

Abstract
A decrease in microbial infection in adolescents is implicated with an increase in the incidence of asthma and allergic diseases in adulthood, indicating that the microbiome plays a critical role in asthma. However, the microbial composition of the lower respiratory tract remains unclear, hindering the further exploration of the pathogenesis of asthma. This study aims to explore the microbial distribution and composition in the lungs of normal rats and rats with allergic asthma via 16S rDNA sequencing.
The DNA of the pulmonary microbiome was extracted from the left lungs collected from normal control group (NC), saline control group (SC), and allergic asthma group (AA) under aseptic conditions. After the 16s rDNA V4—V5 region was amplified, the products were sequenced using Illumina high-throughput technology and subjected to operational taxonomic unit (OTU) cluster and taxonomy analysis.
The OTU values of AA increased significantly compared with those of NC and SC. Microbiome structure analysis showed that the dominant phylum of the pulmonary microbiome changed from Proteobacteria in NC to Firmicutes in AA. Linear discriminant analysis indicated that the key microbiomes involved in the three groups varied.
Numerous microbiomes stably settled in the lungs of the rats in NC and AA. The structure and diversity of the pulmonary microbiome in AA differed from those in NC.
Keywords: 16S rDNA, Allergic asthma, Illumina sequencing, Pulmonary microbiome, Rats
Introduction
Allergic asthma is a chronic airway inflammation in which various immune cells, especially mast cells, eosinophils, and T lymphocytes, participate, and airway hyperreactivity is the main pathological change.1, 2 Approximately 30 million people in China suffer from asthma. The prevalence rate of asthma is 1.24% among e people over 14 years of age, and this rate increases every year.3 In Sweden, the current prevalence of allergic asthma increased from 5.0% in 1996 to 7.3% in 2016.4 The present situation of asthma is unfavorable. Although multiple studies have explored the mechanism in immune,5 genetic,6 microbial,7 and other aspects, the specific mechanism of asthma has not been elucidated. The relationship between microbiome and asthma has acquired wide attention. However, traditional culture technology cannot comprehensively analyze bacterial composition, thereby limiting studies on pulmonary microbiome.8 The emergence of 16S rDNA sequencing technology has provided a powerful tool to explore the composition and evolution of pulmonary microbiome under normal and disease conditions.9 With new technologies, studies have found that bacteria are closely related to the immunological environment in airways related to the occurrence and development of asthma.10, 11
Normal microbiome refers to the microbial community settling on the mucous membrane and surface of human beings and animals. The Human Microbiome Project, launched by the National Institutes of Health in 2008, proposes to consider the genes of normal microbiome as the second genome of human beings.12 This project has provided a basis for studying normal microbiome at molecular and gene levels. Pulmonary microbiome, a primordial microbial community settled in the lungs, is related to a number of respiratory diseases, including asthma,7 pulmonary cystic fibrosis,13 lung cancer,14 and chronic obstructive pulmonary disease.15 The pulmonary microbiome interplays with allergic asthma. Olszak et al.16 showed an increased incidence of allergic asthma in germ-free mice compared with that of pathogen-free mice. Sverrild A et al.17 demonstrated that an altered bacterial abundance profile occurs in patients with asthma with the lowest eosinophil levels.
At present, the composition of pulmonary microbiome in normal rats and rats with allergic asthma has not been studied, but the interactive relationship between pulmonary microbiome and asthma should be further investigated to address the research gap due to limitations on materials and experimental basis. In this study, a model of allergic asthma was established by ovalbumin (OVA) challenge, and the pulmonary microbiome of the rats with asthma was investigated through 16S rDNA sequencing to widen the experimental basis of research.
Materials and methods
Materials
Animals and groups
Twelve specific pathogen-free male rats with SD background were provided by the Experimental Animal Center of Chongqing Medical University (SCXK-[Chongqing] 2012-0001) with a body mass of 200 ± 20 g. The rats were 4–6 weeks old. They were provided access to clean food and water ad libitum and fed under daylight-simulating light for 12 h, followed by 12 h of darkness at room temperature (23 ± 2 °C), relative humidity of 50%–70%, and with natural ventilation. The 12 rats were randomly divided into three groups with four rats in each group: normal control group (NC), saline control group (SC), and allergic asthma group (AA).
Reagents
Grade II and V OVA were purchased from Sigma (MO, USA). Al(OH)3, IgE, IL-2, and IL-4 ELISA kits were acquired from Elisa biotech (Shanghai, China). Bacterial genome extraction kit and DNA marker were obtained from Takara (Dalian, China). FastPfu PCR mix was procured from Transgene Biotech (Beijing, China). Tris, agarose, EDTA2, H2O, and PCR primers were obtained from Sangon Biotech (Shanghai, China). DNA gel purification kit and AxyPrep DNA gel recovery kit were purchased from Axygen Biosciences (CA, USA). Four kinds of fluorescent dNTPs were acquired from Shanghai Meiji Biotech (Shanghai, China). QuantiFluor-ST blue fluorescence quantitative system was procured from Promega (Wisconsin, USA). Rat nose poisoning apparatus was obtained from Hope Co., Ltd., (Tianjin, China).
