

Abstract
Alzheimer’s disease (AD) is the most common type of neurodegenerative disease. Its typical pathology consists of extracellular amyloid-β (Aβ) plaques and intracellular tau neurofibrillary tangles. Mutations in the APP, PSEN1, and PSEN2 genes increase Aβ production and aggregation, and thus cause early onset or familial AD. Even with this strong genetic evidence, recent studies support AD to result from complex etiological alterations. Among them, aging is the strongest risk factor for the vast majority of AD cases: Sporadic late onset AD (LOAD). Accumulation of DNA damage is a well-established aging factor. In this regard, a large amount of evidence reveals DNA damage as a critical pathological cause of AD. Clinically, DNA damage is accumulated in brains of AD patients. Genetically, defects in DNA damage repair resulted from mutations in the BRAC1 and other DNA damage repair genes occur in AD brain and facilitate the pathogenesis. Abnormalities in DNA damage repair can be used as diagnostic biomarkers for AD. In this review, we discuss the association, the causative potential, and the biomarker values of DNA damage in AD pathogenesis.
Keywords: Alzheimer’s disease pathogenesis, diagnostic biomarkers for Alzheimer disease, DNA damage response, DNA damage repair
1. Introduction
Approximately 50 million people worldwide are affected by dementia (World Health Organization/WHO, 2019). With around 10 million new cases each year (WHO, https://www.who.int/news-room/fact-sheets/detail/dementia), dementia is estimated to affect 75.6 million and 135.5 million people worldwide by 2030 and 2050 (WHO) respectively. Among the neurodegenerative diseases that cause dementia, Alzheimer’s disease (AD) is the primary cause, accounting for 60%–70% of dementia cases (WHO). AD is a major health issue and the predominant burden on the healthcare system with respect to dementia.
AD was first described by Alois Alzheimer more than one century ago on a patient named Auguste D [1]; the typical pathological features include extensive cerebral amyloid plaques and neurofibrillary tangles [2,3]. The latter is caused by intraneuronal aggregation of hyperphosphorylated tau; abnormal tau phosphorylation is attributable to several kinases including CDK5 [4,5,6,7]. Additionally, CDK5 phosphorylates the amyloid precursor protein (APP) at threonine 668 (T668), which stimulates β-amyloid (Aβ) peptide accumulation [8]. Aggregation of Aβ peptides, particularly Aβ1-42, causes amyloid (senile) plaques in AD brain [9,10,11]. Furthermore, CDK5 activities affect DNA damage response [12], supporting a linkage of DNA damage with AD [13,14].
Aβ peptides are directly produced by sequential cleavages of APP by β- and γ-secretase; the proteolytic c-terminal fragment of APP (CTFβ) of β-secretase is cleaved within the cell membrane by γ-secretase to generate neurotoxic Aβ peptides with length ranging from 38-43 residues [15,16,17,18,19]. The 40 residue Aβ peptide (Aβ1-40) is the major product, accounting for more than 50% of Aβ peptides [9,17]. Although the Aβ1-42 peptide consists of less than 10% of Aβ peptides [16,17], it is more neurotoxic and associates with a higher risk of AD because of its propensity of aggregation [9]. The importance of Aβ in AD pathogenesis is illustrated by mutations of APP, PSEN1, and PREN2 genes in familial AD with the latter two encoding the presenilin 1 and presenilin 2 subunit of γ-secretase. Individuals with these mutations develop early onset dementia in an autosomal-dominant manner [20]; the typical onset starts between 30 and 50 years of age in PSEN1 mutant carriers with some being affected at in their 20s [20,21]. γ-secretase with mutant presenilin 1 or presenilin 2 subunit favors Aβ1-42 production [16,17]. These genetic observations led to formation of the amyloid cascade hypothesis, in which abnormal Aβ drives AD pathogenesis via regulating other pathological events including tau pathology [22,23,24,25,26,27]. This hypothesis is supported by the similar pathological features between familial AD and sporadic late onset AD (LOAD) [20]. Although familial AD constitutes less than 1% of AD cases [28,29], the hypothesis has been widely accepted to guide research in advancing the understanding of both familial AD and LOAD in the past two decades [30,31].
Nonetheless, it is becoming increasingly clear that complex pathological etiology instead of the amyloid-cascade hypothesis is involved in LOAD, which accounts for more than 90% of AD cases in patients over 65 year old [32,33]. This concept is supported by the lack of success in all phase III clinical trials on drugs developed to remove cerebral Aβ in attempt to slow down cognitive decline in patients with either mild cognitive impairment (MCI) or AD dementia [31,34]. In sporadic LOAD cases, aging is the strongest risk factor [30,35]. Aging is clearly a complex process; nonetheless, accumulative evidence reveals a central role of DNA damage in aging [36]. Of note, DNA damage is clearly linked to AD and other neurodegenerative diseases. The topic of “DNA and neurodegenerative diseases” has been previously reviewed [14,30,37]. We will update the recent developments from 2014 onward; key publications prior to 2014 will be covered. Data used in this review were extracted from PubMed and selected according to the PRISMA Guidelines [38,39] (Figure 1).
Figure 1.

Systemic literature search and selection of articles for review.
2. Association of DNA Damage with Alzheimer’s Disease
The association of DNA damage with aging is well studied and established with 7086 articles under “DNA damage” AND “aging” listed in PubMed as of January 17, 2020. Genome rearrangement and double strand breaks (DSBs) increase in aging mice and senescing human cells [40,41]. DNA lesions are accumulated in AD brains. Elevations in γH2AX, a well-established marker of DSB [42], were detected in 11 of 13 AD brains in astrocytes of hippocampus and cerebral cortex [43]. Using two independent cohorts (n = 13 and n = 23), significant increases in γH2AX were recently demonstrated in astrocytes and neurons in the hippocampus and frontal cortex of AD brains [44]; the increase occurred in brains with mild cognitive impairment (MCI) [44], a preclinical stage of AD [45,46], suggesting an important role of DSB in AD pathogenesis. Furthermore, DSBs and single strand breaks (SSBs) were demonstrated at the DNA level in hippocampi of AD brains [47].
The detection of both DSBs and SSBs in AD brains supports the knowledge that endogenous reactive oxygen species (ROS) is the major source of DNA damage in AD brains [48], considering the brain being protected from external or environmental genotoxins through the blood–brain barrier. Post-mitotic neurons of the central nervous system have a high rate of metabolism [49]; the human brain accounts for 2% of body mass and consumes 20% of oxygen [50]. In this regard, accumulation of DNA oxidization was observed in AD brains [51,52]; the lesion was also detected in brains with MCI [53,54,55] and preclinical AD (PCAD) [55,56]. PCAD subjects are clinically normal, i.e., without overt manifestation of AD, but with pathological features of AD [56,57]. The proceeding of oxidative DNA lesions in MCI and PCAD conditions supports a causative contribution of DNA damage induced by ROS in AD. The predominant base adduct of DNA lesion caused by ROS is 8-hydroxyguanine (8-OHG); 8-hydroxy-2’-deoxyguanosine (8-OHdG) is a widely used marker of DNA oxidation. Increase of 8-OHdG in ventricular cerebrospinal fluid (CSF) was observed in AD brains, suggesting a biomarker value of 8-OHdG in AD diagnosis [58].
Collectively, evidence suggests a role of DNA damage with respect to DNA oxidation and the resultant DSBs and SSBs in AD pathogenesis. The accumulation of these DNA lesions is in part attributable to reductions in the repair of these DNA damages. By using autoradiographic methods to examine DNA damage (SSB) and repair capacity in neurons, an inverse association of accumulation of nuclear DNA (nDNA) breaks and nDNA repair was documented in aging mouse brain [59,60]; accumulation of nDNA SSB was detected in mouse cerebellar granule cells as well as hippocampal pyramidal and granule cells [61], which underlines a causative role of DNA damage in AD pathogenesis.
3. Link of DSB with AD
Brain is particularly vulnerable to impairment in DNA repair [62], which is likely attributable to the high levels of metabolism and transcription activities in neurons of the central nervous system (CNS) [48,49,63]. In addition to ROS as a source of DSBs in neurons of CSN, recent development reveals an essential association of transcription with DSB [64]. Stimulation or performance of variety of physiological tasks generate DSBs in neurons [63,65,66]. DSBs occur in the promoters of several early response genes and contribute to their transcription [65]; early response genes function in learning and memory [67,68] suggesting an association of DSB accumulation with cognitive decline, a major defect in patients with AD. Consistent with these observations, the number of DSB was increased in transgenic mice of hAPP (human APP mutant) [63]. While the underlying mechanisms responsible for DSB generation in neurons performing physiological activities remain to be determined, it is likely that topoisomerase IIβ (TOP2β) produces these DSBs [65,69,70,71,72]. It was first reported by Ju et al. in 2006 that DSB generation by the TOP2β-PARP1 complex in the pS2 promoter is required for estrogen-induced transcription of the target gene in MCF7 cells [69]. Subsequently, glucocorticoid receptor was also reported to transactivate genes via TOP2β-produced DSBs [73]. Additionally, TOP2β is expressed in differentiation cells and neurons [74,75]; the expression of long-transcripts linked to autism is facilitated by TOP2β [76]. While it is indeed intriguing for an essential role of DSBs at least in a subset of gene transcription, it is clear that DSBs need to be effectively repaired regardless of their sources of generation owning to their toxic impact on genome stability [77]. Reduction in DSB repair will lead to DSB accumulation, neuron loss, cognitive decline, and thus AD [44,78,79].
DSBs in mammalian cells are repaired by either homologous recombination (HR) or non-homologous end joining (NHEJ) [80]. DSBs result in activation of ATM (ataxia-telangiectasia mutated) and DNA-PK (DNA-dependent protein kinase) kinases which play essential roles in DSB repair (Figure 2) [81,82,83]. Recruiting ATM to DSB is mediated by the MRN complex; ATM action leads to BRCA1 recruitment, a commitment step for utilization of HR to repair DSBs (Figure 2) [80,84,85]. On the other hand, DSBs also recruit DNA-PK though the Ku70/Ku80 complex, followed by 53BP1 recruitment, which commits to NHEJ-mediated DSBs repair (Figure 2) [86]. The recruitment of BRCA1 and 53BP1 are mutually exclusive (Figure 2).
Figure 2.

A model showing double strand break (DSB) repair pathways. For the homologous recombination (HR) and non-homologous end joining (NHEJ) pathways, DSBs are first recognized by either the MRN or Ku70/80 complex, followed by the recruitment of ATM or DNA-PKCS (the catalytic subunit of DNA-PK) as indicated. ATM and DNA-PK then phosphorylate H2AX at serine 139 to generate γH2AX (event 1), which initiates event 2: recruiting either BRCA1 or 53BP1; recruitment of either inhibits the recruitment of another. 53BP1: p53-binding protein 1; ATM: ataxia-telangiectasia mutated; BRCA1: breast cancer type 1 susceptibility protein; DSB: double strand DNA break; DNA-PK: DNA-dependent protein kinase; HR: homologous recombination; MRN: the complex of MRE11-NBS1-RAD50; NHEJ: non-homologous end joining.
3.1. Association of HR in AD
Evidence suggests a contribution of decreases in HR to AD. ATM is the apical kinase in mediating DSB repair by HR (Figure 2). Ataxia telangiectasia (A−T) is caused by mutations in the ATM gene; the disease manifests defects in multiple system including its typical clinical presentation of ataxia due to progressive neurodegeneration that occurs in early childhood [87]. In comparison to age-matched control subjects, reductions in both the ATM protein and its mRNA were detected in the front cortex of AD brains (n = 9, 5 males and 4 females) [88]. Downregulation of ATM was also suggested in 3 mouse models for AD, transgenic mice expressing mutants of APP, PSEN1/APP, or PSEN1/APP/MAPT (encoding tau) [88].
BRCA1 plays an essential role in DSB repair using the HR process and recruitment of BRCA1 to DSBs commits cells to HR (Figure 2) [80,86]. Reductions in BRCA1’s ability in managing DSB repair are suggested in AD brains. Decreases in BRCA1 expression were detected in the hippocampal neurons of not only AD but also MCI brains [89], indicating a causative role of BRCA1 in AD. In this respect, transgenic expression of hAPP in mice leads to decrease in BRCA1 expression; knockdown of BRCA1 in these mice increases neuronal DSBs and apoptosis with concurrent impairment of learning and memory [89]. On the other hand, exploratory activities induced by change in environment upregulate BRCA1 in the dentate gyrus of mouse hippocampus [89]. These results are well in accordance with the concept that physiological neuronal activities increase transcription of early response genes through processes promoted by DSBs [65]. It is thus intriguing to envisage a role of BRCA1 in the repair of those transcription-utilized DSBs. This possibility is further supported by induction of the HR protein RAD52 by nascent mRNAs in terminally differentiated neurons and Aβ inhibits RAD52 expression [90]. In the APP/PSEN1 mouse model for AD, a decrease in the HR protein RAD51 co-exists with DSB accumulation [91]. Additionally, the BRCA1 promoter is hypomethylated and the BRCA1 protein is increased in AD brains; however, the protein was found to be mis-located into the cytoplasm as a highly non-soluble protein through association with tau; BRCA1 in AD brains is thus dysfunctional with respect to DSB repair [92]. This BRCA1 dysfunctionality can be directly induced by Aβ peptides [92]. The association of BRCA1 with phosphorylated tau in AD brain was also observed [93], consistent with tau hyperphosphorylation causing tau aggregation. In neurons differentiated from induced pluripotent stem cells (iPSC) that were reprogrammed from fibroblasts of familial AD patients, location of BRCA1 to cytosol is also demonstrated [89]. Collectively, evidence favors a critical role of BRCA1 in DSB repair in CNS neurons and its dysfunction contributes to DSB accumulation and AD pathogenesis.
The ATM-BRCA1 pathway of HR not only plays a role in AD through the management of DSB repair but also may affect AD indirectly. FE65 is an adaptor protein with its expression enriched in the brain [94,95]. It binds the APP intracellular domain (AICD), the c-terminal fragment of Aβ, contributes to ACID-derived transcription activities in mice, and likely plays a role in AD [94,96]. FE65 interacts with Tip60 and has an important role in DNA damage response (DDR) in SK-N-SH neuroblastoma cells [97]. Tip60 has a key function in DSB-induced ATM activation through acetylation of ATM at lysine 3016 [98,99]; mice deficient in FE65 are more sensitive to DNA damage [96]. Evidence thus supports a connection between FE65 and ATM in DSB repair in neurons. This concept is supported by recent discoveries of FE65 being an ATM target during DDR [100,101].
While evidence supports a role of HR in the repair of DSBs in post-mitotic neurons, many issues remain unclear. HR requires DNA replication and thus takes place in late S and G2 phase [80,86]; the mechanisms underlying HR in DSB repair of terminally differentiated neurons are still unclear. In studies using neurons differentiated from iPSCs reprogrammed from patients with familial AD, ATM and BRCA1 signaling were found to associate with the expression of cell cycle elements [88,89]; this suggests cell cycle re-entry as a potential mechanism for utilization of HR in post-mitotic neurons. Nonetheless, more evidence is needed to uncover the full mechanism at play.
3.2. Link of NHEJ in AD
NHEJ makes a major contribution to DSB repair because it does not rely on cell division and is the primary choice for differentiated cells to repair DSBs [80,86]. In this regard, downregulation of NHEJ is likely a contributor to DSB accumulation in the AD brain. Herpes simplex virus-type 1 (HSV-1) infection is a well-recognized risk factor of AD [102,103]. HSV-1 induces AD via affecting multiple AD events, such as through enhancing Aβ1−42 production [104]. The virus causes DSB accumulation in rat cortical neuron through downregulation of Ku80, an essential component of NHEJ (Figure 2), and thus impairs NHEJ in cortical neurons [105].
