

Abstract
Summary
To elucidate mutation spectrum and genotype-phenotype correlations in Japanese patients with OI, we conducted comprehensive genetic analyses using NGS, as this had not been analyzed comprehensively in this patient population. Most mutations were located on COL1A1 and COL1A2. Glycine substitutions in COL1A1 resulted in the severe phenotype.
Introduction
Most cases of osteogenesis imperfecta (OI) are caused by mutations in COL1A1 or COL1A2, which encode α chains of type I collagen. However, mutations in at least 16 other genes also cause OI. The mutation spectrum in Japanese patients with OI has not been comprehensively analyzed, as it is difficult to identify using classical Sanger sequencing. In this study, we aimed to reveal the mutation spectrum and genotype-phenotype correlations in Japanese patients with OI using next-generation sequencing (NGS).
Methods
We designed a capture panel for sequencing 15 candidate OI genes and 19 candidate genes that are associated with bone fragility or Wnt signaling. Using NGS, we examined 53 Japanese patients with OI from unrelated families.
Results
Pathogenic mutations were detected in 43 out of 53 individuals. All mutations were heterozygous. Among the 43 individuals, 40 variants were identified including 15 novel mutations. We found these mutations in COL1A1 (n = 30, 69.8%), COL1A2 (n = 12, 27.9%), and IFITM5 (n = 1, 2.3%). Patients with glycine substitution on COL1A1 had a higher frequency of fractures and were more severely short-statured. Although no significant genotype-phenotype correlation was observed for bone mineral density, the trabecular bone score was significantly lower in patients with glycine substitutions.
Conclusion
We identified pathogenic mutations in 81% of our Japanese patients with OI. Most mutations were located on COL1A1 and COL1A2. This study revealed that glycine substitutions on COL1A1 resulted in the severe phenotype among Japanese patients with OI.
Electronic supplementary material
The online version of this article (10.1007/s00198-019-05076-6) contains supplementary material, which is available to authorized users.
Keywords: Fracture, Genotype-phenotype correlation, Next-generation sequencing, Osteogenesis imperfecta, Short stature, Type I collagen
Introduction
Osteogenesis imperfecta (OI) is an inheritable disorder characterized by bone fragility. The fragility of the bones leads to fractures after even mild trauma and subsequent growth restriction. The severity of this condition is highly variable. Patients with OI also suffer from some extraskeletal symptoms including blue or gray discoloration of the sclerae, hearing loss, defective tooth formation (dentinogenesis imperfecta), scoliosis, macrocephaly, barrel chest, and ligamentous laxity [1].
OI is generally categorized into 4 types according to the Sillence classification, which is based on clinical and radiographic features [2]. Type I is the mildest, whereas type II is lethal during the neonatal period owing to skeletal deformities and respiratory compromise. Type III is known as the progressive deforming type. Type IV is a moderately severe form, and a type V classification has recently been defined, which is associated with hypertrophic calluses at fracture or surgery sites and intraosseous calcification [3].
It has been reported that about 85–90% of OI cases are caused by structural or quantitative mutations in COL1A1 and COL1A2, which code for the α1 (I) and α2 (I) chains of type I collagen, respectively. OI type I is related to a quantitative deficiency of structurally normal type I collagen [4], whereas types II, III, and IV are related to structural defects in either of the 2 chains that form the type I collagen heterodimer [5]. However, the genotype-phenotype relationship in patients with OI was not completely understood.
Currently, at least 16 genes other than COL1A1 and COL1A2 have been reported to be disease-causing genes for OI, including FKBP10, SERPINH1, IFITM5, SERPINF1, CRTAP, P3H1, PPIB, SP7, PLOD2, BMP1, CREB3L1, TMEM38B, WNT1, SPARC, and MBTPS2 [3, 6–8]. Because of the large number of genes involved in the pathogenesis of OI and of the large size of COL1A1 and COL1A2, the primary genes causing OI, it is difficult to comprehensively analyze pathogenic mutations using classical Sanger sequencing. Although some reports have described mutations in Japanese patients with OI [9, 10], genotype-phenotype studies have focused only on COL1A1 and COL1A2 in Japan.
In this study, we conducted a comprehensive genetic analysis of Japanese patients with OI using a designed panel of OI-related genes and bone volumes to elucidate the mutation spectrum in this patient population. We also analyzed the mutations and clinical features to clarify the genotype-phenotype relationship.
Subjects and methods
Subjects
This study included all individuals with a typical OI phenotype who were evaluated at Osaka University Hospitals, Osaka Women’s and Children’s Hospital, and Mino City Hospital between 2010 and 2017. Clinical diagnoses of OI were based on a history of more than one fracture following minor trauma, low bone mineral density (BMD), blue sclerae, and a familial history of OI. One of the authors (T.Ki., T.Ku., T.M., Ka.Y., T.Y., N.N., or K.O.) assessed each patient clinically and assigned classifications according to the Sillence classification system.
All 53 individuals reported here were of Japanese descent from unrelated families, residing in Japan, and of Asian ethnicity.
The study was approved by the Institutional Review Board of Osaka University, Osaka Women’s and Children’s Hospital, and Mino City Hospital. Written informed consent was obtained from patients aged 16 years or older. For individuals under 16 years of age, parental consent was obtained, as well as assent from participants over 8 years old.
Clinical analysis
Patients’ heights were measured at each hospital and were converted to age- and sex-specific standard deviation score (SDS) based on reference data from the Japanese Society for Pediatric Endocrinology. The annual fracture rate prior to the initiation of bisphosphonate (BP) treatment was calculated as follows:
BMD at the lumbar spine was measured using dual X-ray absorptiometry (DXA). Areal BMD values were converted to age- and sex-specific SDS using the data of previous studies [11–13].
To calculate the trabecular bone score (TBS), the raw data from the DXA were extracted using TBS iNsight software (v1.9, Medimaps SA, France). TBS was calculated as the mean value of the measurements for L1–L4 at the same region of interest (ROI) as the lumbar spine BMD.
Genetic analysis
Targeted next-generation sequencing (NGS) was performed using the Ion Torrent System (Thermo Fisher Scientific, Waltham, MA, USA). A capture panel of targeted DNA was designed to include 15 OI candidate genes, including COL1A1, COL1A2, IFITM5, SERPINF1, CRTAP, LEPRE1, PPIB, SERPINH1, FKBP10, BMP1, SP7, TMEM38B, WNT1, CREB3L1, and PLOD2. This panel also included 19 candidate genes that are associated with bone fragility or Wnt signaling, including LRP5, LRP6, TNFRSF11A, TNFRSF11B, CBS, MTHFR, MTR, WNT4, CTNB1, WNT16, DKK1, LRP4, SOST, WLS, SFRP4, WNT5B, AXIN1, RSPO3, and TNFSF11. Custom primers were designed using the Ion AmpliSeq™ Designer (Thermo Fisher Scientific, Waltham, MA, USA) to generate 643 amplicons covering 95.84% of the whole exons of the 34 genes. DNA from blood was amplified to enrich the target exons in the 34 genes in a multiplex polymerase chain reaction (PCR) using the Ion AmpliSeq Library Kit 2.0. The library was prepared by ligating the PCR amplicons into adapters with the addition of barcodes. The library concentration and amplicon sizes were determined using an Agilent BioAnalyzer kit (Agilent Technologies, Santa Clara, CA, USA). Multiplexed barcoded libraries were enriched by clonal amplification using emulsion PCR and were loaded on an Ion 318 Chip (Thermo Fisher Scientific, Waltham, MA, USA). Massively parallel sequencing was carried out on the Ion PGM sequencer (Thermo Fisher Scientific, Waltham, MA, USA). Data analysis and variant calling were performed using Torrent Suite and Ion Reporter software (Thermo Fisher Scientific, Waltham, MA, USA). All of the candidate mutations detected by NGS as well as the uncovered and unread regions with this panel in candidate genes were examined by Sanger sequencing. The amplicons generated with the designed primers were Sanger sequenced using a 3730 DNA analyzer (Applied Biosystems, Foster City, CA, USA).
Statistical analysis
Differences between two groups were analyzed using Student’s t test. Differences among more than three groups were evaluated using one-way analysis of variance (ANOVA), followed by multiple comparisons using the Tukey-Kramer method. The difference in familial history and the proportion of types of mutations in COL1A1 and COL1A2 according to the Sillence classification were analyzed using χ2 test or Fisher’s exact test. Correlations between annual fracture rate, SDS of height, and L1–L4 BMD as well as the number of exons where the mutations were located were examined by simple linear regression analysis. Outliers detected in a robust regression were excluded. All statistical analyses were conducted using JMP Pro software version 13.0.0 (SAS Institute Inc., Cary, NC, USA). p < 0.05 was considered significant.
