenberger et al. (2002; calyx of Held synapse), normalized EPSC amplitudes were plotted versus the cumulative normalized EPSC (Fig. 7D). The first 4 points of the normalized EPSC amplitudes were extrapolated to the x-axis. The x-intercept corresponds to RRPEQ (arrows in Fig. 7D), and PrEQ equals EPSC0 divided by RRPEQ (Elmqvist and Quastel, 1965; Lu and Trussell, 2016; Fig. 7D–F). Both methods give close estimates of RRP and Pr, but the estimates are not necessarily identical, due to their different assumptions about replenishment. The train method assumes that once the RRP has been depleted by repetitive stimulation, the remaining responses rely on a replenishment of the RRP (Schneggenburger et al., 1999; Thanawala and Regehr, 2013). Thus, the rate of replenishment will correspond to the slope of the linear fit back-extrapolated to the y-axis (Fig. 7A,I) and the y-intercept of the linear fit represents the cumulative amount of release minus the release by newly recruited vesicles, which is then taken as an estimate of the RRPtrain (Schneggenburger et al., 1999; Thanawala and Regehr, 2013; Lu and Trussell, 2016). In contrast, the EQ method relies primarily on EPSCs early in the train and assumes that the effects of replenishment are small and can be ignored (Elmqvist and Quastel, 1965; Taschenberger et al., 2002; Thanawala and Regehr, 2013; Lu and Trussell, 2016). Because the Pr may decrease during the stimulus train (Wu and Borst, 1999), the EQ method measures the initial RRP that may be released with higher Pr.
Figure 7.

Synaptic vesicle pool size, release and replenishment differ between WT and GluA3 KO mice. In KO mice, the readily releasable pool (RRP) is decreased, the probability of vesicle release (Pr) is increased, and the rate of replenishment is slower. A, Example of estimation of the RRP size in a WT (blue) and a KO (red) neuron using the train method (Schneggenburger et al., 1999). The relative size of RRPtrain corresponds to the y-intercept of a linear regression line back-extrapolated from the last 5 points of cumulative normalized EPSC amplitudes during 300 Hz stimulation (open circles). Arrows indicate the y-intercept (RRPtrain) for the two example neurons (WT: RRPtrain = 2.98; KO: RRPtrain = 2.03). B, Relative RRPtrain of the population (WT: n = 22; KO: n = 15). C, Estimates of the relative probability of vesicle release (Prtrain) from the same populations of neurons as in B, calculated by diving the first EPSC in the train by the RRPtrain. D, Example of estimation of the RRP in the same neurons as in A using the EQ method (RRPEQ; Elmqvist and Quastel, 1965). The relative size of RRPEQ corresponds to the x-intercept of a linear regression line extrapolated from the first 4 points of cumulative normalized EPSC amplitudes during 300 Hz stimulation (open circles) plotted versus the normalized EPSC amplitudes. Arrows indicate the x-intercept (RRPEQ) for the two example neurons (WT: RRPEQ = 3.89; KO: RRPEQ = 2.03). E–F, RRP estimates (E) and Pr estimates (F) obtained by the EQ method (RRPEQ and PrEQ, same population as in B and C). The size of the RRP (B,E) is significantly smaller in KO than in WT mice [2-way RM ANOVA/Holm Sidak method: F(1,35) = 11.563, p = 0.002, genotype; F(1,35) = 19.065, p < 0.001, method; F(1,35) = 2.916, p = 0.097, interaction; WT vs KO, t = 4.366, p < 0.001]. Pr (C,F) is significantly higher in KO than in WT mice [F(1,35) = 16.604, p < 0.001, genotype; F(1,35) = 32.661, p < 0.001, method; F(1,35) = 0.641, p = 0.429, interaction; WT vs KO, t = 4.366, p < 0.001]. G, H, Comparison of RRP estimates (G) and Pr estimates (H) obtained by the two methods for 300 Hz trains during desensitization blockade with cyclothiazide (WT, cyan: n = 6 neurons; KO, orange: n = 5 neurons). Compared with WT mice, KO mice have smaller RRP (G) [F(1,9) = 18.307, p = 0.002, genotype; WT vs KO, RRPtrain: t = 2.580, p = 0.025; RRPEQ: t = 5.512, p < 0.001]) and higher Pr (H) [F(1,9) = 13.473, p = 0.005, genotype; WT vs KO, Prtrain: t = 2.905, p = 0.016; PrEQ: t = 4.321, p = 0.002]. In WT mice, RRPEQ is significantly higher than RRPtrain (G) [F(1,9) = 20.301, p = 0.001, genotype × method interaction; RRPEQ vs RRPtrain, t = 6.964, p < 0.001], whereas in KO mice, RRPEQ is identical to RRPtrain (t = 0.257, p = 0.8). I, The relative rate of replenishment corresponds to the slope of the linear fit back-extrapolated using the train method (A). The relative rate of replenishment is slower in KO than WT mice (two-tailed unpaired t test: t = 2.806, df = 9, p = 0.02). *p < 0.05, ***p < 0.001.
To evaluate whether RRP and Pr estimates differed between genotype (WT vs KO) and method (train vs EQ) two-way repeated-measures ANOVAs (one for each independent variable: RRP and Pr) where performed (factors: genotype, method, genotype × method; one-factor repetition; repeated factor: method), followed by post hoc comparisons (Holm–Sidak method; all pairwise multiple comparisons; Fig. 7A–F). The same analysis was repeated for the population of neurons under the effect of CTZ to control for desensitization (WT, n = 6 neurons; KO, n = 5 neurons; Fig. 7G,H). Replenishment rate was estimated from the slope of the linear fit (train method) for responses under the effect of CTZ (Fig. 7I; same population of neurons as Fig. 7G,H). Comparison for replenishment rate between WT and KO was performed using two tailed t test. Synaptic vesicle size was measured in both genotypes (WT: n = 260 at P20, n = 54 at P44, n = 91 at P100; KO: n = 262, n = 56 at P44, n = 55 at P100; Fig. 8). ImageJ software (1.43u, NIH, https://imagej.nih.gov/ij/) was used for the morphometric analyses of the vesicle sizes. The radius from inner to inner leaflet of the bilayer membrane was measured. Delineation of the larger and minor diameter of the synaptic vesicles was performed while being blinded to the age and genotype of the mice. The diameter of the synaptic vesicles was calculated by dividing the sum of the major and minor diameter by 2. A two-way ANOVA (factors: genotype, age, genotype × age) was performed to determine whether there were differences in vesicle size between genotypes (WT vs KO) and age (P20 vs P44 vs P100), and/or if there were interactions between these two factors. The “histfit” function in MATLAB was used to plot the histograms and superimpose the fitted normal distributions of the vesicle diameters data (Fig. 8C). The histfit function uses the function “fitdist” to create a probability distribution object to fit a probability distribution to the data. The χ2 goodness-of-fit test was used to test the null hypothesis that the vesicle diameters data came from a normal distribution with a mean and variance estimated from these data (“chi2gof” function in MATLAB).
Figure 8.

The lack of GluA3 arrests the developmental increase of synaptic vesicle size. A, Electron micrographs of endbulb-bushy cell synapses of WT (left column) and KO mice (right column) at P20 (upper row) and P44 (lower row) postnatal days. AN, Auditory nerve; BC, Bushy cell. Scale bar: 200 nm. B, Vesicle size as well as their age-dependent increase was different between WT and KO mice [2-Way ANOVA/Holm Sidak method; F(2,772) = 104.249, p < 0.001, interaction]. In WT mice (blue), synaptic vesicle size increases with age (from P20 to P44; P20 vs P44, t = 14.626, p < 0.001; P44 vs P100, t = 0.135, p = 0.892; P20 vs P100, t = 17.767, p < 0.001). In KO mice (red), synaptic vesicle size does not change significantly with age (P20 vs P44, t = 1.728, p = 0.162; P44 vs P100, t = 2.014, p = 0.127; P20 vs P100, t = 0.863, p = 0.388), and is smaller than in the WT at all ages tested (P20: t = 4.478, p < 0.001; P44: t = 14.857, p < 0.001; P100: t = 14.216, p < 0.001). C, Vesicle size distributions with superimposed fitted normal distributions for WT (blue) and KO (red) mice at P20 (left panel), P44 (middle panel) and P100 (right panel) postnatal days. Sigma symbols (σ) as insets represent the SD of the normal distributions. Except for KO mice at P20, all distributions assume a normal distribution (WT: P20, χ2 = 6.9, df = 6, p = 0.334; P44, χ2 = 3.3, df = 3, p = 0.348; P100, χ2 = 2.3, df = 5, p = 0.808; KO: P20, χ2 = 20, df = 6, p = 0.003; P44, χ2 = 5.8, df = 4, p = 0.214; P100, χ2 = 5.4, df = 4, p = 0.249). ***p < 0.001.
