

Abstract
Degenerative disc disease is a highly prevalent, global health problem that represents the primary cause of back pain and is associated with neurological disorders, including radiculopathy, myelopathy, and paralysis, resulting in worker disability and socioeconomic burdens. The intervertebral disc is the largest avascular organ in the body, and degeneration is suspected to be linked to nutritional deficiencies. Autophagy, the process through which cells self‐digest and recycle damaged components, is an important cell survival mechanism under stress conditions, especially nutrient deprivation. Autophagy is negatively controlled by the mammalian target of rapamycin (mTOR) signaling pathway. mTOR is a serine/threonine kinase that detects nutrient availability to trigger the activation of cell growth and protein synthesis pathways. Thus, resident disc cells may utilize autophagy and mTOR signaling to cope with harsh low‐nutrient conditions, such as low glucose, low oxygen, and low pH. We performed rabbit and human disc cell and tissue studies to elucidate the involvement and roles played by autophagy and mTOR signaling in the intervertebral disc. In vitro serum and nutrient deprivation studies resulted in decreased disc cell proliferation and metabolic activity and increased apoptosis and senescence, in addition to increased autophagy. The selective RNA interference‐mediated and pharmacological inhibition of mTOR complex 1 (mTORC1) was protective against inflammation‐induced disc cellular apoptosis, senescence, and extracellular matrix catabolism, through the induction of autophagy and the activation of the Akt‐signaling network. Although temsirolimus, a rapamycin derivative with improved water solubility, was the most effective mTORC1 inhibitor tested, dual mTOR inhibitors, capable of blocking multiple mTOR complexes, did not rescue disc cells. In vivo, high levels of mTOR‐signaling molecule expression and phosphorylation were observed in human intermediately degenerated discs and decreased with age. A mechanistic understanding of autophagy and mTOR signaling can provide a basis for the development of biological therapies to treat degenerative disc disease.
Keywords: aging, autophagy, disc degeneration, intervertebral disc, mTOR signaling, spine
Schematic illustration of disc cellular autophagy. Under stress conditions, e.g. nutrient deprivation, the mammalian target of rapamycin (mTOR), a signal integrator that detects nutrients to signal the execution of cell growth and division, is suppressed, which initiates autophagy through the activation of autophagy‐related (Atg) genes and proteins. High‐mobility group box 1 (HMGB1), which is involved in stress response, translocates from the nucleus to the cytoplasm and directly interacts with Beclin1 (Atg6 homolog). The Beclin1–class‐III phosphatidylinositol 3‐kinase (PI3K) complex initiates autophagosome formation by developing the isolation membrane. Autophagosome maturation is completed by the growth and closure of the isolation membrane, driven by the conjugation of phosphatidylethanolamine with light chain 3 (LC3) (Atg8 homolog), leading to the formation of the autophagosome‐membrane‐bound form LC3‐II. Then, p62/sequestosome 1 (p62/SQSTM1) and p62/SQSTM1‐bound polyubiquitinated proteins become incorporated into completed autophagosomes. The completed autophagosome fuses with the lysosome to form the autolysosome (which can be inhibited by chloroquine), where the enclosed cargo is degraded, and its constituents are released and recycled. Understanding of autophagy requires monitoring this dynamic, multi‐step process of autophagic flux. In our previous time‐course observational study, the graded supply of serum and nutrients decreased proliferation and metabolic activity and increased autophagy, apoptosis, and senescence in rabbit disc annulus fibrosus cells.
1. INTRODUCTION
1.1. Socioeconomic impacts, symptoms, and treatments of intervertebral disc disease
Back pain is a global health problem, with a lifetime prevalence of 70% to 85%,1 resulting in a socioeconomic burden of $102.0 billion/year in the US.2 The causes of back pain are multifactorial; however, a large‐scale twin study identified intervertebral disc degeneration to be an independent cause of back pain.3 Furthermore, disc degeneration can also present as neurological disorders, including radicular pain, numbness, muscle weakness, and paralysis.4 Despite the successful outcomes of conservative treatment protocols for disc disease, using medication and physiotherapy,5 nonresponders must undergo surgery.6 Current surgical interventions consist primarily of symptomatic disc excision, resulting in the loss of function, immobilization, and potential additional complications, due to altered biomechanics.7 Therefore, the development of new biological therapies to treat disc degeneration is an urgent issue.
1.2. Pathophysiology of the intervertebral disc
The intervertebral disc has a complex morphological structure, with the nucleus pulposus (NP) encapsulated by the annulus fibrosus (AF) and endplates.4 Although the AF originates from the mesenchyme,8, 9 the NP comes from the notochord.10 Notochordal cells only exist during the first 10 years of human life and are subsequently replaced by non‐notochordal, chondrocyte‐like cells of unknown provenance.8, 9 A more recent article has found that chondrocyte‐like cells are derived from disc NP cells during degeneration, the phenotype of which represents a terminal stage of differentiation preceding the loss of NP cells and disc collapse.11 Notochordal cell‐conditioned medium protects non‐notochordal cells from apoptosis and inflammation.12 In addition, notochordal cells produce larger amounts of proteoglycans than non‐notochordal cells13 and can stimulate non‐notochordal cells to produce proteoglycans.14
The collagenous, laminar AF surrounds the central, gelatinous NP, maintaining the pressurization of the NP, providing support during compressive loading, and facilitating multidimensional spinal movement.15 Disc AF and NP cells have chondrocytic phenotypes,16 producing matrix components, including proteoglycans (principally aggrecan) and collagens (predominantly types I and II in the AF and NP, respectively).4 Matrix metabolism is regulated by the balance between catabolic enzymes, matrix metalloproteinases (MMPs) and disintegrins and metalloproteinases with thrombospondin motifs (ADAMTSs), and anti‐catabolic inhibitors, such as tissue inhibitors of metalloproteinases (TIMPs).17 Increased MMP and ADAMTS levels, relative to TIMP levels, have been observed in discs during human clinical18, 19, 20 and rodent experimental degeneration.21, 22, 23 Discs are the largest immune‐privileged, low‐nutrient, avascular organs in the body.24 Compared with peripheral disc AF cells, central disc NP cells depend on diffusion from blood vessels at the disc margins to obtain nutrients.25 Therefore, decreased blood supplies, subchondral bone sclerosis, and endplate calcification, which occur during mechanical stress, injury, smoking, and aging, can reduce the transport of nutrients to discs and is suspected to contribute to disc degeneration.25
In humans, intervertebral disc degeneration begins during early childhood,26, 27 and is generally more severe in the NP than in the AF.27 In the NP, obvious clefts and radial tears can occur between ages 11‐16 years.27 Aggrecan biosynthesis and type II procollagen content peak at ages ≤5 years and diminish between ages 5 and 15 years, whereas the percentage of denatured type II collagen increases after 5 years of age.26 Approximately 40% of people under 30 years of age and 90% of those over 55 years of age present with lumbar disc degeneration.28 A reduction in cell numbers, another major characteristic of disc degeneration, primarily results from programmed cell death, or apoptosis.29 Apoptotic cells increase substantially between the ages of 11 and 16 years, associated with the disappearance of notochordal cells and chondrocyte proliferation,27 suggesting a potential link between apoptosis and notochordal cell disappearance during the pathogenesis of disc degeneration.8 Furthermore, a notably high incidence of apoptosis has been observed in human aged and degenerated discs30 and rodent degenerative discs by static compression.23, 31 The incidence of irreversible cell growth arrest due to aging, or senescence,32 also increases during human disc degeneration.33, 34 Understanding of this unique, harsh, low‐glucose, low‐oxygen, low‐pH and high‐osmolality environment and the load fluctuations that disc cells are exposed to is essential for designing new biological therapies to prevent disc aging and degeneration.
1.3. Autophagy and mTOR signaling
Autophagy, the intracellular process during which cells degrade and recycle damaged components, is an important cell survival mechanism that sustains metabolism and prevents the accumulation of damaged, toxic proteins and organelles under stress conditions, especially nutrient deprivation.35, 36, 37, 38 Autophagy involves autophagy‐related (Atg) genes and proteins.36, 37 Under typical physiological conditions, Atg proteins are expressed at relatively low levels; however, basal autophagy serves as the quality‐control machinery for cellular renovation and homeostasis.35, 38 Under stress conditions, the activation of Atg proteins results in the formation and maturation of the autophagosome, which captures damaged organelles, misfolded proteins, and invading microorganisms in induced autophagy.35, 36, 37 The completed autophagosome then fuses with the lysosome, to form the autolysosome, which degrades the enclosed cargo and releases its constituents for reuse.35, 36, 37 The microtubule‐associated protein 1 light chain 3 (LC3) (a mammalian homolog of yeast Atg8) is a ubiquitin‐like protein with cytosolic (LC3‐I) and phosphatidylethanolamine‐conjugated (LC3‐II) forms.39, 40 LC3‐II is the only protein that remains attached to the autophagosome membrane after formation, making it in a robust marker for ongoing autophagy.39, 40 Measuring LC3‐II protein expression by Western blotting and/or counting LC3 puncta during immunofluorescence can be used to determine autophagic activity, as increased LC3‐II expression and LC3 puncta indicate an increased number of autophagosomes.40 High‐mobility group box 1 (HMGB1) is a nuclear DNA‐binding protein and an extracellular damage‐associated molecular pattern molecule.41 In response to stress, HMGB1 translocates from the nucleus to the cytoplasm, before being released extracellularly.41 Cytoplasmic HMGB1 directly interacts with Beclin1 (Atg6 homolog), resulting in autophagosome formation.40, 41 p62/sequestosome 1 (p62/SQSTM1) is a ubiquitin‐binding protein that acts as a link between LC3 and ubiquitinated substrates.40 p62/SQSTM1 and p62/SQSTM1‐bound polyubiquitinated proteins become incorporated into the completed autophagosome and are degraded in the autolysosome; therefore, their expression levels inversely correlate with autophagosome degradation levels.40 Monitoring this dynamic, sequential process, known as autophagic flux, is essential to understand the roles played by autophagy (Figure 1).
