

Redox regulation of STAT3 is targeted by direct inhibition of TrxR1, illuminating a promising link for therapeutic development.
Abstract
Because of its key role in cancer development and progression, STAT3 has become an attractive target for developing new cancer therapeutics. While several STAT3 inhibitors have progressed to advanced stages of development, their underlying biology and mechanisms of action are often more complex than would be expected from specific binding to STAT3. Here, we have identified and optimized a series of compounds that block STAT3-dependent luciferase expression with nanomolar potency. Unexpectedly, our lead compounds did not bind to cellular STAT3 but to another prominent anticancer drug target, TrxR1. We further identified that TrxR1 inhibition induced Prx2 and STAT3 oxidation, which subsequently blocked STAT3-dependent transcription. Moreover, previously identified inhibitors of STAT3 were also found to inhibit TrxR1, and likewise, established TrxR1 inhibitors block STAT3-dependent transcriptional activity. These results provide new insights into the complexities of STAT3 redox regulation while highlighting a novel mechanism to block aberrant STAT3 signaling in cancer cells.
INTRODUCTION
Signal transducer and activator of transcription 3 (STAT3) is a cytosolic transcription factor that is activated in response to cytokine and growth factor stimulation (1). STAT3’s activity is mediated by posttranslational modifications including phosphorylation, acetylation, and reduction/oxidation (redox) processes (2). In healthy cells, these posttranslational modifications ensure that STAT3 signaling is tightly regulated, resulting in transient STAT3 activation under physiological conditions (3). However, STAT3 signaling is commonly deregulated in cancer cells leading to constitutive STAT3 activation (phosphorylation of Tyr705) and overexpression of STAT3 target genes. Among many upstream regulators of STAT3 activity, aberrant STAT3 signaling is commonly linked to activating mutations in tyrosine kinases and/or from an abundance of cytokines and growth factors in the tumor microenvironment (4, 5). Tyr705 phosphorylation induces the formation of the transcriptionally active phosphorylated STAT3 (pSTAT3) dimer, which translocates to the nucleus and binds consensus DNA sequences to initiate target gene expression (3).
In cancer cells, aberrant STAT3 activity drives the expression of genes that promote the cancer phenotype, including proliferation, metabolic changes, apoptosis avoidance, angiogenesis, and immune system evasion (6, 7). Elevated STAT3 activity is critical for cancer cells, which become addicted to high levels of protumorigenic and antiapoptotic factors, making them sensitive to disruptions in STAT3 signaling (8). The requirement for STAT3 activation is specific to cancer cells, as healthy cells can survive in the absence of STAT3 signaling (6, 8). These characteristics have contributed to STAT3’s popularity as a target for developing novel cancer therapeutics.
While STAT3 is a promising anticancer drug target, it is a very difficult protein to inhibit using traditional drug-like molecules. This is because STAT3 lacks an enzyme active site where inhibitors would normally bind (9). Instead, STAT3’s activity is mediated by protein-protein and protein-DNA interactions that involve large, relatively flat regions of the protein’s surface (3). Compared to more traditional drug targets that contain well-defined binding pockets, the development of inhibitors that selectively bind to STAT3’s interaction interfaces is a formidable challenge (10).
Despite this, a wide range of small molecules have been published as direct binders of STAT3 protein (3, 10). In general, these inhibitors can interfere with purified STAT3 protein in biochemical assays, such as the fluorescence polarization assay, electrophoretic mobility shift assay, and enzyme-linked immunosorbent assay (ELISA) (3, 11, 12). Many of these compounds also inhibit STAT3 signaling in cells, as typically demonstrated by characterizing changes in STAT3 phosphorylation, blocking STAT3-dependent gene expression, or inhibiting other STAT3-related cellular processes (3, 11). However, for some STAT3 inhibitors, it is becoming more and more apparent that direct STAT3 binding in vitro and inhibition of STAT3-dependent gene expression in cells may not be directly linked. Although many STAT3 inhibitors can effectively bind pure recombinant STAT3 protein, the effects in cells may be more complex than relating to specific binding to cellular STAT3 protein. As with any drug target, carefully validating that experimental inhibitors bind to the intended target in cells and tissues is critically important for understanding their underlying mechanism of action. While, classically, this has been a difficult challenge for drug discovery researchers, the expanded use of target engagement techniques in the drug discovery process has greatly facilitated this process (13).
Many early STAT3 inhibitors, including Stattic (14), BP1-102 (15, 16), and S3i-201 (17), claimed to bind to STAT3’s Src homology 2 (SH2) domain, a key functional domain that mediates interactions with activated receptors and the formation of the pSTAT3 dimer. These early inhibitors have been broadly used to explore STAT3 biology. However, several recent publications have highlighted that they are reactive compounds, capable of covalently modifying several cysteine (Cys) residues on STAT3 protein and possibly on other targets (18–20). Although they are reactive compounds, whether they engage STAT3 in cells remains uncertain, BP1-102, Stattic, and S3i-201 all inhibit STAT3 signaling in cancer cells (14–17), thus suggesting a possible link between electrophilic compounds and STAT3 biology.
Electrophilic small molecules are proposed to react with exposed Cys residues on STAT3 (18), which have important roles in controlling STAT3’s transcriptional activity (2). Redox regulation of STAT3 also occurs via oxidation and reduction of these Cys residues, through a redox relay involving peroxiredoxin-2 (Prx2) and thioredoxin-1 (Trx1) (21). Prx2 is an important messenger protein for oxidative stress and can modify several downstream protein targets by oxidizing exposed Cys residues (21). Trx1 has an opposing role and reduces oxidized cellular components modulating their activity (21). STAT3 was recently identified as a downstream target of Prx2 signaling and was found to be rapidly oxidized by Prx2 following induction of oxidative stress using H2O2 (21). Oxidation of STAT3 induces the formation of nonphosphorylated, disulfide-linked STAT3 dimer, which is transcriptionally inactive (21). To regain its transcriptional activity, oxidized STAT3 dimers must be reduced by Trx1, which regenerates reduced STAT3 monomers and oligomers that are not linked via disulfide bonds (21). The reduced Trx1 pool is maintained by the cytosolic selenoprotein thioredoxin reductase 1 (TrxR1), which uses NADPH (reduced form of nicotinamide adenine dinucleotide phosphate) to reduce oxidized Trx1 (22). Notably, the selenocysteine (Sec) residue of TrxR1 is up to three orders of magnitude more nucleophilic than a typical Cys residue, making it highly reactive toward electrophilic species (22).
In cancer cells, inhibition of TrxR1 results in increased oxidative stress and accumulation of oxidized Prx2 and STAT3 (21), which blocks STAT3-dependent transcription. Similar to STAT3, TrxR1 is essential for cancer cell survival, with cancer cells relying on up-regulated Trx and glutathione (GSH) pathways to combat the oxidative stress induced by their replicative drive and enhanced metabolic rate (23). In healthy cells, the GSH system can compensate for inhibition of the Trx system, while in cancer cells, both of these systems are required (24, 25). Thus, TrxR1 seems to be essential in cancer cells but dispensable in healthy cells, thereby making it a promising target for novel anticancer drug discovery (22, 23).
The present study explores the effects of novel inhibitors of STAT3-dependent gene expression that were identified from a high-throughput screen (HTS) (26). Optimization of hit compounds produced potent inhibitors of STAT3-dependent luciferase expression with top compounds having IC50 (half maximal inhibitory concentration) values below 1 μM. A fluorescently tagged analog of the top compounds was used to identify TrxR1 (not STAT3) as the main cellular target of these inhibitors, which was confirmed in complementary and orthogonal assays. In agreement with the current understanding of STAT3 redox regulation (21), TrxR1 inhibition resulted in the inactivation of STAT3 through Cys oxidation and formation of the oxidized STAT3 dimer. This inhibitory mechanism is extended beyond our class of compounds and was found to include some other STAT3 inhibitors with electrophilic tendencies, such as Stattic (14, 18), which was also found to inhibit TrxR1 activity and compete for binding TrxR1 with our fluorescent probe.
RESULTS
Small-molecule inhibitors of STAT3 transcriptional activity
To identify inhibitors of STAT3 transcriptional activity, a cell-based STAT3-dependent luciferase assay was used to screen 28,000 compounds from the Enamine diversity set [reported previously; (26)]. Stably transfected A4wt (A4 with wild-type STAT3 reconstituted) cells with a STAT-inducible SIE (sis-inducible element) reporter construct (A4wt-SIE) were stimulated with interleukin-6 (IL6) to activate STAT3-dependent luciferase transcription. In addition to the previously reported inhibitors (26), this HTS also identified a series of 4,5-dichloropyridazinone compounds as inhibitors of STAT3-dependent gene expression. Among the top compounds in this series were three compounds with sub–10 μM IC50 values in the luciferase assay—DG-1, DG-2, and DG-3—depicted in Fig. 1A.
Fig. 1. 4,5-dichloropyridazinone compounds as inhibitors of STAT3-dependent transcription.
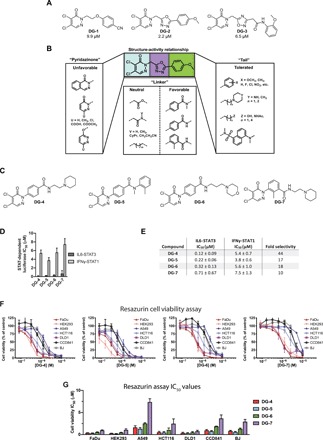
(A) Top compounds from the HTS campaign having the 4,5-dichloropyridazinone core structure. Luciferase IC50 values are reported as an average of two experiments conducted in triplicate. (B) Summary of structure-activity relationship (SAR) study to explore the activity of top compounds. Modifications to the “pyridazinone” moiety (blue) were generally unfavorable, as these compounds lost STAT3 inhibitory capacity, whereas variations on the linker (purple) could increase the potency of the compounds, and several different linkers were tolerated. The tail moiety (green) was quite versatile and could incorporate a large range of functionalities. (C) Four of the most potent compounds from the SAR study, DG-4 to DG-7. (D) Bar graphs of IC50 values for the top four DG compounds for STAT3- and STAT1-driven luciferase assays. IFNγ, interferon γ. (E) Table describing the IC50 values shown in (D), together with fold selectivity for each compound. (F) Resazurin cell viability assays of top inhibitors against several cancer and noncancerous (CCD841 and BJ) cell lines. Compounds were incubated with cells for 72 hours at a concentration range of 0.78 to 100 μM (twofold dilutions); then, resazurin (0.02 mg/ml) was added, and resofurin fluorescence was measured after an additional 5 hours of incubation. Fluorescence values were normalized to DMSO (dimethyl sulfoxide) and media controls, and the resulting points were fit to a nonlinear variable slope curve (four parameters). HEK293, human embryonic kidney–293. (G) IC50 values from the dose-response cell viability experiments shown in (F).
