

Abstract
The present study posits that Alzheimer’s disorder is a “fast” disease. This is in sharp contrast to a view, prevailing until now, that Alzheimer’s Disease (AD) is a quintessential “slow” disease that develops throughout the life as one prolonged process. According to this view, beta-amyloid (Aβ) is produced and secreted solely by the beta-amyloid precursor protein (βAPP) proteolytic/secretory pathway. As its extracellular levels increase, it triggers neurodegeneration starting relatively early in life. Damages accumulate and manifest, late in life in sporadic Alzheimer’s Disease (SAD) cases, as AD symptoms. In familial AD (FAD) cases, where mutations in βAPP gene or in presenilins increase production of either common Aβ isoform or of its more toxic isoforms, neurodegeneration reaches critical threshold sooner and AD symptoms occur earlier in life, mostly in late 40s and 50s. There are currently no preventive AD therapies but if they were available, according to this viewpoint it would be largely futile to intervene late in life in case of potential SAD or at mid-age in cases of FAD because, although AD symptoms have not yet manifested, the damage has already occurred during the preceding decades. In this paradigm, to be effective, preventive therapeutic intervention should be initiated early in life. The outlook suggested by the present study is radically different. According to it, Alzheimer’s disease evolves in two stages. The first stage is a slow process of intracellular beta-amyloid accumulation. It occurs via βAPP proteolytic/secretory pathway and cellular uptake of secreted Aβ common to Homo sapiens, including healthy humans, and to non-human mammals, and results neither in significant damage, nor in manifestation of the disease. The second stage occurs exclusively in humans, commences shortly before symptomatic onset of the disease, sharply accelerates the production and increases intracellular levels of Aβ that is not secreted but is retained intracellularly, generates significant damages, triggers AD symptoms, and is fast. It is driven by an Aβ generation pathway qualitatively and quantitatively different from βAPP proteolytic process and entirely independent of beta-amyloid precursor protein, and results in rapid and substantial intracellular accumulation of Aβ, consequent significant neurodegeneration, and symptomatic AD. In this paradigm, a preventive therapy for AD, an AD “statin”, would be effective when initiated at any time prior to commencement of the second stage. Moreover, there are good reasons to believe that with a drug blocking βAPP-independent Aβ production pathway in the second stage, it would be possible not only to preempt the disease but also to stop and to reverse it even when early AD symptoms have already manifested. The present study posits a notion of AD as a Fast Disease, offers evidence for the occurrence of the AD-specific Aβ production pathway, describes cellular and molecular processes constituting an engine that drives Alzheimer’s disease, and explains why non-human mammals are not susceptible to AD and why only a subset of humans develop the disease. It establishes that Alzheimer’s disease is preventable by therapeutic intervention initiated even late in life, details a powerful mechanism underlying the disease, suggests that Aβ produced in the βAPP-independent pathway is retained intracellularly, elaborates why neither BACE inhibition nor Aβ immunotherapy are effective in treatment of AD and why intracellularly retained beta-amyloid could be the primary agent of neuronal death in Alzheimer’s disease, necessitates generation of a novel animal AD model capable of producing Aβ via βAPP-independent pathway, proposes therapeutic targets profoundly different from previously pursued components of the βAPP proteolytic pathway, and provides conceptual rationale for design of drugs that could be used not only preemptively but also for treatment and reversal of the early stages of the disease.
Keywords: Alzheimer’s disease, Precursor-independent generation of beta-amyloid, Asymmetric RNA-dependent beta-APP mRNA amplification, Mitochondrial dysfunction-related stresses
Introduction
Amyloid Cascade Hypothesis (ACH), the widely accepted theory of Alzheimer’s Disease (AD), postulates that, upon reaching a critical threshold, an increased production and consequent accumulation of beta-amyloid (Aβ) trigger and drive a cascade of molecular and cellular events resulting in the disease. Within conceptual framework of the ACH, the Alzheimer’s disorder is viewed as an epitope of a “slow” disease; although AD symptoms manifest themselves late in life in sporadic form of the disease and in 40s-50s in its hereditary type, the damage presumably occurs starting early in life and builds up for decades. To attain better perspective of cellular events involved in the origination and development of AD, it is instructive to address the apparent exclusivity of the disease to Homo sapiens. Two features are of key importance in humans versus non-human mammals comparison. First, all non-human mammals tested to date accumulate Aβ as they age but do not develop AD. This is true for small-sized, short-lived animals such as mice, and for large-sized, long-lived non-human mammals such as elephants. In contrast, humans do both. It appears, therefore, that the extent of increase in beta-amyloid levels sufficient to trigger amyloid cascade culminating in AD can be reached physiologically in humans but not in non-human mammals. This discordance could be easily explained if different pathways of generation of beta-amyloid were employed in these instances. This is not the case, however: The same Aβ production pathway, proteolysis of beta-amyloid precursor protein described in the following section, is utilized in non-human mammals and in healthy humans, as well as in AD patients. Second feature of key importance is that when the disease is forced, at least symptomatically, upon non-human mammals by genetic manipulations that drastically increase beta-amyloid production by the normally employed precursor protein proteolysis, treatment with agents blocking Aβ-generating proteolytic pathway, designed and developed within the conceptual framework of ACH, mitigate and even reverse both neurodegeneration and the AD symptoms. Conversely, the same treatments are completely ineffective in human Alzheimer’s patients. Taken together, the above considerations leave us with an alternative explanation: In AD, in addition to beta-amyloid production pathway common to non-human mammals and healthy humans, another, qualitatively different, pathway of Aβ generation is in operation, and it is this pathway that drives Alzheimer’s disease.
Pathway of Beta-Amyloid Generation Common to Humans and Non-Human Mammals
Beta-amyloid, the peptide associated with and widely believed to have a pivotal early role in etiology of Alzheimer’s disease, was shown to be generated, both in humans and in non-human mammals, by proteolytic cleavages of a much larger molecule, beta-amyloid precursor protein, βAPP. This precursor can be cleaved by three enzymes designated alpha- beta- and gamma-secretases, each at its specific position. The cleavages can proceed in two separate pathways, with only one generating Aβ. In the non-amyloidogenic proteolytic pathway, a cleavage by the alpha-secretase enzyme occurs within the Aβ-containing segment of βAPP, thus preventing the generation of beta-amyloid. In the amyloidogenic proteolytic pathway, two sequential cleavages of βAPP are involved in the production of Aβ. The first is a cleavage of βAPP by the beta-secretase enzyme. It occurs between residues 671 and 672 of the βAPP molecule (isoform 770 numbering), generating the N-terminus of Aβ, yielding the 12kDa membrane-bound C-terminal fragment, C99 (residues 672–770), releasing a large ectodomain of βAPP, soluble sAPPβ (residues 1–671), and precluding activity of alpha-secretase which cleaves βAPP within its Aβ-containing segment but cannot cut within C99 or Aβ [1–3]. The second cleavage, by gamma-secretase activity, occurs at one of closely clustered multiple sites within C99 around 40 amino acids downstream from its N-terminus and generates the C-terminus of Aβ. Thus released, Aβ is secreted from the cell. The size of Aβ ranges from 36 to 43 amino acids, with Aβ40 being the most abundant species normally formed. Studies of the inherited forms of the disease, FAD (Familial Alzheimer’s Disease), strongly indicated that cerebral Aβ accumulation is essential for and underlies the etiology of the disease [4–6]. This notion, formalized in a theory of AD known as “Amyloid Cascade Hypothesis”, [7–12], has become the dominant model of AD pathogenesis and has guided the development of potential treatments. Most therapeutic strategies attempted to date have been based on this model and virtually all preclinical tests and clinical trials discussed below have been designed within the framework of ACH. Over two hundred autosomal dominant mutations associated with FAD have been identified in genes for βAPP and presenilins, the components of gamma-secretase complex [6]. In βAPP gene, most of the mutations cluster around alpha-, beta-, and gamma-secretases cleavage sites and increase either the production of total Aβ or the relative proportion of a more neurotoxic 42-residue form of Aβ, Aβ42. In terms of the ACH, there is little doubt that abnormal processing of βAPP and increased production of total Aβ or its 42-amino acid isoform are pivotal events in the pathogenesis of FAD. Although the number of individuals affected by FAD is substantial, in relative terms this form of the disease is quite rare, representing less than 5%, in fact less than 1% by some estimates, of the total Alzheimer’s disease burden [5,14,15]. Since the pathological lesions and symptoms in the non-hereditary form of the disease, SAD (Sporadic Alzheimer’s Disease), are analogous to those seen in the familial forms, it has been assumed that abnormal amyloidogenic proteolytic processing of βAPP of a type seen in FAD also underlies the pathogenesis of SAD [4,5]. The assumption that ACH applies to both forms of AD implied that any therapeutic approach effective in FAD would also be successful in treatment of SAD.
Evidence that in Alzheimer’s Disease Aβ is Produced by an Additional Pathway, Absent in Non-Human Mammals and in Healthy Humans, and Independent of βAPP
Success of beta-secretase inhibitors in preclinical tests
The elucidation of βAPP proteolytic pathway resulting in generation of Aβ suggested rational design of a treatment for Alzheimer’s disease. Indeed, in light of the above discussion, beta-secretase activity was viewed as a strategic target of choice: Inhibit beta-secretase cleavage and there is no beta-amyloid. Moreover, such inhibition would shift the equilibrium between alpha- and beta-secretase cleavages toward the former, thus augmenting its efficacy. Therefore, since the identification of beta-site APP-cleaving enzyme (BACE) as beta-secretase [16–18], it became the primary therapeutic target for treatment of AD. Designing BACE-inhibiting agents presented major challenges of cell penetration, oral bioavailability, metabolic clearance, and brain access, but intense efforts, mainly by the pharmaceutical industry, led to development of a number of brain-penetrant small-molecule BACE inhibitors that have been vigorously investigated. The results obtained in the early investigations of BACE inhibition, first appearing around 2007 [19–27], are truly striking. As an example, Merck researchers reported in 2012 the discovery of “compound 16”, which robustly reduced cortex and CSF levels of Aβ when administered orally to rats [28]. Continuous efforts to improve upon “compound 16” culminated in the development of verubecestat (MK-8931). Preclinical tests of this agent achieved dramatic results [29]. Levels of Aβ and sAPPβ were reduced by up to 90% in plasma, brain, and CSF after even a single administration of verubecestat to healthy subjects including rats, monkeys, and humans [29]. The acute reduction of over 80% in CSF and cortical Aβ and sAPPβ produced by verubecestat was maintained after chronic administration for nine months in monkeys [29]. Because of its favorable initial safety profile and its ability to markedly reduce cerebral and CSF Aβ and sAPPβ concentrations, verubecestat was the first BACE inhibitor to progress to phase III clinical trials. Preclinical evaluation of a number of independently developed BACE inhibitors, such as BI 1181181, LY2811376, LY 2886721, AZD3293 (lanabacestat, LY3314814), CNP520, E2609 (elenbacestat), JNJ-54861911, CTS-21166, HPP854, PF-05297909, RG7129, TAK-070, VTP-37948 yielded similarly impressive results in animals and healthy volunteers and all these agents have entered clinical trials.