Results
Characteristics of the study population
This study included 53 individuals with a clinical diagnosis of OI from unrelated non-consanguineous families. A total of 28 male and 25 female patients were included. The patients’ ages ranged from 1 month to 41.7 years at the time of assessment. Familial histories were positive in 29 patients. Patient classification was as follows: type I (N = 34), type III (N = 9), and type IV (N = 10), according to the Sillence classification. Familial histories were positive in 68.8%, 28.6%, and 55.6% of patients with type I, III, and IV OI, respectively, and there was no significant difference according to χ2 test or Fisher’s exact test. The patients’ ages were 10.8 ± 12.4, 1.17 ± 1.62, and 3.61 ± 4.19 years for types I, III, and IV, respectively. Although it was not significant, patients with type III at the time of assessment tended to be younger than those with type I according to ANOVA (p = 0.060). The SDS of height prior to BP treatment was low (− 1.92 ± 2.20) for all study patients and was significantly lower in type III patients (− 5.53 ± 1.57) than in type I (− 0.96 ± 1.41, p < 0.0001) and type IV (− 2.43 ± 1.78, p < 0.001) patients. Furthermore, type IV patients were significantly shorter than type I patients (p < 0.05). Although the L1–L4 BMD SDS was low (− 2.64 ± 1.55) in all study patients, there was no significant difference among the Sillence classification types. The annual fracture rate was 2.78 ± 6.00 among all study patients, occurring significantly more often in type III patients (10.78 ± 10.37) than in type I (0.54 ± 0.36, p < 0.0001) and type IV (2.88 ± 5.10, p < 0.01) patients. Blue sclerae and dentinogenesis imperfecta were positive in 82.4% (42/51) and 30.0% (15/50) of patients, respectively (Table 1).
Table 1.
Characteristics of the study population
| Sillence classification | ||||
|---|---|---|---|---|
| I | III | IV | Total | |
| Sex | ||||
| Male/female | 19/15 | 4/5 | 5/5 | 28/25 |
| Family history | ||||
| Positive/total | 22/32 (68.8%) | 2/7 (28.6%) | 5/9 (55.6%) | 29/48 (60.4%) |
| Age | ||||
| Years, mean ± SD |
10.8 ± 12.4 (N = 34) |
1.17 ± 1.62a (N = 8) |
3.61 ± 4.19 (N = 9) |
8.00 ± 11.0 (N = 51) |
| Height SDS | ||||
| Mean ± SD |
− 0.96 ± 1.41 (N = 31) |
− 5.53 ± 1.57⁎⁎ (N = 7) |
− 2.43 ± 1.78⁎ †† (N = 9) |
− 1.92 ± 2.20 (N = 47) |
| L1–L4 BMD SDS | ||||
| Mean ± SD |
− 2.33 ± 1.47 (N = 28) |
− 3.51 ± 2.06 (N = 3) |
− 3.42 ± 1.43 (N = 8) |
− 2.64 ± 1.55 (N = 39) |
| Annual fracture rate | ||||
| /Year, mean ± SD |
0.54 ± 0.36 (N = 29) |
10.78 ± 10.4⁎⁎ (N = 8) |
2.88 ± 5.10† (N = 9) |
2.78 ± 6.00 (N = 46) |
| Blue sclerae | ||||
| Positive/total | 30/34 (88.2%) | 8/8 (100%) | 4/9 (44.4%) | 42/51 (82.4%) |
| DI | ||||
| Positive/total | 4/33 (12.1%) | 7/8 (87.5%) | 4/9 (44.4%) | 15/50 (30.0%) |
DI dentinogenesis imperfecta, SD standard deviation, SDS standard deviation score
ap = 0.060 vs. type I, ⁎p < 0.05 vs. type I, ⁎⁎p < 0.0001 vs. type I, †p < 0.01 vs. type III, ††p < 0.001 vs. type III
Genetic analysis
Targeted NGS was performed as the initial diagnostic methodology in 52 patients. In 1 patient, whole-exome NGS was used as the initial diagnostic methodology. Pathogenic or likely pathogenic variants were found in 41 individuals. After the initial diagnostic methodology, whole-exome NGS was performed in 6 individuals in whom the target NGS had not detected disease-causing variants, and it revealed pathogenic mutations in 2 individuals. Overall, pathogenic mutations were detected in 43 out of 53 individuals, all of which were heterozygous. Among the 43 individuals, 40 variants in total were identified, including 28 substitutions (70%), 7 deletions (17%), and 5 duplications (13%). No insertion nor insertion/deletion variants were found in this study. These variants resulted in 12 missense (30%), 7 nonsense (17%), 10 splice site (25%), 7 frameshift (17%), 3 in-frame insertion (8%), and 1 new start codon (3%) mutations. There were no in-frame deletion mutations. Fifteen mutations were novel (10 and 5 in COL1A1 and COL1A2, respectively, comprising 2 missense, 2 nonsense, 5 splice site, 4 frameshift, and 2 in-flame insertion mutations; shown in Table 2), and 25 were known mutations including 7 variants that occurred at the previously reported nucleotide or amino acid but associated with different substitutions (Fig. 1) (OI database: https://www.le.ac.uk/ge/collagen/). Three mutations were shared among unrelated families (COL1A1: c.2299G>A (p.Gly767Ser), c.2461G>A (p.Gly821Ser), and c.2829+1G>A).
Table 2.
Novel variants detected by NGS
| Gene | Nucleotide change | Amino acid change | Mutation type | Sillence type |
|---|---|---|---|---|
| COL1A1 | c.387delT | p.Pro129Profs*30 | Frameshift | I |
| COL1A1 | c.495T>G | p.Tyr165* | Nonsense | I |
| COL1A1 | c.1615-2A>T | Splice site | III | |
| COL1A1 | c.2347G>T | p.Glu783* | Nonsense | I |
| COL1A1 | c.2451+2T>G | Splice site | I | |
| COL1A1 | c.2574delT | p.Pro859Leufs*249 | Frameshift | I |
| COL1A1 | c.2716_2717dupCG | p.Gly906Alafs*40 | Frameshift | I |
| COL1A1 | c.3112delG | p.Glu1038fs*70 | Frameshift | I |
| COL1A1 | c.3262-2A>G | Splice site | I | |
| COL1A1 | c.3904C>T | p.Pro1302Ser | Missense | I |
| COL1A2 | c.395G>A | p.Arg132His | Missense | I |
| COL1A2 | c.1252-7delT | Splice site | I | |
| COL1A2 | c.1503+12_14delCAC | Splice site | IV | |
| COL1A2 | c.2419_2427dup | p.Pro807_Pro809dup | In-frame insertion | IV |
| COL1A2 | c.2952_2960dup | p.Gly985_Val987dup | In-frame insertion | I |
Fig. 1.

Mutation spectrum. a Frequency of disease-causing variants according to variant type. b Frequency of disease-causing variants according to mutation effect. c Frequency of novel disease-causing variants according to mutation effect. “Reported” includes variants that are completely matched with the reported one. “Same site variant” represents variants in which other base changes at the same site have been reported. d Frequency of disease-causing variants according to affected genes. e Frequency of OI according to the Sillence classification for each mutation type on COL1A1 and COL1A2. N.D., not detected; GS, glycine substitution group; TG, truncating group; NTG, non-truncating group; OS, other missense group
Among the 43 individuals in whom mutations were detected, mutations were found on the COL1A1 (n = 30, 69.8%), COL1A2 (n = 12, 27.9%), and IFITM5 (n = 1, 2.3%) genes. No pathogenic mutations were found in 10 individuals (Fig. 1). A mutation on IFITM5 (c.-14C>T), which has been reported to create a new upstream-of-start codon, was reported in one patient [14–16].
Genotype-phenotype correlation in COL1A1 and COL1A2 mutations
Among 30 individuals harboring the COL1A1 mutation, 8 glycine substitution (27%), 7 nonsense (23%), 8 splice site (27%), 6 frameshift (20%), and 1 other type of substitution (3%) mutations were detected. No in-frame insertions nor deletions were found on COL1A1. Of the COL1A2 mutations, 5 were glycine substitutions (42%), 3 were splice site mutations (25%), 3 were in-frame insertions (25%), and 1 was another substitution (8%) (Fig. 2). No nonsense, frameshift, or in-frame deletion mutations were found on COL1A2 [17] (Table 3).
