

Abstract
Modelling of protein structures based on backbone chemical shifts, using programs such as CS-ROSETTA, is becoming increasingly popular, especially for systems where few restraints are available or where homologous structures are already known. While the reliability of CS-ROSETTA calculations can be improved by incorporation of some additional backbone NMR data such as those afforded by residual dipolar couplings or minimal NOE data sets involving backbone amide protons, the sidechain conformations are largely modelled by statistical energy terms. Here, we present a simple method based on methyl residual dipolar couplings that can be used to determine the rotameric state of the three-fold symmetry axis of methyl groups that occupy a single rotamer, determine rotameric distributions, and identify regions of high flexibility. The method is demonstrated for methyl side chains of a deletion variant of the human chaperone DNAJB6b.
Keywords: sidechain conformation, residual dipolar couplings, methyl NMR, protein structure refinement, CS-ROSETTA
In the last two decades, advances in NMR spectroscopy have significantly extended the upper limits of molecular size that can be studied by NMR (Rosenzweig and Kay, 2014; Anthis and Clore, 2015). At the same time, new computational methods for protein structure prediction using threading protocols have emerged (Leaver-Fay et al., 2011; Nerli et al., 2018). The combination of NMR-derived structural restraints with the newly developed structure calculation approaches has yielded structural models for a number of challenging systems where only a few NMR restraints could be obtained (Shen et al., 2008; Raman et al., 2010; Sgourakis et al., 2014; Nerli et al., 2018). One such approach, termed CS-ROSETTA (Shen et al., 2008), makes use of backbone chemical shifts to select appropriate protein fragments which are then assembled using an energy function (Leaver-Fay et al., 2011). The reliability of such structural models can be further improved by incorporation of additional (sparse) NMR restraints derived, for example, from backbone and/or methyl NOEs or residual dipolar couplings (RDC) (Shen et al., 2008; Raman et al., 2010). Sidechains are then modelled using an energy function that makes use of a knowledge-based term (Leaver-Fay et al., 2011) to enforce appropriate sidechain orientations. Thus, even though the backbone fold can be predicted from chemical shift data, especially when supplemented by a small number of other NMR-derived structural restraints, side chain conformations and fine-tuning of their torsion angles is primarily guided by the ROSETTA energy function (Leaver-Fay et al., 2011). While this approach will generally result in good structural statistics, it may fail for biochemically important side chains that may, for example, deviate from ideal staggered conformations or populate several rotameric states.
Previous attempts to refine sidechain orientations using NMR data involved, among others, the measurements of three-bond J couplings (Chou et al., 2003). This approach, however, has not, to the best of our knowledge, found widespread use in structure calculations as it is complicated both in terms of data acquisition and analysis. NOE pattern recognition in combination with methine RDCs measured for Leu and Val sidechains has been tested on protein samples where complete resonance assignments are available (Tang et al., 2005), while 13Cβ−1Hβ RDCs have been used to determine sidechain χ1 angles (Mittermaier and Kay, 2001). More recently, the chemical shifts of methyl 13C nuclei have been analysed to gain access to the rotameric states of methyl-bearing sidechains (Hansen et al., 2010a; Hansen et al., 2010b; Hansen and Kay, 2011). At present, however, the sidechain chemical shifts cannot be used directly in structure refinement protocols.
Here, we present a simple approach for determining sidechain orientations of methyl-bearing residues based on methyl 1H-13C RDCs (1DCH) obtained from a series of J-modulated heteronuclear single quantum coherence (HSQC) spectra (Ottiger et al., 1998b) using a sample with methyl isotopic labeling. Complications in structural interpretation of sidechain NMR data arising from increased sidechain mobility are addressed by a semi-quantitative estimation of order parameters squared of the methyl group three-fold symmetry axis, , a measure of the amplitude of methyl motions, obtained from the same experiment. Thus, our approach yields structural information from RDC data and, at the same time, provides a semi-quantitative measure of relative methyl group dynamics. While the RDCs of effectively ‘rigid’ methyl sites can be used for determination of sidechain conformations, those of (partially) flexible methyl groups can be explained by allowing for multiple rotameric states of a methyl-bearing sidechain.
The methodology was applied to the CS-ROSETTA structure of the deletion variant (AST) of the human chaperone DNAJB6b, ΔST-DNAJB6b (Karamanos et al., 2019). The latter consists of a helical J domain (JD), in which internal sidechains are well packed in a hydrophobic core, and a more dynamic C-terminal β-strand domain (CTD) (Karamanos et al., 2019). This system provides an ideal test bed for our approach, as ΔST-DNAJB6b contains numerous methyl groups with variable degrees of flexibility.
Figure 1 shows typical interferograms obtained from a series of J-modulated 1H-13C constant time HSQC experiments (Ottiger et al., 1998b) (see Supplementary Information, SI, for the pulse scheme used in this work; Figure S1) recorded on a {U-[15N, 2H]; Ileδ1-[l3CH3]; Leu, Val-[13CH3,12CD3]}-labeled sample of ΔST-DNAJB6b aligned in a 4.2 % (v/v) pentaethylene glycol monodocecyl ether (C12E5 PEG):hexanol medium. This methyl labeling strategy, where only one of the methyl groups of isopropyl moieties of Leu and Val is enriched in 13C and protonated (Tugarinov and Kay, 2004), is commonly used in NMR studies of high molecular weight proteins (Tugarinov and Kay, 2005). In the context of the present work, this labeling scheme ensures elimination of additional couplings (three-bond 13C-1H scalar couplings, and 13C-1H RDCs between 13C spins of one methyl group and 1H spins of another methyl within isopropyl moieties of Leu and Val).
Figure 1.