To further explore this class of compounds, we performed a structure-activity relationship (SAR) study to optimize their potency in the STAT3-dependent luciferase assay. As summarized in Fig. 1B, a large range of modifications were tolerated within this series, especially at the “linker” and “tail” moieties (highlighted in purple and green, respectively). The most marked increase in activity was seen when groups with electron-withdrawing functionality were appended to the nitrogen atom at the 2-position of the 4,5-dichloropyridazinone ring. The 4,5-dichloropyridazinone moiety was found to be essential for STAT3 inhibitory activity, as modifications to this group were not tolerated. Top compounds with sub–1 μM IC50 values in the STAT3-dependent luciferase assay are depicted in Fig. 1C (DG-4 to DG-7). Lead compounds were also counterscreened to ensure that they did not directly inhibit the luciferase enzyme or exert toxic effects in cells during the short (5-hour) time course of the luciferase assay experiments (fig. S1, A to C). Inhibition of STAT1-dependent luciferase stimulated with interferon-γ (IFNγ) in STAT3-deficient A4-SIE cells was also assessed (Fig. 1D and fig. S1D). Top compounds more potently inhibited STAT3-dependent luciferase, with a selectivity between 10- and 44-fold compared to the STAT1-dependent luciferase assay (Fig. 1E).
To determine whether these compounds displayed cytotoxicity in cancer cell lines, top compounds were analyzed using a resazurin cell viability assay. Compounds DG-4 to DG-7 were incubated for 72 hours with several human cancer cell lines—FaDu, human embryonic kidney (HEK) 293, A549, HCT116, and DLD1—as well as noncancerous CCD841 colon epithelial cells and BJ fibroblasts (Fig. 1, F and G). DG-4 and DG-5 were potently cytotoxic in these cell lines, followed by DG-6 and, last, DG-7, similar to their efficacy profiles in the luciferase assay. The cytotoxicity toward the different cell lines was similar for all four compounds. CCD841 and BJ cells were less sensitive compared to most cancer cell lines; however, the A549 cells were the least sensitive to the compounds.
Under the suspicion that the 4,5-dichloropyridazinone core may impart electrophilic reactivity to these compounds, we assessed their ability to react with pure GSH in vitro using a 5′-5-dithiobis-(2-nitrobenzoic acid) (DTNB) reporter assay (fig. S1E). Compounds were incubated with GSH for the time points indicated, followed by DTNB addition to assess the remaining amount of free thiols. DTNB reacts with free thiols to produce free 5-thio-2-nitrobenzoic acid (TNB), which can be detected by absorbance at 412 nm (A412). DG-4 to DG-7 all decreased the levels of free thiols in a time-dependent manner, suggesting that they could indeed react with GSH (fig. S1E). This reactivity was confirmed using liquid chromatography–mass spectrometry (LC-MS) experiments, which confirmed that GSH could replace both of the chlorine atoms on the 4,5-dichloropyridazinone ring, as illustrated in fig. S1F.
On the basis of this reactivity, we were concerned that these compounds may nonspecifically react with multiple cellular components and thus induce toxic effects in cells by nonspecific mechanisms. This was especially pertinent, as analogs of the 4,5-dichloropyridazinone core that were not electrophilic failed to inhibit STAT3 activity in the luciferase assay (Fig. 1B).
To further investigate the electrophilic nature of this series and to probe the selectivity of lead agents, we synthesized an analog of our top compounds that contained a fluorescent dansyl moiety (DG-8; Fig. 2A). DG-8 also had potent activity in the STAT3 luciferase reporter assay (IC50 = 980 nM; Fig. 2B). Thus, we next aimed to use DG-8 as a fluorescent probe to identify any prominent cellular interaction partner(s) for the 4,5-dichloropyridazinone series of compounds.
Fig. 2. DG-8, a fluorescent 4,5-dichloropyridazinone probe for target identification and specificity.
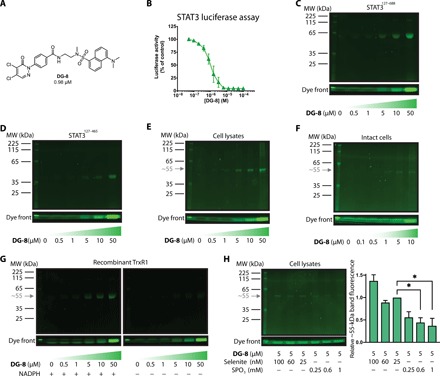
(A) The chemical structure of DG-8, which incorporates many characteristics of the top compounds from the SAR study. (B) STAT3-dependent luciferase assay data showing DG-8 is a potent inhibitor of STAT3-dependent transcription (IC50 = 0.98 μM). (C and D) DG-8 (0.5 to 50 μM) was incubated with 5 μg of two recombinant STAT3 protein truncations STAT3127–688 (C) and STAT3127–465 (D) for 30 min, then run on an SDS–polyacrylamide gel electrophoresis (SDS-PAGE) gel, and analyzed for dansyl fluorescence. The dye front is representative of the amount of DG-8 used in each sample. (E) DG-8 (0.5 to 50 μM) was incubated with A549 cell lysates for 30 min, and the protein content (30 μg) was analyzed by SDS-PAGE and dansyl fluorescence. A single fluorescent band was detected with a molecular weight (MW) of approximately 55 kDa (gray arrow). (F) DG-8 (0.1 to 10 μM) was incubated with A549 cells in culture for 30 min. Cells were then collected and lysed, and 30 μg of the resultant protein lysate was analyzed by SDS-PAGE and dansyl fluorescence. Again, a single fluorescent band was detected at approximately 55 kDa (gray arrow). (G) Recombinant TrxR1 protein (5 μg) was incubated with DG-8 (0.5 to 50 μM) in the presence or absence of NADPH (7.5 μg) as indicated. Following 30 min incubation, samples were analyzed by SDS-PAGE and dansyl fluorescence under reducing conditions. A ~55-kDa fluorescent band is detected only in the presence of NADPH, indicating that DG-8 reactivity with TrxR1 is dependent on NADPH, which is consistent with binding to the Sec residue of TrxR1 (23). (H) To assess whether the ~55-kDa band might be a Sec-containing protein, A549 cells were incubated for 72 hours with sodium selenite (25 to 100 nM) to promote Sec incorporation into cellular selenoproteins or SPO3 (0.25 to 1 mM) to induce Sec-to-Cys substitution (29). The cells were then lysed and treated with DG-8 (5 μM) for 30 min at room temperature. The resulting sample containing 30 μg of protein lysates was run on an SDS-PAGE gel (reducing conditions) and analyzed for dansyl fluorescence. Bands occurring at ~55 kDa were quantified and plotted as bar graphs (*P < 0.05, n = 2). Band intensities were normalized to the sample containing 25 nM selenite, as this was the concentration used throughout this work to ensure adequate selenium supplementation.
Protein target engagement using a fluorescently tagged compound
First, to investigate whether DG-8 could interact with STAT3 protein in vitro, we incubated it with recombinant STAT3 proteins that contained or excluded the SH2 domain [STAT3127–688 and STAT3127–465, respectively (12)]. These truncations were used because of difficulties producing and working with recombinant full-length protein and to assess whether the compound was able to bind preferentially with the SH2 domain of STAT3, which is a primary target for many known STAT3 inhibitors (12). The resulting mixtures were analyzed by SDS–polyacrylamide gel electrophoresis (SDS-PAGE), and the dansyl-tagged proteins could be detected under ultraviolet (UV) excitation, as illustrated in Fig. 2 (C and D). DG-8 could covalently modify both recombinant STAT3127–465 and STAT3127–688 in a concentration-dependent manner, indicating that DG-8 could directly derivatize pure STAT3 in vitro.
To next assess whether STAT3 was the primary target of these compounds in cells, we sought to use DG-8 to fluorescently tag any interaction partner(s) in cell lysates and intact cells. A549 cell lysates were treated with increasing concentrations of DG-8, and the cellular protein content was subsequently run on an SDS-PAGE gel, with any derivatized proteins visualized using the dansyl fluorescence under broad UV excitation. A549 cells were selected because they were the least sensitive cell line for our inhibitors and thus were more likely to tolerate exposure to higher concentrations of the probe. Somewhat unexpectedly, DG-8 appeared to bind a single detectable protein target with a molecular mass of ~55 kDa, noticeably distinct from endogenous STAT3 protein, which runs at ~85 kDa (Fig. 2E). Treatment of live A549 cells in culture also led to the appearance of a single band at ~55 kDa but no noticeable band at ~85 kDa that would have corresponded to endogenous STAT3 (Fig. 2F). In an attempt to identify the protein corresponding to the ~55-kDa band, we isolated the band from the gel, performed a tryptic digest, and analyzed the contents using MS. Unfortunately, no masses could be identified that would correspond to peptides with the added molecular weight of DG-8. Thus, we turned our attention to the scientific literature and performed extensive searches based on the structures identified from our HTS. These searches identified a highly similar compound that is a known TrxR1 inhibitor (having a 4,5-dichloropyridazinone group, appended to an oxadiazole linker, and an aromatic group at the tail position, as shown in fig. S1G) (23, 27). This compound was nearly identical to DG-2 and highly similar to the other compounds from our SAR study. TrxR1, furthermore, has a molecular weight of ~55 kDa and has a Sec residue that is highly nucleophilic (22). TrxR1 peptide fragments were also detected in the sample taken from the fluorescent band, although we could not detect the mass of the Sec-containing peptide or any other TrxR1-derived peptides with the added weight of DG-8.
We therefore tested the ability of DG-8 to modify recombinant TrxR1 in its reduced and oxidized states. NADPH is required to reduce the selenenylsulfide in oxidized TrxR1, which unleashes the nucleophilic activity of the Sec residue (22). Without reduction by NADPH, the Sec residue of TrxR1 is locked in a nonreactive state where it cannot be derivatized with electrophilic compounds. DG-8 could bind recombinant TrxR1 in the presence of NADPH but failed to bind in the absence of NADPH, as shown in Fig. 2G, using SDS-PAGE and visualization of the protein-bound dansyl fluorescence.