Methods
OVA-induced allergic asthma model
On days 0 and 8, the four male rats with a body mass of 200 ± 20 g in AA were injected with 1 mg of OVA and 200 mg of Al (OH)3, which were emulsified in aseptic saline in a total volume of 1 ml. In particular, 0.4 ml of the emulsified treatments was intraperitoneally injected, 0.2 ml was injected to the two anterior toes, and 0.4 ml was injected to the bilateral groins. On day 15, all of the rats in AA were exposed to aerosolized grade II OVA (1% wt/vol diluted in a saline solution) for 60 min daily for 2 weeks. For the rats in SC, all of the solutions used for AA were replaced by aseptic saline. Lung tissues in NC were recovered on day 0 as described in the following section of sample collection. In SC and AA, the tissues were collected on day 29. All procedures were approved by the Ethics Committee of ChongQing Medical University (NO. 20180113) on March 20, 2018. During the experiment, one rat in SC died upon injection, and another died during asthma modeling. None of them were supplemented.
Sample collection
Alcohol (75%) was used to sterilize the skin on the chest and abdomen of the rats after they were anesthetized with avertin overdose. After blood was collected by cardiac puncture, they were killed by cervical dislocation and then dissected under aseptic conditions. The left, upper right, and lower right lobes were recovered and stored at −80 °C. The left lobes were used for microbiome analysis. The upper right lobes were used to determine IL-2 and IL-4 concentrations, and the lower right lobes were preserved in 10% formalin solution for hematoxylin and eosin (H&E) staining. The blood were centrifuged at 3000 rpm at 4 °C for 10 min, and the serum was collected and stored at −80 °C until analysis.
Determination of IgE, IL-2, and IL-4 levels
The serum samples were removed from storage and thawed at room temperature. The right upper lobes were homogenized via bead beating (FastPrep bead matrixE, MP Biomedicals, Santa Ana, CA, USA) with 1000 μl of aseptic saline and then centrifuged for 10 min at 4 °C. The supernatant was collected. The concentrations of IgE in the serum and IL-2 and IL-4 in the supernatant were measured using IgE, IL-2, and IL-4 ELISA kits (Elisa Biotech, Shanghai, China) in accordance with the manufacturer's protocol. Optical absorbance at 450 nm was evaluated with a MultiskanGO plate reader (Thermo, Waltham, MA, USA).
H&E staining
Lung tissues were fixed in 10% formalin solution, embedded in paraffin, sectioned at a thickness of 3 μm, and stained with H&E. Afterward, the pathological changes in lung tissues were observed under a light microscope.
Bacterial DNA extraction
The frozen samples were thawed at room temperature and then homogenized via bead beating (FastPrep bead matrixE, MP Biomedicals, Santa Ana, CA, USA) with 500 μl of aseptic saline. Aseptic saline was added until a volume of 1000 μl was obtained, and bacterial DNA was extracted with a bacterial genomic DNA extraction kit in accordance with the manufacturer's instructions. After its concentration was determined, 1 μl of DNA was diluted to 100 ng/μl with ultra-pure water and then stored at −20 °C.
PCR amplification of 16S rDNA V4—V5 region
A specific primer with its barcode was synthesized to amplify the 16s rDNA V4—V5 region. The sequences of forward primer and reversed primer were 5′-GTGCAGCMGCCGCGG-3′ and 5′-CCGTCAATTCMTTTRAGTTT-3′, respectively. In brief, PCR consisted of 4 μl of 5 × FastPfu Buffer, 2 μl of dNTPs (2.5 mM), 0.8 μl of forward primer (5 μM), 0.8 μl of reverse primer (5 μM), 0.4 μl of FastPfu polymerase, 10 ng of template DNA, and ultra-pure water added to obtain a volume of 20 μl. The reaction conditions were as follows: 5 min at 95 °C, 27 cycles of 30 s each at 95 °C and 53 °C and 45 s at 72 °C, and 10 min at 72 °C. The amplification products were analyzed through Illumina high-throughput sequencing (Illumina PE250). The sequence information was submitted to the NCBI's sequence read archive with the accession number PRJNA504591. Finally, microbial diversity was analyzed on the basis of the sequencing results.
Statistical analysis
Data were assessed using SPSS version 23 (SPSS Inc., Chicago, IL, USA). Statistical significance was determined by ANOVA. When ANOVA indicated a significant difference, Fisher's least significant difference (LSD) test was applied to conduct multiple comparisons. A p value of <0.05 was considered statistically significant.
Results
Model evaluation
Behavioral changes
After inhaling aseptic saline, the rats in SC and NC had normal movement, stable breathing, and shiny hair. Conversely, the rats in AA manifested various symptoms, such as restlessness, hair erection, shortness of breath, nasal scratching, incontinence, and abdominal breathing.
IgE, IL-2, and IL-4 concentrations
The IgE, IL-2, and IL-4 concentrations in the three groups were determined (Fig. 1). The results reflected no significant difference between NS and SC, but the decreased IL-2 concentration and the increased IL-4 and IgE concentrations in AA compared with those in NC were significantly different (PIL-2 < 0.05; PIL-4 < 0.01; PIgE < 0.01).
Figure 1.

IgE, IL-2, and IL-4 concentrations in the three groups. The serum and supernatant of homogenized lung tissues were analyzed through ELISA. (a) IgE concentration in serum (n = 4); (b) IL-2 concentration in lung tissues (n = 3); (c) IL-4 concentration in lung tissues (n = 3). *p < 0.05, **p < 0.01, ***p < 0.001.