Fig. 2.

Gene maps of aCOL1A1 and bCOL1A2 exons, with identified mutations and corresponding protein product domains. The numbered box and line between the boxes represent the corresponding exon and intron of the gene. Black, red, and green indicate patients with Sillence types I, III, and IV, respectively
Table 3.
Type of COL1A1 and COL1A2 mutations
|
COL1A1 N = 30 (69.8%) |
COL1A2 N = 12 (27.9%) |
|
|---|---|---|
| Glycine substitution | 8 (27%) | 5 (42%) |
| Nonsense | 7 (23%) | 0 (0%) |
| Splice site | 8 (27%) | 3 (25%) |
| Frameshift | 6 (20%) | 0 (0%) |
| In-frame insertion | 0 (0%) | 3 (25%) |
| Other substitution | 1 (3%) | 1 (8%) |
To evaluate the contributions of mutations to the phenotype, we distinguished glycine substitutions (GS) from other amino acid substitutions (OS), as glycine substitutions result in severe phenotypes [5]. We also classified nonsense and frameshift mutations as a truncating group (TG), which has been reported to result in haploinsufficiency and mild phenotypes [5, 18]. We classified splice site and in-frame insertions as a non-truncating group (NTG), which are known to produce type I collagen with an incomplete structure [5, 18].
With respect to GS, 2, 7, and 4 patients had Sillence types I, III, and IV, respectively. In the TG, no patient had type III nor IV, but 13 patients had type I. In the NTG, 10, 2, and 2 patients had types I, III, and IV, respectively. Both patients in OS were diagnosed with type I OI. No mutations were detected in 7 and 3 patients with Sillence types I and IV, respectively, and no patient was classified as having type III in this group. The proportion of Sillence type I patients in the entire group was significantly lower in GS than in TG or NTG according to χ2 test or Fisher’s exact test (Fig. 1).
The annual fracture rates prior to the initiation of BP treatment were 2.83 ± 2.18, 0.47 ± 0.29, 0.71 ± 0.38, and 0.20 ± 0.28 in GS, TG, NTG, and OS, respectively. GS resulted in a significantly greater fracture rate than TG and NTG. We evaluated this in both the COL1A1 and COL1A2 mutation groups. Although there were no significant differences in the COL1A2 mutation group (GS 3.06 ± 2.95, NTG 0.65 ± 0.26, and OS 0.39 ± 0), GS caused a significantly greater fracture rate than that the TG, NTG, and OS within the COL1A1 mutation group (GS 2.59 ± 1.38, TG 0.47 ± 0.29, NTG 0.76 ± 0.48, OS 0 ± 0). There were no differences between COL1A1 and COL1A2 mutations in all mutations (COL1A1 1.01 ± 1.12, COL1A2 1.72 ± 2.27), in GS (COL1A1 2.59 ± 1.38, COL1A2 3.06 ± 2.95), nor in NTG (COL1A1 0.76 ± 0.48, COL1A2 0.65 ± 0.26) (Supplemental Fig. 1).
GS resulted in significantly lower SDS of height than TG and NTG before BP treatment (− 4.07 ± 2.19, − 0.93 ± 1.24, − 1.88 ± 2.27, and − 1.73 ± 0.47 in GS, TG, NTG, and OS, respectively). Although no differences were observed in the COL1A2 mutation group (GS − 3.30 ± 3.17, NTG − 2.90 ± 2.56, OS − 2.06 ± 0), GS on COL1A1 resulted in a greater severity of short stature than TG and NTG (GS − 4.51 ± 1.53, TG − 0.93 ± 1.24, NTG − 1.00 ± 1.71, OS − 1.40 ± 0). No significant differences were observed between COL1A1 and COL1A2 in all mutations (COL1A1 − 1.89 ± 2.09, COL1A2 − 2.97 ± 2.53), in GS (COL1A1 − 4.51 ± 1.53, COL1A2 − 3.30 ± 3.17), or in NTG (COL1A1 − 1.00 ± 1.71, COL1A2 − 2.90 ± 2.56) (Supplemental Fig. 2).
Although the L1–L4 BMD SDS prior to the initiation of BP treatment was low in all types of mutations in COL1A1 and COL1A2 (GS − 3.12 ± 1.39, TG − 2.32 ± 1.01, NTG − 3.13 ± 1.75, OS − 1.38 ± 0), there was no difference among these mutations. No significant differences were observed even when they were analyzed on each gene (COL1A1: GS − 2.71 ± 1.26, TG − 2.32 ± 1.01, NTG − 2.81 ± 2.07; COL1A2: GS − 3.53 ± 1.65, NTG − 3.64 ± 1.05, OS − 1.38 ± 0). No gene effect was observed in L1–L4 BMD SDS in all mutations (COL1A1 − 2.56 ± 1.47, COL1A2 − 3.35 ± 1.34), in GS (COL1A1 − 2.71 ± 1.26, COL1A2 − 3.53 ± 1.65), or in NTG (COL1A1 − 2.81 ± 2.07, COL1A2 − 3.64 ± 1.05). The L1–L4 TBS was also evaluated before the initiation of BP treatment, and it was revealed that GS resulted in a significantly lower TBS than NTG (GS 1.11 ± 0.05, TG 1.22 ± 0.10, NTG 1.31 ± 0.09, and OS 1.29 ± 0.10) (Supplemental Fig. 3).
Among 42 individuals harboring mutations in COL1A1 or COL1A2, 5, 35, and 2 mutations were located in the N-terminal pro-peptide, triple helix portion, and C-terminal pro-peptide, respectively. No mutations were located in the signal peptide, N-terminal telopeptide, or C-terminal telopeptide. Among 30 individuals with mutations in COL1A1, 5, 23, and 2 mutations were located in the N-terminal pro-peptide, triple helix portion, and C-terminal pro-peptide, respectively. All of the mutations in COL1A2 were located in the triple helix portion (N = 12) (Supplemental Table 1).
The annual fracture rates were 0.57 ± 0.38 (N = 4), 1.39 ± 1.69 (N = 28), and 0 ± 0 (N = 1) in patients with mutations located in the N-terminal pro-peptide, triple helix portion, and C-terminal pro-peptide, respectively. No significant differences in the location of mutations were observed with respect to the annual fracture rate when analyzing COL1A1 and COL1A2 together or COL1A1 alone. There were no significant differences in gene effects with mutations at the triple helix (COL1A1 1.18 ± 1.21, N = 17; COL1A2 1.72 ± 2.27, N = 11). There was no correlation between exon number, mutation location, and fracture rate (Supplemental Fig. 4).
The SDS of height was − 0.38 ± 1.30 (N = 4), − 2.50 ± 2.30 (N = 32), and − 1.18 ± 0.32 (N = 2) among patients with mutations located in the N-terminal pro-peptide, triple helix portion, and C-terminal pro-peptide, respectively. There were no significant differences in the location of mutations analyzed on COL1A1 and COL1A2 together or on COL1A1 alone, nor was there any gene effect for mutations in the triple helix (COL1A1 − 2.25 ± 2.19, N = 21; COL1A2 − 2.97 ± 2.53, N = 11). There was no correlation between exon number and SDS of height (Supplemental Fig. 5).
The L1–L4 BMD SDS was − 1.33 ± 0.86 for the N-terminal pro-peptide (N = 3) and − 2.96 ± 1.43 for the triple helix (N = 27). No significant differences were observed between these groups. There were significant correlations between L1–L4 BMD SDS and exon numbers analyzed on COL1A1 and COL1A2 together (r2 = 0.176, p = 0.02) or on COL1A2 alone (r2 = 0.82, p = 0.0008) (Supplemental Fig. 6).
Discussion
The results of this study revealed the mutation spectrum in Japanese patients with OI. The targeted NGS of genomic DNA identified disease-causing mutations in 43 out of 53 individuals (81%). We identified 40 variants including 15 novel variants. Although some papers have analyzed COL1A1 and COL1A2 mutations in Japanese patients with OI [9, 10], to the best of our knowledge, our study was the largest and first to comprehensively analyze not only COL1A1 and COL1A2 but also other genes associated with OI in Japanese patients.
Consistent with some previous reports, COL1A1 and COL1A2 mutations were dominant in our study (97.7%) [19–21]. We detected only 1 mutation on IFITM5. Although similar to previous reports indicating that the c.-14C>T mutation in IFITM5 was the major variant other than the COL1A1 and COL1A2 mutations, the prevalence of this in our population was less than that reported previously [19, 21].