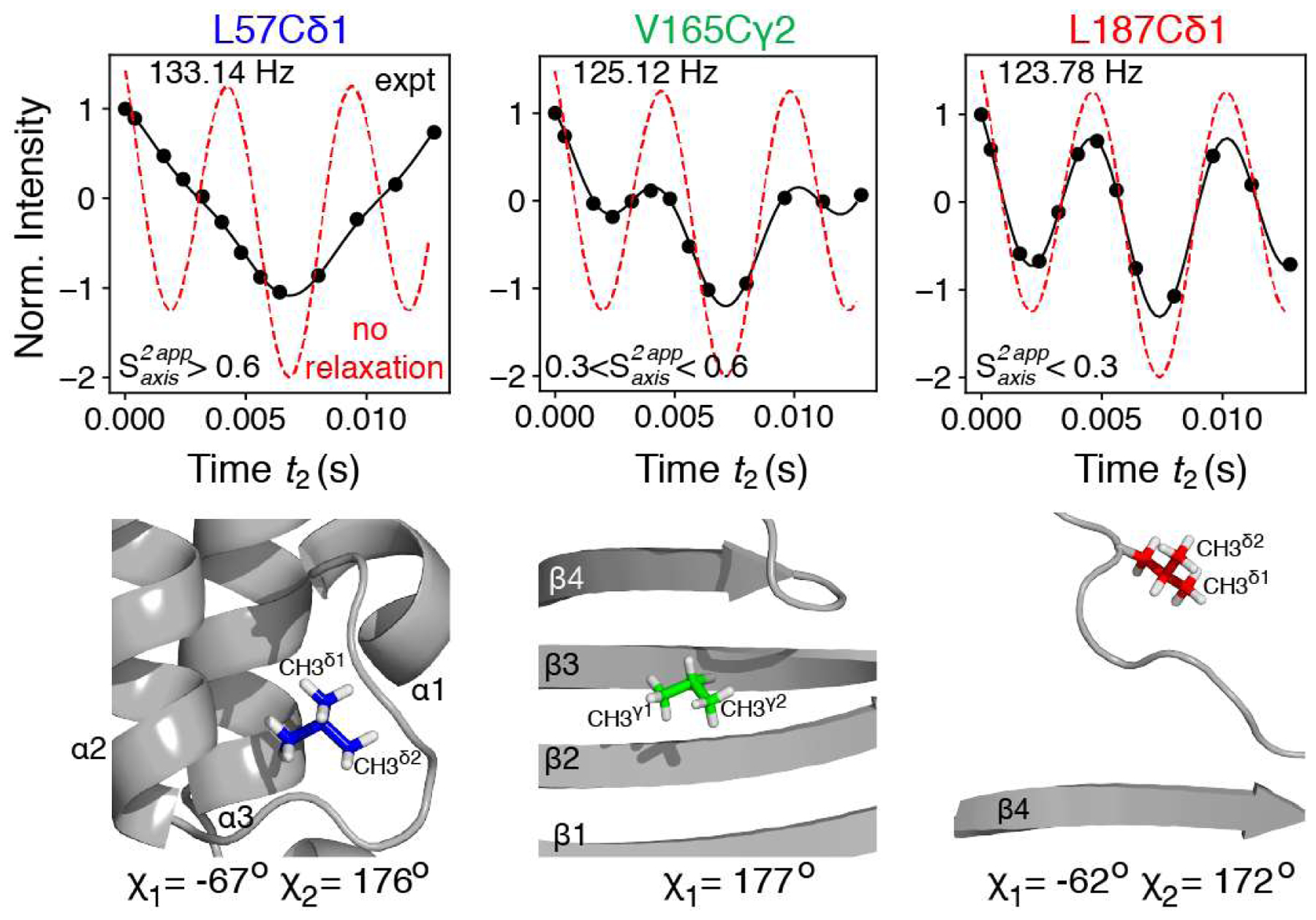
Measurement of methyl lH-13C RDCs in {U-[15N,2H]; Ileδ1-[13CH3]; Leu, Val-[13CH3,12CD3]}-labeled ΔST-DNAJB6b (600 MHz; 25 °C). (Top row) Typical interferograms showing the modulation of methyl cross-peak intensities as a function of the J-evolution period (t2) for methyl groups with varying degree of flexibility. The intensities are normalized to those at t2 = 0. The interferograms in the absence of relaxation (Eq. (1)) are shown with red dashed lines. Black lines represent the best-fits to Eq. (2). The extracted values of (JCH + DCh) are shown in top left corner of each panel. The constant-time (CT) period T was set to 28 ms (see SI, Figure S1 for the pulse-scheme). The sample was 200 μM in protein concentration, containing 20 mM sodium phosphate buffer, pH = 7.0 (uncorrected), 50 mM NaCl, dissolved in 90% H2O/10% D2O (v/v), and aligned in a 4.2% (v/v) PEG/hexanol mixture (Rückert and Otting, 2000). Stereospecific assignments of methyl groups were performed using the method of (Neri et al., 1989). (Bottom row) The location of the corresponding methyl-containing sidechains in the structure of ΔST-DNAJB6b (PDB ID: 36UR, 36US (Karamanos et al., 2019)), with the calculated χ1 and χ2 torsion angles indicated underneath.
In the absence of relaxation, the modulation of the methyl cross-peak intensity I as a function of the J dephasing delay t2 in the J-modulation experiment (SI, Figure S1) follows the relationship (Ottiger et al., 1998b),
| (1) |
where I0 is initial intensity of the methyl signal (at t2 = 0) and 1JCH is a one-bond scalar 13C-1H coupling in a methyl group. The first cosine term in (Eq. 1) describes the evolution of the outer 13C transitions, while the second cosine term corresponds to the inner 13C transitions, with their intensity ratios 3:1 in the absence of relaxation. Significant deviations from the expected ‘relaxation-free’ modulation profiles (shown with red dashed lines in Figure 1) are observed for a subset of methyl groups in ΔST-DNAJB6b - primarily those located in the JD domain (e.g. Leu57, leftmost panel of Figure 1). These deviations arise from differential relaxation of the inner (slow-relaxing) and outer (fast-relaxing) methyl 13C transitions during the constant-time period of the experiment (Figure S1).