Lung histology
The lung histology (Fig. 2) revealed the successful preparation of the asthma showing inflammatory cell infiltration, widened alveoli, and dilated alveoli. Such changes did not occur in the two other groups.
Figure 2.

Lung sections of rats in the three groups. Lung sections from NC, SC, and AA were stained with H&E. (a) In NC, the structure of the alveolar wall was clear and complete without inflammatory cell infiltration in the lungs. (b) In SC, the bronchial wall and the smooth muscle were slightly thickened, and few inflammatory cells infiltrated the interstitium and alveoli of the lung; (c) In AA, the bronchial wall of the rats with asthma was damaged invasively by numerous inflammatory cells, which infiltrated the whole layer of the bronchial wall. The pulmonary alveoli collapsed, the intervals between pulmonary alveoli were widened, and a part of the pulmonary alveoli were dilated.
Microbiome analysis
Evaluation of sequencing depth
The Shannon value of random sampling was calculated by Mothur, and the Shannon-Wiener curve (Fig. 3) was drawn using the R programming language to evaluate the sequencing depth. A total of 440,343 sequences were detected in all of the samples. These sequences covered 285–695 OTUs and showed a high community richness. All of the curves were flat, indicating that the sequencing depth was sufficient to cover more than 97% of the microbiome in the samples and reflected the information of the major bacteria present in the samples.
Figure 3.

Shannon–Wiener curve. Curves of different colors show the Shannon index (X-axis) of each sample at different numbers of the sampled reads (Y-axis). When the curve tended to be flat, the amount of the sequenced data was large enough to reflect the majority of the microbial data in the samples.
Microbial diversity analysis (alpha-diversity)
The Chao index, coverage index, Shannon index, and Simpson index of each sample are listed in Table 1. A value of 0.97 indicates a similarity level of 0.97. The ANOVA results showed no significant difference (PChao > 0.05) in the Chao index among the three groups, suggesting that the abundance remained the same among the three groups. The differences in Shannon and Simpson indexes were statistically significant (PShannon < 0.001; PSimpson < 0.001). No differences were detected between NC and SC, but differences between the two groups and AA (P < 0.01) were significant. The diversity of the pulmonary microbiome in the rats with asthma increased. The coverage index of each sample was greater than 0.9985, implying that the coverage of each sample was greater than 99.85%, which could fully reflect the true situation of the microbiome in the samples.
Table 1.
Analysis of community diversity.
| Sample ID | Reads | 0.97 |
||||
|---|---|---|---|---|---|---|
| OTU | chao | coverage | shannon | simpson | ||
| A1 | 44,046 | 308 | 325 (315,350) | 0.999,342 | 2.01 (1.99,2.03) | 0.4295 (0.4239,0.4351) |
| A2 | 30,276 | 285 | 319 (300,359) | 0.998,679 | 2.02 (2,2.05) | 0.4055 (0.399,0.412) |
| A3 | 38,855 | 316 | 336 (324,365) | 0.999,202 | 2.39 (2.37,2.42) | 0.2652 (0.2614,0.2689) |
| A4 | 49,925 | 306 | 337 (319,379) | 0.999,339 | 1.86 (1.84,1.88) | 0.4699 (0.4645,0.4753) |
| B1 | 47,023 | 438 | 460 (448,486) | 0.999,149 | 2.82 (2.8,2.85) | 0.2539 (0.2496,0.2582) |
| B2 | 45,011 | 318 | 345 (330,377) | 0.999,111 | 2.09 (2.07,2.11) | 0.3083 (0.3048,0.3118) |
| B3 | 32,615 | 385 | 402 (392,424) | 0.998,866 | 2.35 (2.32,2.37) | 0.3052 (0.3,0.3103) |
| C1 | 51,460 | 695 | 738 (719,772) | 0.998,504 | 3.86 (3.84,3.88) | 0.063 (0.062,0.064) |
| C2 | 51,839 | 353 | 361 (355,382) | 0.999,749 | 4.55 (4.53,4.56) | 0.0414 (0.0404,0.0424) |
| C3 | 49,293 | 574 | 590 (581,615) | 0.999,412 | 4.66 (4.65,4.68) | 0.0422 (0.0411,0.0433) |
The Chao index was used to reflect the community richness of the microbiome. The higher the Chao index was, the more abundant the microbiome would be. The Shannon and Simpson indices were used to analyze the community diversity in the samples. The higher the Shannon index was, the higher the diversity of the microbiome would be. The higher the Simpson index was, the lower the diversity of the microbiome would be. The coverage index was determined to reflect whether the sequencing results represented the true situation of the microbiome in the sample. The higher the values were, the higher the probabilities of the sequences to be detected and the lower probabilities of not being detected would be.
Community-composition analysis
The RDP classifier Bayes algorithm was used to analyze OTU at the similarity level of 97%. The OTU sequences were then compared with the Silva 16s ribosomal database of bacteria and ancient bacteria. The community composition of the samples was examined at phylum, family, and genus levels, and a multi-sample histogram was drawn.
OTU distribution
The total and particular OTU sequences in the three groups were calculated using the R programming language. A total of 304 OTUs were shared by the three groups. A total of 68, 59, and 352 particular OTUs were present in NC, SC, and AA (Fig. 4), respectively. This finding indicated that the number of lung-specific OTUs in AA was much higher than that in NC and SC.
Figure 4.
