

Abstract
Introduction
Apolipoprotein E (APOE) ε2 and ε4 alleles encoded by rs7412 and rs429358 polymorphisms, respectively, are landmark contra and pro “risk” factors for Alzheimer's disease (AD).
Methods
We examined differences in linkage disequilibrium (LD) structures between (1) AD‐affected and unaffected subjects and (2) older AD‐unaffected and younger subjects in the 19q13.3 region harboring rs7412 and rs429358.
Results
AD is associated with sex‐nonspecific heterogeneous patterns of decreased and increased LD of rs7412 and rs429358, respectively, with other polymorphisms from five genes in this region in AD‐affected subjects. The LD patterns in older AD‐unaffected subjects resembled those in younger individuals. Polarization of the ε4‐ and ε2 allele–related heterogeneous LD clusters differentiated cell types and implicated specific tissues in AD pathogenesis.
Discussion
Protection and predisposition to AD is characterized by an interplay of rs7412 and rs429358, with multiple polymorphisms in the 19q13.3 region in a tissue‐specific manner, which is not driven by common evolutionary forces.
Keywords: Alzheimer's disease, apolipoprotein E, linkage disequilibrium
1. BACKGROUND
The strongest evidence for genetic predisposition to Alzheimer's disease (AD) was reported for the apolipoprotein E (APOE)/ translocase of outer mitochondrial membrane 40 (TOMM40) region 19q13.3 with the APOE ε4 allele as the strongest genetic risk factor for AD development in various populations1 and the APOE ε2 allele as a protective factor against AD.2, 3 However, even the pathogenic role of the ε4 allele in AD remains poorly understood, consistent with the inefficiency of AD clinical trials4 and finding of cognitively normal homozygous ε4 carriers among centenarians.5 Understanding the protective role of the ε2 allele has lagged behind the ε4 research because of, in part, seemingly smaller effects of this allele on AD.3
Mainstream research considers the effects of risk alleles in genetics of such complex traits as AD as a result of incomplete penetrance.6 We emphasize inherent heterogeneity in the effects of the same alleles on AD. This view is supported by evolutionary biology, which argues that the conceptual problem in the genetics of traits that make bodies vulnerable to disease(s) in post‐reproductive life, called age‐related traits, is an uncertain role of evolution in establishing their molecular mechanisms.7 Increased human life expectancy8 and changes in the environment9, 10, 11, 12 contribute to this problem. Accordingly, in the framework of evolutionary biology, age‐related traits are viewed as the results of indirect mechanisms such as co‐evolution with fast‐evolving pathogens, mismatch with environments, reproductive success at the expense of health, and so on,7 that increase heterogeneity.
Following the framework of evolutionary biology, we examined the molecular signatures of AD in the APOE region, represented by 32 single nucleotide polymorphisms (SNPs) from five genes (BCAM, NECTIN2, TOMM40, APOE, and APOC1), as differences in linkage disequilibrium (LD) patterns in mega‐samples of 2673 AD‐affected and 16,246 AD‐unaffected subjects of European ancestry. We emphasized protective and detrimental heterogeneous signatures involving the APOE ε2 and ε4 alleles, encoded by rs7412 and rs429358, respectively. We show that susceptibility to AD is the result of a complex interplay of these SNPs with SNPs from other genes in the APOE region, which is not driven by common evolutionary forces characteristic for the general (AD‐unaffected) population.
2. METHODS
2.1. Data availability
This article was prepared using limited access data sets obtained though dbGaP (accession numbers phs000007.v28.p10, phs000287.v5.p1, phs000428.v1.p1, and phs000168.v2.p2) and the University of Michigan. Phenotypic Health and Retirement Study (HRS) data are available publicly and through restricted access from http://hrsonline.isr.umich.edu/index.php?p=data.
RESEARCH IN CONTEXT
Systematic review: Recently, we reported significant molecular signatures of Alzheimer's disease (AD) in the apolipoprotein E (APOE) region, which excluded the ε2 and ε4 alleles. A literature review (PubMed and Google Scholar) identified few other publications, which reported significant associations of linkage disequilibrium (LD) structures with AD. These relevant publications are appropriately cited.
Interpretation: Susceptibility to AD is the result of a complex interplay of the ε2 and ε4 alleles with other alleles from different genes in the APOE region, which is not driven by common evolutionary forces. Accordingly, this interplay is the result of AD‐specific exposures, which, therefore, can be amendable to AD preventive interventions even with natural, for example, lifestyle, factors.
Future directions: This work suggests an approach to examine the potential role of complex genotypes/haplotypes in the AD etiology in loci with complex LD structures. Further work should be focused on elucidating personalized, that is, more homogeneous, group‐specific, polygenic profiles of AD risk and protection.