Figure 1.
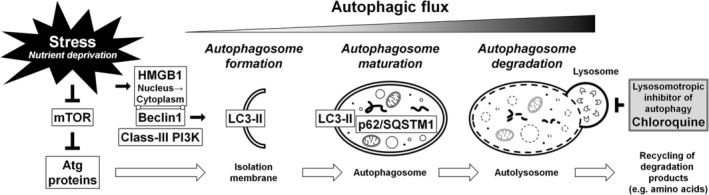
Schematic illustration of disc cellular autophagy. Under stress conditions, for example, nutrient deprivation, the mammalian target of rapamycin (mTOR), a signal integrator that detects nutrients to signal the execution of cell growth and division, is suppressed, which initiates autophagy through the activation of autophagy‐related (Atg) genes and proteins. High‐mobility group box 1 (HMGB1), which is involved in stress response, translocates from the nucleus to the cytoplasm and directly interacts with Beclin1 (Atg6 homolog). The Beclin1–class‐III phosphatidylinositol 3‐kinase (PI3K) complex initiates autophagosome formation by developing the isolation membrane. Autophagosome maturation is completed by the growth and closure of the isolation membrane, driven by the conjugation of phosphatidylethanolamine with light chain 3 (LC3) (Atg8 homolog), leading to the formation of the autophagosome‐membrane‐bound form LC3‐II. Then, p62/sequestosome 1 (p62/SQSTM1) and p62/SQSTM1‐bound polyubiquitinated proteins become incorporated into completed autophagosomes. The completed autophagosome fuses with the lysosome to form the autolysosome (which can be inhibited by chloroquine), where the enclosed cargo is degraded, and its constituents are released and recycled. Understanding of autophagy requires monitoring this dynamic, multi‐step process of autophagic flux. In our previous time‐course observational study, the graded supply of serum and nutrients decreased proliferation and metabolic activity and increased autophagy, apoptosis, and senescence in rabbit disc annulus fibrosus cells
Autophagy is negatively regulated by the mammalian target of rapamycin (mTOR).42 mTOR is a serine/threonine kinase that integrates nutrients, growth factors, energy, and stress signals to trigger the activation cell growth and division.42 mTOR exists in two complexes: mTOR complex 1 (mTORC1), which contains regulatory‐associated protein of mTOR (RAPTOR), and mTOR complex 2 (mTORC2), which contains rapamycin‐insensitive companion of mTOR (RICTOR).42 Downstream mTORC1 effectors, including p70/ribosomal S6 kinase (p70/S6K), regulate cell proliferation, messenger RNA (mRNA) translation, and protein synthesis.42 mTORC1 is regulated upstream by Akt, an essential prosurvival mediator that suppresses apoptosis.43 Moreover, Akt phosphorylation has been associated with class‐I phosphatidylinositol 3‐kinase (PI3K) and mTORC2.42, 43 Because mTOR is the central signal integrator for nutrition,42 the extensive modulation of mTOR signaling would be harmful, and homozygous mTOR deletion results in embryonic lethality.44 Therefore, identifying which subunit(s) of mTOR exert effects on target cells is necessary (Figure 2A,B).
Figure 2.
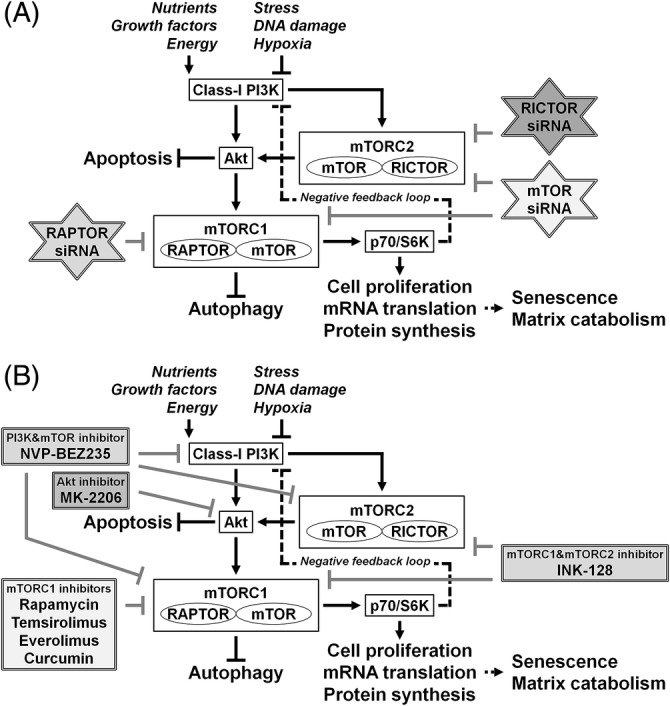
Schematic illustration of the RNA interference (RNAi)‐mediated and pharmacological modulation of disc cellular mTOR signaling. The mammalian target of rapamycin (mTOR) is a serine/threonine kinase that detects nutrients to signal the execution of cell growth and division. mTOR exists in two complexes: mTOR complex 1 (mTORC1), which contains regulatory‐associated protein of mTOR (RAPTOR), and mTOR complex 2 (mTORC2), which contains rapamycin‐insensitive companion of mTOR (RICTOR). mTORC1 acts as a signal integrator for nutrients, growth factors, energy, stress, DNA damage, and hypoxia. Down‐stream effectors of mTORC1, including p70/ribosomal S6 kinase (p70/S6K), regulate cell proliferation, messenger RNA (mRNA) translation, and protein synthesis. Autophagy, an intracellular degradation system, is under the tight negative regulation of mTORC1. mTORC1 can be regulated upstream by Akt, an essential prosurvival mediator that suppresses apoptotic cell death. Akt phosphorylation is governed by the class‐I phosphatidylinositol 3‐kinase (PI3K) and mTORC2. Furthermore, a negative feedback loop exists between p70/S6K and the class‐I PI3K. (A) In our previous study to modulate mTOR signaling by RNAi, small interfering RNAs (siRNAs) against mTOR, RAPTOR, and RICTOR were applied. In human disc nucleus pulposus cells, RNAi treatments clarified mTOR‐dependent senescent cell aging and extracellular matrix catabolism. The selective suppression of mTORC1/RAPTOR, but not the extensive suppression of mTORC1/mTORC2/mTOR and mTORC2/RICTOR, protected against inflammation‐induced disc cellular apoptosis, senescence, and matrix catabolism through the induction of autophagy and Akt. (B) In our previous study to pharmacologically modulate mTOR signaling, mTORC1 inhibitors, such as rapamycin, temsirolimus, everolimus, and curcumin, a dual mTORC1 and mTORC2 inhibitor, INK‐128, a dual PI3K and mTOR inhibitor, NVP‐BEZ235, and/or an allosteric Akt inhibitor, MK‐2206, were examined. In human disc nucleus pulposus cells, protective effects were observed for mTORC1, but not mTORC2, inhibitors against inflammation‐induced disc cellular apoptosis, senescence, and matrix catabolism through the induction of autophagy and Akt
1.4. Autophagy and mTOR signaling in the intervertebral disc
Autophagy has previously been studied in articular chondrocytes. In the mouse knee cartilage, spontaneous aging and experimental osteoarthritis induced by the progressive transection of the medial meniscotibial and collateral ligaments both reduced Atg protein expression.45 Traumatic impaction injury also suppressed autophagy.46 Mechanical damages have been partially rescued by the administration of rapamycin, an autophagy inducer through mTORC1 inhibition.46, 47 In human chondrocytes, protective effects of rapamycin against inflammation‐induced apoptosis and catabolic gene expression have been shown, in vitro.48 Compared with femoral head cartilage derived from surgically treated femoral neck fracture patients (nondegenerated), LC3 expression increased in lateral femoral condyle cells (mildly degenerated) and decreased in medial femoral condyle cells (severely degenerated) derived from patients with varus‐type knee osteoarthritis, in vivo.48 Osteoarthritis severity‐dependent decreases in Atg protein expression have been observed during a different human articular cartilage study.45 These may be caused by the stress‐response induction of autophagy.36, 37 Chondrocytes may induce autophagy in response to stress, such as inflammation and injury, during earlier osteoarthritis stages. However, cells are thought to lose the reaction potential of autophagy during advanced osteoarthritis, requiring arthroplasty.
Compared with osteoarthritis, the involvement of autophagy during degenerative disc disease has not been fully clarified. In human49 and rat discs,50, 51 autophagy activation has been confirmed by electron microscopy. In rat disc NP tissues, Atg protein expression increased with spontaneous aging.50 In chemically induced type‐I diabetic rat discs, the accelerated development of mild degeneration, associated with stress‐response autophagy, apoptosis, and senescence induction, has been observed.51 Differential autophagy levels between disc and cartilage tissues may be caused by the presence of NP notochordal cells in rodents.8, 9 Because of their markedly slow proliferation and prolonged maintenance,52 notochordal cells may require autophagy induction with aging and in response to disease stress. Furthermore, the upregulation of Atg gene expression was observed in human discs with the Pfirrmann53 grades 4 to 5, compared with grades 1 to 349; however, the confirmation of autophagic flux at either the protein or organelle levels, using LC3‐I to LC3‐II conversion and p62/SQSTM1 degradation, was not performed in these cells. Disc cellular autophagy has gained increasing interest, including the activation in both disc NP and AF cells in response to a variety of stress conditions, such as nutrient deprivation,54, 55, 56, 57 oxidative stress,58, 59, 60 compression overload,61 inflammation,62 hyperlactatemia,63 hyperosmolarity,64 and hypoxia.65 Meanwhile, evidence regarding mTOR signaling in disc cells is still limited.64, 65, 66 Therefore, mechanistic investigations to clarify the roles played by autophagy and mTOR signaling must be conducted. Furthermore, the clinical relevance of autophagy and mTOR signaling during degenerative disc disease remains largely unexplored.