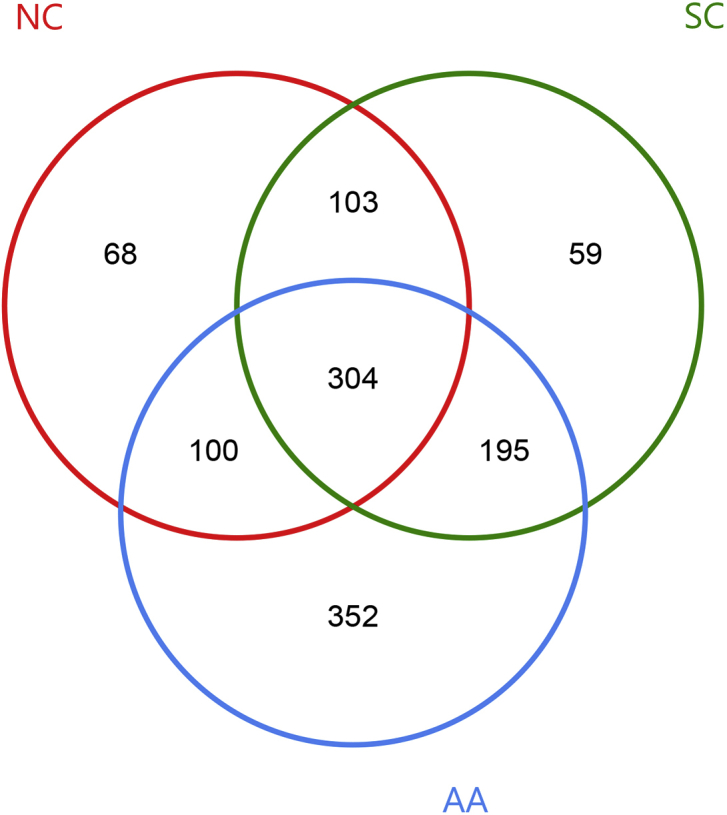
OTU distribution in the three groups. The red, green, and blue circles represented NC, SC, and AA, respectively. The overlapping part denoted the OTU that each group had.
Distribution of the microbiome at phylum, family, and genus levels
At the phylum level, 24 phyla were identified in 10 samples (Table 2). Six phyla, including Actinobacteria, Firmicutes, and Proteobacteria, were detected in all of the samples. The hierarchical cluster analysis combined with the microbiome distribution of the phylum is shown in Fig. 5. NC, SC, and AA could be divided into two groups (groups A and B). NC, SC, and AA1 were included in group A, and the remaining AA samples were clustered to group B. However, the distribution of AA1 was different from that of NC and SC at the phylum level. Proteobacteria was the most dominant phylum in NC. In AA, the most dominant phylum changed into Firmicutes. This change was consistent in all of the samples between the two groups. All of the samples could be well expanded in the cluster tree, indicating that some similarities were present between SC and NC. A total of 158 families were detected in all of the samples. At the genus level, the dominant bacteria basically remained the same in NC and SC, although some differences existed. The species and abundance of the dominant bacteria in AA were significantly different from those in NA and SC. The dominant family and genus of each sample are shown in Table 3, and the distribution of the microbiome structure is presented in Fig. 6.
Table 2.
Structural analysis of microbiome at phylum level.
| Phylum type | Comparative result | Detected sample |
|---|---|---|
| Identical bacteria | Actinobacteria | All samples |
| Bacteroidetes | All samples | |
| Cyanobacteria | All samples | |
| Firmicutes | All samples | |
| Proteobacteria | All samples | |
| Spirochaetae | All samples | |
| Different bacteria | Acidobacteria | A1A2A3A4B1B3C1C2 |
| Armatimonadetes | A2 | |
| Atribacteria | A1A2A3A4 B1B2B3 | |
| Chloroflexi | A1A2A3A4B1B2B3C1 | |
| Cloacimonetes | A1A2A3A4 B1B2B3 C1 | |
| Deferribacteres | A1A2A3A4B1B3C1C2C3 | |
| Deinococcus-Thermus | A3B2 | |
| Elusimicrobia | C3 | |
| Fibrobacteres | A1A2B1C1C2C3 | |
| Fusobacteria | B1B2 | |
| Gemmatimonadetes | B2 C1 | |
| Nitrospirae | C2 | |
| Planctomycetes | A1A4 B1 C1 | |
| Synergistetes | A1A3A4 B1B3 | |
| TM6 (Dependentiae) | C1 | |
| Tenericutes | A1A2A4B1B2B3C1C2C3 | |
| WS1 | A1A4B2 | |
| Verrucomicrobia | A1B1B3 | |
| Unclassified | A1A2A3A4B1B2B3C1 |
The phyla detected in all of the samples are listed in “identical bacteria,” whereas the phyla not detected in all of the samples are listed in “different bacteria.”
Figure 5.

Microbial community barplot with a cluster tree. Bar charts showing the relative abundance of the phyla detected in the lung tissues collected from the rats of NC (NC1–NC4), SC (SC1–SC3), and AA (AA1–AA3). The identities of microbial phyla were shown with color blocks on the right, and the length of the color blocks denoted the relative abundance. An Hcluster tree was placed on the left side of the graph. The length of the branches indicated the distance between the samples.
Table 3.