TrxR1 as the target of 4,5-dichloropyridazinone compounds
To investigate whether the cellular ~55-kDa band was a Sec-containing protein, we used incubation of the cells with thiophosphate (SPO3) to promote Cys insertion at Sec-encoding UGA codons in A549 cells. This approach has previously been used to drive Cys insertion in place of Sec in TrxR1 and thereby reduce its nucleophilic character (28, 29). As expected, culturing of the cells in higher concentrations of SPO3 decreased the binding of the fluorescent probe to the ~55-kDa band in the corresponding cell lysates (Fig. 2H). This finding strongly suggested that this band represents a Sec-containing protein, as non–Sec-containing proteins would not be altered upon incubation with SPO3. Notably, the intensity of the fluorescent band was not increased with statistical significance upon the incubation with higher sodium selenite concentrations, typically used to ensure that adequate levels of selenium for selenoprotein synthesis are present in the cell culture media. This indicated that Sec incorporation was already at a maximum under our standard cell culture conditions (Fig. 2H). The only selenoproteins that were detected in the mass spectrometric analysis of the fluorescent band were cytosolic TrxR1 and mitochondrial TrxR2 (both proteins having similar molecular weights), suggesting that either one or both of these two selenoproteins had been derivatized.
To confirm that our top compounds could also bind to this ~55-kDa band, we next performed competition assays with our top inhibitors in A549 cell lysates (Fig. 3, A to C). DG-4 and DG-6 were both able to potently outcompete the fluorescent probe in binding to the ~55-kDa band; however, DG-7 only outcompeted the probe at the highest concentration (50 μM). In line with these results and under the suspicion that this band may correspond to TrxR1, we tested whether the previously described TrxR1 inhibitors TRi-1, TRi-2, TRi-3, and auranofin (23) could also compete with DG-8 for binding. While TRi-1 and auranofin potently outcompeted the probe, TRi-2 was unable to do so, and TRi-3 only decreased the fluorescence at high concentrations (50 μM) similar to DG-7 (Fig. 3, D to G).
Fig. 3. DG-8 competition assays in A549 cell lysates.
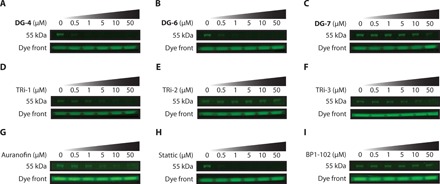
(A to C) To investigate whether top DG compounds shared the same target as DG-8, A549 protein lysates (30 μg) were incubated with DG compounds (0.5 to 50 μM) for 30 min and then with DG-8 (5 μM) for 30 min. Samples were then run on an SDS-PAGE gel, and DG-8 fluorescence was measured using a Gel Doc EZ Gel Documentation System with UV tray. Both DG-4 and DG-5 outcompeted DG-8 for binding at low micromolar concentrations, whereas DG-7 was much less potent. (D to G) Established TrxR1 inhibitors TRi-1, TRi-2, TRi-3, and auranofin were assessed in the DG-8 competition assay. TRi-3 is highly similar to our lead series of compounds and has a 4,5-dichloropyridazinone group, suggesting that it could have the same molecular target(s) as our top compounds. TRi-1 and auranofin inhibit TrxR1 through binding to its Sec residue, whereas TRi-2 is thought to function by a non-Sec binding mechanism. Hence, TRi-1, TRi-3, and auranofin could all compete with DG-8, suggesting that this band may correspond to the 55-kDa selenoprotein TrxR1 by covalently reacting with its Sec residue. (H and I) Two popular STAT3 inhibitors Stattic and BP1-102 were assessed for DG-8 competition. While both Stattic and BP1-102 claim to be direct binders of STAT3 protein in cells, Stattic showed competition with DG-8, indicating that it may share some common STAT3 inhibitory effects as our top DG compounds. BP1-102 was not able to compete with DG-8 for binding to the ~55-kDa band. All SDS-PAGE gels were run under reducing conditions, and the fluorescence of the dye front was used to ensure regular loading.
We subsequently sought to analyze whether other known STAT3 inhibitors might also compete with DG-8 for binding to the ~55-kDa protein in cell lysates. We used two classic STAT3 inhibitors that have recently been highlighted for their reactivity and ability to bind Cys residues on STAT3: Stattic (14) and BP1-102 (15). While Stattic could compete with DG-8 for binding, BP1-102 was not able to compete with the probe (Fig. 3, H and I), suggesting that Stattic, but not BP1-102, may inhibit STAT3 signaling with the same mechanism as demonstrated with our top inhibitors. It was unexpected to us that Stattic could compete with DG-8, as this band cannot be STAT3 because of its molecular weight and would rather be TrxR1 (or TrxR2). The ~55-kDa band in cell lysates also overlaid nicely with TrxR1 when comparing the SDS-PAGE fluorescence with immunoblotting for TrxR1 (fig. S4, A to C).
Compounds inhibit TrxR1 function
Cellular TrxR1 inhibitory activity was next analyzed using the TrxR1-specific Trx1-linked insulin disulfide reduction end point assay, which does not detect TrxR2 activity because TrxR2 cannot reduce Trx1 (30). Briefly, cultured cells were treated with inhibitors for 3 hours and then lysed, and TrxR1 activity was assessed in the protein lysates. TrxR1 uses NADPH to reduce Trx1, which, in turn, reduces the disulfide bonds in exogenously added insulin, which is detected using DTNB. If TrxR1 is inhibited, then insulin will not be reduced and will thereby not react with DTNB to produce TNB− anions, which are detected by A412. This assay demonstrated that DG-4 and DG-5 were potent inhibitors of cellular TrxR1 activity following incubation of the cells with the compounds (1 μM) (Fig. 4A). DG-6 and Stattic inhibited approximately 40% of the cellular TrxR1 activity, while DG-7 and BP1-102 did not appreciably alter the TrxR1 activity. This mirrored the ability of these compounds to compete with DG-8 for binding to the ~55-kDa band well.
Fig. 4. Top DG compounds inhibit TrxR1 activity and contribute to TrxR1 inhibitory effects in cells.
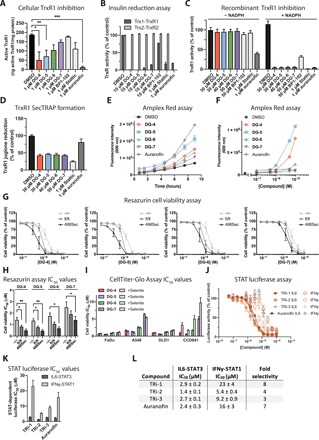
(A) Cellular TrxR1 activity was analyzed using the insulin end point assay (23) following incubation of the indicated compounds (1 μM) in cultured FaDu cells for 3 hours. *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, n = 3. (B) Inhibition of recombinant TrxR1 and TrxR2 proteins were assessed in vitro using an insulin reduction assay, where insulin was reduced by Trx1 and Trx2, respectively. (C) Inhibition of TrxR1 activity was assessed in vitro using an enzymatic DTNB assay after 90 min of incubation (23). In the absence of NADPH, the Sec residue of TrxR1 forms a Sec-cysteine (Cys) bond and is incapable of reacting with electrophilic compounds; therefore, no inhibitory activity is observed with any of the tested compounds without NADPH. Addition of NADPH reduces the Sec-Cys bond, releasing the Sec residue so that it can react with electrophilic compounds. Thus, when NADPH is present, strong inhibition of TrxR1 activity is detected. (D) Irreversible binding of the Sec residue of TrxR1 leads to the formation of selenium-compromised TrxR–derived apoptotic proteins (SecTRAPs), which can be measured by juglone reduction independent of the activity of the Sec residue. Redox cycling of juglone occurs at a distinct site from TrxR1 and will continue even in the absence of Sec redox activity. Under the same conditions that generated complete inhibition of TrxR1 activity in the DTNB assay, TrxR1 retained its ability to reduce juglone, indicating the formation of SecTRAPs. (E and F) Cellular H2O2 production was measured using the Amplex Red assay. Treatment of FaDu cells with top DG compounds led to a time-dependent increase (E) [compounds (0.5 μM)] and concentration-dependent (F) increase in cellular H2O2 levels. (G) Top DG compounds were assessed in mouse embryonic fibroblast (MEF) cells with altered mouse TrxR1 gene expression (Trxnd1). Resazurin cell viability was assessed following 72 hours of compound treatment and additional 5 hours of exposure to resazurin. TrxR1 knockout cells (−/−) were less sensitive to the top compounds compared to wild type (fl/fl). Overexpression of TrxR1 (498Sec) also increased their sensitivity to top compounds. (H) IC50 values for the viability curves shown in (G). (I) IC50 values for CellTiter-Glo cell viability of top DG compounds cultured without or with sodium selenite (100 nM) supplemented medium. Cell viability was assessed following 72 hours of compound treatment. Diminished compound activity by sodium selenite (100 nM) was only detected in noncancerous CCD841 cells, while for cancer cells, no changes were observed. (J) Known TrxR1 inhibitors TRi-1, TRi-2, TRi-3, and auranofin were analyzed in the STAT3- and STAT1-dependent luciferase assay. To measure STAT-dependent transcription, A4wt-SIE cells were stimulated with IL6 (50 ng/ml) and sIL6R (100 ng/ml) for 1 hour, while A4-SIE cells were stimulated with IFNγ (40 IU/ml) for 1 hour, and then, compounds were added for an additional 5 hours, followed by luciferase measurement. (K and L) IC50 values from the experiments described in (J) displayed as a bar graph (K) and table containing fold selectivity comparing STAT3 to STAT1 inhibition (L).
Since both TrxR1 and TrxR2 were detected in the fluorescent band MS, inhibition of both TrxR1 and TrxR2 was also assessed using recombinant proteins in enzymatic activity assays in the presence of NADPH, where Trx1 (for TrxR1) or Trx2 (for TrxR2) were used to reduce insulin (Fig. 4B). Reduction of the corresponding Trx substrate is completely dependent on the Sec-containing active site in both TrxR1 and TrxR2. The top DG compounds, Stattic and auranofin, potently inhibited the activity of TrxR1 in this assay. However, auranofin was the only compound that inhibited TrxR2 under these conditions. BP1-102 did not fully inhibit the enzymatic activity of TrxR1, in agreement with its inability to outcompete the fluorescent probe (Fig. 3I).
Furthermore, compounds were incubated with TrxR1 either in the presence or in the absence of NADPH. Following incubation with inhibitors, TrxR1 activity was assessed using DTNB as a direct substrate of TrxR1 (30). Prior reduction of TrxR1 by NADPH was necessary for the compounds to inhibit the enzyme (Fig. 4C), suggesting targeting of its NADPH-reduced active site residue(s), most likely the highly nucleophilic Sec residue.
Covalent modification of the Sec residue leads to the inactivation of the C-terminal active site of TrxR1. However, TrxR1 also has another active site with a flavin adenine dinucleotide (FAD) moiety and a redox active disulfide/dithiol motif, which can display NADPH oxidase activity and redox cycle with substrates such as juglone when the Sec residue has been compromised in the enzyme (31). Inhibiting the Sec-containing active site, but not the FAD-containing active site, can thereby convert TrxR1 into a SecTRAP (selenium-compromised TrxR–derived apoptotic protein), which can lead to additional production of reactive oxygen species in cells (23, 31). Following incubation of TrxR1 with inhibitors and NADPH, TrxR1 lost its ability to reduce DTNB, indicating a loss of function of its Sec-containing active site (Fig. 4C). However, under these conditions, TrxR1 still maintained its ability to consume NADPH coupled to redox cycling with juglone, thus indicating the formation of SecTRAPs (Fig. 4D).