1.5. Hypothesis
We hypothesized that resident cells would utilize autophagy and mTOR signaling to cope with low‐nutrient, stressful conditions of the disc.25 However, little evidence exists regarding the effects of autophagy and mTOR signaling on disc cell and tissue homeostasis. Therefore, we performed in vitro and in vivo studies of human and rabbit disc cells and tissues, modulated by the graded supply of nutrition, RNA interference (RNAi), and pharmacological agents, to elucidate the involvement and roles played by autophagy and mTOR signaling in the intervertebral disc. This brief review introduces our study findings and interpretations, discussing the future applications of therapeutically modulating biological autophagy and/or mTOR signaling to treat degenerative disc disease.
2. SERUM AND NUTRIENT DEPRIVATION IN RABBIT DISC CELLS
Published studies have not confirmed or mechanistically examined autophagic flux in intervertebral disc cells. Furthermore, many in vitro studies have been performed using conventional cell‐culture conditions, such as 10% to 20% serum supplementation and normoxia, which differ considerably from the in vivo disc environment.25 Therefore, as a preliminary study, we carefully and systematically examined intervertebral disc cell fate, in vitro.67 We conducted time‐course experiments to measure autophagic flux in rabbit disc AF cells under varying degrees of nutrient withdrawal by applying media with various serum concentrations. In this study, a conventional monolayer cell culture system was used due to the difficulty observing detailed changes in intracellular signaling networks and autophagic flux using 3D cell culture systems. Although in vitro monolayer cultures may limit physiological relevance to in vivo situations,68 the laminar AF is better simulated by monolayers than the gelatinous NP. All experiments were performed in 5% O2 to simulate the physiological environment of disc AF cells, as the concentration of oxygen in the bone marrow, whose vasculature supplies discs, is approximately 4% to 7%.69 We used rabbit cells due to reduced phenotypic variability, including age, sex, and degeneration grade, compared with surgically obtained human cells. Hence, the objective of our first study was to clarify the fundamental relationships between nutrient supply and autophagy, apoptosis, and senescence levels in rabbit disc AF cells (Figure 1).67
2.1. Decreased disc cell proliferation and metabolic activity under reduced serum and nutrient conditions
To characterize disc cellular responses to changes in serum‐related nutrient supply, we investigated cell proliferation in Hank's balanced salt solution (HBSS) or Dulbecco's modified Eagle's medium (DMEM), containing 0% to 10% fetal bovine serum (FBS). Cell numbers decreased in HBSS and 0% FBS‐supplemented DMEM, remained unchanged in 1% FBS‐supplemented DMEM, and increased in 10% FBS‐supplemented DMEM. Although serum deprivation reduced cell numbers, the nutrients supplied by DMEM containing 1% FBS were sufficient to maintain AF disc cell numbers.
To evaluate disc cell metabolism, we measured cell dehydrogenase activity and DNA amount in HBSS or DMEM, containing 0% to 20% FBS. Total dehydrogenase activity and DNA amount both decreased under limited‐serum and limited‐nutrient conditions. Normalized cell metabolic activity (dehydrogenase activity/DNA amount) remained relatively unchanged in DMEM containing 3% to 20% FBS but decreased in DMEM containing 0% to 1% FBS. Normalized cell metabolic activity showed dropped sharply in HBSS. Disc AF cells responded to nutrient deprivation by reducing cell proliferation and metabolism. Further investigations aim to identify the optimal culture conditions for rabbit disc AF cells in 5% O2, although DMEM containing 5% FBS resulted in the highest normalized cell metabolic activity.67
2.2. Increased disc cellular autophagy under reduced serum and nutrient conditions
To monitor autophagic flux in disc AF cells, we performed time‐course LC3 puncta counting and nuclear and cytoplasmic HMGB1 measurements, using imaging cytometry. Although the number of LC3 puncta per cell peaked after 12 hours in DMEM containing 1% to 10% FBS, more pronounced increases in LC3 puncta were observed in HBSS and 0% FBS‐supplemented DMEM, peaking at 24 to 48 hours. Nuclear HMGB1 intensity peaked at 12 to 24 hours, whereas cytoplasmic HMGB1 intensity increased over time, in HBSS and DMEM containing 0% to 1% FBS. Thus, the degree of LC3 puncta and cytoplasmic HMGB1 changes corresponded to the severity of nutrient withdrawal. Reduced serum and nutrient conditions activate the early (cytoplasmic HMGB1 translocation) and middle (LC3 puncta formation) phases of disc cellular autophagic flux (Figure 1).
We also performed Western blotting for LC3, HMGB1, and a selective autophagy substrate, p62/SQSTM1. Western blotting results demonstrated time‐dependent increases in LC3‐II in 0% FBS‐supplemented DMEM, transient increases at 12 hours in 1% FBS‐supplemented DMEM, and no obvious increases in 10% FBS‐supplemented DMEM. Likewise, total (nuclear + cytoplasmic) HMGB1 increased markedly at 12 hours and time‐dependent p62/SQSTM1 decreases were observed in DMEM containing 0% FBS. Reduced serum concentrations further activated the late phase (p62/SQSTM1 degradation) of disc cellular autophagic flux (Figure 1).
An autophagy inhibitor, chloroquine, used to induce the autophagosome (LC3‐II) accumulation by blocking lysosomal acidification,40 increased LC3‐II levels for all serum concentrations, even nutrient‐rich 10% FBS, indicating the presence of basal physiological autophagy. Furthermore, differences in LC3‐II levels with and without chloroquine were consistent with the severity of FBS withdrawal, indicating the induction of stress‐response autophagy during nutrient deprivation in disc AF cells (Figure 1).
2.3. Increased disc cellular apoptosis, senescence, and autophagy under reduced serum conditions
To examine other mechanisms that occur in disc AF cells under nutrient deprivation conditions, we measured apoptosis and senescence. In staining assays, the percentage of apoptotic terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL)70 ‐positive cells and nuclear cleaved caspase‐371 ‐positive cells increased with decreasing FBS concentrations. The percentage of senescence‐associated β‐galactosidase (SA‐β‐gal)72 ‐positive cells and nuclear p16/INK4A73 ‐positive cells also increased with diminishing FBS concentrations.
Western blotting also showed increased LC3‐II, HMGB1, cleaved caspase‐3, and p16/INK4A levels and decreased p62/SQSTM1 levels with FBS withdrawal, indicating the concurrent induction of disc cellular autophagy, apoptosis, and senescence under limited nutrition conditions.
Subsequently, we examined the co‐localization of autophagy and senescence markers in cells positive for an apoptotic marker by multi‐color immunofluorescence. After the withdrawal of FBS, “apoptotic” nuclear cleaved caspase‐3‐positive cells presented a higher percentage of immunopositivity for “autophagic” cytoplasmic LC3 than for “senescent” nuclear p16/INK4A. “Non‐apoptotic” but “senescent” cells negative for nuclear cleaved caspase‐3 but positive for p16/INK4A also showed “autophagic” accumulation of LC3 puncta; however, the degree of LC3 accumulation was markedly lower than in “apoptotic” cells positive for nuclear cleaved caspase‐3. These disc AF‐cell findings indicate the involvement of autophagy during both apoptosis and senescence. Then, the degree of autophagic response in apoptosis might be greater than in senescence. However, the observed severity of autophagic response would depend on conditions of cell passage number, nutrient abundance, oxygen concentration, and treatment strength and duration. Further studies for intervertebral disc cell fate are warranted.
2.4. Cytoplasmic HMGB1 as an autophagy marker
Compared with well‐established LC3‐II and p62/SQSTM1 to monitor autophagy,40 time‐dependent changes in the expression and localization of HMGB1 observed by independent nuclear and cytoplasmic HMGB1 measurements is noteworthy, suggesting the usefulness of HMGB1 as an autophagy sensor in adult disc AF cells, similar to embryonic fibroblasts and cancer cells.41 Western blotting for total (nuclear + cytoplasmic) HMGB1 expression supported stress‐reactive cytoplasmic translocation, followed by extracellular release, of HMGB1 nuclear protein. However, in this study, nuclear HMGB1 levels changed under limited nutrition unlike embryonic and cancer cells.41 Therefore, we examined the extent to which markers of autophagy were concurrently present in apoptotic cells.
Multicolor immunofluorescence detected that many “apoptotic” nuclear cleaved caspase‐3‐positive cells simultaneously showed marked nuclear HMGB1 elevation as well as LC3 puncta formation and cytoplasmic HMGB1 release. Nuclear retention of HMGB1 occurs during apoptosis because of posttranslational modifications that affect chromatin binding.74 Thus, roles of HMGB1 in disc disease need to be further studied. However, these findings are the first to support cytoplasmic HMGB1 as an autophagy marker in disc AF cells.
2.5. Summary
Serum and nutrient deprivation decreased rabbit disc AF cell proliferation and metabolic activity and increased autophagy, apoptosis, and senescence.67 Serum deprivation‐induced autophagy in disc cells is consistent with prior reports.54, 55, 56, 57 Our findings support the understanding of disc cell fate and the monitoring of autophagic flux. The prosurvival effects of autophagy are well‐known among diverse cell types,75 including disc cells57, 64, 76 and chondrocytes.46, 47, 48 However, further mechanistic investigations of the intersection between autophagy, apoptosis, and senescence under clinically relevant conditions remain necessary.