2.2. Study cohorts and phenotypes
We used data from five studies. Data for older individuals were drawn from the Framingham Heart Study (FHS) original (FHS_C1) and offspring (FHS_C2) cohorts,13 Cardiovascular Health Study (CHS),14 Health and Retirement Study (HRS),15 and the National Institute on Aging (NIA) Late‐Onset Alzheimer's Disease Family Based Study (LOADFS)16 for individuals of Caucasian ancestry. In LOADFS, FHS, and CHS, AD was defined based on diagnoses made according to National Institute of Neurological Disorders and Stroke and the Alzheimer's Disease and Related Disorders Association. A diagnosis of AD in HRS was defined based on ICD‐9:331.0x codes in Medicare service use files. Individuals with AD constituted the case group, n = 2673, and those without AD constituted the non‐case group, n = 16,246 (Table 1). Data from the FHS third‐generation cohort (FHS_C3) and Coronary Artery Risk Development in Young Adults (CARDIA) cohort (Table 1) were used in comparative analyses of LD patterns in younger and older individuals.
Table 1.
Basic characteristics of the genotyped participants in the selected studies
| Cohort | N | AD cases (%) | Men (%) | Birth year mean (SD) | Age at baseline mean (SD), years | Age at DNA mean (SD), years | Age at the end of follow‐up mean (SD), years | Follow‐up through |
|---|---|---|---|---|---|---|---|---|
| LOADFS | 3715 | 1850 (49.8) | 1395 (37.6) | 1928.5 (12.5) | 73.5 (12.5) | 73.5 (12.5) | 77.3 (10.9) | 2015a |
| HRS | 7226 | 263 (3.6) | 3129 (43.3) | 1934.2 (8.4) | 60.6 (8.7) | 73.2 (8.4) | 79.1 (8.1) | 2012 |
| CHS | 4326 | 252 (5.8) | 1884 (43.6) | 1914.1 (5.7) | 72.8 (5.6) | 73.5 (5.7) | 83.5 (5.4) | 2002 |
| FHS_C1 | 631 | 205 (32.5) | 210 (33.3) | 1911.8 (4.2) | 35.7 (4.2) | 84.1 (4.3) | 91.4 (4.8) | 2012 |
| FHS_C2 | 3021 | 103 (3.4) | 1383 (45.8) | 1935.8 (9.6) | 34.7 (9.7) | 60.3 (9.7) | 72.2 (9.2) | 2012 |
| FHS_C3 | 3980 | NA | 1862 (46.8) | 1960.5 (8.9) | 40.2 (8.8) | 40.2 (8.7) | 47.8 (9.0) | 2012 |
| CARDIA | 1941 | NA | 909 (46.8) | 1957.5 (3.5) | 25.0 (3.6) | 25.0 (3.6) | 40.4 (3.8) | 2011 |
AD denotes Alzheimer's disease and related dementias.
N denotes genotyped sample after excluding individuals with missingness for SNPs >5% and missing information on AD.
Large proportion of AD cases in LOADFS is due to case‐control design.
Large proportion of AD cases in FHS is due to older age of participants of this cohort at the end of follow‐up (mean age for total sample is 91.4 years) and larger proportion of women (66.7%) who are at higher risk of AD.
CHS, Cardiovascular Health Study; FHS_C1, Framingham Heart Study (FHS) original cohort; FHS_C2, FHS offspring cohort; FHS_C3, FHS third generation cohort; HRS, Health and Retirement Study; LOADFS, NIA Late‐Onset Alzheimer's Disease Family Study; CARDIA, Coronary Artery Risk Development in Young Adults cohort; NA, not applicable; SD, standard deviation.
Information on age at onset of AD in LOADFS was not known for all cases.
2.3. Genotypes
Genotyping was performed using the same customized Illumina iSelect array (the IBC‐chip, ≈50 K SNPs) in the FHS and CHS cohorts, Affymetrix 500 K in the FHS, Illumina HumanCNV370v1 chip (370 K SNPs) in the CHS, Illumina HumanOmni 2.5 Quad chip (≈2.5 M SNPs) in the HRS, and Illumina Human 610Quadv1_B Beadchip (≈610 K SNPs) in the LOADFS.
Thirty‐two SNPs representing the BCAM‐NECTIN2‐TOMM40‐APOE‐APOC1 locus (Table S1) were not in perfect LD (r2 < 0.8) and directly genotyped in at least two cohorts.
We excluded individuals with >5% missingness. For cross‐platform comparisons, we selected directly genotyped target SNPs or their proxies (r2 > 0.8 in the 1000 Genomes Project, CEU population) using all available arrays for each study. Non‐genotyped SNPs were imputed (IMPUTE217) according to the 1000 Genomes Project Phase 3 integrated variant set release (SHAPEIT2) in the NCBI build 37 (hg19) coordinate. Retaining SNPs with high imputation quality (info > 0.8), rs11668536 in FHS/FHSO (info < 0.66) was excluded (details in Table S1).