In line with these findings, we investigated whether treatment with our inhibitors led to increased H2O2 production in FaDu cells using the Amplex Red assay (Fig. 4, E and F). Treatment with DG-4 or DG-5 led to increasing levels of H2O2, as detected in the culture medium, in a time- and concentration-dependent manner. Treatment with DG-6 induced quite low levels of H2O2 production, and DG-7 induced the lowest levels of H2O2 in this assay setting.
To assess the importance of TrxR1 expression for the effects of the compounds on cell viability, we tested mouse embryonic fibroblast (MEF) cells expressing wild-type TrxR1 (Txnrd1fl/fl), having a TrxR1 genetic knockout (Txnrd1−/−) and overexpressing a Sec-containing active variant of the enzyme (Txnrd1498Sec) (32). Treatment of these cell lines gave a similar overall trend in the cell viability assays for all four compounds (Fig. 4, G and H). Txnrd1fl/fl cells were more sensitive to inhibitor treatment than the Txnrd1−/− cells, indicating that TrxR1 is important for the activity of these compounds. The Txnrd1498Sec-overexpressing cells were the most sensitive to the compounds. This also supports a SecTRAP-dependent mechanism, where the additional Txnrd1 is likely converted to the prooxidant SecTRAP enzyme species that induce further oxidative stress and can kill the cells. DG-7 was less potent than other inhibitors in these experiments, in accordance with the cellular TrxR1 inhibition and H2O2 production results.
Last, to ensure that sufficient levels of selenium were present in the cell viability experiments, the effects of adding 100 nM sodium selenite were investigated in combination with the top DG compounds. The additional selenium did not affect the viability of FaDu, A549, and DLD1 cells with the DG compounds (Fig. 4I and fig. S5), similar to the finding with DG-8 (Fig. 2H). However, the additional selenium had a moderate protective effect in the noncancerous CCD841 cell line with approximately two- to fourfold higher IC50 values upon selenium supplementation (Fig. 4I). These results are in agreement with the high basal oxidative stress generation in cancer cells compared to healthy cells (33). Selenium supplementation and, thereby, higher activity of the Trx system were likely able to compensate the additional oxidative stress induction by the top DG compounds in the noncancerous CCD841 cells, while in cancer cells, the Trx system is already substantially encumbered under the oxidative stress in cancer cells and thereby did not benefit from the additional available selenium.
Established TrxR1 inhibitors selectively block STAT3-driven transcription
To further explore the relationship between TrxR1 and STAT3 signaling, we assessed whether the previously identified TrxR1 inhibitors (TRi-1, TRi-2, TRi-3, and auranofin) that outcompeted DG-8 (Fig. 3, D to G) could also inhibit STAT1- and STAT3-driven transcription. All four TrxR1 inhibitors, indeed, potently blocked STAT3-dependent luciferase expression, with IC50 values between 1 to 3 μM (Fig. 4J). In comparison, these compounds inhibited the STAT1-dependent luciferase expression with IC50 values between 5 and 23 μM, resulting in three- to eightfold selectivity for STAT3 over STAT1 (Fig. 4, J to L).
DG compounds induce oxidative stress leading to Prx2/STAT3 oxidation and cell death
To explore the underlying mechanisms leading to STAT3 inhibition, HEK293 cells were treated with the top compounds and analyzed for STAT3 and Prx2 oxidation. Previous studies with HEK293 cells have demonstrated their ability to produce oxidized STAT3 dimers following H2O2 treatment in a process dependent on Prx2 (21). Treatment of the cells with 10 μM DG-4, DG-5, and DG-6 indeed induced formation of oxidized STAT3 dimers (Fig. 5A). Somewhat unexpectedly, treatment with DG-7, which is also a potent STAT3 inhibitor in the luciferase assay (IC50 = 709 nM), failed to induce STAT3 dimer formation. In a similar fashion, Prx2 was oxidized upon exposure to DG-4, DG-5, and DG-6 but only slightly with DG-7, supporting that STAT3 dimer formation is likely driven by an oxidative stress response (Fig. 5A). All these protein dimers were resolved by the addition of the reducing agent dithiothreitol (DTT) to the samples (Fig. 5B), thus confirming that the dimers were mediated by disulfide bond formation.
Fig. 5. Top DG compounds affect cellular redox balance.
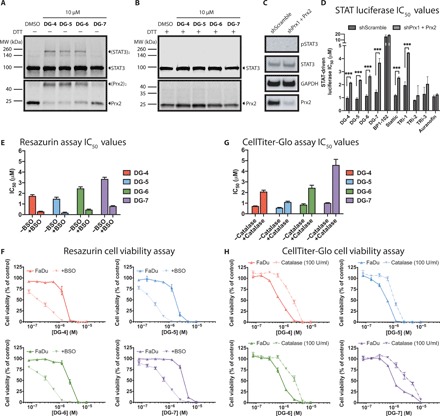
(A) Western blot analyses of HEK293 cells treated with the top DG compounds. Following a 30-min exposure to compounds, STAT3 and Prx2 were oxidized to form dimers in the absence of reducing agents such as DTT. (B) Similar to the experiment shown in (A), however, the addition of a reducing agent (DTT) during protein sample preparation reduces the inter- and intraprotein disulfide interactions, eliminating the bands corresponding to oligomeric STAT3 and Prx2. (C) Western blot analyses of pSTAT3/STAT3 and Prx2 expression in HEK293-shScramble and HEK293–shPrx1 + Prx2 cells. (D) IC50 values of indicated compounds for STAT-driven luciferase inhibition curves in HEK293-shScramble and HEK293–shPrx1 + Prx2 cells stimulated with IL6 (50 ng/ml) and sIL6R (100 ng/ml). Prx1 + Prx2 knockdown could rescue STAT-dependent transcription leading for our top DG compounds and for TRi-1 and Stattic. (E) Resazurin viability IC50 values for FaDu cells incubated with top DG compounds for 72 hours. Cells were preincubated with or without buthionine sulfoximine (BSO) (100 μM) for 24 hours before the addition of top DG compounds. (F) Cell viability curves for the data described in (E). (G) CellTiter-Glo viability IC50 values for FaDu cells incubated with top DG compounds for 72 hours. Cells were preincubated with or without catalase (100 U/ml) for 4 hours before the addition of top inhibitors. (H) Cell viability curves for the data described in (G).
To assess the importance of Prxs, we evaluated STAT-dependent luciferase transcription in HEK293-shScramble and shPrx1 + Prx2 cells. Both cell lines did not display any detectable basal endogenous STAT3 activation, as assessed by its phosphorylation status, and Prx2 levels were visibly diminished in the HEK293–shPrx1 + Prx2 cells (Fig. 5C). HEK293 cells express both STAT3 and STAT1; therefore, it is expected that the SIE reporter construct would be driven by activation of STAT3 and STAT1 when stimulated with IL6. Nevertheless, we were able to detect significant differences in IC50 values comparing the inhibition of STAT-driven luciferase expression using both our top compounds and the established TrxR1 inhibitors (Fig. 5D and fig. S7A). Top DG compounds, Stattic and TRi-1, more potently inhibited luciferase expression in the control cells than in the Prx1 + Prx2 knockdown cells. These results were not linked with general disturbances in the STAT pathway activation patterns since IL6-dependent induction of luciferase was similar in both cell lines (fig. S7B).
To further probe whether the impairment of viability could be linked to oxidative stress, we investigated cytotoxic activities of the top DG compounds in combination with buthionine sulfoximine (BSO), an irreversible inhibitor of γ-glutamylcysteine synthetase that induces GSH depletion in cells. Cells rely on the combined antioxidant activities of the GSH and Trx systems to maintain redox balance (24). Thus, a blockade of GSH synthesis should potentiate the effects of TrxR1 inhibition. Supporting this mechanism of action, incubation of FaDu cells with 100 μM BSO induced an approximate fivefold potentiation of DG-4, DG-5, DG-6, and DG-7, further supporting TrxR1 as the primary target of these compounds (Fig. 5, E and F). DG-7 was the least potent of the top DG compounds, consistent with its failure to induce oxidized STAT3 and Prx2 dimers and increased cellular H2O2 levels, as well as its poor ability to compete with DG-8 for TrxR1 binding. However, BSO treatment still potentiated the cytotoxicity of DG-7, indicating that there may be additional factors that lead to an enhanced cytotoxicity of these compounds in conjunction with BSO treatment.
The mechanism of action was further confirmed to be related to increased H2O2 levels and oxidative stress upon assessing the cytotoxicity of the top DG compounds in combination with catalase, an enzyme that catalyzes the decomposition of H2O2 to water and oxygen. Thereby, catalase should lower the cytotoxicity of the top DG compounds if it would be linked to H2O2 production. This was indeed confirmed in both FaDu and A549 cells, where catalase addition increased the IC50 values of all DG compounds (Fig. 5, G and H, and fig. S8, A and B). These robust effects were, however, not seen in DLD1 cells, where catalase had no effect on viability (fig. S8, A and B), suggesting that excess H2O2 production was not the sole cause of the triggered cell death.
Compounds induce Nrf2 activation
The possible specificity of TrxR1 inhibition on STAT3 pathway inhibition was further explored by assessing the effects of the compounds on nuclear factor erythroid 2-related factor 2 (Nrf2) and nuclear factor κB (NFκB) activation patterns. HEK293 cells were transfected with the plasmid for transcription factor reporter activation based on fluorescence (pTRAF) vector, which uses expression of different fluorescent proteins as reporters for Nrf2 and NFκB activities (fig. S9A) (34). The pTRAF vector can also be used to investigate hypoxia-inducible factor (HIF), but in our cellular models, HIF activation was minimal, so this parameter was not evaluated. All top DG compounds and Stattic had little effect on NFκB activation but clearly triggered Nrf2 activation (fig. S9B), again in agreement with TrxR1 inhibition, as that often leads to Nrf2 activation. BP1-102 did not activate Nrf2, which agrees with its inability to outcompete DG-8 and its poor inhibition of TrxR1 activity (Figs. 3I and 4, B and C).