3. RNAI‐MEDIATED MTOR SIGNALING MODULATION IN HUMAN DISC CELLS
The substantial involvement of autophagy in disc cells suggests the potential for autophagy‐modulating therapies during degenerative disc disease. Autophagy is negatively controlled by intracellular mTOR signaling,42 and many autophagy‐inducing and autophagy‐inhibiting agents exert their effects through mTOR signaling.40 However, due to cross‐talk and redundancy in the mTOR‐signaling network,42, 77 the specific pharmacological induction and inhibition of mTOR signaling remain difficult. The chronic administration of rapamycin, a selective mTORC1 inhibitor and autophagy inducer, can inhibit mTORC2.78 A PI3K inhibitor, 3‐methyladenine, effectively blocks the early phase of autophagy, through class‐III PI3K inhibition‐mediated Beclin1 inactivation,40 and also inhibits class‐I PI3K, promoting autophagy even at suboptimal concentrations during long‐term experiments.79 Therefore, we utilized RNAi to knockdown specific mTOR‐signaling cascades,80 using small interfering RNAs (siRNAs) against mTOR, RAPTOR, and RICTOR. To exclude off‐target effects of RNAi,81 findings were confirmed using at least two siRNAs, with different sequences, for all assays.
In this study, to assess clinical relevance, we utilized human disc NP cells obtained from patients who underwent lumbar interbody fusion surgeries to treat degenerative disease. To simulate the physiologically hypoxic disc NP environment,25 experiments were performed in 2% O2. Understanding of disc NP‐cell fate and intracellular signaling is essential as the pathology of degenerative disc disease is widely thought to be originated from NP.27 Moreover, compared with disc AF cells, NP cells are susceptible against the environmental changes in nutrient deprivation, hypoxia, and inflammation,82 thereby suggesting a deeper involvement of autophagy and mTOR signaling. The inclusion of two different cell types, NP and AF, would further strength the experimental results in this review.
In addition, interleukin‐1 beta (IL‐1β) was used to simulate a clinically relevant inflammatory disease condition.83 IL‐1β is a proinflammatory cytokine closely linked to the pathogenesis83 and severity of disc degeneration.84 In fact, there are complex cross‐talk mechanisms between mTOR signaling and inflammation, for example, IL‐1β and tumor necrosis factor‐alpha.85 Akt promotes nuclear factor kappa‐light‐chain‐enhancer of activated B cells (NF‐κB) inhibitor kinase‐dependent regulation of NF‐κB signaling through mTORC1.86, 87 In our observation, IL‐1β administration stimulated mTORC1 and p70/S6K but also induced autophagy presented with increased LC3‐II and decreased p62/SQSTM1 in human disc NP cells.80 Effects of inflammation on disc cellular autophagy and mTOR signaling are subjects to be studied in the future.
The objective of our second study was to elucidate the involvement and roles played by mTOR signaling in human disc NP cells (Figure 2A).80
3.1. Consistent disc cellular autophagy and differential Akt activation by the selective interference of mTOR signaling
First, we performed Western blotting on disc cell protein extracts, confirming specific decreases in the expression levels of mTOR, RAPTOR, and RICTOR proteins caused by the corresponding siRNAs. Western blotting further showed that mTOR and RICTOR siRNAs decreased downstream p70/S6K and upstream Akt phosphorylation, whereas RAPTOR siRNA decreased p70/S6K phosphorylation but increased Akt phosphorylation. Akt activation occurs through the negative feedback loop between p70/S6K and class‐I PI3K.88, 89, 90 Class‐I PI3K is responsible for various cellular functions related to the activation of Akt.42 Therefore, the suppression of mTORC1 and p70/S6K by RAPTOR RNAi resulted in the loss of class‐I PI3K feedback and subsequent Akt activation.89 In greater details, at baseline, the mTORC1/p70/S6K axis suppresses the upstream PI3K/Akt signaling pathway through the phosphorylation of insulin receptor substrate 1 (IRS1),90, 91 a second messenger from insulin‐like growth factor 1 receptor to PI3K.88 Then, mTORC1 inhibition‐mediated relief of this negative feedback loop activates IRS1, PI3K, Akt, and ERK in some cell types.92, 93 This feedback system has been found in a variety of cancers90, 91, 92, 93, 94, 95, 96, 97 and also non‐malignant intervertebral disc cells (Figure 2A).80, 98
Western blotting results demonstrated that all three siRNAs increased LC3‐II and decreased p62/SQSTM1 levels, which are consistent with enhanced autophagy induction,40 indicating successful RNAi‐mediated mTORC1 suppression (Figure 2A).42
3.2. Differential effects on disc cell proliferation, viability, apoptosis, senescence, and matrix metabolism by the selective interference of mTOR signaling
To examine disc cellular responses to RNAi, we measured cell proliferation and dehydrogenase activity. Cell numbers decreased in response to mTOR and RICTOR siRNAs, but not RAPTOR siRNA. Total dehydrogenase‐based cell viability also decreased following mTOR and RICTOR siRNAs, but not RAPTOR siRNA. Cell cycle and proliferation are positively controlled by translation‐stimulating p70/S6K, which is positively regulated by mTORC1 and negatively regulated by translation‐repressing eukaryotic translation initiation factor 4E‐binding protein 1 (4E‐BP1), which is, in turn, negatively regulated by mTORC1.42 Therefore, in this study, all RNAi treatments should facilitate the negative regulation of disc cell cycle and proliferation through mTORC1 suppression. However, the relatively unchanged disc cell proliferation and viability following mTORC1/RAPTOR RNAi may be due to differences in Akt phosphorylation levels. Akt increases cell proliferation through mTORC1 induction (p70/S6K induction and 4E‐BP1 inhibition) and other pathways, including the inhibition of negative cell‐cycle regulators p27/KIP1 and p21/CIP1.43 Thus, increased Akt phosphorylation after mTORC1/RAPTOR RNAi, associated with the negative feedback loop between p70/S6K and class‐I PI3K,88, 89, 90 resulted in differential effects on disc cell proliferation and viability from mTORC1/mTORC2/mTOR and mTORC2/RICTOR RNAi.
We then assessed RNAi effects on disc cell death, aging, and matrix metabolism. TUNEL staining showed that proinflammatory cytokine IL‐1β‐induced apoptotic cells decreased after RAPTOR siRNA only. Western blotting showed that IL‐1β‐induced apoptotic markers were decreased by RAPTOR siRNA only. Differential anti‐apoptotic effects associated with mTORC1/RAPTOR RNAi, compared with mTORC1/mTORC2/mTOR and mTORC2/RICTOR RNAi, may also be due to increased Akt phosphorylation. A major role of Akt is to enhance cell survival, by blocking proapoptotic proteins, for example, Bcl‐2‐associated death promoter protein, and through effects on transcription factors, for example, forkhead box O and p53.43 Collectively, Akt‐mediated survival and proliferation strongly support mTORC1/RAPTOR interference as a potential therapeutic application for intervertebral disc disease.
Western blotting results showed that IL‐1β‐induced senescent markers were decreased by all mTOR, RAPTOR, and RICTOR siRNAs. Similar findings were observed during SA‐β‐gal staining. These consistent anti‐senescent RNAi findings are likely caused by mTORC1 suppression, resulting in p70/S6K inhibition and 4E‐BP1 induction. 4E‐BP1 is required for dietary restriction‐induced lifespan extensions,99 and the deletion of p70/S6K also extends lifespans.100 mTOR‐hypomorphic mice increase lifespans with reduced senescent p16/INK4A expression.101 Hence, our findings of cellular senescence suggested the disruption of mTORC1‐mediated proliferation. In this review, we try to provide a possible interpretation that mTORC1/RAPTOR interference vs mTORC1/mTORC2/mTOR and mTORC2/RICTOR interference exerts different responses in apoptosis and similar responses in senescence under proinflammatory IL‐1β stimulation; however, the complexity between disc cell fate and intracellular signaling warrants further mechanistic clarification.
Western blotting showed that the IL‐1β‐induced release of catabolic MMP‐3 and MMP‐13 into cultured supernatants was markedly decreased by mTOR, RAPTOR, and RICTOR siRNAs. Gelatin zymography also showed that the IL‐1β‐induced activation of MMP‐2 and MMP‐9 were effectively decreased by RAPTOR siRNA. Real‐time reverse transcription‐polymerase chain reaction (RT‐PCR) further demonstrated the IL‐1β‐induced upregulation of catabolic MMP‐3, MMP‐13, and ADAMTS‐4 genes was suppressed by RAPTOR siRNA. IL‐1β‐induced the downregulation of anabolic ACAN, encoding aggrecan, and COL2A1, encoding collagen type II alpha 1 chain, was not completely rescued by RAPTOR siRNA; however, RAPTOR siRNA treatment resulted in a trend toward the upregulation of these anabolic genes. Only a few reports have described the regulation and mechanisms underlying matrix metabolism by mTOR. Articular cartilage‐specific mTOR deletions have been shown to be protective against destabilized medial meniscus‐induced osteoarthritis in mice, based on increased autophagy, decreased apoptosis, and decreased catabolic MMP‐13 expression.102 Our anti‐catabolic RNAi findings suggested that the expression and activation of MMPs potentially depend on mTORC1‐mediated mRNA translation and protein synthesis, although the possibility of other regulatory mechanisms including RNA transcription and mRNA stability cannot be excluded. Further investigations are required; however, mTORC1‐mediated proliferation and translation provide a plausible interpretation for reduced disc cell aging and matrix catabolism in response to mTOR, RAPTOR, and RICTOR RNAi treatments.