Results
Altered spontaneous synaptic transmission in GluA3 KO mice
To better understand the role of AMPAR subunits in synaptic transmission at the endbulb synapse, we performed whole-cell patch-clamp recordings from BCs in brain slices from WT and GluA3 KO mice (P17 to P21). We first evaluated spontaneous neurotransmitter release by analyzing AMPAR-mediated sEPSCs (Fig. 1). We found that the absence of GluA3 in KO mice changed the amplitude, half-width, 10–90% rise time, decay tau, and interevent intervals of sEPSCs (Fig. 1B–F; two-sample Kolmogorov–Smirnov test). The distribution of sEPSC amplitudes from GluA3 KO mice was significantly shifted to the left (Fig. 1B; two-sample Kolmogorov–Smirnov test), indicating an increase in the proportion of smaller amplitude events in KO mice (median amplitude: WT = 104.37 pA, KO = 80.57 pA). Compared with WT mice, sEPSCs in KO mice also had slower kinetics as indicated by a right shift of the distributions of their half-widths, rise times, and decay constants (Fig. 1C–E; two-sample Kolmogorov–Smirnov test; median half-width: WT = 0.30 ms, KO = 0.34 ms; median rise-time: WT = 0.13 ms, KO = 0.14 ms; median decay tau: WT = 0.20 ms, KO = 0.25 ms). Finally, compared with WT mice, in KO mice sEPSCs occurred at a higher frequency as indicated by a left shift of the distribution of sEPSC interevent intervals (Fig. 1F; two-sample Kolmogorov–Smirnov test; median interevent intervals: WT = 234.37 ms, KO = 192.90 ms).
In summary, sEPSCs in WT mice were large and brief, resembling those previously described in mice at a similar age (Lu et al., 2007; Wright et al., 2014). Absence of GluA3 subunits in AMPA receptors resulted in events that were smaller, occurred at higher frequencies, and had slower kinetics, mostly due to a slower decay than WT animals (Fig. 1). Because during development, sEPSCs in endbulb synapses become progressively larger (Bellingham et al., 1998; Brenowitz and Trussell, 2001) and briefer (Brenowitz and Trussell, 2001; Lu et al., 2007) our results may reflect a developmental delay in the maturation of sEPSCs in GluA3 KO mice.
Evoked synaptic transmission is slower in GluA3 KO mice
To examine whether a lack of GluA3 altered evoked transmission in endbulb synapses, we recorded single fiber-evoked EPSCs in BCs using a minimal stimulation protocol (Fig. 2A). Stimulating the WT endbulbs elicited large and brief events, resembling those previously described (Cao and Oertel, 2010; Chanda and Xu-Friedman, 2010; Wang et al., 2010; Fig. 2B). Lack of GluA3 had no significant effect on EPSC peak amplitudes (Fig. 2C; Wilcoxon's rank-sum test) and rise times (Fig. 2E; Wilcoxon's rank-sum test). However, it significantly slowed EPSC kinetics as revealed by their longer half-widths (Fig. 2D; Wilcoxon's rank-sum test) caused by longer decay time constants (Fig. 2F; Wilcoxon's rank-sum test).
The unchanged amplitudes of evoked EPSCs in GluA3 KO mice (Fig. 2C) despite smaller quantal size (Fig. 1B) suggests that in KO mice, more vesicles were released per action potential due to an increased probability of release, which is in line with the higher frequency of sEPSC in these mice (Fig. 1A,F). Because the fast decay of EPSCs in endbulb synapses is largely determined by the fast desensitization of postsynaptic AMPA receptors (Gardner et al., 2001; Cao and Oertel, 2010; Chanda and Xu-Friedman, 2010), we next investigated whether the slower decay of evoked EPSCs in KO mice (Fig. 2D) is due to slower AMPAR desensitization.
Higher proportion of flip subunits confers slower desensitization of AMPARs in GluA3 KO mice
AMPAR desensitization is greatly regulated by the relative proportion of flip and flop splice variant subunits (Sommer et al., 1990; Mosbacher et al., 1994; Koike et al., 2000). Flip subunits have slower desensitization than flop subunits, resulting in slower decays of AMPAR-mediated currents (Trussell and Fischbach, 1989; Lawrence and Trussell, 2000; Gardner et al., 2001; Quirk et al., 2004; Pei et al., 2007). Therefore, we tested whether an enhanced proportion of flip-containing subunits in postsynaptic AMPARs may explain the slower EPSC kinetics in KO mice. To this end, we measured changes in desensitization caused by the application of cyclothiazide (CTZ), an AMPAR desensitization blocker with pronounced preference for flip isoforms (Partin et al., 1994; Fleck et al., 1996; Kessler et al., 2000). The magnitude of the effect of CTZ on desensitization (reflected by EPSC duration) will thus be a function of the proportion of flip subunits in the synapse (Fig. 3). As expected, in KO mice, CTZ prolonged the duration of EPSCs more strongly than in WT mice (Fig. 3; 2-way RM ANOVAs/Holm–Sidak comparisons), by prolonging EPSC half-widths (Fig. 3D) and decay times (Fig. 3F). In contrast, in either genotype CTZ had no effect on EPSC amplitudes (Fig. 3C), and slightly increased EPSC rise times (Fig. 3E).
The weak modulation by CTZ in the WT is consistent with the predominant expression of fast desensitizing flop subunits in mature endbulb synapses (Mosbacher et al., 1994; Koike et al., 2000; Lawrence and Trussell, 2000; Gardner et al., 2001). The stronger modulation by CTZ in KO mice compared with WT mice indicates that the slower kinetics of endbulb AMPAR currents (Figs. 1, 2, 3) in KO mice is due to a higher portion of slow desensitizing flip subunits. As AMPARs at endbulb synapses undergo a developmental enrichment of flop subunits (Lawrence and Trussell, 2000; Gardner et al., 2001), the high portion of flip subunits in KO mice may indicate that GluA3 plays a role in the maturation of endbulb synapses.
Enhanced short-term depression in GluA3 KO mice
Some of the changes in synaptic transmission in GluA3 KO mice may also reflect changes in the presynaptic endbulb terminal. For example, the higher sEPSC frequency we observed in KO mice (Fig. 1A,F) could reflect a higher probability of vesicle release (Pr). If this is the case, we expect to see differences in short-term synaptic depression at the endbulb synapse between WT and KO mice (Oleskevich et al., 2000, 2004; Wang and Manis, 2008; Yang and Xu-Friedman, 2008; Wright et al., 2014). In the endbulb synapse (and the majority of synapses in the CNS; Zucker and Regehr, 2002; Taschenberger et al., 2002) short-term depression is primarily due to a depletion of presynaptic vesicles (Wang and Manis, 2008; Yang and Xu-Friedman, 2008; Cao and Oertel, 2010). The time course of short-term depression largely depends on the initial Pr: the larger the Pr, the faster the depletion of vesicles and the faster the speed of depression (Abbott et al., 1997; Abbott and Regehr, 2004; Regehr, 2012). Therefore, if Pr is increased in KO mice, we expect that short-term depression is faster in KO mice. To address this hypothesis, we evoked EPSCs using stimulus trains at 10, 50, 100, and 300 Hz, which correspond to firing rate frequencies that occur in the auditory nerve in vivo (Joris et al., 1994; Taberner and Liberman, 2005; Figs. 4, 5, 6). Consistent with previous studies in WT mice (Wang and Manis, 2008; Cao and Oertel, 2010; Chanda and Xu-Friedman, 2010), EPSC amplitudes decreased over the course of the train indicating short-term depression, which was present at all frequencies tested in both genotypes (Fig. 4).