2.4. Statistical analysis
Associations between AD and each selected SNP were evaluated using an additive genetic model, with the minor allele as an effect allele. Given limited information on AD age at onset in the LOADFS, the associations in this study were characterized using a logistic model with AD as a binary outcome and random effects to adjust for potential familial clustering (gee package in R). Associations in the other studies were evaluated using the Cox proportional hazard mixed‐effects regression model (coxme package in R) to adjust for familial clustering. The time variable in the Cox model was the age at onset of AD or the age at right censoring in 2002 (CHS) and 2012 (FHS and HRS). All statistical tests were adjusted for (all studies) age, sex; (CHS) field center; (FHS) whether the DNA samples had been subject to whole‐genome amplification; and (HRS) HRS cohorts. Meta‐statistics were evaluated using METAL.18
2.5. Linkage disequilibrium analysis
We have used methods detailed in Ref. 19. In brief, LD was characterized by the correlation coefficient r using haplotype‐based and genotype‐based methods. Differences in their LD estimates indicate deviation from Hardy‐Weinberg equilibrium (HWE). This information is important because HWE in the entire sample does not guarantee HWE in subsamples and/or at the haplotype level (see below), and thus, the observed deviation from HWE may be biologically plausible. Significance of the LD estimates was characterized using chi‐square statistics, defined as χ2 = r2N, where N = 2n is the number of gametes and n is the sample size. Given the potential loss of power because of inferring haplotypes from genotypes, we used a more conservative estimate, with n instead of N. We employed an LD contrast test20 to compare the LD estimates between the AD‐affected and unaffected groups. This test was used to characterize the significance of the differences in pairwise estimates of LD between these two groups. Significance of the r2 estimates and the differences in the pairwise estimates of LD were corrected for multiple testing. For the 32 SNPs examined, this represented 496 (=32 × 31/2) tests. We adopted a conservative Bonferroni correction for significance, P ≤ 10−4, despite some correlation between these SNPs. Asymptotically valid confidence intervals were constructed using asymptotic variance adapted from.21
2.6. Functional annotation
Potential regulatory functions of the selected SNPs were annotated using the Ensembl genome browser (https://www.ensembl.org/), RegulomeDB (http://www.regulomedb.org/), and HaploReg (http://archive.broadinstitute.org) databases. Information on expression quantitative trait loci (eQTLs) was obtained from the GTEx (v7 release) portal (https://www.gtexportal.org/).
3. RESULTS
3.1. Study overview
Molecular signatures of AD were examined as the difference of LD patterns in mega‐samples of AD‐affected and unaffected subjects of Caucasian ancestry, with men and women combined and separately pooled from four independent studies comprising five cohorts: LOADFS, HRS, CHS, FHS_C1, and FHS_C2 (Table 1). LD patterns were characterized by 32 non‐proxy SNPs (defined as LD with r2 < 0.8), representing the BCAM, NECTIN2, TOMM40, APOE, and APOC1 genes in the 19q13.3 region (Table S1) including two SNPs, rs429358 and rs7412 SNPs, whose minor alleles encode the APOE ε4 and ε2 alleles, respectively. We examined the potential role of survival selection in the AD signatures by contrasting LD patterns between older AD‐unaffected individuals from those five cohorts (who were at exponentially increased mortality risk) and younger individuals (who were at negligible mortality risk), enriched by subjects from two additional cohorts, FHS_C3 and CARDIA (Table 1).
Unless explicitly stated, the results of LD analyses are presented using a haplotype‐based method (details in Materials and Methods).
Of the examined 32 SNPs, the minor allele of rs429358 was associated with the highest risks of AD development, β = 1.26, P = 8.05 × 10−130, whereas the minor allele of rs7412 showed the strongest protective effect, β = −0.59, P = 1.02 × 10−9 (Table S1). The effect directions were consistent in all studies for rs429358, but not for rs7412. The largest magnitude of effects for these SNPs was observed in LOADFS (β = 1.78, P = 9.33 × 10−97 for rs429358 and β = −1.50, P = 2.77 × 10−14 for rs7412) and the smallest in FHS_C1 (β = 0.51, P = 1.87 × 10−3 for rs429358 and β = 0.10, P = 6.48 × 10−1 for rs7412) (Figure 1).
Figure 1.
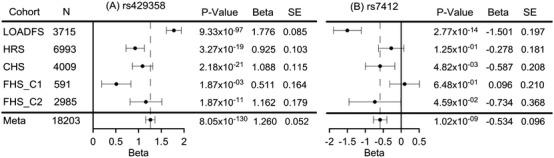
Forest plots for the associations of (A) rs429358 (ε4‐coding SNP) and (B) rs7412 (ε2‐coding SNP) with Alzheimer's disease (AD). LOADFS, NIA Late‐Onset Alzheimer's Disease Family Study; HRS, Health and Retirement Study; CHS, Cardiovascular Health Study; FHS_C1, Framingham Heart Study (FHS) original cohort; FHS_C2, FHS offspring cohort; SE, standard error; N, sample size. Meta indicates the results from the meta‐analysis. Horizontal bars show 95% confidence intervals
3.2. Molecular signature of Alzheimer's disease
We contrasted LD patterns of the entire APOE region between AD‐affected and unaffected individuals (Table S2) and found that they differed significantly (P < 2 × 10−4). The pattern of the difference represents a molecular signature of AD illustrated by a heat map for Δr = rcases − rnon‐cases (Figure 2). Figure 2 shows the complex rearrangement of LD in AD cases compared with non‐cases spanning the entire region. Our analysis identified 193 of 496 (=32 × 31/2) SNP pairs (38.9%) with Δr values significant at the Bonferroni‐adjusted level: P ≤ PBonf = 10−4. For 33 additional SNP pairs, we observed suggestive significances: PBonf < P < 10−3.
Figure 2.