3.3. Summary
This is the first mechanistic study to demonstrate cascade‐dependent, differential roles for mTOR signaling in human disc NP cells.80 Although the extensive RNAi‐mediated suppression of mTORC1/mTORC2/mTOR and mTORC2/RICTOR was not always advantageous to disc cells, the selective suppression of mTORC1/RAPTOR protected against inflammation‐induced disc cellular apoptosis, senescence, and matrix catabolism.80 The beneficial effects of mTORC1/RAPTOR interference were dependent on the activation of autophagy and Akt.80
4. PHARMACOLOGICAL MTOR SIGNALING MODULATION IN HUMAN DISC CELLS
Although the RNAi‐mediated modulation of mTOR signaling identified protective effects for mTORC1/RAPTOR suppression in human disc NP cells,80 pharmacological modulation is favorable compared with gene‐silencing therapy in clinical practice due to safety issues. Rapamycin, an mTORC1 inhibitor, extends mammalian lifespan103 and plays protective roles in chondrocytes.46, 47, 48 Despite the effectiveness of rapamycin,46, 47, 48, 103 rapamycin is not widely used clinically due to poor water solubility, which limits administration routes, and serious adverse effects, including immunosuppression.104 Rapamycin derivatives, such as everolimus and temsirolimus, have been developed.105 Everolimus is only available orally,105 whereas temsirolimus is a prodrug that can be administered both intravenously and orally.105 Including rapamycin, these agents represent first‐generation mTOR inhibitors.105 A natural polyphenol from the rhizomes of turmeric, curcumin, has also been reported to inhibit mTOR signaling in various cancer cells.106 Therefore, we performed an additional in vitro study to identify the most suitable mTORC1 inhibitor for human degenerative disc disease treatment.98 We also tested more recently developed second‐generation mTOR inhibitors: a dual mTORC1 and mTORC2 inhibitor, INK‐128, and a dual PI3K and mTOR inhibitor, NVP‐BEZ235.105 Both are in clinical trials for advanced solid tumor treatment.105 To clarify the role of increased Akt phosphorylation during mTORC1/RAPTOR interference in human disc NP cells, an allosteric Akt inhibitor, MK‐2206, was tested, which is also in clinical trials for renal cell carcinoma (Figure 2B).107
4.1. Consistent disc cellular autophagy induction and Akt activation by pharmacological mTORC1 inhibition
To identify drug toxicity, we assessed dose‐dependent disc cell viability and determined that a 100 nM dose was effective but non‐toxic for rapamycin, temsirolimus, everolimus, and curcumin.
Western blotting demonstrated that mTORC1‐inhibiting rapamycin, temsirolimus, and everolimus successfully decreased mTOR and downstream p70/S6K phosphorylation but increased upstream Akt phosphorylation; however, curcumin did not affect phosphorylation. These mTORC1 inhibition‐induced changes in mTOR and related feedback signaling molecules88, 89, 90 were consistent with the mTORC1/RAPTOR RNAi results (Figure 2B).80
Western blotting results showed that rapamycin, temsirolimus, and everolimus increased LC3‐II and decreased p62/SQSTM1 levels, indicating enhanced autophagy activation.40 Additional Western blotting following chloroquine treatments40 revealed that autophagic LC3‐II accumulation was most prominent in temsirolimus‐treated cells (Figure 2B).
4.2. Protective effects of pharmacological mTORC1 inhibition against disc cellular apoptosis, senescence, and matrix catabolism
Western blotting, TUNEL staining, and SA‐β‐gal staining showed that IL‐1β‐induced apoptosis and senescence were suppressed by mTORC1 inhibitors, which was consistent with the mTORC1/RAPTOR RNAi results.80 This trend was distinct for rapamycin, temsirolimus, and everolimus treatments compared with curcumin treatments.
Western blotting in cultured supernatants showed that the IL‐1β‐induced release of catabolic MMP‐2, MMP‐3, MMP‐9, and MMP‐13 was decreased by rapamycin, temsirolimus, and everolimus. Real‐time RT‐PCR further demonstrated that IL‐1β‐induced downregulation of ACAN and COL2A1 expression was reversed by first‐generation mTOR inhibitors. The pharmacological mTORC1 inhibition results were all similar to those for RNAi‐mediated suppression of mTORC1/RAPTOR.80
The tested mTORC1 inhibitors induced anti‐apoptosis, anti‐senescence, and anti‐matrix catabolism in disc NP cells. Rapamycin, temsirolimus, and everolimus showed similar mTORC1 inhibition effects; however, temsirolimus appeared to be the most effective, possibly as a result of improved water solubility, which also enables intravenous administration, whereas the other inhibitors are limited to oral administration.105, 106 In contrast, curcumin was not toxic but was less effective, and poor aqueous solubility, low bioavailability, and intense color‐staining are limitations associated with curcumin.108
4.3. Nonprotective effects of pharmacological dual mTOR inhibition against disc cellular apoptosis, senescence, and matrix catabolism
We additionally tested second‐generation mTOR inhibitors, INK‐128 and NVP‐BEZ235, which block multiple mTOR‐signaling molecules. Based on cell viability results, 100 nM was used to compare effects at the same dose as the first‐generation mTORC1 inhibitors.
Western blotting showed that INK‐128 and NVP‐BEZ235 both decreased mTOR, p70/S6K, and Akt phosphorylation, indicating successful mTOR inhibition. However, unlike mTORC1 inhibitors, dual mTOR inhibitors did not induce autophagy, despite mTORC1 inhibition (Figure 2B).
Furthermore, INK‐128 and NVP‐BEZ235 did not suppress IL‐1β‐induced apoptotic and senescent effects. In contrast, both agents accelerated apoptosis and senescence. Western blotting showed that INK‐128 and NVP‐BEZ235 promoted IL‐1β‐induced catabolic MMP‐2, MMP‐3, MMP‐9, and MMP‐13 release to cultured supernatants. Thus, second‐generation mTOR inhibitors did not protect human disc NP cells against inflammation, consistent with the results of mTORC1/mTORC2/mTOR and mTORC2/RICTOR RNAi.80
4.4. Akt‐dependent effects of pharmacological mTORC1 inhibition on disc cellular proautophagy, anti‐apoptosis, anti‐senescence, and anti‐matrix catabolism
We examined the Akt inhibitor MK‐2206 at 5 μM, based on cell viability assay results and prior reports.109
Western blotting showed that MK‐2206, either with or without temsirolimus, decreased mTOR, p70/S6K, and Akt phosphorylation, indicating successful Akt inhibition. Temsirolimus did but MK‐2206 did not enhance autophagy. Despite the negative regulation of autophagy by mTORC1 followed by Akt,42 dual mTOR inhibitors and MK‐2206 with extensive suppression of PI3K/Akt/mTOR signaling did not activate autophagy in human disc cells. Similar responses are observed in human lymphoblasts, in which accelerated autophagy at early time points but switched autophagy to apoptosis are observed by MK‐2206 supplementation.110 Further studies are required to disclose the presence of other cross talks through PI3K, mTORC2, and Akt to mTORC1 and autophagy (Figure 2B).
Under IL‐1β stimulation, apoptosis and senescence were suppressed by temsirolimus but exacerbated by MK‐2206. Similarly, IL‐1β‐induced catabolic MMP release was suppressed by temsirolimus but stimulated by MK‐2206. Based on the proapoptotic, prosenescent, and procatabolic findings associated with the Akt inhibition by MK‐2206, the protective effects of temsirolimus against inflammation primarily depend on Akt activation in human disc NP cells. Akt1 regulates osteophyte formation in osteoarthritis as well as endochondral ossification during skeletal growth in mice.111 Accordingly, Akt is the key regulator of mTOR signaling in human disc cells.
4.5. Summary
In human disc NP cells, mTORC1 inhibitors, but not dual mTOR inhibitors, protected against inflammation‐induced apoptosis, senescence, and matrix catabolism.98 The beneficial effects of mTORC1 inhibitors depended on the induction of autophagy and Akt.98 Based on these results, we propose temsirolimus, with improved water solubility, to be a candidate mTORC1 inhibitor for human disc disease treatment.
5. CLINICAL RELEVANCE OF MTOR SIGNALING IN HUMAN DISC TISSUES
We examined the in vivo involvement of mTOR signaling in human disc NP surgical specimens. Western blotting showed the expression of mTOR, RAPTOR, RICTOR, p70/S6K, and Akt and the phosphorylation of mTOR, p70/S6K, and Akt in all patient samples with 19 to 81 in ages and 2 to 5 in degeneration grades.80, 98 Regression analysis demonstrated age‐dependent decreases in Akt expression and phosphorylation levels.80 Furthermore, mTOR, p70/S6K, and Akt expression and phosphorylation were increased in Pfirrmann53 grade‐3 discs.98 Despite the elevated interest in mTOR signaling,42 its involvement in disc degeneration remains largely unknown. The observed findings of mTOR‐signaling molecules associated with aging and degeneration severity potentially support mTOR involvement in disc health. However, due to variations among in vivo expression and phosphorylation for intracellular signaling molecules, future examinations with larger sample sizes are necessary to find conclusive evidence.
6. FUTURE CLINICAL APPLICATIONS OF AUTOPHAGY‐MODULATING AND/OR MTOR SIGNALING‐MODULATING THERAPIES FOR DEGENERATIVE DISC DISEASE
We found the in vivo involvement of mTOR signaling in human degenerative disc NP tissues from patients. Therefore, we propose gene therapy, targeting mTORC1/RAPTOR disruption, and pharmacological therapy, using mTORC1‐inhibiting temsirolimus, as future treatment strategies for degenerative disc disease. During gene therapy, safety is a primary concern. However, the disc NP is a unique, encapsulated, isolated structure that may be suitable for local delivery,112 and the prolonged >20‐week RNAi of a target gene, without adverse effects, has been reported.113 During drug therapy, local administration to the disc is preferable. The systemic administration of mTORC1 inhibitors is potentially risky due to serious adverse effects, including immunosuppression.114 Therefore, intradiscal gene and pharmacological therapies targeting mTORC1/RAPTOR suppression are potential biological treatments for degenerative disc disease. More specifically, based on by the consistent anti‐apoptotic, anti‐senescent, and anti‐catabolic effects of mTORC1/RAPTOR RNAi and mTORC1‐inhibiting temsirolimus administration, the application to prevent the chronic progression of disc degeneration, for example, by gene therapy, and also to treat the acute phase of painful discs, for example, by pharmaceuticals, would be suggested after careful animal testing and human clinical trials.