No biallelic mutations associated with recessive OI were observed in our study. This is totally different from the findings in a report by Essawi et al., which showed that 61% of individuals in their study were affected with autosomal recessive OI in a Palestinian population [22]. The prevalence of recessive OI may depend on geographical area, as recessive disorders can be more frequent where consanguinity is common.
As reported previously [8, 23, 24], in our results, GS on COL1A1 can be clinically distinguishable from other types of mutations since the annual fracture rates and short-stature severity were greater in patients with GS. Although the cause of the short stature derived from the COL1A1 mutation is not completely understood [25], it may be secondary to vertebral and long-bone fractures. Alternatively, the mutations may have a direct effect on growth plate activity, as OI is related to disturbances in bone remodeling and regulatory proteins [26, 27]. Scheiber et al. reported that GS in COL1A2 induced endoplasmic reticulum stress in the growth plate hypertrophic chondrocytes and contributed to growth deficiency in a mouse model [28]. These data suggest that a genetic diagnosis is informative and beneficial in patients with OI to appropriately manage their healthcare.
The initial diagnosis of OI is largely based on clinical and radiographic findings that include BMD [24]. In our study, the L1–L4 BMDs among our patients with OI were lower than those of age-matched healthy children and adolescents in previous reports [29]. Although BMD provides useful information for diagnosing OI, it is difficult to assess bone strength using BMD [30], as bone strength is derived from not only BMD but also bone quality, which includes the characteristics of the bone microarchitecture [31, 32]. The TBS is based on gray-level measurements derived from DXA images. TBS is known to strongly correlate with 3-dimensional microstructure parameters [33] and predict vertebral and major osteoporotic fractures in patients with osteoporosis, independent of the BMD [34, 35]. Kocijan et al. reported that adult patients with OI types III and IV had significantly lower TBS values than those with OI type I [36]. Rehberg et al. reported that TBS increased significantly after treatment with denosumab in children with OI [37]. Our data suggest that BMD cannot predict bone fragility in patients with OI. However, TBS may be a useful tool for predicting bone strength and fracture risk in patients with OI. More studies are required to clarify this.
Rauch et al. reported that the position of glycine mutations within the α1 and α2 triple helical domains had no obvious relationship with fractures or deformities at birth [38]. In our study, consistent with previous reports, no relationship was observed between mutation position, annual fracture rate, and SDS of height. Interestingly, among patients with α2 triple helical domain mutations, a significant negative relationship was observed between the position of mutation and the L1–L4 BMD, similar to findings reported by Rauch et al. [39]. However, it is still difficult to anticipate the severity of bone fragility on the basis of the position of mutations in COL1A1 and COL1A2.
There are some limitations to this study. First, we could not identify any pathogenic mutations in 10 individuals. They may still have a genetic cause for their bone fragility, especially in cases of positive familial histories. As we did not perform the Multiplex Ligation-dependent Probe Amplification method in this study, it is possible that we failed to identify some deletion or duplication mutations. Second, it is possible that we failed to identify intronic mutations located far from a splice site that could have led to splicing defects [40]. Recently, some variants in several genes including SPARC and MBTPS2 have been identified as disease-causing genes with respect to OI [7, 41]. We failed to identify pathogenic variants in these genes, as we have not included them. In this study, whole-exome NGS was performed in 6 individuals in whom the target NGS had not detected disease-causing variants, and it revealed pathogenic mutations in 2 individuals. They harbored c.441dupC and c.3262-2A>G in COL1A1. Targeted NGS could fail to detect single-nucleotide duplication or deletion such as c.441dupC mutation, which could be detected by whole-exome NGS. To resolve these issues, we need to update our panel or perform whole-exome sequencing in order to improve accuracy. Finally, few reports have described the Japanese OI mutation spectrum; hence, it is possible that there are unknown gene mutations that can result in OI pathology.
Conclusions
We identified disease-causing mutations in 81% of our Japanese patients with OI. Most mutations were located on COL1A1 and COL1A2 in these patients. No significant difference in L1–L4 BMD SDS was observed among mutations; nonetheless, GS resulted in a significantly greater fracture rate than other mutations. These findings suggest that bone qualities in patients harboring GS were worse than those with other mutations. Considering that TBS was lower in patients with GS, it may be important to measure TBS in order to evaluate their bone qualities. As there is such genotype-phenotype correlation with respect to the severity of bone fragility, comprehensive genetic diagnosis can be useful in treatment and management decision-making for patients with OI.
Electronic supplementary material
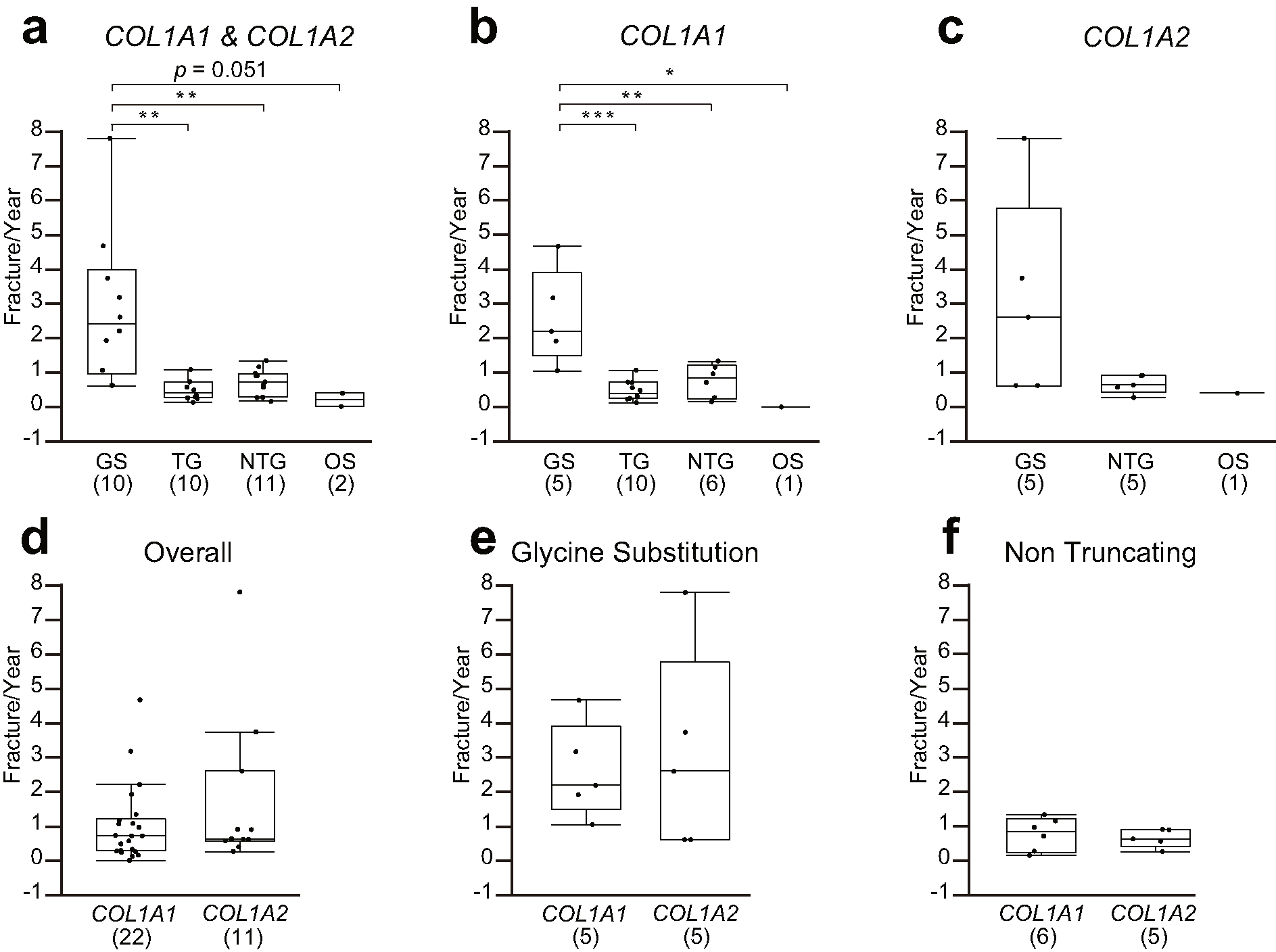{kind=link}