As expected (Wang and Manis, 2008; Yang and Xu-Friedman, 2009), the decay of amplitudes (normalized to the first response) was well fitted by single exponentials (Fig. 4C; adjusted r2 at 10, 50, 100, and 300 Hz: WT, 0.38, 0.41, 0.61, and 0.85, respectively; KO, 0.61, 0.63, 0.76, and 0.92, respectively), suggesting that release is primarily mediated by a population of vesicles with similar release probability (Thanawala and Regehr, 2016). The high percentage of variance explained by the exponential fits (high adjusted r2) at 100 and 300 Hz is also consistent with theoretical models that predict an exponential decay of responses when depression is caused by vesicle depletion (Hennig, 2013), which is the case of the WT endbulb at these frequencies (Wang et al., 2010; at frequencies <100 Hz other factors than depletion may contribute to depression). Overall, the percentage of explained variance was higher in KO mice, even at frequencies <100 Hz, suggesting that the lack of GluA3 enhanced vesicle depletion. Consistent with this, depression was more pronounced in KO than in WT mice as indicated by significantly lower amplitudes during the final steady-state plateau of depression (gray dashed line in Fig. 4C; steady state, [WT, KO]: 10 Hz, [0.60, 0.52]; 50 Hz, [0.52, 0.42]; 100 Hz, [0.36, 0.27]; 300 Hz, [0.13, 0.10]; WT vs KO, different for all frequencies, 95% CI).
Furthermore, depression was significantly faster at high stimulation frequencies in KO than in WT mice (Figs. 4C, 6A; decay constant, [WT, KO]: 10 Hz, [−0.44, −0.40]; 50 Hz, [−0.34, −0.44]; 100 Hz, [−0.30, −0.60]; 300 Hz, [−0.44, −0.95]; different at 100 Hz and 300 Hz, 95% CI). Finally, whereas the speed of depression was stimulus frequency-invariant in WT mice (blue traces in Figs. 4C, 6A; decay constant, [10 Hz, 50 Hz, 100 Hz, 300 Hz]: [−0.44, −0.34, −0.30, −0.44]; not different between frequencies, 95% CI), in KO mice, the speed of depression increased with increasing stimulation frequency, leading to very fast depression at high frequencies (red traces in Figs. 4C, 6A; decay constant, [10 Hz, 50 Hz, 100 Hz, 300 Hz]: [−0.40, −0.44, −0.60, −0.95]; significantly different between frequencies, 95% CI). To the best of our knowledge, the stimulus frequency-invariant speed of depression in WT endbulb synapses was not mentioned in previous studies, as opposed to the steady-state that is thought to be frequency-dependent at high rates (Wang and Manis, 2008; Cao and Oertel, 2010; Chanda and Xu-Friedman, 2010; >100 Hz). However, these studies (and our study) used a rather small number of stimuli (≤40), and it is possible that steady-state depression is also frequency-invariant for very long stimulus trains similar to what has been observed in other synapses (Yang et al., 2009; Turecek et al., 2017).
While these results support a higher probability of neurotransmitter release and faster vesicle depletion in GluA3 KO mice, they cannot exclude the possibility that differences in AMPAR desensitization also contribute to the differences in depression between genotypes. This possibility may be especially relevant in respect to the speed of depression that is sensitive to receptor desensitization as faster desensitization leads to faster depression (Yang and Xu-Friedman, 2008). To test for this possibility, we compared the speed of depression at high-frequency rates (100 and 300 Hz) with and without blocking desensitization with CTZ (Figs. 5, 6B). Consistent with previous studies, we found that desensitization contributed to the speed of depression but not to the steady-state of depression (Yang and Xu-Friedman, 2008; Cao and Oertel, 2010; Fig. 5). Specifically, CTZ significantly slowed depression at 100 Hz in KO mice (Fig. 5C; decay constant, [CTR, CTZ]: KO, [−0.66, −0.28], different, 95% CI; WT, [−0.27, −0.21], not different, 95% CI), and at 300 Hz in both genotypes (Fig. 5D; decay constant, [CTR, CTZ]: KO, [−0.92, −0.50]; WT, [−0.42, −0.22], all different, 95% CI). In contrast, CTZ did not change the steady-state depression in either genotype and at either frequency (Fig. 5C,D; steady-state, [CTR, CTZ]: WT, 100 Hz [0.35, 0.39], 300 Hz [0.14, 0.12]; KO, 100 Hz [0.23, 0.25], 300 Hz [0.08, 0.08]; not different, 95% CI).
More importantly, differences in AMPAR desensitization cannot fully account for the stronger depression in KO mice. First, when AMPAR desensitization was blocked by CTZ, KO mice still had significantly faster and stronger depression than WT mice (Figs. 5, 6B; decay constant and steady-state; different, 95% CI). Second, CTZ also did not abolish the frequency-dependency of the speed of depression in the KO, which was still significantly faster at 300 than at 100 Hz during CTZ application, indicating that frequency-dependent speed of depression is not entirely mediated by AMPAR desensitization (Figs. 5C, 6B; decay constant, [100 Hz, 300 Hz]: [−0.28, −0.50], different, 95% CI). This is again in sharp contrast to WT mice in which the speed of depression remained frequency-invariant in the presence of CTZ (Figs. 5D, 6B; decay constant, [100 Hz, 300 Hz]: [−0.21, - 0.22], not different, 95% CI).
Together, these data indicate that AMPAR desensitization plays a rather minor role in the depression of synaptic transmission in endbulb synapses in either WT or KO mice and thus does not contribute to the differences in depression between both genotypes. Therefore, both in WT as well as KO mice, depression is primarily due to presynaptic mechanisms of vesicle release (Bellingham and Walmsley, 1999; Taschenberger et al., 2002; Wang and Manis, 2008; Yang and Xu-Friedman, 2008).
Presynaptic changes underlie the enhanced depression in GluA3 KO mice
To test whether alterations in vesicle dynamics and release underlie the enhanced depression of the endbulb synapses in GluA3 KO mice, we used two independent methods to estimate the size of the RRP and the Pr: the train method (Schneggenburger et al., 1999; Thanawala and Regehr, 2013; Lu and Trussell, 2016; Fig. 7A–C; RRPtrain and Prtrain) and the EQ method (Elmqvist and Quastel, 1965; Taschenberger and von Gersdorff, 2000; Lu and Trussell, 2016; Fig. 7D–F; RRPEQ and PrEQ). Both methods provide good estimates of synaptic parameters when applied to highly depressing synapses with high Pr and prominent vesicle depletion (Thanawala and Regehr, 2016; Kaeser and Regehr, 2017), such as the endbulb (Wang and Manis, 2008; Yang and Xu-Friedman, 2008; Ngodup et al., 2015).
Both methods indicate that GluA3 KO mice have a higher Pr and a smaller RRP than WT mice (Fig. 7A–F; 2-way RM ANOVAs/Holm–Sidak comparisons), which can explain the enhanced speed (due to higher Pr) and steady-state level (smaller RRP) of depression in KO mice. However, these estimates are influenced by AMPAR desensitization, which would cause an underestimation of the RRP size that could mask the actual differences between genotypes. To address this issue, we estimated RRP and Pr for responses recorded in the presence of CTZ (Fig. 7G,H). Even under these conditions, GluA3 KO synapses still have smaller RRP and higher Pr (Fig. 7G,H; 2-way RM ANOVAs/Holm–Sidak comparisons). Regarding the differences between methods, in WT mice, RRPEQ is significantly higher than RRPtrain (cyan dots in Fig. 7G), whereas in KO mice, RRPEQ is identical to RRPtrain (orange dots in Fig. 7G). The fact that in WT mice, the RRP obtained by the EQ method is higher than the RRP obtained by the train method (Fig. 7G) likely reflects the different assumptions about replenishment used by the two methods (Thanawala and Regehr, 2013). The train method assumes that the rate of replenishment is constant throughout the train, when in fact replenishment likely is smallest at the beginning of the train and highest at the end. Thus, by determining replenishment at the end the train, the train method overestimates replenishment. Because the RRP obtained by the train method corresponds to the cumulative EPSCs minus the replenished vesicles, the overestimated replenishment causes an underestimation of the real RRP (Thanawala and Regehr, 2013). In contrast, the EQ method assumes complete absence of replenishment at the beginning of the train, when likely there is some replenishment, which results in an overestimation of the RRP (Thanawala and Regehr, 2013). Because replenishment occurs at the WT endbulb synapse (Wang and Manis, 2008; Yang and Xu-Friedman, 2008; Chanda and Xu-Friedman, 2010), our results are in line with the prediction that RRPtrain provides a lower-bound estimate of the RRP while RRPEQ provides an upper-bound estimate of the RRP (Thanawala and Regehr, 2013). Why is the expected difference in RRPtrain and RRPEQ not present in the KO mice? Because the rate of replenishment significantly contributes to the discrepancy between RRPtrain and RRPEQ a lower rate of replenishment in KO mice would decrease this discrepancy. Our data indicate that this is indeed the case, because the slopes of the linear fits obtained with the train method, which correspond to the rate of replenishment, are significantly shallower in KO mice than in WT mice (Fig. 7I; two-tailed unpaired t test). This is also in close accordance with the lower steady-state plateau of depression observed in KO mice compared with WT mice (gray dashed lines in Figs. 4C, 5C,D).