DISCUSSION
The development of STAT3 inhibitors is a highly active field of research, which continues to produce novel inhibitors at a rapid rate. These compounds are diverse in chemical structure and may block STAT3 signaling through a variety of different mechanisms to exert their anti-STAT3 and anticancer effects. While demonstrating impaired STAT3 signaling in cells is a relatively straightforward task (using STAT3 phosphorylation or STAT3-dependent gene expression as readouts), it is traditionally very difficult to determine whether these phenotypes are due to direct binding of STAT3 in cells or whether they may be an indirect consequence of inhibiting other targets that influence STAT3 signaling. Therefore, determining the cellular interaction partners of experimental inhibitors should be a top priority in the drug discovery and development process for STAT3 inhibitors. Not only will this streamline the optimization of inhibitors, but also it can lead to new discoveries related to fundamental biological phenomena.
In this light, we demonstrated that our compounds could covalently bind to recombinant STAT3 protein truncations in vitro and that they could also block STAT3-dependent luciferase expression in cells. While this could suggest that these compounds bind directly to STAT3 to block its transcriptional activity in cells, this was clearly not the case for this group of compounds.
Using a chemical biology approach, we demonstrated that these inhibitors bind to TrxR1 to inhibit its function. This induces oxidative stress and produces oxidized Prx2 and STAT3 dimers, which prevents the transcriptional activation of STAT3. This is in line with recently described redox regulation of STAT3 signaling by Prx2, Trx1, and TrxR1 (21), as summarized in Fig. 6. The activities of STAT1, the closest family member of STAT3, as well as NFκB, were left relatively unaffected, while Nrf2 was activated by the compounds.
Fig. 6. Proposed model of STAT3 inhibition mediated by TrxR1.
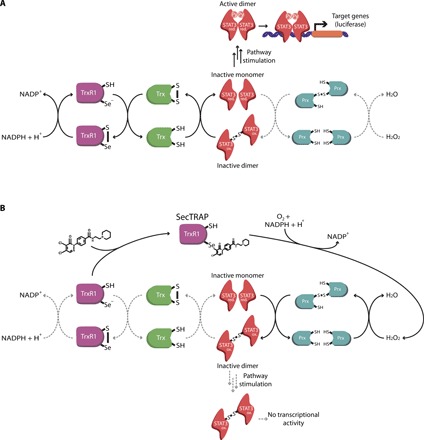
Black arrows indicate predominantly occurring activities, whereas gray dashed arrows indicate infrequent or inhibited pathways. (A) A general model of the TrxR1 relay system: H2O2 levels are sensed and managed by Prx, which relays oxidative equivalents to STAT3, Trx, and/or other targets. High levels of reduced Trx are maintained by TrxR1, which passes reducing equivalents to Trx from NADPH. Upon cytokine stimulation (for example, with IL6), reduced STAT3 monomers or oligomers will be phosphorylated on Tyr705 to form the transcriptionally active pSTAT3 homodimer, which can interact with specific DNA sequences to promote STAT3-dependent gene expression. (B) Targeting the Sec residue of TrxR1 with electrophilic small-molecule inhibitors leads to the formation of SecTRAPs and disruption of the TrxR1 relay system. SecTRAP formation induces H2O2 production and consumption of NADPH by the second TrxR1 active site containing an FAD moiety and a redox active disulfide/dithiol motif. Increased cellular H2O2 levels cause oxidation of Prx, which relays oxidative equivalents to STAT3 and/or other downstream targets (including directly to Trx). This leads to the formation of disulfide-linked STAT3 dimers (and potentially other oxidized monomeric species) that are not transcriptionally active. SecTRAP formation also blocks the antioxidant function of TrxR1, which will no longer be able to supply reducing equivalents from NADPH to Trx, which prevents the subsequent reduction of Trx’s downstream targets.
While inhibition of STAT3 transcription was observed upon DG compound treatment, other markers of TrxR1 inhibition were seen, including its direct derivatization, the selenium dependence of the effects, the cellular TrxR1 activity inhibition, the increased oxidative stress as assessed by H2O2 measurements using Amplex Red, and the Nrf2 activation. It is likely that both the increased oxidative stress from SecTRAP formation and inhibition of cellular STAT3 activity contribute to the impaired viability of the cells upon exposure to the DG compounds.
TrxR1 is an important target for the development of novel cancer therapies in its own right, and there have been advanced preclinical studies highlighting TrxR1 inhibitors as potential cancer therapeutic agents (22, 23, 35). An impactful finding of our work is that STAT3 transcriptional activity may be an important biomarker of TrxR1 inhibition, as the known TrxR1 inhibitors (TRi-1, TRi-2, TRi-3, and auranofin) all inhibited STAT3-dependent luciferase activity. While our experiments clearly highlighted this phenomenon in cell-based assays, further in vivo investigations into this relationship, to assess the general importance of TrxR1 for STAT3 inhibition in cancer treatment, are warranted.
Because STAT3 lacks a traditional binding site for small-molecule inhibitors, developing compounds that bind directly to STAT3 protein is a very difficult task. By contrast, the Sec residue of TrxR1 is highly reactive, well defined, and solvent exposed, which facilitates TrxR1 inhibitor development. Our TrxR1 inhibitors have a delicate balance in reactivity, as they are reactive enough to covalently modify the Sec residue of TrxR1 but seem stable enough not to indiscriminately bind multiple cellular targets. This molecular “sweet spot” works well for cell-based assays; however, these compounds will need to also be metabolically stable if they are to function in vivo, which will be a major translational challenge.
In addition to the promising TrxR1 inhibitors, we identified a selective fluorescent probe, DG-8, that enabled us to visualize TrxR1 target engagement in A549 cells. While this visualization may be, in part, due to A549 cells expressing high levels of TrxR1, we propose that the herein described competition assay may be a valuable tool for further studies of TrxR1 inhibitors, especially if they are suspected to behave as covalent modifiers of the TrxR1 Sec residue.
Stattic represents one of the first STAT3 inhibitors to have been found and is widely used in biological studies as a presumed selective inhibitor of STAT3 signaling (14). Notably, Stattic was used as a positive control in our prior HTS for STAT3 inhibitors (26). While we here had intended to use Stattic and BP1-102 as negative controls in the DG-8 competition assay, we found that Stattic also competed with the probe, suggesting that it also may target TrxR1 in cells as a bona fide mechanism of action. While this was an unexpected result to us, further investigation has revealed other reports of Stattic’s ability to also bind another selenoprotein with TrxR activity, namely, the thioredoxin glutathione reductase of the parasite Schistosoma mansoni (36).
The described 4,5-dichloropyridazinone compounds are generally reactive toward thiol species in vitro, as demonstrated by their ability to bind GSH. This reactivity was initially perceived as a liability for these compounds, and we had hoped to eliminate their thiol reactivities with maintained STAT3 inhibitory activity through structural modifications. The SAR study, however, identified that this 4,5-dichloropyridazinone moiety was critical for the anti-STAT3 activity. Further optimization led to the discovery of potent, nanomolar inhibitors of STAT3-dependent luciferase activity. While both active and inactive compounds in the luciferase assay could bind to GSH in biochemical experiments, only our top STAT3 inhibitors showed inhibition of TrxR1 in cell-based assays. This suggests that the mechanism of action for these inhibitors is not merely due to their inherent reactivity toward GSH but instead rather strongly points to selective inhibition of TrxR1 as their primary mechanism of action in cells. Notably, however, the STAT3 inhibitor BP1-102, which has electrophilic tendencies, was not a potent inhibitor of TrxR1, suggesting that not all electrophilic compounds inhibiting cellular STAT3 function are de facto TrxR1 inhibitors.
While there are previous studies that have linked the cellular functions of STAT3 and TrxR1 (21, 37, 38), our findings are the first to firmly support that inhibition of STAT3-dependent gene expression can be induced by targeting TrxR1 in cancer cells. These results are further supported by previous investigations where the pan-TrxR inhibitor auranofin was shown to block IL6-induced Janus kinase 1 and STAT3 phosphorylation (37) and, more recently, a previously unidentified TrxR1 inhibitor was shown to inhibit tumorigenesis by down-regulation of programmed death ligand 1 via the STAT3 pathway (38). Several anticancer drugs have separately been shown to inhibit TrxR1 in conjunction with their principal targets including cisplatin, chlorambucil, carmustine, and melphalan (39). This suggests that inhibition of TrxR1, and potentially STAT3, may contribute to the clinical efficacy of these drugs, and this should be an important consideration when evaluating the underlying mechanisms of known or novel cancer therapeutics.
Inhibition of TrxR1 and the specific formation of SecTRAPs can broadly influence the cellular redox state (31). While we have demonstrated that TrxR1 inhibition induces Prx2 and STAT3 oxidation, the wider consequences on redox-sensitive pathways in cells remain an active area of interest. Since STAT3 activity is mediated by serine and tyrosine phosphorylation, the effect of SecTRAPs on redox-sensitive protein tyrosine phosphatases (PTPs), such as PTP1B (40), may also contribute to changes in STAT3 signaling and transcriptional activity. Several PTPs are known to deactivate pSTAT3 via removal of its phosphate groups, including those that are sensitive to oxidative stress (PTP1B) and those that are not, such as the SH2 domain–containing phosphatases and others (41). Further elucidating this complex relationship should be of interest.
The herein described compounds represent novel potent inhibitors of STAT3-dependent luciferase activity in cells and were shown to directly target TrxR1. These compounds will be further explored as potential cancer therapeutics and will be used as probes to study the relationship between TrxR1 and the STAT3 signaling cascade. We further hope that the link between STAT3 and TrxR1 can be exploited to generate novel cancer therapies involving concomitant inhibition of TrxR1 and STAT3.
MATERIALS AND METHODS
Cell culture and treatment conditions
Cell cultures were grown at 37°C and 5% CO2 in medium containing 10% fetal bovine serum, 2 mM l-glutamine, and penicillin-streptomycin (100 μg/ml). Experiments were performed in duplicate or triplicate, as indicated. Media were supplemented with 25 nM sodium selenite in all experiments, excluding those shown in Figs. 1 (A to C) and 3, unless otherwise indicated. All compounds were diluted in dimethyl sulfoxide (DMSO) to maximally 1% final concentration (as indicated). Unless otherwise noted, DMSO controls were exposed to the same amount of DMSO as the highest concentration of treatment.
Sublines of human adenocarcinoma cell line DLD1 A4 (with homologous deletion of STAT3) and A4wt were a gift of Z. Wang, Genetics and Case Comprehensive Cancer Center, Case Western Reserve University (Cleveland, OH). Both lines that stably expressed a STAT-inducible luciferase reporter (A4-SIE and A4wt-SIE) were previously generated in the laboratory and were grown on hygromycin (1 mg/ml) during selection (26). BJ and CCD841 cell lines were provided by the Thomas Helleday Lab, Karolinska Institutet, Department of Oncology and Pathology. HEK293-shScramble and HEK293–shPrx1 + Prx2 were gifted by the lab of T. Dick, German Cancer Research Center, Heidelberg, Germany.