Accounting for relaxation in these measurements in a rigorous manner is problematic as the relaxation rates of 13C transitions depend on the micro-dynamics parameters of methyl groups which are (a) not known a-priori, and (b) vary from one methyl-containing sidechain to another. An approximate, semi-quantitative approach adopted here, consists of fitting the methyl cross-peak intensities to the function,
| (2) |
where is the transverse spin relaxation rate of the slow-relaxing 13C transitions, is the transverse spin relaxation rate of the fast-relaxing 13C transitions, and T is the constant-time period during which spin relaxation is active. The coefficient ‘a’ varies from 3 in the absence of relaxation to practically 0 for very large proteins, and is pre-calculated as described in the (SI Figure S2A). To reduce the number of free variable parameters the fast relaxation rate . is assumed to be proportional to , ,. where ~1 < b < ~8 and is estimated as described in the (SI Figure S2B). The set of free variable parameters of the fit is thus comprised of: (1JCH + Dch) couplings, I0 and the relaxation rates .
Excellent fits of experimental modulation profiles to (Eq. 2) are obtained for all methyl sites of ΔST-DNAJB6b (black curves in Figure 1). Importantly, the derived (1JCH + DCH) couplings are essentially independent of the exact values of the coefficients ‘a’ and ‘b’ as the accuracy of the (1JCH + DCH) determination is predicated on a sufficient number of zero-crossings in the interferograms (two or more). The relaxation rates can be subsequently converted to ‘apparent’ order parameters squared, , using standard relationships (Ollerenshaw et al., 2003; Tugarinov et al., 2003; Tugarinov and Kay, 2013) (see SI, Eq. (S3)). The rationale behind using the fast rates for re-calculation of is based on smaller relative contributions of mechanisms other than methyl 1H-13C dipolar relaxation to the rates (such as dipolar interactions with external 1H spins and methyl 13C chemical shift anisotropy; see SI). Empirically, we found that similar distributions of in ΔST-DNAJB6b are obtained for a wide range of b values from ~6 and ~8. Plots of values obtained for ΔST-DNAJB6b, with a = 2.7 and b = 7, are shown in the (SI Figure S3). Although accurate (correct) values of cannot be obtained in this manner, the estimation of the relative mobility of the methyl three-fold symmetry axes of different methyl sites is readily achieved. For example, the methyl sites in the hydrophobic core of the JD domain have (Leu57; Figure 1), while those located in the β-strands of the CTD domain (but not packed against other residues) are characterized by in the range between 0.3 and 0.6 (Vall65; Figure 1), and for the methyl sites located in flexible regions such as the C-terminal residues, (Leul87; Figure 1). We note that, very recently, we devised NMR methodology for isolating transitions belonging to I = 1/2 manifolds of 13CH3 methyl groups (Tugarinov et al. 2020). Although this approach effectively reduces the modulations of the signal in Figure 1 to a simple I0cos[π(JCH + DCH)t2] function, thus eliminating relaxation from the picture, it does not allow one to obtain an estimate of methyl side-chain mobility.
Figure 2A shows that, as expected, the values of 1DCH derived from the J-modulated experiment, are related to 13C-13C RDCs ( in Val and in Leu/Ile), measured directly from cross-peak splittings in the 13C dimension of well-resolved 2D HMQC spectra of ΔST-DNAJB6b, by , where γj is the gyromagnetic ratio of spin j. rCH and rCC are l3C-1H and 13C-13C distances in methyl groups, respectively, P2(x) = (3×2 −1) / 2, and θ is the tetrahedral angle of a methyl group (Ottiger and Bax, 1999; Mittermaier and Kay, 2002). For larger proteins, where sensitivity limitations do not permit the determination of 1DCH from the indirect (13C) dimension of the J-modulated experiments, RDCs can be measured in the directly detected (1H) dimension using the in-phase/anti-phase (IPAP) (Yang and Nagayama, 1996; Ottiger et al., 1998a) experiment developed by Sprangers and Kay (2007). Figure 2B compares the values of 1DCH obtained for ΔST-DNAJB6b using the approach described above with those measured using the experiment of Sprangers and Kay (2007). Good agreement between the two sets of dipolar couplings implies that even for a moderately sized protein such as ΔST-DNAJB6b, the scheme of Sprangers and Kay (2007) represents a good alternative for RDC measurements as long as the isotope labeling scheme with only one methyl 13CH3-labeled is used for Leu/Val in an otherwise highly deuterated background.
Figure 2.