In summary, our electrophysiological experiments support the hypothesis that the presynaptic mechanisms that enhance depression in GluA3 KO mice are caused by an increase in Pr, a decrease in RRP, and a decrease in replenishment (Fig. 7). Furthermore because depression of WT endbulb synapses is reduced during development (Brenowitz and Trussell, 2001), these findings argue in favor of an abnormal synaptic development of GluA3 KO endbulb synapses. Additionally, our findings link the absence of GluA3, a postsynaptic AMPAR subunit, to alterations in the presynaptic terminal, suggesting that GluA3 participates in a retrograde mechanism that influences short-term plasticity in WT endbulb synapses (Petersen et al., 1997; Haghighi et al., 2003; Lindskog et al., 2010; Tracy et al., 2011; Haj-Dahmane et al., 2017). Our results are unexpected because a previous study using GluA3 KO mice report only small changes in synaptic transmission in calyx of Held synapses (Yang et al., 2011) suggesting that GluA4 is the main determinant for fast synaptic transmission in calyx synapses. Thus, it appears that GluA4 and GluA3 have different roles in calyx vs endbulb synapses, although we cannot exclude the possibility that loss of GluA4 would produce strong effects in endbulb synapses as well.
Synaptic vesicle size is smaller in GluA3 KO mice
The smaller quantal size in GluA3 KO mice (Fig. 1A,B) may reflect a smaller vesicle size (Sulzer and Edwards, 2000; Karunanithi et al., 2002). It should be noted that quantal size variation cannot always be predicted by synaptic vesicle size (Taschenberger et al., 2002). Other sources, such as vesicular glutamate concentration as well as the number of AMPARs and their clustering in the postsynaptic density contribute to quantal size variation (Edwards, 2007; Wu et al., 2007). Here, we measured synaptic vesicle size of endbulb synapses at different ages (P20, P44, and P100 postnatal days) using electron microscopy (Fig. 8). In line with their smaller quantal sizes (Fig. 1A,B), KO mice had significantly smaller vesicle size than WT mice at all ages tested (Fig. 8B; 2-Way ANOVA/Holm Sidak comparisons). Furthermore, in WT mice, vesicle size increased with age until it reached maturity at ∼P44 (Fig. 8B,C), but in KO mice vesicle size did not increase with age (Fig. 8B,C). This indicates that GluA3 is necessary for the normal developmental increase in presynaptic vesicle size (Fig. 8B) as well as quantal size (Fig. 1A,B; Bellingham et al., 1998) that occurs in the WT endbulb.
Discussion
In this study, we demonstrate novel roles of the glutamate receptor subunit GluA3 in synaptic transmission and short-term plasticity at endbulb synapses between auditory nerve fibers and BCs in the cochlear nucleus. At the postsynaptic site, GluA3 promotes the insertion of postsynaptic AMPARs that contain flop subunits thereby contributing to the generation of ultrafast glutamatergic currents (Lawrence and Trussell, 2000; Gardner et al., 2001; Pei et al., 2007). Unexpectedly, GluA3 is also required for the normal function and maturation of the presynaptic terminal, whose properties are primarily responsible for shaping short-term plasticity. In GluA3 KO mice, we observed faster and more pronounced short-term depression, which is likely caused by an increased Pr and a decreased size and speed of replenishment of the RRP of synaptic vesicles. Finally, we observed that in WT mice the speed of synaptic depression was rate-invariant, a feature that was lost in KO mice. We propose that the slower speed of depression and its rate-invariance in WT mice circumvents the nonlinearities of synaptic transmission imposed by short-term depression (Tsodyks and Markram, 1997; Abbott and Regehr, 2004). This allows preservation of rate information over a wider range of spike frequencies, aiding the encoding of sound intensity and sound localization in binaural brainstem nuclei, which are the main projection targets of spherical BCs.
GluA3 promotes the insertion of flop subunits in postsynaptic AMPARs
Our results indicate that a lower proportion of flop subunits in AMPARs leads to slower kinetics of glutamatergic synaptic currents at endbulb synapses in GluA3 KO mice. Because flop subunits increase gradually during development to become the predominant isoform in mature synapses (Monyer et al., 1991; Lawrence and Trussell, 2000; Gardner et al., 2001; Sugden et al., 2002; Pei et al., 2007), it is likely that our results from young adult KO mice reflect impaired synaptic development. The mechanisms that regulate the developmental increase of flop subunits are largely unknown. It has been proposed that in endbulb synapses flop subunit insertion is influenced by signals released from developing endbulb terminals, because nucleus magnocellularis neurons (avian homologues of spherical BCs) grown in cell culture without endbulb synapses fail to transition to flop-dominated AMPARs (Lawrence and Trussell, 2000). Results from the present study point to a critical role of GluA3 in mediating the insertion of flop-containing AMPAs in BCs because flop-containing AMPAs were significantly reduced in GluA3 KO mice (Fig. 3). Because in BCs, GluA1 and GluA2 subunits are not or only weakly expressed, it is likely that in KO mice, GluA3 is replaced by GluA4, which shares with GluA3 the fast kinetics and high expressions levels in BCs (Wang and Kaczmarek, 1998; Petralia et al., 2000; Gardner et al., 2001; Schmid et al., 2001). In this scenario, AMPARs in BC of KO mice are predominantly GluA4 homomers, which can only be trafficked to the membrane as GluA4 flip subunits (Rossmann et al., 2011; Coleman et al., 2016; GluA4 flop homomers are retained at the endoplasmic reticulum). Thus, we propose that lack of GluA3 and loss of the ability to enable trafficking of GluA4 flop subunits as heterodimeric GluA3/GluA4 flop receptors in WT animals (GluA3 is an obligatory heteromer; Coleman et al., 2016) leads to the expression of homomeric GluA4/GluA4 flip receptors in mutant mice. Although a previous study using freeze-fracture immunogold labeling failed to observe an increase in GluA4 subunit in GluA3 KO mice (Rubio et al., 2017), such an increase may have remained undetected due to technical limitations. Freeze-fracture can only detect one subunit per AMPAR (Tanaka et al., 2005) and thus will only detect one GluA4 subunit per GluA4-containing AMPAR. This limitation would leave additional GluA4 subunits associated with a switch from GluA3/GluA4flops to GluA4/GluA4flips undetected. It is also possible that GluA2 subunits are increased in KO mice, which would result in a reduction in the AMPAR-mediated postsynaptic Ca2+influx. Although the present study did not address this, it has been shown that Ca2+ influx via AMPARs play an important role in the maturation of calyx synapses (Lujan et al., 2019). If this scenario also applies to endbulb synapses, a decrease in calcium influx could also have contributed to the delayed synaptic development in KO mice.