Top three dominant families and genera in each sample.
| Sample | Dominant family | Relative abundance (%) |
Dominant genus | Relative abundance (%) |
|---|---|---|---|---|
| A1 | Burkholderiaceae | 65.04 | Ralstonia | 64.99 |
| Bradyrhizobiaceae | 7.58 | Bradyrhizobium | 4.83 | |
| Ruminococcaceae | 4.17 | Enterobacter | 3.38 | |
| A2 | Burkholderiaceae | 63.00 | Ralstonia | 62.94 |
| Bradyrhizobiaceae | 10.58 | Bradyrhizobium | 7.14 | |
| Lachnospiraceae | 3.74 | Lactobacillus | 3.44 | |
| A3 | Burkholderiaceae | 45.05 | Ralstonia | 45.05 |
| Bradyrhizobiaceae | 27.07 | Bradyrhizobium | 23.94 | |
| Sphingomonadaceae | 3.67 | Sphingomonas | 3.64 | |
| A4 | Burkholderiaceae | 68.12 | Ralstonia | 68.07 |
| Bradyrhizobiaceae | 7.75 | Bosea | 3.95 | |
| Lachnospiraceae | 4.67 | Bradyrhizobium | 3.79 | |
| B1 | Burkholderiaceae | 49.41 | Ralstonia | 49.37 |
| Lachnospiraceae | 12.68 | Mycoplasma | 5.82 | |
| Ruminococcaceae | 7.96 | Blautia | 4.58 | |
| B2 | Mycoplasmataceae | 47.66 | Mycoplasma | 47.66 |
| Burkholderiaceae | 28.13 | Ralstonia | 28.12 | |
| Bradyrhizobiaceae | 3.26 | Bradyrhizobium | 2.49 | |
| B3 | Burkholderiaceae | 52.72 | Ralstonia | 52.72 |
| Bradyrhizobiaceae | 18.38 | Bradyrhizobium | 15.18 | |
| Lactobacillaceae | 5.31 | Lactobacillus | 5.31 | |
| C1 | Lactobacillaceae | 21.82 | Lactobacillus | 21.82 |
| Chloroplast_norank | 15.36 | Chloroplast_norank | 15.36 | |
| Burkholderiaceae | 14.59 | Ralstonia | 14.58 | |
| C2 | Ruminococcaceae | 22.31 | Lactobacillus | 16.75 |
| Lactobacillaceae | 16.75 | Ralstonia | 15.72 | |
| Burkholderiaceae | 15.72 | Ruminococcaceae UCG-014 | 7.29 | |
| C3 | Lactobacillaceae | 26.96 | Lactobacillus | 26.95 |
| Ruminococcaceae | 24.90 | Ruminococcaceae UCG-014 | 11.71 | |
| Lachnospiraceae | 10.89 | Bacteroidales S24-7 group_norank |
8.58 |
The table lists the top three dominant families and genera in each sample with their relative abundance.
Figure 6.

Microbial community barplot at the genus level. Bar charts showing the relative abundance of all genera detected in the lung tissues collected from the rats in NC (NC1–NC4), SC (SC1–SC3), and AA (AA1–AA3). The identities of the microbiome were shown with color blocks on the right.
Intergroup diversity analysis (beta-diversity)
Distance matrix was obtained by calculating the distance between two samples using the statistical algorithm Bray-Curtis, and β-diversity analysis and visual statistical analysis were performed. The distance matrix was represented by a heat map to show the difference distribution between the samples. LEfSE (linear discriminant analysis [LDA] effect size) was used to analyze the differences among various groups, and LDA was conducted to estimate the effect of each species’ abundance on the differential effect.
Variance analysis of samples
The beta diversity distance matrix was represented by a heat map (Fig. 7). The values represented by different colors were the coefficient of difference between two samples. The smaller the coefficient was, the smaller the difference in species diversity would be. The difference between the individual samples of NC and SC was obscure, but the difference between the individual samples of NC and AA was significant, indicating that the lung microbiome was evidently altered under asthmatic conditions.
Figure 7.

Heatmap analysis of distance matrix. The heatmap plot depicts the discrepancy of each sample by color intensity. Higher values of the color key shown below the figure indicate a higher discrepancy between two samples.
LEfSE analysis
Fig. 8 shows the cladogram of the LEfSE analysis. The red, green, and blue parts represented the samples of NC, SC, and AA, respectively. The red, green, and blue nodes in the cladogram denoted the bacteria playing a critical role in NC, SC, and AA respectively. The yellow nodes corresponded to the bacteria that did not have an important role in each group. Fig. 9 shows the LDA score obtained through LDA, indicating that the bacteria with a major role were Proteobacteria, Ralstonia, and Burkholderiales in NC; Mollicutes, Tenericutes, and Proteus in SC; and Lactobacillaceae, Bacilli, and Lactobacillales in AA.
Figure 8.

Cladogram of the three groups. The circle radiating inside-out demonstrated the classification from the phylum to the genus. Each small circle at different classifications represented a taxon, the diameter of the circle was proportional to the relative abundance. Red, green, and blue dots denoted the core bacterial populations in each respective group. The histogram of the LDA score (below) showed the biomarkers with statistical differences between groups.
Figure 9.

LEfSe result. The graph shows the LDA scores obtained from linear regression analysis of the significant microorganism groups in the two groups. When the default LDA value is more than 2 and the P value is less than 0.05, the result corresponds to a differential species. The differential group is the one with high abundance in three groups.
ANOSIM analysis
ANOSIM, a nonparametric test, was performed to determine whether the grouping was meaningful or not. Table 4 indicates that the differences between the groups were greater than the differences within the groups. Thus, the grouping was meaningful.