BACE inhibition rescues functional impairments in animal models of Alzheimer’s disease
With the ability to significantly reduce the production and lower the levels of Aβ thus established, the question remained whether such a reduction would translate into a “treatment” of the disease. This question was answered resolutely and convincingly, with animal models bioengineered to mimic FAD and overproducing Aβ solely by βAPP proteolytic pathway [86], in two recent studies using different approaches to inhibit beta-secretase activity. One study utilized BACE inhibitor NB-360 [30]. It was based on a previous study [31] showing NB-360 to be a potent, brain penetrable BACE inhibitor capable of completely blocking Aβ deposition in the brains of βAPP transgenic mice, as well as of rats and dogs. Moreover, this inhibitor blocked accumulation of activated inflammatory cells in the brains of βAPP transgenic mice. The more recent study with NB-360 [30] further assessed the notion that suppression of Aβ production can have beneficial downstream effects on the progression of Alzheimer’s disease. Using histochemistry, in vivo imaging, and behavioral analyses in a mouse model of AD, the authors demonstrated that along with reducing prefibrillary Aβ surrounding plaques, the inhibition of BACE activity rescued neuronal hyperactivity, impaired long-range circuit function and memory defects. That all these effects were due to inhibition of Aβ production was strongly indicated by the observation that functional neuronal impairments reappeared after infusion of soluble Aβ [30].
In the second study [32], mimicking BACE1 inhibition in adult organisms, the authors generated BACE1 conditional knockout (BACE1fl/fl) mice and bred them with ubiquitin-Cre mice to induce deletion of BACE1 after passing early developmental stages. Strikingly, sequential deletions of BACE1 in an adult AD mouse model were capable of reversing amyloid deposition and resulted in significant improvement in gliosis and neuritic dystrophy. Moreover, in correlation with amyloid plaque reversal, it also significantly improved synaptic functions, as was determined by long-term potentiation and contextual fear conditioning experiments. These studies offered great hope that sustained inhibition of BACE1 activity can constitute a treatment, or at least be beneficial, for AD patients. This assumption was tested in several clinical trials.
BACE inhibition is completely ineffective in Alzheimer’s disease
The results of clinical trials of BACE inhibitors in humans, however, do not support this assumption. All BACE inhibitor clinical trials that ended to date, ended in failure. Some trials, such as that of BI 1181181, LY2811376, LY2886721 and RG7129, were terminated because of technical and safety issues. On the other hand, there were no such issues in the trials of the verubecestat (MK-8931). This agent was shown to be very efficient in suppressing Aβ production in preclinical tests and was proven safe in clinical trials. Yet, its Phase III, 2000 patient-strong “EPOCH” trial in mild to moderate AD patients was terminated prematurely for the lack of efficacy, with an interim analysis by an external data-monitoring committee giving the trial “virtually no chance of finding a positive effect”. Similarly, a separate large Phase III clinical trial of verubecestat in prodromal AD patients, the “APECS”, was too terminated prematurely and for the same reason; lack of efficacy. The clinical trials of several other BACE inhibitors are still in progress but the verubecestat results do not inspire confidence in their successful outcome.
Results of clinical trials in humans can be explained by βAPP-independent and therefore BACE inhibition-insensitive generation of beta-amyloid in Alzheimer’s disease
One rational explanation for the strikingly different effects of BACE inhibition in animal models and healthy human subjects and in Alzheimer’s disease is that in AD, in addition to conventional βAPP/beta-secretase-dependent component of Aβ production that operates in non-human mammals, healthy humans and in AD models described above, there is another, unconventional, Aβ-generating component in operation, possibly facilitated or enabled by epigenetic changes associated with the disease [33], which is βAPP–independent and thus bypasses the requirement for beta-secretase activity. In these AD cases, administration of effective BACE inhibitors would suppress the βAPP-dependent component, but would have no effect on the second, βAPP- and beta-secretase-independent, component. The extent of suppression of total Aβ production by BACE inhibitors would depend on the relative input of two components in the generation of Aβ, and if the input of the second significantly exceeds that of the first component, BACE inhibitors would be ineffective both in lowering Aβ levels and in the treatment of Alzheimer’s disease.
To summarize, taken together, the results of BACE inhibition studies and trials indicate the following: (a) Only one Aβ production pathway, that of βAPP proteolysis by beta- and gamma-secretases, is operating in non-human mammals and in healthy humans; (b) In addition to βAPP proteolytic pathway common to animals and healthy humans, another pathway is in operation in Alzheimer’s disease; (c) The additional Aβ production pathway operating in AD is qualitatively different in that it is βAPP-independent; and (d) This additional pathway is also quantitatively different in that it is the predominant process of beta-amyloid production in Alzheimer’s disease.
Additional βAPP-independent Aβ Production Pathway Operating in Alzheimer’s Disease: Asymmetric amplification of βAPP mRNA leading to generation of C99 fragment of βAPP
In considering a mechanism underlying the postulated additional pathway of Aβ production in AD, one requirement clearly stands out. Since it is presumably βAPP- and beta-secretase-independent, the N-terminus of the resulting polypeptide should be absolutely precisely that of Aβ (it can be longer than Aβ at its C-end and be trimmed to size by gamma-secretase cleavage). This is an exceedingly tall order yet, as described below, it can be accomplished by a process known as “chimeric RNA-dependent mammalian mRNA amplification” [34,48,49].
“Chimeric” pathway of mammalian RNA-dependent mRNA amplification
De novo production of RNA on an RNA template, a process known as RNA-dependent RNA synthesis (RdRs) and the enzymatic activity conducting it, RNA-dependent RNA polymerase (RdRp), were initially considered to be exclusively virus-specific. Eventually, however, the occurrence of RdRs and the ubiquitous presence of conventional RdRp were demonstrated in numerous eukaryotic organisms [39]. The evidence that the enzymatic machinery capable of RdRs is present in mammalian cells was derived from studies of viruses, such as hepatitis delta virus, HDV, that do not encode RdRp, yet undergo a robust RNA replication once inside the mammalian host [39–41]; thus firmly establishing its occurrence and functionality. Moreover, it became clear that RdRp activity, apparently in a non-conventional form [42–44], is constitutively present in most, if not in all, mammalian cells. Because such activity was shown to produce short transcripts, because of its apparent involvement in RNA interference phenomena, and because double-stranded RNA is known to trigger cellular responses leading to its degradation, it was generally assumed that its role in mammalian cells is restricted to a regulatory function. However, at the same time, an enzymatic activity capable of generating complete antisense RNA complements of mRNAs was discovered in mammalian cells undergoing terminal differentiation [45]. Moreover, observations of widespread synthesis of antisense RNA initiating at the 3’poly(A) of mRNAs in human cells [46] suggested an extensive cellular utilization of mammalian RdRp activity. These results led to the development of a model of RdRp-facilitated and antisense RNA-mediated amplification of mammalian mRNA [47–49]. Recent detection of the major model-predicted identifiers, chimeric RNA intermediates containing both sense and antisense RNA strands covalently joined in a rigorously predicted and uniquely defined manner [49,50], as well as the identification of a putative chimeric RNA end product of this process [49], validated the proposed model.
The process of RNA-dependent amplification of mammalian mRNA is described in detail elsewhere [48,49]. Here, it is diagrammed in Figure 1 below and can be briefly summarized as follows. The amplification process occurs in the cytoplasm and starts with transcription of the antisense complement from a conventional spliced mRNA template, initiating at the 3’poly(A), possibly with the help of a uridilated protein, as seen in viral RdRs [51] (Figure 1, Step 1), and terminating at the 3’end with the “C”, a transcript of the capG of mRNA [48–50]. Generation of a complete antisense transcript requires the presence of an eligible RNA template and a compatible polymerase activity. The only major prerequisite for a potential RNA template appears to be the presence of the poly(A) segment at its 3’ terminus [46–49]. The compatible polymerase activity is RdRp. The RdRp activity in mammalian cells appears to be non-conventional; two possible candidates for this role are the RNA polymerase II complex or its components [42,43] and RdRp activity of the TERT complex [44], both ubiquitously present in all cells.
Figure 1: Projected stages of the chimeric pathway of RdRp-facilitated, antisense RNA-mediated amplification of mammalian mRNA.
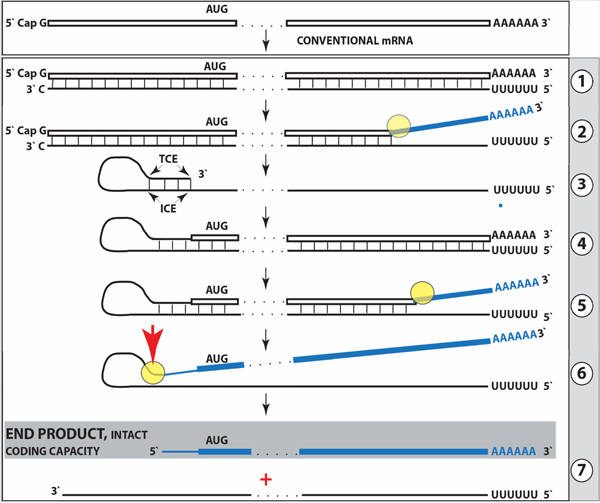
Top panel: Conventional, genome-originated mRNA molecule. Bottom panel: Projected stages of antisense RNA-mediated mRNA amplification. Boxed line – sense strand RNA. Single line – antisense strand RNA. “AUG” – functional translation initiation codon (could be other than “AUG”). “TCE”– 3’-terminal complementary element; “ICE”– internal complementary element, both on the antisense RNA strand. Yellow circle – helicase/modifying activity complex. Blue lines (both single and boxed) – RNA strand modified and separated from its complement by a helicase complex. Red arrowhead – position of cleavage of the chimeric intermediate. Step 1: Synthesis of antisense strand; step 2: Strand separation; step 3: Folding of antisense strand into self-priming configuration; step 4: Extension of self-primed antisense RNA; step 5: Strand separation; step 6: Cleavage of the chimeric intermediate; step 7: End-products of amplification. Note that chimeric RNA end product retains the intact coding capacity of conventional mRNA.