DNA-PK consists of the Ku70/Ku80 subunits and the catalytic subunit DNA-PKcs [106,107]. In a retrospective study using post-mortem AD brains, reductions in both Ku and DNA-PKcs in cortex were suggested [108]. However, reductions in DNA-PK expression did not reach a level of statistical significance [109]. In immunodeficient mice lacking DNA-PK, hippocampal CA1 and CA3 neurons are vulnerable to DNA damage [110]. In vitro, Aβ peptides including Aβ1−42 reduce DNA-PKcs expression and compromises DSB repair in PC12 cells [111].
In consideration of the unique characteristics of NHEJ [80,86], reductions in the DNA-PK-dependent classical NHEJ (c-NHEJ) are conceptually the primary cause of DSB accumulation in AD brains in comparison to HR. Additionally, recent evidence shows DSB generation to be an essential process in transcription initiation [69] and elongation [112] in which DSBs are managed by TOP2β complexed with DNA-PK [69]; the involvement of DNA-PK in the process is likely for dynamic repair of DSBs. As high transcription activity in post-mitotic neurons remains a major source of DSBs associated with neuron activities and transcription [63,65,66], it seems logical for defects in NHEJ being favored over HR leading to DSB accumulation in AD. Nonetheless, the reverse seems evident based on the available studies (see Section 3.1). Furthermore, in a direct comparison study using APP/PSEN1 transgenic mice, reductions in RAD51 but not 53 BP1 expression are evident in the hippocampus of both young and aging mice [91]. RAD51 and 53BP1 are essential in HR and NHEJ respectively (Figure 2). Evidence thus supports a major contribution of HR defects in DSB accumulation in AD.
4. Defects in Base Excision Repair (BER) in AD
Cells are more prone to SSB compared to DSB; mammalian cells are estimated to have approximately 104 oxidized base and SSB [113] in comparison to approximately 50 DSBs daily [114]. This concept is supported by a large amount of evidence revealing ROS as a major source of DNA damage in terminally differentiated neurons of CNS [48]. Oxidized bases are repaired via BER [49]. Repair starts with the recognition and removal of an oxidized base by a DNA glycosylase, including 8-oxoguanine (8-oxoG) DNA glycosylase/OGG1, NHT1, NEIL1, or NEIL2 (Figure 3). The end products are modified by AP (apurine/apirimidine) endonuclease 1 (APE-1) or polynucleotide kinase phosphatase (PNKP) to make the ends competent for DNA synthesis (Figure 3). The gap is filled by DNA polymerase β (POLβ), followed by ligation with DNA ligase IIIα (LIG3α) with assistance from the X-ray repair cross-complementing protein 1 (XRCC1) (Figure 3); this process of gap filling by incorporation of single nucleotide is known as short-patch BER (SP-BER). Alternatively, the gap is filled by long-patch BER (LP-BER) with the incorporation of 2-8 nucleotides (Figure 3). ROS can produce SSB, which is recognized by poly(ADP) ribose polymerase 1 (PARP1); the ends are then processed by PNKP, ataxia with oculomotor apraxia (APTX), or tyrosyl-DNA phosphodiesterase 1 (TDP1), followed by gap filling with either SP- or LP-BER (Figure 3) [49,115,116,117].
Figure 3.

An illustration demonstrating BER. ROS induces oxidized base lesion or SSB. The oxidized bases are removed by DNA glycosylase OGG1 and NHT1 or NEIL1 and NEIL2; the ends are then processed, followed by filling the gap with synthesis of a single nucleotide or 2-8 nucleotides; ligation via a ligase will then complete the repair. SSB was first recognized by PARP1; different ends produced by end processing are accordingly modified by the indicated proteins, followed by gap filling using either the SP-BER or the LP-BER pathway. APE-1: AP (apurine/apirimidine) endonuclease 1; APTX: ataxia with oculomotor apraxia; BER: base excision repair; dRP: 3’ phosphor-α,β-unsaturated aldehyde; FEN1: Figure 1. LIG: DNA ligase; NEIL1: Nei like DNA glycosylase 1; NEIL2: Nei like DNA glycosylase 2; NTH1: Nth like DNA glycosylase; OGG1: DNA glycosylase; PARP1: poly(ADP) ribose polymerase 1; PNKP: polynucleotide kinase phosphatase; POLβ: DNA polymerase β; ROS: reactive oxygen species; SSB: single strand DNA break; TDP1: tyrosyl-DNA phosphodiesterase 1; XRCC1: X-ray repair cross-complementing protein 1.
4.1. Reductions of DNA Glycosylase Activity in AD
Decreases in 8-oxoguanine (8-oxoG) DNA glycosylase activities occur in the neurons of hippocampus and other regions of AD brains (n = 10) compared to age-matched brains (n = 8) [118]. Reductions in DNA glycosylase activities and POLβ expression were also observed in AD brains (n = 10) in comparison to age-matched control brains (n = 10) [119]. Downregulation of mitochondrial BER activity in AD brain was also reported [120]. Furthermore, impairment of BER also occurs in MCI brains (n = 9) [119], indicating an important role of BER capacity decreases in AD pathogenesis.
Deletion of codon C796 of OGG1 was detected in 2 AD brains out of 14 samples; protein encoded by the deletion leads to complete loss of 8-oxoG DNA glycosylase activity [121]. Mutations leading to change of alanine 53 to threonine (A53T) and A288V (valine) were detected in AD brains [121]; the substitutions result in significant decreases in 8-oxoG DNA glycosylase activity (Table 1) [121]. Both A53T and A288V variants decrease interaction with PARP1 and XRCC1; while A53T reduces binding to substrates, A288V compromises AP lyase activity [122]. Significant decreases of the NEIL1 DNA glycosylase protein expression were detected in AD brains (n = 6) compared to control brains (n = 6) [120]. NEIL1 null mice are defective in memory retention in a water maze test [123]. NEIL1 and NEIL2 are members of the Nei family of DNA glycosylase [124,125,126,127]; their expressions are increased during rat brain development [128], suggesting the importance of NEIL1 and NEIL2 in maintaining genome integrity of CNS. Taken together, evidence supports that declines in NEIL1 expression in brain facilitate AD.
Table 1.
Downregulation of BER in Alzheimer’s disease (AD).
| Factors | Observation | Cohort (n) | Ref. |
|---|---|---|---|
| DNA glycosylase | Activity decrease in AD | 20 | [118,119] |
| OGG1 | Deletion of codon C796; loss of 8-oxoG DG activity | 2/14 | [121] |
| OGG1 | A53T and A288V; significant reduction in 8-oxoG DG activity | 1/14 for each mutation | [121] |
| NEIL1 | Decrease protein expression in AD | 6 | [120] |
| PARP1 | Activity increase in AD | 20 | [129] |
| POLβ | Downregulation | 10 | [119] |
OGG1: DNA glycosylase; NEIL1: Nei like DNA glycosylase 1; PARP1: poly(ADP) ribose polymerase 1; POLβ: DNA polymerase β.
4.2. Reductions of PARP1 Activity in AD
The impact of PARP1 on AD is complex. By ADP-ribosylation of target proteins though utilization of nicotinamide adenine dinucleotide+ (NDP+), PARP1 regulates metabolism; persistent PARP1 activation results in depletion of cellular NDP+, which leads to decreases in ATP production and alterations in other cellular events including induction of apoptosis [130,131]. Aβ peptides activate PARP1 via induction of oxidative stress in the hippocampus of adult rats [132], which likely contributes to AD via affecting brain metabolism [133]. Elevations in PARP1 activity and increases of PAR level in neurons were observed in AD brains compared to controls (Table 1) [129,133]. Evidence suggests a toxic impact of high levels of PARP1 activity in part through NAD+ depletion, as administration of exogenous NAD+ reduces Aβ-caused toxic effects in primary rat cortical neurons [134]. Concurrently, addition of NAD+ also protects neurons from Aβ-induced DNA damage [134], consistent with the concept that defects in PARP1-derived DNA repair contribute to AD. Of note, decreases in nucleolar PARP1 in hippocampal neurons of AD brains (n = 8) were recently reported [135]. Evidence thus indicates an insufficient level of NAD+ in AD brains as a cause of PARP1 dysfunction. In the 3xTgAD (transgenic co-expression of the Swedish APP mutations KM670/671NL, PSEN1 mutation M146V, and Tau mutation P301L) mice, reductions of NAD+ in cerebrum occur. In these AD mice, normalization of NAD+ by nicotinamide riboside (NR) treatment attenuate the Tau pathology but not Aβ accumulation with concurrent reductions in DNA damage and improvement of cognitive function [136]. These benefits are greater in 3xTgAD/POLβ+/− mice than in 3xTgAD mice [136], supporting that the NR treatment improves BER. Additionally, Aβ23−35 and Aβ1−42 increase oxidative DNA damage, SSB, and DSB in rat cortical neurons in vitro and addition of nicotinamide adenine dinucleotide (NAD) reduces all these toxic events [134]. Collectively, these studies highlight a possible clinical management of AD patients using NAD-related approaches. Polymorphisms of PARP1 are associated with AD risk [137,138]; nonetheless, the impact of these polymorphisms on PARP1 activity remains unclear.
4.3. A Major Contribution of Decreases in POLβ to AD
POLβ plays a role in AD [119]. The protein is expressed at reduced levels during aging in multiple tissues, including brain, kidney, liver, spleen, and testes in mice [139] and in the aging neurons of rat [140]. Loss of POLβ leads to senescence, indicating a major role of POLβ in anti-aging [141]. This concept is supported by a major contribution of defects in BER caused by POLβ reduction to Down syndrome (DS) [142,143], a typical process of precocious aging [144]. These observations comprehensively support a critical role of POLβ in regulating aging progression. As aging is the strongest risk factor of AD, it is conceivable that POLβ may play an important role in AD. This hypothesis is in accordance with the observation that almost all patients with DS developing AD pathology by their 40s [145,146,147,148].
Decreases in DNA synthesis-dependent gap filling activities occur in MCI and AD brains, which is in part attributable to a significant reduction of POLβ expression (Table 1) [119]. Mice with genetic downregulation of POLβ (POLβ+/−) in the 3xTgAD background display neuron death and worsen memory and synaptic plasticity with concurrent increases in DNA damage in comparison to age-matched 3xTgAD mice [149]. In comparison to 3xTgAD mice, 3xTgAD/ POLβ+/−mice exacerbate olfactory deficit in part via attenuation of neuron generation by neural progenitor cells [150].
While these data clearly reveal a critical role of POLβ in AD pathogenesis, reduction of POLβ alone is not sufficient to initiate AD; POLβ+/− mice express POLβ at a comparable level at 6 month old and a reduced level at 14 month old compared to 3xTgAD mice, the latter genotype mice develop AD pathology [149].
4.4. Coordination of BER Defects in AD
The high rate of metabolism and oxygen utilization as well as a low ratio of antioxidant to pro-oxidant enzymes in post-mitotic neurons of CNS [48] make these cells rely on BER to manage oxidative DNA damage. This knowledge agrees with an important contribution of BER capacity reductions to AD pathogenesis. However, it seems not all major components of BER as illustrated in Figure 3 are clearly affected in AD. For instance, the polymorphism of R194W (arginine 194 to tryptophan 194) of XRCC1 does not appear to be a significant risk factor of AD [151,152]. While reagent limitations restrained the detection of APE1 in post-mortem brain by immunohistochemistry [153], the APE1 mRNA level is decreased in entorhinal cortex in AD brains compared to controls (Figure 4) [154]. It thus remains to be determined whether BER dysfunction in AD is resulted through a coordinated manner. A recent report has systemically determined the expression of OGG1, APE1, PARP1, and POLβ in a cohort of 42 ADs and 9 controls using quantitative real-time PCR [154]. Consistent with the discussions in Section 4.2, PARP1 mRNA expression is increased in hippocampus and entorhinal cortex (Figure 4) [154]; while APE1 mRNA is only reduced in entorhinal cortex, POLβ is significantly reduced in frontal cortex, hippocampus, and entorhinal cortex (Figure 4) [154], suggesting a possibility for alterations in more than one factor to impair BER during AD pathogenesis.
Figure 4.

Summary of expression of the indicated BER genes in AD brains compared to age-matched controls. Arrows indicate upregulation and downregulation respectively. APE-1: AP (apurine/apirimidine) endonuclease 1; ND: no differences; OGG1: DNA glycosylase; PARP1: poly(ADP) ribose polymerase 1; and POLβ: DNA polymerase β.
5. Other AD Risk Factors Affecting DNA Repair
5.1. Role of CDK5 Abnormalities in AD Via Affecting DNA Damage
Unlike of majority of the cyclin-dependent kinase (CDK) family, CDK5 is most well-studied for its neuron-specific functions owing to the identification of its neuron-specific activators p35 and p39 in 1994 and 1995 [155,156,157]. Accumulative studies in the past 2 decades established essential roles of CDK5 in CNS development in mice, including synaptic plasticity and memory [158], the major neuronal functions that are compromised in AD. In this regard, abnormalities in CDK5 activation play major roles in AD pathogenesis [159]. In response to increases in calcium concentration, p35 is cleaved to p25 which is more stable and causes CDK5 hyper-activation; additionally, CDK5/p25 complex has changes in substrate specificity and cellular localization [160]. Abnormal CDK5 activity promotes the major AD pathology: Extracellular senile plaque and intracellular neurofibrillary tangles (NFTs) through facilitating Aβ production and tau phosphorylation [7,159,161,162,163], and thus impairs synaptic plasticity and induces neuronal cell death [159]. Abnormal CDK5 activation leads to ROS accumulation in neuronal cells including Neuro-2a cells [164,165], suggesting a role of CDK5 dysfunction in DNA damage in neurons. Of note, induction of transgenic expression of p25 in postnatal mouse forebrain results in AD progression, Aβ accumulation, tau neurofibrillary tangles, synaptic density reduction, neuron loss, and accumulation of DSBs [8,166].
Mechanistically, CDK5 contributes to DNA damage in neurons likely via multiple pathways. In line with HSV-1 infection as a well-recognized risk factor of AD [102,103] in part via inducing DSB accumulation in neurons [105], the virus stimulates CDK5 activation, changes its subcellular location, and induces γH2AX nuclear foci (DSB marker) in infected mouse neurons [167]. CDK5 facilitates DSB-induced DDR via enhancing ATM activation in primary cerebellar granule neurons isolated from rats (Figure 5) [168]. How this action contributes to DSB accumulation remains unclear, as ATM activation is required for HR-mediated repair of DSBs. Nonetheless, CDK5 induces neuronal cell death partly via ATM activation [168] (Figure 5) and the connection of CDK5/p25-ATM is a cause of DDR-induced neurodegeneration in a mouse model for fragile-X-associated tremor/ataxia syndrome (FXTAS) [169]. Protein phosphatase 4 (PP4) dephosphorylates 53BP1 in late mitosis, an event required for 53BP1 recruitment to DSB in G1 phase for NHEJ-mediated repair of DSBs. CDK5 phosphorylates PP4R3β, the PP4 regulatory subunit, which facilitates PP4 action and contributes to cell proliferation-associated NHEJ [170]. However whether this function is active in post-mitotic neurons remains to be determined.
Figure 5.