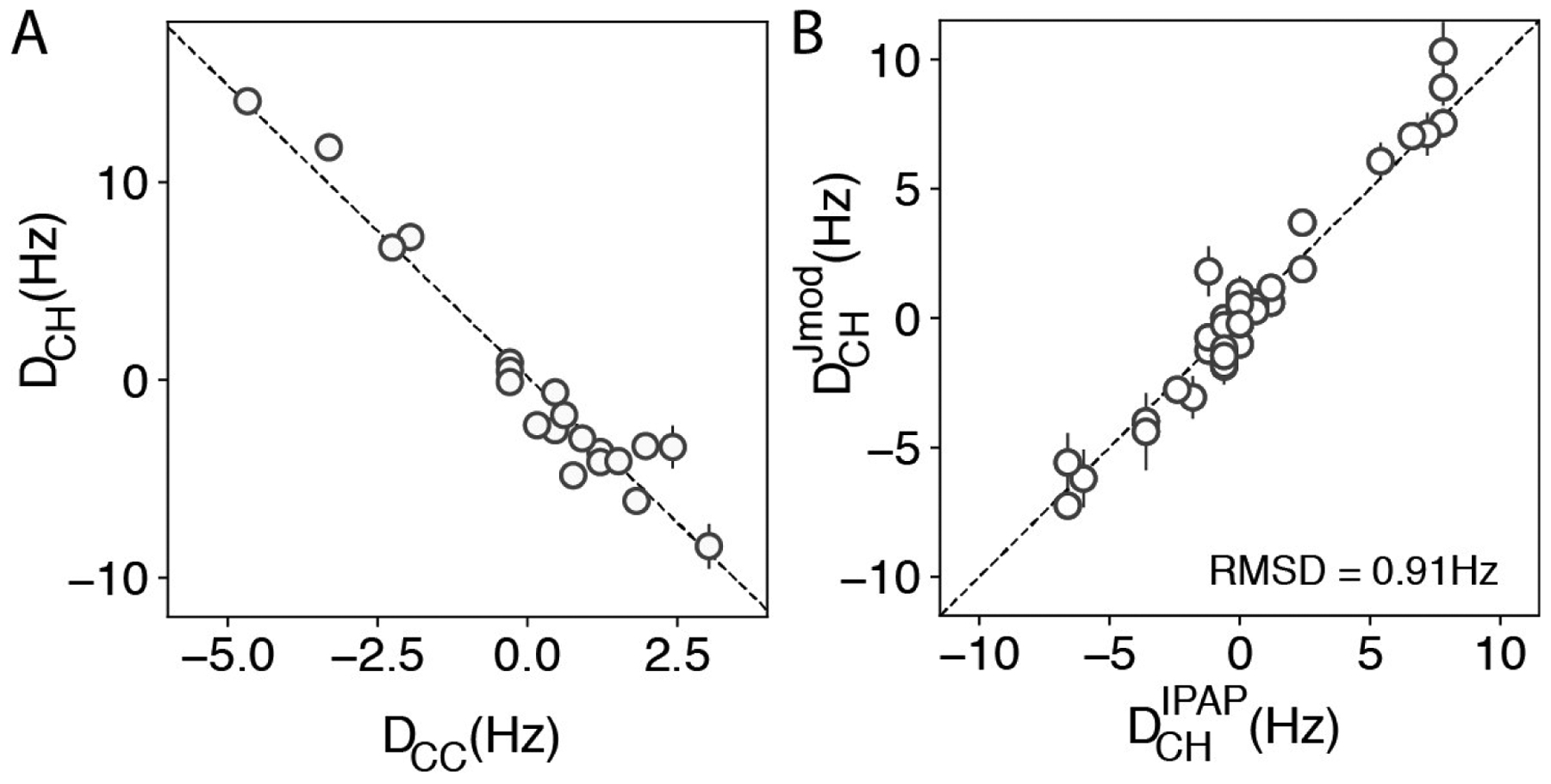
Robustness of the 1DCH values measured for ΔST-DNAJB6b. (A) Correlation between the 1H-13C RDC values derived from the J-modulated experiment and 13C-13C RDCs (DCC) measured from cross-peak splitting in well resolved HMQC spectra using a 120 μM sample of {U-[15N,13C,2H]; Ileδ1-[13CH3]; Leu, Val-[13CH3,12CD3]}-ΔST-DNAJB6b aligned in 12 mg/mL pf1 bacteriophage (600 MHz; 25 °C). The average DCH/DCC ratio for the residues shown in (A) is −3.0 ± 0.2. The correlation coefficient between the two datasets is −0.98. (B) Correlation between and the 1H-13C methyl RDCs measured with the scheme of Sprangers and Kay (2007), , using the sample described in Figure 1 (correlation coefficient 0.96). The dashed line has a slope of 1.03.
The values of 1DCH back-calculated from the CS-ROSETTA structure of the JD domain show only modest agreement with their measured experimental values, providing R factors (Clore and Garrett, 1999) of 48 % and 51 % for the samples aligned in PEG/hexanol and pf1 bacteriophage media, respectively (Figure 3A). Since essentially all methyl sites in the JD domain are predicted to be ‘rigid’ (Figure 3B), a simple refinement approach, implemented in the program Xplor-NIH (Schwieters et al., 2018), can be used to determine the conformations of methyl three-fold symmetry axes using a single-conformer (N = 1) representation of the sidechain. Amide 1H-15N RDCs measured on the same samples of ΔST-DNAJB6b were used to determine the alignment tensors for each medium, yielding the following values for the magnitude of the principal component of the alignment tensor Da (in units of Hz) and rhombicity η: 12.8 Hz and 0.50, and 10.4 Hz and 0.66 for the JD and CTD domains in pf1 phage, respectively; and 15.4 Hz and 0.56, and 1.1 Hz and 0 for the JD and CTD domains in PEG/hexanol, respectively. Of note, no scaling of 1DCH values by was attempted in this work, as the order parameters estimated from relaxation as described above are (a) inaccurate, and (b) not necessarily good indicators of the ordering of the methyl three-fold symmetry axis affecting the values of methyl RDCs. Excellent agreement with experimental methyl RDC data was obtained upon refinement, with R factors of 27 % and 17 % for the pf1 and PEG/hexanol media, respectively. Methyl groups in the JD domain occupy predominantly a single rotameric state resulting in high values because their motion is restricted by packing against other sidechains in the hydrophobic core of the JD structure. In the case of ΔST-DNAJB6b, the CS-ROSETTA calculations generated the correct rotameric state for all the “rigid” methyl sites, and only slight changes in orientation (up to ~12°) occurred upon refinement. Nevertheless, in other proteins where CS-ROSETTA may fail to accurately predict the rotameric states of methyl-bearing sidechains, the refinement procedure described here would be expected to correct errors in side-chain orientations.
Figure 3.