Under regular circumstances, the RdRp activity in mammalian cells produces only short antisense RNA transcripts. For example, a widespread synthesis of diverse short antisense RNA transcripts initiating at the 3’poly(A) of mRNA was observed in human cells [46]. On the other hand, RdRp activity isolated from rabbit reticulocytes [45] was able to produce, in assays, long antisense RNA transcripts. Subsequent studies identified full-length antisense transcripts of globin mRNA in erythroid cells [47–49]. It could be argued that the component responsible for the production of long antisense transcripts in mammalian cells is a processivity conferring co-factor of RdRp activity that is induced under special circumstances when overproduction of specific proteins is required [48–50]. The notion of a processivity co-factor is strongly supported by studies of HDV replication in “normal” (i.e. apparently lacking processivity co-factor) mammalian cells [39–41]. Within the framework of the above considerations, the ability of RdRp-deficient viruses to use RdRp activity of mammalian cell for their replication implies that they should encode a processivity co-factor of cellular RdRp. In case of HDV, it appears to be hepatitis delta antigen HDAg, the only protein encoded by HDV. HDAg is essential both for production of long transcripts by cellular RdRp, and for viral replication [41]. In its absence only short transcripts are generated [41]. These observations provide a proof of concept for the notion of RdRp processivity co-factor, central for our understanding of mammalian mRNA amplification. Identification of a cellular homolog of HDAg, DIPA [52,53] suggests directions for a search for the cellular RdRp processivity co-factor.
The resulting double stranded sense/antisense structure is then separated into single-stranded molecules by a helicase activity that mounts the poly(A) segment of the 3’poly(A)-containing strand (the sense-oriented strand) of the double helical structure and proceeds along this strand, modifying, on average, every fifth nucleotide in the process [48,49] (Figure 1, Step 2). Only purines, the “A” and the “G” appear to be modified in the separation/modification process [48,49]. The 5’ poly(U)-containing antisense strand remains unmodified during and after the separation [47–49]; this being essential, as described below, for the production of a new sense strand since modifications were shown to interfere with and would preclude complementary interactions required in this process [48,49].
The vast majority of mammalian mRNA species contains 3’-terminal poly(A) segments. The notion that many, or possibly most, of them could be eligible templates for RdRp was suggested in our previous studies [47–49]. Subsequent observations by Kapranov et al. [46] showed a widespread synthesis of antisense RNA initiating, apparently indiscriminately, at the 3’ poly(A) of mRNA in human cells. This, seemingly undiscerning, RdRp template eligibility of the bulk of mammalian mRNA species raises questions with regard to mechanisms underlying the manifestly stringent specificity of the mRNA amplification process [47–49]. The specificity of mRNA amplification appears to be determined at the 3’ terminus of an antisense transcript by its ability, or inability, to support production of a complementary sense strand RNA molecule, the end product of the amplification process.
The generation of a sense strand on an antisense template occurs via the extension of the 3’ terminus of a self-primed antisense template and requires the presence within the antisense transcript of two spatially independent complementary elements [48,49,54]. One of these is the strictly 3’-Terminal Complementary Element (TCE), the other is the Internal Complementary Element (ICE). These elements (Figure 1, Step 3) must be complementary to a sufficient extent to form a priming structure but may contain mismatches and utilize unconventional G/U pairings. The generation of a sense strand also requires the thermodynamic feasibility, enhanced/enabled by the occurrence of two complementary and topologically compatible elements, of the antisense strand folding into a self-priming configuration.
Provided that a self-priming structure is formed, the 3’ end of the folded antisense strand is extended by RdRp into a sense-oriented molecule terminating with the poly(A) at the 3’end (Figure 1, Step 4), thus generating a hairpin-structured chimeric intermediate consisting of covalently joined sense and antisense strands. The double stranded portion of the resulting structure is separated by a helicase activity invoked above, which mounts the 3’poly(A) of a newly synthesized sense strand component of the chimeric intermediate and proceeds along this strand in the 5’ direction, modifying the molecule as it advances (Figure 1, Step 5). When the helicase activity reaches a single stranded portion of the hairpin structure, it, or associated activities, cleave the molecule either within the TCE, at a TCE/ICE mismatch, or immediately upstream of the TCE; the cleavage occurs between the 5’ hydroxyl group and the 3’ phosphate [48,49] (red arrowhead, Figure 1, Step 6).
Strand separation, in conjunction with the cleavage, produces two single-stranded molecules (Figure 1, Step 7) one of which is a chimeric mRNA, the functional mRNA end product of amplification and the basis for defining this pathway as the “chimeric”. The chimeric nature of this end product is due to the presence at its 5’ end of a 3’-terminal segment of the antisense strand consisting, depending on the site of cleavage of the chimeric intermediate, of either the entire TCE or a portion thereof covalently attached, in a 5’ to 3’ orientation, to the 5’-truncated sense strand. This chimeric molecule is modified and 3’ polyadenylated. In contrast to conventional mRNA that can be repeatedly used as RdRp template [48,49], it cannot be further amplified because its antisense complement would be lacking the TCE but can be translated into the conventional mRNA-encoded polypeptide [48,49]. In the chimeric pathway of mRNA amplification, the cleavage of the chimeric intermediate following the strand separation and the associated modification of its poly(A)-containing component of the double-stranded structure is the ultimate act in the generation of the chimeric mRNA end product. Consequently, it is formed already modified and is never present in the unmodified form. Therefore, because the modified amplified RNA is resistant to reverse transcription [48,49], it cannot be detected by conventional reverse transcription-based sequencing methods.
The chimeric RNA-dependent mRNA amplification process, illustrated in Figure 1 above, results in an mRNA molecule containing the entire protein-coding region of a conventional, genome-transcribed, mRNA that can be translated into the original, conventional mRNA-encoded, polypeptide as was described elsewhere [48,49]. If this process were to apply to βAPP mRNA, it would result in the complete βAPP polypeptide. However, in the proposed scenario of βAPP-independent generation of beta-amyloid in Alzheimer’s disease, the expected translational outcome is only a segment of amyloid precursor protein, its C-Terminal Fragment (CTF). Such an outcome can indeed be achieved in an asymmetric RNA-dependent mRNA amplification pathway.
Asymmetric RNA-dependent mRNA amplification pathway
Above, a scenario was discussed where both complementary elements required for an appropriate folding and self-priming of the antisense strand, TCE and ICE, are located within its segment corresponding to the 5’UTR of a conventional genome-encoded mRNA. In such a situation, depicted in steps 3 trough 7 of Figure 2, the chimeric RNA end product contains the entire protein coding region of a conventional mRNA and can be translated into the original, conventional mRNA-encoded, polypeptide. In the chimeric mRNA amplification pathway, the position of the TCE within the antisense molecule is fixed; it is always strictly 3’-terminal. In contrast, the intramolecular location of the ICE is variable, and potentially it can be positioned within a segment of the antisense strand corresponding to the coding portion of an mRNA, a scenario diagrammed in steps 3’ trough 7’ of Figure 2. In this scenario, the chimeric RNA end product would consist of a 3’-terminal segment of the antisense strand (the TCE or its fraction) and a 3’ portion of a conventional mRNA progenitor with a 5’-truncated coding region. In such a case, the translational outcome would be decided by the position of the first functional (capable of initiation of translation) AUG or other translation initiation-competent codon. If it were in-frame with the protein-encoding information content of conventional mRNA, translation would result in the C-terminal fragment, CTF, of a conventionally encoded polypeptide. This variant of RNA-dependent mRNA amplification pathway would be asymmetric. Indeed, only one end, a 3-terminal portion, of conventional mRNA would be amplified, and its translation would produce only one end of conventional genome-encoded polypeptide, its C-terminal fragment. With regard to possible βAPP-independent production of beta-amyloid in AD, three crucial questions are: (1) Is human βAPP mRNA eligible for RNA-dependent amplification? (2) If it is, does amplification occur asymmetrically? (3) If affirmative, would the N-terminus of the resulting polypeptide be precisely that of Aβ?
Figure 2: RNA-dependent mRNA amplification can result in a 5’-truncated molecule encoding C-terminal fragment of a conventionally encoded polypeptide.
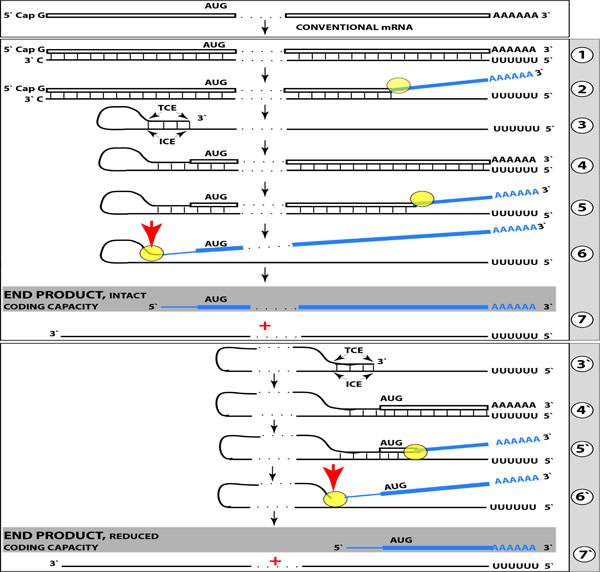
Boxed line-sense strand RNA. Single line-antisense strand RNA. “AUG”- functional translation initiation codon (could be other than AUG). “TCE”–3’-terminal complementary element; “ICE”– internal complementary element, both on the antisense RNA strand. Yellow circle – helicase/ modifying activity complex. Blue lines (both single and boxed) – RNA strand modified and separated from its complement by a helicase complex. Red arrow – position of cleavage of the chimeric intermediate. Step 1: Synthesis of antisense strand; step 2: Strand separation; step 3: Folding of antisense strand into self-priming configuration; step 4: Extension of self-primed antisense RNA; step 5: Strand separation; step 6: Cleavage of the chimeric intermediate; step 7: End-products of RNA amplification. Steps 3’–7’ correspond to steps 3–7. Top panel: Conventional, genome-transcribed mRNA molecule. Middle panel: Projected stages of RNA-dependent mRNA amplification. “ICE” is located within a segment of antisense RNA corresponding to the 5’UTR of conventional mRNA; the chimeric RNA end product contains the entire coding content of conventional mRNA. Bottom panel: “ICE” is located within a segment of antisense RNA corresponding to the coding region of conventional mRNA. The amplified chimeric end product contains a 5’-truncated coding region of conventional mRNA. The translational outcome is decided by position of the first functional translation initiation codon; if in-frame, a CTF of conventional polypeptide is produced.