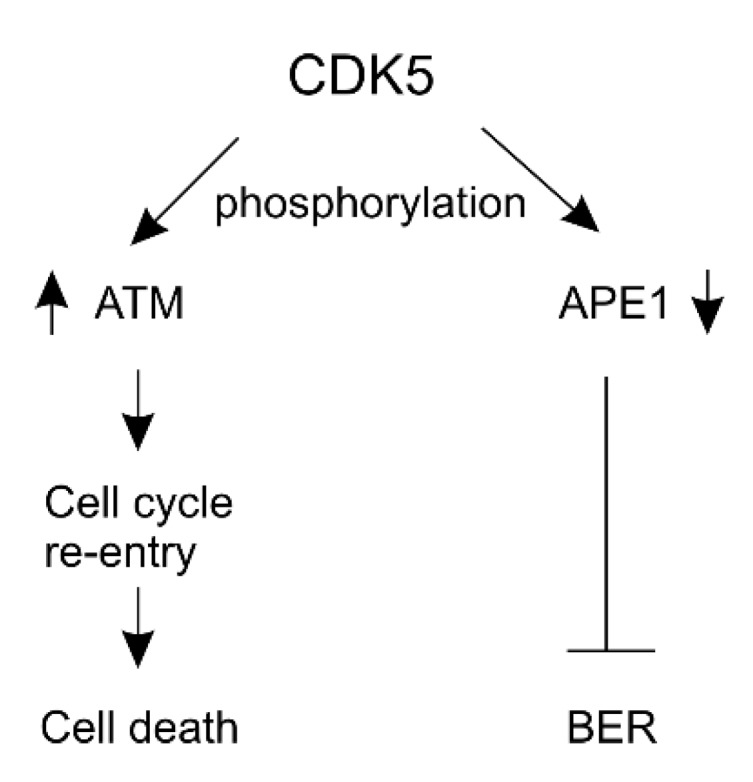
An illustration shows the effects of CDK5 in regulating DNA repair in neurons. Arrows indicate enhancing (upward direction) and reducing (downward direction) the protein’s functions respectively. CDK5 activity towards ATM in neurons is likely via p25. APE-1: AP (apurine/apirimidine) endonuclease 1; ATM: ataxia-telangiectasia mutated; BER: base excision repair; CDK5: cyclin-dependent kinase 5.
CDK5 reduces BER in neurons. CDK5 interacts with APE1 and phosphorylates it at threonine 232 (T232), which reduces APE1’s activity in cleavage of abasic (AP) sites and thus inhibits BER in mouse cortical neurons (Figure 5) [171].
5.2. Downregulation of Sirtuine 6 (SIRT6) Facilitating AD in Part Via Decreases in DNA Repair
SIRT6 is an emerging risk factor of AD [172] with 12 articles listed in PubMed under “SIRT6” AND “Alzheimer’s disease” (January 22, 2020). SIRT6 is a longevity gene [172,173]. The anti-aging activities of SIRT6 can be attributable to its NAD+ dependent histone deacetylase activity that functions in energy metabolism, inflammation, telomere maintenance, genome integrity and DNA repair [172,174,175,176,177,178]. SIRT6 null mice display genome instability and premature aging [176]. Mice with brain-specific SIRT6 deficiency showed impaired learning by the age of 4 months, elevated DNA damage, and development of tau phosphorylation-dependent toxicity [179]. Importantly, SIRT6 expression is significantly reduced at the protein level in AD brains (n = 7) compared to controls (n = 7) [179,180]; at the mRNA level, decreases in SIRT6 expression are associated with AD progression from Braak stage iii (n = 11) to v (n = 11) or vi (n = 23) [179]. In a cohort consisting of postmortem AD brains (n = 32) and control subjects (n = 47), reductions of SIRT6 occur in temporal cortex and hippocampus [179]. In the 5XFAD mouse model for AD [expressing three APP mutants (Swedish K670N/M671L, Florida I716V, and London V717I) and 2 PSEN1 (M146L and L286V) mutants], SIRT6 expression is reduced in hippocampus and frontal cortex [180]. Aβ42 reduces SIRT6 expression and induces DNA damage in mouse hippocampal neurons; overexpression of SIRT6 prevents the DNA damage [180]. SIRT6 contributes to BER and DSB repair [172]. SIRT6-mediated protection of neuron injury is inhibited by miR-34a-derived SIRT6 downregulation [181]. Collectively, evidence reveals SIRT6 to show protective effect for AD in part via enhancing DNA repair. Intriguingly, SIRT6 was reported to promote hippocampal neurogenesis in SIRT6 overexpressing adult mice [182], implying a role of SIRT6 in facilitating progenitor-derived neurogenesis in part via repairing oxidative stress-induced DNA damage.
5.3. A role of DNA Damage in AD Via Affecting Neurogenesis
Neurons of CNS are regenerated in the adult brain via a process that is mediated by neural stem cells (NSCs); compromising neurogenesis is a cause of neurodegeneration diseases, including AD [183,184]. Oxidative DNA damage and its repairing process affect NSC proliferation and differentiation [184]. For instance, SIRT6 exerts anti-AD function in part via facilitating neurogenesis of hippocampal neurons via enhancing BER and DSB repair [182]. Maintenance of the stemness of NSC requires BMI1 (B lymphoma Mo-MLV insertion region 1) [185,186,187,188,189,190]; the process is partly mediated by suppression of the INK4A/ARF locus [191], which encodes two important tumor suppressors p16INK4A and p14ARF (or p19ARF in mice) [192,193]. BMI1 is emerging to facilitate DSB repair through both the HR and NHEJ pathways [194,195,196,197]. BMI1 protein expression is reduced in post-mortem AD brains (n = 2) in comparison to age-matched controls (n = 2) [198]; the downregulation is detected in LOAD brain but not in early-onset familial AD (FAD) [199]. BMI1 heterozygous mice develop cognitive deficiency with accumulation of tau phosphorylation, Aβ plaques, and neuron loss [198]; the animals also display reductions in DDR [198]. Additionally, knockout of BMI1 in post-mitotic neurons induces Aβ deposition and tau hyperphosphorylation [199]. Collectively, evidence suggests a role of BMI1 in reducing AD via facilitating neurogenesis partially through DNA repair.
BRCA1 plays an essential role in HR-mediated repair of DSB [80,84,85,200]. In neurons produced from iPSCs that were reprogrammed from FAD patients, elevations of BRCA1 activity are associated with events of cell cycle progression (phosphorylation of CDC25C at serine 216) with concurrent increases in Aβ, suggesting an association of cell cycle re-entry with the utilization of HR in post-mitotic neurons that are regenerated [89]. Cell cycle re-entry induces apoptosis in neurons [201].
In NSCs, Aβ42 oligomers (AβO) induces ROS and DSBs with concomitant impairment of NHEJ-mediated DSB repair; enhancing DNA-PK function protects NSCs from AβO-induced toxicity [202]. Taken together, evidence supports a need to maintain genome integrity in neuron regeneration, which plays an anti-AD role.
5.4. Contributions of Chromosome Instability to AD
Chromosome instability (CIN) is a typical outcome of defects in DDR and activates DDR [203,204]. Accumulative evidence reveals increases in somatic aneuploidy as a feature of aging [205]. The rate of mosaic CIN increases from 0.23% for cancer-free individual under 50 years to 1.91% for those with ages of 75-79 years [205]. Brain has a high level of aneuploidy and the frequency increases with aging [206]. CIN is associated with neurodegeneration [207]. X chromosome aneuploidy is associated with AD [207,208]. Down syndrome is associated with chromosome 21 trisomy and all DS patients develop AD [145,146,147,148]. The existence of the A4 gene encoding APP in chromosome 21 is likely a contributing factor for the high rate of AD in DS patients [209]. However, this does not exclude a major role of CIN-associated genome instability in AD pathogenesis; in addition to chromosome 21, AD has a higher rate of aneuploidy in chromosomes 18 and X [208,210].
6. Systemic Alterations of DNA Repair Genes in AD Patients
In addition to changes in genes functioning HR and BER in AD brains, polymorphisms in these genes have been detected in blood and are associated with AD risk [211,212]. Using peripheral blood mononuclear cells (PBMCs) isolated from AD patients (n = 22) and healthy individuals (n = 13), profiling of mRNA expression identified 593 differentially expressed genes (DEGs) in AD subjects with 428 DEGs upregulated and 165 DEGs downregulated. These DEGs are enriched in pathways regulating inflammation, DDR, cell cycle, and neuronal processes [211]. Interestingly, DNA lesions are elevated in PBMCs of AD patients [213]. Compared to control subjects (n = 40), γH2AX, indicative of DSBs, is increased in lymphocytes of AD patients [214]. The increase can stratify AD patients from control individuals with an area under the curve (AUC) value of 0.91, sensitivity of 0.85 and specificity of 0.92 [214], suggesting a potential biomarker value of DSB in AD diagnosis. Clearly, the results need to be confirmed, as the cohort used in this study is quite small. In a similar study, increases in γH2AX were observed in lymphocytes of MCI subjects compared to controls [215].
Consistent with BER playing an essential role in repair oxidative DNA damage in neurons of CSN, there are numerous investigation of changes in BER genes in the circulation of AD patients [216]. In a study of PBMCs from 105 AD patients and 130 controls, the polymorphisms of c.580C>T and c.1196A>G of XRCC1 and c.977C>G of OGG1 are significantly associated with AD risk (Table 2) [216]; both XRCC1 and OGG1 function in BER (Figure 3). In comparison to 110 controls, significant increases in 8-oxoG DNA content with concomitant downregulations of 8-oxoG DNA glycosylase OGG1 and MUTYH [217], other DNA glycosylase NEIL1, APE1, and PARP1 were demonstrated in the PBMCs of AD patients (n = 100) (Table 2) [218,219]. Elevations in serum 8-OHdG was reported in AD patients (n = 30) compared to age-matched controls (n = 30) [220]. Polymorphisms of XRCC1 rs25487 (c.1196A > G; https://www.ncbi.nlm.nih.gov/snp/rs25487) (odds ratio/OR 3.72, 95% confidence interval/CI 1.739-7.891) and PARP1 rs1136410 (OR 4.159, 95% CI; 1.978−8.745) are significantly associated with AD risk [212]. The expression of the catalytic subunit POLD1 of DNA polymerase δ is significantly reduced in PBMCs of AD patients (n = 60) compared to controls (n = 40) (Table 2) [221]. Decreases in the expression of these BER genes in PBMCs of AD patients (Table 2) are not likely due to promoter methylation. In a study of LOAD patients (n = 56) and controls (n = 55) using PBMCs, no difference in promoter methylation was detected for OGG1, PARP1, BRCA1, MRELLA, MLH1, and MGMT [222]. Collectively, evidence supports biomarker values of alterations of BER genes in AD diagnosis (Table 2).
Table 2.
Alteration of BER genes in PBMCs of AD patients.
| BER Genes | Changesi | AD Patients | Controls | Ref |
|---|---|---|---|---|
| OGG1 | Reduction c.977C>G |
n = 100 n = 105 |
n = 110 n = 130 |
[218,219] [216] |
| MUTYH | reduction | n = 100 | n = 110 | [219] |
| NEIL1 | reduction | n = 100 | n = 110 | [219] |
| APE1 | reduction | n = 100 | n = 110 | [219] |
| PARP1 | Reduction rs1136410 |
n = 100 n = 120 |
n = 110 n = 110 |
[219] [212] |
| XCCR1 | c.580C>T, c.1196A>G rs25487 (c.1196A>G) |
n = 105 n = 120 |
n = 130 n = 110 |
[216] [212] |
| POLD1 | reduction | n = 60 | n = 40 | [221] |
i: AD vs control; AD: Alzheimer’s disease; APE-1: AP (apurine/apirimidine) endonuclease 1; OGG1: DNA glycosylase; MUTYH: MYH glycosylase; NEIL1: Nei like DNA glycosylase 1; PARP1: poly(ADP) ribose polymerase 1; POLD1: the catalytic subunit of DNA polymerase δ; XRCC1: X-ray repair cross-complementing protein 1.
7. Conclusions
AD, particularly sporadic LOAD, is a multi-factorial disease, including metabolic dysfunction, insulin resistance, and others [223]. Among these factors, DNA damage is clearly an important one. In view of the typical AD pathology of Aβ-based senile plaques and hyperphosphorylated tau-formed neurofibrillary tangles, both Aβ peptides and tau affect genome instability. For example, nuclear tau plays a role in maintaining pericentromeric heterochromatin (PCH); loss of tau disrupts PCH, leading accumulation of DNA breaks [224]. Tau aggregation in the cytosol will deplete the nuclear pool of tau, abolishing tau-derived protection of genome integrity [225]. This concept is supported by the inability of hyperphosphorylated tau in the protection of DNA from thermal denaturation [226] and the loss of heterochromatin in tau transgenic Drosophila and mice, which contributes to neurodegeneration [227]. These associations of Aβ peptides and tau pathology with DNA damage clear strengthen the importance of DNA damage in AD pathogenesis. Accumulative evidence reviewed here collectively reveals the critical contributions of abnormalities in DNA lesion repair for both DSB and SSB in AD pathogenesis. In addition to neurons, DNA damage also occurs in oligodendrocytes, which has a major impact on AD [228,229]. Oligodendrocytes are exclusively responsible to produce myelin ensheathing axons of CNS [230,231]. Myelination is essential for the high speed transmission in the neural network; myelinated fibers have at minimum 10-fold faster conduct velocity than unmyelinated fibers with the same diameter [232]. Loss in myelination impairs performance of CNS, leads to neurodegeneration, and is among the earliest abnormalities during AD pathogenesis (see review [233]). Oligodendrocytes are vulnerable to oxidative DNA damage, which contributes to loss of neurons and onset of AD (see review [228]). This concept is in accordance with the observed declines of brain white matter, of which myelin constitutes 50%–60% [234], during aging [235,236].
8. Future Perspectives
For many years, AD research and drug development have been largely focused on the amyloid-cascade hypothesis which places Aβ at the apical position in AD pathogenesis. Because of fails in all phase 3 clinical trials on drug targeting the Aβ process, it is apparent that the hypothesis misses some major components particularly for LOAD cases. For instance, tau pathology seems to show higher match with AD development than with Aβ pathology [31]. Based on a study using human NSC line hNS1, Aβ42 at low concentration promotes hNSCs proliferation without compromising neuronal differentiation [237]. DNA damage clearly is a major contributing factor to AD. The pathogenicity of DNA damage is not only strengthened by its association with the typical AD pathological factors (Aβ peptide and tau pathology, see Section 7) but also impairment of DNA damage repair occurs prior to the onset of AD [55,78,79,238].
Considering aging being the strongest risk factor of AD and accumulative DNA damage as a well-established influence on aging, the concept of DNA damage abnormalities as the major contributor of AD is appealing. Although this concept is supported by a large amount of preclinical and clinical evidence as reviewed here, the pathological cause of DNA damage in AD remains to be demonstrated. This task is not only challenging but also should be cautiously considered. AD is a progressive neurodegenerative disease and likely also a systemic disease, in which abnormalities in DNA damage are an aspect. Nonetheless, evidence supports a major contribution of DSB accumulation in AD-associated loss of memory and neuron; for instance knockdown of BRCA1 in mice causes DSB, loss of neurons, as well as deficits in learning and memory [239]. Additionally, DSBs may occur prior to the onset of AD [78,79]. In view of AD being a progressive neurodegenerative disease, the current knowledge indicates a utility of early disease intervention through prevention or reduction of DSBs or other type of DNA damage. This strategy could delay the systemic alterations that progressively occur during AD development.