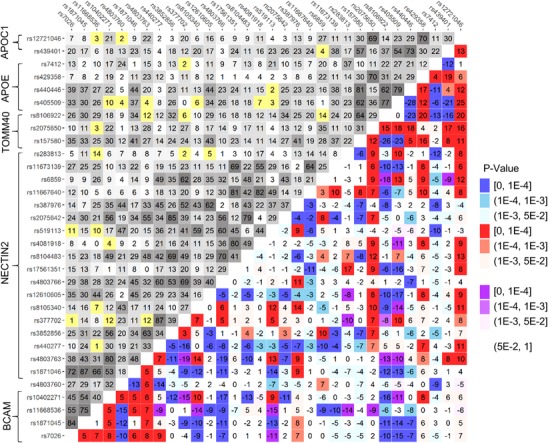
Molecular signature of Alzheimer's disease (AD). Upper‐left triangle: Linkage disequilibrium (LD) pattern (r, %) in the pooled sample from all studies, non‐cases, for 32 single nucleotide polymorphisms (SNPs). Lower‐right triangle: Heat map for Δr = rcases−rnon‐cases representing the molecular signature of AD. Red denotes rcases > rnon‐cases and blue denotes rcases < rnon‐cases. Purple and yellow show the estimates with opposite signs of rcases and rnon‐cases. For convenience, positive sign of rnon‐cases has been selected. Legend on the right shows color‐coded P‐values. The heat map shows that LD changes for the vast majority of SNPs in the entire region spanning all five genes. Numerical estimates are shown in Table S2
Molecular signatures of AD estimated using the genotype‐based method (Table S3) were qualitatively the same as those estimated using the haplotype‐based method, with significant differences observed between cases and non‐cases (P < 2 × 10−4). The genotype‐based method provided 153 SNP pairs significant at P < PBonf and 33 additional SNP pairs with suggestive significance (PBonf < P < 10−3).
For 149 SNP pairs, the estimates of Δr were significant at P ≤ PBonf in both the haplotype‐ and genotype‐based methods. Given that all SNPs in the large sample of non‐cases were in HWE at P HW > 10−3, the discordant estimates of Δr for 44 SNP pairs between these two methods indicated SNPs with a plausible biological role because the deviation from HWE occurs in cases (Table S1) and/or at the haplotype level, that is, when (see Materials and Methods). Accordingly, important biologically plausible information can be missed using the genotype‐based method alone.
3.3. The APOE ε2 (rs7412) and ε4 (rs429358) coding SNPs are parts of the molecular signature of AD
In non‐cases, rs7412 and rs429358 SNPs were in significant LD between each other, r = 11.6%, P = 7.95 × 10−94, and with most of the other SNPs (Table S2). The strongest LD for rs429358 was observed with rs2075650 (r = 70%, TOMM40) and rs12721046 (r = 69%, APOC1) SNPs. For rs7412, the strongest LD of r = 37% was with rs283813 (NECTIN2).
Rearrangement of LD between AD cases and non‐cases was characterized by a significant increase in LD of rs429358 with 13 SNPs, including rs7412 (Figure 3B), and decrease in LD of rs7412 with 8 SNPs (Figure 3A). Although the change in LD was somewhat larger for rs429358 with nearby SNPs from the TOMM40‐APOE‐APOC1 locus (Figure 3), LD changed regardless of genomic distance between the other SNP pairs. LD of rs429358 and rs7412 SNPs changed in opposite directions with the same four SNPs (rs8106922, rs405509, rs440446, and rs439401) from the TOMM40‐APOE‐APOC1 locus. LD for rs429358 and rs7412 with SNPs from the BCAM‐NECTIN2 locus also changed in opposite directions but for non‐overlapping SNPs. Significant changes in LD between rs7412 and 8 SNPs as well as between rs429358 and 13 SNPs were not explained by LD between those 8 or 13 SNPs. This is because LD between these 8 or 13 SNPs can be very small (Figure 3, brackets) and, therefore, it cannot be explained by clustering of specific alleles from different SNPs in the same subjects. The latter implies genetic heterogeneity. The changes in LD between AD cases and non‐cases observed for rs7412 and rs429358 in the mega sample of pooled studies were consistent in independent studies (Table S4). Consistency of changes in LD for other SNPs in independent studies was reported in Ref. 22.
Figure 3.
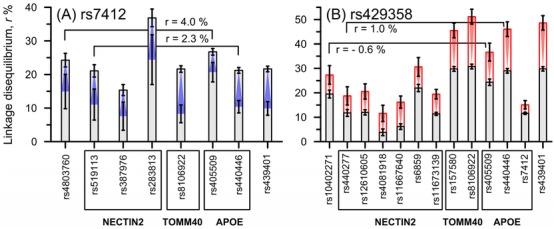
Significant ε2‐ and ε4‐related molecular signatures of Alzheimer's disease (AD). (A) The ε2‐related signature is characterized by a significant decrease (blue) in linkage disequilibrium (LD) for rs7412 with eight single nucleotide polymorphisms (SNPs) in AD cases compared with non‐cases. (B) The ε4‐related signature is characterized by a significant increase (red) for LD of rs429358 with 13 SNPs, including rs7412, in AD cases compared with non‐cases. Insets show examples of small LD between SNPs indicated by brackets. Vertical lines show 95% confidence intervals. Numerical estimates are shown in Table S2
3.4. Molecular signatures of AD in men and women
We evaluated LD structure for the selected 32 SNPs in AD‐affected and unaffected men and women separately (Table S5). The 95% confidence intervals for Δr in men and women well overlapped for all SNP pairs, implying no significant difference in Δr between these sexes.