GluA3 is required for the functional development of the presynaptic terminal
GluA3 KO mice exhibited profound and unexpected changes in the physiological characteristics of the presynaptic endbulb, including smaller vesicle content as indicated by reduced quantal size and vesicle diameter (Figs. 1, 8), reduced RRP (Fig. 7), and higher Pr (Fig. 7). These properties in young adult KO mice resemble the properties of endbulbs in immature WT animals (Bellingham et al., 1998; Brenowitz and Trussell, 2001; McKay and Oleskevich, 2007), again suggesting that a lack of GluA3 leads to a delayed or an arrested synaptic development. Because GluA3 is also expressed in the hair cell to spiral ganglion cell synapse (Safieddine and Eybalin, 1992; Knipper et al., 1997; Sebe et al., 2017), it is possible that the observed changes reflect developmental impairments originating in spiral ganglion neurons and perhaps arise from abnormal afferent activity. One possibility is that excitability of the nerve terminal in KO mice has a more “immature phenotype”. For example, the increased Pr in KO mice (Fig. 7) could partly be caused by an immature, broader action potential waveform (Kandler and Friauf, 1995; Taschenberger and von Gersdorff, 2000), which could lead to a larger presynaptic Ca2+ influx (Taschenberger and von Gersdorff, 2000). Consistent with this, auditory brainstem responses in adult KO mice, indicate impaired transmission already in the auditory nerve (García-Hernández et al., 2017), although it remains to be shown when exactly these changes appear during development.
Another interesting possibility is that AMPARs in BCs participate in a retrograde homeostatic signaling mechanism that adjusts glutamate release in endbulb terminals (Klug et al., 2012; Yang and Xu-Friedman, 2012), as demonstrated for postsynaptic AMPARs and other glutamatergic receptors at other synapses (Petersen et al., 1997; Haghighi et al., 2003; Lindskog et al., 2010; Tracy et al., 2011; Haj-Dahmane et al., 2017). Such homeostatic mechanism can compensate for reduced AMPAR activation by increasing presynaptic glutamate release (Petersen et al., 1997; Haghighi et al., 2003; Lindskog et al., 2010), which can be triggered by an increase in presynaptic Ca2+ current (Delvendahl et al., 2019). Because the number of synaptic AMPARs is reduced in GluA3 KO mice (Rubio et al., 2017), the increased Pr in KO mice (Fig. 7) could reflect a form of retrograde homeostatic compensation in endbulb synapses that leads to a recovery of synaptic strength (Figs. 2, 7). Retrograde signaling is thought to underlie the “coordinated plasticity” that coregulates Pr (and short-term plasticity) of different endbulbs that converge on the same BC (Yang and Xu-Friedman, 2009, 2012). Because the similarity between converging endbulbs initiates shortly after the onset of hearing, activity-dependent homeostatic mechanisms may underlie this 'coordinated plasticity' (Klug et al., 2012; Yang and Xu-Friedman, 2012). Postsynaptic regulation of presynaptic Pr has been found in numerous brain regions suggesting that it is a general mechanism regulating short-term plasticity in the brain (Neocortex: Markram et al., 1998; Cerebellum and cochlear nucleus: Yang and Xu-Friedman, 2012; Avian cochlear nucleus: Ahn and MacLeod, 2016; Cerebellum: Delvendahl et al., 2019). Finally, a reduced number of AMPARs in KO mice was correlated with a smaller RRP (Fig. 7), similarly to what has been described previously in developing hippocampal neurons (Tracy et al., 2011), indicating that postsynaptic AMPARs influence the presynaptic release machinery at functionally very diverse synapses in the brain.
GluA3 is required for slower and rate-invariant speed of synaptic depression
The increased Pr in KO mice strongly accelerated short-term depression during high-frequency train stimulation (Abbott et al., 1997; Abbott and Regehr, 2004). In addition, whereas in WT mice the speed of synaptic depression was rate-invariant, in KO mice the speed of depression increased with the stimulation rate (Figs. 4, 5, 6). Our recordings were made in the most anterior region of the VCN, which primarily contains small spherical BCs in mice (Willard and Ryugo, 1983; Wright et al., 2014). In mice, a high-frequency hearing animal, an important function of BCs is to encode sound intensity as a rate code, which plays an important role to compute interaural sound level differences in the major target nucleus of BCs, the lateral superior olive (Sanes and Rubel, 1988; Cant, 1991; Smith et al., 1993; Doucet and Ryugo, 2003; Tollin, 2003; Gómez-Álvarez and Saldaña, 2016). Because synaptic depression at endbud synapses quickly leads to high failure rates in eliciting postsynaptic spikes, it is still unknown how BCs accurately transmit rate information to the lateral superior olive (Yang and Xu-Friedman, 2009; Wang et al., 2010). It should be noted, however, that the external calcium concentration of 2 mm commonly used in slices and in our experiments likely is higher than that in vivo (∼1.2 mm, Borst, 2010), which would result in a higher Pr and an overestimation of depression in slice recordings. Because synaptic depression varies between individual BCs in vitro it has been proposed that less-depressing BCs are suitable for rate coding of sound intensities (Yang and Xu-Friedman, 2009). However, even strongly depressing synapses can transmit rate information, for as long as the speed of depression is slow enough to allow computation of spike rates during the initial (transient) response window (Tsodyks and Markram, 1997). If this also holds true for BCs, the faster depression speed in BCs of KO mice (Figs. 4, 5, 6) may interfere with rate coding of sound intensities.
We also observed that the speed of depression in WT mice was rate invariant over a stimulus frequency range of 10–300 Hz (Fig. 6), indicating that the transient response needed for rate coding is preserved over a range of firing rates that naturally occur in the auditory nerve in vivo (Joris et al., 1994; Taberner and Liberman, 2005). We propose that the rate-invariance of depression speed in WT mice (Fig. 6) extends the dynamic range of rate encoding of BCs to allow rate-invariant transmission, which is a fundamental property of synapses that transmit information via rate codes (MacLeod et al., 2007; Bagnall et al., 2008; McElvain et al., 2015). We suggest that in endbulb synapses, rate-invariant speed of depression is likely achieved by their large RRP and by the activity-dependent rate of replenishment both of which counteract vesicle depletion (Wang and Kaczmarek, 1998; Wang and Manis, 2008; Yang and Xu-Friedman, 2008). This is also supported by our finding that the faster and rate-dependent speed of depression in GluA3 KO mice correlated with slower replenishment (Figs. 6, 7). By adjusting replenishment in an activity-dependent manner, the endbulb synapse can achieve rate-invariant transmission, similarly to what occurs at synapses in the avian intensity pathway (MacLeod et al., 2007) and other nonauditory pathways (Turecek et al., 2016, 2017) where facilitation occurs together with depression to produce rate-invariant transmission.
Footnotes
This work was supported by the National Institute on Deafness and Other Communication Disorders (Grant DC013048 to M.E.R.). We thank Dr. Tzounopoulos and members of his laboratory for help in the initial stages of the project and the members of the Kandler and Sadagopan, laboratories for helpful discussions. GluA3 KO mice were kindly provided by Dr. Sakimura. We thank Lester Torres, Hou-Ming Cai, and Brian Brockway for technical assistance.
The authors declare no competing financial interests.