The common detection of alterations of the BER genes in circulation (Table 2) and their potential in stratification of AD risk support their diagnostic applications. Although more research is clearly needed for these applications, their potential should certainly be carefully investigated, particularly considering the lack of biochemistry-based means in current AD diagnosis (https://www.mayoclinic.org/diseases-conditions/alzheimers-disease/diagnosis-treatment/drc-20350453) and the non-invasive nature of obtaining PBMCs. The presence of alterations of the BER genes in circulation is in line with the hypothesis of AD being a systemic disease. The concept of AD, particularly LOAD cases (the vast majority of AD), as a systemic disease is consistent with DNA damage being a major factor of aging. In this regard, there is a need to develop a bona fide animal model for aging in which DNA damage accumulates in the brain during the aging process in order to systemically study AD. Owing to its unique anti-aging function, SIRT6-based animal models will be appealing. The mutation rate increases following aging in mouse liver but not in the animal brain [40], thus mouse models of AD likely have major limitations in studying aging process of LOAD.
Abbreviations
| 8-OHdG | 8-hydroxy-2’-deoxyguanosine |
| 8-OHG | 8-hydroxyguanine |
| AD | Alzheimer’s disease |
| Aβ | amyloid-β |
| AICD | APP intracellular domain |
| APE-1 | AP (apurine/apirimidine) endonuclease 1 |
| APP | amyloid precursor protein |
| APTX | ataxia with oculomotor apraxia |
| ATM | ataxia-telangiectasia mutated |
| AUC | area under the curve |
| BER | base excision repair |
| CDK | cyclin-dependent kinase |
| CIN | Chromosome instability |
| CNS | central nervous system |
| CSF | cerebrospinal fluid |
| DEGs | differentially expressed genes |
| DSBs | double strand breaks |
| FEN1 | FLAP endonuclease 1 |
| FXTAS | fragile-X-associated tremor/ataxia syndrome |
| HR | homologous recombination |
| HSV-1 | herpes simplex virus-type 1 |
| iPSC | induced pluripotent stem cells |
| LOAD | sporadic late onset AD |
| MCI | mild cognitive impairment |
| MUTYH | MYH glycosylase |
| NDP | adenine dinucleotide |
| NHEJ | non-homologous end joining |
| NEIL1 | Nei like DNA glycosylase 1 |
| NTH1 | Nth like DNA glycosylase |
| OGG1 | DNA glycosylase |
| PARP1 | poly(ADP) ribose polymerase 1 |
| PBMCs | peripheral blood mononuclear cells |
| PCAD | preclinical AD |
| PNKP | polynucleotide kinase phosphatase |
| POLβ | DNA polymerase β |
| POLD1 | the catalytic subunit of DNA polymerase δ |
| PP4 | Protein phosphatase 4 |
| ROS | reactive oxygen species |
| SSBs | single strand breaks |
| TDP1 | tyrosyl-DNA phosphodiesterase 1 |
| TOP2β | topoisomerase Iiβ |
| WHO | World Health Organization |
| XRCC1 | X-ray repair cross-complementing protein 1. |
Author Contributions
Conceptualization, X.L., A.K., Y.G. and D.T.; material preparation, X.L., Y.G., M.J.C., J.P., and K.Z.; writing—original draft preparation, X.L., Y.G., and D.T.; writing—review, all authors; writing editing—Y.G., X.L., and D.T.; supervision, D.T. All authors have read and agreed to the published version of the manuscript.
Funding
D. Tang is supported by grants from Cancer Research Society, Canadian Cancer Society (grant #: 319412), CIHR and by funds from Urological Cancer Center for Research and Innovation (UCCRI). Y.G. is supported by Studentship provided by Ontario Graduate Scholarships and Research Institute of St Joe’s Hamilton.
Conflicts of Interest
The authors declare no conflict of interest.
References
- 1.Maurer K., Volk S., Gerbaldo H. Auguste D and Alzheimer’s disease. Lancet. 1997;349:1546–1549. doi: 10.1016/S0140-6736(96)10203-8. [DOI] [PubMed] [Google Scholar]
- 2.Graeber M.B., Kosel S., Grasbon-Frodl E., Moller H.J., Mehraein P. Histopathology and APOE genotype of the first Alzheimer disease patient, Auguste D. Neurogenetics. 1998;1:223–228. doi: 10.1007/s100480050033. [DOI] [PubMed] [Google Scholar]
- 3.Graeber M.B., Mehraein P. Reanalysis of the first case of Alzheimer’s disease. Eur. Arch. Psychiatry Clin. Neurosci. 1999;249(Suppl. 3):10–13. doi: 10.1007/PL00014167. [DOI] [PubMed] [Google Scholar]
- 4.Baumann K., Mandelkow E.M., Biernat J., Piwnica-Worms H., Mandelkow E. Abnormal Alzheimer-like phosphorylation of tau-protein by cyclin-dependent kinases cdk2 and cdk5. Febs Lett. 1993;336:417–424. doi: 10.1016/0014-5793(93)80849-P. [DOI] [PubMed] [Google Scholar]
- 5.Wilkaniec A., Czapski G.A., Adamczyk A. Cdk5 at crossroads of protein oligomerization in neurodegenerative diseases: Facts and hypotheses. J. Neurochem. 2016;136:222–233. doi: 10.1111/jnc.13365. [DOI] [PubMed] [Google Scholar]
- 6.Castro-Alvarez J.F., Uribe-Arias S.A., Kosik K.S., Cardona-Gomez G.P. Long- and short-term CDK5 knockdown prevents spatial memory dysfunction and tau pathology of triple transgenic Alzheimer’s mice. Front. Aging Neurosci. 2014;6:243. doi: 10.3389/fnagi.2014.00243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kimura T., Tsutsumi K., Taoka M., Saito T., Masuda-Suzukake M., Ishiguro K., Plattner F., Uchida T., Isobe T., Hasegawa M., et al. Isomerase Pin1 stimulates dephosphorylation of tau protein at cyclin-dependent kinase (Cdk5)-dependent Alzheimer phosphorylation sites. J. Biol. Chem. 2013;288:7968–7977. doi: 10.1074/jbc.M112.433326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cruz J.C., Tseng H.C., Goldman J.A., Shih H., Tsai L.H. Aberrant Cdk5 activation by p25 triggers pathological events leading to neurodegeneration and neurofibrillary tangles. Neuron. 2003;40:471–483. doi: 10.1016/S0896-6273(03)00627-5. [DOI] [PubMed] [Google Scholar]
- 9.Teixeira J.P., de Castro A.A., Soares F.V., da Cunha E.F.F., Ramalho T.C. Future Therapeutic Perspectives into the Alzheimer’s Disease Targeting the Oxidative Stress Hypothesis. Molecules. 2019;24:4410. doi: 10.3390/molecules24234410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Burdick D., Soreghan B., Kwon M., Kosmoski J., Knauer M., Henschen A., Yates J., Cotman C., Glabe C. Assembly and aggregation properties of synthetic Alzheimer’s A4/beta amyloid peptide analogs. J. Biol. Chem. 1992;267:546–554. [PubMed] [Google Scholar]
- 11.Gravina S.A., Ho L., Eckman C.B., Long K.E., Otvos L., Jr., Younkin L.H., Suzuki N., Younkin S.G. Amyloid beta protein (A beta) in Alzheimer’s disease brain. Biochemical and immunocytochemical analysis with antibodies specific for forms ending at A beta 40 or A beta 42. J. Biol. Chem. 1995;270:7013–7016. doi: 10.1074/jbc.270.13.7013. [DOI] [PubMed] [Google Scholar]
- 12.Liu W., Li J., Song Y.S., Li Y., Jia Y.H., Zhao H.D. Cdk5 links with DNA damage response and cancer. Mol. Cancer. 2017;16:60. doi: 10.1186/s12943-017-0611-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Huang E., Qu D., Park D.S. Cdk5: Links to DNA damage. Cell Cycle. 2010;9:3142–3143. doi: 10.4161/cc.9.16.12955. [DOI] [PubMed] [Google Scholar]
- 14.Madabhushi R., Pan L., Tsai L.H. DNA damage and its links to neurodegeneration. Neuron. 2014;83:266–282. doi: 10.1016/j.neuron.2014.06.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vassar R. BACE1: The beta-secretase enzyme in Alzheimer’s disease. J. Mol. Neurosci. 2004;23:105–114. doi: 10.1385/JMN:23:1-2:105. [DOI] [PubMed] [Google Scholar]
- 16.Selkoe D.J., Wolfe M.S. Presenilin: Running with scissors in the membrane. Cell. 2007;131:215–221. doi: 10.1016/j.cell.2007.10.012. [DOI] [PubMed] [Google Scholar]
- 17.Thinakaran G., Koo E.H. Amyloid precursor protein trafficking, processing, and function. J. Biol. Chem. 2008;283:29615–29619. doi: 10.1074/jbc.R800019200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chow V.W., Savonenko A.V., Melnikova T., Kim H., Price D.L., Li T., Wong P.C. Modeling an anti-amyloid combination therapy for Alzheimer’s disease. Sci. Transl. Med. 2010;2:13ra1. doi: 10.1126/scitranslmed.3000337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang H., Ma Q., Zhang Y.W., Xu H. Proteolytic processing of Alzheimer’s beta-amyloid precursor protein. J. Neurochem. 2012;120(Suppl. 1):9–21. doi: 10.1111/j.1471-4159.2011.07519.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bateman R.J., Aisen P.S., De Strooper B., Fox N.C., Lemere C.A., Ringman J.M., Salloway S., Sperling R.A., Windisch M., Xiong C. Autosomal-dominant Alzheimer’s disease: A review and proposal for the prevention of Alzheimer’s disease. Alzheimer‘s Res. Ther. 2011;3:1. doi: 10.1186/alzrt59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Snider B.J., Norton J., Coats M.A., Chakraverty S., Hou C.E., Jervis R., Lendon C.L., Goate A.M., McKeel D.W., Jr., Morris J.C. Novel presenilin 1 mutation (S170F) causing Alzheimer disease with Lewy bodies in the third decade of life. Arch. Neurol. 2005;62:1821–1830. doi: 10.1001/archneur.62.12.1821. [DOI] [PubMed] [Google Scholar]
- 22.Hardy J., Allsop D. Amyloid deposition as the central event in the aetiology of Alzheimer’s disease. Trends Pharmacol. Sci. 1991;12:383–388. doi: 10.1016/0165-6147(91)90609-V. [DOI] [PubMed] [Google Scholar]
- 23.Selkoe D.J. The molecular pathology of Alzheimer’s disease. Neuron. 1991;6:487–498. doi: 10.1016/0896-6273(91)90052-2. [DOI] [PubMed] [Google Scholar]
- 24.Hardy J.A., Higgins G.A. Alzheimer’s disease: The amyloid cascade hypothesis. Science. 1992;256:184–185. doi: 10.1126/science.1566067. [DOI] [PubMed] [Google Scholar]
- 25.Hardy J., Selkoe D.J. The amyloid hypothesis of Alzheimer’s disease: Progress and problems on the road to therapeutics. Science. 2002;297:353–356. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- 26.Selkoe D.J., Hardy J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 2016;8:595–608. doi: 10.15252/emmm.201606210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.De Strooper B., Karran E. The Cellular Phase of Alzheimer’s Disease. Cell. 2016;164:603–615. doi: 10.1016/j.cell.2015.12.056. [DOI] [PubMed] [Google Scholar]
- 28.Lane C.A., Hardy J., Schott J.M. Alzheimer’s disease. Eur. J. Neurol. 2018;25:59–70. doi: 10.1111/ene.13439. [DOI] [PubMed] [Google Scholar]
- 29.Migliore L., Coppede F. Genetics, environmental factors and the emerging role of epigenetics in neurodegenerative diseases. Mutat. Res. 2009;667:82–97. doi: 10.1016/j.mrfmmm.2008.10.011. [DOI] [PubMed] [Google Scholar]
- 30.Tse K.H., Herrup K. Re-imagining Alzheimer’s disease—The diminishing importance of amyloid and a glimpse of what lies ahead. J. Neurochem. 2017;143:432–444. doi: 10.1111/jnc.14079. [DOI] [PubMed] [Google Scholar]
- 31.van der Kant R., Goldstein L.S.B., Ossenkoppele R. Amyloid-beta-independent regulators of tau pathology in Alzheimer disease. Nat. Rev. Neurosci. 2020;21:21–35. doi: 10.1038/s41583-019-0240-3. [DOI] [PubMed] [Google Scholar]
- 32.Coppede F., Migliore L. DNA damage in neurodegenerative diseases. Mutat. Res. 2015;776:84–97. doi: 10.1016/j.mrfmmm.2014.11.010. [DOI] [PubMed] [Google Scholar]
- 33.Masters C.L., Bateman R., Blennow K., Rowe C.C., Sperling R.A., Cummings J.L. Alzheimer’s disease. Nat. Rev. Dis. Primers. 2015;1:15056. doi: 10.1038/nrdp.2015.56. [DOI] [PubMed] [Google Scholar]
- 34.Caruso A., Nicoletti F., Gaetano A., Scaccianoce S. Risk Factors for Alzheimer’s Disease: Focus on Stress. Front. Pharmacol. 2019;10:976. doi: 10.3389/fphar.2019.00976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Riedel B.C., Thompson P.M., Brinton R.D. Age, APOE and sex: Triad of risk of Alzheimer’s disease. J. Steroid Biochem. Mol. Biol. 2016;160:134–147. doi: 10.1016/j.jsbmb.2016.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Petr M.A., Tulika T., Carmona-Marin L.M., Scheibye-Knudsen M. Protecting the Aging Genome. Trends Cell Biol. 2020:S0962–8924. doi: 10.1016/j.tcb.2019.12.001. [DOI] [PubMed] [Google Scholar]
- 37.Hou Y., Song H., Croteau D.L., Akbari M., Bohr V.A. Genome instability in Alzheimer disease. Mech. Ageing Dev. 2017;161(Pt A):83–94. doi: 10.1016/j.mad.2016.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shamseer L., Moher D., Clarke M., Ghersi D., Liberati A., Petticrew M., Shekelle P., Stewart L.A., Group P.-P. Preferred reporting items for systematic review and meta-analysis protocols (PRISMA-P) 2015: Elaboration and explanation. BMJ. 2015;350:g7647. doi: 10.1136/bmj.g7647. [DOI] [PubMed] [Google Scholar]
- 39.Moher D., Shamseer L., Clarke M., Ghersi D., Liberati A., Petticrew M., Shekelle P., Stewart L.A., Group P.-P. Preferred reporting items for systematic review and meta-analysis protocols (PRISMA-P) 2015 statement. Syst. Rev. 2015;4:1. doi: 10.1186/2046-4053-4-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dolle M.E., Giese H., Hopkins C.L., Martus H.J., Hausdorff J.M., Vijg J. Rapid accumulation of genome rearrangements in liver but not in brain of old mice. Nat. Genet. 1997;17:431–434. doi: 10.1038/ng1297-431. [DOI] [PubMed] [Google Scholar]
- 41.Sedelnikova O.A., Horikawa I., Zimonjic D.B., Popescu N.C., Bonner W.M., Barrett J.C. Senescing human cells and ageing mice accumulate DNA lesions with unrepairable double-strand breaks. Nat. Cell Biol. 2004;6:168–170. doi: 10.1038/ncb1095. [DOI] [PubMed] [Google Scholar]