Table 4.
ANOSIM analysis.
| Name of Test Statistic | R |
| Sample size | 10 |
| Number of groups | 3 |
| Test statistic | 0.58,080,808,080,808,077 |
| p-value | 0.001 |
| Number of permutations | 999 |
In theory, R ranges from −1 to 1. In practice, R ranges from 0 to 1. When R is close to 1, the differences between groups are greater than those within groups. Conversely, the differences between and within groups are not different when R is close to 0. The reliability of the statistical analysis is expressed by P value.
Discussion
The complex interplay between the body and the microbial community determines the state of human health and the occurrence and development of diseases. In the respiratory system, the lower respiratory tract, especially the lungs, is aseptic when it is healthy. However, recent studies have revealed the diversity and dynamics of microbes in the lower respiratory tract, supporting the view of the microbiome setting in the lungs.18, 19 Further studies on the human microbiome demonstrated that the microbiome existed not only in the lungs of healthy people but also in the lungs with various diseases, such as asthma.20
In the current study, lung tissue sampling under aseptic condition was conducted to avoid pollution from the upper respiratory tract, and 16S rDNA sequencing was performed to observe the microbiome of rats with asthma, which could reliably reflect the alteration of the pulmonary microbiome in the lungs. The lung microbiome diversity of AA was higher than that of NC, and this observation was consistent with the clinical results of Huang et al.21 The sequencing of the pulmonary microbiome in the lungs affected by bronchial asthma has shown multiple pathogens, especially Haemophilus, in the lower respiratory tract.22 These studies have demonstrated that patients with asthma possess an imbalance of the lung microbiome, an increased pathogenic microbial diversity, and a decreased beneficial microbial diversity. Thus, the increase in microbial diversity might be closely related to the increase in the population of pathogenic bacteria.
Further analysis of the sequencing data showed that the structure of the microbiome in the lungs of the rats in AA was significantly different from that in NC. The most dominant and the second-most dominant phyla in the lung microbiome of the normal rats were Proteobacteria and Firmicutes, and this finding was consistent with the report of Liu et al.23 In AA, however, Firmicutes was the most dominant phylum instead of Proteobacteria, and this observation was contrary to that of the bacteria dominant in the human body. Blainey et al.24 confirmed that five major bacterial phyla exist in the respiratory tract of healthy people: Firmicutes, Bacteroidetes, Proteobacteria, Actinobacteria, and Fusobacteria. Their respective proportions decreased in succession. Thus, Firmicutes is the most dominant phylum in normal adults rather than in patients with asthma. In comparison with healthy people, patients with asthma have a lung microbiome with an increased abundance of Proteobacteria.21, 22 In the current study, Burkholderiaceae, the dominant bacterial family in NC, was replaced by Lactobacillaceae in AA. Ralstonia was replaced by Lactobacillus in AA. However, in the lower respiratory tract of normal adults, Prevotellaceae and Sreptococcaceae are the dominant families.25 This finding might be due to species differences, which should be further explored.
Immune imbalance is one of the important mechanisms of asthma. Changes in the local inflammatory environment can affect the composition of the pulmonary microbiome.19 Therefore, we detected the concentrations of IL-2 and IL-4 in lung tissues and the concentrations of IgE in serum through ELISA. Our results showed a statistically significant decrease in IL-2 and an increase in IL-4. IL-2, a representative of Th1 cytokines, inhibits inflammation, whereas IL-4, a representative of Th2 cytokines, promotes inflammation. Th1/Th2 imbalance, which is the main mechanism of asthma, has been found in lung tissues.26, 27 In the current study, the increased serum IgE concentration was also statistically significant. This finding is attributed to IL-4 promoting the secretion of B plasma cells. IgE triggers mast cells to secrete histamine. PGD2 is an important cytokine that participates in allergic reactions and causes the symptoms of an allergy. In the current study, H&E staining demonstrated numerous inflammatory cells infiltrating the lungs. The inflammation caused by this imbalance was closely related to the structure of the pulmonary microbiome in asthmatic rats. Herbst et al.28 found that the degree of inflammatory responses in germ-free mice is more serious than that in specific pathogen-free mice, indicating that the lung microbiome and inflammation demonstrate a close interplay. However, whether local inflammation or pulmonary microbiome initiates asthma remains unclear and requires further exploration.
In a healthy state, pulmonary microbiome composition is similar to that of the upper respiratory tract.29, 30 The abundance and composition of the microbiome are affected by many factors, including birth mode, feeding type, genetics, and smoking.31 In a diseased state, the local environment of the lung plays an important role in the alteration of pulmonary microbiome. The local environment involves relative blood perfusion, relative alveolar ventilation, deposition of inhaled particles, and concentration and behavior of inflammatory cells.19 Relative blood perfusion affects the microbiome, and the increased IgE observed in our results could increase vascular permeability, thereby altering the pulmonary microbiome.26 In airway remodeling, the clearance of the microbiome is affected, influencing the settlement of the microbiome.31 In addition to pulmonary inflammation, alteration in the pulmonary microbiome may be associated with changes in the local metabolic environment. In a mouse model of LPS-induced lung injury, the increased abundance of Stenotrohomonas maltophilia (Xanthomonadaceae) and Ochrobactrum anthropi (Brucellaceae) is connected closely with the increased presence of bacterial substrates suitable for their survival.32 In asthma, multiple metabolic pathways and metabolites are disrupted.33, 34, 35 Adult bronchial microbiome sequencing and functional analysis demonstrating the predicted bacterial functions revealed that the microbiome is involved in the metabolism of amino acids and carbohydrates, especially short-chain fatty acids, playing a remarkable role in regulating inflammation.36 Therefore, pulmonary microbiome, metabolism, and inflammation closely interplay, indicating that the alteration of pulmonary microbiome in asthma is an extremely complicated process that requires further exploration, which we intend to do.