References
- Abbott LF, Varela JA, Sen K, Nelson SB (1997) Synaptic depression and cortical gain control. Science 275:220–224. 10.1126/science.275.5297.221 [DOI] [PubMed] [Google Scholar]
- Abbott LF, Regehr WG (2004) Synaptic computation. Nature 431:796–803. 10.1038/nature03010 [DOI] [PubMed] [Google Scholar]
- Ahn J, MacLeod KM (2016) Target-specific regulation of presynaptic release properties at auditory nerve terminals in the avian cochlear nucleus. J Neurophysiol 115:1679–1690. 10.1152/jn.00752.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allam SL, Bouteiller JM, Hu EY, Ambert N, Greget R, Bischoff S, Baudry M, Berger TW (2015) Synaptic efficacy as a function of ionotropic receptor distribution: a computational study. PLoS One 10:e0140333. 10.1371/journal.pone.0140333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagnall MW, McElvain LE, Faulstich M, du Lac S (2008) Frequency-independent synaptic transmission supports a linear vestibular behavior. Neuron 60:343–352. 10.1016/j.neuron.2008.10.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellingham MC, Walmsley B (1999) A novel presynaptic inhibitory mechanism underlies paired pulse depression at a fast central synapse. Neuron 23:159–170. 10.1016/S0896-6273(00)80762-X [DOI] [PubMed] [Google Scholar]
- Bellingham MC, Lim R, Walmsley B (1998) Developmental changes in EPSC quantal size and quantal content at a central glutamatergic synapse in rat. J Physiol 511:861–869. 10.1111/j.1469-7793.1998.861bg.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borst JG. (2010) The low synaptic release probability in vivo. Trends Neurosci 33:259–266. 10.1016/j.tins.2010.03.003 [DOI] [PubMed] [Google Scholar]
- Brenowitz S, Trussell LO (2001) Maturation of synaptic transmission at end-bulb synapses of the cochlear nucleus. J Neurosci 21:9487–9498. 10.1523/JNEUROSCI.21-23-09487.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cant NB. (1991) Projections to the lateral and medial superior olivary nuclei from the spherical and globular bushy cells of the anteroventral cochlear nucleus. In: Neurobiology of hearing: the central auditory system (Altschuler RA, Hoffman DW, Bobbin RB, Clopton B, eds), pp 99–119. New York: Raven. [Google Scholar]
- Cao XJ, Oertel D (2010) Auditory nerve fibers excite targets through synapses that vary in convergence, strength, and short-term plasticity. J Neurophysiol 104:2308–2320. 10.1152/jn.00451.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao XJ, Shatadal S, Oertel D (2007) Voltage-sensitive conductances of bushy cells of the mammalian ventral cochlear nucleus. J Neurophysiol 97:3961–3975. 10.1152/jn.00052.2007 [DOI] [PubMed] [Google Scholar]
- Cathala L, Holderith NB, Nusser Z, DiGregorio DA, Cull-Candy SG (2005) Changes in synaptic structure underlie the developmental speeding of AMPA receptor-mediated EPSCs. Nat Neurosci 8:1310–1318. 10.1038/nn1534 [DOI] [PubMed] [Google Scholar]
- Chanda S, Xu-Friedman MA (2010) A low-affinity antagonist reveals saturation and desensitization in mature synapses in the auditory brain stem. J Neurophysiol 103:1915–1926. 10.1152/jn.00751.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarkson C, Antunes FM, Rubio ME (2016) Conductive hearing loss has long-lasting structural and molecular effects on presynaptic and postsynaptic structures of auditory nerve synapses in the cochlear nucleus. J Neurosci 36:10214–10227. 10.1523/JNEUROSCI.0226-16.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coleman SK, Hou Y, Willibald M, Semenov A, Möykkynen T, Keinänen K (2016) Aggregation limits surface expression of homomeric GluA3 receptors. J Biol Chem 291:8784–8794. 10.1074/jbc.M115.689125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delvendahl I, Kita K, Müller M (2019) Rapid and sustained homeostatic control of presynaptic exocytosis at a central synapse. Proc Natl Acad Sci U S A 116:23783–23789. 10.1073/pnas.1909675116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doucet JR, Ryugo DK (2003) Axonal pathways to the lateral superior olive labeled with biotinylated dextran amine injections in the dorsal cochlear nucleus of rats. J Comp Neurol 461:452–465. 10.1002/cne.10722 [DOI] [PubMed] [Google Scholar]
- Edwards RH. (2007) The neurotransmitter cycle and quantal size. Neuron 55:835–858. 10.1016/j.neuron.2007.09.001 [DOI] [PubMed] [Google Scholar]
- Elmqvist D, Quastel DM (1965) A quantitative study of end-plate potentials in isolated human muscle. J Physiol 178:505–529. 10.1113/jphysiol.1965.sp007639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleck MW, Bähring R, Patneau DK, Mayer ML (1996) AMPA receptor heterogeneity in rat hippocampal neurons revealed by differential sensitivity to cyclothiazide. J Neurophysiol 75:2322–2333. 10.1152/jn.1996.75.6.2322 [DOI] [PubMed] [Google Scholar]
- Freche D, Pannasch U, Rouach N, Holcman D (2011) Synapse geometry and receptor dynamics modulate synaptic strength. PLoS One 6:e25122. 10.1371/journal.pone.0025122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- García-Hernández S, Abe M, Sakimura K, Rubio ME (2017) Impaired auditory processing and altered structure of the endbulb of Held synapse in mice lacking the GluA3 subunit of AMPA receptors. Hear Res 344:284–294. 10.1016/j.heares.2016.12.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardner SM, Trussell LO, Oertel D (2001) Correlation of AMPA receptor subunit composition with synaptic input in the mammalian cochlear nuclei. J Neurosci 21:7428–7437. 10.1523/JNEUROSCI.21-18-07428.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geiger JR, Melcher T, Koh DS, Sakmann B, Seeburg PH, Jonas P, Monyer H (1995) Relative abundance of subunit mRNAs determines gating and Ca2+ permeability of AMPA receptors in principal neurons and interneurons in rat CNS. Neuron 15:193–204. 10.1016/0896-6273(95)90076-4 [DOI] [PubMed] [Google Scholar]
- Gómez-Álvarez M, Saldaña E (2016) Different tonotopic regions of the lateral superior olive receive a similar combination of afferent inputs. J Comp Neurol 524:2230–2250. 10.1002/cne.23942 [DOI] [PubMed] [Google Scholar]
- Haghighi AP, McCabe BD, Fetter RD, Palmer JE, Hom S, Goodman CS (2003) Retrograde control of synaptic transmission by postsynaptic CaMKII at the drosophila neuromuscular junction. Neuron 39:255–267. 10.1016/S0896-6273(03)00427-6 [DOI] [PubMed] [Google Scholar]
- Haj-Dahmane S, Béïque JC, Shen RY (2017) Glua2-lacking AMPA receptors and nitric oxide signaling gate spike-timing-dependent potentiation of glutamate synapses in the dorsal raphe nucleus. eNeuro 4ENEURO.0116–17.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hennig MH. (2013) Theoretical models of synaptic short term plasticity. Front Comput Neurosci 7:45. 10.3389/fncom.2013.00045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isaacson JS, Walmsley B (1996) Amplitude and time course of spontaneous and evoked excitatory postsynaptic currents in bushy cells of the anteroventral cochlear nucleus. J Neurophysiol 76:1566–1571. 10.1152/jn.1996.76.3.1566 [DOI] [PubMed] [Google Scholar]
- Joris PX, Carney LH, Smith PH, Yin TC (1994) Enhancement of neural synchronization in the anteroventral cochlear nucleus. I. Responses to tones at the characteristic frequency. J Neurophysiol 71:1022–1036. 10.1152/jn.1994.71.3.1022 [DOI] [PubMed] [Google Scholar]
- Kaeser PS, Regehr WG (2017) The readily releasable pool of synaptic vesicles. Curr Opin Neurobiol 43:63–70. 10.1016/j.conb.2016.12.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kandler K, Friauf E (1995) Development of electrical membrane properties and discharge characteristics of superior olivary complex neurons in fetal and postnatal rats. Eur J Neurosci 7:1773–1790. 10.1111/j.1460-9568.1995.tb00697.x [DOI] [PubMed] [Google Scholar]
- Karunanithi S, Marin L, Wong K, Atwood HL (2002) Quantal size and variation determined by vesicle size in normal and mutant Drosophila glutamatergic synapses. J Neurosci 22:10267–10276. 10.1523/JNEUROSCI.22-23-10267.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kessler M, Rogers G, Arai A (2000) The norbornenyl moiety of cyclothiazide determines the preference for flip-flop variants of AMPA receptor subunits. Neurosci Lett 287:161–165. 10.1016/S0304-3940(00)01180-0 [DOI] [PubMed] [Google Scholar]