All media and other cell culture reagents were purchased from Gibco (Thermo Fisher Scientific). A4, A4wt, and DLD1 cells were cultured in Dulbecco’s modified Eagle medium (DMEM). HEK293, BJ, and CCD841 were cultured in DMEM + GlutaMAX (without additional l-glutamine). FaDu cells were cultured in DMEM + GlutaMAX (without additional l-glutamine) with 0.02 M Hepes buffer, 1 mM sodium pyruvate, and 1× nonessential amino acids. HCT116 and A549 cells were cultured in RPMI 1640 media. All remaining cell lines were purchased from the American Type Culture Collection. Mycoplasma testing was conducted every time a new stock vial was taken up from storage.
Compound screening STAT3-dependent luciferase assay
Luciferase assay screening was conducted as previously reported (26). Briefly, 4000 A4wt cells per well were seeded in white opaque 384-well plates (PerkinElmer, Sweden) using a multidrop microplate dispenser (Thermo Fisher Scientific). The following day, cells were initially treated with IL6 and its soluble receptor (sIL6R, PeproTech Inc.) at 50 and 100 ng/ml, respectively, and after 1 hour of incubation, test compounds were added. After an additional 5 hours of incubation with compounds, steadylite plus reporter gene assay system (PerkinElmer, Netherlands) was added to the cells, and luminescence per well was measured on a Spark 10M microplate reader (Tecan Group Ltd.).
Compound analogs were either purchased from Enamine Ltd. or synthesized in-house. Compounds were serially diluted and consecutively added to the cells using a Bravo Automated Liquid Handling Platform (Agilent Technologies) or by hand. BP1-102 (Merck Millipore) was used as a positive control. All cytokines and compounds were stored according to the recommendations from the manufacturers.
Counterscreen for assay inhibition
The assay was run similarly to the normal luciferase assay; however, compounds were added right before the addition of the steadylite reagent and consecutive luciferase measurement to assess direct effects on steadylite or luciferase enzyme activity.
Counterscreen for cell viability
The assay was run similarly to the normal luciferase assay. However, no IL6 and sIL6R was added before compound addition. In addition, instead of the steadylite reagent, CellTiter-Glo (Promega) reagent was added to measure cell viability after 5 hours of compound treatment using a luciferase readout.
STAT1-dependent luciferase assay
The assay was run similarly to the STAT3 luciferase assay. However, A4 cells (STAT3 knockout) that stably expressed the STAT-inducible luciferase reporter were used. A4 cells were stimulated with IFNγ (40 IU/ml), and after 1 hour of incubation, compounds were added.
STAT-dependent luciferase in HEK293-shScramble and HEK293–shPrx1 + Prx2
The assay was run similarly to the STAT3 luciferase assay. However, HEK293 cells were seeded at 2000 cells per well. Cells were grown in medium supplemented with 100 nM sodium selenite at least 72 hours before seeding. The following day, cells were transfected with 25 ng of the pGL4-SIE reporter construct together with 20 nl of Viromer Red and 480 nl of Buffer Red in Opti-MEM to a total volume of 5 μl per well. After an additional 24 hours, cells were stimulated with IL6 (50 ng/ml) and sIL6R (100 ng/ml), and after 1 hour of incubation, compounds were added.
Resazurin cell viability assays
The resazurin assay was performed as previously reported (42). Briefly, 2000 cells per well were plated in 96-well plates in media. The following day, compounds were added and incubated for 72 hours and then treated with resazurin cell viability reagent (0.02 mg/ml), and plates were incubated at 37°C for an additional 5 hours. Resorufin fluorescence was measured at 544-nm excitation and 590-nm emission with a Hidex Sense fluorescent plate reader. Cell viability was normalized to DMSO controls and blank wells containing media but no cells.
CellTiter-Glo cell viability assays
Cells were seeded at a density of 750 cells per well in white opaque 384-well plates using a multidrop microplate dispenser. The following day, compounds were added and incubated for 72 hours, followed by addition of CellTiter-Glo (10 μl per well) (Promega), and luminescence was measured using an Infinite M200 PRO plate reader (Tecan Group Ltd.). Cell viability was normalized to DMSO controls.
Cell viability with catalase addition
The assay was run similarly to the CellTiter-Glo assay. However, catalase (10 μl per well) (C30, Sigma-Aldrich) to a final concentration of 100 U/ml was added to cells 4 hours before addition of the compounds.
Cell viability with selenite addition
The assay was run exactly as the CellTiter-Glo assay. However, cells were grown at least 72 hours with the addition of 100 nM sodium selenite in their respective growth medium before cell seeding of the experiment.
DTNB GSH reactivity assay
Similar to previously reported methods (43), 100 μM compound was incubated with 200 μM GSH in tris-EDTA (TE) buffer for 12 hours. At 0, 1, 2, 4, 6, 8, and 12 hours, 180 μl of 1 mM DTNB was added to 20 μl of the reaction to assess the concentration of reduced GSH remaining in the reaction. TNB production was measured as A412. The reduced GSH concentration in the reaction was derived from a GSH standard curve from 0 to 200 μM.
GSH reactivity assay by high-performance LC-MS
DG-3 was assessed for its ability to react with GSH in vitro using a slightly modified procedure from previously reported methods (18–20). From 10 mM stock solutions in DMSO, 5 μl of aliquots were transferred to an analytical high-performance LC (HPLC) vial containing 495 μl of reaction buffer (50 mM Hepes, buffered to pH 7.4) and with or without 10.0 mM reduced GSH. Samples were taken at 30 min and 18 hours for a Hewlett Packard Series 1100 analytical HPLC-MS system using an ACE C8 column (50 mm by 3.0 mm) with 3-μm particles and flow rate of 1 ml/min. Samples were eluted using a gradient of A, 0.1% trifluoroacetic acid in H2O, and B, acetonitrile, 10 to 90% B over 1.5 min and an additional minute at 90% B. Changes in the absorbance profile at 254 nm were observed, and the HPLC peak areas corresponding to the decay of the parent compound were recorded. Metabolite identification was based on mass/charge ratio values and reported alongside their retention time (tR).
SDS-PAGE and dansyl fluorescence analysis using DG-8
Recombinant proteins
Five micrograms of human recombinant protein (TrxR1, STAT3127–465, and STAT3127–688) was incubated with the indicated concentrations of dansyl-tagged analog (DG-8) for 30 min. Recombinant TrxR1 experiments were also supplemented with 7.5 μg of NADPH, as indicated. Samples were mixed with NuPAGE LDS Sample Buffer (6.25 μl) and 100 mM DTT (2.5 μl), heated at 95°C for 5 min, and then loaded onto 10% bis-tris gels (NP0316BOX, Thermo Fisher Scientific). Gels were run at 180 V. The fluorescent signal was imaged using a Gel Doc EZ Gel Documentation System with UV tray.
Intact cells and cell lysates
To assess binding of the dansyl-tagged compound in live cells, 500,000 A549 cells per well were seeded in six-well plates. The following day, cells or cell lysates (prepared as described below) were treated with a concentration gradient DG-8 for 30 min. Intact cells were then harvested, washed with phosphate-buffered saline (PBS), and lysed in a modified radioimmunoprecipitation assay (RIPA) buffer [50 mM tris-HCl (pH 7.4), 150 nM NaCl, 1 mM EDTA, 1% NP-40, and 1% glycerol]. Samples were then prepared as above, by combining 30 μg of protein lysate with 6.25 μl of NuPAGE LDS Sample Buffer and 100 mM DTT. Again, samples were heated to 95°C for 5 min, run on the gels, and analyzed according to the conditions described above.
Competition experiments
Experimental inhibitors (0.5 to 50 μM) were incubated in cell lysates for 30 min, followed by 30 min of incubation with DG-8 (5 μM). Samples were prepared and analyzed as described above.
Band fluorescence intensity with differential and SPO3 levels
A549 cells were incubated for 72 hours with sodium selenite (25 to 100 nM) to promote Sec incorporation into cellular selenoproteins or SPO3 (0.25 to 1 mM) to induce Sec-to-Cys substitution (29). The cells were then harvested, washed with PBS, and lysed. Protein lysates (30 μg) were treated with DG-8 (5 μM) for 30 min at room temperature. Lysates were processed and run as mentioned above.
Cellular TrxR activity assay
Following a protocol reported previously (23), FaDu cells were seeded at a density of 400,000 cells per well in six-well plates. After 24 hours, compounds were added and incubated at 37°C for 3 hours. Cells were then lysed in a modified RIPA buffer [50 mM tris-HCl (pH 7.4), 150 nM NaCl, 1 mM EDTA, 1% NP-40, and 1% glycerol]. Protein concentrations were determined using the bicinchoninic acid assay. TrxR1 activity was determined using an insulin end point assay coupled with Trx1. Lysates were incubated together with 0.16 mM insulin, 0.33 mM NADPH, 16 μM Trx1 in TE buffer [50 mM tris-HCl and 2 mM EDTA (pH 7.5)]. Samples were incubated at 37°C for 0, 30, 60, and 90 min, respectively; at each time point, 7.2 M guanidine-HCl (pH 8.0) with 2.5 mM DTNB was added to the reactions, and A412 was measured. Cellular TrxR activity was determined from the increase in A412 over time and using a standard curve with pure enzymes.
Recombinant TrxR1 and TrxR2 activity measurement
Insulin reduction assay
Sec-dependent TrxR1 and TrxR2 activity was assessed by the reduction of insulin by Trx1 and Trx2, respectively. Compounds were incubated in a reaction respectively containing 10 nM TrxR1 or 10 nM TrxR2, with bovine serum albumin (BSA) (0.1 mg/ml) and 250 μM NADPH. After 75 min, 25 μM Trx1 or 25 μM Trx2, respectively, together with 160 μM insulin, were added, and NADPH consumption was measured at A340 for 20 min.
Juglone reduction assay
Formation of SecTRAPs was assessed using a juglone reduction assay (23). Compounds were incubated for 90 min in a reaction containing 300 nM TrxR1, 250 μM NADPH, and BSA (0.1 mg/ml), completely inhibiting Sec-dependent TrxR1 function. Reactions were then added to a buffer containing 100 μM juglone and 250 μM NADPH, and NADPH consumption was measured at A340 for 10 to 15 min.
Cellular H2O2 production assay
Cellular H2O2 production was assessed using the Amplex Red assay (A22188, Invitrogen), as previously reported (23). FaDu cells were plated at 5000 cells per well in black 96-well plates in media and incubated overnight. Cells were then washed with Krebs-Ringer bicarbonate (KRPG) buffer, and compounds were added in KRPG buffer. Immediately, 20 μM Amplex Red and horseradish peroxidase (0.01 U/ml) were added. Fluorescence measurements (550-nm excitation and 600-nm emission) were taken at the indicated time points and compared to matched untreated control wells to subtract background signal.