Potentially pivotal role of the AUG encoding Met671 in conventional human βAPP mRNA in βAPP-independent generation of Aβ in Alzheimer’s disease
The answer to question (3) above is suggested by the primary structure of the human βAPP mRNA. In this molecule, the Aβ-encoding segment is preceded immediately, contiguously, and in-frame by the AUG codon normally encoding methionine in position 671 of the βAPP (isoform 770 numbering). If translation were initiated at this position, it would produce 12kDa C-terminal βAPP fragment (C99, after the removal of methionine by the N-terminal methionine aminopeptidase) independently of βAPP. Interestingly, the AUG in question is situated within a nucleotide context optimal for the initiation of translation (an “A” in position −3 and a “G” in position +4 relative to the “A” of the AUG codon). In fact, of the twenty AUG codons encoding methionine residues in the human βAPP mRNA, only the AUG encoding Met671 (not even the AUG encoding Met1) is located within an optimal translation initiation context. Such favorable positioning of the AUG encoding Met671 of βAPP was the basis for a proposal that in Alzheimer’s disease, C99 fragment may be generated independently from βAPP by the internal initiation of translation of the intact βAPP mRNA [37]. Such precursor-independent generation of C99 would be an efficient way to overproduce Aβ. This is because (a) C99 is not susceptible to the alpha-secretase cleavage [1–3], and (b) cleavage by gamma-secretase was shown to be not the rate-limiting step in the production of Aβ [1–3]. The possibility of internal initiation of translation, proposed by Breimer and Denny [37], has been, however, subsequently ruled out by experiments of Citron and co-investigators [38].
On the other hand, the implications of the occurrence of an AUG codon in such a position for βAPP-independent generation of Aβ are clear: If human βAPP mRNA is eligible for RNA-dependent amplification, if it is amplified in an asymmetric manner in AD, and if in the resulting 5’-truncated chimeric mRNA the first, 5’-most, functional translation initiation codon were the AUG encoding Met671 in the conventional βAPP mRNA, the translational outcome would be the C99 fragment of amyloid precursor protein produced independently of βAPP and containing Aβ at its N-terminus. Is such an outcome feasible?
Projected pathway of asymmetric amplification of βAPP mRNA resulting in chimeric mRNA encoding C99 fragment of amyloid precursor protein
With regard to questions (1) and (2) above, to determine if an mRNA species of interest can potentially be a subject of RNA-dependent mRNA amplification (provided that cellular RdRs machinery is activated), one needs to assess whether its antisense complement contains both TCE and ICE and is capable of folding into a self-priming configuration. If it were, the position of the ICE would indicate the possible translational outcome. Such an assessment can be conducted in a model experiment where an mRNA of interest serves as a template for synthesis of cDNA, initiating at the 3’-terminal poly(A), and is subsequently removed by RNAse H activity present in a preparation of reverse transcriptase used. If an mRNA were fully transcribed, if complementary elements were present within the antisense strand (cDNA), if one of them were 3’-terminal, and if they were topologically compatible, self-priming and the extension synthesis of a segment of the sense strand would occur. The junction between the antisense and sense components would define the site of self-priming and facilitate identification of the TCE and ICE. Just such an experiment was inadvertently carried out with human βAPP mRNA [55]. The results of this experiment, misinterpreted and eventually dismissed by the authors as an artifact [56], indicated the occurrence of topologically compatible TCE and ICE within the antisense strand of βAPP mRNA and defined their sequence, as well as the position of self-priming. Based on these results, the TCE/ICE-guided folding of the antisense strand of βAPP mRNA [34–36,57] can be depicted as shown in Figure 3.
Figure 3: Projected topology of RNA-dependent generation of 5’-truncated mRNA encoding Aβ-containing C-terminal fragment of human beta-amyloid precursor protein.
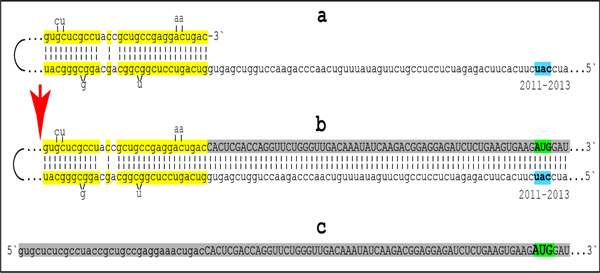
Lowercase letters -- nucleotide sequence of the antisense RNA. Uppercase letters -- nucleotide sequence of the sense RNA. Double-stranded portions highlighted in yellow: TCE (top) and ICE (bottom) elements of the antisense RNA. Note that the TCE and ICE are separated by about 2000 nucleotides. “2011–2013”: nucleotide positions on the antisense RNA (starting from the complement of the AUG encoding Met1 of the βAPP) of the “uac” (highlighted in blue) corresponding to the “AUG” (highlighted in green) encoding Met671 in the βAPP mRNA. a: TCE/ICE-guided folding of the antisense βAPP RNA. 3’-terminal “c” corresponds to one of multiple transcription start sites of βAPP mRNA located 149 nucleotides upstream from its AUG initiation codon [83]; note that such folding configuration would accommodate an additional 3’C, a transcript of the capG of βAPP mRNA (not shown). b: Extension of self-primed antisense RNA into sense RNA and cleavage (red arrow; may also occur at one of the TCE/ICE mismatches), after strand separation, of the chimeric intermediate. c: Chimeric RNA end product contains 5’terminal antisense segment extending into severely 5’-truncated βAPP mRNA. Its translation initiates from the “AUG” (highlighted in green and encoding Met671 in conventional βAPP mRNA) immediately preceding the beta amyloid-encoding region.
Approximately 30 nucleotide-long 3’-terminal segment of the antisense strand of βAPP mRNA constitutes the TCE. Its counterpart, the ICE, is separated by nearly 2000 nucleotides, yet these elements are topologically compatible and the folding of the antisense molecule results in a self-priming configuration capable to accommodate an additional 3’C if the capG of βAPP mRNA is transcribed [48–50] (Figure 3a). The TCE serves as a primer and is extended; thus generating the sense strand as shown in the Figure 3b. Strands are then separated as illustrated in Steps 5’ and 6’ of Figure 2, and cleavage occurs either at the mismatches within the TCE or immediately upstream from it as indicated by the arrow in Figure 3b. The resulting chimeric RNA end product, shown in Figure 3c, consists of an antisense segment (TCE or its portion) continued into a senseorientation molecule. The translational outcome is decided by the first, 5’-most, initiation-competent AUG codon. As shown in Figure 3b and c, the first AUG codon is located 58 nucleotides downstream from the TCE portion of the chimeric RNA end-product and it is, in fact, the AUG encoding Met671 in the intact βAPP mRNA! Translation from this position would produce beta amyloid-containing CTF, the C99 fragment of amyloid precursor protein, in the βAPP-independent manner. The major prediction of such a mechanism is a complete inefficiency of beta-secretase inhibition in Alzheimer’s disease. This prediction was, in fact, borne out in several massive stage III clinical trials [57].
Exclusivity of AD to Homo Sapiens: In Animals, in Contrast to Humans, βAPP mRNA is Ineligible for RNA-Dependent Amplification Process, A Presumed Driver of the Disease
As discussed above, presence of poly(A) at the 3’ terminus of an RNA makes the molecule an eligible RdRp template. It does not make it eligible for amplification. For this, the antisense RNA strand should be capable of forming a stable structure where its 3’ terminus can be extended into a segment of conventional mRNA molecule. Folding of the antisense RNA in such a self-priming configuration requires, in turn, the occurrence of two complementary and topologically compatible elements, one of which is strictly 3’-terminal. It appears that this requirement is not met in antisense RNA complements of βAPP mRNA in non-human mammals. In animals, βAPP antisense RNA segments corresponding to TCE and ICE elements of human βAPP antisense RNA show little, if any, complementarity; moreover, 3’-terminal segment of animal antisense βAPP RNA has no extensive complementarity with the rest of the molecule. Thus, in non-human mammals, βAPP mRNA, although suitable RdRp template, is not eligible for the amplification process. Therefore, if, as suggested above, the extent of increase in beta-amyloid levels, sufficient to trigger amyloid cascade culminating in AD, cannot be reached by βAPP proteolytic pathway alone and requires the activation of βAPP mRNA amplification pathway, Alzheimer’s disease can occur in humans but not in animals, a conclusion consistent with observations in the field.
In Humans, only a Subset of βAPP mRNA Transcripts is Eligible for RNA-Dependent Amplification Process: TSSs Utilization may Contribute to Susceptibility or Resistance to AD
The presence of a regulatory element known as a “TATA-box” is characteristic for a large class of mammalian genes. Usually, it occurs about 30 nucleotides upstream from the transcription start site (TSS) and rigidly defines its position. The βAPP gene belongs to a class of TATA-less genes that are characterized by multiple transcription start sites. There are at least five and possibly more positions where transcription of human βAPP mRNA can be initiated [34,83]. Of those, only one, 149 nucleotides upstream from the AUG translation initiation codon, corresponding to the 3’-terminal “C” of the antisense RNA shown in Figure 3, results in mRNA molecule eligible for RNA-dependent mRNA amplification process because only for this transcript the position of TCE on its antisense strand would be strictly 3’-terminal [34], and an additional 3’C, a transcript of the capG of βAPP mRNA [48–50], could be accommodated in the antisense self-priming structure (Figure 3). Utilization of human βAPP transcription start sites can be, therefore, one of the factors that define susceptibility or resistance to Alzheimer’s disease. The ability to regulate the usage of βAPP TSSs, or even to shift it at will, could open powerful therapeutic applications.
Engine that Drives Beta-Amyloid Overproduction and Alzheimer’s Disease
Cellular machinery capable of RNA-dependent mRNA amplification is activated by certain stresses
As discussed above, the core enzymatic machinery required for RNA-dependent mRNA amplification appears to be constitutively present, albeit in a non-conventional form [42–44], in all mammalian cells. Under regular circumstances, this core RdRp activity produces only short antisense transcripts due to the lack of a processivity co-factor [48,49]. Our current understanding indicates that mammalian core RdRp activity is constitutively expressed and that its processivity co-factor is inducible [48,49]. It doesn’t inform us on the status of other RdRs components involved such as helicase/modifying activity and single-strand cleaving activity [48,49]. Inducible components of mammalian RdRp complex appear to be expressed under special circumstances requiring a substantial overproduction of specific proteins. Their induction is likely triggered by certain cellular stresses. One possible example of such regulation is RNA-dependent amplification of mRNA encoding secreted extracellular matrix proteins.
Previously, we demonstrated the occurrence of RNA-dependent amplification of mRNA encoding all three chains of laminin in mouse tissue producing very large quantities of this protein [50]. It was suggested [48–50] that the initial ER stress resulting from increased transcription and subsequent translation of conventional mRNA encoding a secreted protein could be one of potentially multiple cellular events, which may trigger mRNA amplification. In this case, one can envision that conventional overproduction of a secreted protein induces ER stress and activates multiple transcription factors [58,59] that complement cellular core RdRp activity and enable RNA-dependent mRNA amplification process. It could be argued that this can further exacerbate the ER stress and trigger cell death. However, the amplified and heavily modified mRNA could behave in ways that are different spatially, qualitatively and quantitatively from those of conventional mRNA. One cellular response to ER stress appears to be a shift of translation and secretion outside the ER [60]. Therefore, nucleotide modifications of amplified mRNA may direct its translation and secretion of the resulting protein via pathways bypassing the ER, despite the presence of a signal peptide sequence. In such a case, mRNA amplification triggered by ER stress would eventually relieve the stress because not only the modified chimeric RNA end product but also conventional mRNA molecules, used as templates for the production of antisense RNA and modified during strand separation (Figure 1, step 2), would be translationally processed outside the ER.
