

Abstract
α2δ proteins (Cacna2d1-4) are auxiliary subunits of voltage-dependent calcium channels that also drive synapse formation and maturation. Because cerebellar Purkinje cells (PCs) predominantly, if not exclusively, express one isoform of this family, α2δ-2 (Cacna2d2), we used PCs as a model system to examine roles of α2δ in excitatory synaptic function in male and female Cacna2d2 knock-out (KO) mice. Whole-cell recordings of PCs from acute cerebellar slices revealed altered climbing fiber (CF)-evoked complex spike generation, as well as increased amplitude and faster decay of CF-evoked EPSCs. CF terminals in the KO were localized more proximally on PC dendrites, as indicated by VGLUT2+ immunoreactive puncta, and computational modeling demonstrated that the increased EPSC amplitude can be partly attributed to the more proximal location of CF terminals. In addition, CFs in KO mice exhibited increased multivesicular transmission, corresponding to greater sustained responses during repetitive stimulation, despite a reduction in the measured probability of release. Electron microscopy demonstrated that mutant CF terminals had twice as many vesicle release sites, providing a morphologic explanation for the enhanced glutamate release. Though KO CFs evoked larger amplitude EPSCs, the charge transfer was the same as wild-type as a result of increased glutamate reuptake, producing faster decay kinetics. Together, the larger, faster EPSCs in the KO explain the altered complex spike responses, which degrade information transfer from PCs and likely contribute to ataxia in Cacna2d2 KO mice. Our results also illustrate the multidimensional synaptic roles of α2δ proteins.
SIGNIFICANCE STATEMENT α2δ proteins (Cacna2d1-4) regulate synaptic transmission and synaptogenesis, but coexpression of multiple α2δ isoforms has obscured a clear understanding of how various α2δ proteins control synaptic function. We focused on roles of the α2δ-2 protein (Cacna2d2), the deletion of which causes cerebellar ataxia and epilepsy in mice and humans. Because cerebellar Purkinje cells (PCs) only express this single isoform, we studied excitatory climbing fiber synaptic function onto PCs in Cacna2d2 KO mice. Using optical and electrophysiological analysis, we provide a detailed description of the changes in PCs lacking α2δ-2, and provide a comprehensive mechanistic explanation for how functional synaptic phenotypes contribute to the altered cerebellar output.
Keywords: alpha2delta proteins, CACNA2D2, calcium channel, climbing fiber, Purkinje cell
Introduction
Synapses are indispensable to neural circuit function, yet our understanding of synapse formation and physiology in health and neurological disease is incomplete. Recently, α2δ proteins (Cacna2d1-4) have been recognized as important regulators of synapse formation and plasticity (Dolphin, 2012). In addition to their roles in synaptic transmission as auxiliary subunits of voltage-dependent calcium channels (VDCCs) (Cantí et al., 2005; Hoppa et al., 2012), these proteins have multiple postsynaptic roles in driving synapse formation, trans-synaptic communication, and glutamate receptor function (Eroglu et al., 2009; Kurshan et al., 2009; Fell et al., 2016; Wang et al., 2016; Brockhaus et al., 2018; Chen et al., 2018; Geisler et al., 2019). Mutations in human α2δ genes have been associated with epilepsy, movement disorders, and schizophrenia (Edvardson et al., 2013; Pippucci et al., 2013), and α2δ-1/2 proteins are the primary targets of the widely prescribed antiepileptic and analgesic, gabapentin (Gee et al., 1996; Brown and Gee, 1998; Gao et al., 2000). However, the roles of α2δ proteins in controlling synaptic and network function at any given synapse are still elusive, due in part to expression of multiple isoforms in many neurons.
Purkinje cells (PCs), the primary output pathway from the cerebellar cortex, appear to exclusively and abundantly express the α2δ-2 isoform (Cacna2d2) (Barclay et al., 2001; Cole et al., 2005; Lein et al., 2007; Dolphin, 2012), thus providing an opportunity to determine how this protein contributes to synaptic transmission. Like the spontaneous 'ducky' mouse mutants (du2j/du2j or du/du), targeted deletion of α2δ-2 causes cerebellar ataxia, epilepsy and premature death (Barclay et al., 2001; Brodbeck et al., 2002; Ivanov et al., 2004; Donato et al., 2006), indicative of its importance in neurological function.
The global loss of α2δ-2 likely affects synapses throughout the brain. However, the climbing fiber (CF) to PC synapse has multiple distinctive features, including the mono-innervation of mature PCs by a single CF (Hashimoto et al., 2009) and presynaptic expression of vesicular glutamate transporter 2 (VGLUT2), allowing for the visualization of CF terminals and making this an ideal site to determine how α2δ-2 contributes to synaptic function. In contrast to PCs, the inferior olivary (IO) cells (from which CFs arise) predominantly express the α2δ-1 isoform, with low expression of α2δ-2/3 isoforms (Cole et al., 2005; Lein et al., 2007). Finally, CF activity drives complex spike (CpS) generation in PCs, which produces a high-fidelity error prediction signal important to the processing of motor coordination and learning (Yang and Lisberger, 2014; Heffley et al., 2018).
We combined structural and electrophysiological analysis of the CF to PC synapse in Cacna2d2 KO mice (Ivanov et al., 2004) to elucidate the contribution of α2δ-2 to excitatory synapse formation and transmission. Contrary to positive regulation of excitatory synaptogenesis by the α2δ-1 isoform (Li et al., 2004; Eroglu et al., 2009; Chen et al., 2018; Risher et al., 2018), loss of α2δ-2 increased functional synaptic innervation by CFs. α2δ-2 KO CFs had elevated glutamate release and clearance compared with wild-type (WT), resulting in profound deficiencies in the generation of CpS spikelets. Together, our studies demonstrate the critical role of α2δ-2 in proper CF–PC synapse organization and network function, and allude to the wide versatility of α2δ proteins in synaptic transmission.
Materials and Methods
Animals.
Cacna2d2 KO mice (Cacna2d2tm1Svi, MGI = 3055290; generously supplied by Drs. Sergey Ivanov and Lino Tessarollo) were obtained as cryopreserved sperm and rederived via in vitro fertilization on a C57B/6J background. Breeding mice were kept heterozygous, and genotyping was performed using the following primers: forward 5′-ACTGGTGGGCATCTTACAGC-3′, reverse mutant 5′-AAAGAACGGAGCCGGTTG-3′, reverse WT 5′-TGTTAGCGGGGAGGTCACTA-3′. This produced a ∼700 bp product from the WT allele and a ∼550 bp product from the mutant allele. Mice were born in the Mendelian ratio of 1:2:1; with WT mice having two copies of the intact Cacna2d2 gene, and KO mice as homozygous mutants. Because ∼50% of KO mice die prematurely (Ivanov et al., 2004), all experiments used male and female mice between p21–p30, when CF–PC synapses have reached maturity, but before significant loss of KO mice.
Cacna2d2 deletion was validated, and Cacna2d isoform abundance were measured, using quantitative PCR from fresh-frozen tissues. Regions of the IO and the PC layer (PCL), including the proximal molecular layer, were microdissected by laser capture for pre- vs postsynaptic comparative analysis. Briefly, mice were deeply anesthetized by inhalation of 4% isoflurane followed by injection of 0.8 ml of 2% avertin i.p. (Sigma-Aldrich) and rapidly decapitated. Brains were rapidly dissected, mounted in OCT medium (Tissue-Tek) on dry ice, and placed at −20°C for cryosectioning. 25 μm coronal sections from cerebellum and brainstem were collected on RNase-treated PEN membrane slides (Zeiss). Slides were then dehydrated through a succession of EtOH rinses (70% EtOH 30 s, 100% EtOH 30 s), and nuclei stained using 1 mg/ml Cresyl Violet in 100% EtOH (1 min). IO and PCL regions were identified based on anatomic criteria, and isolated using laser microdissection (Zeiss). Samples were resuspended in 20 μl of QIAzol Lysis Reagent (Qiagen) for 20 min at RT and stored at −80°C. RNA isolation and qPCR were performed by the Oregon Health & Science University Gene Profiling/RNA and DNA Services Shared Resource. In brief, RNA was isolated using the Trizol/RNeasy hybrid protocol with QIAcube automation. SuperScript IV VILO Master Mix (Invitrogen) was used for reverse transcription with 636 pg of input RNA per 20 μl reaction. Following reverse transcription, cDNA was quantified and normalized for 7500 ng of cDNA input for preamplification in 14 PCR cycles. A 1:4 dilution of the pre-amp reaction was used as input for qPCR. The TaqMan Universal master mix (Life Technologies) was used for the PCR, using a single master mix per TaqMan probe set for Cacna2d1 (Mm00486607_m1), Cacna2d2 (Mm01230564_g1), Cacna2d3 (Mm00486641_m1), Cacna2d4 (Mm01190080_m1), and ACTB (Mm04394036_g1) as the endogenous control. Data were acquired using Applied Biosystems QuantStudio 12K Flex Software v1.2.2 (Life Technologies) with settings set to default. Measurements are reported as either ΔCt values (difference in cycle time for gene of interest relative to ACTB, in which a higher ΔCt indicates lower relative expression) or mean fold difference, as determined by 2−(Ct target − Ct ACTB target) − (Ct reference − Ct ACTB reference) ± SEM.
Mice were maintained in facilities fully accredited by the Association for Assessment and Accreditation of Laboratory Animal Care and veterinary care was provided by Oregon Health & Science University's Department of Comparative Medicine. All animal care and experiments were performed in accordance with state and federal guidelines, and all protocols were approved by the OHSU Institutional Animal Care and Use Committee.
Immunohistochemistry.
Following deep anesthesia as described above, p21 Cacna2d2 KO and WT littermates were transcardially perfused with 5 ml of PBS followed by 4% paraformaldehyde-PBS (PFA-PBS). Mice were decapitated, and brains were removed and postfixed for 24 h in 4% PFA-PBS. Cerebella were cut in sagittal sections at 50 μm on a vibratome and stored in PBS at 4°C. Sections containing vermis lobe VI were permeabilized with 0.4% Triton-PBS containing 10% normal goat serum for 1 h at room temperature, then stained with mouse anti-Calbindin 1:20 (Neuromab #73-452) and guinea pig anti-VGLUT2 1:200 (Synaptic Systems #135404) at 4°C overnight. After rinsing, corresponding fluorescently labeled secondaries (Invitrogen) were applied at 1:400 and glass coverslips were mounted on glass slides with Fluoromount G (Sigma-Aldrich).
To image VGLUT2+ synapses, 6 μm z-stack images from the vermis lobe VI were acquired at 0.2 μm intervals with a Zeiss LSM780 laser scanning confocal microscope at 40× magnification and 1024 × 1024 pixel density using ZEN software. This produced images of the entire thickness of the molecular layer, including the PC somata. Images were then analyzed in FIJI/ImageJ. VGLUT2+ puncta distribution/density were quantified from maximum projection images as distinct VGLUT2+ puncta of at least 0.2 μm2, after subtracting the background for increased contrast. For each punctum, the y-distance from the top of the PCL to the terminal was measured. Per image, a minimum of 100 μm of linear PCL was quantified. VGLUT2+ puncta distributions were then binned into 10 μm distances. VGLUT2+ puncta size and a second measure of density (from whole volume of tissue) were calculated using a masking feature in FIJI/ImageJ that captures puncta between 0.1–5 μm2, and this was normalized to the length of PCL imaged to produce a measure of density per μm of PCL. Punctum distribution, density and size data were averaged from two or three sections per animal using five or six animals of each genotype. Slides were coded before imaging, and image acquisition and analysis were performed by investigators blinded to genotype. Sections were stained side by side with the same antibody mixtures, and imaging parameters were kept constant between samples.
Slice preparation and electrophysiology.