Western blot analyses
After compound treatment, cells were washed with PBS. Free thiols were blocked with 100 mM Pierce N-ethylmaleimide (#23030, Thermo Fisher Scientific) for 10 min. Cells were lysed by addition of 1% NP-40 and scraping. Reduced samples were mixed with NuPAGE LDS Sample Buffer (NP0008, Thermo Fisher Scientific) and 100 mM DTT and heated at 95°C for 5 min. Nonreduced samples were mixed with Pierce LDS Sample Buffer, Non-Reducing (#84788, Thermo Fisher Scientific). Protein (15 μg) was loaded onto 10% TGX gels (#4568033, Bio-Rad). Proteins were transferred to nitrocellulose membranes and blocked for 30 min in Odyssey blocking buffer 1:1 diluted with tris-buffered saline with 0.1% Tween 20 (Merck Millipore, Germany). Primary antibodies were incubated overnight at 4°C with mild agitation. Secondary antibodies were incubated for an hour at room temperature (IRDye 800CW goat anti-rabbit conjugated). Blots were imaged using an Odyssey FC Imaging System (LI-COR Biotechnology). Anti-pSTAT3 (Y705) (#D3A7; 1:1000) and anti-STAT3 (#79D7; 1:2000) were purchased from Cell Signaling Technology, anti-Prx2 (EPR5154; 1:20,000) was purchased from Abcam, and anti–glyceraldehyde-3-phosphate dehydrogenase (sc-25778; 1:1000) was purchased from Santa Cruz Biotechnology.
Cellular GSH depletion
Similar to previously reported methods (23), FaDu cells were plated at 2000 cells per well in 96-well plates. After 24 hours, 100 μM BSO (B2515, Sigma-Aldrich) was added to the cells with matched untreated controls. After an additional 24 hours, compounds were added and incubated for 72 hours. After treatment, resazurin cell viability reagent was added to each well at 0.02 mg/ml, and plates were incubated at 37°C for an additional 5 hours. Resorufin fluorescence was measured as described above. Cell viability was normalized to DMSO controls and blank wells.
Nrf2 and NFκB transcriptional activity analysis
Nrf2 and NFκB transcriptional activities were measured using the pTRAF tool as previously described (34). Briefly, HEK293 cells were seeded on black clear-bottom collagen I–coated 96-well plates (Biocoat 354649, BD Biosciences) at 20,000 cells per well a day before transfection with the pTRAF-Nrf2/HIF/NFκB vector. For transfection, 50 ng of DNA with 0.1 μl of Turbofect transfection reagent in 5 μl of Opti-MEM was added in 55 μl of Eagle’s minimum essential medium (EMEM) per well and incubated overnight. Cells were then exposed for 24 hours to either only the indicated compound (1 μM) or in combination with either auranofin (1 μM) or tumor necrosis factor–α (20 ng/ml) as positive controls for Nrf2 and NFκB, respectively. For microscopy, cell nuclei were stained using Hoechst (40 ng/ml) for 30 min, and an Operetta High-Content Imaging System (PerkinElmer) with the Columbus System was used to image and analyze the fluorescent signal per well.
Statistical analyses
For the quantified fluorescent band comparison with differential concentrations of sodium selenite and SPO3, significant differences were determined using repeated-measures analysis of variance (ANOVA) with Dunnett’s multiple comparisons test. For the cellular TrxR1 activity, differences were compared using repeated-measures ANOVA with Dunnett’s multiple comparisons test. For the MEF viability experiments, IC50 values were compared using repeated-measures ANOVA with Tukey’s multiple comparisons test. For the luciferase experiments in HEK293-shScramble and HEK293–shPrx1 + Prx2, IC50 values were compared using unpaired t tests with the Holm-Sidak method correction for multiple comparisons. Generated P values are displayed as *P ≤ 0.05, **P ≤ 0.01, and ***P ≤ 0.001.
Supplementary Material
Acknowledgments
We thank H. Axelsson and H. Almqvist of the Chemical Biology Consortium of Sweden (Karolinska Intsitutet) for assistance with regard to screening. In addition, we thank A. Vegvari of Proteomics Biomedicum (Karolinska Institutet) for running and analyzing MS experiments. Part of this work was facilitated by the Protein Science Facility at Karolinska Institutet (http://ki.se/psf). Last, we thank D. Talwar and T. Dick for the gift of HEK293-shScramble and HEK293-shPrx1 + Prx2. We would like to dedicate this article to D.G. who sadly passed away before this manuscript was submitted. In his memory, we have named the compounds produced within this project after him with the “DG” acronym. Funding: We acknowledge funding from Karolinska Institutet (D.G., E.S.J.A., and B.D.G.P.), the Swedish Research Council (2013-765, 2014-2603, and 2017-01872 to E.S.J.A.), the Swedish Cancer Society (2015/238 and 2018/333 to E.S.J.A. and 2015/698 to D.G.), the Knut and Alice Wallenberg Foundation (KAW 2015.0063 to E.S.J.A.), the Swedish Society for Medical Research (SSMF) (B.D.G.P.), and the King Gustaf Jubilee Fund (174122 to D.G.). KID funding from Karolinska Institutet was granted to this project (D.G.). B.D.G.P. is a Michael Smith Foundation for Health Research Scholar (18292). Author contributions: S.B., I.K., D.G., K.P.T., and B.D.G.P. conceived the project. S.B., I.K., M.D., D.G., E.S.J.A., K.P.T., and B.D.G.P. designed experiments. M.H., W.Q., L.J., and B.D.G.P. synthesized compounds. S.B., I.K., B.E., S.A., and J.L. performed experiments. S.B. analyzed the data. S.B. and B.D.G.P. wrote the manuscript. Competing interests: E.S.J.A. is an inventor on three patents submitted by E.S.J.A. and the United States as represented by the Secretary, Department of Health and Human Services Office of Technology Transfer, for protection of intellectual property concerning the TrxR1 inhibitors (TRi-1, TRi-2, and TRi-3) used in Figs. 3 and 4 in this study (no. WO2017027357A8, filed 7 August 2015, published 20 April 2017; no. WO2017027358A8, filed 7 August 2015, published 11 May 2017; no. WO2017027359A8, filed 7 August 2015, published 20 April 2017). B.D.G.P. is an inventor on a patent submitted by the Governing Council of the University of Toronto, Toronto, Canada; UTI Limited Partnership, Calgary, Canada; and Indiana University Research and Technology Corporation, Indianapolis, USA, which relates to the STAT3 inhibitor BP1-102, which was used in Figs. 3 and 4 of this study (no. US10377780B2, filed 21 September 2017, published 13 August 2019). The authors declare no other competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/12/eaax7945/DC1
Supplementary Material and Methods
Fig. S1. Compounds inhibit STAT3-dependent luciferase and covalently interact with GSH.
Fig. S2. Whole gel images of competition experiments of Fig. 3.
Fig. S3. Whole gel images of competition experiments of Fig. 3.
Fig. S4. SDS-PAGE fluorescent band matches TrxR1 protein on Western blot.
Fig. S5. CellTiter-Glo cell viability with or without selenium supplementation.
Fig. S6. Whole blot images of Western blot replicates.
Fig. S7. STAT-driven luciferase inhibition in Prx1 + Prx2 knockdown cells.
Fig. S8. Catalase decreases H2O2 production affecting cell cytotoxicity of top DG compounds.
Fig. S9. Compound effects on alternative transcription factors.
REFERENCES AND NOTES
- 1.Zhong Z., Wen Z., Darnell J. E. Jr., Stat3: A STAT family member activated by tyrosine phosphorylation in response to epidermal growth factor and interleukin-6. Science 264, 95–98 (1994). [DOI] [PubMed] [Google Scholar]
- 2.Linher-Melville K., Singh G., The complex roles of STAT3 and STAT5 in maintaining redox balance: Lessons from STAT-mediated xCT expression in cancer cells. Mol. Cell. Endocrinol. 451, 40–52 (2017). [DOI] [PubMed] [Google Scholar]
- 3.Furtek S. L., Backos D. S., Matheson C. J., Reigan P., Strategies and approaches of targeting STAT3 for cancer treatment. ACS Chem. Biol. 11, 308–318 (2016). [DOI] [PubMed] [Google Scholar]
- 4.Bournazou E., Bromberg J., Targeting the tumor microenvironment: JAK-STAT3 signaling. JAKSTAT 2, e23828 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Quintás-Cardama A., Verstovsek S., Molecular pathways: Jak/STAT pathway: Mutations, inhibitors, and resistance. Clin. Cancer Res. 19, 1933–1940 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Demaria M., Camporeale A., Poli V., STAT3 and metabolism: How many ways to use a single molecule? Int. J. Cancer 135, 1997–2003 (2014). [DOI] [PubMed] [Google Scholar]
- 7.Lee H., Pal S. K., Reckamp K., Figlin R. A., Yu H., STAT3: A target to enhance antitumor immune response. Curr. Top. Microbiol. Immunol. 344, 41–59 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lee H.-J., Zhuang G., Cao Y., Du P., Kim H.-J., Settleman J., Drug resistance via feedback activation of Stat3 in oncogene-addicted cancer cells. Cancer Cell 26, 207–221 (2014). [DOI] [PubMed] [Google Scholar]
- 9.Becker S., Groner B., Müller C. W., Three-dimensional structure of the Stat3beta homodimer bound to DNA. Nature 394, 145–151 (1998). [DOI] [PubMed] [Google Scholar]
- 10.Lai P.-S., Rosa D. A., Ali A. M., Gómez-Biagi R. F., Ball D. P., Shouksmith A. E., Gunning P. T., A STAT inhibitor patent review: Progress since 2011. Expert Opin. Ther. Pat. 25, 1397–1421 (2015). [DOI] [PubMed] [Google Scholar]
- 11.Furtek S. L., Matheson C. J., Backos D. S., Reigan P., Evaluation of quantitative assays for the identification of direct signal transducer and activator of transcription 3 (STAT3) inhibitors. Oncotarget 7, 77998–78008 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Desroses M., Busker S., Astorga-Wells J., Attarha S., Kolosenko I., Zubarev R. A., Helleday T., Grandér D., Page B. D. G., STAT3 differential scanning fluorimetry and differential scanning light scattering assays: Addressing a missing link in the characterization of STAT3 inhibitor interactions. J. Pharm. Biomed. Anal. 160, 80–88 (2018). [DOI] [PubMed] [Google Scholar]