Mitochondrial dysfunction and related stresses are possible activators of RNA-dependent βAPP mRNA amplification in Alzheimer’s disease
In the case of Alzheimer’s disease, a probable trigger of asymmetric RNA-dependent βAPP mRNA amplification, resulting in 5’truncated mRNA encoding Aβ-containing C99 fragment in a precursor-independent manner, is stress of metabolic mitochondrial dysfunction. Mitochondria are dynamic ATP-generating organelles which contribute to many cellular functions, including bioenergetic processes, intracellular calcium regulation, alteration of reduction-oxidation potential of cells, free radical scavenging and activation of caspase-mediated cell death. There is mounting evidence showing that mitochondrial damage plays an important role in Alzheimer disease. Increased oxygen species generation and deficient mitochondrial dynamic balance have been suggested to be the reason as well as the consequence of Alzheimer-related pathology. In AD, mitochondrial functions can be negatively affected by beta-amyloid, which can interact with mitochondria and cause mitochondrial dysfunction and consequent oxidative stress. This, in turn, mediates an increased production of Aβ and thus drives the disease.
Mitochondrial dysfunction is a prominent and early feature of AD [61], with reduced energy metabolism as one of the best documented early abnormalities [62]. Key mitochondrial enzymes of oxidative metabolism (i.e., cytochrome C oxidase, KGDHC, and PDHC) are deficient in AD [63,64]. Early deficits in synaptic mitochondria were also detected in an AD mouse model [65]. In addition, damaged mitochondrial DNA (mtDNA), including DNA mutation and DNA defects, are also found to be involved in AD [66]. Both beta-amyloid precursor protein and Aβ were localized in mitochondria [67–71], and Aβ not only contributes to significant oxidative damage of mtDNA, but leads also to impaired mtDNA gene expression [72,73]. More recent studies suggest that abnormal mitochondrial dynamics, including excessive mitochondrial fragmentation and abnormal mitochondrial distribution, plays a critical role in mitochondrial dysfunction in AD [74–81]. It appears that beta-amyloid is taken up by the organelle from elsewhere inside the cell. In fact, it was demonstrated that Aβ could be internalized by cells from an extracellular source and then imported into the mitochondria via the translocase of the outer membrane complex before accumulating in the mitochondrial cristae [71].
It should be emphasized that there is a strong probability that beta-amyloid, translated in a possibly compartmentalized manner from the modified end product of βAPP mRNA amplification not encoding a signal peptide, does not follow a migratory pattern described above but rather is not secreted and is retained intracellularly. In such a case, measured levels of Aβ in cerebrospinal fluid and in plasma of AD patients would reflect only beta-amyloid produced and secreted in βAPP proteolytic pathway but not that generated in βAPP mRNA amplification process. This scenario explains the observed inefficiency of beta-amyloid immunotherapy in human clinical trials; indeed, if the bulk of Aβ produced in patients is retained inside the cell, it would be inaccessible to and unaffected by the antibodies. It also rationalizes successes of beta-amyloid immunotherapy in animal AD models where Aβ is generated solely by βAPP proteolysis, exported from the cell and exposed to the antibodies.
How stresses associated with mitochondrial dysfunction enhance the production of Aβ in Alzheimer’s disease remains to be elucidated. Recently, it was proposed that they activate the expression of RdRp processivity co-factor and of other inducible components of the RdRp complex and thus enable asymmetric RNA-dependent amplification of human βAPP mRNA, the overproduction of the C99 fragment, and, consequently, the overproduction of Aβ [86]. This is consistent with a notion articulated above that a substantial overproduction of Aβ, to the extent sufficient to activate beta-amyloid cascade and trigger Alzheimer’s disease, cannot occur without βAPP mRNA amplification. This also implies that mitochondrial dysfunction, on its own, cannot trigger Alzheimer’s disease, as supported by the observation that in mice, where RNA-dependent amplification of βAPP mRNA cannot take place because of its ineligibility, mitochondrial dysfunction, even in long terms, results in neither a substantial Aβ accumulation, nor in excessive beta-amyloid plaque formation and neurodegeneration [82]. One of mitochondrial components, a microprotein PIGBOS, was shown to interact with the ER in mitigating the Unfolded Protein Response (UPR) [84]. It is feasible that mitochondrial dysfunction suppresses the occurrence and/or functionality of PIGBOS; this, in turn, would lead to suppression of UPR and, consequently, to ER stress. It is possible, therefore that mitochondrial dysfunction triggers the expression of inducible components of RdRp complex by initiating ER stress, implicated in activation of RNA-dependent mRNA amplification pathway [50].
Cause-and-effect relationships between mitochondrial dysfunction and Alzheimer’s disease
The cause-and-effect relationships between mitochondrial dysfunction and Alzheimer’s disease appear to be very different, in fact diametrically opposite, in FAD and SAD. In FAD, increased levels or more toxic species of Aβ, resulting from the mutations-mediated abnormal proteolysis of βAPP, trigger metabolic mitochondrial dysfunction, augmented ROS levels and ER stress. This, in turn, activates βAPP mRNA amplification, causes the additional increase in production of Aβ, and reinforces the cycle. Thus in FAD, mitochondrial dysfunction is an intrinsic component of the amyloid cascade. The reverse sequence is true in SAD where aging-related mitochondrial dysfunction, mitochondria-derived reactive oxygen species, and, possibly ER stress connected to mitochondrial dysfunction activate RNA-dependent amplification of βAPP mRNA and enhanced production of Aβ. This causes further mitochondrial dysfunction, the cycle repeats and degeneration increases. Thus in SAD, the initial mitochondrial dysfunction arises prior to the disease, independently of and upstream from the increased Aβ production. In other words, in SAD, in contrast to FAD, mitochondrial pathology hierarchically supersedes Aβ pathology. This is the primary reason for the formulation of the Mitochondrial Cascade Hypothesis [13,14]. But even in terms of the MCH, the core of the disease is the amyloid cascade as defined in the ACH [7–12]. The role of mitochondrial dysfunction in relation to this core is causative in the case of SAD and auxiliary in FAD.
Engine that drives Alzheimer’s disease: Self-perpetuating mutual Aβ overproduction/mitochondrial dysfunction feedback cycles
The initial increases in levels of Aβ are attained differently in different forms of Alzheimer’s disease. In FAD, it is caused by mutations in and, consequently, abnormal proteolysis of βAPP and occurs relatively early in life. In SAD, it is stimulated by an aging-contingent component and takes place, accordingly, late in life. Regardless of their origin, however, upon reaching a certain threshold, elevated levels of beta-amyloid initiate, in both forms of AD, mechanistically identical self-perpetuating mutual Aβ overproduction/mitochondrial dysfunction feedback cycles that drive, via RNA-dependent βAPP mRNA amplification, Aβ overproduction and are an essential element of the amyloid cascade leading to Alzheimer’s disease. This explains drastic differences in the age of onset, yet profound pathological and symptomatic similarities in the progression, of familial and sporadic forms of Alzheimer’s disease. These relationships are diagrammatically summarized in Figure 4; it depicts the mutual feedback cycles as a two-stroke engine, an engine that drives beta-amyloid overproduction and, consequently, Alzheimer’s disease. If beta-amyloid produced by the mitochondrial dysfunction-enabled βAPP mRNA amplification pathway is retained intracellularly, once the βAPP mRNA amplification process is activated, cellular levels of Aβ will sharply increase and so will the extent of Aβ-induced mitochondrial dysfunction, thus further intensifying mutual feedback cycles.
Figure 4: The engine that drives AD: Self-propagating mutual feedback cycles of mitochondrial dysfunction-mediated overproduction of beta-amyloid and vice versa in Alzheimer’s disease.

FAD: Familial Alzheimer’s disease; SAD: Sporadic Alzheimer’s disease; Highlighted in grey: Initial stimuli of the increased production of Aβ (different in FAD and SAD); Highlighted in blue: asymmetric RNA-dependent βAPP mRNA amplification, a molecular basis of beta-amyloid overproduction in Alzheimer’s disease (note a requirement for βAPP TSS utilization discussed in main text above); Horizontal arrows: the initial Aβ overproduction cycle; Arched arrows: Mutual feedback cycles; Blue arrows: Aβ-mediated induction of mitochondrial dysfunction and, possibly, ER stress; Red arrows: Mitochondrial dysfunction (and, possibly, ER stress)-mediated asymmetric RNA-dependent amplification of βAPP mRNA resulting in overproduction of Aβ. Note that in FAD, mitochondrial dysfunction is an intrinsic component of the amyloid cascade whereas in SAD, the initial mitochondrial pathology hierarchically supersedes and triggers Aβ pathology, where self-perpetuating mutual Aβ overproduction/mitochondrial dysfunction feedback cycles are, as in FAD, a central component of the amyloid cascade. In FAD, the initial increased production of Aβ results from mutations-driven abnormal proteolysis of βAPP and occurs relatively early in life, whereas in SAD, it is compelled by an aging-dependent component; hence drastic temporal difference in the age of onset yet profound pathological and symptomatic similarity in the progression of familial and sporadic Alzheimer’s disease, reflecting mechanistically identical nature of feedback cycles in both forms of AD.
Therapeutic Approaches for Alzheimer’s Disease: Targeting the Engine
Regardless of the initial cause-and-effect relationships between mitochondrial dysfunction and different forms of AD, neither SAD nor FAD can occur without mitochondrial dysfunction-related stresses that enable RNA-dependent amplification of βAPP mRNA. The RNA-dependent βAPP mRNA amplification-enabling potential denotes mitochondrial dysfunction as possible therapeutic target; this is especially important in view of the fact that success with pursuing other presumptive targets associated with the production of beta-amyloid has proved to be elusive. Moreover, if the aim is to pursue therapeutically RNA-dependent mRNA amplification process as the plausible molecular basis of beta-amyloid overproduction in Alzheimer’s disease, mitochondrial dysfunction, an ostensive trigger of this process, is the target of choice. Indeed, systemically targeting any component of the enzymatic machinery of RNA-dependent RNA synthesis would interfere with normal physiological functions of this process, such as, for example, specific mRNAs amplification in both erythroid differentiation [47–49] and the deposition of extracellular matrix proteins [50], and would have highly deleterious consequences. On the other hand, successfully targeting mitochondrial dysfunction would potentially result in two highly beneficial outcomes, namely, switching off an abnormal RNA-dependent βAPP mRNA amplification process and repairing metabolic defects. In this respect, a recent study with C.elegans [85] is very encouraging. In this study beta-amyloid-induced mitochondrial dysfunction was modeled and achieved by expressing human Aβ specifically in neurons (GRU102). Importantly, treatment with an anti-diabetes drug, metformin, reversed Aβ-induced metabolic defects, reduced protein aggregation and normalized lifespan of GRU102 thus establishing metabolic mitochondrial dysfunction as a promising and feasible therapeutic intervention target.