KO or WT littermates were deeply anesthetized as described above, and transcardially perfused with ice-cold sucrose-based solution containing the following (in mm): 87 NaCl, 2.5 KCl, 1.25NaH2PO4, 0.4 ascorbate, 2 Na pyruvate, 25 d-glucose, 25 NaHCO3, 75 Sucrose, 7 MgCl2, 0.5 CaCl2 (osmolarity adjusted to 300–305 mOsm) and equilibrated with 95% O2 and 5% CO2 gas mixture. Acute 300 μm sagittal slices were cut from cerebellum using a vibratome (VT1200; Leica Microsystems), and incubated for 30 min in standard artificial CSF (aCSF) at 34°C.
Whole-cell recordings were obtained using 1–3 MΩ borosilicate glass pipettes filled with either K-gluconate or CsCl2-based internal solution. For current-clamp experiments, internal solution contained the following (in mm): 135 K-gluconate, 10 HEPES, 10 NaCl, 1 MgCl2, 0.1 BAPTA, 0.1 CaCl2, 2 ATP-Mg, and 10 phosphocreatine, pH 7.28 adjusted with KOH (osmolarity adjusted to 289 mOsm). For voltage-clamp recordings, the internal solution contained the following (in mm): 100 CsMeSO4, 35 CsCl, 15 TEA-Cl, 1 MgCl2, 15 HEPES, 0.2 EGTA, 2 ATP-Mg, 0.3 TrisGTP, 10 phosphocreatine, and 2 QX-314, pH 7.3 adjusted with CsOH (osmolarity adjusted to 295 mOsm). External solution contained the following (in mm): 125 NaCl, 25 NaHCO3, 1.25 NaH2PO4, 3 KCl, 25 Dextrose, 2 CaCl2, 1 MgCl2 (osmolarity adjusted to 300 mOsm) and was continuously perfused via roller pump.
PCs from the vermis, lobe VI, were identified by soma size and location in the PCL using live infrared differential contrast microscopy on an upright Olympus microscope. Inhibition was blocked in all experiments by 10 μm SR95531 (Tocris Bioscience). Whole-cell patch-clamp recordings were obtained in voltage-clamp mode with cell capacitance, series resistance and input resistance monitored in real time using intermittent −10 mV voltage steps. Signals were amplified with a MultiClamp 700B (Molecular Devices) amplifier and pipette capacitance was compensated for using MultiClamp software. Signals were low-pass filtered at 6 kHz and sampled at 10 kHz, and digitized with a National Instruments analog-to-digital board. All recordings were acquired and analyzed using IgorPro-based (Wavemetrics) software. All recordings were performed at room temperature.
Climbing fiber-mediated EPSCs were evoked using theta or monopolar glass electrode stimulation in the granule cell layer (0.1 ms, 0–100 V square pulses; 0.1 or 0.05 Hz stimulation frequency), placed ∼50 μm from the PC, and the stimulation electrode position was adjusted as needed to obtain a CF response. CF responses were identified as large all-or-none EPSCs that appeared during incremental increases in stimulation intensity, with paired-pulse depression when stimulated with 2 pulses 50 ms apart. All cells were first recorded in voltage-clamp mode, where a hyperpolarizing step of −10 mV was applied to monitor cell capacitance, series resistance and input resistance. Series resistance was not compensated; cells with series resistance >10 MΩ, or a >2 MΩ change in series resistance over the course of the experiment, were excluded.
For current-clamp experiments, PC resting membrane potential was measured by holding the cell in zero current mode, then a small amount of bias current (∼−130 pA) was injected to keep the cell near −60 mV for complex spike and current step experiments. Bridge balance was applied to compensate for pipette and series resistance throughout the recording. First, CFs were stimulated at 0.1 Hz and 10 consecutive sweeps of evoked complex spikes were recorded. Then with CF stimulation off, 500 ms steps of current injection from −200 pA to 1 nA were delivered at least 5 times per step. Cells were returned to voltage-clamp mode to assess recording stability. For complex spike analysis, sweeps were averaged to measure the initial spike amplitude and rise rate. Spikelets were classified as rapid depolarizations (>1000 V/s) that reached +20 mV from baseline, from which spikelet trough-to-peak amplitudes were measured. Area under the curve was measured by integrating the trace during the first 100 ms following stimulation.
For most voltage-clamp experiments, cells were held at −70 mV in the presence of 0.5 μm NBQX (Tocris Bioscience) to maintain voltage clamp during CF EPSCs. Brief hyperpolarizing steps (−10 mV) were delivered to monitor PC access and input resistance preceding each CF-evoked EPSC. 10 CF EPSC trials were averaged, and these averages were used to determine peak amplitude, 20–80% rise time and tau of decay (from single-exponential fits of the EPSC decay). Then, 10 paired-pulse responses with an interstimulus interval of 50 ms were collected, followed by 10 Hz trains of CF stimulation or drug wash-in experiments. For DL-TBOA experiments, baseline EPSCs were acquired at 0.05 Hz before 50 μm DL-TBOA (Tocris Bioscience) was added to the bath. For kynurenic acid (KYN) experiments, aCSF excluded NBQX and PCs were held at −20 mV to maintain voltage-clamp during 0.1 Hz CF stimulation. After acquiring baseline EPSCs, 1 mm KYN (Sigma-Aldrich) was added to the perfusate. Quantification of EPSC peak amplitude and tau of decay from drug wash-in experiments used averages from 10 sweeps before wash-on compared with 10 sweeps after 10 min of exposure to drug (For KYN effect: (EPSCControl − EPSCKYN) * 100; For TBOA effect: (τTBOA − τControl) * 100). For asynchronous EPSC (aEPSC) experiments, aCSF was composed of 1.3 mm Sr2+ in replacement of Ca2+, 3.3 mm Mg2+, and NBQX was omitted. PCs were held at −70 mV and CFs were stimulated at 0.05 Hz. ∼10 trials were used for quantification of aEPSCs, which were sampled from a 500 ms window starting at 150 ms from CF stimulation, and selected manually. For data presentation, aEPSC traces were off-line box-filtered at 1 kHz. To estimate the readily releasable pool, cumulative CF response amplitudes were plotted, and the last third of the train was fit with a linear regression that was extrapolated to time 0 (Schneggenburger et al., 1999).
NEURON computational PC model.
CF simulations were performed with NEURON version 7.7.0, using source code generously supplied by Dr. Michael Häusser (Roth and Häusser, 2001). The following model parameters were kept constant across all simulations: Rm = 120.2 kΩ cm2, Cm = 0.64 μF/cm2 and a residual uncompensated series resistance of 1 MΩ. Because the model is based on recordings and dimensions of a p21 rat PC (Roth and Häusser, 2001), we normalized our WT measurements for CF distribution to the model cell as control, and adjusted the dendritic innervation by KO CFs based on our VGLUT2+ distribution as a relative decrease in length of coverage (i.e., 0.7× CF length of control). CF EPSCs were simulated using 500 inputs of 1 nS peak conductance (simulated as the sum of two exponentials for rise and decay) with a reversal potential of 0 mV and a constant density per dendritic length distribution. Simulation time step was 10 μs for integration. Waveforms were created in IgorPro8 from simulation output to measure EPSC decay time constants by fitting with a single exponential.
Transmission electron microscopy (TEM).
Animals were deeply anesthetized with isoflurane and avertin, as described above, and then transcardially perfused with 10 ml of ice-cold heparin (1k USP/ml; Novaplus) followed by freshly prepared 2% glutaraldehyde/2% paraformaldehyde in 0.1 m PB solution filtered with #3 filter paper (VWR) and pH to 7.4. Brains were dissected and cerebella were blocked and postfixed for 30 min in 4% paraformaldehyde. Tissue was transferred to 0.1 m PB for storage at 4°C and 40 μm sagittal slices of vermis lobe VI were made using a Leica microtome. Slices were collected in separate wells to assure TEM would be from slices >100 μm apart. PFA, glutaraldehyde and microtome blades were all from Electron Microscopy Sciences.
Tissue samples were coded before processing for TEM by a blinded investigator. Briefly, sections were incubated in 1% osmium tetroxide in PB for 30 min, dehydrated through a graded series of ethanols, placed into propylene oxide for 30 min, and then placed in 1:1 propylene oxide:EMBed resin (Electron Microscopy Sciences) rotating at room temperature overnight. Sections were then incubated in 100% resin for 2 h, embedded between sheets of Aclar plastic, and incubated at 60°C for 48 h. Cerebellum sections were then glued to resin blocks and ultrathin sections (50 nm) were collected onto 400 mesh copper grids (Electron Microscopy Sciences). The ultrathin sections were then counterstained with uranyl acetate and Reynolds lead citrate and examined using an FEI Tecnai 12 electron microscope and images were captured using an Advanced Microscopy Techniques digital camera.
CF terminals were imaged from the most highly innervated region of the molecular layer (20–60 μm from PC somata) and identified at 6800× magnification by the following criteria: proximity (<3 μm) to PC primary dendrites, dense-packing of round vesicles, and when synaptic contacts were present, asymmetric Gray's type 1 excitatory synaptic markers. Images were taken at 18500× magnification and CF terminals with clearly delineated membranes that met the criteria described above were analyzed using Fiji/ImageJ software by a separate blinded investigator. Terminal area, total SV density, the length and number of synaptic contacts made by each terminal (as determined by the opposing postsynaptic density), and the number of SVs within 100 nm of each synaptic contact as a proxy for the readily releasable pool were measured. Quantifications from ∼10–15 CF images per animal were averaged (n = animals).
Statistics.
Data were tested for Gaussian distribution using the Kolmogorov–Smirnov normality test with Dallal–Wilkinson–Lilliefor p-value. Differences between genotypes in VGLUT2+ puncta distribution and TEM total synaptic contacts/terminal were tested for significance using Kolmogorov–Smirnov test. For other nonparametric data, the Mann–Whitney test was used (i.e., CpS spikelet number). Immunofluorescence, TEM, and electrophysiology data with normal distributions were analyzed using Student's t tests. Normalized and cumulative amplitude responses during repetitive CF stimulation were compared between genotypes using multiple t tests with Holm–Sidak correction. For morphology data, multiple images per animal were averaged where n = the number of mice. For electrophysiology, all experiments used at least four animals per genotype, where n = the number of cells. Data were graphed in Prism GraphPad version 8 and are reported as the mean ± SEM (*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001).
Results
α2δ-2 controls PC spiking patterns in response to climbing fiber activation
To examine how the loss of α2δ-2 affects cerebellar output, we performed whole-cell recordings from PCs in acutely prepared cerebellar slices from α2δ-2 KO mice and their WT littermates at p21–p30, after CF innervation has reached maturity (Hashimoto et al., 2009). We focally stimulated CFs in the granule cell layer and unitary CF-mediated EPSCs were identified by their large amplitude, all-or-nothing nature and paired-pulse depression (Dittman and Regehr, 1998; Hashimoto and Kano, 1998; Liu and Friel, 2008; Rudolph et al., 2011). CF-evoked CpSs were then recorded in current-clamp mode.
The voltage envelope of CpS waveforms was comparable between WT and KO (Fig. 1A; Integral of CpS between time 0–100 ms: WT = 0.96 ± 0.06 V · s, n = 10; KO = 0.81 ± 0.06 V · s, n = 11; p = 0.1; unpaired Student's t test), but the number of CpS spikelets was substantially reduced in KO PCs (Fig. 1B; WT = 3.2 ± 0.5, n = 10; KO = 1.2 ± 0.4, n = 11; p = 0.002; Mann–Whitney). In addition, the few spikelets that did occur during the KO CpS were of lower trough-to-peak amplitude. All first spikelets in the WT were >30 mV compared with 75% in the KO (Fig. 1C), which is consistent with a lower probability of CpS transmission (Khaliq and Raman, 2005) in the α2δ-2 KO. Moreover, though subsequent spikelets were present in 2 of 11 KO cells, none reached 30 mV trough-to-peak amplitude (Mean KO spikelet2 = 16.5 ± 5.7 mV, n = 2; spikelet3 = 7.6 mV, n = 1; spikelet4 = 7.2 mV, n = 1; spikelet5 = 6.5 mV, n = 1). Despite the differences in CpS spikelet generation between genotypes, initial spike amplitudes (Fig. 1D) and rise rates were unchanged between WT and KO (Spike slope: WT = 1790 ± 230 V/s, n = 10; KO = 1660 ± 240 V/s, n = 11; p = 0.69; unpaired Student's t test).