Comparison of annual fracture rate. (a) Comparison of fracture rate among the 4 groups with COL1A1 and COL1A2 mutations. (b) Comparison of fracture rate among the 4 groups with COL1A1 mutations only. (c) Comparison of fracture rate among the 3 groups with COL1A2 mutations only. (d) Comparison of fracture rate between COL1A1 and COL1A2 mutations with all groups of mutations. (e) Comparison of fracture rate between COL1A1 and COL1A2 mutations with GS. (f) Comparison of fracture rate between COL1A1 and COL1A2 mutations with non-truncating mutations. The numbers in parentheses represent the sample numbers. GS: glycine substitution group; TG: truncating group; NTG: non-truncating group; OS: other missense group, ⁎: p < 0.05, ⁎⁎: p < 0.01, ⁎⁎⁎: p < 0.001 (PNG 33 kb)
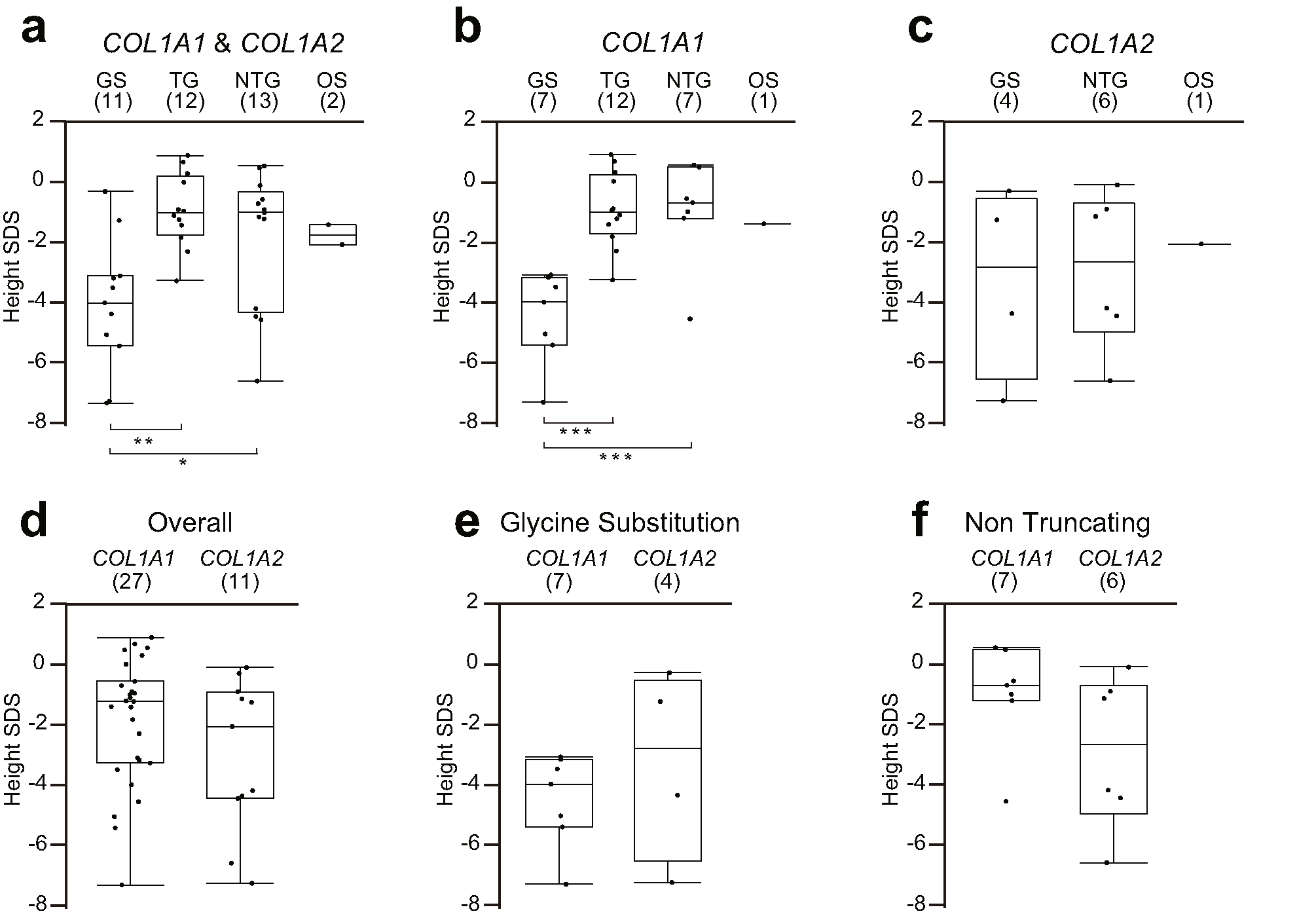{kind=link}
Comparison of height. (a) Comparison of SDS of height among the 4 groups with COL1A1 and COL1A2 mutations. (b) Comparison of SDS of height among the 4 groups with COL1A1 mutations only. (c) Comparison of SDS of height among the 3 groups with COL1A2 mutations only. (d) Comparison of SDS of height between COL1A1 and COL1A2 mutations with all groups of mutations. (e) Comparison of SDS of height between COL1A1 and COL1A2 mutations with glycine substitutions. (f) Comparison of SDS of height between COL1A1 and COL1A2 mutations with non-truncating mutations. The numbers in parentheses represent the sample numbers. GS: glycine substitution group; TG: truncating group; NTG: non-truncating group; OS: other missense group; SDS: standard deviation score, ⁎: p < 0.05, ⁎⁎: p < 0.01, ⁎⁎⁎: p < 0.001 (PNG 32 kb)
{kind=link}
Comparison of L1-L4 BMD and TBS. (a) Comparison of SDS of L1-L4 BMD among the 4 groups with COL1A1 and COL1A2 mutations. (b) Comparison of SDS of L1-L4 BMD among the 4 groups with COL1A1 mutations only. (c) Comparison of SDS of L1-L4 BMD among the 3 groups with COL1A2 mutation only. (d) Comparison of SDS of L1-L4 BMD between COL1A1 and COL1A2 mutations with all groups of mutations. (e) Comparison of SDS of L1-L4 BMD between COL1A1 and COL1A2 mutations with glycine substitutions. (f) Comparison of SDS of L1-L4 BMD between COL1A1 and COL1A2 mutations with non-truncating mutations. The numbers in parentheses represent the sample numbers, GS: glycine substitution group; TG: truncating group; NTG: non-truncating group; OS: other missense group; SDS: standard deviation score. ⁎: p < 0.05 (PNG 40 kb)
{kind=link}
Positional effect of mutations on fracture rate. (a) Comparison of fracture rates among N-terminal pro-peptide, triple helix, and C-terminal pro-peptide mutations in COL1A1 and COL1A2. (b) Comparison of fracture rates among N-terminal pro-peptide, triple helix, and C-terminal pro-peptide mutations in COL1A1 only. (c) Comparison of fracture rates between COL1A1 and COL1A2 mutations located on the triple helix. (d) Relationship between fracture rate and exon number for mutations located in COL1A1 and COL1A2. (e) Relationship between fracture rate and exon number for mutations located in COL1A1 only. (f) Relationship between fracture rate and exon number for mutations located in COL1A2 only. The numbers in parentheses represent the sample numbers. N: N-terminal pro-peptide; Helix: triple helix; C: C-terminal pro-peptide. No significant differences nor correlations were observed in these analyses (PNG 31 kb)
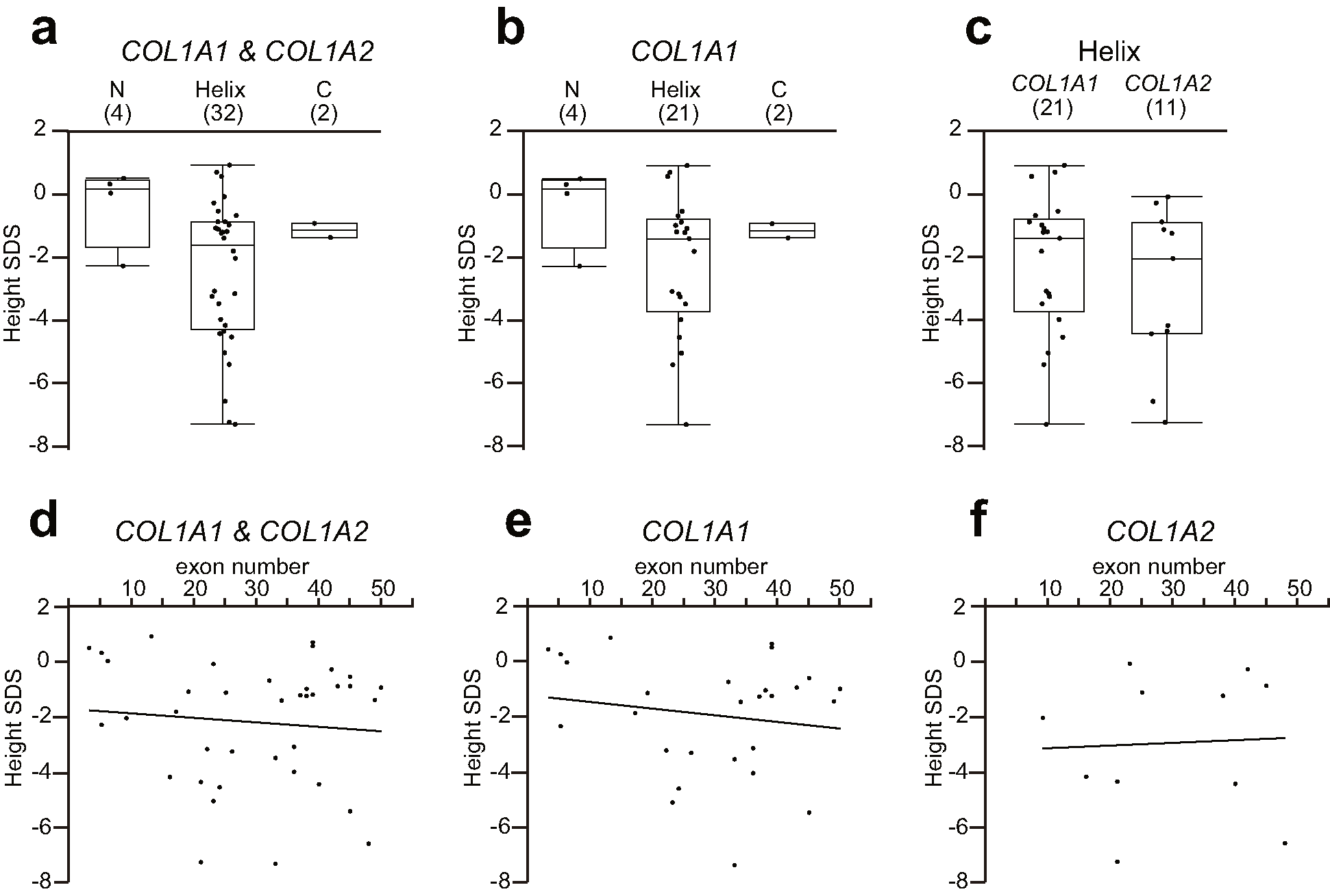{kind=link}
Positional effect of mutations on height. (a) Comparison of SDS of height among N-terminal pro-peptide, triple helix, and C-terminal pro-peptide mutations in COL1A1 and COL1A2. (b) Comparison of SDS of height among N-terminal pro-peptide, triple helix, and C-terminal pro-peptide mutation in COL1A1 only. (c) Comparison of SDS of height between COL1A1 and COL1A2 mutations located in the triple helix. (d) Relationship between SDS of height and exon number for mutations located in COL1A1 and COL1A2. (e) Relationship between SDS of height and exon number for mutations located in COL1A1 only. (f) Relationship between SDS of height and exon number for mutations located in COL1A2 only. The numbers in parentheses represent the sample numbers. N: N terminal pro-peptide; Helix: triple helix; C: C terminal pro-peptide. No significant differences nor correlations were observed in these analyses (PNG 30 kb)
{kind=link}
Positional effect of mutations on L1-L4 BMD. (a) Comparison of SDS of L1-L4 BMD among N-terminal pro-peptide, triple helix, and C-terminal pro-peptide mutations in COL1A1 and COL1A2. (b) Comparison of SDS of L1-L4 BMD among N-terminal pro-peptide, triple helix, and C-terminal pro-peptide mutations in COL1A1 only. (c) Comparison of SDS of L1-L4 BMD between COL1A1 and COL1A2 mutations located in the triple helix. (d) Relationship between SDS of L1-L4 BMD and exon number for mutations located in COL1A1 and COL1A2. (e) Relationship between SDS of L1-L4 BMD and exon number for mutations located in COL1A1 only. (f) Relationship between SDS of L1-L4 BMD and exon number for mutations located in COL1A2 only. A significant correlation was observed between L1-L4 BMD and location of mutations in COL1A2 (r2= 0.74, p = 0.003). The numbers in parentheses represent the sample numbers. N: N-terminal pro-peptide; Helix: triple helix; C: C-terminal pro-peptide. No significant differences nor correlations were observed in (a-e) (PNG 33 kb)
(DOCX 12 kb)
Acknowledgments
We wish to thank the patients and their families for participating in this study.
Funding information
This study was financially supported by grants from Japan Agency for Medical Research and Development, titled “Development and application of innovative drug-screening technology using patient derived iPS cells for intractable bone and cartilage disease,” “Creation of a network for skeletal dysplasia research and care to develop clinical guidelines,” and “Initiative on Rare and Undiagnosed Disease.”
Compliance with ethical standards
Ethical approval
All procedures were performed in accordance with the ethical standards of the institutional research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards.
Conflicts of interest
None.
Footnotes
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Change history