- 42.Lin X., Gu Y., Tang D. BMI1, ATM and DDR. Oncoscience. 2015;2:665–666. doi: 10.18632/oncoscience.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Myung N.H., Zhu X., Kruman I.I., Castellani R.J., Petersen R.B., Siedlak S.L., Perry G., Smith M.A., Lee H.G. Evidence of DNA damage in Alzheimer disease: Phosphorylation of histone H2AX in astrocytes. Age. 2008;30:209–215. doi: 10.1007/s11357-008-9050-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shanbhag N.M., Evans M.D., Mao W., Nana A.L., Seeley W.W., Adame A., Rissman R.A., Masliah E., Mucke L. Early neuronal accumulation of DNA double strand breaks in Alzheimer’s disease. Acta Neuropathol. Commun. 2019;7:77. doi: 10.1186/s40478-019-0723-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kirova A.M., Bays R.B., Lagalwar S. Working memory and executive function decline across normal aging, mild cognitive impairment, and Alzheimer’s disease. Biomed. Res. Int. 2015;2015:748212. doi: 10.1155/2015/748212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Korolev I.O., Symonds L.L., Bozoki A.C. Alzheimer’s Disease Neuroimaging, I. Predicting Progression from Mild Cognitive Impairment to Alzheimer’s Dementia Using Clinical, MRI, and Plasma Biomarkers via Probabilistic Pattern Classification. PLoS ONE. 2016;11:e0138866. doi: 10.1371/journal.pone.0138866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Adamec E., Vonsattel J.P., Nixon R.A. DNA strand breaks in Alzheimer’s disease. Brain Res. 1999;849:67–77. doi: 10.1016/S0006-8993(99)02004-1. [DOI] [PubMed] [Google Scholar]
- 48.Canugovi C., Misiak M., Ferrarelli L.K., Croteau D.L., Bohr V.A. The role of DNA repair in brain related disease pathology. DNA Repair. 2013;12:578–587. doi: 10.1016/j.dnarep.2013.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hegde M.L., Mantha A.K., Hazra T.K., Bhakat K.K., Mitra S., Szczesny B. Oxidative genome damage and its repair: Implications in aging and neurodegenerative diseases. Mech. Ageing Dev. 2012;133:157–168. doi: 10.1016/j.mad.2012.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Shulman R.G., Rothman D.L., Behar K.L., Hyder F. Energetic basis of brain activity: Implications for neuroimaging. Trends Neurosci. 2004;27:489–495. doi: 10.1016/j.tins.2004.06.005. [DOI] [PubMed] [Google Scholar]
- 51.Gabbita S.P., Lovell M.A., Markesbery W.R. Increased nuclear DNA oxidation in the brain in Alzheimer’s disease. J. Neurochem. 1998;71:2034–2040. doi: 10.1046/j.1471-4159.1998.71052034.x. [DOI] [PubMed] [Google Scholar]
- 52.Lyras L., Cairns N.J., Jenner A., Jenner P., Halliwell B. An assessment of oxidative damage to proteins, lipids, and DNA in brain from patients with Alzheimer’s disease. J. Neurochem. 1997;68:2061–2069. doi: 10.1046/j.1471-4159.1997.68052061.x. [DOI] [PubMed] [Google Scholar]
- 53.Lovell M.A., Markesbery W.R. Oxidative DNA damage in mild cognitive impairment and late-stage Alzheimer’s disease. Nucleic Acids Res. 2007;35:7497–7504. doi: 10.1093/nar/gkm821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wang J., Markesbery W.R., Lovell M.A. Increased oxidative damage in nuclear and mitochondrial DNA in mild cognitive impairment. J. Neurochem. 2006;96:825–832. doi: 10.1111/j.1471-4159.2005.03615.x. [DOI] [PubMed] [Google Scholar]
- 55.Bradley-Whitman M.A., Timmons M.D., Beckett T.L., Murphy M.P., Lynn B.C., Lovell M.A. Nucleic acid oxidation: An early feature of Alzheimer’s disease. J. Neurochem. 2014;128:294–304. doi: 10.1111/jnc.12444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lovell M.A., Soman S., Bradley M.A. Oxidatively modified nucleic acids in preclinical Alzheimer’s disease (PCAD) brain. Mech. Ageing Dev. 2011;132:443–448. doi: 10.1016/j.mad.2011.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Schmitt F.A., Davis D.G., Wekstein D.R., Smith C.D., Ashford J.W., Markesbery W.R. “Preclinical” AD revisited: Neuropathology of cognitively normal older adults. Neurology. 2000;55:370–376. doi: 10.1212/WNL.55.3.370. [DOI] [PubMed] [Google Scholar]
- 58.Lovell M.A., Gabbita S.P., Markesbery W.R. Increased DNA oxidation and decreased levels of repair products in Alzheimer’s disease ventricular CSF. J. Neurochem. 1999;72:771–776. doi: 10.1046/j.1471-4159.1999.0720771.x. [DOI] [PubMed] [Google Scholar]
- 59.Brasnjevic I., Hof P.R., Steinbusch H.W., Schmitz C. Accumulation of nuclear DNA damage or neuron loss: Molecular basis for a new approach to understanding selective neuronal vulnerability in neurodegenerative diseases. DNA Repair. 2008;7:1087–1097. doi: 10.1016/j.dnarep.2008.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Korr H., Thorsten Rohde H., Benders J., Dafotakis M., Grolms N., Schmitz C. Neuron loss during early adulthood following prenatal low-dose X-irradiation in the mouse brain. Int. J. Radiat. Biol. 2001;77:567–580. doi: 10.1080/09553000010028467. [DOI] [PubMed] [Google Scholar]
- 61.Rutten B.P., Schmitz C., Gerlach O.H., Oyen H.M., de Mesquita E.B., Steinbusch H.W., Korr H. The aging brain: Accumulation of DNA damage or neuron loss? Neurobiol. Aging. 2007;28:91–98. doi: 10.1016/j.neurobiolaging.2005.10.019. [DOI] [PubMed] [Google Scholar]
- 62.McKinnon P.J. Genome integrity and disease prevention in the nervous system. Genes Dev. 2017;31:1180–1194. doi: 10.1101/gad.301325.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Suberbielle E., Sanchez P.E., Kravitz A.V., Wang X., Ho K., Eilertson K., Devidze N., Kreitzer A.C., Mucke L. Physiologic brain activity causes DNA double-strand breaks in neurons, with exacerbation by amyloid-beta. Nat. Neurosci. 2013;16:613–621. doi: 10.1038/nn.3356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Marnef A., Cohen S., Legube G. Transcription-Coupled DNA Double-Strand Break Repair: Active Genes Need Special Care. J. Mol. Biol. 2017;429:1277–1288. doi: 10.1016/j.jmb.2017.03.024. [DOI] [PubMed] [Google Scholar]
- 65.Madabhushi R., Gao F., Pfenning A.R., Pan L., Yamakawa S., Seo J., Rueda R., Phan T.X., Yamakawa H., Pao P.C., et al. Activity-Induced DNA Breaks Govern the Expression of Neuronal Early-Response Genes. Cell. 2015;161:1592–1605. doi: 10.1016/j.cell.2015.05.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Crowe S.L., Movsesyan V.A., Jorgensen T.J., Kondratyev A. Rapid phosphorylation of histone H2A.X following ionotropic glutamate receptor activation. Eur. J. Neurosci. 2006;23:2351–2361. doi: 10.1111/j.1460-9568.2006.04768.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.West A.E., Greenberg M.E. Neuronal activity-regulated gene transcription in synapse development and cognitive function. Cold Spring Harb. Perspect. Biol. 2011;3:a005744. doi: 10.1101/cshperspect.a005744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Cholewa-Waclaw J., Bird A., von Schimmelmann M., Schaefer A., Yu H., Song H., Madabhushi R., Tsai L.H. The Role of Epigenetic Mechanisms in the Regulation of Gene Expression in the Nervous System. J. Neurosci. Off. J. Soc. Neurosci. 2016;36:11427–11434. doi: 10.1523/JNEUROSCI.2492-16.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ju B.G., Lunyak V.V., Perissi V., Garcia-Bassets I., Rose D.W., Glass C.K., Rosenfeld M.G. A topoisomerase IIbeta-mediated dsDNA break required for regulated transcription. Science. 2006;312:1798–1802. doi: 10.1126/science.1127196. [DOI] [PubMed] [Google Scholar]
- 70.Bunch H., Lawney B.P., Lin Y.F., Asaithamby A., Murshid A., Wang Y.E., Chen B.P., Calderwood S.K. Transcriptional elongation requires DNA break-induced signalling. Nat. Commun. 2015;6:10191. doi: 10.1038/ncomms10191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Haffner M.C., Aryee M.J., Toubaji A., Esopi D.M., Albadine R., Gurel B., Isaacs W.B., Bova G.S., Liu W., Xu J., et al. Androgen-induced TOP2B-mediated double-strand breaks and prostate cancer gene rearrangements. Nat. Genet. 2010;42:668–675. doi: 10.1038/ng.613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Williamson L.M., Lees-Miller S.P. Estrogen receptor alpha-mediated transcription induces cell cycle-dependent DNA double-strand breaks. Carcinogenesis. 2011;32:279–285. doi: 10.1093/carcin/bgq255. [DOI] [PubMed] [Google Scholar]
- 73.Trotter K.W., King H.A., Archer T.K. Glucocorticoid Receptor Transcriptional Activation via the BRG1-Dependent Recruitment of TOP2beta and Ku70/86. Mol. Cell. Biol. 2015;35:2799–2817. doi: 10.1128/MCB.00230-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Austin C.A., Lee K.C., Swan R.L., Khazeem M.M., Manville C.M., Cridland P., Treumann A., Porter A., Morris N.J., Cowell I.G. TOP2B: The First Thirty Years. Int. J. Mol. Sci. 2018;19:2765. doi: 10.3390/ijms19092765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Harkin L.F., Gerrelli D., Gold Diaz D.C., Santos C., Alzu’bi A., Austin C.A., Clowry G.J. Distinct expression patterns for type II topoisomerases IIA and IIB in the early foetal human telencephalon. J. Anat. 2016;228:452–463. doi: 10.1111/joa.12416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.King I.F., Yandava C.N., Mabb A.M., Hsiao J.S., Huang H.S., Pearson B.L., Calabrese J.M., Starmer J., Parker J.S., Magnuson T., et al. Topoisomerases facilitate transcription of long genes linked to autism. Nature. 2013;501:58–62. doi: 10.1038/nature12504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Gorbunova V., Seluanov A. DNA double strand break repair, aging and the chromatin connection. Mutat. Res. 2016;788:2–6. doi: 10.1016/j.mrfmmm.2016.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Su J.H., Deng G., Cotman C.W. Neuronal DNA damage precedes tangle formation and is associated with up-regulation of nitrotyrosine in Alzheimer’s disease brain. Brain Res. 1997;774:193–199. doi: 10.1016/S0006-8993(97)81703-9. [DOI] [PubMed] [Google Scholar]
- 79.Sheng J.G., Mrak R.E., Griffin W.S. Progressive neuronal DNA damage associated with neurofibrillary tangle formation in Alzheimer disease. J. Neuropathol. Exp. Neurol. 1998;57:323–328. doi: 10.1097/00005072-199804000-00003. [DOI] [PubMed] [Google Scholar]
- 80.Ceccaldi R., Rondinelli B., D’Andrea A.D. Repair Pathway Choices and Consequences at the Double-Strand Break. Trends Cell Biol. 2016;26:52–64. doi: 10.1016/j.tcb.2015.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Zhou B.B., Elledge S.J. The DNA damage response: Putting checkpoints in perspective. Nature. 2000;408:433–439. doi: 10.1038/35044005. [DOI] [PubMed] [Google Scholar]
- 82.Smith J., Tho L.M., Xu N., Gillespie D.A. The ATM-Chk2 and ATR-Chk1 pathways in DNA damage signaling and cancer. Adv. Cancer Res. 2010;108:73–112. doi: 10.1016/B978-0-12-380888-2.00003-0. [DOI] [PubMed] [Google Scholar]
- 83.Cassimere E.K., Mauvais C., Denicourt C. p27Kip1 Is Required to Mediate a G1 Cell Cycle Arrest Downstream of ATM following Genotoxic Stress. PLoS ONE. 2016;11:e0162806. doi: 10.1371/journal.pone.0162806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Lin X., Yan J., Tang D. ERK kinases modulate the activation of PI3 kinase related kinases (PIKKs) in DNA damage response. Histol. Histopathol. 2013;28:1547–1554. doi: 10.14670/HH-28.1547. [DOI] [PubMed] [Google Scholar]
- 85.Jin M.H., Oh D.Y. ATM in DNA repair in cancer. Pharmacol. Ther. 2019;203:107391. doi: 10.1016/j.pharmthera.2019.07.002. [DOI] [PubMed] [Google Scholar]
- 86.Trenner A., Sartori A.A. Harnessing DNA Double-Strand Break Repair for Cancer Treatment. Front. Oncol. 2019;9:1388. doi: 10.3389/fonc.2019.01388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Biton S., Barzilai A., Shiloh Y. The neurological phenotype of ataxia-telangiectasia: Solving a persistent puzzle. DNA Repair. 2008;7:1028–1038. doi: 10.1016/j.dnarep.2008.03.006. [DOI] [PubMed] [Google Scholar]
- 88.Shen X., Chen J., Li J., Kofler J., Herrup K. Neurons in Vulnerable Regions of the Alzheimer’s Disease Brain Display Reduced ATM Signaling. eNeuro. 2016;3:ENEURO.0124-15.2016. doi: 10.1523/ENEURO.0124-15.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Wezyk M., Szybinska A., Wojsiat J., Szczerba M., Day K., Ronnholm H., Kele M., Berdynski M., Peplonska B., Fichna J.P., et al. Overactive BRCA1 Affects Presenilin 1 in Induced Pluripotent Stem Cell-Derived Neurons in Alzheimer’s Disease. J. Alzheimer’s Dis. JAD. 2018;62:175–202. doi: 10.3233/JAD-170830. [DOI] [PubMed] [Google Scholar]
- 90.Welty S., Teng Y., Liang Z., Zhao W., Sanders L.H., Greenamyre J.T., Rubio M.E., Thathiah A., Kodali R., Wetzel R., et al. RAD52 is required for RNA-templated recombination repair in post-mitotic neurons. J. Biol. Chem. 2018;293:1353–1362. doi: 10.1074/jbc.M117.808402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Yu H., Harrison F.E., Xia F. Altered DNA repair; an early pathogenic pathway in Alzheimer’s disease and obesity. Sci. Rep. 2018;8:5600. doi: 10.1038/s41598-018-23644-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Mano T., Nagata K., Nonaka T., Tarutani A., Imamura T., Hashimoto T., Bannai T., Koshi-Mano K., Tsuchida T., Ohtomo R., et al. Neuron-specific methylome analysis reveals epigenetic regulation and tau-related dysfunction of BRCA1 in Alzheimer’s disease. Proc. Natl. Acad. Sci. USA. 2017;114:E9645–E9654. doi: 10.1073/pnas.1707151114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Nakamura M., Kaneko S., Dickson D.W., Kusaka H. Aberrant Accumulation of BRCA1 in Alzheimer Disease and Other Tauopathies. J. Neuropathol. Exp. Neurol. 2020;79:22–33. doi: 10.1093/jnen/nlz107. [DOI] [PubMed] [Google Scholar]