- Klug A, Borst JG, Carlson BA, Kopp-Scheinpflug C, Klyachko VA, Xu-Friedman MA (2012) How do short-term changes at synapses fine-tune information processing? J Neurosci 32:14058–14063. 10.1523/JNEUROSCI.3348-12.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knipper M, Köpschall I, Rohbock K, Köpke AK, Bonk I, Zimmermann U, Zenner H (1997) Transient expression of NMDA receptors during rearrangement of AMPA-receptor-expressing fibers in the developing inner ear. Cell Tissue Res 287:23–41. 10.1007/s004410050729 [DOI] [PubMed] [Google Scholar]
- Koike M, Tsukada S, Tsuzuki K, Kijima H, Ozawa S (2000) Regulation of kinetic properties of GluR2 AMPA receptor channels by alternative splicing. J Neurosci 20:2166–2174. 10.1523/JNEUROSCI.20-06-02166.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence JJ, Trussell LO (2000) Long-term specification of AMPA receptor properties after synapse formation. J Neurosci 20:4864–4870. 10.1523/JNEUROSCI.20-13-04864.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindner B, Gangloff D, Longtin A, Lewis JE (2009) Broadband coding with dynamic synapses. J Neurosci 29:2076–2088. 10.1523/JNEUROSCI.3702-08.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindskog M, Li L, Groth RD, Poburko D, Thiagarajan TC, Han X, Tsien RW (2010) Postsynaptic GluA1 enables acute retrograde enhancement of presynaptic function to coordinate adaptation to synaptic inactivity. Proc Natl Acad Sci U S A 107:21806–21811. 10.1073/pnas.1016399107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu HW, Trussell LO (2016) Spontaneous Activity Defines Effective Convergence Ratios in an Inhibitory Circuit. J Neurosci 36:3268–3280. 10.1523/JNEUROSCI.3499-15.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Y, Harris JA, Rubel EW (2007) Development of spontaneous miniature EPSCs in mouse AVCN neurons during a critical period of afferent-dependent neuron survival. J Neurophysiol 97:635–646. 10.1152/jn.00915.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lujan B, Dagostin A, von Gersdorff H (2019) Presynaptic diversity revealed by Ca2+-permeable AMPA receptors at the calyx of Held synapse. J Neurosci 39:2981–2994. 10.1523/JNEUROSCI.2565-18.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacLeod KM, Horiuchi TK, Carr CE (2007) A role for short-term synaptic facilitation and depression in the processing of intensity information in the auditory brain stem. J Neurophysiol 97:2863–2874. 10.1152/jn.01030.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markram H, Wang Y, Tsodyks M (1998) Differential signaling via the same axon of neocortical pyramidal neurons. Proc Natl Acad Sci U S A 95:5323–5328. 10.1073/pnas.95.9.5323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McElvain LE, Faulstich M, Jeanne JM, Moore JD, du Lac S (2015) Implementation of linear sensory signaling via multiple coordinated mechanisms at central vestibular nerve synapses. Neuron 85:1132–1144. 10.1016/j.neuron.2015.01.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKay SM, Oleskevich S (2007) The role of spontaneous activity in development of the endbulb of Held synapse. Hear Res 230:53–63. 10.1016/j.heares.2007.05.006 [DOI] [PubMed] [Google Scholar]
- Monyer H, Seeburg PH, Wisden W (1991) Glutamate-operated channels: Developmentally early and mature forms arise by alternative splicing. Neuron 6:799–810. 10.1016/0896-6273(91)90176-Z [DOI] [PubMed] [Google Scholar]
- Mosbacher J, Schoepfer R, Monyer H, Burnashev N, Seeburg PH, Ruppersberg JP (1994) A molecular determinant for submillisecond desensitization in glutamate receptors. Science 266:1059–1062. 10.1126/science.7973663 [DOI] [PubMed] [Google Scholar]
- Ngodup T, Goetz JA, McGuire BC, Sun W, Lauer AM, Xu-Friedman MA (2015) Activity-dependent, homeostatic regulation of neurotransmitter release from auditory nerve fibers. Proc Natl Acad Sci U S A 112:6479–6484. 10.1073/pnas.1420885112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oertel D. (1983) Synaptic responses and electrical properties of cells in brain slices of the mouse anteroventral cochlear nucleus. J Neurosci 3:2043–2053. 10.1523/JNEUROSCI.03-10-02043.1983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oleskevich S, Clements J, Walmsley B (2000) Release probability modulates short-term plasticity at a rat giant terminal. J Physiol 524:513–523. 10.1111/j.1469-7793.2000.00513.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oleskevich S, Youssoufian M, Walmsley B (2004) Presynaptic plasticity at two giant auditory synapses in normal and deaf mice. J Physiol 560:709–719. 10.1113/jphysiol.2004.066662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Partin KM, Patneau DK, Mayer ML (1994) Cyclothiazide differentially modulates desensitization of alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor splice variants. Mol Pharmacol 46:129–138. [PubMed] [Google Scholar]
- Pei W, Huang Z, Niu L (2007) GluR3 flip and flop: differences in channel opening kinetics. Biochemistry 46:2027–2036. 10.1021/bi062213s [DOI] [PubMed] [Google Scholar]
- Petersen SA, Fetter RD, Noordermeer JN, Goodman CS, DiAntonio A (1997) Genetic analysis of glutamate receptors in Drosophila reveals a retrograde signal regulating presynaptic transmitter release. Neuron 19:1237–1248. 10.1016/S0896-6273(00)80415-8 [DOI] [PubMed] [Google Scholar]
- Petralia RS, Rubio ME, Wang YX, Wenthold RJ (2000) Differential distribution of glutamate receptors in the cochlear nuclei. Hear Res 147:59–69. 10.1016/S0378-5955(00)00120-9 [DOI] [PubMed] [Google Scholar]
- Quirk JC, Siuda ER, Nisenbaum ES (2004) Molecular determinants responsible for differences in desensitization kinetics of AMPA receptor splice variants. J Neurosci 24:11416–11420. 10.1523/JNEUROSCI.2464-04.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raman IM, Trussell LO (1992) The kinetics of the response to glutamate and kainate in neurons of the avian cochlear nucleus. Neuron 9:173–186. 10.1016/0896-6273(92)90232-3 [DOI] [PubMed] [Google Scholar]
- Regehr WG. (2012) Short-term presynaptic plasticity. Cold Spring Harb Perspect Biol 4:a005702. 10.1101/cshperspect.a005702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossmann M, Sukumaran M, Penn AC, Veprintsev DB, Babu MM, Greger IH (2011) Subunit-selective N-terminal domain associations organize the formation of AMPA receptor heteromers. EMBO J 30:959–971. 10.1038/emboj.2011.16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubio ME, Matsui K, Fukazawa Y, Kamasawa N, Harada H, Itakura M, Molnár E, Abe M, Sakimura K, Shigemoto R (2017) The number and distribution of AMPA receptor channels containing fast kinetic GluA3 and GluA4 subunits at auditory nerve synapses depend on the target cells. Brain Struct Funct 222:3375–3393. 10.1007/s00429-017-1408-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Safieddine S, Eybalin M (1992) Co-expression of NMDA and AMPA/kainate receptor mRNAs in cochlear neurones. Neuroreport 3:1145–1148. 10.1097/00001756-199212000-00029 [DOI] [PubMed] [Google Scholar]
- Sanes DH, Rubel EW (1988) The ontogeny of inhibition and excitation in the gerbil lateral superior olive. J Neurosci 8:682–700. 10.1523/JNEUROSCI.08-02-00682.1988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmid S, Guthmann A, Ruppersberg JP, Herbert H (2001) Expression of AMPA receptor subunit flip/flop splice variants in the rat auditory brainstem and inferior colliculus. J Comp Neurol 430:160–171. [DOI] [PubMed] [Google Scholar]
- Schneggenburger R, Meyer AC, Neher E (1999) Released fraction and total size of a pool of immediately available transmitter quanta at a calyx synapse. Neuron 23:399–409. 10.1016/S0896-6273(00)80789-8 [DOI] [PubMed] [Google Scholar]
- Sebe JY, Cho S, Sheets L, Rutherford MA, von Gersdorff H, Raible DW (2017) Ca2+-permeable AMPARs mediate glutamatergic transmission and excitotoxic damage at the hair cell ribbon synapse. J Neurosci 37:6162–6175. 10.1523/JNEUROSCI.3644-16.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith PH, Joris PX, Yin TC (1993) Projections of physiologically characterized spherical bushy cell axons from the cochlear nucleus of the cat: evidence for delay lines to the medial superior olive. J Comp Neurol 331:245–260. 10.1002/cne.903310208 [DOI] [PubMed] [Google Scholar]