7. CONCLUSION
This brief review describes the current understanding of autophagy and mTOR signaling in the intervertebral disc. In vitro serum and nutrient deprivation reduced rabbit disc AF cell proliferation and metabolic activity, induced the dynamic activation of disc cellular autophagy, and increased apoptosis and senescence.67 The selective RNAi‐mediated mTORC1/RAPTOR suppression and the pharmacological mTORC1 inhibition were both protective against inflammation‐induced apoptosis, senescence, and matrix catabolism and were associated with Akt and autophagy induction in human disc NP cells.80, 98 These effects were primarily Akt phosphorylation‐dependent, resulting from negative feedback following mTORC1 suppression.98 Temsirolimus, a rapamycin derivative with improved water solubility, was the most effective among the tested mTORC1 inhibitors.98 However, the extensive interference of mTORC1/mTORC2/mTOR and mTORC2/RICTOR and the pharmacological inhibition of multiple mTOR complexes did not protect human disc NP cells against inflammation.80, 98 In vivo, mTOR‐signaling molecules showed marked expression and phosphorylation in human degenerated discs, which decreased with age.80, 98 A mechanistic understanding of autophagy and mTOR signaling provides a basis for the development of biological therapies to treat degenerative disc disease.
CONFLICT OF INTEREST
The authors have no conflicts of interest to declare.
AUTHOR CONTRIBUTIONS
T.Y. drafted the article. M.I., Y.K., R.K. and K.K. revised it critically for important intellectual content. All authors approved the final version to be published.
ACKNOWLEDGMENTS
This work was supported by Japan Society for the Promotion of Science KAKENHI (Grant Numbers JP26893151 [T.Y.]; JP15H03033 [T.Y, K.K.]; JP15K10406 [T.Y., K.K.]; and JP16K20051 [T.Y.]), the Japan Orthopaedics and Traumatology Research Foundation, Inc. (Grant Number 312 [T.Y.]), and the Kobe University School of Medicine Alumni Association Shinryokukai (T.Y.). We thank Lisa Giles, PhD, from Edanz Group (http://www.edanzediting.com/ac) for editing a draft of this manuscript.
Yurube T, Ito M, Kakiuchi Y, Kuroda R, Kakutani K. Autophagy and mTOR signaling during intervertebral disc aging and degeneration. JOR Spine. 2020;3:e1082 10.1002/jsp2.1082
Funding information Japan Orthopaedics and Traumatology Research Foundation, Grant/Award Number: 312; Japan Society for the Promotion of Science, Grant/Award Numbers: JP15H03033, JP15K10406, JP16K20051, JP26893151; Kobe University School of Medicine Alumni Association Shinryokukai
REFERENCES
- 1. Andersson GB. Epidemiological features of chronic low‐back pain. Lancet. 1999;354:581‐585. [DOI] [PubMed] [Google Scholar]
- 2. Martin BI, Deyo RA, Mirza SK, et al. Expenditures and health status among adults with back and neck problems. JAMA. 2008;299:656‐664. [DOI] [PubMed] [Google Scholar]
- 3. Livshits G, Popham M, Malkin I, et al. Lumbar disc degeneration and genetic factors are the main risk factors for low back pain in women: the UK Twin Spine Study. Ann Rheum Dis. 2011;70:1740‐1745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Urban JP, Roberts S. Degeneration of the intervertebral disc. Arthritis Res Ther. 2003;5:120‐130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bydon M, De la Garza‐Ramos R, Macki M, Baker A, Gokaslan AK, Bydon A. Lumbar fusion versus nonoperative management for treatment of discogenic low back pain: a systematic review and meta‐analysis of randomized controlled trials. J Spinal Disord Tech. 2014;27:297‐304. [DOI] [PubMed] [Google Scholar]
- 6. Saal JA, Saal JS. Nonoperative treatment of herniated lumbar intervertebral disc with radiculopathy. An outcome study. Spine (Phila PA 1976). 1989;14:431‐437. [DOI] [PubMed] [Google Scholar]
- 7. Ghiselli G, Wang JC, Bhatia NN, Hsu WK, Dawson EG. Adjacent segment degeneration in the lumbar spine. J Bone Joint Surg Am. 2004;86:1497‐1503. [DOI] [PubMed] [Google Scholar]
- 8. Hunter CJ, Matyas JR, Duncan NA. The notochordal cell in the nucleus pulposus: a review in the context of tissue engineering. Tissue Eng. 2003;9:667‐677. [DOI] [PubMed] [Google Scholar]
- 9. Alini M, Eisenstein SM, Ito K, et al. Are animal models useful for studying human disc disorders/degeneration? Eur Spine J. 2008;17:2‐19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Choi KS, Cohn MJ, Harfe BD. Identification of nucleus pulposus precursor cells and notochordal remnants in the mouse: implications for disk degeneration and chordoma formation. Dev Dyn. 2008;237:3953‐3958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Mohanty S, Pinelli R, Pricop P, Albert TJ, Dahia CL. Chondrocyte‐like nested cells in the aged intervertebral disc are late‐stage nucleus pulposus cells. Aging Cell. 2019;18:e13006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Erwin WM, Islam D, Inman RD, Fehlings MG, Tsui FW. Notochordal cells protect nucleus pulposus cells from degradation and apoptosis: implications for the mechanisms of intervertebral disc degeneration. Arthritis Res Ther. 2011;13:R215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Cappello R, Bird JL, Pfeiffer D, Bayliss MT, Dudhia J. Notochordal cell produce and assemble extracellular matrix in a distinct manner, which may be responsible for the maintenance of healthy nucleus pulposus. Spine (Phila PA 1976). 2006;31:873‐882. discussion 883. [DOI] [PubMed] [Google Scholar]
- 14. Erwin WM, Inman RD. Notochord cells regulate intervertebral disc chondrocyte proteoglycan production and cell proliferation. Spine (Phila PA 1976). 2006;31:1094‐1099. [DOI] [PubMed] [Google Scholar]
- 15. Hartman RA, Yurube T, Ngo K, et al. Biological responses to flexion/extension in spinal segments ex‐vivo. J Orthop Res. 2015;33:1255‐1264. [DOI] [PubMed] [Google Scholar]
- 16. Poiraudeau S, Monteiro I, Anract P, Blanchard O, Revel M, Corvol MT. Phenotypic characteristics of rabbit intervertebral disc cells. Comparison with cartilage cells from the same animals. Spine (Phila PA 1976). 1999;24:837‐844. [DOI] [PubMed] [Google Scholar]
- 17. Vo NV, Hartman RA, Yurube T, Jacobs LJ, Sowa GA, Kang JD. Expression and regulation of metalloproteinases and their inhibitors in intervertebral disc aging and degeneration. Spine J. 2013;13:331‐341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kanemoto M, Hukuda S, Komiya Y, Katsuura A, Nishioka J. Immunohistochemical study of matrix metalloproteinase‐3 and tissue inhibitor of metalloproteinase‐1 human intervertebral discs. Spine (Phila PA 1976). 1996;21:1‐8. [DOI] [PubMed] [Google Scholar]
- 19. Roberts S, Caterson B, Menage J, Evans EH, Jaffray DC, Eisenstein SM. Matrix metalloproteinases and aggrecanase: their role in disorders of the human intervertebral disc. Spine (Phila PA 1976). 2000;25:3005‐3013. [DOI] [PubMed] [Google Scholar]
- 20. Pockert AJ, Richardson SM, Le Maitre CL, et al. Modified expression of the ADAMTS enzymes and tissue inhibitor of metalloproteinases 3 during human intervertebral disc degeneration. Arthritis Rheum. 2009;60:482‐491. [DOI] [PubMed] [Google Scholar]
- 21. Yurube T, Nishida K, Suzuki T, et al. Matrix metalloproteinase (MMP)‐3 gene up‐regulation in a rat tail compression loading‐induced disc degeneration model. J Orthop Res. 2010;28:1026‐1032. [DOI] [PubMed] [Google Scholar]
- 22. Yurube T, Takada T, Suzuki T, et al. Rat tail static compression model mimics extracellular matrix metabolic imbalances of matrix metalloproteinases, aggrecanases, and tissue inhibitors of metalloproteinases in intervertebral disc degeneration. Arthritis Res Ther. 2012;14:R51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hirata H, Yurube T, Kakutani K, et al. A rat tail temporary static compression model reproduces different stages of intervertebral disc degeneration with decreased notochordal cell phenotype. J Orthop Res. 2014;32:455‐463. [DOI] [PubMed] [Google Scholar]
- 24. Nishida K, Kang JD, Gilbertson LG, et al. Modulation of the biologic activity of the rabbit intervertebral disc by gene therapy: an in vivo study of adenovirus‐mediated transfer of the human transforming growth factor beta 1 encoding gene. Spine (Phila PA 1976). 1999;24:2419‐2425. [DOI] [PubMed] [Google Scholar]
- 25. Urban JP, Smith S, Fairbank JC. Nutrition of the intervertebral disc. Spine (Phila PA 1976). 2004;29:2700‐2709. [DOI] [PubMed] [Google Scholar]
- 26. Antoniou J, Steffen T, Nelson F, et al. The human lumbar intervertebral disc: evidence for changes in the biosynthesis and denaturation of the extracellular matrix with growth, maturation, ageing, and degeneration. J Clin Invest. 1996;98:996‐1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Boos N, Weissbach S, Rohrbach H, Weiler C, Spratt KF, Nerlich AG. Classification of age‐related changes in lumbar intervertebral discs: 2002 Volvo award in basic science. Spine (Phila PA 1976). 2002;27:2631‐2644. [DOI] [PubMed] [Google Scholar]
- 28. Cheung KM, Karppinen J, Chan D, et al. Prevalence and pattern of lumbar magnetic resonance imaging changes in a population study of one thousand forty‐three individuals. Spine (Phila PA 1976). 2009;34:934‐940. [DOI] [PubMed] [Google Scholar]