- 94.Guenette S., Chang Y., Hiesberger T., Richardson J.A., Eckman C.B., Eckman E.A., Hammer R.E., Herz J. Essential roles for the FE65 amyloid precursor protein-interacting proteins in brain development. EMBO J. 2006;25:420–431. doi: 10.1038/sj.emboj.7600926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Kesavapany S., Banner S.J., Lau K.F., Shaw C.E., Miller C.C., Cooper J.D., McLoughlin D.M. Expression of the Fe65 adapter protein in adult and developing mouse brain. Neuroscience. 2002;115:951–960. doi: 10.1016/S0306-4522(02)00422-0. [DOI] [PubMed] [Google Scholar]
- 96.Minopoli G., Gargiulo A., Parisi S., Russo T. Fe65 matters: New light on an old molecule. Iubmb Life. 2012;64:936–942. doi: 10.1002/iub.1094. [DOI] [PubMed] [Google Scholar]
- 97.Ryu S., Teles F., Minopoli G., Russo T., Rosenfeld M.G., Suh Y. An epigenomic role of Fe65 in the cellular response to DNA damage. Mutat. Res. 2015;776:40–47. doi: 10.1016/j.mrfmmm.2015.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Sun Y., Jiang X., Chen S., Fernandes N., Price B.D. A role for the Tip60 histone acetyltransferase in the acetylation and activation of ATM. Proc. Natl. Acad. Sci. USA. 2005;102:13182–13187. doi: 10.1073/pnas.0504211102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Sun Y., Xu Y., Roy K., Price B.D. DNA damage-induced acetylation of lysine 3016 of ATM activates ATM kinase activity. Mol. Cell. Biol. 2007;27:8502–8509. doi: 10.1128/MCB.01382-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Jowsey P.A., Blain P.G. Fe65 Ser228 is phosphorylated by ATM/ATR and inhibits Fe65-APP-mediated gene transcription. Biochem. J. 2015;465:413–421. doi: 10.1042/BJ20140656. [DOI] [PubMed] [Google Scholar]
- 101.Langlands H., Blain P.G., Jowsey P.A. Fe65 Is Phosphorylated on Ser289 after UV-Induced DNA Damage. PLoS ONE. 2016;11:e0155056. doi: 10.1371/journal.pone.0155056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Itzhaki R.F., Wozniak M.A. Herpes simplex virus type 1 in Alzheimer’s disease: The enemy within. J. Alzheimer’s Dis. JAD. 2008;13:393–405. doi: 10.3233/JAD-2008-13405. [DOI] [PubMed] [Google Scholar]
- 103.Ball M.J., Lukiw W.J., Kammerman E.M., Hill J.M. Intracerebral propagation of Alzheimer’s disease: Strengthening evidence of a herpes simplex virus etiology. Alzheimer’s Dement. J. Alzheimer’s Assoc. 2013;9:169–175. doi: 10.1016/j.jalz.2012.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Ezzat K., Pernemalm M., Palsson S., Roberts T.C., Jarver P., Dondalska A., Bestas B., Sobkowiak M.J., Levanen B., Skold M., et al. The viral protein corona directs viral pathogenesis and amyloid aggregation. Nat. Commun. 2019;10:2331. doi: 10.1038/s41467-019-10192-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.De Chiara G., Racaniello M., Mollinari C., Marcocci M.E., Aversa G., Cardinale A., Giovanetti A., Garaci E., Palamara A.T., Merlo D. Herpes Simplex Virus-Type1 (HSV-1) Impairs DNA Repair in Cortical Neurons. Front. Aging Neurosci. 2016;8:242. doi: 10.3389/fnagi.2016.00242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Wei F., Yan J., Tang D., Lin X., He L., Xie Y., Tao L., Wang S. Inhibition of ERK activation enhances the repair of double-stranded breaks via non-homologous end joining by increasing DNA-PKcs activation. Biochim. Biophys. Acta. 2013;1833:90–100. doi: 10.1016/j.bbamcr.2012.10.016. [DOI] [PubMed] [Google Scholar]
- 107.Jette N., Lees-Miller S.P. The DNA-dependent protein kinase: A multifunctional protein kinase with roles in DNA double strand break repair and mitosis. Prog. Biophys. Mol. Biol. 2015;117:194–205. doi: 10.1016/j.pbiomolbio.2014.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Davydov V., Hansen L.A., Shackelford D.A. Is DNA repair compromised in Alzheimer’s disease? Neurobiol. Aging. 2003;24:953–968. doi: 10.1016/S0197-4580(02)00229-4. [DOI] [PubMed] [Google Scholar]
- 109.Simpson J.E., Ince P.G., Haynes L.J., Theaker R., Gelsthorpe C., Baxter L., Forster G., Lace G.L., Shaw P.J., Matthews F.E., et al. Population variation in oxidative stress and astrocyte DNA damage in relation to Alzheimer-type pathology in the ageing brain. Neuropathol. Appl. Neurobiol. 2010;36:25–40. doi: 10.1111/j.1365-2990.2009.01030.x. [DOI] [PubMed] [Google Scholar]
- 110.Culmsee C., Bondada S., Mattson M.P. Hippocampal neurons of mice deficient in DNA-dependent protein kinase exhibit increased vulnerability to DNA damage, oxidative stress and excitotoxicity. Brain Res. Mol. Brain Res. 2001;87:257–262. doi: 10.1016/S0169-328X(01)00008-0. [DOI] [PubMed] [Google Scholar]
- 111.Cardinale A., Racaniello M., Saladini S., De Chiara G., Mollinari C., de Stefano M.C., Pocchiari M., Garaci E., Merlo D. Sublethal doses of beta-amyloid peptide abrogate DNA-dependent protein kinase activity. J. Biol. Chem. 2012;287:2618–2631. doi: 10.1074/jbc.M111.276550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Garcia-Muse T., Aguilera A. Transcription-replication conflicts: How they occur and how they are resolved. Nat. Rev. Mol. Cell Biol. 2016;17:553–563. doi: 10.1038/nrm.2016.88. [DOI] [PubMed] [Google Scholar]
- 113.Lindahl T. Instability and decay of the primary structure of DNA. Nature. 1993;362:709–715. doi: 10.1038/362709a0. [DOI] [PubMed] [Google Scholar]
- 114.Vilenchik M.M., Knudson A.G. Endogenous DNA double-strand breaks: Production, fidelity of repair, and induction of cancer. Proc. Natl. Acad. Sci. USA. 2003;100:12871–12876. doi: 10.1073/pnas.2135498100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Hazra T.K., Das A., Das S., Choudhury S., Kow Y.W., Roy R. Oxidative DNA damage repair in mammalian cells: A new perspective. DNA Repair. 2007;6:470–480. doi: 10.1016/j.dnarep.2006.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Wallace S.S. Base excision repair: A critical player in many games. DNA Repair. 2014;19:14–26. doi: 10.1016/j.dnarep.2014.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Akbari M., Morevati M., Croteau D., Bohr V.A. The role of DNA base excision repair in brain homeostasis and disease. DNA Repair. 2015;32:172–179. doi: 10.1016/j.dnarep.2015.04.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Lovell M.A., Xie C., Markesbery W.R. Decreased base excision repair and increased helicase activity in Alzheimer’s disease brain. Brain Res. 2000;855:116–123. doi: 10.1016/S0006-8993(99)02335-5. [DOI] [PubMed] [Google Scholar]
- 119.Weissman L., Jo D.G., Sorensen M.M., de Souza-Pinto N.C., Markesbery W.R., Mattson M.P., Bohr V.A. Defective DNA base excision repair in brain from individuals with Alzheimer’s disease and amnestic mild cognitive impairment. Nucleic Acids Res. 2007;35:5545–5555. doi: 10.1093/nar/gkm605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Canugovi C., Shamanna R.A., Croteau D.L., Bohr V.A. Base excision DNA repair levels in mitochondrial lysates of Alzheimer’s disease. Neurobiol. Aging. 2014;35:1293–1300. doi: 10.1016/j.neurobiolaging.2014.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Mao G., Pan X., Zhu B.B., Zhang Y., Yuan F., Huang J., Lovell M.A., Lee M.P., Markesbery W.R., Li G.M., et al. Identification and characterization of OGG1 mutations in patients with Alzheimer’s disease. Nucleic Acids Res. 2007;35:2759–2766. doi: 10.1093/nar/gkm189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Jacob K.D., Noren Hooten N., Tadokoro T., Lohani A., Barnes J., Evans M.K. Alzheimer’s disease-associated polymorphisms in human OGG1 alter catalytic activity and sensitize cells to DNA damage. Free Radic. Biol. Med. 2013;63:115–125. doi: 10.1016/j.freeradbiomed.2013.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Canugovi C., Yoon J.S., Feldman N.H., Croteau D.L., Mattson M.P., Bohr V.A. Endonuclease VIII-like 1 (NEIL1) promotes short-term spatial memory retention and protects from ischemic stroke-induced brain dysfunction and death in mice. Proc. Natl. Acad. Sci. USA. 2012;109:14948–14953. doi: 10.1073/pnas.1204156109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Bandaru V., Sunkara S., Wallace S.S., Bond J.P. A novel human DNA glycosylase that removes oxidative DNA damage and is homologous to Escherichia coli endonuclease VIII. DNA Repair. 2002;1:517–529. doi: 10.1016/S1568-7864(02)00036-8. [DOI] [PubMed] [Google Scholar]
- 125.Hazra T.K., Izumi T., Boldogh I., Imhoff B., Kow Y.W., Jaruga P., Dizdaroglu M., Mitra S. Identification and characterization of a human DNA glycosylase for repair of modified bases in oxidatively damaged DNA. Proc. Natl. Acad. Sci. USA. 2002;99:3523–3528. doi: 10.1073/pnas.062053799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Hazra T.K., Kow Y.W., Hatahet Z., Imhoff B., Boldogh I., Mokkapati S.K., Mitra S., Izumi T. Identification and characterization of a novel human DNA glycosylase for repair of cytosine-derived lesions. J. Biol. Chem. 2002;277:30417–30420. doi: 10.1074/jbc.C200355200. [DOI] [PubMed] [Google Scholar]
- 127.Takao M., Kanno S., Shiromoto T., Hasegawa R., Ide H., Ikeda S., Sarker A.H., Seki S., Xing J.Z., Le X.C., et al. Novel nuclear and mitochondrial glycosylases revealed by disruption of the mouse Nth1 gene encoding an endonuclease III homolog for repair of thymine glycols. EMBO J. 2002;21:3486–3493. doi: 10.1093/emboj/cdf350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Englander E.W., Ma H. Differential modulation of base excision repair activities during brain ontogeny: Implications for repair of transcribed DNA. Mech. Ageing Dev. 2006;127:64–69. doi: 10.1016/j.mad.2005.09.008. [DOI] [PubMed] [Google Scholar]
- 129.Love S., Barber R., Wilcock G.K. Increased poly (ADP-ribosyl)ation of nuclear proteins in Alzheimer’s disease. Brain A J. Neurol. 1999;122(Pt 2):247–253. doi: 10.1093/brain/122.2.247. [DOI] [PubMed] [Google Scholar]
- 130.Alano C.C., Garnier P., Ying W., Higashi Y., Kauppinen T.M., Swanson R.A. NAD+ depletion is necessary and sufficient for poly (ADP-ribose) polymerase-1-mediated neuronal death. J. Neurosci. Off. J. Soc. Neurosci. 2010;30:2967–2978. doi: 10.1523/JNEUROSCI.5552-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Martire S., Fuso A., Mosca L., Forte E., Correani V., Fontana M., Scarpa S., Maras B., d’Erme M. Bioenergetic Impairment in Animal and Cellular Models of Alzheimer’s Disease: PARP-1 Inhibition Rescues Metabolic Dysfunctions. J. Alzheimer’s Dis. JAD. 2016;54:307–324. doi: 10.3233/JAD-151040. [DOI] [PubMed] [Google Scholar]
- 132.Strosznajder J.B., Jesko H., Strosznajder R.P. Effect of amyloid beta peptide on poly (ADP-ribose) polymerase activity in adult and aged rat hippocampus. Acta Biochim. Pol. 2000;47:847–854. doi: 10.18388/abp.2000_4003. [DOI] [PubMed] [Google Scholar]
- 133.Strosznajder J.B., Czapski G.A., Adamczyk A., Strosznajder R.P. Poly (ADP-ribose) polymerase-1 in amyloid beta toxicity and Alzheimer’s disease. Mol. Neurobiol. 2012;46:78–84. doi: 10.1007/s12035-012-8258-9. [DOI] [PubMed] [Google Scholar]
- 134.Wu M.F., Yin J.H., Hwang C.S., Tang C.M., Yang D.I. NAD attenuates oxidative DNA damages induced by amyloid beta-peptide in primary rat cortical neurons. Free Radic. Res. 2014;48:794–805. doi: 10.3109/10715762.2014.907889. [DOI] [PubMed] [Google Scholar]
- 135.Zeng J., Libien J., Shaik F., Wolk J., Hernandez A.I. Nucleolar PARP-1 Expression Is Decreased in Alzheimer’s Disease: Consequences for Epigenetic Regulation of rDNA and Cognition. Neural Plast. 2016;2016:8987928. doi: 10.1155/2016/8987928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Hou Y., Lautrup S., Cordonnier S., Wang Y., Croteau D.L., Zavala E., Zhang Y., Moritoh K., O’Connell J.F., Baptiste B.A., et al. NAD+ supplementation normalizes key Alzheimer’s features and DNA damage responses in a new AD mouse model with introduced DNA repair deficiency. Proc. Natl. Acad. Sci. USA. 2018;115:E1876–E1885. doi: 10.1073/pnas.1718819115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Infante J., Llorca J., Mateo I., Rodriguez-Rodriguez E., Sanchez-Quintana C., Sanchez-Juan P., Fernandez-Viadero C., Pena N., Berciano J., Combarros O. Interaction between poly(ADP-ribose) polymerase 1 and interleukin 1A genes is associated with Alzheimer’s disease risk. Dement. Geriatr. Cogn. Disord. 2007;23:215–218. doi: 10.1159/000099471. [DOI] [PubMed] [Google Scholar]
- 138.Liu H.P., Lin W.Y., Wu B.T., Liu S.H., Wang W.F., Tsai C.H., Lee C.C., Tsai F.J. Evaluation of the poly(ADP-ribose) polymerase-1 gene variants in Alzheimer’s disease. J. Clin. Lab. Anal. 2010;24:182–186. doi: 10.1002/jcla.20379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Cabelof D.C., Raffoul J.J., Yanamadala S., Ganir C., Guo Z., Heydari A.R. Attenuation of DNA polymerase beta-dependent base excision repair and increased DMS-induced mutagenicity in aged mice. Mutat. Res. 2002;500:135–145. doi: 10.1016/S0027-5107(02)00003-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Rao K.S., Annapurna V.V., Raji N.S. DNA polymerase-beta may be the main player for defective DNA repair in aging rat neurons. Ann. N. Y. Acad. Sci. 2001;928:113–120. doi: 10.1111/j.1749-6632.2001.tb05641.x. [DOI] [PubMed] [Google Scholar]
- 141.Ahmed A.A., Smoczer C., Pace B., Patterson D., Cress Cabelof D. Loss of DNA polymerase beta induces cellular senescence. Environ. Mol. Mutagenes. 2018;59:603–612. doi: 10.1002/em.22206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Athanasiou K., Sideris E.G., Bartsocas C. Decreased repair of x-ray induced DNA single-strand breaks in lymphocytes in Down’s syndrome. Pediatr. Res. 1980;4(Pt 1):336–338. doi: 10.1203/00006450-198004000-00015. [DOI] [PubMed] [Google Scholar]