Figure 1.
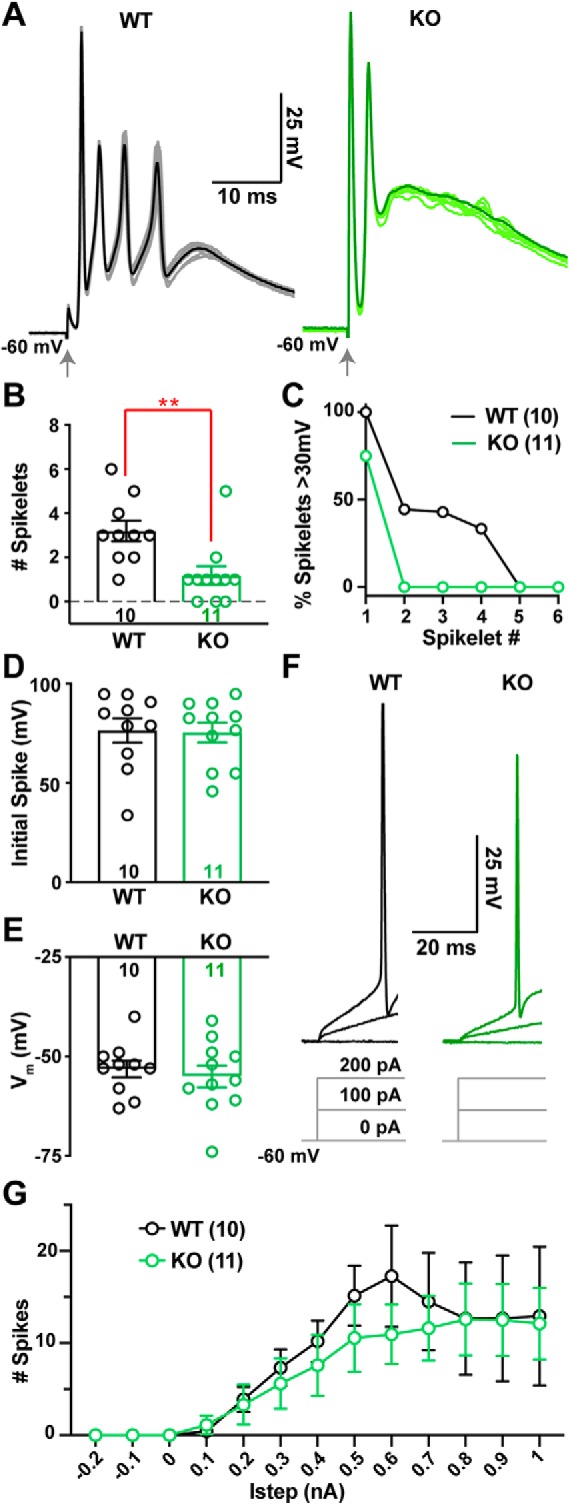
CF-evoked complex spikes (CpSs) are altered in the Cacna2d2 KO, but intrinsic PC excitability is unchanged. A, Representative CF-evoked complex spikes in WT (left) and KO (right) PCs, arrow indicates CF stimulation. Each trace consists of 10 overlaid traces (lighter color) and the corresponding CpS average (dark color). B, Average number of spikelets per CpS in WT and KO PCs; **p < 0.01 (Mann–Whitney). C, Percentage of spikelets exceeding 30 mV trough-to-peak amplitude, ordered by spikelet number. D, Average CpS initial spike amplitude; p = 0.89 (NS). E, Average PC membrane potential when in zero current mode; p = 0.60 (NS). F, Representative single traces of membrane voltage responses to current injection, showing steps of 0, 100, 200 pA injections from Vm = −60 mV; left, WT (black); right, KO (green). Average Istep to initiate spiking WT = 200 ± 39 pA, n = 10; KO = 289 ± 82 pA, n = 9; p = 0.33 (NS). G, Average spike count during current steps from −0.2 to 1 nA, Vm = −60 mV. WT (black) and KO (green); p = 0.63 (NS; Two-way ANOVA with repeated measures, F(1,19) = 0.23). Data shown ± SEM, n = cells; unpaired Student's t test unless otherwise indicated.
The altered CpS in KO PCs were not associated with changes in the intrinsic excitability of α2δ-2 KO PCs as measured by their resting membrane potentials (Fig. 1E), input resistance (WT 140 ± 20 MΩ, n = 8; KO 122 ± 7.8 MΩ, n = 11; p = 0.38, unpaired Student's t test), or response to current injection (Fig. 1F,G). Thus, we explored whether altered CpS patterns might represent differences in CF-mediated synaptic currents.
α2δ-2 KO mice have larger CF-evoked EPSCs with accelerated decay kinetics
The kinetics of CF-evoked EPSCs influence the CpS shape, such that a slower EPSC decay increases the likelihood of spikelet generation (Rudolph et al., 2011). Spikelet generation is also sensitive to the peak synaptic conductance, as an increased phasic conductance can result in depolarization block and failure to generate spikelets (Davie et al., 2008). Thus, we performed whole-cell recordings of CF EPSCs in the presence of low concentrations of the α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor (AMPAR) antagonist, NBQX (0.5 μm), to facilitate voltage-clamp control (as in Dittman and Regehr, 1998; Liu and Friel, 2008; Rudolph et al., 2011).
Peak EPSC amplitudes were 37% larger in α2δ-2 KO mice (Fig. 2A,B; WT = 703 ± 63 pA, n = 17; KO = 961 ± 86 pA, n = 19, p = 0.03; unpaired Student's t test). WT and KO EPSC decays were well fit by a single exponential curve with KO EPSCs exhibiting faster decay kinetics (Fig. 2C,D; WT τdecay = 20.6 ± 1.1 ms, n = 17; KO τdecay = 13.2 ± 0.8 ms, n = 20, p < 0.0001; unpaired Student's t test). Despite these two alterations, the EPSC in the KO had an equivalent charge transfer to that of WT EPSCs (Fig. 2E), and 20–80% rise times were similar in WT and KO PCs (Fig. 2F,G).
Figure 2.
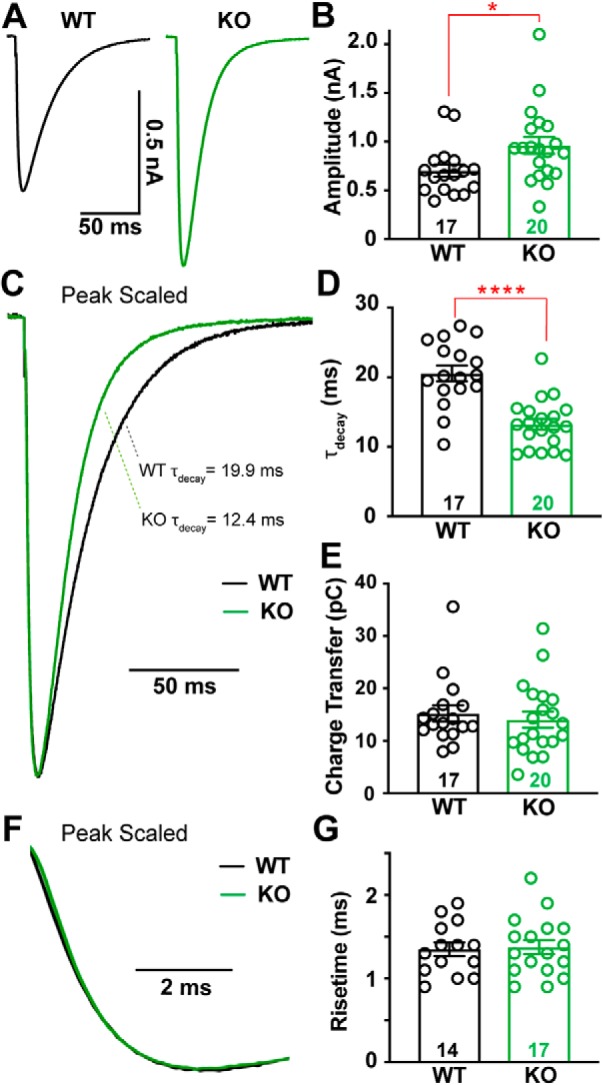
CF-evoked EPSCs are larger and faster in Cacna2d2 KO mice, but total charge transfer is conserved. A, Representative CF-evoked EPSCs. Left, WT average (black); right, KO average (green). B, Average peak CF EPSC amplitude; *p < 0.05. C, Peak scaled EPSCs, demonstrating the relative decay time constants for these example traces (τdecay) based on single exponential fits; WT (black) and KO (green). D, τdecay (ms) for CF EPSCs in WT vs KO PCs; ****p < 0.0001. E, Average charge transfer within the first 100 ms of EPSC; p = 0.58 (NS). F, Peak scaled EPSCs, expanded to display risetime kinetics; WT (black) and KO (green). G, Average CF EPSC 20–80% risetime (ms); p = 0.83 (NS). Data are shown as ± SEM, n = cells; unpaired Student's t test.
Proximal redistribution of CF synapses in the α2δ-2 KO partially contributes to larger CF-evoked EPSCs
To examine the larger EPSC in KO mice, we isolated quantal events at CF synapses by desynchronizing CF evoked release. We replaced extracellular calcium with strontium (Rudolph et al., 2011; Zhang et al., 2015), and measured the amplitudes of CF-derived asynchronous quantal release events (aEPSCs). The average aEPSC was 24% larger in KO compared with WT PCs (Fig. 3A–C; WT = 25.6 ± 1.0 pA, n = 8; KO = 31.8 ± 1.6 pA, n = 8, p = 0.004; unpaired Student's t test), indicating that part, but not all, of the increased CF-evoked EPSC amplitude could be accounted for by a larger unitary response.
Figure 3.
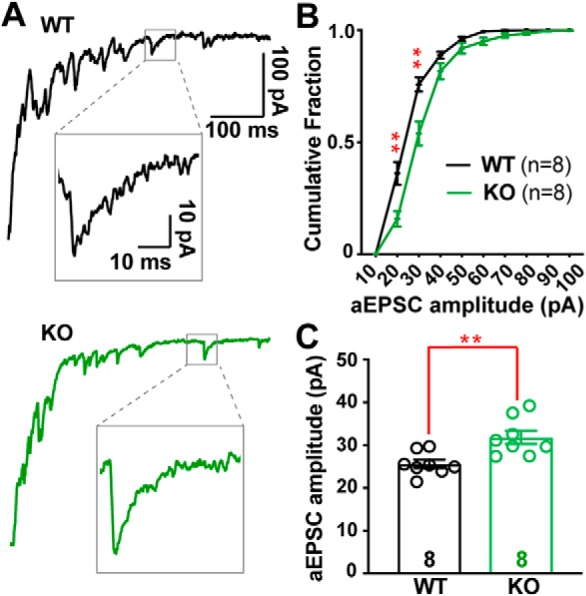
Desynchronized CF-evoked vesicle release reveals larger quantal responses in Cacna2d2 KO. A, Representative CF-evoked EPSCs in the presence of 1.3 mm Sr2+; top, WT EPSC (black) and example asynchronous EPSC (aEPSC; inset); bottom, KO EPSC (green) and aEPSC (inset). B, Cumulative aESPC amplitude distribution graphed in 10 pA bins; WT (black) and KO (green); **p < 0.01 for 20 and 30 pA bins, all others NS (multiple t tests with Holm–Sidak correction for multiple comparisons). C, Average aEPSC amplitudes; **p < 0.01. Data are shown as ± SEM, n = cells; unpaired Student's t test.