Conflict of interest
The authors declare no conflict of interest.
Acknowledgments
This research was funded by Key Items of Scientific Research and Innovation Experiment Project of Chongqing Medical University in 2017, grant number 201710” and The Project of Tutorial System of Medical Undergraduate in Lab Teaching & Management Center in Chongqing Medical University, grant number LTMCMTS201805. The following individuals are gratefully acknowledged: Yanqin Ran,Weilai Hao and Yinde Huang for their technical assistance; the Innovation Laboratory of Chongqing Medical University for their excellent research environment.
Footnotes
Peer review under responsibility of Chongqing Medical University.
Contributor Information
Yang Xiong, Email: cqmu_xy@163.com.
Sen Hu, Email: husanwmuym@163.com.
Hongyao Zhou, Email: ZhouHongyaoZHY@163.com.
Hui Zeng, Email: emmazengh@gmail.com.
Xuan He, Email: HeXuanghh@163.com.
Dongni Huang, Email: xfx1412@126.com.
Xiaoyu Li, Email: lixiaoyu.lin@gmail.com.
References
- 1.Mims J.W. Asthma: definitions and pathophysiology. Int Forum Allergy Rhinol. 2015;5:S2–S6. doi: 10.1002/alr.21609. [DOI] [PubMed] [Google Scholar]
- 2.Robinson D., Humbert M., Buhl R. Revisiting Type 2-high and Type 2-low airway inflammation in asthma: current knowledge and therapeutic implications. Clin Exp Allergy. 2017;47:161–175. doi: 10.1111/cea.12880. [DOI] [PubMed] [Google Scholar]
- 3.Hua W., Huang H., Shen H. Interpretation of 2016 asthma management and prevention guideline. Zhejiang Da Xue Xue Bao Yi Xue Ban. 2016;45:447–452. doi: 10.3785/j.issn.1008-9292.2016.09.01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Backman H., Räisänen P., Hedman L. Increased prevalence of allergic asthma from 1996 to 2006 and further to 2016 - results from three population surveys. Clin Exp Allergy. 2017;47:1426–1435. doi: 10.1111/cea.12963. [DOI] [PubMed] [Google Scholar]
- 5.Panettieri R.A., Jr. Neutrophilic and pauci-immune phenotypes in severe asthma. Immunol Allergy Clin N AM. 2016;36:569–579. doi: 10.1016/j.iac.2016.03.007. [DOI] [PubMed] [Google Scholar]
- 6.Bonnelykke K., Ober C. Leveraging gene-environment interactions and endotypes for asthma gene discovery. J Allergy Clin Immunol. 2016;137:667–679. doi: 10.1016/j.jaci.2016.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Durack J., Huang Y.J., Nariya S. Bacterial biogeography of adult airways in atopic asthma. Microbiome. 2018;6:104. doi: 10.1186/s40168-018-0487-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kahn F.W., Jones J.M. Diagnosing bacterial respiratory infection by bronchoalveolar lavage. J Infect Dis. 1987;155:862–869. doi: 10.1093/infdis/155.5.862. [DOI] [PubMed] [Google Scholar]
- 9.Suárez Moya A. Microbiome and next generation sequencing. Rev Española Quimioter. 2017;30:305–311. [PubMed] [Google Scholar]
- 10.Gensollen T., Iyer S.S., Kasper D.L., Blumberg R.S. How colonization by microbiota in early life shapes the immune system. Science. 2016;352:539–544. doi: 10.1126/science.aad9378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pattaroni C., Watzenboeck M.L., Schneidegger S. Early-Life formation of the microbial and immunological environment of the human airways. Cell Host Microbe. 2018;24:857–865. doi: 10.1016/j.chom.2018.10.019. e4. [DOI] [PubMed] [Google Scholar]
- 12.Human Microbiome Jumpstart Reference Strains Consortium, Nelson K.E., Weinstock G.M. A catalog of reference genomes from the human microbiome. Science. 2010;328:994–999. doi: 10.1126/science.1183605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.O'Toole G.A. Cystic fibrosis airway microbiome: overturning the old, opening the way for the new. J Bacteriol. 2018 Jan 24;200(4) doi: 10.1128/JB.00561-17. pii: e00561-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mao Q., Jiang F., Yin R. Interplay between the lung microbiome and lung cancer. Cancer Lett. 2018;415:40–48. doi: 10.1016/j.canlet.2017.11.036. [DOI] [PubMed] [Google Scholar]
- 15.Wang L., Hao K., Yang T., Wang C. Role of the lung microbiome in the pathogenesis of chronic obstructive pulmonary disease. Chin Med J (Engl). 2017;130:2107–2111. doi: 10.4103/0366-6999.211452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Olszak T., An D., Zeissig S. Microbial exposure during early life has persistent effects on natural killer T cell function. Science. 2012;336:489–493. doi: 10.1126/science.1219328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sverrild A., Kiilerich P., Brejnrod A. Eosinophilic airway inflammation in asthmatic patients is associated with an altered airway microbiome. J Allergy Clin Immunol. 2017;140:407–417. doi: 10.1016/j.jaci.2016.10.046. [DOI] [PubMed] [Google Scholar]