- 143.Cabelof D.C., Patel H.V., Chen Q., van Remmen H., Matherly L.H., Ge Y., Taub J.W. Mutational spectrum at GATA1 provides insights into mutagenesis and leukemogenesis in Down syndrome. Blood. 2009;114:2753–2763. doi: 10.1182/blood-2008-11-190330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Esbensen A.J. Health conditions associated with aging and end of life of adults with Down syndrome. Int. Rev. Res. Ment. Retard. 2010;39:107–126. doi: 10.1016/S0074-7750(10)39004-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Hartley D., Blumenthal T., Carrillo M., DiPaolo G., Esralew L., Gardiner K., Granholm A.C., Iqbal K., Krams M., Lemere C., et al. Down syndrome and Alzheimer’s disease: Common pathways, common goals. Alzheimer’s Dement. J. Alzheimer’s Assoc. 2015;11:700–709. doi: 10.1016/j.jalz.2014.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Mann D.M. The pathological association between Down syndrome and Alzheimer disease. Mech. Ageing Dev. 1988;43:99–136. doi: 10.1016/0047-6374(88)90041-3. [DOI] [PubMed] [Google Scholar]
- 147.Wisniewski K.E., Wisniewski H.M., Wen G.Y. Occurrence of neuropathological changes and dementia of Alzheimer’s disease in Down’s syndrome. Ann. Neurol. 1985;17:278–282. doi: 10.1002/ana.410170310. [DOI] [PubMed] [Google Scholar]
- 148.Lai F., Williams R.S. A prospective study of Alzheimer disease in Down syndrome. Arch. Neurol. 1989;46:849–853. doi: 10.1001/archneur.1989.00520440031017. [DOI] [PubMed] [Google Scholar]
- 149.Sykora P., Misiak M., Wang Y., Ghosh S., Leandro G.S., Liu D., Tian J., Baptiste B.A., Cong W.N., Brenerman B.M., et al. DNA polymerase beta deficiency leads to neurodegeneration and exacerbates Alzheimer disease phenotypes. Nucleic Acids Res. 2015;43:943–959. doi: 10.1093/nar/gku1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Misiak M., Vergara Greeno R., Baptiste B.A., Sykora P., Liu D., Cordonnier S., Fang E.F., Croteau D.L., Mattson M.P., Bohr V.A. DNA polymerase beta decrement triggers death of olfactory bulb cells and impairs olfaction in a mouse model of Alzheimer’s disease. Aging Cell. 2017;16:162–172. doi: 10.1111/acel.12541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Dogru-Abbasoglu S., Aykac-Toker G., Hanagasi H.A., Gurvit H., Emre M., Uysal M. The Arg194Trp polymorphism in DNA repair gene XRCC1 and the risk for sporadic late-onset Alzheimer’s disease. Neurol. Sci. Off. J. Ital. Neurol. Soc. Ital. Soc. Clin. Neurophysiol. 2007;28:31–34. doi: 10.1007/s10072-007-0744-x. [DOI] [PubMed] [Google Scholar]
- 152.Qian Y., Chen W., Wu J., Tao T., Bi L., Xu W., Qi H., Wang Y., Guo L. Association of polymorphism of DNA repair gene XRCC1 with sporadic late-onset Alzheimer’s disease and age of onset in elderly Han Chinese. J. Neurol. Sci. 2010;295:62–65. doi: 10.1016/j.jns.2010.05.002. [DOI] [PubMed] [Google Scholar]
- 153.Marcon G., Tell G., Perrone L., Garbelli R., Quadrifoglio F., Tagliavini F., Giaccone G. APE1/Ref-1 in Alzheimer’s disease: An immunohistochemical study. Neurosci. Lett. 2009;466:124–127. doi: 10.1016/j.neulet.2009.09.039. [DOI] [PubMed] [Google Scholar]
- 154.Lillenes M.S., Rabano A., Stoen M., Riaz T., Misaghian D., Mollersen L., Esbensen Y., Gunther C.C., Selnes P., Stenset V.T., et al. Altered DNA base excision repair profile in brain tissue and blood in Alzheimer’s disease. Mol. Brain. 2016;9:61. doi: 10.1186/s13041-016-0237-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Lew J., Huang Q.Q., Qi Z., Winkfein R.J., Aebersold R., Hunt T., Wang J.H. A brain-specific activator of cyclin-dependent kinase 5. Nature. 1994;371:423–426. doi: 10.1038/371423a0. [DOI] [PubMed] [Google Scholar]
- 156.Tsai L.H., Delalle I., Caviness V.S., Jr., Chae T., Harlow E. p35 is a neural-specific regulatory subunit of cyclin-dependent kinase 5. Nature. 1994;371:419–423. doi: 10.1038/371419a0. [DOI] [PubMed] [Google Scholar]
- 157.Tang D., Yeung J., Lee K.Y., Matsushita M., Matsui H., Tomizawa K., Hatase O., Wang J.H. An isoform of the neuronal cyclin-dependent kinase 5 (Cdk5) activator. J. Biol. Chem. 1995;270:26897–26903. doi: 10.1074/jbc.270.45.26897. [DOI] [PubMed] [Google Scholar]
- 158.Sananbenesi F., Fischer A., Wang X., Schrick C., Neve R., Radulovic J., Tsai L.H. A hippocampal Cdk5 pathway regulates extinction of contextual fear. Nat. Neurosci. 2007;10:1012–1019. doi: 10.1038/nn1943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Liu S.L., Wang C., Jiang T., Tan L., Xing A., Yu J.T. The Role of Cdk5 in Alzheimer’s Disease. Mol. Neurobiol. 2016;53:4328–4342. doi: 10.1007/s12035-015-9369-x. [DOI] [PubMed] [Google Scholar]
- 160.Su S.C., Tsai L.H. Cyclin-dependent kinases in brain development and disease. Annu. Rev. Cell Dev. Biol. 2011;27:465–491. doi: 10.1146/annurev-cellbio-092910-154023. [DOI] [PubMed] [Google Scholar]
- 161.Liu F., Su Y., Li B., Zhou Y., Ryder J., Gonzalez-DeWhitt P., May P.C., Ni B. Regulation of amyloid precursor protein (APP) phosphorylation and processing by p35/Cdk5 and p25/Cdk5. Febs Lett. 2003;547:193–196. doi: 10.1016/S0014-5793(03)00714-2. [DOI] [PubMed] [Google Scholar]
- 162.Zheng Y.L., Kesavapany S., Gravell M., Hamilton R.S., Schubert M., Amin N., Albers W., Grant P., Pant H.C. A Cdk5 inhibitory peptide reduces tau hyperphosphorylation and apoptosis in neurons. EMBO J. 2005;24:209–220. doi: 10.1038/sj.emboj.7600441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Piedrahita D., Hernandez I., Lopez-Tobon A., Fedorov D., Obara B., Manjunath B.S., Boudreau R.L., Davidson B., Laferla F., Gallego-Gomez J.C., et al. Silencing of CDK5 reduces neurofibrillary tangles in transgenic alzheimer’s mice. J. Neurosci. Off. J. Soc. Neurosci. 2010;30:13966–13976. doi: 10.1523/JNEUROSCI.3637-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Park J., Choi H., Min J.S., Kim B., Lee S.R., Yun J.W., Choi M.S., Chang K.T., Lee D.S. Loss of mitofusin 2 links beta-amyloid-mediated mitochondrial fragmentation and Cdk5-induced oxidative stress in neuron cells. J. Neurochem. 2015;132:687–702. doi: 10.1111/jnc.12984. [DOI] [PubMed] [Google Scholar]
- 165.Weishaupt J.H., Kussmaul L., Grotsch P., Heckel A., Rohde G., Romig H., Bahr M., Gillardon F. Inhibition of CDK5 is protective in necrotic and apoptotic paradigms of neuronal cell death and prevents mitochondrial dysfunction. Mol. Cell. Neurosci. 2003;24:489–502. doi: 10.1016/S1044-7431(03)00221-5. [DOI] [PubMed] [Google Scholar]
- 166.Kim D., Frank C.L., Dobbin M.M., Tsunemoto R.K., Tu W., Peng P.L., Guan J.S., Lee B.H., Moy L.Y., Giusti P., et al. Deregulation of HDAC1 by p25/Cdk5 in neurotoxicity. Neuron. 2008;60:803–817. doi: 10.1016/j.neuron.2008.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Mostafa H.H., van Loben Sels J.M., Davido D.J. Herpes simplex virus 1 upregulates p35, alters CDK-5 localization, and stimulates CDK-5 kinase activity during acute infection in neurons. J. Virol. 2015;89:5171–5175. doi: 10.1128/JVI.00106-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Tian B., Yang Q., Mao Z. Phosphorylation of ATM by Cdk5 mediates DNA damage signalling and regulates neuronal death. Nat. Cell Biol. 2009;11:211–218. doi: 10.1038/ncb1829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Robin G., Lopez J.R., Espinal G.M., Hulsizer S., Hagerman P.J., Pessah I.N. Calcium dysregulation and Cdk5-ATM pathway involved in a mouse model of fragile X-associated tremor/ataxia syndrome. Hum. Mol. Genet. 2017;26:2649–2666. doi: 10.1093/hmg/ddx148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Zheng X.F., Acharya S.S., Choe K.N., Nikhil K., Adelmant G., Satapathy S.R., Sharma S., Viccaro K., Rana S., Natarajan A., et al. A mitotic CDK5-PP4 phospho-signaling cascade primes 53BP1 for DNA repair in G1. Nat. Commun. 2019;10:4252. doi: 10.1038/s41467-019-12084-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Huang E., Qu D., Zhang Y., Venderova K., Haque M.E., Rousseaux M.W., Slack R.S., Woulfe J.M., Park D.S. The role of Cdk5-mediated apurinic/apyrimidinic endonuclease 1 phosphorylation in neuronal death. Nat. Cell Biol. 2010;12:563–571. doi: 10.1038/ncb2058. [DOI] [PubMed] [Google Scholar]
- 172.Mohamad Nasir N.F., Zainuddin A., Shamsuddin S. Emerging Roles of Sirtuin 6 in Alzheimer’s Disease. J. Mol. Neurosci. 2018;64:157–161. doi: 10.1007/s12031-017-1005-y. [DOI] [PubMed] [Google Scholar]
- 173.Khan R.I., Nirzhor S.S.R., Akter R. A Review of the Recent Advances Made with SIRT6 and its Implications on Aging Related Processes, Major Human Diseases, and Possible Therapeutic Targets. Biomolecules. 2018;8:44. doi: 10.3390/biom8030044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Xiao C., Wang R.H., Lahusen T.J., Park O., Bertola A., Maruyama T., Reynolds D., Chen Q., Xu X., Young H.A., et al. Progression of chronic liver inflammation and fibrosis driven by activation of c-JUN signaling in Sirt6 mutant mice. J. Biol. Chem. 2012;287:41903–41913. doi: 10.1074/jbc.M112.415182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Michishita E., McCord R.A., Berber E., Kioi M., Padilla-Nash H., Damian M., Cheung P., Kusumoto R., Kawahara T.L., Barrett J.C., et al. SIRT6 is a histone H3 lysine 9 deacetylase that modulates telomeric chromatin. Nature. 2008;452:492–496. doi: 10.1038/nature06736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Mostoslavsky R., Chua K.F., Lombard D.B., Pang W.W., Fischer M.R., Gellon L., Liu P., Mostoslavsky G., Franco S., Murphy M.M., et al. Genomic instability and aging-like phenotype in the absence of mammalian SIRT6. Cell. 2006;124:315–329. doi: 10.1016/j.cell.2005.11.044. [DOI] [PubMed] [Google Scholar]
- 177.Kaidi A., Weinert B.T., Choudhary C., Jackson S.P. Human SIRT6 promotes DNA end resection through CtIP deacetylation. Science. 2010;329:1348–1353. doi: 10.1126/science.1192049. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 178.Mao Z., Hine C., Tian X., Van Meter M., Au M., Vaidya A., Seluanov A., Gorbunova V. SIRT6 promotes DNA repair under stress by activating PARP1. Science. 2011;332:1443–1446. doi: 10.1126/science.1202723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Kaluski S., Portillo M., Besnard A., Stein D., Einav M., Zhong L., Ueberham U., Arendt T., Mostoslavsky R., Sahay A., et al. Neuroprotective Functions for the Histone Deacetylase SIRT6. Cell Rep. 2017;18:3052–3062. doi: 10.1016/j.celrep.2017.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Jung E.S., Choi H., Song H., Hwang Y.J., Kim A., Ryu H., Mook-Jung I. p53-dependent SIRT6 expression protects Abeta42-induced DNA damage. Sci. Rep. 2016;6:25628. doi: 10.1038/srep25628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Chua C.E.L., Tang B.L. miR-34a in Neurophysiology and Neuropathology. J. Mol. Neurosci. 2019;67:235–246. doi: 10.1007/s12031-018-1231-y. [DOI] [PubMed] [Google Scholar]
- 182.Okun E., Marton D., Cohen D., Griffioen K., Kanfi Y., Illouz T., Madar R., Cohen H.Y. Sirt6 alters adult hippocampal neurogenesis. PLoS ONE. 2017;12:e0179681. doi: 10.1371/journal.pone.0179681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Bacigaluppi M., Sferruzza G., Butti E., Ottoboni L., Martino G. Endogenous neural precursor cells in health and disease. Brain Res. 2019;1730:146619. doi: 10.1016/j.brainres.2019.146619. [DOI] [PubMed] [Google Scholar]
- 184.Kieron M., Zekanowski C., Falk A., Wezyk M. Oxidative DNA Damage Signalling in Neural Stem Cells in Alzheimer’s Disease. Oxidative Med. Cell. Longev. 2019;2019:2149812. doi: 10.1155/2019/2149812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Park I.K., Morrison S.J., Clarke M.F. Bmi1, stem cells, and senescence regulation. J. Clin. Investig. 2004;113:175–179. doi: 10.1172/JCI200420800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Park I.K., Qian D., Kiel M., Becker M.W., Pihalja M., Weissman I.L., Morrison S.J., Clarke M.F. Bmi-1 is required for maintenance of adult self-renewing haematopoietic stem cells. Nature. 2003;423:302–305. doi: 10.1038/nature01587. [DOI] [PubMed] [Google Scholar]
- 187.Molofsky A.V., He S., Bydon M., Morrison S.J., Pardal R. Bmi-1 promotes neural stem cell self-renewal and neural development but not mouse growth and survival by repressing the p16Ink4a and p19Arf senescence pathways. Genes Dev. 2005;19:1432–1437. doi: 10.1101/gad.1299505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Molofsky A.V., Pardal R., Iwashita T., Park I.K., Clarke M.F., Morrison S.J. Bmi-1 dependence distinguishes neural stem cell self-renewal from progenitor proliferation. Nature. 2003;425:962–967. doi: 10.1038/nature02060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Bruggeman S.W., Valk-Lingbeek M.E., van der Stoop P.P., Jacobs J.J., Kieboom K., Tanger E., Hulsman D., Leung C., Arsenijevic Y., Marino S., et al. Ink4a and Arf differentially affect cell proliferation and neural stem cell self-renewal in Bmi1-deficient mice. Genes Dev. 2005;19:1438–1443. doi: 10.1101/gad.1299305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Oguro H., Iwama A., Morita Y., Kamijo T., van Lohuizen M., Nakauchi H. Differential impact of Ink4a and Arf on hematopoietic stem cells and their bone marrow microenvironment in Bmi1-deficient mice. J. Exp. Med. 2006;203:2247–2253. doi: 10.1084/jem.20052477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Lopez-Arribillaga E., Rodilla V., Pellegrinet L., Guiu J., Iglesias M., Roman A.C., Gutarra S., Gonzalez S., Munoz-Canoves P., Fernandez-Salguero P., et al. Bmi1 regulates murine intestinal stem cell proliferation and self-renewal downstream of Notch. Development. 2015;142:41–50. doi: 10.1242/dev.107714. [DOI] [PubMed] [Google Scholar]