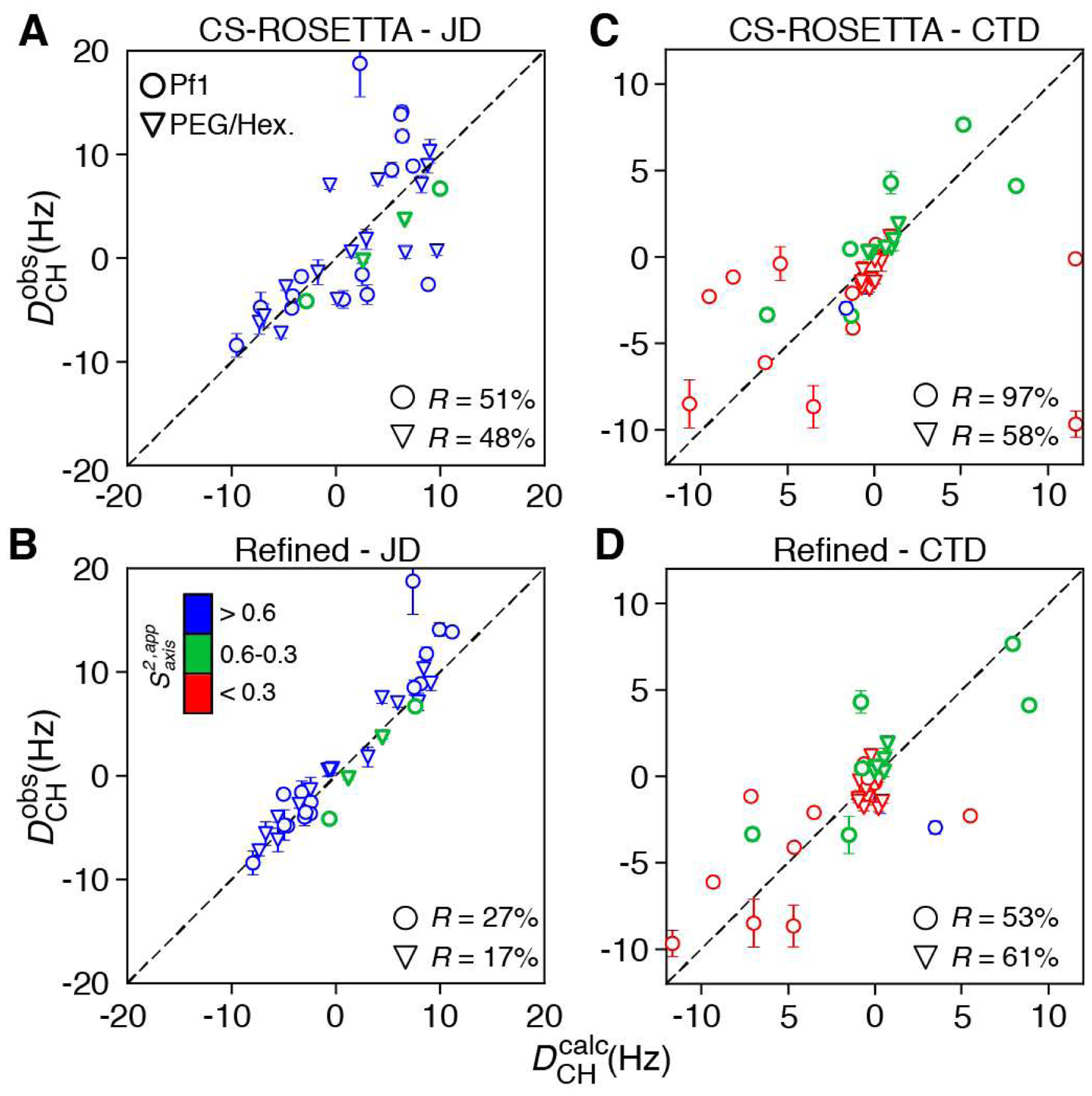
Determination of methyl sidechain conformations using methyl 1H-13C RDCs. Comparison of experimental versus back-calculated RDCs . measured in pf1 bacteriophage (open circles) or a PEG/hexanol mixture (open inverted triangles) for (A, B) the JD domain of ΔST-DNAJB6b and (C, D) the CTD domain of ΔST-DNAJB6b. In panels A and C, was calculated from the lowest energy CS-ROSETTA model (Karamanos et al., 2019), while in (B, D) was calculated post-refinement using a single sidechain conformation (N = 1). The residual dipolar coupling R factors, R = [< (Dobs − Dcalc)2 >/(2 < Dobs2 >)]1/2 (Clore and Garrett, 1999), are indicated in each panel. Data points are colored according to their values as indicated in panel B (red for ; green for in the range between 0.3 and 0.6; blue for ).
Inclusion of additional rotameric states for the ‘rigid’ methyl sites of the JD domain results in a slightly (statistically insignificantly) better fit (R = 21 % and R = 18 % for the pf1 phage and PEG/hexanol alignment media, respectively, compared to R = 27 % and R = 17 % for the two alignment media in Figure 3B), with very low populations for the two additional rotamers. These improvements cannot be justified either statistically, or on the grounds of obtaining a more accurate structure. On the other hand, when the methyl axis orientations of all three conformers are fixed to their ideal values and only their populations are allowed to vary in the calculation, R = 47% and R = 35 % for the pf1 phage and PEG/hexanol alignment media, respectively, representing a relatively modest improvement compared to R = 51 % and R = 48 % for the two alignment media before refinement (Figure 3A). These observations taken together, indicate that extensions of the ‘single-conformer’ model for the ‘rigid’ methyl sites of the JD domain of ΔST-DNAJB6b (where only the methyl symmetry axis orientation of the preferred rotameric state is adjusted) are not warranted.