Therefore, we asked whether there might also be an increase in the number of CF synapses onto PCs using an immunohistochemical approach. CF terminals can be selectively identified as discrete puncta along the primary dendrites of PCs through their expression of VGLUT2 (Miyazaki et al., 2004; Zhang et al., 2015). As is apparent in Figure 4A, VGLUT2+ puncta were closer to PC somata in KO mice than in WTs. CF terminals were a mean distance of 40.8 ± 0.5 μm from the PC soma in WT, whereas α2δ-2 KO mice had a mean CF terminal distance of 30.6 ± 0.3 μm (Fig. 4A,C; n = 3–5 images from 5 mice of each genotype; p < 0.0001; Kolmogorov–Smirnov test). Moreover, distal CF innervation (beyond 50 μm from the PC soma; Fig. 4B) accounted for 23% of puncta in WT, but only 6% in KO. This shift in CF terminal distribution in KO animals was not associated with a change in VGLUT2+ puncta size (Fig. 4D) or overall number of puncta within the molecular layer (Fig. 4E; Mean number of puncta normalized to length of PCL: WT = 1.91 ± 0.21puncta/μmPCL; KO = 1.81 ± 0.14 puncta/μmPCL, n = 3–5 images from 5 mice of each genotype; p = 0.7; unpaired Student's t test). There was no change in the molecular layer width, and the density of PCs was unchanged (Mean molecular layer width; WT = 110 ± 3.8 μm; KO = 106 ± 4.8 μm, p = 0.43; Mean PC density; WT = 0.62 ± 0.08 cells/μmPCL; KO = 0.65 ± 0.06 cells/μmPCL, p = 0.72; n = 8 images from 3 mice of each genotype; unpaired Student's t test).
Figure 4.
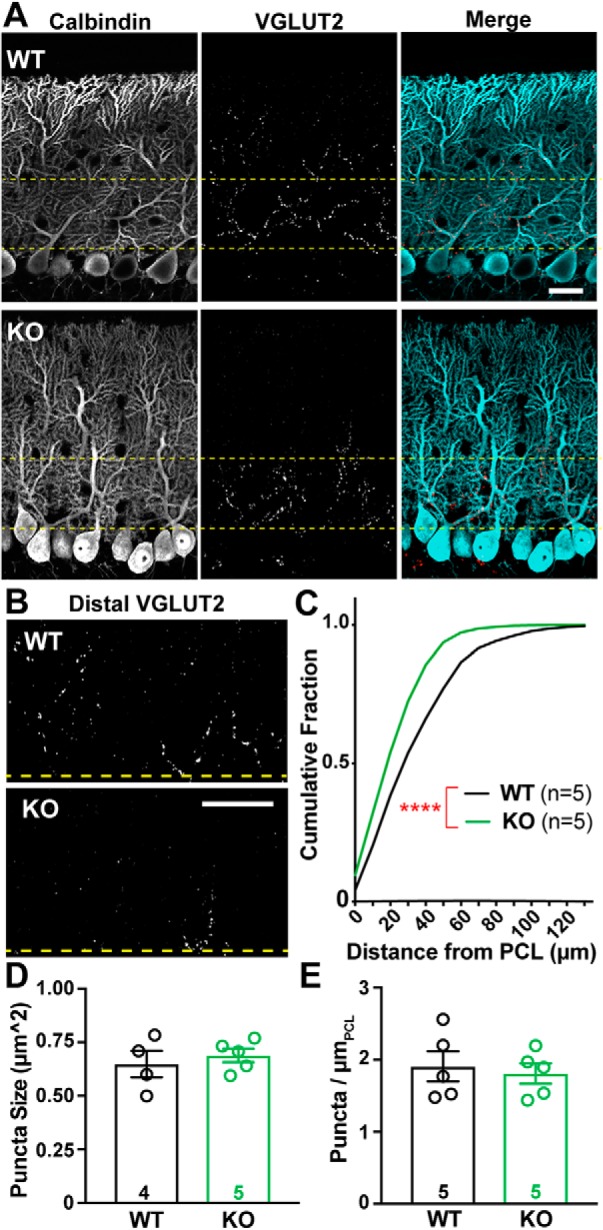
CF terminal distribution, but not number, is altered in Cacna2d2 KO cerebellum. A, Representative images from p21 WT (above) and KO (below) tissue, depicting the PCL. Calbindin (left/blue in merge) marks PCs, and VGLUT2 immunoreactivity (middle/red in merge) marks CF terminals. Yellow lines demarcate the 50 μm most proximal to PC somata and is the region most highly innervated by climbing fibers. Scale bar, 20 μm. B, VGLUT2-immunoreactive CF terminals in the outer molecular layer, cropped at the distal yellow line (50 μm), illustrate differences in CF innervation of distal PC dendrites in WT (top) and KO (below) PCs. Scale bar, 20 μm. C, Cumulative distribution of VGLUT2+ puncta relative to PC somas in WT (black, n = 5 animals) and KO (green, n = 5 animals); ****p < 0.0001 (Kolmogorov–Smirnov test). D, Average VGLUT2+ punctum size was not significantly different between WT and KO terminals; p = 0.55 (NS). E, Average VGLUT2+ puncta density per length of PCL (puncta/ μmPCL) was not significantly different between WT and KO; p = 0.72 (NS). Unless otherwise stated, Data are shown as ± SEM, n = animal; unpaired Student's t test.
The more proximal location of CF inputs in the KO could contribute to both the increased EPSC amplitude and decay rate, due to decreased dendritic filtering (Roth and Häusser, 2001). To ask whether changes in CF synapse localization alone were sufficient to account for the altered EPSC amplitude and kinetics, we modified the Roth and Häusser (2001) computational model of dendritic integration of CF inputs onto PCs to match our data. This model, based on morphological reconstructions of single PCs and empirical measurements of dendritic filtering (Roth and Häusser, 2001), allowed us to simulate how redistribution of CF inputs would affect the ensemble CF EPSC. Using this model (Fig. 5A; For KO, “model CF70% Control” simulates the empirically observed VGLUT2+ distribution from Fig. 4C), a shift from the WT to the KO distribution of CF inputs produced a 16% increase in simulated EPSC amplitude and a 15% decrease in the decay time constant (Fig. 5B). Thus, the proximal distribution of CF inputs in the KO augment the quantal response (Fig. 5C), to account for the increase in evoked CF EPSC amplitude.
Figure 5.
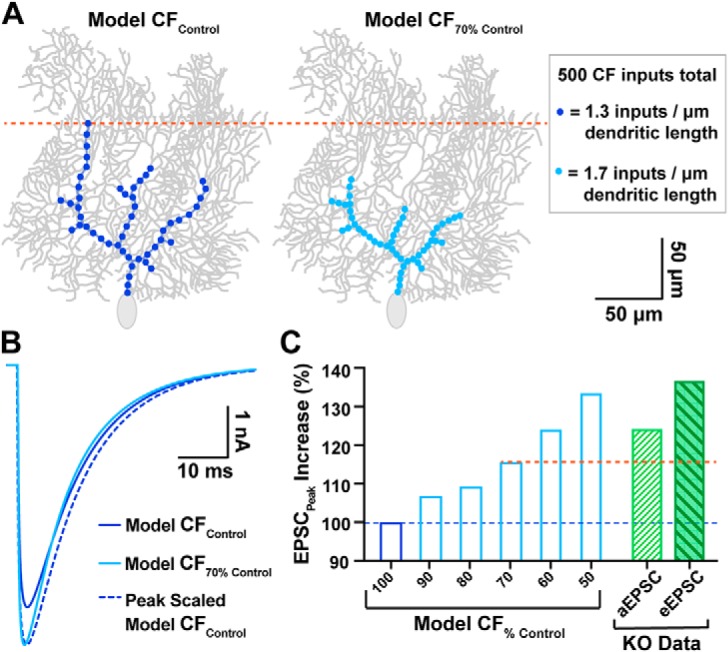
Computational PC model simulates the impact of proximally shifted CF inputs on EPSC waveform. A, Left, model CF input distribution similar to control PCs (dark blue; “model CFControl”) vs a similar PC with CF inputs shifted 30% more proximal (right, light blue, “model CF70% Control”), which matches the degree of proximal shift in WT vs KO innervation, respectively. All models conserved the total number of CF quantal inputs (500 inputs with 1 nS conductance), though input density was adjusted to accommodate the shortened region of CF innervation (see inset). B, Overlay of EPSC output waveforms from model CFControl simulations (dark blue; 4.7 nA), model CF70% Control (light blue; 5.4 nA), and peak scaled model CFControl to compare decay kinetics. For tau of decay; model CFControl τdecay = 12.0 ms; model CF70% Control τdecay = 10.2 ms). C, Predicted increase in EPSC peak amplitude for various degrees of proximally shifted model CFs (light blue bars, restricted to a zone 100–50% the width of control CFs, all including 500 quantal inputs) compared with model CFControl (dark blue bar). For comparison, the empirically determined increase in quantal EPSC (aEPSC; hatched green bar) and evoked EPSC (eEPSC; filled-hatched dark green bar) amplitudes in KO PCs are also displayed (derived from Figs. 3 and 2, respectively). The orange dotted line demarcates the predicted EPSC increase from the model based on the observed shift in CF location.
Increased multivesicular release from α2δ-2 KO CFs
CF synapses exhibit multivesicular release, increasing the synaptic glutamate concentration (Wadiche and Jahr, 2001; Rudolph et al., 2011). To determine whether KO mice displayed altered multivesicular release, we used the low affinity, competitive AMPAR antagonist, kynurenic acid (KYN), to assay synaptic glutamate concentrations. Because KYN binds and unbinds AMPARs throughout the duration of the CF-evoked glutamate transient, KYN inhibition of the AMPAR-mediated current is inversely proportional to the concentration of glutamate present at postsynaptic receptors (Wadiche and Jahr, 2001). KYN (1 μm) inhibited WT EPSC peak amplitudes by 65% (Fig. 6A,B; EPSCPeak amplitude control vs KYN; WTControl = 1.77 ± 0.41 nA vs WTKYN = 0.62 ± 0.16 nA, n = 6, p = 0.007; paired Student's t test), whereas KO EPSCs were inhibited by only 40% (EPSCPeak amplitude control vs KYN; KOControl = 2.63 ± 0.31 nA vs KOKYN = 1.57 ± 0.21 nA, n = 8, p = 0.0009; paired Student's t test; relative change in WT vs KO, p = 0.001; unpaired Student's t test), demonstrating enhanced multivesicular release from KO CFs.
Figure 6.