- 29. Fuchs Y, Steller H. Programmed cell death in animal development and disease. Cell. 2011;147:742‐758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Gruber HE, Hanley EN Jr. Analysis of aging and degeneration of the human intervertebral disc. Comparison of surgical specimens with normal controls. Spine (Phila PA 1976). 1998;23:751‐757. [DOI] [PubMed] [Google Scholar]
- 31. Yurube T, Hirata H, Kakutani K, et al. Notochordal cell disappearance and modes of apoptotic cell death in a rat tail static compression‐induced disc degeneration model. Arthritis Res Ther. 2014;16:R31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lombard DB, Chua KF, Mostoslavsky R, Franco S, Gostissa M, Alt FW. DNA repair, genome stability, and aging. Cell. 2005;120:497‐512. [DOI] [PubMed] [Google Scholar]
- 33. Gruber HE, Ingram JA, Norton HJ, Hanley EN Jr. Senescence in cells of the aging and degenerating intervertebral disc: immunolocalization of senescence‐associated beta‐galactosidase in human and sand rat discs. Spine (Phila PA 1976). 2007;32:321‐327. [DOI] [PubMed] [Google Scholar]
- 34. Le Maitre CL, Freemont AJ, Hoyland JA. Accelerated cellular senescence in degenerate intervertebral discs: a possible role in the pathogenesis of intervertebral disc degeneration. Arthritis Res Ther. 2007;9:R45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Mizushima N. The pleiotropic role of autophagy: from protein metabolism to bactericide. Cell Death Differ. 2005;12(suppl 2):1535‐1541. [DOI] [PubMed] [Google Scholar]
- 36. Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell. 2008;132:27‐42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Mizushima N, Levine B, Cuervo AM, Klionsky DJ. Autophagy fights disease through cellular self‐digestion. Nature. 2008;451:1069‐1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Mizushima N, Komatsu M. Autophagy: renovation of cells and tissues. Cell. 2011;147:728‐741. [DOI] [PubMed] [Google Scholar]
- 39. Kabeya Y, Mizushima N, Ueno T, et al. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000;19:5720‐5728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Klionsky DJ, Abdelmohsen K, Abe A, et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy. 2016;12:1‐222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Tang D, Kang R, Livesey KM, et al. Endogenous HMGB1 regulates autophagy. J Cell Biol. 2010;190:881‐892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Zoncu R, Efeyan A, Sabatini DM. mTOR: from growth signal integration to cancer, diabetes and ageing. Nat Rev Mol Cell Biol. 2011;12:21‐35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Manning BD, Cantley LC. AKT/PKB signaling: navigating downstream. Cell. 2007;129:1261‐1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Murakami M, Ichisaka T, Maeda M, et al. mTOR is essential for growth and proliferation in early mouse embryos and embryonic stem cells. Mol Cell Biol. 2004;24:6710‐6718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Carames B, Taniguchi N, Otsuki S, Blanco FJ, Lotz M. Autophagy is a protective mechanism in normal cartilage, and its aging‐related loss is linked with cell death and osteoarthritis. Arthritis Rheum. 2010;62:791‐801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Carames B, Taniguchi N, Seino D, Blanco FJ, D'Lima D, Lotz M. Mechanical injury suppresses autophagy regulators and pharmacologic activation of autophagy results in chondroprotection. Arthritis Rheum. 2012;64:1182‐1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Carames B, Hasegawa A, Taniguchi N, Miyaki S, Blanco FJ, Lotz M. Autophagy activation by rapamycin reduces severity of experimental osteoarthritis. Ann Rheum Dis. 2012;71:575‐581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Sasaki H, Takayama K, Matsushita T, et al. Autophagy modulates osteoarthritis‐related gene expression in human chondrocytes. Arthritis Rheum. 2012;64:1920‐1928. [DOI] [PubMed] [Google Scholar]
- 49. Gruber HE, Hoelscher GL, Ingram JA, Bethea S, Hanley EN Jr. Autophagy in the degenerating human intervertebral disc: in vivo molecular and morphological evidence, and induction of autophagy in cultured annulus cells exposed to proinflammatory cytokines‐implications for disc degeneration. Spine (Phila PA 1976). 2015;40:773‐782. [DOI] [PubMed] [Google Scholar]
- 50. Ye W, Xu K, Huang D, et al. Age‐related increases of macroautophagy and chaperone‐mediated autophagy in rat nucleus pulposus. Connect Tissue Res. 2011;52:472‐478. [DOI] [PubMed] [Google Scholar]
- 51. Jiang L, Zhang X, Zheng X, et al. Apoptosis, senescence, and autophagy in rat nucleus pulposus cells: implications for diabetic intervertebral disc degeneration. J Orthop Res. 2013;31:692‐702. [DOI] [PubMed] [Google Scholar]
- 52. Kim JH, Deasy BM, Seo HY, et al. Differentiation of intervertebral notochordal cells through live automated cell imaging system in vitro. Spine (Phila PA 1976). 2009;34:2486‐2493. [DOI] [PubMed] [Google Scholar]
- 53. Pfirrmann CW, Metzdorf A, Zanetti M, Hodler J, Boos N. Magnetic resonance classification of lumbar intervertebral disc degeneration. Spine (Phila PA 1976). 2001;26:1873‐1878. [DOI] [PubMed] [Google Scholar]
- 54. Shen C, Yan J, Jiang LS, Dai LY. Autophagy in rat annulus fibrosus cells: evidence and possible implications. Arthritis Res Ther. 2011;13:R132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Ni BB, Li B, Yang YH, et al. The effect of transforming growth factor beta1 on the crosstalk between autophagy and apoptosis in the annulus fibrosus cells under serum deprivation. Cytokine. 2014;70:87‐96. [DOI] [PubMed] [Google Scholar]
- 56. Chen JW, Ni BB, Zheng XF, Li B, Jiang SD, Jiang LS. Hypoxia facilitates the survival of nucleus pulposus cells in serum deprivation by down‐regulating excessive autophagy through restricting ROS generation. Int J Biochem Cell Biol. 2015;59:1‐10. [DOI] [PubMed] [Google Scholar]
- 57. Miyazaki S, Kakutani K, Yurube T, et al. Recombinant human SIRT1 protects against nutrient deprivation‐induced mitochondrial apoptosis through autophagy induction in human intervertebral disc nucleus pulposus cells. Arthritis Res Ther. 2015;17:253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Park EY, Park JB. High glucose‐induced oxidative stress promotes autophagy through mitochondrial damage in rat notochordal cells. Int Orthop. 2013;37:2507‐2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Chen JW, Ni BB, Li B, Yang YH, Jiang SD, Jiang LS. The responses of autophagy and apoptosis to oxidative stress in nucleus pulposus cells: implications for disc degeneration. Cell Physiol Biochem. 2014;34:1175‐1189. [DOI] [PubMed] [Google Scholar]
- 60. Kong CG, Park JB, Kim MS, Park EY. High glucose accelerates autophagy in adult rat intervertebral disc cells. Asian Spine J. 2014;8:543‐548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Ma KG, Shao ZW, Yang SH, et al. Autophagy is activated in compression‐induced cell degeneration and is mediated by reactive oxygen species in nucleus pulposus cells exposed to compression. Osteoarthr Cartil. 2013;21:2030‐2038. [DOI] [PubMed] [Google Scholar]
- 62. Xu K, Chen W, Wang X, et al. Autophagy attenuates the catabolic effect during inflammatory conditions in nucleus pulposus cells, as sustained by NF‐kappaB and JNK inhibition. Int J Mol Med. 2015;36:661‐668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Wu W, Zhang X, Hu X, et al. Lactate down‐regulates matrix systhesis and promotes apoptosis and autophagy in rat nucleus pulposus cells. J Orthop Res. 2014;32:253‐261. [DOI] [PubMed] [Google Scholar]
- 64. Jiang LB, Cao L, Yin XF, et al. Activation of autophagy via Ca(2+)‐dependent AMPK/mTOR pathway in rat notochordal cells is a cellular adaptation under hyperosmotic stress. Cell Cycle. 2015;14:867‐879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Choi H, Merceron C, Mangiavini L, et al. Hypoxia promotes noncanonical autophagy in nucleus pulposus cells independent of MTOR and HIF1A signaling. Autophagy. 2016;12:1631‐1646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Ouyang ZH, Wang WJ, Yan YG, Wang B, Lv GH. The PI3K/Akt pathway: a critical player in intervertebral disc degeneration. Oncotarget. 2017;8:57870‐57881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Yurube T, Buchser WJ, Moon HJ, et al. Serum and nutrient deprivation increase autophagic flux in intervertebral disc annulus fibrosus cells: an in vitro experimental study. Eur Spine J. 2019;28:993‐1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Knight E, Przyborski S. Advances in 3D cell culture technologies enabling tissue‐like structures to be created in vitro. J Anat. 2015;227:746‐756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Kofoed H, Sjontoft E, Siemssen SO, Olesen HP. Bone marrow circulation after osteotomy. Blood flow, pO2, pCO2, and pressure studied in dogs. Acta Orthop Scand. 1985;56:400‐403. [DOI] [PubMed] [Google Scholar]
- 70. Gavrieli Y, Sherman Y, Ben‐Sasson SA. Identification of programmed cell death in situ via specific labeling of nuclear DNA fragmentation. J Cell Biol. 1992;119:493‐501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Kamada S, Kikkawa U, Tsujimoto Y, Hunter T. Nuclear translocation of caspase‐3 is dependent on its proteolytic activation and recognition of a substrate‐like protein(s). J Biol Chem. 2005;280:857‐860. [DOI] [PubMed] [Google Scholar]