The same strategy proved ineffective for the more mobile methyl sites of the CTD domain (Figures 3C and D) implying that these sidechains populate more than one rotameric state. The large conformational space commonly sampled by protein sidechains is vastly reduced for methyl-bearing sidechains of Ile, Leu and Val, which can sample only a small number of χ1/χ2 rotamers (Kuszewski et al., 1996; Lovell et al., 2000; Clore and Kuszewski, 2002). Valine samples all three rotamers: g+ (χ1 = 63°), t (χ1 =175°) and g− (χ1 = −63°); leucine samples two χ1/χ2 rotamers: t/g+ (χ1 = −177°, χ2 = 65°) and g−/t (χ1 = −65°, χ2 = 175°); and isoleucine predominantly samples two χ2 rotamers: g− (χ2 = −60°) which is associated with a g− χ1 angle (−65°), and t (χ2 = 170°) where χ1 can be g+ (62°), g− (−65°) or t (−177°). Rotameric jumps between these states contribute to increased mobility of the methyl three-fold symmetry axis and can be captured by methyl 1H-13C RDCs which are sensitive to motions on the pico-to-millisecond timescale (Chou et al., 2003). Considering that for Val and Leu sidechains, two independent measurements of 1DCH for the two prochiral methyl groups are available, it is possible to determine the populations of each rotamer (ptrans, pg+, pg−) even from a single alignment medium. The situation is more complex, however, for Ile sidechains where the measured 1DCH values do not report exclusively on the χ2 conformation, but depend also on the χ1 angle, with this dependence maximized when the three-fold symmetry axis of a methyl group is oriented at ~45° with respect to the principal axis of the alignment tensor. Therefore, methyl RDCs can be used to determine unambiguously the χ1 angles of Val sidechains, the χ2 angles of Leu sidechains (yielding at the same time the χ1 angle), and only in favourable cases (when the methyl three-fold symmetry axis is close to being either collinear or orthogonal to the principal axis of the alignment tensor), the χ2 angle of Ile sidechains.
To fit the 1DCH data of the CTD domain, we performed a N = 2 (for Ile/Leu sidechains) or N = 3 (for Val sidechains) calculation, where each ensemble member was forced to occupy a different rotameric state (allowing a ±10° deviation from each ideally staggered rotamer (Lovell et al., 2000)), while keeping the coordinates of the backbone fixed. The DCH values back-calculated for each χ2 angle (, for Leu; , for Ile) or χ1 angle (, , for Val) were subsequently used in a grid search to determine the populations of each rotamer (Figure 4) by minimizing the following set of target functions, F:
| (3) |
for Leu,
| (4) |
for Ile (where a g− χ1 conformation was assumed for the trans χ2 rotamer), and
| (5) |
for Val. Using this approach, good agreement between observed and calculated 1DCH values is obtained for moderately mobile methyls of the CTD domain (; Figure 4A). These methyl sites are characterized by significant populations of all allowed rotamers (Figure 4B), suggesting that their decreased values arise from jumps between different rotameric states. In fact, rotameric averaging may be expected for these sidechains since, despite their location in β-strands, they are not restricted by packing against other elements of the CTD structure. Good agreement is also observed between the rotameric distributions obtained from fitting of the 1DCH data and those predicted from methyl 13C chemical shifts (Hansen et al., 2010a; Hansen and Kay, 2011) (Figure 4B), with the two datasets sharing a linear correlation coefficient of 0.75. Some disagreements can be noted for Ile 152 and especially Ile 157, which have their three-fold symmetry axes oriented at 104° and 60°, respectively, with respect to the principal axis of the alignment tensor.
Figure 4.
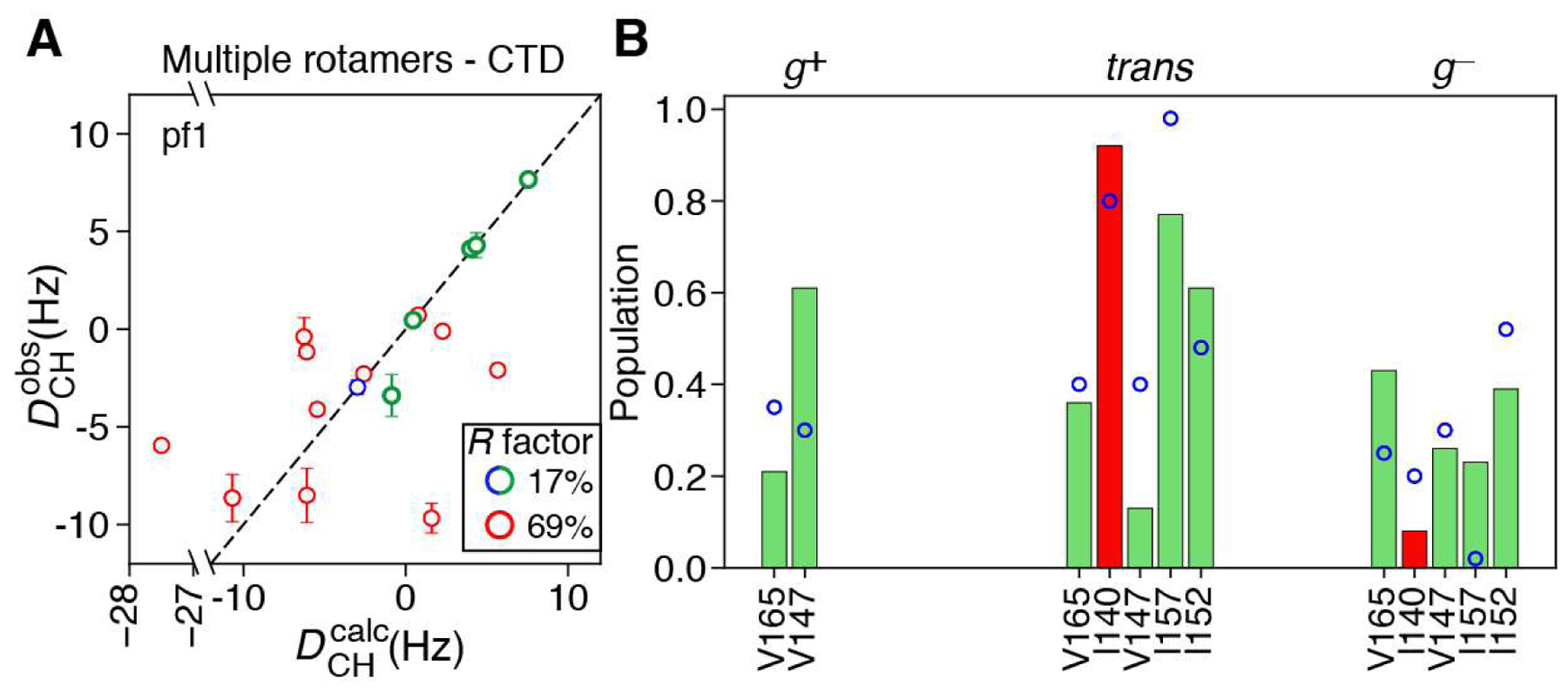
Determination of rotamer populations using methyl 1H−13C RDCs. (A) Agreement between measured and back-calculated RDCs obtained in pf1 bacteriophage for the CTD domain of ΔST-DNAJB6b (Note PEG/hexanol alignment data were not used for the CTD domain due to the very small degree of alignment). Data points are colored according to their values using the same color-coding as in Figure 3. (B) Bar plots showing the rotamer populations for the χ2 angles of Leu and Ile, and the χ1 angle of Val, determined by minimization of the target functions in (Eqs. 3)–(5), for residues of ΔST-DNAJB6b where good agreement between and is achieved. Rotamer populations calculated based on the chemical shifts of methyl 13C resonances (Hansen et al., 2010a; Hansen and Kay, 2011) are shown as open blue circles. Ilel40 has an estimated value of ~0.28, but is located in a ‘rigid’ β-strand in the structure of ΔST-DNAJB6b. By way of an exception, its Dch values can be explained by a combination of different rotameric states.