AD “Statin”: Alzheimer’s Disorder is a “Fast” Disease Preventable by Therapeutic Intervention Initiated Even Late in Life, and Treatable and Reversible at the Early Stages
The above considerations radically change the outlook on the dynamics of Alzheimer’s disease. Until now, it was presumed, at least in terms of the ACH, that the development of AD occurs as one prolonged process where extracellular Aβ levels steadily increase and damages slowly accumulate throughout the life and culminate in manifestation of the disease (Figure 5A and B). This viewpoint implied that preemptive therapeutic intervention late in life in case of potential SAD, or mid-age in case of FAD, is largely futile since the damage has already occurred over the preceding decades. To be effective, according to this view, intervention should start early in life. The outlook suggested by the present study is drastically different (Figure 5C and D). According to it, the disease evolves in two stages. The first stage is slow process of intracellular beta-amyloid accumulation via βAPP proteolytic/secretory pathway and cellular uptake of secreted Aβ. It occurs in humans and non-human mammals and results neither in significant damage nor in the manifestation of the disease. The second stage, which occurs only in humans and produces significant damages, is fast. It commences, shortly before symptomatic onset of the disease, with the activation of βAPP mRNA amplification, sharply increases the rate of beta-amyloid generation, and results in rapid intracellular accumulation of Aβ, neurodegeneration and AD. This means that preventive therapeutic intervention, an AD “statin”, can be initiated and be effective at any time prior to the second stage, i.e. late in life in case of potential SAD (Figure 5E). As for possible treatment strategies, they should address not only mitochondrial dysfunction and/or ER stress but also the regulation of utilization of βAPP transcription start sites, with the aim of preventing the production of βAPP mRNA eligible for RNA-dependent amplification process. Moreover, βAPP TSSs profile may serve as a prognostic indicator of susceptibility to AD.
Figure 5: Dynamics of Alzheimer’s disease in two paradigms.

Left panels: Dynamics of Aβ production; Right panels: Dynamics of neurodegeneration. Blue lines: Levels of beta-amyloid; Red lines: Extent of neurodegeneration; Black lines: Indicator lines, no significant neurodegeneration; Red blocks: Manifestation of Alzheimer’s disease. T: Threshold of symptomatic manifestation of AD (reflects levels of Aβ and consequent extent of neurodegeneration); T1: Threshold of activation of RNA-dependent βAPP mRNA amplification (reflects not only Aβ levels but also the extent of Aβ−coupled mitochondrial dysfunction and related stresses, possibly including ER stress); Numerous genetic factors such as, for example, the occurrence of various alleles of ApoE gene, as well as certain epigenetic factors, influence the age when thresholds T and T1 are reached, hence, the fanning lines. T2: Threshold of symptomatic occurrence of AD (reflects levels of Aβ and consequent extent of neurodegeneration). Panels A, B: View of the dynamics of AD prevailing until now (A: Dynamics of SAD; B: Dynamics of FAD). Levels of Aβ increase, neurodegeneration starts early and accumulates throughout the life. When threshold T is reached, AD symptoms manifest. Panels C, D: The outlook on the dynamics of Alzheimer’s disease envisioned in the present study (C: Dynamics of SAD; D: Dynamics of FAD). Levels of Aβ increase, the extent of mitochondrial dysfunction and related stresses reach threshold T1 and RNA-dependent βAPP mRNA amplification is activated. There is no significant neurodegeneration until after a lag period (when amplified RNA encoding C99 fragment of βAPP accrues and Aβ further accumulates) following the crossing of T1 threshold and activation of βAPP mRNA amplification; when the extent of neurodegeneration reaches threshold T2, AD symptoms manifest. Panel E: Dynamics of Aβ production and neurodegeneration in non-human mammals in light of the present study: T1 threshold is crossed but βAPPP mRNA is not amplified because it is not eligible for RNA-dependent amplification process. There is no significant neurodegeneration; T2 threshold is not reached, no AD symptoms manifest, no disease occurs. Note: Scenario depicted in panel (E) would occur in humans not susceptible to Alzheimer’s disease due to variations in βAPP TSSs utilization or when activation of βAPP mRNA amplification is blocked by therapeutic intervention.
Numerous genetic factors such as, for example, the occurrence of various alleles of ApoE gene, as well as certain epigenetic factors, influence the age when thresholds T (Figure 5A and B) and T1 (Figure 5C and D) are reached and, consequently, when AD symptoms manifest. Statistically, sporadic AD occurs predominantly after age of 65 (only 3% of AD cases, probably most if not all of them FAD, occur prior to age of 65). The bulk of FAD cases are diagnosed starting from age of mid-40s. Practically, it means that when AD “statin” is available, it should be taken preventively starting at age of 60–65 for general population and 40–45 for carriers of FAD mutations. Moreover, there are good reasons to believe that with a drug blocking RNA-dependent βAPP mRNA amplification, via mitigation of stresses associated with mitochondrial dysfunction or otherwise, it would be possible to stop and to reverse the disease even when early AD symptoms have already manifested. This assumption is based on observations in mouse models where AD symptoms, as well as neurodegeneration, were reversed by suppressing, via BACE inhibition, overproduction of beta-amyloid in mouse models generating Aβ solely by βAPP proteolytic pathway [30–32].
Conclusion
The present study posits that Alzheimer’s disorder is a Fast Disease. This is in sharp contrast to a view, prevailing until now, that Alzheimer’s disease is a quintessential “slow” disease that develops throughout the life as one prolonged process. According to this view, beta-amyloid is produced and secreted solely by the beta-amyloid precursor protein proteolytic/secretory pathway. As its extracellular levels increase, it triggers neurodegeneration starting relatively early in life. Damages accumulate and manifest, late in life in sporadic cases, as AD symptoms. In familial AD cases, where mutations in βAPP gene or in presenilins increase production of either common Aβ isoform or of its more toxic isoforms, neurodegeneration reaches critical threshold sooner and AD symptoms occur earlier in life, mostly in late 40s and 50s. There are currently no preventive AD therapies but if they were available, according to this viewpoint it would be largely futile to intervene late in life in case of potential SAD or at mid-age in cases of FAD because, although AD symptoms have not yet manifested, the damage has already occurred during the preceding decades. In this paradigm, to be effective, preventive therapeutic intervention should be initiated early in life.
The outlook suggested by the present study is radically different. According to it, Alzheimer’s disease evolves in two stages. The first stage is a slow process of intracellular beta-amyloid accumulation. It occurs via βAPP proteolytic/secretory pathway and cellular uptake of secreted Aβ common to Homo sapiens, including healthy humans, and to non-human mammals and results neither in significant damage, nor in manifestation of the disease. The second stage occurs exclusively in humans, commences shortly before symptomatic onset of the disease, sharply increases the rate of Aβ generation and its intracellular levels, produces significant damages, triggers AD symptoms, and is fast. It is driven by asymmetric βAPP mRNA amplification pathway of Aβ production that is activated by mitochondrial dysfunction-related stresses and results in beta-amyloid that is not secreted but is retained intracellularly. It is qualitatively and quantitatively different from βAPP proteolytic process, entirely independent of beta-amyloid precursor protein, and results in rapid and substantial intracellular accumulation of Aβ, consequent significant neurodegeneration, and symptomatic AD. In this paradigm, a preventive therapy for Alzheimer’s disease, an AD “statin”, would be effective when initiated at any time prior to commencement of the second stage. Moreover, there are good reasons to believe that with a drug blocking βAPP-independent Aβ production pathway, RNA-dependent βAPP mRNA amplification, via mitigation of stresses associated with mitochondrial dysfunction or otherwise, it would be possible not only to prevent but also to stop and to reverse the disease even when early AD symptoms have already manifested.
As discussed above, there is a strong probabilty that beta-amyloid, translated in a possibly compartmentalized manner from the modified end product of βAPP mRNA amplification not encoding a signal peptide, is not secreted but is retained intracellularly. In such a case, measured levels of Aβ in cerebrospinal fluid and in plasma of AD patients would reflect only beta-amyloid produced and secreted in βAPP proteolytic pathway but not that generated in βAPP mRNA amplification pathway. This scenario explains the observed inefficiency of therapeutic antibodies to beta-amyloid in human clinical trials. Indeed, if the bulk of beta-amyloid produced in patients is retained inside the cell, it would be inaccessible to and unaffected by the antibodies, thus rendering Aβ immunotherapeutic approach unfeasible. This scenario also rationalizes successes of beta-amyloid immunotherapy in animal AD models where Aβ is generated solely by βAPP proteolysis, exported from the cell and exposed to the antibodies.
Until now, toxicity of beta-amyloid in Alzheimer’s disease was associated with its extracellular localization. First, it was considered that beta-amyloid plaques cause cellular damage and mediate neuronal death. When it transpired that beta-amyloid plaques are not harmful, the toxic function was assigned to Aβ in fibrillar and oligomeric forms, both acting from outside the cell. However, it is clear that intracellular beta-amyloid is toxic to cells, at least by virtue of its ability to cause mitochondrial dysfunction. If beta-amyloid produced by the mitochondrial dysfunction-enabled βAPP mRNA amplification pathway is retained intracellularly, once the βAPP mRNA amplification process is activated, cellular levels of beta-amyloid will sharply increase and so will the extent of Aβ-induced mitochondrial dysfunction, potentially triggering, or at least facilitating, cell death. It appears that intracellular beta-amyloid could be the primary agent of neuronal death in Alzheimer’s disease. Whether this is the case remains to be elucidated.
The present study posits a notion of AD as a Fast Disease, defines cellular and molecular processes constituting an engine that drives Alzheimer’s disease, establishes that it is preventable by therapeutic intervention initiated even late in life, details a powerful mechanism underlying the disease, suggests that Aβ produced in the βAPP-independent pathway is retained intracellularly, elaborates why neither BACE inhibition nor Aβ immunotherapy are effective in treatment of AD and why intracellularly retained beta-amyloid could be the primary agent of neuronal death in Alzheimer’s disease, necessitates generation of a novel animal AD model capable of producing Aβ via βAPP-independent pathway, proposes therapeutic targets profoundly different from previously pursued components of the βAPP proteolytic pathway, and provides conceptual rationale for design of drugs that could be used not only preemptively but also for treatment and reversal of the early stages of Alzheimer’s disease.