- 192.Sherr C.J. Tumor surveillance via the ARF-p53 pathway. Genes Dev. 1998;12:2984–2991. doi: 10.1101/gad.12.19.2984. [DOI] [PubMed] [Google Scholar]
- 193.Robertson K.D., Jones P.A. Tissue-specific alternative splicing in the human INK4a/ARF cell cycle regulatory locus. Oncogene. 1999;18:3810–3820. doi: 10.1038/sj.onc.1202737. [DOI] [PubMed] [Google Scholar]
- 194.Ismail I.H., Andrin C., McDonald D., Hendzel M.J. BMI1-mediated histone ubiquitylation promotes DNA double-strand break repair. J. Cell Biol. 2010;191:45–60. doi: 10.1083/jcb.201003034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Facchino S., Abdouh M., Chatoo W., Bernier G. BMI1 confers radioresistance to normal and cancerous neural stem cells through recruitment of the DNA damage response machinery. J. Neurosci. Off. J. Soc. Neurosci. 2010;30:10096–10111. doi: 10.1523/JNEUROSCI.1634-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Chagraoui J., Hebert J., Girard S., Sauvageau G. An anticlastogenic function for the Polycomb Group gene Bmi1. Proc. Natl. Acad. Sci. USA. 2011;108:5284–5289. doi: 10.1073/pnas.1014263108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Lin X., Ojo D., Wei F., Wong N., Gu Y., Tang D. A Novel Aspect of Tumorigenesis-BMI1 Functions in Regulating DNA Damage Response. Biomolecules. 2015;5:3396–3415. doi: 10.3390/biom5043396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.El Hajjar J., Chatoo W., Hanna R., Nkanza P., Tetreault N., Tse Y.C., Wong T.P., Abdouh M., Bernier G. Heterochromatic genome instability and neurodegeneration sharing similarities with Alzheimer’s disease in old Bmi1+/− mice. Sci. Rep. 2019;9:594. doi: 10.1038/s41598-018-37444-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Flamier A., El Hajjar J., Adjaye J., Fernandes K.J., Abdouh M., Bernier G. Modeling Late-Onset Sporadic Alzheimer’s Disease through BMI1 Deficiency. Cell Rep. 2018;23:2653–2666. doi: 10.1016/j.celrep.2018.04.097. [DOI] [PubMed] [Google Scholar]
- 200.Aparicio T., Baer R., Gautier J. DNA double-strand break repair pathway choice and cancer. DNA Repair. 2014;19:169–175. doi: 10.1016/j.dnarep.2014.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Frade J.M., Ovejero-Benito M.C. Neuronal cell cycle: The neuron itself and its circumstances. Cell Cycle. 2015;14:712–720. doi: 10.1080/15384101.2015.1004937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Lu J., Li Y., Mollinari C., Garaci E., Merlo D., Pei G. Amyloid-beta Oligomers-induced Mitochondrial DNA Repair Impairment Contributes to Altered Human Neural Stem Cell Differentiation. Curr. Alzheimer Res. 2019;16:934–949. doi: 10.2174/1567205016666191023104036. [DOI] [PubMed] [Google Scholar]
- 203.Bernal A., Tusell L. Telomeres: Implications for Cancer Development. Int. J. Mol. Sci. 2018;19:294. doi: 10.3390/ijms19010294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Tahmasebi-Birgani M., Ansari H., Carloni V. Defective mitosis-linked DNA damage response and chromosomal instability in liver cancer. Biochim. Biophys. Acta. Rev. Cancer. 2019;1872:60–65. doi: 10.1016/j.bbcan.2019.05.008. [DOI] [PubMed] [Google Scholar]
- 205.Jacobs K.B., Yeager M., Zhou W., Wacholder S., Wang Z., Rodriguez-Santiago B., Hutchinson A., Deng X., Liu C., Horner M.J., et al. Detectable clonal mosaicism and its relationship to aging and cancer. Nat. Genet. 2012;44:651–658. doi: 10.1038/ng.2270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Andriani G.A., Vijg J., Montagna C. Mechanisms and consequences of aneuploidy and chromosome instability in the aging brain. Mech. Ageing Dev. 2017;161 Pt A(Pt A):19–36. doi: 10.1016/j.mad.2016.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Yurov Y.B., Vorsanova S.G., Iourov I.Y. Chromosome Instability in the Neurodegenerating Brain. Front. Genet. 2019;10:892. doi: 10.3389/fgene.2019.00892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Yurov Y.B., Vorsanova S.G., Liehr T., Kolotii A.D., Iourov I.Y. X chromosome aneuploidy in the Alzheimer’s disease brain. Mol. Cytogenet. 2014;7:20. doi: 10.1186/1755-8166-7-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Oyama F., Cairns N.J., Shimada H., Oyama R., Titani K., Ihara Y. Down’s syndrome: Up-regulation of beta-amyloid protein precursor and tau mRNAs and their defective coordination. J. Neurochem. 1994;62:1062–1066. doi: 10.1046/j.1471-4159.1994.62031062.x. [DOI] [PubMed] [Google Scholar]
- 210.Geller L.N., Potter H. Chromosome missegregation and trisomy 21 mosaicism in Alzheimer’s disease. Neurobiol. Dis. 1999;6:167–179. doi: 10.1006/nbdi.1999.0236. [DOI] [PubMed] [Google Scholar]
- 211.Leandro G.S., Evangelista A.F., Lobo R.R., Xavier D.J., Moriguti J.C., Sakamoto-Hojo E.T. Changes in Expression Profiles Revealed by Transcriptomic Analysis in Peripheral Blood Mononuclear Cells of Alzheimer’s Disease Patients. J. Alzheimer’s Dis. JAD. 2018;66:1483–1495. doi: 10.3233/JAD-170205. [DOI] [PubMed] [Google Scholar]
- 212.Kwiatkowski D., Czarny P., Galecki P., Bachurska A., Talarowska M., Orzechowska A., Bobinska K., Bielecka-Kowalska A., Pietras T., Szemraj J., et al. Variants of Base Excision Repair Genes MUTYH, PARP1 and XRCC1 in Alzheimer’s Disease Risk. Neuropsychobiology. 2015;71:176–186. doi: 10.1159/000381985. [DOI] [PubMed] [Google Scholar]
- 213.Leandro G.S., Lobo R.R., Oliveira D.V., Moriguti J.C., Sakamoto-Hojo E.T. Lymphocytes of patients with Alzheimer’s disease display different DNA damage repair kinetics and expression profiles of DNA repair and stress response genes. Int. J. Mol. Sci. 2013;14:12380–12400. doi: 10.3390/ijms140612380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Siddiqui M.S., Francois M., Hecker J., Faunt J., Fenech M.F., Leifert W.R. gammaH2AX is increased in peripheral blood lymphocytes of Alzheimer’s disease patients in the South Australian Neurodegeneration, Nutrition and DNA Damage (SAND) study of aging. Mutat. Res. Genet. Toxicol. Environ. Mutagenes. 2018;829:6–18. doi: 10.1016/j.mrgentox.2018.03.001. [DOI] [PubMed] [Google Scholar]
- 215.Francois M., Leifert W.R., Hecker J., Faunt J., Fenech M.F. Guanine-quadruplexes are increased in mild cognitive impairment and correlate with cognitive function and chromosomal DNA damage. DNA Repair. 2016;46:29–36. doi: 10.1016/j.dnarep.2016.08.001. [DOI] [PubMed] [Google Scholar]
- 216.Kwiatkowski D., Czarny P., Toma M., Jurkowska N., Sliwinska A., Drzewoski J., Bachurska A., Szemraj J., Maes M., Berk M., et al. Associations between DNA Damage, DNA Base Excision Repair Gene Variability and Alzheimer’s Disease Risk. Dement. Geriatr. Cogn. Disord. 2016;41:152–171. doi: 10.1159/000443953. [DOI] [PubMed] [Google Scholar]
- 217.Banda D.M., Nunez N.N., Burnside M.A., Bradshaw K.M., David S.S. Repair of 8-oxoG:A mismatches by the MUTYH glycosylase: Mechanism, metals and medicine. Free Radic. Biol. Med. 2017;107:202–215. doi: 10.1016/j.freeradbiomed.2017.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Sliwinska A., Kwiatkowski D., Czarny P., Toma M., Wigner P., Drzewoski J., Fabianowska-Majewska K., Szemraj J., Maes M., Galecki P., et al. The levels of 7,8-dihydrodeoxyguanosine (8-oxoG) and 8-oxoguanine DNA glycosylase 1 (OGG1)—A potential diagnostic biomarkers of Alzheimer’s disease. J. Neurol. Sci. 2016;368:155–159. doi: 10.1016/j.jns.2016.07.008. [DOI] [PubMed] [Google Scholar]
- 219.Sliwinska A., Sitarek P., Toma M., Czarny P., Synowiec E., Krupa R., Wigner P., Bialek K., Kwiatkowski D., Korycinska A., et al. Decreased expression level of BER genes in Alzheimer’s disease patients is not derivative of their DNA methylation status. Prog. Neuro-Psychopharmacol. Biol. Psychiatry. 2017;79(Pt B):311–316. doi: 10.1016/j.pnpbp.2017.07.010. [DOI] [PubMed] [Google Scholar]
- 220.Moslemnezhad A., Mahjoub S., Moghadasi M. Altered plasma marker of oxidative DNA damage and total antioxidant capacity in patients with Alzheimer’s disease. Casp. J. Intern. Med. 2016;7:88–92. [PMC free article] [PubMed] [Google Scholar]
- 221.Gao S., Zhang X., Song Q., Liu J., Ji X., Wang P. POLD1 deficiency is involved in cognitive function impairment in AD patients and SAMP8 mice. Biomed. Pharmacother. Biomed. Pharmacother. 2019;114:108833. doi: 10.1016/j.biopha.2019.108833. [DOI] [PubMed] [Google Scholar]
- 222.Coppede F., Tannorella P., Stoccoro A., Chico L., Siciliano G., Bonuccelli U., Migliore L. Methylation analysis of DNA repair genes in Alzheimer’s disease. Mech. Ageing Dev. 2017;161(Pt A):105–111. doi: 10.1016/j.mad.2016.04.003. [DOI] [PubMed] [Google Scholar]
- 223.De la Monte S.M., Tong M. Brain metabolic dysfunction at the core of Alzheimer’s disease. Biochem. Pharmacol. 2014;88:548–559. doi: 10.1016/j.bcp.2013.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Mansuroglu Z., Benhelli-Mokrani H., Marcato V., Sultan A., Violet M., Chauderlier A., Delattre L., Loyens A., Talahari S., Begard S., et al. Loss of Tau protein affects the structure, transcription and repair of neuronal pericentromeric heterochromatin. Sci. Rep. 2016;6:33047. doi: 10.1038/srep33047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Violet M., Delattre L., Tardivel M., Sultan A., Chauderlier A., Caillierez R., Talahari S., Nesslany F., Lefebvre B., Bonnefoy E., et al. A major role for Tau in neuronal DNA and RNA protection in vivo under physiological and hyperthermic conditions. Front. Cell. Neurosci. 2014;8:84. doi: 10.3389/fncel.2014.00084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Lu Y., He H.J., Zhou J., Miao J.Y., Lu J., He Y.G., Pan R., Wei Y., Liu Y., He R.Q. Hyperphosphorylation results in tau dysfunction in DNA folding and protection. J. Alzheimer’s Dis. JAD. 2013;37:551–563. doi: 10.3233/JAD-130602. [DOI] [PubMed] [Google Scholar]
- 227.Frost B., Hemberg M., Lewis J., Feany M.B. Tau promotes neurodegeneration through global chromatin relaxation. Nat. Neurosci. 2014;17:357–366. doi: 10.1038/nn.3639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Tse K.H., Herrup K. DNA damage in the oligodendrocyte lineage and its role in brain aging. Mech. Ageing Dev. 2017;161(Pt A):37–50. doi: 10.1016/j.mad.2016.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Tse K.H., Cheng A., Ma F., Herrup K. DNA damage-associated oligodendrocyte degeneration precedes amyloid pathology and contributes to Alzheimer’s disease and dementia. Alzheimer’s Dement. J. Alzheimer’s Assoc. 2018;14:664–679. doi: 10.1016/j.jalz.2017.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Cahoy J.D., Emery B., Kaushal A., Foo L.C., Zamanian J.L., Christopherson K.S., Xing Y., Lubischer J.L., Krieg P.A., Krupenko S.A., et al. A transcriptome database for astrocytes, neurons, and oligodendrocytes: A new resource for understanding brain development and function. J. Neurosci. Off. J. Soc. Neurosci. 2008;28:264–278. doi: 10.1523/JNEUROSCI.4178-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Snaidero N., Simons M. Myelination at a glance. J. Cell Sci. 2014;127(Pt 14):2999–3004. doi: 10.1242/jcs.151043. [DOI] [PubMed] [Google Scholar]
- 232.Salami M., Itami C., Tsumoto T., Kimura F. Change of conduction velocity by regional myelination yields constant latency irrespective of distance between thalamus and cortex. Proc. Natl. Acad. Sci. USA. 2003;100:6174–6179. doi: 10.1073/pnas.0937380100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Butt A.M., De La Rocha I.C., Rivera A. Oligodendroglial Cells in Alzheimer’s Disease. Adv. Exp. Med. Biol. 2019;1175:325–333. doi: 10.1007/978-981-13-9913-8_12. [DOI] [PubMed] [Google Scholar]
- 234.Morell P., Norton W.T. Myelin. Sci. Am. 1980;242:88–90. doi: 10.1038/scientificamerican0580-88. [DOI] [PubMed] [Google Scholar]
- 235.Bartzokis G., Beckson M., Lu P.H., Nuechterlein K.H., Edwards N., Mintz J. Age-related changes in frontal and temporal lobe volumes in men: A magnetic resonance imaging study. Arch. Gen. Psychiatry. 2001;58:461–465. doi: 10.1001/archpsyc.58.5.461. [DOI] [PubMed] [Google Scholar]
- 236.Bartzokis G., Cummings J.L., Sultzer D., Henderson V.W., Nuechterlein K.H., Mintz J. White matter structural integrity in healthy aging adults and patients with Alzheimer disease: A magnetic resonance imaging study. Arch. Neurol. 2003;60:393–398. doi: 10.1001/archneur.60.3.393. [DOI] [PubMed] [Google Scholar]
- 237.Bernabeu-Zornoza A., Coronel R., Palmer C., Calero M., Martinez-Serrano A., Cano E., Zambrano A., Liste I. Abeta42 Peptide Promotes Proliferation and Gliogenesis in Human Neural Stem Cells. Mol. Neurobiol. 2019;56:4023–4036. doi: 10.1007/s12035-018-1355-7. [DOI] [PubMed] [Google Scholar]
- 238.Silva A.R., Santos A.C., Farfel J.M., Grinberg L.T., Ferretti R.E., Campos A.H., Cunha I.W., Begnami M.D., Rocha R.M., Carraro D.M., et al. Repair of oxidative DNA damage, cell-cycle regulation and neuronal death may influence the clinical manifestation of Alzheimer’s disease. PLoS ONE. 2014;9:e99897. doi: 10.1371/journal.pone.0099897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Suberbielle E., Djukic B., Evans M., Kim D.H., Taneja P., Wang X., Finucane M., Knox J., Ho K., Devidze N., et al. DNA repair factor BRCA1 depletion occurs in Alzheimer brains and impairs cognitive function in mice. Nat. Commun. 2015;6:8897. doi: 10.1038/ncomms9897. [DOI] [PMC free article] [PubMed] [Google Scholar]