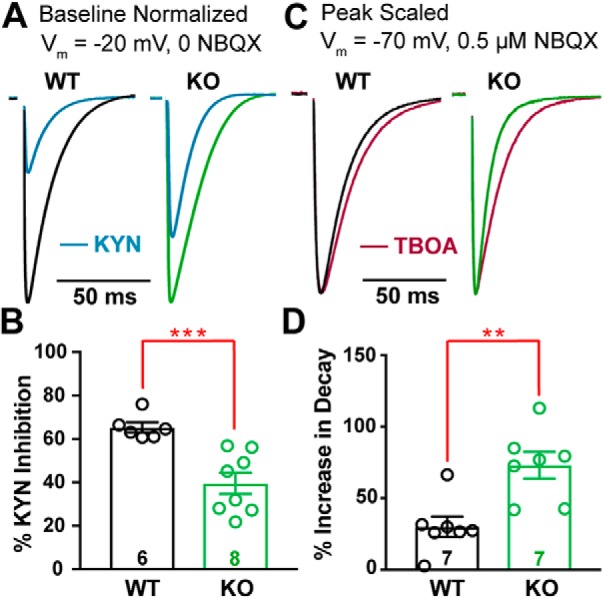
Cacna2d2 KO has increased glutamate release and clearance at CF–PC synapses. A, Representative CF EPSCs recorded at Vm = −20 mV in the absence of NBQX for WT and KO PCs (relative scales; WT = black; KO = green). For each, traces after exposure to 1 mm kynurenic acid (KYN; blue) are shown normalized to baseline EPSC amplitudes. B, Percentage inhibition of ESPC peak amplitude by KYN; ***p < 0.001. C, Representative normalized CF-evoked EPSCs recorded at Vm = −70 mV in the presence of 0.5 μm NBQX; left, WT average (black); right, KO average (green). Overlay average peak-scaled traces after exposure to 50 μm DL-TBOA (TBOA; magenta). D, Average increase in EPSC decay by TBOA; **p < 0.01. Data are shown as ± SEM, n = cells; unpaired Student's t test.
For these experiments, PCs were held at −20 mV to maintain voltage clamp of the CF-evoked EPSC (Harrison and Jahr, 2003; Rudolph et al., 2011) and NBQX was omitted because coapplication of NBQX and KYN facilitates AMPAR-mediated responses (Prescott et al., 2006). Interestingly, at this holding potential, KO EPSCs exhibited slower decay kinetics (for τdecay at baseline Vm = −20 mV; WT−20mV = 11.6 ± 1.12 ms, n = 6; KO−20mV = 18.5 ± 1.32 ms, n = 8, p = 0.002; unpaired Student's t test). Although this is consistent with a larger glutamate transient due to multivesicular release (Paukert et al., 2010), it is in apparent odds with the faster decay kinetics in KO PCs at more hyperpolarized potentials. One explanation for this discrepancy in decay kinetics could be the voltage dependence of glutamate reuptake by PCs. PCs and surrounding Bergmann glia express high levels of glutamate transporters to manage spillover and glutamate clearance, and activity of these transporters shapes the CF-evoked EPSC waveform (Paukert et al., 2010). However, PCs play a dominant role in synaptic glutamate clearance (Auger and Attwell, 2000), and glutamate transporters have decreased efficiency at depolarized voltages (Bergles et al., 1997). In this scenario, the contributions of PC and Bergmann glia glutamate transporters together result in rapid EPSC decay in the KO at hyperpolarized potentials, but prolonged decay expected from multivesicular release dominates the EPSC waveform when KO PCs are held at depolarized potentials.
Faster EPSC decay in α2δ-2 KO due to enhanced glutamate clearance
We further investigated the role of glutamate transporters in shaping the CF EPSC waveform in WT and KO mice while holding PCs at −70 mV in the presence of low NBQX (0.5 μm), as in our prior voltage-clamp experiments (Fig. 2). Block of glutamate transporters with the nonselective transport reuptake inhibitor, DL-TBOA (50 μm), increased WT decay constants by ∼30% (similar to Rudolph et al., 2011) (Fig. 6C; WT τdecay control vs TBOA; WTControl = 13.6 ± 0.9 ms vs WTTBOA = 17.4 ± 0.9 ms, n = 7, p = 0.001; paired Student's t test). In contrast, TBOA increased decay constants of KO EPSCs by 73% (KO τdecay control vs TBOA; KOControl = 11.1 ± 1.1 ms vs KOTBOA = 19.1 ± 2.2 ms, n = 7, p = 0.001; paired Student's t test). After TBOA exposure, WT and KO had similar decay constants (τdecay WT vs KO in TBOA; p = 0.48; unpaired Student's t test). However, the relative effect of TBOA on decay was greater in KO EPSCs (Fig. 6D; percentage increase in τdecay WT vs KO, p = 0.003; unpaired Student's t test) consistent with enhanced glutamate clearance by surrounding glutamate transporters.
Repetitive stimulation of CFs suggests lower release probability in the α2δ-2 KO, though cumulative vesicle release is greater
The enhanced multivesicular release in the α2δ-2 KO (Fig. 6A,B) suggests a presynaptic contribution to the CF EPSC phenotype. CF synapses have high initial probability of release (PR), which is associated with paired-pulse depression at short interstimulus intervals (Hashimoto and Kano, 1998). However, CF-evoked EPSCs from KO mice showed a consistent increase in the paired-pulse ratio when compared with WT CFs (Fig. 7A,B; pulse ratio: WT = 0.41 ± 0.03, n = 17; KO = 0.51 ± 0.02, n = 18, p = 0.01; unpaired Student's t test), suggesting CFs have a lower PR when α2δ-2 is deleted. Therefore, we hypothesized that KO CFs might have a substantially greater readily releasable pool of vesicles to exhibit increased multivesicular release while also having a lower PR.
Figure 7.

Repetitive stimulation of CF synapses reveals a lower probability of release and greater cumulative release in Cacna2d2 KO. A, Representative traces from WT (black) and KO (green) PCs during 50 ms paired-pulse stimulation. Traces are scaled to the first EPSC (EPSC1). Dotted gray line shows paired-pulse depression of the second EPSC (EPSC2) in WT compared with KO. B, Average paired-pulse ratio (EPSC2 /EPSC1); **p < 0.01. C, Representative traces in response to 10 Hz stimulation; WT (black); KO (green). D, Traces from (C) peak scaled to EPSC1 and overlaid, illustrating different relative steady-state EPSC amplitudes during latter portions of the train. E, Summary data of EPSC amplitudes normalized to EPSC1 during 10 Hz stimulation in WT (black) and KO (green). *p < 0.05 (multiple t tests with Holm–Sidak correction). F, EPSC amplitudes during 10 Hz stimulation plotted as cumulative amplitude from WT (black) and KO (green). For comparison of cumulative amplitude between WT and KO at various stimulation numbers (stim #); *p < 0.05, **p < 0.01, ***p < 0.001 (multiple t tests with Holm–Sidak correction). Dotted gray lines illustrate a linear fit to cumulative amplitude between stim # 20–30 from WT and KO trains. Data are shown as ± SEM, n = cells; unpaired Student's t test unless otherwise indicated.
To estimate the relative size of the readily releasable pool in WT and KO, we stimulated CFs with a 10 Hz train. WT and KO CF synapses had strikingly different responses during repetitive stimulation. Both genotypes exhibited a delayed facilitation followed by depression, which may indicate multiple pools of vesicles with differing release probabilities (Lu and Trussell, 2016). However, KO EPSCs were larger at every stimulus throughout the train (Fig. 7C–E), providing further evidence of enhanced vesicle release compared with WT. A linear fit to the last 10 responses of a cumulative amplitude plot produced a steeper slope (Linear regression using 95% Confidence Intervals; WTslope = 78.7 ± 0.6, n = 5; KOslope = 172.2 ± 0.5, n = 5; p < 0.0001; unpaired Student's t test) and a larger y-intercept in KO PCs when compared with WT (Fig. 7F). This analysis provided an estimate of the readily releasable pool (Schneggenburger et al., 1999), which was 29% larger in the KO compared with WT (WT y-intercept = 2.08 ± 0.02 nA, n = 5; KO y-intercept = 2.77 ± 0.01 nA, n = 5; p < 0.0001; unpaired Student's t test). Thus, both a single stimulus (Fig. 6A,B) and repetitive stimulation of CFs produced increased vesicle release in the α2δ-2 KO, suggesting either an enhancement of a low PR pool of vesicles (Lu and Trussell, 2016) and/or more discrete release sites with reduced PR compared with WT.
CF terminals in α2δ-2 KO have more release sites
Because we did not see an increased density of VGLUT2+ puncta by light microscopy, we hypothesized CF terminals in the KO had either a greater number of release sites or vesicles. CF terminals can be identified with electron microscopy (EM) by their high density of round synaptic vesicles (SVs) and contacts onto PC “thorns” along primary PC dendritic shafts (Palay and Chan-Palay, 1974; Miyazaki et al., 2004). While blinded to genotype, we imaged and quantified CF terminals from WT and KO mice (10–15 images/animal; see Materials and Methods). In agreement with VGLUT2+ puncta size determined by confocal microscopy (Fig. 4D), CF terminal cross-sectional area was no different between genotypes when sampled using EM (Fig. 8A,B). WT and KO CF terminals also had an equivalent density of SVs (Fig. 8C) and no change in number of SVs within 100 nm of the synaptic contact, a proxy for the readily releasable pool (WT = 18.5 ± 2.4 SV/μmContact, n = 5 mice; KO = 20.4 ± 2.7 SV/μmContact, n = 6 mice, p = 0.6; unpaired Student's t test).
Figure 8.
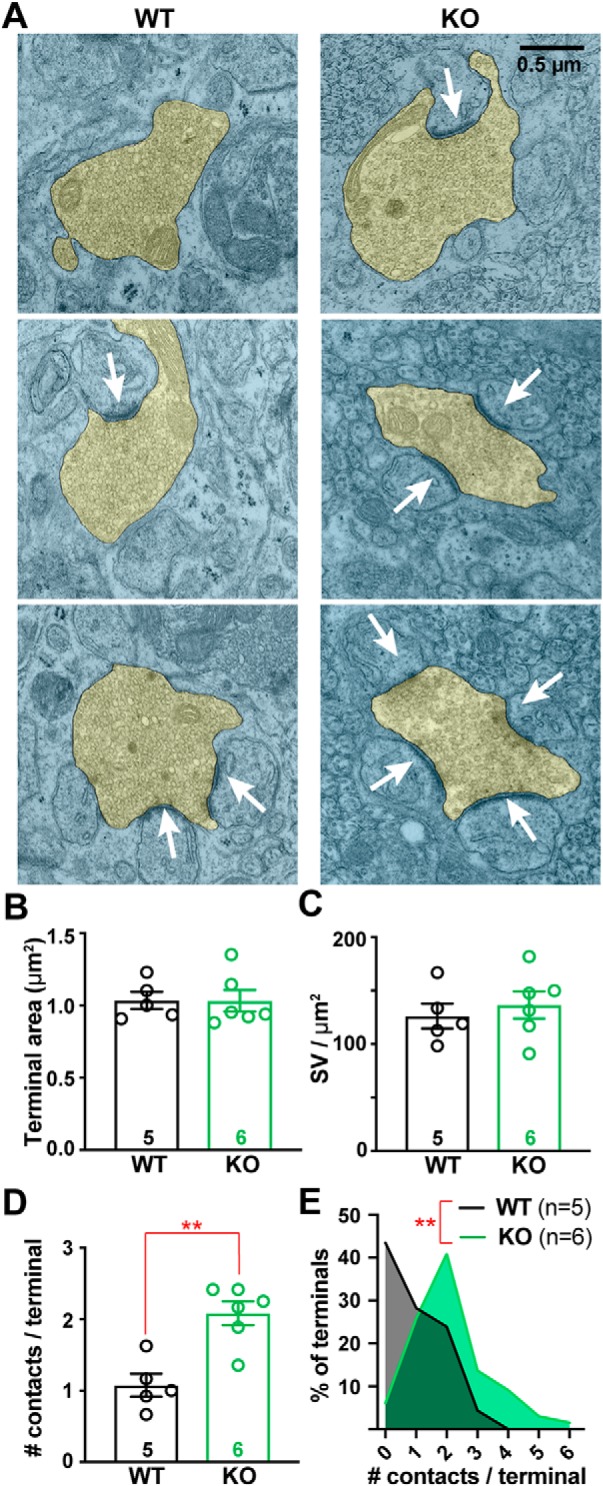
CF terminals have increased numbers of synaptic contacts in Cacna2d2 KO animals. A, Representative transmission electron micrographs of CF terminals (pseudocolored yellow) from p21 WT (left) and KO (right) animals. White arrows indicate postsynaptic densities used to quantify synaptic contacts/terminal. Scale bar, 0.5 μm. B, Average CF terminal area (μm2); p = 0.55 (NS). C, Synaptic vesicle (SV) density (SV/ μm2) was not different between WT and KO animals; p = 0.57 (NS). D, Average number of contacts per CF terminal (# contacts/terminal) was increased in KO animals. **p < 0.01 (Mann–Whitney test). E, Histogram of all CFs analyzed from WT (black) and KO (green) cerebella, displaying the number of contacts per sampled CF terminal normalized to total number of CF terminals; **p < 0.01 (Kolmogorov–Smirnov test). Data are shown as ± SEM, n = animals using 15–20 images/animal; unpaired Student's t test unless otherwise stated.