Acknowledgments
Funding
NIH R21 GM056179; NIH RO1 AR036819
Footnotes
Conflict of Interest
Authors declare no conflict of interest.
References
- 1.Haass C, Lemere C, Capell A, Citron M, Seubert P, Schenk D, et al. The Swedish mutation causes early-onset Alzheimer’s disease by beta-secretase cleavage within the secretory pathway. Nat Med. 1995;1(12):1291–6. [DOI] [PubMed] [Google Scholar]
- 2.Dyrks T, Dyrks E, Monning U, Urmoneit B, Turner J, Beyreuther, K. Generation of beta A4 from the amyloid protein precursor and fragments thereof. FEBS Lett. 1993;335(1):89–93. [DOI] [PubMed] [Google Scholar]
- 3.Iizuka T, Shoji M, Kawarabayashi T, Sato M, Kobayashi T, Tada N, et al. Intracellular generation of amyloid beta-protein from amyloid beta-protein precursor fragment by direct cleavage with beta- and gamma-secretase. Biochem Biophys Res Commun. 1996;218(1):238–42. [DOI] [PubMed] [Google Scholar]
- 4.DeStrooper B, Annaert W. Proteolytic processing and cell biological functions of the amyloid precursor protein. J Cell Sci. 2000;113(11):1857–70. [DOI] [PubMed] [Google Scholar]
- 5.Barber RC. The genetics of Alzheimer’s disease. Scientifica (Cairo). 2012;2012:246210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vassar R. BACE1 inhibitor drugs in clinical trials for Alzheimer’s disease. Alzheimers Res Ther. 2014;6(9):89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Beyreuther K, Masters C. Amyloid precursor protein (APP) and beta A4 amyloid in the etiology of Alzheimer’s disease: precursor-product relationships in the derangement of neuronal function. Brain Pathol. 1991;1(4):241–51. [DOI] [PubMed] [Google Scholar]
- 8.Hardy J, Allsop D. Amyloid deposition as the central event in the aetiology of Alzheimer’s disease. Trends Pharmacol Sci. 1991;12(10):383–8. [DOI] [PubMed] [Google Scholar]
- 9.Selkoe DJ. The molecular pathology of Alzheimer’s disease. Neuron. 1991;6(4):487–98. [DOI] [PubMed] [Google Scholar]
- 10.Hardy JA, Higgins GA. Alzheimer’s disease: the amyloid cascade hypothesis. Science. 1992;256(5054):184–5. [DOI] [PubMed] [Google Scholar]
- 11.Hardy J, Selkoe D. The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science. 2002;297(5580):353–6. [DOI] [PubMed] [Google Scholar]
- 12.Selkoe DJ, Hardy J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol Med. 2016;8(6):595–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Swerdlow RH. Mitochondria and Mitochondrial Cascades in Alzheimer’s Disease. J Alzheimers Dis. 2018;62(3):1403–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hunter S, Brayne C. Understanding the roles of mutations in the amyloid precursor protein in Alzheimer disease. Mol Psychiatry. 2018;23(1):81–93. [DOI] [PubMed] [Google Scholar]
- 15.Zhang S, Wang Z, Cai F, Zhang M, Wu Y, Zhang J, et al. BACE1 Cleavage Site Selection Critical for Amyloidogenesis and Alzheimer’s Pathogenesis. J Neurosci. 2017;37(29):6915–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vassar R, Bennett B, Babu-Khan S, Kahn S, Mendiaz E, Denis P, et al. Beta-secretase cleavage of Alheimer’s amyloid precursor protein by the transmembrane aspartic protease BACE. Science. 1999;286(5440):735–41. [DOI] [PubMed] [Google Scholar]
- 17.Hussain I, Powell D, Howlett DR, Tew DG, Meek TD, Chapman C, et al. Identification of a novel aspartic protease (Asp 2) as beta-secretase. Mol Cell Neurosci. 1999;14(6):419–27. [DOI] [PubMed] [Google Scholar]
- 18.Sinha S, Anderson J, Barbour R, Basl G, Caccavello R, Davis D, et al. Purification and cloning of amyloid precursor protein beta secretase from human brain. Nature. 1999;402(6761):537–40. [DOI] [PubMed] [Google Scholar]
- 19.Wang Y, Strickland C, Voigt J, Kennedy M, Beyer B, Senior MM, et al. Application of fragment-based NMR screening, X-ray crystallography, structure-based design, and focused chemical library design to identify novel μM leads for the development of nM BACE-1 inhibitors. J Med Chem. 2010;53(3):942–50. [DOI] [PubMed] [Google Scholar]
- 20.Zhu Z, Sun Z, Ye Y, Voigt J, Strickland C, Smith E, et al. Discovery of cyclic acylguanidines as highly potent and selective ß-site amyloid cleaving enzyme (BACE) inhibitors: Part I - inhibitor design and validation. J Med Chem. 2010;53(3):951–65. [DOI] [PubMed] [Google Scholar]
- 21.Cumming J, Smith E, Wang L, Misiaszek J, Durkin J, Pan J, et al. Structure based design of iminohydantoin BACE1 inhibitors: identification of an orally available, centrally active BACE1 inhibitor. Bioorg Med Chem Lett. 2012;22(7):2444–9. [DOI] [PubMed] [Google Scholar]
- 22.Edwards P, Albert J, Sylvester M, Aharony D, Andisik D, Callaghan O, et al. Application of fragment-based lead generation to the discovery of novel, cyclic amidine ß-secretase inhibitors with nanomolar potency, cellular activity, and high ligand efficiency. J Med Chem. 2007;50(24):5912–25. [DOI] [PubMed] [Google Scholar]
- 23.Barrow J, Stauffer S, Rittle K, Ngo P, Yang Z, Selnick H, et al. Discovery and X-ray crystallographic analysis of a S-piropiperidine iminohydantoin inhibitor of ß-secretase. J Med Chem. 2008;51(20):6259–62. [DOI] [PubMed] [Google Scholar]
- 24.Malamas M, Erdei J, Gunawan I, Turner J, Hu Y, Wagner E, et al. Design and synthesis of 5,5’-disubstituted aminohydantoins as potent and selective human ß-secretase (BACE1) inhibitors. J Med Chem. 2010;53(3):1146–58. [DOI] [PubMed] [Google Scholar]
- 25.Rueeger H, Rondeau J, McCarthy C, Moebitz H, Tintelnot-Blomley M, Neumann U, et al. Structure based design, synthesis and SAR of cyclic hydroxyethylamine (HEA) BACE-1 inhibitors. Bioorg Med Chem Lett. 2011;21(7):1942–7. [DOI] [PubMed] [Google Scholar]
- 26.Probst G, Xu YZ. Small-molecule BACE1 inhibitors: a patent literature review (2006 – 2011). Expert Opin Ther Pat. 2012;22(5):511–40. [DOI] [PubMed] [Google Scholar]
- 27.May P, Dean R, Lowe S, Martenyi F, Sheehan S, Boggs L, et al. Robust central reduction of amyloid-ß in humans with an orally available, non-peptidic ß-secretase inhibitor. J Neurosci. 2011;31(46):16507–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Stamford A, Scott J, Li S, Babu S, Tadesse D, Hunter R, et al. Discovery of an orally available, brain penetrant BACE1 inhibitor that affords robust CNS Aß reduction. ACS Med Chem Lett. 2012;3(11):897–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kennedy M, Stamford A, Chen X, Cox K, Cumming J, Dockendorf M, et al. The BACE1 inhibitor verubecestat (MK-8931) reduces CNS ß-amyloid in animal models and in Alzheimer’s disease patients. Sci Transl Med. 2016;8(363):363ra150. [DOI] [PubMed] [Google Scholar]
- 30.Keskin A, Kekuš M, Adelsberger H, Neumann U, Shimshek D, Song B, et al. BACE inhibition-dependent repair of Alzheimer’s pathophysiology. Proc Natl Acad Sci U S A. 2017;114(32):8631–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Neumann U, Rueeger H, Machauer R, Veenstra S, Lueoend R, Laue G, et al. A novel BACE inhibitor NB-360 shows a superior pharmacological profile and robust reduction of amyloid-ß and neuroinflammation in APP transgenic mice. Mol Neurodegener. 2015;10:44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hu X, Das B, Hou H, He W, Yan R. BACE1 deletion in the adult mouse reverses preformed amyloid deposition and improves cognitive functions. J Exp Med. 2018;215(3):927–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nativio R, Donahue G, Berson A, Lan Y, Amlie-Wolf A, Tuzer F, et al. Dysregulation of the epigenetic landscape of normal aging in Alzheimer’s disease. Nature Neurosci. 2018;10:1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Volloch V. A mechanism for ß-amyloid overproduction in Alzheimer’s disease: Precursor-independent generation of ß-amyloid via antisense RNA-primed mRNA synthesis. FEBS Lett. 1996;390(2):124–8. [DOI] [PubMed] [Google Scholar]
- 35.Volloch V. Mechanism for ß-amyloid overproduction in sporadic Alzheimer’s Disease: Possible antisense RNA-mediated generation of a 5’-truncated ßAPP mRNA encoding 12 kDa C-terminal fragment of ßAPP, the immediate precursor of Aß. In: Molecular Mechanisms of Dementia. 1997, Wasco W and Tanzi R, Eds. [Google Scholar]
- 36.Volloch V. Possible mechanism for resistance to Alzheimer’s disease (AD) in mice suggests new approach to generate a mouse model for sporadic AD and may explain familial resistance to AD in man. Exp Neurol. 1997;144(1):214–8. [DOI] [PubMed] [Google Scholar]
- 37.Breimer LH, Denny P. Alzheimer amyloid aspects. Nature. 1987;326(6115):749–50. [DOI] [PubMed] [Google Scholar]
- 38.Citron M, Haass C, Selkoe DJ. Production of amyloid-beta-peptide by cultured cells: no evidence for internal initiation of translation at Met596. Neurobiol Aging. 1993;14(6):571–3. [DOI] [PubMed] [Google Scholar]
- 39.Lai M. RNA replication without RNA-dependent RNA polymerase: surprises from hepatitis delta virus. J. Virol. 2005;79:7951–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Taylor J. Replication of human hepatitis delta virus: recent developments. Trends Microbiol. 2003;11: 185–90. [DOI] [PubMed] [Google Scholar]
- 41.Tseng CH, Lai MM. Hepatitis delta virus RNA replication. Viruses. 2009;1(3):818–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lehmann E, Brueckner F, Cramer P. Molecular basis of RNA-dependent RNA polymerase II activity. Nature. 2007;450(7168):445–9. [DOI] [PubMed] [Google Scholar]