3.5. LD patterns in younger and older individuals
We examined the role of survival selection in the molecular signature of AD by contrasting LD patterns in older subjects with no AD who were 55 years and older at biospecimen collection (N = 14,803), and younger subjects who were <55 years at biospecimen collection (N = 6565). We excluded four SNPs from this analysis (rs7026, rs4803760, rs440277, and rs11667640) because they were imputed for most subjects (95.4%) from the young group. The 55‐year cutoff was used to separate younger individuals who were under negligible mortality risk in modern developed countries from those who were under exponentially increasing mortality risk. This choice allowed consideration of LD patterns in the younger group as a proxy for the evolutionary selected LD structure in the APOE genomic region. This analysis did not identify significant differences in LD patterns between these two groups. At the level of individual SNP pairs, only two pairs in the BCAM‐NECTIN2 locus exhibited significant differences (Δryo = ryoumg − rold at P ≤ PBonf = 10−4) in these large samples (Table S6). No significant differences were identified in the TOMM40‐APOE‐APOC1 locus (Figure 4).
Figure 4.
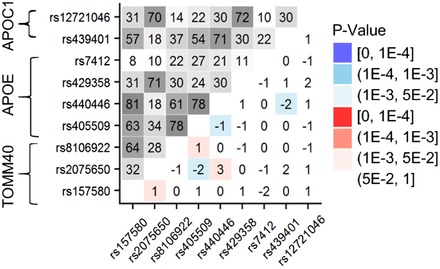
Linkage disequilibrium (LD) patterns in younger and older individuals. Upper‐left triangle: LD pattern (r, %) in younger subjects who were <55 years at biospecimens collection for nine single nucleotide polymorphisms (SNPs) from the TOMM40‐APOE‐APOC1 locus. Lower‐right triangle: Heat map showing Δryo = ryoumg − rold as the difference in LD estimates in younger and older samples. Older sample included subjects with no Alzheimer's disease (AD) who were 55 years and older at biospecimens collection. Numerical estimates are shown in Table S6
3.6. Regulatory architecture in the APOE region across cell types and tissues
Using data from Ensembl, 10 of 32 SNPs were identified as regulatory variants in active expression states in a variety of tissues ranging from one to 63 of the 68 cell types (Table 2). For seven of them, RegulomeDB assigned functionality scores of 1b to 2a corresponding to strong regulatory potential (Table 2). Most SNPs may affect transcription factor (TF) binding ability. Altered motifs for TFs were identified for 28 SNPs in HaploReg (Table S7). The protein motifs at these sites are for known TFs that could contribute to the complex regulation of genes in this region. HaploReg showed that 10 SNPs could affect the binding of various proteins (from one to seven), suggesting that they could be in actively transcribed regions. Five more SNPs affected protein binding according to RegulomeDB (Table S7). Twenty‐six SNPs acted as eQTLs for the nearby protein‐coding genes, according to GTEx, affecting expression in a number of tissues (Table 2 and Table S7).
Table 2.
Functional annotation of 32 SNPs in the APOE region
| ID | SNP ID | LD cluster | Function | Regulatory feature | Gene | Active | Poised | Score | M0&M1 macrophage | CD14+ monocytes | NHA | NHLF | Selected eQTL |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | rs7026 | 3'UTR | PFR | BCAM | 6 | 2 | 5 | NECTIN2 | |||||
| 2 | rs1871045 | Downstream | BCAM | 4 | NECTIN2 | ||||||||
| 3 | rs11668536 | Downstream | BCAM | NECTIN2 | |||||||||
| 4 | rs10402271 | ε4 | Downstream | BCAM | 5 | NECTIN2 | |||||||
| 5 | rs4803760 | ε2 | Intergenic | 6 | BCAM | ||||||||
| 6 | rs1871046 | Intron | Promoter, TFBS | NECTIN2 | 46 | 20 | 2a | No | |||||
| 7 | rs4803763 | Intron | NECTIN2 | 5 | NECTIN2 | ||||||||
| 8 | rs440277 | ε4 | Intron | PFR | NECTIN2 | 5 | 1 | 1f | Yesa | Yesa | Yes | NECTIN2, FOSB | |
| 9 | rs3852856 | Intron | NECTIN2 | NECTIN2 | |||||||||
| 10 | rs377702 | Intron | NECTIN2 | 2b | NECTIN2, FOSB, CLASRP | ||||||||
| 11 | rs8105340 | Intron | NECTIN2 | 6 | NECTIN2 | ||||||||
| 12 | rs12610605 | ε4 | Intron | NECTIN2 | 5 | ||||||||
| 13 | rs4803766 | Intron | NECTIN2 | 5 | NECTIN2 | ||||||||
| 14 | rs17561351 | Intron | PFR | NECTIN2 | 5 | 1 | 1b | NECTIN2a | |||||
| 15 | rs8104483 | Intron | PFR | NECTIN2 | 5 | 1 | 1b | NECTIN2a | |||||
| 16 | rs4081918 | ε4 | Intron | PFR | NECTIN2 | 5 | 1 | 1f | Yesa | Yesa | Yes | NECTIN2a | |
| 17 | rs519113 | ε2 | Intron | NECTIN2 | 1f | BCAM, NECTIN2a | |||||||
| 18 | rs2075642 | Intron | NECTIN2 | 5 | NECTIN2 | ||||||||
| 19 | rs387976 | ε2 | Intron | OCR | NECTIN2 | 1 | 1 | 5 | Yes | NECTIN2 | |||
| 20 | rs11667640 | ε4 | Intron | NECTIN2 | 4 | NECTIN2 | |||||||
| 21 | rs6859 | ε4 | 3'UTR | NECTIN2 | 4 | NECTIN2 | |||||||
| 22 | rs11673139 | ε4 | Intron | NECTIN2 | 4 | NECTIN2, MARK4 | |||||||
| 23 | rs283813 | ε2 | Intron | NECTIN2 | 5 | No | |||||||
| 24 | rs157580 | ε4 | Intron | Promoter | TOMM40 | 63 | 5 | 1f | Yesa | Yesa | Yes | APOE, APOC1, DMPK | |
| 25 | rs2075650 | Intron | TOMM40 | 1f | No | ||||||||
| 26 | rs8106922 | ε2, ε4 | Intron | TOMM40 | 5 | DMPKa | |||||||
| 27 | rs405509 | ε2, ε4 | Upstream | Promoter | APOE | 1f | APOE | ||||||
| 28 | rs440446 | ε2, ε4 | Missense intron | Promoter | APOE | 18 | 32 | 4 | Yesa | Yes | Yes | Yes | APOE, APOC1 |