4/3/2020
The original article has been corrected.
References
- 1.Marini JC (2018) Osteogenesis imperfecta. In: Bilezikian JP (ed) Primer on the metabolic bone diseases and disorders of mineral metabolism. Wiley-Blackwell, pp 871–877
- 2.Sillence DO, Senn A, Danks DM. Genetic heterogeneity in osteogenesis imperfecta. J Med Genet. 1979;16:101–116. doi: 10.1136/jmg.16.2.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bonafe L, Cormier-Daire V, Hall C, Lachman R, Mortier G, Mundlos S, Nishimura G, Sangiorgi L, Savarirayan R, Sillence D, Spranger J, Superti-Furga A, Warman M, Unger S. Nosology and classification of genetic skeletal disorders: 2015 revision. Am J Med Genet A. 2015;167A:2869–2892. doi: 10.1002/ajmg.a.37365. [DOI] [PubMed] [Google Scholar]
- 4.Willing MC, Pruchno CJ, Byers PH. Molecular heterogeneity in osteogenesis imperfecta type I. Am J Med Genet. 1993;45:223–227. doi: 10.1002/ajmg.1320450214. [DOI] [PubMed] [Google Scholar]
- 5.Marini JC, Forlino A, Cabral WA, Barnes AM, San Antonio JD, Milgrom S, Hyland JC, Korkko J, Prockop DJ, De Paepe A, Coucke P, Symoens S, Glorieux FH, Roughley PJ, Lund AM, Kuurila-Svahn K, Hartikka H, Cohn DH, Krakow D, Mottes M, Schwarze U, Chen D, Yang K, Kuslich C, Troendle J, Dalgleish R, Byers PH. Consortium for osteogenesis imperfecta mutations in the helical domain of type I collagen: regions rich in lethal mutations align with collagen binding sites for integrins and proteoglycans. Hum Mutat. 2007;28:209–221. doi: 10.1002/humu.20429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shaheen R, Alazami AM, Alshammari MJ, Faqeih E, Alhashmi N, Mousa N, Alsinani A, Ansari S, Alzahrani F, Al-Owain M, Alzayed ZS, Alkuraya FS. Study of autosomal recessive osteogenesis imperfecta in Arabia reveals a novel locus defined by TMEM38B mutation. J Med Genet. 2012;49:630–635. doi: 10.1136/jmedgenet-2012-101142. [DOI] [PubMed] [Google Scholar]
- 7.Mendoza-Londono R, Fahiminiya S, Majewski J, Care4Rare Canada C. Tetreault M, Nadaf J, Kannu P, Sochett E, Howard A, Stimec J, Dupuis L, Roschger P, Klaushofer K, Palomo T, Ouellet J, Al-Jallad H, Mort JS, Moffatt P, Boudko S, Bachinger HP, Rauch F. Recessive osteogenesis imperfecta caused by missense mutations in SPARC. Am J Hum Genet. 2015;96:979–985. doi: 10.1016/j.ajhg.2015.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Willing MC, Deschenes SP, Slayton RL, Roberts EJ. Premature chain termination is a unifying mechanism for COL1A1 null alleles in osteogenesis imperfecta type I cell strains. Am J Hum Genet. 1996;59:799–809. [PMC free article] [PubMed] [Google Scholar]
- 9.Kataoka K, Ogura E, Hasegawa K, Inoue M, Seino Y, Morishima T, Tanaka H. Mutations in type I collagen genes in Japanese osteogenesis imperfecta patients. Pediatr Int. 2007;49:564–569. doi: 10.1111/j.1442-200X.2007.02422.x. [DOI] [PubMed] [Google Scholar]
- 10.Kanno J, Saito-Hakoda A, Kure S, Fujiwara I. Responsiveness to pamidronate treatment is not related to the genotype of type I collagen in patients with osteogenesis imperfecta. J Bone Miner Metab. 2018;36:344–351. doi: 10.1007/s00774-017-0840-9. [DOI] [PubMed] [Google Scholar]
- 11.Kalkwarf HJ, Zemel BS, Gilsanz V, Lappe JM, Horlick M, Oberfield S, Mahboubi S, Fan B, Frederick MM, Winer K, Shepherd JA. The bone mineral density in childhood study: bone mineral content and density according to age, sex, and race. J Clin Endocrinol Metab. 2007;92:2087–2099. doi: 10.1210/jc.2006-2553. [DOI] [PubMed] [Google Scholar]
- 12.Zemel BS, Kalkwarf HJ, Gilsanz V, Lappe JM, Oberfield S, Shepherd JA, Frederick MM, Huang X, Lu M, Mahboubi S. Revised reference curves for bone mineral content and areal bone mineral density according to age and sex for black and non-black children: results of the bone mineral density in childhood study. J Clin Endocrinol Metab. 2011;96:3160–3169. doi: 10.1210/jc.2011-1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kalkwarf HJ, Zemel BS, Yolton K, Heubi JE. Bone mineral content and density of the lumbar spine of infants and toddlers: influence of age, sex, race, growth, and human milk feeding. J Bone Miner Res. 2013;28:206–212. doi: 10.1002/jbmr.1730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Semler O, Garbes L, Keupp K, Swan D, Zimmermann K, Becker J, Iden S, Wirth B, Eysel P, Koerber F, Schoenau E, Bohlander SK, Wollnik B, Netzer C. A mutation in the 5′-UTR of IFITM5 creates an in-frame start codon and causes autosomal-dominant osteogenesis imperfecta type V with hyperplastic callus. Am J Hum Genet. 2012;91:349–357. doi: 10.1016/j.ajhg.2012.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cho TJ, Lee KE, Lee SK, Song SJ, Kim KJ, Jeon D, Lee G, Kim HN, Lee HR, Eom HH, Lee ZH, Kim OH, Park WY, Park SS, Ikegawa S, Yoo WJ, Choi IH, Kim JW. A single recurrent mutation in the 5′-UTR of IFITM5 causes osteogenesis imperfecta type V. Am J Hum Genet. 2012;91:343–348. doi: 10.1016/j.ajhg.2012.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rauch F, Moffatt P, Cheung M, Roughley P, Lalic L, Lund AM, Ramirez N, Fahiminiya S, Majewski J, Glorieux FH. Osteogenesis imperfecta type V: marked phenotypic variability despite the presence of the IFITM5 c.-14C>T mutation in all patients. J Med Genet. 2013;50:21–24. doi: 10.1136/jmedgenet-2012-101307. [DOI] [PubMed] [Google Scholar]
- 17.de Wet WJ, Pihlajaniemi T, Myers J, Kelly TE, Prockop DJ. Synthesis of a shortened pro-alpha 2(I) chain and decreased synthesis of pro-alpha 2(I) chains in a proband with osteogenesis imperfecta. J Biol Chem. 1983;258:7721–7728. [PubMed] [Google Scholar]
- 18.Forlino A, Marini JC. Osteogenesis imperfecta: prospects for molecular therapeutics. Mol Genet Metab. 2000;71:225–232. doi: 10.1006/mgme.2000.3039. [DOI] [PubMed] [Google Scholar]