- 13.Jensen A. J., Martinez Molina D., Lundbäck T., CETSA: A target engagement assay with potential to transform drug discovery. Future Med. Chem. 7, 975–978 (2015). [DOI] [PubMed] [Google Scholar]
- 14.Schust J., Sperl B., Hollis A., Mayer T. U., Berg T., Stattic: A small-molecule inhibitor of STAT3 activation and dimerization. Chem. Biol. 13, 1235–1242 (2006). [DOI] [PubMed] [Google Scholar]
- 15.Zhang X., Yue P., Page B. D. G., Li T., Zhao W., Namanja A. T., Paladino D., Zhao J., Chen Y., Gunning P. T., Turkson J., Orally bioavailable small-molecule inhibitor of transcription factor Stat3 regresses human breast and lung cancer xenografts. Proc. Natl. Acad. Sci. U.S.A. 109, 9623–9628 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Page B. D. G., Fletcher S., Yue P., Li Z., Zhang X., Sharmeen S., Datti A., Wrana J. L., Trudel S., Schimmer A. D., Turkson J., Gunning P. T., Identification of a non-phosphorylated, cell permeable, small molecule ligand for the Stat3 SH2 domain. Bioorg. Med. Chem. Lett. 21, 5605–5609 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Siddiquee K., Zhang S., Guida W. C., Blaskovich M. A., Greedy B., Lawrence H. R., Yip M. L. R., Jove R., McLaughlin M. M., Lawrence N. J., Sebti S. M., Turkson J., Selective chemical probe inhibitor of Stat3, identified through structure-based virtual screening, induces antitumor activity. Proc. Natl. Acad. Sci. U.S.A. 104, 7391–7396 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Heidelberger S., Zinzalla G., Antonow D., Essex S., Basu B. P., Palmer J., Husby J., Jackson P. J. M., Rahman K. M., Wilderspin A. F., Zloh M., Thurston D. E., Investigation of the protein alkylation sites of the STAT3:STAT3 inhibitor Stattic by mass spectrometry. Bioorg. Med. Chem. Lett. 23, 4719–4722 (2013). [DOI] [PubMed] [Google Scholar]
- 19.Ali A. M., Gómez-Biagi R. F., Rosa D. A., Lai P.-S., Heaton W. L., Park J. S., Eiring A. M., Vellore N. A., de Araujo E. D., Ball D. P., Shouksmith A. E., Patel A. B., Deininger M. W., O’Hare T., Gunning P. T., Disarming an electrophilic warhead: Retaining potency in tyrosine kinase inhibitor (TKI)-resistant CML lines while circumventing pharmacokinetic liabilities. ChemMedChem 11, 850–861 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ball D. P., Lewis A. M., Williams D., Resetca D., Wilson D. J., Gunning P. T., Signal transducer and activator of transcription 3 (STAT3) inhibitor, S3I-201, acts as a potent and non-selective alkylating agent. Oncotarget 7, 20669–20679 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sobotta M. C., Liou W., Stöcker S., Talwar D., Oehler M., Ruppert T., Scharf A. N. D., Dick T. P., Peroxiredoxin-2 and STAT3 form a redox relay for H2O2 signaling. Nat. Chem. Biol. 11, 64–70 (2015). [DOI] [PubMed] [Google Scholar]
- 22.Arnér E. S., Focus on mammalian thioredoxin reductases—Important selenoproteins with versatile functions. Biochim. Biophys. Acta 1790, 495–526 (2009). [DOI] [PubMed] [Google Scholar]
- 23.Stafford W. C., Peng X., Olofsson M. H., Zhang X., Luci D. K., Lu L., Cheng Q., Trésaugues L., Dexheimer T. S., Coussens N. P., Augsten M., Ahlzén H.-S. M., Orwar O., Östman A., Stone-Elander S., Maloney D. J., Jadhav A., Simeonov A., Linder S., Arnér E. S. J., Irreversible inhibition of cytosolic thioredoxin reductase 1 as a mechanistic basis for anticancer therapy. Sci. Transl. Med. 10, eaaf7444 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mandal P. K., Schneider M., Kölle P., Kuhlencordt P., Förster H., Beck H., Bornkamm G. W., Conrad M., Loss of thioredoxin reductase 1 renders tumors highly susceptible to pharmacologic glutathione deprivation. Cancer Res. 70, 9505–9514 (2010). [DOI] [PubMed] [Google Scholar]
- 25.Harris I. S., Treloar A. E., Inoue S., Sasaki M., Gorrini C., Lee K. C., Yung K. Y., Brenner D., Knobbe-Thomsen C. B., Cox M. A., Elia A., Berger T., Cescon D. W., Adeoye A., Brüstle A., Molyneux S. D., Mason J. M., Li W. Y., Yamamoto K., Wakeham A., Berman H. K., Khokha R., Done S. J., Kavanagh T. J., Lam C.-W., Mak T. W., Glutathione and thioredoxin antioxidant pathways synergize to drive cancer initiation and progression. Cancer Cell 27, 211–222 (2015). [DOI] [PubMed] [Google Scholar]
- 26.Kolosenko I., Yu Y., Busker S., Dyczynski M., Liu J., Haraldsson M., Apergi C. P., Helleday T., Tamm K. P., Page B. D. G., Grander D., Identification of novel small molecules that inhibit STAT3-dependent transcription and function. PLOS ONE 12, e0178844 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.E. S. J. Arner, “Pyridazinones and their use in the treatment of cancer,” WO/2017/027357 (2016).
- 28.Xu X.-M., Turanov A. A., Carlson B. A., Yoo M.-H., Everley R. A., Nandakumar R., Sorokina I., Gygi S. P., Gladyshev V. N., Hatfield D. L., Targeted insertion of cysteine by decoding UGA codons with mammalian selenocysteine machinery. Proc. Natl. Acad. Sci. U.S.A. 107, 21430–21434 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Peng X., Xu J., Arner E. S. J., Thiophosphate and selenite conversely modulate cell death induced by glutathione depletion or cisplatin: Effects related to activity and Sec contents of thioredoxin reductase. Biochem. J. 447, 167–174 (2012). [DOI] [PubMed] [Google Scholar]
- 30.Arnér E. S., Zhong L., Holmgren A., Preparation and assay of mammalian thioredoxin and thioredoxin reductase. Methods Enzymol. 300, 226–239 (1999). [DOI] [PubMed] [Google Scholar]
- 31.Anestål K., Prast-Nielsen S., Cenas N., Arnér E. S., Cell death by SecTRAPs: Thioredoxin reductase as a prooxidant killer of cells. PLOS ONE 3, e1846 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mandal P. K., Seiler A., Perisic T., Kölle P., Canak A. B., Förster H., Weiss N., Kremmer E., Lieberman M. W., Bannai S., Kuhlencordt P., Sato H., Bornkamm G. W., Conrad M., System x(c)- and thioredoxin reductase 1 cooperatively rescue glutathione deficiency. J. Biol. Chem. 285, 22244–22253 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Trachootham D., Alexandre J., Huang P., Targeting cancer cells by ROS-mediated mechanisms: A radical therapeutic approach? Nat. Rev. Drug Discov. 8, 579–591 (2009). [DOI] [PubMed] [Google Scholar]
- 34.Kipp A. P., Deubel S., Arnér E. S. J., Johansson K., Time- and cell-resolved dynamics of redox-sensitive Nrf2, HIF and NF-κB activities in 3D spheroids enriched for cancer stem cells. Redox Biol. 12, 403–409 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang J., Zhang B., Li X., Han X., Liu R., Fang J., Small molecule inhibitors of mammalian thioredoxin reductase as potential anticancer agents: An update. Med. Res. Rev. 39, 5–39 (2019). [DOI] [PubMed] [Google Scholar]
- 36.Li T., Ziniel P. D., He P.-Q., Kommer V. P., Crowther G. J., He M., Liu Q., Van Voorhis W. C., Williams D. L., Wang M.-W., High-throughput screening against thioredoxin glutathione reductase identifies novel inhibitors with potential therapeutic value for schistosomiasis. Infect. Dis. Poverty 4, 40 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kim N.-H., Lee M.-Y., Park S.-J., Choi J.-S., Oh M.-K., Kim I.-S., Auranofin blocks interleukin-6 signalling by inhibiting phosphorylation of JAK1 and STAT3. Immunology 122, 607–614 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zou Q., Chen Y.-F., Zheng X.-Q., Ye S.-F., Xu B.-Y., Liu Y.-X., Zeng H.-H., Novel thioredoxin reductase inhibitor butaselen inhibits tumorigenesis by down-regulating programmed death-ligand 1 expression. J. Zhejiang Univ. Sci. B 19, 689–698 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Witte A.-B., Anestål K., Jerremalm E., Ehrsson H., Arnér E. S. J., Inhibition of thioredoxin reductase but not of glutathione reductase by the major classes of alkylating and platinum-containing anticancer compounds. Free Radic. Biol. Med. 39, 696–703 (2005). [DOI] [PubMed] [Google Scholar]
- 40.Frijhoff J., Dagnell M., Godfrey R., Ostman A., Regulation of protein tyrosine phosphatase oxidation in cell adhesion and migration. Antioxid. Redox Signal. 20, 1994–2010 (2014). [DOI] [PubMed] [Google Scholar]
- 41.Kim M., Morales L. D., Jang I.-S., Cho Y.-Y., Kim D. J., Protein tyrosine phosphatases as potential regulators of STAT3 signaling. Int. J. Mol. Sci. 19, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Page B. D. G., Valerie N. C. K., Wright R. H. G., Wallner O., Isaksson R., Carter M., Rudd S. G., Loseva O., Jemth A.-S., Almlöf I., Font-Mateu J., Llona-Minguez S., Baranczewski P., Jeppsson F., Homan E., Almqvist H., Axelsson H., Regmi S., Gustavsson A.-L., Lundbäck T., Scobie M., Strömberg K., Stenmark P., Beato M., Helleday T., Targeted NUDT5 inhibitors block hormone signaling in breast cancer cells. Nat. Commun. 9, 250 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Liu X., Pietsch K. E., Sturla S. J., Susceptibility of the antioxidant selenoenyzmes thioredoxin reductase and glutathione peroxidase to alkylation-mediated inhibition by anticancer acylfulvenes. Chem. Res. Toxicol. 24, 726–736 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/12/eaax7945/DC1
Supplementary Material and Methods
Fig. S1. Compounds inhibit STAT3-dependent luciferase and covalently interact with GSH.
Fig. S2. Whole gel images of competition experiments of Fig. 3.
Fig. S3. Whole gel images of competition experiments of Fig. 3.
Fig. S4. SDS-PAGE fluorescent band matches TrxR1 protein on Western blot.
Fig. S5. CellTiter-Glo cell viability with or without selenium supplementation.
Fig. S6. Whole blot images of Western blot replicates.
Fig. S7. STAT-driven luciferase inhibition in Prx1 + Prx2 knockdown cells.
Fig. S8. Catalase decreases H2O2 production affecting cell cytotoxicity of top DG compounds.
Fig. S9. Compound effects on alternative transcription factors.