- 18.Dickson R.P., Erb-Downward J.R., Freeman C.M. Bacterial topography of the healthy human lower respiratory tract. mBio. 2017;8(1) doi: 10.1128/mBio.02287-16. e02287-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dickson R.P., Erb-Downward J.R., Martinez F.J., Huffnagle G.B. The microbiome and the respiratory tract. Annu Rev Physiol. 2016;78:481–504. doi: 10.1146/annurev-physiol-021115-105238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Faner R., Sibila O., Agustí A. The microbiome in respiratory medicine: current challenges and future perspectives. Eur Respir J. 2017;49:1602086. doi: 10.1183/13993003.02086-2016. [DOI] [PubMed] [Google Scholar]
- 21.Huang Y.J., Nelson C.E., Brodie E.L. Airway microbiota and bronchial hyperresponsiveness in patients with suboptimally controlled asthma. J Allergy Clin Immunol. 2011;127:372–381. doi: 10.1016/j.jaci.2010.10.048. e1-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hilty M., Burke C., Pedro H. Disordered microbial communities in asthmatic airways. PLoS One. 2010;5:e8578. doi: 10.1371/journal.pone.0008578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu T., Yang Z., Zhang X., Han N., Yuan J., Cheng Y. 16S rDNA analysis of the effect of fecal microbiota transplantation on pulmonary and intestinal flora. 3 Biotech. 2017;7:370. doi: 10.1007/s13205-017-0997-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Blainey P.C., Milla C.E., David N.C., Stephen R.Q. Quantitative analysis of the human airway microbial ecology reveals a pervasive signature for cystic fibrosis. Sci Transl Med. 2012;4 doi: 10.1126/scitranslmed.3004458. 153ra130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Charlson E.S., Bittinger K., Haas A.R. Topographical continuity of bacterial populations in the healthy human respiratory tract. Am J Respir Crit Care Med. 2011;184:957–963. doi: 10.1164/rccm.201104-0655OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kubo M. Innate and adaptive type 2 immunity in lung allergic inflammation. Immunol Rev. 2017;278:162–172. doi: 10.1111/imr.12557. [DOI] [PubMed] [Google Scholar]
- 27.Kuo C.S., Pavlidis S., Loza M. T-helper cell type 2 (Th2) and non-Th2 molecular phenotypes of asthma using sputum transcriptomics in U-BIOPRED. Eur Respir J. 2017;49:1602135. doi: 10.1183/13993003.02135-2016. [DOI] [PubMed] [Google Scholar]
- 28.Herbst T., Sichelstiel A., Schär C. Dysregulation of allergic airway inflammation in the absence of microbial colonization. Am J Respir Crit Care Med. 2011;184:198–205. doi: 10.1164/rccm.201010-1574OC. [DOI] [PubMed] [Google Scholar]
- 29.Bassis C.M., Erb-Downward J.R., Dickson R.P. Analysis of the upper respiratory tract microbiotas as the source of the lung and gastric microbiotas in healthy individuals. mBio. 2015;6:e00037. doi: 10.1128/mBio.00037-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Marsh R.L., Kaestli M., Chang A.B. The microbiota in bronchoalveolar lavage from young children with chronic lung disease includes taxa present in both the oropharynx and nasopharynx. Microbiome. 2016;4:37. doi: 10.1186/s40168-016-0182-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Man W.H., de Steenhuijsen Piters W.A., Bogaert D. The microbiota of the respiratory tract: gatekeeper to respiratory health. Nat Rev Microbiol. 2017;15:259–270. doi: 10.1038/nrmicro.2017.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Poroyko V., Meng F., Meliton A. Alterations of lung microbiota in a mouse model of LPS-induced lung injury. Am J Physiol Lung Cell Mol Physiol. 2015;309:L76–L83. doi: 10.1152/ajplung.00061.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Su L., Shi L., Liu J., Huang L., Huang Y., Nie X. Metabolic profiling of asthma in mice and the interventional effects of SPA using liquid chromatography and Q-TOF mass spectrometry. Mol Biosyst. 2017;13:1172–1181. doi: 10.1039/c7mb00025a. [DOI] [PubMed] [Google Scholar]
- 34.Quinn K.D., Schedel M., Nkrumah-Elie Y. Dysregulation of metabolic pathways in a mouse model of allergic asthma. Allergy. 2017;72:1327–1337. doi: 10.1111/all.13144. [DOI] [PubMed] [Google Scholar]
- 35.Wang Z., Gao S., Xie J., Li R. Identification of multiple dysregulated metabolic pathways by GC-MS-based profiling of lung tissue in mice with PM2.5-induced asthma. Chemosphere. 2019;220:1–10. doi: 10.1016/j.chemosphere.2018.12.092. [DOI] [PubMed] [Google Scholar]
- 36.Cait A., Hughes M.R., Antignano F. Microbiome-driven allergic lung inflammation is ameliorated by short-chain fatty acids. Mucosal Immunol. 2018;11:785–795. doi: 10.1038/mi.2017.75. [DOI] [PubMed] [Google Scholar]