- 19.Bardai G, Moffatt P, Glorieux FH, Rauch F. DNA sequence analysis in 598 individuals with a clinical diagnosis of osteogenesis imperfecta: diagnostic yield and mutation spectrum. Osteoporos Int. 2016;27:3607–3613. doi: 10.1007/s00198-016-3709-1. [DOI] [PubMed] [Google Scholar]
- 20.Lindahl K, Astrom E, Rubin CJ, Grigelioniene G, Malmgren B, Ljunggren O, Kindmark A. Genetic epidemiology, prevalence, and genotype-phenotype correlations in the Swedish population with osteogenesis imperfecta. Eur J Hum Genet. 2015;23:1112. doi: 10.1038/ejhg.2015.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu Y, Asan MD, Lv F, Xu X, Wang J, Xia W, Jiang Y, Wang O, Xing X, Yu W, Wang J, Sun J, Song L, Zhu Y, Yang H, Wang J, Li M. Gene mutation spectrum and genotype-phenotype correlation in a cohort of Chinese osteogenesis imperfecta patients revealed by targeted next generation sequencing. Osteoporos Int. 2017;28:2985–2995. doi: 10.1007/s00198-017-4143-8. [DOI] [PubMed] [Google Scholar]
- 22.Essawi O, Symoens S, Fannana M, Darwish M, Farraj M, Willaert A, Essawi T, Callewaert B, De Paepe A, Malfait F, Coucke PJ. Genetic analysis of osteogenesis imperfecta in the Palestinian population: molecular screening of 49 affected families. Mol Genet Genomic Med. 2018;6:15–26. doi: 10.1002/mgg3.331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Forlino A, Cabral WA, Barnes AM, Marini JC. New perspectives on osteogenesis imperfecta. Nat Rev Endocrinol. 2011;7:540–557. doi: 10.1038/nrendo.2011.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Forlino A, Marini JC. Osteogenesis imperfecta. Lancet. 2016;387:1657–1671. doi: 10.1016/S0140-6736(15)00728-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hoyer-Kuhn H, Hobing L, Cassens J, Schoenau E, Semler O. Children with severe osteogenesis imperfecta and short stature present on average with normal IGF-I and IGFBP-3 levels. J Pediatr Endocrinol Metab. 2016;29:813–818. doi: 10.1515/jpem-2015-0385. [DOI] [PubMed] [Google Scholar]
- 26.Rauch F, Travers R, Parfitt AM, Glorieux FH. Static and dynamic bone histomorphometry in children with osteogenesis imperfecta. Bone. 2000;26:581–589. doi: 10.1016/S8756-3282(00)00269-6. [DOI] [PubMed] [Google Scholar]
- 27.Brunetti G, Papadia F, Tummolo A, Fischetto R, Nicastro F, Piacente L, Ventura A, Mori G, Oranger A, Gigante I, Colucci S, Ciccarelli M, Grano M, Cavallo L, Delvecchio M, Faienza MF. Impaired bone remodeling in children with osteogenesis imperfecta treated and untreated with bisphosphonates: the role of DKK1, RANKL, and TNF-alpha. Osteoporos Int. 2016;27:2355–2365. doi: 10.1007/s00198-016-3501-2. [DOI] [PubMed] [Google Scholar]
- 28.Scheiber AL, Guess AJ, Kaito T, Abzug JM, Enomoto-Iwamoto M, Leikin S, Iwamoto M, Otsuru S. Endoplasmic reticulum stress is induced in growth plate hypertrophic chondrocytes in G610C mouse model of osteogenesis imperfecta. Biochem Biophys Res Commun. 2019;509:235–240. doi: 10.1016/j.bbrc.2018.12.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rauch F, Land C, Cornibert S, Schoenau E, Glorieux FH. High and low density in the same bone: a study on children and adolescents with mild osteogenesis imperfecta. Bone. 2005;37:634–641. doi: 10.1016/j.bone.2005.06.007. [DOI] [PubMed] [Google Scholar]
- 30.Reinus WR, McAlister WH, Schranck F, Chines A, Whyte MP. Differing lumbar vertebral mineralization rates in ambulatory pediatric patients with osteogenesis imperfecta. Calcif Tissue Int. 1998;62:17–20. doi: 10.1007/s002239900387. [DOI] [PubMed] [Google Scholar]
- 31.(2000) Osteoporosis prevention, diagnosis, and therapy. NIH Consens Statement 17:1–45 [PubMed]
- 32.Friedman AW. Important determinants of bone strength: beyond bone mineral density. J Clin Rheumatol. 2006;12:70–77. doi: 10.1097/01.rhu.0000208612.33819.8c. [DOI] [PubMed] [Google Scholar]
- 33.Hans D, Barthe N, Boutroy S, Pothuaud L, Winzenrieth R, Krieg MA. Correlations between trabecular bone score, measured using anteroposterior dual-energy X-ray absorptiometry acquisition, and 3-dimensional parameters of bone microarchitecture: an experimental study on human cadaver vertebrae. J Clin Densitom. 2011;14:302–312. doi: 10.1016/j.jocd.2011.05.005. [DOI] [PubMed] [Google Scholar]
- 34.Hans D, Goertzen AL, Krieg MA, Leslie WD. Bone microarchitecture assessed by TBS predicts osteoporotic fractures independent of bone density: the Manitoba study. J Bone Miner Res. 2011;26:2762–2769. doi: 10.1002/jbmr.499. [DOI] [PubMed] [Google Scholar]
- 35.Pothuaud L, Barthe N, Krieg MA, Mehsen N, Carceller P, Hans D. Evaluation of the potential use of trabecular bone score to complement bone mineral density in the diagnosis of osteoporosis: a preliminary spine BMD-matched, case-control study. J Clin Densitom. 2009;12:170–176. doi: 10.1016/j.jocd.2008.11.006. [DOI] [PubMed] [Google Scholar]
- 36.Kocijan R, Muschitz C, Haschka J, Hans D, Nia A, Geroldinger A, Ardelt M, Wakolbinger R, Resch H. Bone structure assessed by HR-pQCT, TBS and DXL in adult patients with different types of osteogenesis imperfecta. Osteoporos Int. 2015;26:2431–2440. doi: 10.1007/s00198-015-3156-4. [DOI] [PubMed] [Google Scholar]
- 37.Rehberg M, Winzenrieth R, Hoyer-Kuhn H, Duran I, Schoenau E, Semler O. TBS as a tool to differentiate the impact of antiresorptives on cortical and trabecular bone in children with osteogenesis imperfecta. J Clin Densitom. 2018;22:229–235. doi: 10.1016/j.jocd.2018.09.001. [DOI] [PubMed] [Google Scholar]
- 38.Rauch F, Lalic L, Roughley P, Glorieux FH. Genotype-phenotype correlations in nonlethal osteogenesis imperfecta caused by mutations in the helical domain of collagen type I. Eur J Hum Genet. 2010;18:642–647. doi: 10.1038/ejhg.2009.242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rauch F, Lalic L, Roughley P, Glorieux FH. Relationship between genotype and skeletal phenotype in children and adolescents with osteogenesis imperfecta. J Bone Miner Res. 2010;25:1367–1374. doi: 10.1359/jbmr.091109. [DOI] [PubMed] [Google Scholar]