- 43.Wagner SD, Yakovchuk P, Gilman B, Ponicsan SL, Drullinger LF, Kugel JF, et al. RNA polymerase II acts as an RNA-dependent RNA polymerase to extend and destabilize a non-coding RNA. EMBO J. 2013;32(6):781–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Maida Y, Yasukawa M, Masutomi K. De Novo RNA Synthesis by RNA-Dependent RNA Polymerase Activity of Telomerase Reverse Transcriptase. Mol Cell Biol. 2016;36(8):1248–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Downey KM, Byrnes JJ, Jurmark BS, So AG. Reticulocyte RNA-dependent RNA polymerase. Proc Natl Acad Sci U S A. 1973;70(12):3400–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kapranov P, Ozsolak F, Kim SW, Foissac S, Lipson D, Hart C, et al. New class of gene-termini-associated human RNAs suggests a novel RNA copying mechanism. Nature. 2010;466(7306):642–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Volloch V, Schweitzer B, Rits S. Antisense Globin RNA in Murine Erythroid Tissues: Structure, Origin and Possible Function. Proc Natl Acad Sci USA. 1996;93(6):2476–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Volloch V. Protein-encoding RNA to RNA transfer in mammalian cells: Principles of RNA-dependent mRNA amplification. Ann Integr Mol Med. 2019;1(1):1002. [PMC free article] [PubMed] [Google Scholar]
- 49.Rits S, Olsen BR, Volloch V. Protein-encoding RNA to RNA information transfer in mammalian cells: RNA-dependent mRNA amplification. Identification of chimeric RNA intermediates and putative RNA end products. Ann Integr Mol Med. 2019;1(1):23–47. [PMC free article] [PubMed] [Google Scholar]
- 50.Volloch V, Rits S, Olsen BR. RNA-dependent Amplification of Mammalian mRNA Encoding Extracellullar Matrix Proteins: Identification of Chimeric RNA Intermediates for a1, ß1, and ?1 Chains of Laminin. Ann Integr Mol Med. 2019;1(1):1004. [PMC free article] [PubMed] [Google Scholar]
- 51.Richards OC, Ehrenfeld E. Poliovirus RNA replication. Curr Top Microbiol Immunol. 1990;161:89–119. [DOI] [PubMed] [Google Scholar]
- 52.Brazas R, Ganem D A cellular homolog of hepatitis delta antigen: implications for viral replication and evolution. Science. 1996;274(5284):90–4. [DOI] [PubMed] [Google Scholar]
- 53.Huang CR, Lo SJ. Evolution and diversity of the human hepatitis d virus genome. Adv Bioinformatics. 2010;. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Volloch V, Schwetizer B, Rits S. Evolutionarily Conserved Elements in the 5’-untranslated Region of beta-Globin mRNA Mediate Site-specific Priming of a Unique Hairpin Structure during cDNA Synthesis. Nucleic Acids Res. 1994;22(24):5302–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mita S, Sadlock J, Herbert J, Schon E. A cDNA specifying the human amyloid beta precursor protein encodes a 95-kDa polypeptide. Nucleic Acids Res. 1988;16(19):9351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mita S, Sadlock J, Herbert J, Schon E. A cDNA specifying the human amyloid beta precursor protein encodes a 95-kDa polypeptide: Correction. Nucl Acids Res. 1988;16:11402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Volloch V, Rits S. Results of beta secretase-inhibitor clinical trials support amyloid precursor protein-independent generation of beta amyloid in sporadic Alzheimer’s disease. Med Sci (Basel). 2018;6(2):pii: E45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yoshida H, Matsui T, Yamamoto A, Okada T, Mori K. XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell. 2001;107(7):881–91. [DOI] [PubMed] [Google Scholar]
- 59.Jiang S, Zhang E, Zhang R, Li X. Altered activity patterns of transcription factors induced by endoplasmic reticulum stress. BMC Biochem. 2016;17:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Rabouille C. Pathways of unconventional protein secretion. Trends Cell Biol. 2017;27(3):230–40. [DOI] [PubMed] [Google Scholar]
- 61.Zhu X, Perry G, Moreira PI, Aliev G, Cash AD, Hirai K, et al. Mitochondrial abnormalities and oxidative imbalance in Alzheimer disease. J Alzheimers Dis. 2006;9(2):147–53. [DOI] [PubMed] [Google Scholar]
- 62.Blass JP. The mitochondrial spiral. An adequate cause of dementia in the Alzheimer’s syndrome. Ann N Y Acad Sci. 2000;924:170–83. [DOI] [PubMed] [Google Scholar]
- 63.Manczak M, Park BS, Jung Y, Reddy PH. Differential expression of oxidative phosphorylation genes in patients with Alzheimer’s disease: implications for early mitochondrial dysfunction and oxidative damage. Neuromolecular Med. 2004;5(2):147–62. [DOI] [PubMed] [Google Scholar]
- 64.Qin W, Haroutunian V, Katsel P, Cardozo CP, Ho L, Buxbaum JD, et al. PGC-1alpha expression decreases in the Alzheimer disease brain as a function of dementia. Arch Neurol. 2009;66(3):352–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Du H, Guo L, Yan S, Sosunov AA, McKhann GM, Yan SS. Early deficits in synaptic mitochondria in an Alzheimer’s disease mouse model. Proc Natl Acad Sci U S A. 2010;107(43):18670–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lin MT, Simon DK, Ahn CH, Kim LM, Beal MF. High aggregate burden of somatic mtDNA point mutations in aging and Alzheimer’s disease brain. Hum Mol Genet. 2002;11(2):133–45. [DOI] [PubMed] [Google Scholar]
- 67.Calkins M, Manczak M, Mao P, Shirendeb U, Reddy PH. Impaired mitochondrial biogenesis, defective axonal transport of mitochondria, abnormal mitochondrial dynamics and synaptic degeneration in a mouse model of Alzheimer’s disease. Hum Mol Genet. 2011;20(23):4515–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Anandatheerthavarada HK, Biswas G, Robin MA, Avadhani NG. Mitochondrial targeting and a novel transmembrane arrest of Alzheimer’s amyloid precursor protein impairs mitochondrial function in neuronal cells. J Cell Biol. 2003;161(1):41–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Caspersen C, Wang N, Yao J, Sosunov A, Chen X, Lustbader JW, et al. Mitochondrial A-beta: a potential focal point for neuronal metabolic dysfunction in Alzheimer’s disease. FASEB J. 2005;19(14):2040–1. [DOI] [PubMed] [Google Scholar]
- 70.Chen JX, Yan SS. Role of mitochondrial amyloid-beta in Alzheimer’s disease. J Alzheimers Dis. 2010;20:S569–78. [DOI] [PubMed] [Google Scholar]
- 71.Hansson Petersen CA, Alikhani N, Behbahani H, Wiehager B, Pavlov PF, Alafuzoff I, et al. The amyloid beta-peptide is imported into mitochondria via the TOM import machinery and localized to mitochondrial cristae. Proc Natl Acad Sci U S A. 2008;105(35):13145–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.de la Monte SM, Luong T, Neely TR, Robinson D, Wands JR. Mitochondrial DNA damage as a mechanism of cell loss in Alzheimer’s disease. Lab Invest. 2000;80(8):1323–35. [DOI] [PubMed] [Google Scholar]
- 73.Brooks WM, Lynch PJ, Ingle CC, Hatton A, Emson PC, Faull RL, et al. Gene expression profiles of metabolic enzyme transcripts in Alzheimer’s disease. Brain Res. 2007;1127(1):127–35. [DOI] [PubMed] [Google Scholar]
- 74.Wang X, Perry G, Smith MA, Zhu X. Amyloid-beta-derived diffusible ligands cause impaired axonal transport of mitochondria in neurons. Neurodegener Dis. 2010;7(1–3):56–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Wang X, Su B, Fujioka H, Zhu X. Dynamin-like protein 1 reduction underlies mitochondrial morphology and distribution abnormalities in fibroblasts from sporadic Alzheimer’s disease patients. Am J Pathol. 2008;173(2):470–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Wang X, Su B, Lee HG, Li X, Perry G, Smith MA, et al. Impaired balance of mitochondrial fission and fusion in Alzheimer’s disease. J Neurosci. 2009;29(28):9090–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Wang X, Su B, Siedlak SL, Moreira PI, Fujioka H, Wang Y, et al. Amyloidbeta overproduction causes abnormal mitochondrial dynamics via differential modulation of mitochondrial fission/fusion proteins. Proc Natl Acad Sci U S A. 2008;105(49):19318–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Su B, Wang X, Bonda D, Perry G, Smith M, Zhu X. Abnormal mitochondrial dynamics--a novel therapeutic target for Alzheimer’s disease? Mol Neurobiol. 2010;41(2–3):87–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Manczak M, Calkins MJ, Reddy PH. Impaired mitochondrial dynamics and abnormal interaction of amyloid beta with mitochondrial protein Drp1 in neurons from patients with Alzheimer’s disease: implications for neuronal damage. Hum Mol Genet. 2011;20(13):2495–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Leuner K, Schütt T, Kurz C, Eckert SH, Schiller C, Occhipinti A, et al. Mitochondrionderived reactive oxygen species lead to enhanced amyloid beta formation. Antioxidants and Redox Signaling. 2012;16(12):1421–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.LaFerla FM, Green KN, Oddo S. Intracellular amyloid-ß in Alzheimer’s disease. Nat Rev Neurosci. 2007;8(7):499–509. [DOI] [PubMed] [Google Scholar]
- 82.Lapointe J, Hekimi S. Early mitochondrial dysfunction in long-lived Mclk1+/− mice. J Biol Chem. 2008;283(38):26217–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Salbaum JM, Weidemann A, Lemaire HG, Masters CL, Beyreuther K. The promoter of Alzheimer’s disease amyloid A4 precursor gene. EMBO J. 1988;7(9):2807–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Chu Q, Martinez T, Novak S, Donaldson C, Tan D, Chang T, et al. Regulation of the ER stress response by a mitochondrial microprotein. Nat Commun. 2019;10(1):4883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Teo E, Ravi S, Barardo D, Kim H, Fong S, Tan TY, et al. Metabolic stress is a primary pathogenic event in transgenic Caenorhabditis elegans expressing pan-neuronal human amyloid beta. eLife. 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Volloch V, Olsen BR, Rits S. Precursor-Independent Overproduction of Beta-Amyloid in AD: Mitochondrial Dysfunction as Possible Initiator of Asymmetric RNA-Dependent ßAPP mRNA Amplification. An Engine that Drives Alzheimer’s Disease. Ann Integr Mol Med. 2019;1(1):1005. [PMC free article] [PubMed] [Google Scholar]