| 29 | rs429358 | Missense | Coding region, exon 4 | 5 | No | ||||||||
| 30 | rs7412 | Missense | Coding region, exon 4 | 4 | APOE | ||||||||
| 31 | rs439401 | ε2, ε4 | Non coding transcript exon | PFR | APOE‐APOC1 | 13 | 3 | 1b | Yesa | Yesa | Yesa | APOE, APOC1 | |
| 32 | rs12721046 | Intron | APOC1 | 6 | No |
Linkage disequilibrium (LD) cluster indicates SNPs in LD with rs429358 and rs7412 SNPs, whose minor alleles code the APOE ε4 and ε2 alleles, respectively.
Table includes activity levels (active/poised) for 68 cell types (epigenoms).
Column “Score” shows RegulomeDB score based on the integration of multiple high‐throughput datasets with 1a being the highest score and 6 being the lowest score. Note that because RegulomeDB focuses on noncoding SNPs, missense SNPs may not have large scores.
NHA denotes epigenetic signature in normal human astrocytes cells.
NHFL denotes human lung fibroblasts.
eQTLs denote expression quantitative trait loci selected for affected protein‐coding gene in specific cell types.
OCR, open chromatin region; PFR, promoter flanking region; TF, transcription factor; TFBS, TF binding site.
Active state.
The APOE ε4 allele–related LD cluster (Figure 3B) includes five SNPs located in promoter regions of the associated genes, which were active in 5 to 63 of 68 cell types (Table 2). We found that all five regulatory variants shared the same feature, exhibiting the active state in M0 and M1 macrophages from venous blood. Four of them were active (rs440277, rs4081918, and rs157580) or poised (rs440446) in CD14+ monocytes (Table 2). One of five promoter variants, rs439401, was active in normal human astrocytes (NHAs) and four variants (rs440277, rs4081918, rs157580, and rs440446) exhibited a poised epigenetic signature in NHAs. The APOE ε2 allele–related LD cluster (Figure 3A) included two SNPs in promoter regions and rs387976 SNP in open chromatin, which were active in one to 18 of 68 cell types (Table 2). All three variants shared an active (rs439401) or poised (rs440446 and rs387976) expression state in normal human lung fibroblasts (NHLF). Variants in poised expression states can be epigenetically activated at a later stage in development or in response to exogenous stimuli.23, 24
4. DISCUSSION
Unlike few small‐scale prior studies examining associations of LD patterns with AD,25, 26 we found that AD was associated with a highly heterogeneous molecular signature in the APOE region, which included rs7412 and rs429358 encoding the ε2 and ε4 alleles, respectively, and SNPs from all five genes in the BCAM‐APOC1 locus, regardless of genomic distance between them. This signature is represented by the pattern of differences in LD structures between AD‐affected and unaffected subjects (Figure 2). The AD signature is consistent with a haplotype rather than a single allele origin of AD.27, 28, 29 Significant changes in LD indicate complex genetic architecture of AD in this region that is consistent with the view on AD as a continuum, rather than distinct clinically defined entities, driven by multimodal cognitive decline.30 No significant differences between the AD signatures in men and women were identified. Our results show that rs429358 and rs7412 are an inherent part of this signature. This finding indicates that the role of the ε4 and ε2 alleles in AD is dependent on the other SNPs in this locus. Indeed, decreased LD of rs7412 with eight SNPs in this locus in AD‐affected subjects compared with unaffected subjects shows that the larger LD strengthens the protective effect because the large LD is observed in unaffected subjects. Likewise, increased LD of rs429358 with 13 SNPs in AD‐affected subjects shows that the larger LD strengthens the detrimental effect because the larger LD is observed in AD‐affected subjects. Complexity of the molecular signature of AD implies that other SNPs in this locus can indirectly modify the effects of the ε4 and ε2 alleles in AD pathogenesis. Changes in the LD of the ε4‐ or ε2 allele–coding SNPs with the other SNPs in a heterogeneous manner (Figure 3) indicate more homogeneous carrier groups of detrimental or protective polygenic variants. This finding naturally strengthens a gene‐based precision‐medicine approach31 to AD treatment and prevention. The lack of the role of survival selection (Figure 4) in the AD signature implies that the LD pattern for the 32 SNPs in AD‐unaffected subjects was likely evolutionary selected, whereas that in AD‐affected subjects was not driven by the same evolutionary forces. This result offsets potential age‐related bias and is consistent with the uniquely human origin of AD, which is sensitive to the modern environment.32 More detailed analyses are required to better understand driving force of the AD signatures, for example, whether they are the result of AD‐related selection within a given human generation, AD‐related selection across recent generations within families or communities, or AD‐related divergence of ancestral groups.