- 40.Scotti MM, Swanson MS. RNA mis-splicing in disease. Nat Rev Genet. 2016;17:19–32. doi: 10.1038/nrg.2015.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lindert U, Cabral WA, Ausavarat S, Tongkobpetch S, Ludin K, Barnes AM, Yeetong P, Weis M, Krabichler B, Srichomthong C, Makareeva EN, Janecke AR, Leikin S, Rothlisberger B, Rohrbach M, Kennerknecht I, Eyre DR, Suphapeetiporn K, Giunta C, Marini JC, Shotelersuk V. MBTPS2 mutations cause defective regulated intramembrane proteolysis in X-linked osteogenesis imperfecta. Nat Commun. 2016;7:11920. doi: 10.1038/ncomms11920. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Comparison of annual fracture rate. (a) Comparison of fracture rate among the 4 groups with COL1A1 and COL1A2 mutations. (b) Comparison of fracture rate among the 4 groups with COL1A1 mutations only. (c) Comparison of fracture rate among the 3 groups with COL1A2 mutations only. (d) Comparison of fracture rate between COL1A1 and COL1A2 mutations with all groups of mutations. (e) Comparison of fracture rate between COL1A1 and COL1A2 mutations with GS. (f) Comparison of fracture rate between COL1A1 and COL1A2 mutations with non-truncating mutations. The numbers in parentheses represent the sample numbers. GS: glycine substitution group; TG: truncating group; NTG: non-truncating group; OS: other missense group, ⁎: p < 0.05, ⁎⁎: p < 0.01, ⁎⁎⁎: p < 0.001 (PNG 33 kb)
Comparison of height. (a) Comparison of SDS of height among the 4 groups with COL1A1 and COL1A2 mutations. (b) Comparison of SDS of height among the 4 groups with COL1A1 mutations only. (c) Comparison of SDS of height among the 3 groups with COL1A2 mutations only. (d) Comparison of SDS of height between COL1A1 and COL1A2 mutations with all groups of mutations. (e) Comparison of SDS of height between COL1A1 and COL1A2 mutations with glycine substitutions. (f) Comparison of SDS of height between COL1A1 and COL1A2 mutations with non-truncating mutations. The numbers in parentheses represent the sample numbers. GS: glycine substitution group; TG: truncating group; NTG: non-truncating group; OS: other missense group; SDS: standard deviation score, ⁎: p < 0.05, ⁎⁎: p < 0.01, ⁎⁎⁎: p < 0.001 (PNG 32 kb)
Comparison of L1-L4 BMD and TBS. (a) Comparison of SDS of L1-L4 BMD among the 4 groups with COL1A1 and COL1A2 mutations. (b) Comparison of SDS of L1-L4 BMD among the 4 groups with COL1A1 mutations only. (c) Comparison of SDS of L1-L4 BMD among the 3 groups with COL1A2 mutation only. (d) Comparison of SDS of L1-L4 BMD between COL1A1 and COL1A2 mutations with all groups of mutations. (e) Comparison of SDS of L1-L4 BMD between COL1A1 and COL1A2 mutations with glycine substitutions. (f) Comparison of SDS of L1-L4 BMD between COL1A1 and COL1A2 mutations with non-truncating mutations. The numbers in parentheses represent the sample numbers, GS: glycine substitution group; TG: truncating group; NTG: non-truncating group; OS: other missense group; SDS: standard deviation score. ⁎: p < 0.05 (PNG 40 kb)
Positional effect of mutations on fracture rate. (a) Comparison of fracture rates among N-terminal pro-peptide, triple helix, and C-terminal pro-peptide mutations in COL1A1 and COL1A2. (b) Comparison of fracture rates among N-terminal pro-peptide, triple helix, and C-terminal pro-peptide mutations in COL1A1 only. (c) Comparison of fracture rates between COL1A1 and COL1A2 mutations located on the triple helix. (d) Relationship between fracture rate and exon number for mutations located in COL1A1 and COL1A2. (e) Relationship between fracture rate and exon number for mutations located in COL1A1 only. (f) Relationship between fracture rate and exon number for mutations located in COL1A2 only. The numbers in parentheses represent the sample numbers. N: N-terminal pro-peptide; Helix: triple helix; C: C-terminal pro-peptide. No significant differences nor correlations were observed in these analyses (PNG 31 kb)
Positional effect of mutations on height. (a) Comparison of SDS of height among N-terminal pro-peptide, triple helix, and C-terminal pro-peptide mutations in COL1A1 and COL1A2. (b) Comparison of SDS of height among N-terminal pro-peptide, triple helix, and C-terminal pro-peptide mutation in COL1A1 only. (c) Comparison of SDS of height between COL1A1 and COL1A2 mutations located in the triple helix. (d) Relationship between SDS of height and exon number for mutations located in COL1A1 and COL1A2. (e) Relationship between SDS of height and exon number for mutations located in COL1A1 only. (f) Relationship between SDS of height and exon number for mutations located in COL1A2 only. The numbers in parentheses represent the sample numbers. N: N terminal pro-peptide; Helix: triple helix; C: C terminal pro-peptide. No significant differences nor correlations were observed in these analyses (PNG 30 kb)
Positional effect of mutations on L1-L4 BMD. (a) Comparison of SDS of L1-L4 BMD among N-terminal pro-peptide, triple helix, and C-terminal pro-peptide mutations in COL1A1 and COL1A2. (b) Comparison of SDS of L1-L4 BMD among N-terminal pro-peptide, triple helix, and C-terminal pro-peptide mutations in COL1A1 only. (c) Comparison of SDS of L1-L4 BMD between COL1A1 and COL1A2 mutations located in the triple helix. (d) Relationship between SDS of L1-L4 BMD and exon number for mutations located in COL1A1 and COL1A2. (e) Relationship between SDS of L1-L4 BMD and exon number for mutations located in COL1A1 only. (f) Relationship between SDS of L1-L4 BMD and exon number for mutations located in COL1A2 only. A significant correlation was observed between L1-L4 BMD and location of mutations in COL1A2 (r2= 0.74, p = 0.003). The numbers in parentheses represent the sample numbers. N: N-terminal pro-peptide; Helix: triple helix; C: C-terminal pro-peptide. No significant differences nor correlations were observed in (a-e) (PNG 33 kb)
(DOCX 12 kb)