However, the number of synaptic contacts made by CF terminals in α2δ-2 KOs was nearly twice that of WT (Fig. 8A,D; WT = 1.08 ± 0.16 contacts/terminal, n = 5 mice; KO = 2.08 ± 0.17 contacts/terminal, n = 6 mice, p = 0.009; Mann–Whitney test). Moreover, the distribution of the number of contacts made by CF terminals was right-shifted in KO (Fig. 8E; p < 0.002; Kolmogorov–Smirnov test). Although quantifying synaptic contacts per terminal using single EM sections likely underestimates the true number of contacts per terminal, the observed doubling of discrete contacts made by KO CFs is consistent with increased multivesicular release in the α2δ-2 KO.
Differential Cacna2d isoform expression in the IO and PCL
By in situ hybridization, there is robust and exclusive expression of the Cacna2d2 isoform in PCs, and relatively low expression in the inferior olive (IO), which gives rise to climbing fibers (Cole et al., 2005; Lein et al., 2007). Given the presynaptic morphological and physiological changes we observed at Cacna2d2 KO CF–PC synapses, we reexamined expression of α2δ isoforms in the Purkinje cell layer (PCL) and IO using quantitative PCR of fresh-frozen tissue obtained by laser capture microdissection (Fig. 9A). In agreement with prior observations (Cole et al., 2005; Lein et al., 2007), PCL tissue from WT mice expressed the α2δ-2 isoform 3.85 ± 0.96-fold higher than presynaptic IO samples (Fig. 9B; Cacna2d2 ΔCtPCL = 1.62 ± 0.17; ΔCtIO = 3.37 ± 0.38, n = 4, p = 0.03; paired Student's t test). Importantly, analysis of Cacna2d transcripts from IO samples revealed that α2δ-1 is the predominant isoform in that region (Fig. 9C), and is 7.0 ± 2.4-fold more abundant than α2δ-2 in IO samples (Cacna2d1 ΔCtIO = 0.94 ± 0.4; Cacna2d2 ΔCtIO = 3.37 ± 0.38, n = 4, p = 0.04; paired Student's t test).
Figure 9.
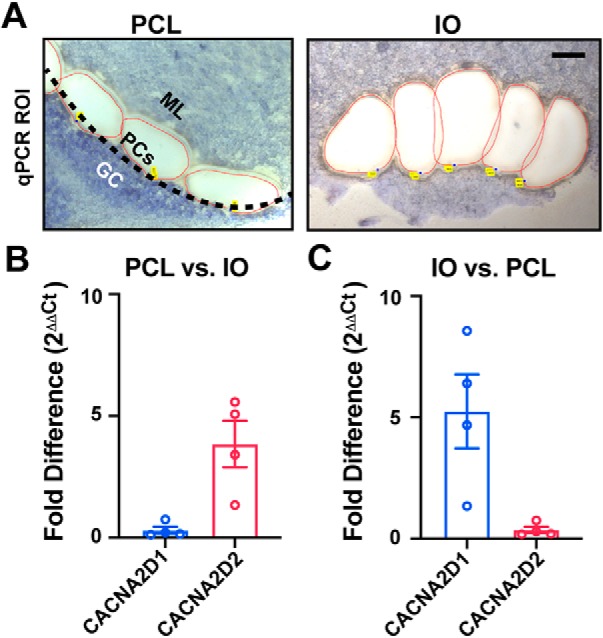
Relative expression of Cacna2d transcripts by qPCR from Purkinje cell layer (PCL) and inferior olive (IO) tissues. A, Example PCL and IO regions of interest isolated by laser capture microdissection from fresh-frozen WT tissue. Left, PCL region of interest; black-dotted line indicates the monolayer of PCs that have been dissected along with regions of the inner molecular layer (ML; granule cells, GC). Right, IO region of interest; one hemisphere from a coronal section of ventral brainstem, scale = 100 μm. (B, C) Fold difference (2−ΔΔCt) expression of Cacna2d isoforms by quantitative PCR comparing (B) PCL versus IO, and (C) IO versus PCL samples. Data are shown as ± SEM, n = 4 animals.
Our PCL samples contained other cell types found adjacent to PCs and within the proximal molecular layer, such as Bergmann glia and molecular layer interneurons. In situ hybridization experiments suggest that these cells, and not PCs, express α2δ-1 and/or α2δ-3 (Cole et al., 2005; Lein et al., 2007), which may explain the detection of low levels of α2δ-1 and a relatively high level of α2δ-3 mRNA in our PCL samples. Additionally, consistent with previous reports, α2δ-4 was undetectable in both the PCL and IO samples.
Discussion
In mice lacking α2δ-2, profound deficiencies in CF-induced complex spike (CpS) generation were associated with altered underlying CF-evoked EPSC amplitude and kinetics. CpSs are initiated by a AMPAR-mediated depolarization that drives an initial sodium spike followed by multiple axonally generated 'spikelets' (Davie et al., 2008). Furthermore, dynamic clamp experiments suggested that increased CF EPSC amplitudes would result in depolarization block of spikelet generation (Davie et al., 2008). Thus, we hypothesize that increased glutamatergic transmission and accelerated EPSC kinetics (Rudolph et al., 2011) shape the CpS in the α2δ-2 KO (Fig. 10).
Figure 10.

Summary of CF–PC phenotypes in Cacna2d2 KO mice. Proximal distribution of CF inputs onto KO PCs enhanced postsynaptic quantal responses to CF glutamate release, and the increased number of synaptic release sites increased total glutamate concentration. Together, this resulted in a 140% EPSC amplitude in the KO compared with control. Counteracting effects included a lower CF PR and enhanced glutamate clearance, which doubled the EPSC decay rate. Ultimately, larger synaptic conductances in the KO likely contribute to depolarization block of CF-evoked spikelet generation.
Each CF terminal in the KO contained more discrete synaptic contacts, leading to increased multivesicular release and consequently larger EPSCs, despite an apparent reduction in the probability of release. Together, these findings seem to contradict observations in which synapse formation is positively regulated by α2δ expression (Li et al., 2004; Eroglu et al., 2009; Chen et al., 2018; Risher et al., 2018), reflecting a more complex and nuanced role of α2δ proteins in the α2δ-2 KO phenotype.
α2δ-2 proteins as auxiliary Ca2+ channel subunits
α2δ proteins were first identified as auxiliary subunits of voltage-dependent Ca2+ channels (VDCCs) that facilitate surface trafficking of VDCCs in heterologous expression systems (Cantí et al., 2005; Cassidy et al., 2014). α2δ-1 knockdown reduces presynaptic vesicle release in cultured hippocampal neurons (Hoppa et al., 2012), and capacitive measurements from inner hair cells of the spontaneous Cacna2d2 mutant du/du show reduced exocytosis (Fell et al., 2016). We also observed a lowered PR at the CF–PC synapse, as determined by paired-pulse ratio, which is consistent with these prior observations and could relate to altered VDCC localization or abundance in CFs. Given that α2δ-1 is the predominant isoform expressed by IO cells (Cole et al., 2005; Lein et al., 2007), if the phenotype we observed is mediated by presynaptic loss of α2δ-2, it suggests isoform-specific functions that cannot be compensated by α2δ-1. Furthermore, this raises the possibility that different α2δ isoforms in individual neurons may have distinct functional roles.
In addition to their abundant α2δ-2 expression, PCs highly express the VDCC, Cav2.1 (Barclay et al., 2001), which localizes to PC somata and primary dendrites in scattered and clustered patterns (Indriati et al., 2013). Dissociated PCs from the spontaneous mutant, du2j/du2j, have ∼30% reduced somatic calcium currents (Barclay et al., 2001). Similarly, CF vesicle release is mostly (70–90%) regulated by Cav2.1 (Regehr and Mintz, 1994). Because CF–PC transmission and development has been extensively characterized in Cav2.1 mutant mice (Matsushita et al., 2002; Miyazaki et al., 2004; Hashimoto et al., 2011), we looked to this literature for clues as to whether our observed phenotypes could reflect altered pre- and/or postsynaptic VDCC function or localization.
For example, the leaner phenotype, in which a Cav2.1 pore mutation reduces PC calcium currents by 60%, is associated with larger, rapidly decaying CF-evoked EPSCs (Liu and Friel, 2008), like the α2δ-2 KO. However, leaner CFs do not exhibit changes in PR (Liu and Friel, 2008). Other Cav2.1 pore mutants show reductions in PC calcium current analogous to α2δ-2 mutants, but display heterogeneity in EPSC phenotypes. Both rolling Nagoya and tottering mutants have 40% reductions in calcium currents, yet rolling Nagoya exhibits larger, slowly decaying EPSCs, whereas tottering EPSCs are unchanged (Matsushita et al., 2002). PC-specific Cav2.1 KO show proximal innervation by CFs (Miyazaki et al., 2012), reminiscent of the CF redistribution we observed in Cacna2d2 KOs. This striking similarity provides evidence for postsynaptic control of CF development. Surprisingly however, global and PC-specific Cav2.1 KOs exhibit normal CF EPSC amplitude and decay kinetics (Miyazaki et al., 2004; Hashimoto et al., 2011). Thus, although some similarities exist between the Cacna2d2 KO and certain VDCC mutant mice, the roles of α2δ-2 at the CF–PC synapse likely involve other effector mechanisms. The phenotype of α2δ-2 loss may be more nuanced than altered VDCC abundance, but perhaps result from differences in VDCC localization, clustering or association with other molecules (including other VDCC subtypes).
Altered presynaptic function could result from PC-driven morphological changes in the Cacna2d2 KO, independent of altered presynaptic VDCC trafficking. For example, alterations in presynaptic morphology alone, rather than altered presynaptic gene expression, can influence measures of apparent PR and the readily releasable pool size by repetitive stimulation (Fekete et al., 2019). It is also possible that PR at individual release sites is unchanged, but an increased number of release sites per terminal could allow for accumulation of [Ca2+]i during repetitive stimulation via intersite crosstalk. In this scenario, repetitive stimuli recruit vesicles from a low PR pool, sustaining release during subsequent stimuli. Currently, there is no definitive way to distinguish between a heterogeneous population of PR at discrete release sites, or whether the vesicle recruitment from low vs high PR pools differs (Kaeser and Regehr, 2017). The possibility of multiple vesicle pools in the CF, as seen by the bimodal responses in Figure 7C, complicates our ability to derive readily releasable pool size and PR at this synapse (Neher, 2015; Lu and Trussell, 2016). Given the various possible effector mechanisms for α2δ-2 that could contribute to the phenotype in mutant mice, further experiments using cell-type selective genetic manipulations will be necessary to fully understand the locus of α2δ-2 action.