The 12 C-terminal residues of the CTD domain have been shown to be predominantly flexible although possessing a propensity for ‘docking’ onto (forming contacts with) the β4 strand (see Leu187δ1 in the rightmost panel of Figure 1), with this part of the protein forming a short β-strand in 5 out of the 10 lowest energy CS-ROSETTA models (Karamanos et al., 2019). These residues are important for the function of DNAJB6b as they are directly implicated in the assembly of DNAJB6b oligomers (Karamanos et al., 2019). This region contains a total of 9 methyl sites, all with . Figure 4A shows that the 1DCH values of these methyl sites (9 out of 11 shown with red circles) cannot be explained by a linear combination of the allowed rotameric states, as observed for the remaining methyl groups of the CTD domain (green/blue circles; Figure 4A), confirming that the C-terminal residues are highly flexible in the open form of ΔST-DNAJB6b and do not adopt a preferred conformational state.
In summary, we describe a simple strategy for determination of sidechain orientations of methyl-bearing residues based on methyl 1H-13C RDCs obtained from a series of J-modulated HSQC spectra. A semi-quantitative approach towards interpretation of J-modulated interferograms of 13CH3 methyl groups in the presence of spin relaxation, allows one to obtain accurate methyl 1H-13C RDCs and, at the same time, estimate approximately the order parameters of methyl three-fold symmetry axis, . This approach thus yields structural information from RDC data and simultaneously provides a relative measure of methyl three-fold axis dynamics. Whereas the RDCs of well-ordered methyl sites can be used directly in determination of sidechain rotameric states, those of moderately mobile methyl groups are accounted for by allowing multiple sidechain rotamers to be populated. The approach was applied to the CS-ROSETTA-derived structure of the deletion variant of the human chaperone DNAJB6b, ΔST-DNAJB6b, where remarkable agreement is achieved for ordered methyl sites (mainly in the JD domain) between observed 1H-13C RDCs and those back-calculated post-refinement.
Supplementary Material
Acknowledgement.
We thank Drs. James Baber, Jinfa Ying and Dan Garrett for technical support. This work was supported by the Intramural Program of the National Institute of Diabetes and Digestive and Kidney Diseases, National Institutes of Health (DK029023 to G.M.C.).
Footnotes
Publisher's Disclaimer: This Author Accepted Manuscript is a PDF file of an unedited peer-reviewed manuscript that has been accepted for publication but has not been copyedited or corrected. The official version of record that is published in the journal is kept up to date and so may therefore differ from this version.
Supplementary Information. The pulse scheme used for recording the J-modulated interferograms (Figure S1). Description of theoretical considerations involved in the interpretation of J-modulated interferograms of 13CH3 methyl groups in the presence of spin relaxation according to Eq. (2) (Figure S2). A plot of values obtained for ΔST-DNAJB6b using the described analysis of J-modulated interferograms (Figure S3). ‘Materials and Methods’ section describing the details of NMR sample preparation, NMR experiments and structure calculation protocols.