Our bioinformatics analysis identified regulatory variants from the APOE ε4‐ and ε2 allele–related LD clusters (Figure 3), which shared the same features within each cluster. A hallmark for regulatory variants from the ε4‐allele LD cluster was an active state in primary macrophages (M0) and pro‐inflammatory M1 macrophages and active or poised expression states in CD14+ monocytes and NHAs. Monocytes that originate in the bone marrow can differentiate into specific tissue macrophages and dendritic cells in response to inflammation/infection. Blood monocyte–derived macrophages, representing innate immunity, can contribute to the immune response in the central nervous system (CNS) along with brain‐resident macrophages (microglia).33 A pro‐inflammatory (M1) macrophage response causes neurotoxicity.34 Enrichment in these specific immune cells is consistent with the role of peripheral monocytes/macrophages, along with microglia, in Aβ clearance and a potential role in AD.33, 35 It is important to note that our results are in line with recent advances implicating monocyte‐specific eQTLs in AD36 and the AD susceptibility alleles as significant eQTLs in CD14+ monocytes.37 Given crosstalk between macrophages/microglia and astrocytes, they show neurotoxic or neuroprotective phenotypes. M1 macrophages particularly induce astrocyte proliferation and a reactive phenotype. The interaction between macrophages and astrocytes plays an important role in the increasing inflammatory response leading to neurodegeneration.38 Astrocytes are implicated in the induction of neuroinflammation and AD, and apoE‐mediated Aβ clearance, which may be impaired by the reactive phenotype.39 Stressed, dysfunctional astrocytes are connected with ε4‐associated AD.40 Thus, the shared features of regulatory variants from the ε4 allele LD cluster highlight its connection with changes in immune response and inflammation in the CNS and the APOE ε4–dependent crosstalk of astrocytes with macrophages in neuroinflammation in AD. This suggests that the ε4 allele LD cluster is the result of rebalancing of neuroinflammatory tolerance mediated by astrocytes and macrophages in an exposure‐dependent manner.
A common feature of regulatory variants in the ε2 allele–related LD cluster is having an active or poised state in NHLFs. Lung fibroblasts play a role in airway inflammation and remodeling. Pulmonary health is important in risk prevention of cognitive decline and dementia.41 In addition, rs4803760 (intergenic NECTIN2‐BCAM) and rs519113 (NECTIN2) are eQTLs for BCAM in lung. The ε4‐ and ε2‐allele LD clusters have two common promoter variants (rs440446 and rs439401). Of interest, rs439401 is located in the APOE‐APOC1 intergenic region, which includes a specific macrophage, adipocyte, and astrocyte enhancer for the APOE gene,42 and the peroxisome proliferator–activated receptor γ (PPARγ) regulatory region,42 which may simultaneously affect transcriptional regulation. PPARγ is implicated in the regional transcriptional regulation of chr19q13.32 with the highest increase in expression observed for APOE messenger RNA (mRNA).42, 43 It plays a role in determining anti‐inflammatory macrophage (M2) phenotype,44 astrocyte inflammatory brain pathology,45 and airway and lung inflammation.46
Thus, polarization of the ε4‐ and ε2‐allele–related heterogeneous LD clusters differentiates cell types and implicates specific tissues in AD pathogenesis. These clusters can be a result of alteration in functional properties of complex regulatory networks in specific cell/tissue types linked with activation and function of immune cells (ie, pro‐[M1] and anti‐inflammatory [M2] macrophages) directed by the tissue‐specific micro‐environmental effects and other factors.47 Specifically, the detrimental ε4 allele LD cluster highlights the simultaneous effects of macrophage and astrocytes, whereas the protective ε2 allele LD cluster is implicated in non‐brain tissue. Our results support the idea that the effect of even the strongest genetic risk factor of AD, the APOE ε4 allele, can be naturally altered by changing the epigenetic landscape earlier in life by lifestyle and environmental interventions to decrease negative epigenetic changes in the APOE region and macrophage‐driven “inflamm‐aging.”48, 49 However, they indicate the critical role of heterogeneity and show that it can be informatively dissected as directed by molecular signatures of AD in the APOE region.