α2δ-2 proteins as synaptic organizers
Recently, roles for α2δ proteins independent of VDCCs are supported by their ability to regulate synapse formation despite pharmacological block or deletion of VDCCs (Eroglu et al., 2009; Kurshan et al., 2009). α2δ-1 induces excitatory synapse formation in response to glial-secreted thrombospondin (Eroglu et al., 2009), which is dependent on postsynaptic signaling through N-methyl-d-aspartate receptors (NMDARs) (Risher et al., 2018). Though NMDARs are transiently expressed in newborn rat PCs (Rosenmund et al., 1992), they are not detected at mouse CF–PC synapses until late adulthood (Piochon et al., 2007; Renzi et al., 2007), making this interaction unlikely to contribute to the Cacna2d2 KO phenotype.
Studies in C. elegans suggest trans-synaptic roles for α2δ proteins, as presynaptic α2δ-3 and binds to postsynaptic neurexin to control neuromuscular synaptic function (Tong et al., 2017). In mammals, neurexins are presynaptically expressed (Missler et al., 2003; Zhang et al., 2015) and interact trans-synaptically with postsynaptic neuroligins. Interestingly, similar to our observations in the Cacna2d2 KO, the neuroligin triple KO mouse (Zhang et al., 2015) also has a proximally shifted CF distribution, without a change in VGLUT2+ puncta size or overall density (Zhang et al., 2015). Although CF–PC synaptic function is unchanged in the neuroligin tKO (Zhang et al., 2015), this finding suggests that α2δ-2 may act in parallel with neurexin-neuroligin to coordinate features such as synaptic localization, whereas the functional components that depend on α2δ-2 involve other effector mechanisms.
The stereotyped development of the CF–PC synapse has made it an attractive model to study numerous important molecules that share mutant phenotypes, yet have quite disparate functions. For example, the CF innervation pattern is modulated by postsynaptically expressed neuroligins, TrkB, Cav2.1, GluRδ2 (exclusively expressed in PCs), cerebellins, and myosin Va (for review, see Bosman and Konnerth, 2009), revealing CF redistribution as a sensitive indicator of underlying dysfunction, and demonstrating postsynaptic control of synaptic innervation. Although Cacna2d2 KO mice share this phenotype, additional functional alterations at the CF–PC synapse are dissimilar from other mutant mice. Thus, our data suggest that the contributions of α2δ-2 to the CF–PC synapse may comprise both VDCC-dependent and -independent mechanisms.
Impacts of α2δ-2 loss on cerebellar function
Of the four α2δ isoforms (Cacna2d1-4), α2δ-2 loss has the most severe phenotype, as mice and humans with Cacna2d2 mutations have ataxia, epilepsy and motor control deficits (Barclay et al., 2001; Brodbeck et al., 2002; Ivanov et al., 2004; Donato et al., 2006; Pippucci et al., 2013). Because PCs provide the output from the cerebellum, some of these neurologic phenotypes likely reflect PC dysfunction. PC spike-rate and plasticity are instructed by CpSs, providing error prediction information for motor coordination, presumably graded by the number of spikelets generated (Rasmussen et al., 2013; Yang and Lisberger, 2014; Burroughs et al., 2017). As CpSs generated by Cacna2d2 KO PCs had fewer spikelets, we predict PC information transfer is degraded, implicating direct influence of α2δ-2 loss in cerebellar dysfunction.
Particularly surprising was the variety of alterations at the Cacna2d2 KO CF–PC synapse, including enhanced glutamate reuptake. It is likely that some phenotypes developed as compensation for a primary derangement directly related to α2δ-2 loss. Delayed conditional deletion of α2δ may help to clarify this issue. Likewise, deletion of α2δ-2 may have differential effects on other synapses and functions, either onto PCs or other cell types. As many neurons coexpress multiple α2δ isoforms, it will be important to understand the role of each isoform at individual synapses, as well as address whether postsynaptic α2δ proteins instruct presynaptic development or function (or vice versa), using cell-specific targeted rescue or deletion. Overall, our results underscore the critical roles of α2δ-2 in both proper organization and transmission at the CF–PC synapse.
Footnotes
This work was supported in part by the Department of Veterans Affairs, Veterans Health Administration, Office of Research and Development, Biomedical Laboratory Research and Development Merit Review Award I01-BX002949 (E.S.), a Department of Defense CDMRP Award W81XWH-18–1-0598 (ES), NIH T32NS007466 (K.A.B.), NIH R01-NS080979 (G.L.W.), NINDS 1R21NS102948 (Dr. Ines Koerner and E.S.), NIH P30NS061800 (Dr. Sue Aicher), and OHSU Innovation Fund (E.S.) awards. The electron microscope was purchased with funds from the Murdock Charitable Trust awarded to Dr. Aicher. We thank Dr. Stefanie Kaech-Petrie of the OHSU Advanced Light Microscopy Core for assistance with imaging; Dr. Gail Mandel and Dr. John Sinnamon for expertise and use of laser capture equipment; the OHSU Gene Profiling/RNA and DNA Services Shared Resource for support with qPCR experiments; and Dr. Sue Aicher, James Carroll, and Jo Hill for the electron microscopy expertise. We would like to acknowledge Drs. Sergey Ivanov and Lino Tessarollo for generously providing the Cacna2d2 KO mice; Drs. Michael Häusser and Arnd Roth for necessary files and permission to use the NEURON model; and research associates Arielle Isakharov and Ada Zhang for outstanding technical support. We also thank Dr. Christopher Vaaga and Dr. Laurence Trussell for constructive comments on the manuscript. The contents of this manuscript do not represent the views of the U.S. Department of Veterans Affairs or the United States Government.
The authors declare no competing financial interests.
References
- Auger C, Attwell D (2000) Fast removal of synaptic glutamate by postsynaptic transporters. Neuron 28:547–558. 10.1016/S0896-6273(00)00132-X [DOI] [PubMed] [Google Scholar]
- Barclay J, Balaguero N, Mione M, Ackerman SL, Letts VA, Brodbeck J, Canti C, Meir A, Page KM, Kusumi K, Perez-Reyes E, Lander ES, Frankel WN, Gardiner RM, Dolphin AC, Rees M (2001) Ducky mouse phenotype of epilepsy and ataxia is associated with mutations in the Cacna2d2 gene and decreased calcium channel current in cerebellar Purkinje cells. J Neurosci 21:6095–6104. 10.1523/JNEUROSCI.21-16-06095.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergles DE, Dzubay JA, Jahr CE (1997) Glutamate transporter currents in bergmann glial cells follow the time course of extrasynaptic glutamate. Proc Natl Acad Sci U S A 94:14821–14825. 10.1073/pnas.94.26.14821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosman LW, Konnerth A (2009) Activity-dependent plasticity of developing climbing fiber-Purkinje cell synapses. Neuroscience 162:612–623. 10.1016/j.neuroscience.2009.01.032 [DOI] [PubMed] [Google Scholar]
- Brockhaus J, Schreitmüller M, Repetto D, Klatt O, Reissner C, Elmslie K, Heine M, Missler M (2018) α-neurexins together with α2δ-1 auxiliary subunits regulate Ca2+ influx through Cav2.1 channels. J Neurosci 38:8277–8294. 10.1523/JNEUROSCI.0511-18.2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brodbeck J, Davies A, Courtney JM, Meir A, Balaguero N, Canti C, Moss FJ, Page KM, Pratt WS, Hunt SP, Barclay J, Rees M, Dolphin AC (2002) The ducky mutation in Cacna2d2 results in altered Purkinje cell morphology and is associated with the expression of a truncated α2δ-2 protein with abnormal function. J Biol Chem 277:7684–7693. 10.1074/jbc.M109404200 [DOI] [PubMed] [Google Scholar]
- Brown JP, Gee NS (1998) Cloning and deletion mutagenesis of the alpha2delta calcium channel subunit from porcine cerebral cortex. J Biol Chem 273:25458–25465. 10.1074/jbc.273.39.25458 [DOI] [PubMed] [Google Scholar]
- Burroughs A, Wise AK, Xiao J, Houghton C, Tang T, Suh CY, Lang EJ, Apps R, Cerminara NL (2017) The dynamic relationship between cerebellar Purkinje cell simple spikes and the spikelet number of complex spikes. J Physiol 595:283–299. 10.1113/JP272259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantí C, Nieto-Rostro M, Foucault I, Heblich F, Wratten J, Richards MW, Hendrich J, Douglas L, Page KM, Davies A, Dolphin AC (2005) The metal-ion-dependent adhesion site in the von Willebrand factor-A domain of α2δ subunits is key to trafficking voltage-gated Ca2+ channels. Proc Natl Acad Sci U S A 102:11230–11235. 10.1073/pnas.0504183102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cassidy JS, Ferron L, Kadurin I, Pratt WS, Dolphin AC (2014) Functional exofacially tagged N-type calcium channels elucidate the interaction with auxiliary alpha2delta-1 subunits. Proc Natl Acad Sci U S A 111:8979–8984. 10.1073/pnas.1403731111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Li L, Chen SR, Chen H, Xie JD, Sirrieh RE, MacLean DM, Zhang Y, Zhou MH, Jayaraman V, Pan HL (2018) The α2δ-1-NMDA receptor complex is critically involved in neuropathic pain development and gabapentin therapeutic actions. Cell Rep 22:2307–2321. 10.1016/j.celrep.2018.02.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole RL, Lechner SM, Williams ME, Prodanovich P, Bleicher L, Varney MA, Gu G (2005) Differential distribution of voltage-gated calcium channel alpha-2 delta (α2δ) subunit mRNA-containing cells in the rat central nervous system and the dorsal root ganglia. J Comp Neurol 491:246–269. 10.1002/cne.20693 [DOI] [PubMed] [Google Scholar]
- Davie JT, Clark BA, Häusser M (2008) The origin of the complex spike in cerebellar Purkinje cells. J Neurosci 28:7599–7609. 10.1523/JNEUROSCI.0559-08.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dittman JS, Regehr WG (1998) Calcium dependence and recovery kinetics of presynaptic depression at the climbing fiber to Purkinje cell synapse. J Neurosci 18:6147–6162. 10.1523/JNEUROSCI.18-16-06147.1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolphin AC. (2012) Calcium channel auxiliary alpha2delta and beta subunits: trafficking and one step beyond. Nat Rev Neurosci 13:542–555. 10.1038/nrn3311 [DOI] [PubMed] [Google Scholar]
- Donato R, Page KM, Koch D, Nieto-Rostro M, Foucault I, Davies A, Wilkinson T, Rees M, Edwards FA, Dolphin AC (2006) The ducky(2J) mutation in Cacna2d2 results in reduced spontaneous Purkinje cell activity and altered gene expression. J Neurosci 26:12576–12586. 10.1523/JNEUROSCI.3080-06.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edvardson S, Oz S, Abulhijaa FA, Taher FB, Shaag A, Zenvirt S, Dascal N, Elpeleg O (2013) Early infantile epileptic encephalopathy associated with a high voltage gated calcium channelopathy. J Med Genet 50:118–123. 10.1136/jmedgenet-2012-101223 [DOI] [PubMed] [Google Scholar]