- 72. Dimri GP, Lee X, Basile G, et al. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc Natl Acad Sci U S A. 1995;92:9363‐9367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Krishnamurthy J, Torrice C, Ramsey MR, et al. Ink4a/Arf expression is a biomarker of aging. J Clin Invest. 2004;114:1299‐1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Scaffidi P, Misteli T, Bianchi ME. Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature. 2002;418:191‐195. [DOI] [PubMed] [Google Scholar]
- 75. Maiuri MC, Zalckvar E, Kimchi A, Kroemer G. Self‐eating and self‐killing: crosstalk between autophagy and apoptosis. Nat Rev Mol Cell Biol. 2007;8:741‐752. [DOI] [PubMed] [Google Scholar]
- 76. Jiang W, Zhang X, Hao J, et al. SIRT1 protects against apoptosis by promoting autophagy in degenerative human disc nucleus pulposus cells. Sci Rep. 2014;4:7456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Manning BD, Toker A. AKT/PKB signaling: navigating the network. Cell. 2017;169:381‐405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Schreiber KH, Ortiz D, Academia EC, Anies AC, Liao CY, Kennedy BK. Rapamycin‐mediated mTORC2 inhibition is determined by the relative expression of FK506‐binding proteins. Aging Cell. 2015;14:265‐273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Klionsky DJ, Abdalla FC, Abeliovich H, et al. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy. 2012;8:445‐544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Ito M, Yurube T, Kakutani K, et al. Selective interference of mTORC1/RAPTOR protects against human disc cellular apoptosis, senescence, and extracellular matrix catabolism with Akt and autophagy induction. Osteoarthr Cartil. 2017;25:2134‐2146. [DOI] [PubMed] [Google Scholar]
- 81. Echeverri CJ, Beachy PA, Baum B, et al. Minimizing the risk of reporting false positives in large‐scale RNAi screens. Nat Methods. 2006;3:777‐779. [DOI] [PubMed] [Google Scholar]
- 82. Guehring T, Wilde G, Sumner M, et al. Notochordal intervertebral disc cells: sensitivity to nutrient deprivation. Arthritis Rheum. 2009;60:1026‐1034. [DOI] [PubMed] [Google Scholar]
- 83. Risbud MV, Shapiro IM. Role of cytokines in intervertebral disc degeneration: pain and disc content. Nat Rev Rheumatol. 2014;10:44‐56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Le Maitre CL, Freemont AJ, Hoyland JA. The role of interleukin‐1 in the pathogenesis of human intervertebral disc degeneration. Arthritis Res Ther. 2005;7:R732‐R745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Xu L, Brink M. mTOR, cardiomyocytes and inflammation in cardiac hypertrophy. Biochim Biophys Acta. 1863;2016:1894‐1903. [DOI] [PubMed] [Google Scholar]
- 86. Dan HC, Adli M, Baldwin AS. Regulation of mammalian target of rapamycin activity in PTEN‐inactive prostate cancer cells by I kappa B kinase alpha. Cancer Res. 2007;67:6263‐6269. [DOI] [PubMed] [Google Scholar]
- 87. Dan HC, Cooper MJ, Cogswell PC, Duncan JA, Ting JP, Baldwin AS. Akt‐dependent regulation of NF‐{kappa}B is controlled by mTOR and Raptor in association with IKK. Genes Dev. 2008;22:1490‐1500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Manning BD. Balancing Akt with S6K: implications for both metabolic diseases and tumorigenesis. J Cell Biol. 2004;167:399‐403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Harrington LS, Findlay GM, Lamb RF. Restraining PI3K: mTOR signalling goes back to the membrane. Trends Biochem Sci. 2005;30:35‐42. [DOI] [PubMed] [Google Scholar]
- 90. O'Reilly KE, Rojo F, She QB, et al. mTOR inhibition induces upstream receptor tyrosine kinase signaling and activates Akt. Cancer Res. 2006;66:1500‐1508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Shi Y, Yan H, Frost P, Gera J, Lichtenstein A. Mammalian target of rapamycin inhibitors activate the AKT kinase in multiple myeloma cells by up‐regulating the insulin‐like growth factor receptor/insulin receptor substrate‐1/phosphatidylinositol 3‐kinase cascade. Mol Cancer Ther. 2005;4:1533‐1540. [DOI] [PubMed] [Google Scholar]
- 92. Carracedo A, Ma L, Teruya‐Feldstein J, et al. Inhibition of mTORC1 leads to MAPK pathway activation through a PI3K‐dependent feedback loop in human cancer. J Clin Invest. 2008;118:3065‐3074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Soares HP, Ni Y, Kisfalvi K, Sinnett‐Smith J, Rozengurt E. Different patterns of Akt and ERK feedback activation in response to rapamycin, active‐site mTOR inhibitors and metformin in pancreatic cancer cells. PLoS One. 2013;8:e57289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Wan X, Harkavy B, Shen N, Grohar P, Helman LJ. Rapamycin induces feedback activation of Akt signaling through an IGF‐1R‐dependent mechanism. Oncogene. 2007;26:1932‐1940. [DOI] [PubMed] [Google Scholar]
- 95. Marone R, Erhart D, Mertz AC, et al. Targeting melanoma with dual phosphoinositide 3‐kinase/mammalian target of rapamycin inhibitors. Mol Cancer Res. 2009;7:601‐613. [DOI] [PubMed] [Google Scholar]
- 96. Deng W, Gopal YN, Scott A, Chen G, Woodman SE, Davies MA. Role and therapeutic potential of PI3K‐mTOR signaling in de novo resistance to BRAF inhibition. Pigment Cell Melanoma Res. 2012;25:248‐258. [DOI] [PubMed] [Google Scholar]
- 97. Kwong LN, Davies MA. Navigating the therapeutic complexity of PI3K pathway inhibition in melanoma. Clin Cancer Res. 2013;19:5310‐5319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Kakiuchi Y, Yurube T, Kakutani K, et al. Pharmacological inhibition of mTORC1 but not mTORC2 protects against human disc cellular apoptosis, senescence, and extracellular matrix catabolism through Akt and autophagy induction. Osteoarthr Cartil. 2019;27:965‐976. [DOI] [PubMed] [Google Scholar]
- 99. Zid BM, Rogers AN, Katewa SD, et al. 4E‐BP extends lifespan upon dietary restriction by enhancing mitochondrial activity in drosophila. Cell. 2009;139:149‐160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Selman C, Tullet JM, Wieser D, et al. Ribosomal protein S6 kinase 1 signaling regulates mammalian life span. Science. 2009;326:140‐144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Wu JJ, Liu J, Chen EB, et al. Increased mammalian lifespan and a segmental and tissue‐specific slowing of aging after genetic reduction of mTOR expression. Cell Rep. 2013;4:913‐920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Zhang Y, Vasheghani F, Li YH, et al. Cartilage‐specific deletion of mTOR upregulates autophagy and protects mice from osteoarthritis. Ann Rheum Dis. 2015;74:1432‐1440. [DOI] [PubMed] [Google Scholar]
- 103. Harrison DE, Strong R, Sharp ZD, et al. Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature. 2009;460:392‐395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Cravedi P, Ruggenenti P, Remuzzi G. Sirolimus to replace calcineurin inhibitors? Too early yet. Lancet. 2009;373:1235‐1236. [DOI] [PubMed] [Google Scholar]
- 105. Zaytseva YY, Valentino JD, Gulhati P, Evers BM. mTOR inhibitors in cancer therapy. Cancer Lett. 2012;319:1‐7. [DOI] [PubMed] [Google Scholar]
- 106. Zhou H, Luo Y, Huang S. Updates of mTOR inhibitors. Anticancer Agents Med Chem. 2010;10:571‐581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Jonasch E, Hasanov E, Corn PG, et al. A randomized phase 2 study of MK‐2206 versus everolimus in refractory renal cell carcinoma. Ann Oncol. 2017;28:804‐808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Mendonca LM, Machado Cda S, Teixeira CC, Freitas LA, Bianchi ML, Antunes LM. Comparative study of curcumin and curcumin formulated in a solid dispersion: evaluation of their antigenotoxic effects. Genet Mol Biol. 2015;38:490‐498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Hirai H, Sootome H, Nakatsuru Y, et al. MK‐2206, an allosteric Akt inhibitor, enhances antitumor efficacy by standard chemotherapeutic agents or molecular targeted drugs in vitro and in vivo. Mol Cancer Ther. 2010;9:1956‐1967. [DOI] [PubMed] [Google Scholar]
- 110. Cheng Y, Zhang Y, Zhang L, et al. MK‐2206, a novel allosteric inhibitor of Akt, synergizes with gefitinib against malignant glioma via modulating both autophagy and apoptosis. Mol Cancer Ther. 2012;11:154‐164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Fukai A, Kawamura N, Saito T, et al. Akt1 in murine chondrocytes controls cartilage calcification during endochondral ossification under physiologic and pathologic conditions. Arthritis Rheum. 2010;62:826‐836. [DOI] [PubMed] [Google Scholar]
- 112. Nishida K, Suzuki T, Kakutani K, et al. Gene therapy approach for disc degeneration and associated spinal disorders. Eur Spine J. 2008;17(suppl 4):459‐466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Suzuki T, Nishida K, Kakutani K, et al. Sustained long‐term RNA interference in nucleus pulposus cells in vivo mediated by unmodified small interfering RNA. Eur Spine J. 2009;18:263‐270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Thomson AW, Turnquist HR, Raimondi G. Immunoregulatory functions of mTOR inhibition. Nat Rev Immunol. 2009;9:324‐337. [DOI] [PMC free article] [PubMed] [Google Scholar]