References
- Anthis NJ, Clore GM (2015) Visualizing transient dark states by NMR spectroscopy. Quart Rev Biophys 48:1–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou JJ, Case DA, Bax A (2003) Insights into the mobility of methyl-bearing side chains in proteins from 3JCC and 3Jcn couplings. J Am Chem Soc 125:8959-8966 [DOI] [PubMed] [Google Scholar]
- Clore GM, Garrett DS (1999) R-factor, free R, and complete cross-validation for dipolar coupling refinement of NMR structures. J Am Chem Soc 121:9008–9012 [Google Scholar]
- Clore GM, Kuszewski J (2002) χ1 rotamer populations and angles of mobile surface side chains are accurately predicted by a torsion angle database potential of mean force. J Am Chem Soc 124:2866–2867 [DOI] [PubMed] [Google Scholar]
- Hansen DF, Kay LE (2011) Determining valine sidechain rotamer conformations in proteins from methyl 13C chemical shifts: Application to the 360 kda half-proteasome. J Am Chem Soc 133:8272–8281 [DOI] [PubMed] [Google Scholar]
- Hansen DF, Neudecker P, Kay LE (2010) Determination of isoleucine sidechain conformations in ground and excited states of proteins from chemical shifts. J Am Chem Soc 132:7589–7591 [DOI] [PubMed] [Google Scholar]
- Hansen DF, Neudecker P, Vallurupalli P, Mulder FAA, Kay LE (2010) Determination of Leu sidechain conformations in excited protein states by NMR relaxation dispersion. J Am Chem Soc 132:42-43 [DOI] [PubMed] [Google Scholar]
- Karamanos TK, Tugarinov V, Clore GM (2019) Unraveling the structure and dynamics of the human DNAJB6b chaperone by NMR reveals insights into HSP40-mediated proteostasis. Proc Natl Acad Sci USA 43:21529–21538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuszewski J, Gronenborn AM, Clore GM (1996) Improving the quality of NMR and crystallographic protein structures by means of a conformational database potential derived from structure databases. Protein Sci 5:1067–1080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leaver-Fay A et al. (2011) Rosetta3: An object-oriented software suite for the simulation and design of macromolecules. Meth Enzymol 487:545–574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovell SC, Word JM, Richardson JS, Richardson DC (2000) The penultimate rotamer library. Proteins 40:389–408 [PubMed] [Google Scholar]
- Mittermaier A, Kay LE (2001) χ1 torsion angle dynamics in proteins from dipolar couplings. J Am Chem Soc 123:6892–6903 [DOI] [PubMed] [Google Scholar]
- Mittermaier A, Kay LE (2002) Effect of deuteration on some structural parameters of methyl groups in proteins as evaluated by residual dipolar couplings. J Biomol NMR 23:35–45 [DOI] [PubMed] [Google Scholar]
- Neri D, Szyperski T, Otting G, Senn H, Wüthrich K (1989) Stereospecific nuclear magnetic resonance assignments of the methyl groups of valine and leucine in the DNA-binding domain of the 434 repressor by biosynthetically directed fractional carbon-13 labeling. Biochemistry 28:7510–7516 [DOI] [PubMed] [Google Scholar]
- Nerli S, McShan AC, Sgourakis NG (2018) Chemical shift-based methods in NMR structure determination. Progr Nucl Magn Res Spec 106–107:1-25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ollerenshaw JE, Tugarinov V, Kay LE (2003) Methyl TROSY: Explanation and experimental verification. Magn Reson Chem 41:843–852 [Google Scholar]
- Ottiger M, Bax A (1999) How tetrahedral are methyl groups in proteins? A liquid crystal NMR study. J Am Chem Soc 121:4690–4695 [Google Scholar]
- Ottiger M, Delaglio F, Bax A (1998a) Measurement of J and dipolar couplings from simplified two-dimensional NMR spectra. J Magn Reson 131:373–378 [DOI] [PubMed] [Google Scholar]
- Ottiger M, Delaglio F, Marquardt JL, Tjandra N, Bax A (1998b) Measurement of dipolar couplings for methylene and methyl sites in weakly oriented macromolecules and their use in structure determination. J Magn Reson 134:365–369 [DOI] [PubMed] [Google Scholar]
- Raman S et al. (2010) NMR structure determination for larger proteins using backbone-only data. Science 327:1014–1018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenzweig R, Kay LE (2014) Bringing dynamic molecular machines into focus by methyl-TROSY NMR. Annu Rev Biochem 83:291–315 [DOI] [PubMed] [Google Scholar]
- Rückert M, Otting G (2000) Alignment of biological macromolecules in novel nonionic liquid crystalline media for NMR experiments. J Am Chem Soc 122:7793–7797 [Google Scholar]
- Schwieters CD, Bermejo GA, Clore GM (2018) Xplor-NIH for molecular structure determination from NMR and other data sources. Protein Science 27: 26–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sgourakis NG et al. (2014) The structure of mouse cytomegalovirus M04 protein obtained from sparse NMR data reveals a conserved fold of the M02-M06 viral immune modulator family. Structure 22:1263–1273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen Y et al. (2008) Consistent blind protein structure generation from NMR chemical shift data. Proc Natl Acad Sci USA 105:4685–4690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sprangers R, Kay LE (2007) Probing supramolecular structure from measurement of methyl 1H–13C residual dipolar couplings. J Am Chem Soc 129:12668–12669 [DOI] [PubMed] [Google Scholar]
- Tang C, Iwahara J, Clore GM (2005) Accurate determination of leucine and valine sidechain conformations using U-[15N/13C/2H]/[1H-(methine/methyl)-Leu/Val] isotope labeling, NOE pattern recognition, and methine Cγ-Hγ/Cβ-Hβ residual dipolar couplings: Application to the 34-kDa enzyme IIAchitobiose. J Biomol NMR 33:105–121 [DOI] [PubMed] [Google Scholar]
- Tugarinov V, Hwang PM, Ollerenshaw JE, Kay LE (2003) Cross-correlated relaxation enhanced 1H–13C NMR spectroscopy of methyl groups in very high molecular weight proteins and protein complexes. J Am Chem Soc 125:10420–10428 [DOI] [PubMed] [Google Scholar]
- Tugarinov V, Kay LE (2004) An isotope labeling strategy for methyl TROSY spectroscopy. J Biomol NMR 28:165–172 [DOI] [PubMed] [Google Scholar]
- Tugarinov V, Kay LE (2005) Methyl groups as probes of structure and dynamics in NMR studies of high-molecular-weight proteins. ChemBiochem 6:1567–1577 [DOI] [PubMed] [Google Scholar]
- Tugarinov V, Kay LE (2013) Estimating sidechain order in [U-2H; 13CH3]-labeled high molecular weight proteins from analysis of HMQC/HSQC spectra. J Phys Chem B 117:3571–3577 [DOI] [PubMed] [Google Scholar]
- Tugarinov V, Karamanos TK, Ceccon A, Clore GM (2020) Optimized NMR experiments for the isolation of I=1/2 manifold transitions in methyl groups of proteins. ChemPhysChem, epub ahead of print doi: 10.1002/cphc.201900959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang D, Nagayama K (1996) A sensitivity-enhanced method for measuring heteronuclear long-range coupling constants from the displacement of signals in two 1D subspectra. J Magn Reson Ser A 118:117–121 [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.