- Eroglu C, Allen NJ, Susman MW, O'Rourke NA, Park CY, Ozkan E, Chakraborty C, Mulinyawe SB, Annis DS, Huberman AD, Green EM, Lawler J, Dolmetsch R, Garcia KC, Smith SJ, Luo ZD, Rosenthal A, Mosher DF, Barres BA (2009) Gabapentin receptor alpha2delta-1 is a neuronal thrombospondin receptor responsible for excitatory CNS synaptogenesis. Cell 139:380–392. 10.1016/j.cell.2009.09.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fekete A, Nakamura Y, Yang YM, Herlitze S, Mark MD, DiGregorio DA, Wang LY (2019) Underpinning heterogeneity in synaptic transmission by presynaptic ensembles of distinct morphological modules. Nat Commun 10:826. 10.1038/s41467-019-08452-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fell B, Eckrich S, Blum K, Eckrich T, Hecker D, Obermair GJ, Münkner S, Flockerzi V, Schick B, Engel J (2016) α2δ2 controls the function and trans-synaptic coupling of Cav1.3 channels in mouse inner hair cells and is essential for normal hearing. J Neurosci 36:11024–11036. 10.1523/JNEUROSCI.3468-14.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao B, Sekido Y, Maximov A, Saad M, Forgacs E, Latif F, Wei MH, Lerman M, Lee JH, Perez-Reyes E, Bezprozvanny I, Minna JD (2000) Functional properties of a new voltage-dependent calcium channel. J Biol Chem 275:12237–12242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gee NS, Brown JP, Dissanayake VU, Offord J, Thurlow R, Woodruff GN (1996) The novel anticonvulsant drug, gabapentin (Neurontin), binds to α2δ subunit of a calcium channel. J Biol Chem 271:5768–5776. 10.1074/jbc.271.10.5768 [DOI] [PubMed] [Google Scholar]
- Geisler S, Schöpf CL, Stanika R, Kalb M, Campiglio M, Repetto D, Traxler L, Missler M, Obermair GJ (2019) Presynaptic α2δ-2 calcium channel subunits regulate postsynaptic GABAA receptor abundance and axonal wiring. J Neurosci 39:2581–2605. 10.1523/JNEUROSCI.2234-18.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison J, Jahr CE (2003) Receptor occupancy limits synaptic depression at climbing fiber synapses. J Neurosci 23:377–383. 10.1523/JNEUROSCI.23-02-00377.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto K, Kano M (1998) Presynaptic origin of paired-pulse depression at climbing fibre-Purkinje cell synapses in the rat cerebellum. J Physiol 506:391–405. 10.1111/j.1469-7793.1998.391bw.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto K, Ichikawa R, Kitamura K, Watanabe M, Kano M (2009) Translocation of a “winner” climbing fiber to the Purkinje cell dendrite and subsequent elimination of “losers” from the soma in developing cerebellum. Neuron 63:106–118. 10.1016/j.neuron.2009.06.008 [DOI] [PubMed] [Google Scholar]
- Hashimoto K, Tsujita M, Miyazaki T, Kitamura K, Yamazaki M, Shin HS, Watanabe M, Sakimura K, Kano M (2011) Postsynaptic P/Q-type Ca2+ channel in Purkinje cell mediates synaptic competition and elimination in developing cerebellum. Proc Natl Acad Sci U S A 108:9987–9992. 10.1073/pnas.1101488108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heffley W, Song EY, Xu Z, Taylor BN, Hughes MA, McKinney A, Joshua M, Hull C (2018) Coordinated cerebellar climbing fiber activity signals learned sensorimotor predictions. Nat Neurosci 21:1431–1441. 10.1038/s41593-018-0228-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoppa MB, Lana B, Margas W, Dolphin AC, Ryan TA (2012) α2δ expression sets presynaptic calcium channel abundance and release probability. Nature 486:122–125. 10.1038/nature11033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Indriati DW, Kamasawa N, Matsui K, Meredith AL, Watanabe M, Shigemoto R (2013) Quantitative localization of Cav2.1 (P/Q-type) voltage-dependent calcium channels in Purkinje cells: somatodendritic gradient and distinct somatic coclustering with calcium-activated potassium channels. J Neurosci 33:3668–3678. 10.1523/JNEUROSCI.2921-12.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanov SV, Ward JM, Tessarollo L, McAreavey D, Sachdev V, Fananapazir L, Banks MK, Morris N, Djurickovic D, Devor-Henneman DE, Wei MH, Alvord GW, Gao B, Richardson JA, Minna JD, Rogawski MA, Lerman MI (2004) Cerebellar ataxia, seizures, premature death, and cardiac abnormalities in mice with targeted disruption of the Cacna2d2 Gene. Am J Pathol 165:1007–1018. 10.1016/S0002-9440(10)63362-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaeser PS, Regehr WG (2017) The readily releasable pool of synaptic vesicles. Curr Opin Neurobiol 43:63–70. 10.1016/j.conb.2016.12.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khaliq ZM, Raman IM (2005) Axonal propagation of simple and complex spikes in cerebellar Purkinje neurons. J Neurosci 25:454–463. 10.1523/JNEUROSCI.3045-04.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurshan PT, Oztan A, Schwarz TL (2009) Presynaptic α2δ-3 is required for synaptic morphogenesis independent of its Ca2+-channel functions. Nat Neurosci 12:1415–1423. 10.1038/nn.2417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lein ES, Hawrylycz MJ, Ao N, Ayres M, Bensinger A, Bernard A, Boe AF, Boguski MS, Brockway KS, Byrnes EJ, Chen L, Chen L, Chen TM, Chin MC, Chong J, Crook BE, Czaplinska A, Dang CN, Datta S, Dee NR, et al. (2007) Genome-wide atlas of gene expression in the adult mouse brain. Nature 445:168–176. 10.1038/nature05453 [DOI] [PubMed] [Google Scholar]
- Li CY, Song YH, Higuera ES, Luo ZD (2004) Spinal dorsal horn calcium channel alpha2delta-1 subunit upregulation contributes to peripheral nerve injury-induced tactile allodynia. J Neurosci 24:8494–8499. 10.1523/JNEUROSCI.2982-04.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S, Friel DD (2008) Impact of the leaner P/Q-type Ca2+ channel mutation on excitatory synaptic transmission in cerebellar Purkinje cells. J Physiol 586:4501–4515. 10.1113/jphysiol.2008.156232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu HW, Trussell LO (2016) Spontaneous activity defines effective convergence ratios in an inhibitory circuit. J Neurosci 36:3268–3280. 10.1523/JNEUROSCI.3499-15.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsushita K, Wakamori M, Rhyu IJ, Arii T, Oda S, Mori Y, Imoto K (2002) Bidirectional alterations in cerebellar synaptic transmission of tottering and rolling Ca2. J Neurosci 22:4388–4398. 10.1523/JNEUROSCI.22-11-04388.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Missler M, Zhang W, Rohlmann A, Kattenstroth G, Hammer RE, Gottmann K, Südhof TC (2003) Alpha-neurexins couple Ca21 channels to synaptic vesicle exocytosis. Nature 423:939–948. 10.1038/nature01755 [DOI] [PubMed] [Google Scholar]
- Miyazaki T, Hashimoto K, Shin HS, Kano M, Watanabe M (2004) P/Q-type Ca2+ channel α1A regulates synaptic competition on developing cerebellar Purkinje cells. J Neurosci 24:1734–1743. 10.1523/JNEUROSCI.4208-03.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyazaki T, Yamasaki M, Hashimoto K, Yamazaki M, Abe M, Usui H, Kano M, Sakimura K, Watanabe M (2012) Cav2.1 in cerebellar Purkinje cells regulates competitive excitatory synaptic wiring, cell survival, and cerebellar biochemical compartmentalization. J Neurosci 32:1311–1328. 10.1523/JNEUROSCI.2755-11.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neher E. (2015) Merits and limitations of vesicle pool models in view of heterogeneous populations of synaptic vesicles. Neuron 87:1131–1142. 10.1016/j.neuron.2015.08.038 [DOI] [PubMed] [Google Scholar]
- Palay SL, Chan-Palay V (1974) Cerebellar cortex: cytology and organization. Berlin: Springer. [Google Scholar]
- Paukert M, Huang YH, Tanaka K, Rothstein JD, Bergles DE (2010) Zones of enhanced glutamate release from climbing fibers in the mammalian cerebellum. J Neurosci 30:7290–7299. 10.1523/JNEUROSCI.5118-09.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piochon C, Irinopoulou T, Brusciano D, Bailly Y, Mariani J, Levenes C (2007) NMDA receptor contribution to the climbing fiber response in the adult mouse Purkinje cell. J Neurosci 27:10797–10809. 10.1523/JNEUROSCI.2422-07.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pippucci T, Parmeggiani A, Palombo F, Maresca A, Angius A, Crisponi L, Cucca F, Liguori R, Valentino ML, Seri M, Carelli V (2013) A novel null homozygous mutation confirms CACNA2D2 as a gene mutated in epileptic encephalopathy. PLoS One 8:e82154. 10.1371/journal.pone.0082154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prescott C, Weeks AM, Staley KJ, Partin KM (2006) Kynurenic acid has a dual action on AMPA receptor responses. Neurosci Lett 402:108–112. 10.1016/j.neulet.2006.03.051 [DOI] [PubMed] [Google Scholar]
- Rasmussen A, Jirenhed DA, Zucca R, Johansson F, Svensson P, Hesslow G (2013) Number of spikes in climbing fibers determines the direction of cerebellar learning. J Neurosci 33:13436–13440. 10.1523/JNEUROSCI.1527-13.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Regehr WG, Mintz IM (1994) Participation of multiple calcium channel types in transmission at single climbing fiber to Purkinje cell synapses. Neuron 12:605–613. 10.1016/0896-6273(94)90216-X [DOI] [PubMed] [Google Scholar]
- Renzi M, Farrant M, Cull-Candy SG (2007) Climbing-fibre activation of NMDA receptors in Purkinje cells of adult mice. J Physiol 585:91–101. 10.1113/jphysiol.2007.141531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Risher WC, Kim N, Koh S, Choi JE, Mitev P, Spence EF, Pilaz LJ, Wang D, Feng G, Silver DL, Soderling SH, Yin HH, Eroglu C (2018) Thrombospondin receptor alpha2delta-1 promotes synaptogenesis and spinogenesis via postsynaptic Rac1. J Cell Biol 217:3747–3765. 10.1083/jcb.201802057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenmund C, Legendre P, Westbrook GL (1992) Expression of NMDA channels on cerebellar Purkinje cells acutely dissociated from newborn rats. J Neurophysiol 68:1901–1905. 10.1152/jn.1992.68.5.1901 [DOI] [PubMed] [Google Scholar]
- Roth A, Häusser M (2001) Compartmental models of rat cerebellar Purkinje cells based on simultaneous somatic and dendritic patch-clamp recordings. J Physiol 535:445–472. 10.1111/j.1469-7793.2001.00445.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudolph S, Overstreet-Wadiche L, Wadiche JI (2011) Desynchronization of multivesicular release enhances Purkinje cell output. Neuron 70:991–1004. 10.1016/j.neuron.2011.03.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneggenburger R, Meyer AC, Neher E (1999) Released fraction and total size of a pool of immediately available transmitter quanta at a calyx synapse. Neuron 23:399–409. 10.1016/S0896-6273(00)80789-8 [DOI] [PubMed] [Google Scholar]
- Tong XJ, López-Soto EJ, Li L, Liu H, Nedelcu D, Lipscombe D, Hu Z, Kaplan JM (2017) Retrograde synaptic inhibition is mediated by alpha-neurexin binding to the alpha2delta subunits of N-type calcium channels. Neuron 95:326–340.e5. 10.1016/j.neuron.2017.06.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wadiche JI, Jahr CE (2001) Multivesicular release at climbing fiber-Purkinje cell synapses. Neuron 32:301–313. 10.1016/S0896-6273(01)00488-3 [DOI] [PubMed] [Google Scholar]
- Wang T, Jones RT, Whippen JM, Davis GW (2016) α2δ-3 is required for rapid transsynaptic homeostatic signaling. Cell Rep 16:2875–2888. 10.1016/j.celrep.2016.08.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Lisberger SG (2014) Purkinje-cell plasticity and cerebellar motor learning are graded by complex-spike duration. Nature 510:529–532. 10.1038/nature13282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B, Chen LY, Liu X, Maxeiner S, Lee SJ, Gokce O, Südhof TC (2015) Neuroligins sculpt cerebellar Purkinje-cell circuits by differential control of distinct classes of synapses. Neuron 87:781–796. 10.1016/j.neuron.2015.07.020 [DOI] [PMC free article] [PubMed] [Google Scholar]