- Sommer B, Keinänen K, Verdoorn TA, Wisden W, Burnashev N, Herb A, Köhler M, Takagi T, Sakmann B, Seeburg PH (1990) Flip and flop: a cell-specific functional switch in glutamate-operated channels of the CNS. Science 249:1580–1585. 10.1126/science.1699275 [DOI] [PubMed] [Google Scholar]
- Sugden SG, Zirpel L, Dietrich CJ, Parks TN (2002) Development of the specialized AMPA receptors of auditory neurons. J Neurobiol 52:189–202. 10.1002/neu.10078 [DOI] [PubMed] [Google Scholar]
- Sulzer D, Edwards R (2000) Vesicles: equal in neurotransmitter concentration but not in volume. Neuron 28:5–7. 10.1016/s0896-6273(00)00077-5 [DOI] [PubMed] [Google Scholar]
- Taberner AM, Liberman MC (2005) Response properties of single auditory nerve fibers in the mouse. J Neurophysiol 93:557–569. 10.1152/jn.00574.2004 [DOI] [PubMed] [Google Scholar]
- Tanaka JI, Matsuzaki M, Tarusawa E, Momiyama A, Molnar E, Kasai H, Shigemoto R (2005) Number and density of AMPA receptors in single synapses in immature cerebellum. J Neurosci 25:799–807. 10.1523/JNEUROSCI.4256-04.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taschenberger H, von Gersdorff H (2000) Fine-tuning an auditory synapse for speed and fidelity: developmental changes in presynaptic waveform, EPSC kinetics, and synaptic plasticity. J Neurosci 20:9162–9173. 10.1523/JNEUROSCI.20-24-09162.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taschenberger H, Leão RM, Rowland KC, Spirou GA, von Gersdorff H (2002) Optimizing synaptic architecture and efficiency for high-frequency transmission. Neuron 36:1127–1143. 10.1016/S0896-6273(02)01137-6 [DOI] [PubMed] [Google Scholar]
- Thanawala MS, Regehr WG (2013) Presynaptic calcium influx controls neurotransmitter release in part by regulating the effective size of the readily releasable pool. J Neurosci 33:4625–4633. 10.1523/JNEUROSCI.4031-12.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thanawala MS, Regehr WG (2016) Determining synaptic parameters using high-frequency activation. J Neurosci Methods 264:136–152. 10.1016/j.jneumeth.2016.02.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tolbert LP, Morest DK (1982) The neuronal architecture of the anteroventral cochlear nucleus of the cat in the region of the cochlear nerve root: Golgi and Nissl methods. Neuroscience 7:3013–3030. 10.1016/0306-4522(82)90227-5 [DOI] [PubMed] [Google Scholar]
- Tollin DJ. (2003) The lateral superior olive: a functional role in sound source localization. Neuroscientist 9:127–143. 10.1177/1073858403252228 [DOI] [PubMed] [Google Scholar]
- Tracy TE, Yan JJ, Chen L (2011) Acute knockdown of AMPA receptors reveals a trans-synaptic signal for presynaptic maturation. EMBO J 30:1577–1592. 10.1038/emboj.2011.59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trussell LO, Fischbach GD (1989) Glutamate receptor desensitization and its role in synaptic transmission. Neuron 3:209–218. 10.1016/0896-6273(89)90034-2 [DOI] [PubMed] [Google Scholar]
- Tsodyks MV, Markram H (1997) The neural code between neocortical pyramidal neurons depends on neurotransmitter release probability. Proc Natl Acad Sci U S A 94:719–723. 10.1073/pnas.94.2.719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turecek J, Jackman SL, Regehr WG (2016) Synaptic specializations support frequency-independent Purkinje cell output from the cerebellar cortex. Cell Rep 17:3256–3268. 10.1016/j.celrep.2016.11.081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turecek J, Jackman SL, Regehr WG (2017) Synaptotagmin 7 confers frequency invariance onto specialized depressing synapses. Nature 551:503–506. 10.1038/nature24474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Yin G, Rogers K, Miralles C, De Blas AL, Rubio ME (2011) Monaural conductive hearing loss alters the expression of the GluA3 AMPA and glycine receptor α1 subunits in bushy and fusiform cells of the cochlear nucleus. Neuroscience 199:438–451. 10.1016/j.neuroscience.2011.10.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang LY, Kaczmarek LK (1998) High-frequency firing helps replenish the readily releasable pool of synaptic vesicles. Nature 394:384–388. 10.1038/28645 [DOI] [PubMed] [Google Scholar]
- Wang Y, Manis PB (2006) Temporal coding by cochlear nucleus bushy cells in DBA/2J mice with early onset hearing loss. J Assoc Res Otolaryngol 7:412–424. 10.1007/s10162-006-0052-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Manis PB (2008) Short-term synaptic depression and recovery at the mature mammalian endbulb of held synapse in mice. J Neurophysiol 100:1255–1264. 10.1152/jn.90715.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Ren C, Manis PB (2010) Endbulb synaptic depression within the range of presynaptic spontaneous firing and its impact on the firing reliability of cochlear nucleus bushy neurons. Hear Res 270:101–109. 10.1016/j.heares.2010.09.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang YX, Wenthold RJ, Ottersen OP, Petralia RS (1998) Endbulb synapses in the anteroventral cochlear nucleus express a specific subset of AMPA-type glutamate receptor subunits. J Neurosci 18:1148–1160. 10.1523/JNEUROSCI.18-03-01148.1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weisz CJ, Rubio ME, Givens RS, Kandler K (2016) Excitation by axon terminal GABA spillover in a sound localization circuit. J Neurosci 36:911–925. 10.1523/JNEUROSCI.1132-15.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whiting B, Moiseff A, Rubio ME (2009) Cochlear nucleus neurons redistribute synaptic AMPA and glycine receptors in response to monaural conductive hearing loss. Neuroscience 163:1264–1276. 10.1016/j.neuroscience.2009.07.049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willard FH, Ryugo DK (1983) Anatomy of the central auditory system. In: The auditory psychobiology of the mouse (Willott JF, ed), pp 201–304. Springfield, IL: Thomas. [Google Scholar]
- Wright S, Hwang Y, Oertel D (2014) Synaptic transmission between end bulbs of Held and bushy cells in the cochlear nucleus of mice with a mutation in Otoferlin. J Neurophysiol 112:3173–3188. 10.1152/jn.00522.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu LG, Borst JG (1999) The reduced release probability of releasable vesicles during recovery from short-term synaptic depression. Neuron 23:821–832. 10.1016/S0896-6273(01)80039-8 [DOI] [PubMed] [Google Scholar]
- Wu SH, Oertel D (1987) Maturation of synapses and electrical properties of cells in the cochlear nuclei. Hear Res 30:99–110. 10.1016/0378-5955(87)90187-0 [DOI] [PubMed] [Google Scholar]
- Wu XS, Xue L, Mohan R, Paradiso K, Gillis KD, Wu LG (2007) The origin of quantal size variation: vesicular glutamate concentration plays a significant role. J Neurosci 27:3046–3056. 10.1523/JNEUROSCI.4415-06.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H, Xu-Friedman MA (2008) Relative roles of different mechanisms of depression at the mouse endbulb of Held. J Neurophysiol 99:2510–2521. 10.1152/jn.01293.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H, Xu-Friedman MA (2009) Impact of synaptic depression on spike timing at the endbulb of Held. J Neurophysiol 102:1699–1710. 10.1152/jn.00072.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H, Xu-Friedman MA (2012) Emergence of coordinated plasticity in the cochlear nucleus and cerebellum. J Neurosci 32:7862–7868. 10.1523/JNEUROSCI.0167-12.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H, Xu-Friedman MA (2015) Skipped-stimulus approach reveals that short-term plasticity dominates synaptic strength during ongoing activity. J Neurosci 35:8297–8307. 10.1523/JNEUROSCI.4299-14.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang YM, Aitoubah J, Lauer AM, Nuriya M, Takamiya K, Jia Z, May BJ, Huganir RL, Wang LY (2011) GluA4 is indispensable for driving fast neurotransmission across a high-fidelity central synapse. J Physiol 589:4209–4227. 10.1113/jphysiol.2011.208066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z, Hennig MH, Postlethwaite M, Forsythe ID, Graham BP (2009) Wide-band information transmission at the calyx of Held. Neural Comput 21:991–1017. 10.1162/neco.2008.02-08-714 [DOI] [PubMed] [Google Scholar]
- Zucker RS, Regehr WG (2002) Short-term synaptic plasticity. Annu Rev Physiol 64:355–405. 10.1146/annurev.physiol.64.092501.114547 [DOI] [PubMed] [Google Scholar]