CONFLICTS OF INTEREST
The authors have no conflicts of interest related to this study.
Supporting information
Table S1. Basic information on SNPs and the associations of these SNPs with AD in the combined sample and each cohort separately.
Table S2. Linkage disequilibrium estimates using haplotype‐based method in the pooled sample of all cohorts.
Table S3. Linkage disequilibrium estimates using genotype‐based method in the pooled sample of all cohorts.
Table S4. Linkage disequilibrium estimates using haplotype‐based method in the pooled sample of all cohorts and each cohort separately for rs7412 and rs429358.
Table S5A. Linkage disequilibrium estimates using haplotype‐based method in the pooled sample of all cohorts for women.
Table S5B. Linkage disequilibrium estimates using haplotype‐based method in the pooled sample of all cohorts for men.
Table S6. Linkage disequilibrium estimates using haplotype‐based method in younger and older individuals.
Table S7. Functional annotation of potential regulatory SNPs in the APOE region.
ACKNOWLEDGMENTS
This research was supported by grants No. P01 AG043352, R01 AG047310, and R01 AG061853 from the National Institute on Aging. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. This manuscript was prepared using limited access data sets obtained though dbGaP (accession numbers phs000007.v28.p10, phs000287.v5.p1, phs000428.v1.p1, phs000168.v2.p2) and the University of Michigan. Phenotypic HRS data are available publicly and through restricted access from http://hrsonline.isr.umich.edu/index.php?p=data. The authors declare no competing interests.
The Framingham Heart Study (FHS) is conducted and supported by the National Heart, Lung, and Blood Institute (NHLBI) in collaboration with Boston University (Contract No. N01‐HC‐25195 and HHSN268201500001I). This manuscript was not prepared in collaboration with investigators of the FHS and does not necessarily reflect the opinions or views of the FHS, Boston University, or NHLBI. Funding for SHARe Affymetrix genotyping was provided by NHLBI Contract N02‐HL‐64278. SHARe Illumina genotyping was provided under an agreement between Illumina and Boston University. Funding for CARe genotyping was provided by NHLBI Contract N01‐HC‐65226. Funding support for the Framingham Dementia dataset was provided by NIH/NIA grant R01 AG08122.
The Cardiovascular Health Study (CHS) was supported by contracts HHSN268201200036C, HHSN268200800007C, N01‐HC‐85079, N01‐HC‐85080, N01‐HC‐85081, N01‐HC‐85082, N01‐HC‐85083, N01‐HC‐85084, N01‐HC‐85085, N01‐HC‐85086, N01‐HC‐35129, N01 HC‐15103, N01 HC‐55222, N01‐HC‐75150, N01‐HC‐45133, and N01‐HC‐85239; grant numbers U01 HL080295 and U01 HL130014 from the NHLBI, and R01 AG‐023629 from the National Institute on Aging, with additional contribution from the National Institute of Neurological Disorders and Stroke. A full list of principal CHS investigators and institutions can be found at https://chs-nhlbi.org/pi. This manuscript was not prepared in collaboration with CHS investigators and does not necessarily reflect the opinions or views of CHS, or the NHLBI. Additional support for infrastructure was provided by HL105756 and additional genotyping among the African American cohort was supported in part by HL085251. DNA handling and genotyping at Cedars‐Sinai Medical Center was supported in part by the National Center for Research Resources grant UL1RR033176, now at the National Center for Advancing Translational Technologies CTSI grant UL1TR000124; in addition to the National Institute of Diabetes and Digestive and Kidney Diseases grant DK063491 to the Southern California Diabetes Endocrinology Research Center.
The Health and Retirement Study (HRS) genetic data are sponsored by the Genetics Resource with HRS April 21, 2010, version G Page 5 of 7 National Institute on Aging (grant numbers U01AG009740, RC2AG036495, and RC4AG039029) and was conducted by the University of Michigan. This manuscript was not prepared in collaboration with HRS investigators and does not necessarily reflect the opinions or views of HRS.
Funding support for the Late‐Onset Alzheimer's Disease Family Study (LOADFS) was provided through the NIA Division of Neuroscience. The LOADFS includes a genome‐wide association study funded as part of the NIA Division of Neuroscience. Assistance with phenotype harmonization and genotype cleaning, as well as with general study coordination, was provided by Genetic Consortium for Late Onset Alzheimer's Disease. This manuscript was not prepared in collaboration with LOADFS investigators and does not necessarily reflect the opinions or views of LOADFS.
The authors thank Arseniy Yashkin for help in preparation of phenotypes in HRS.
AUTHOR CONTRIBUTIONS
AMK conceived and designed the experiment and wrote the paper; LS coded statistical tests and performed statistical analyses; YL and LH prepared data and coded statistical tests; AN, KA, and SU, prepared the data; AY contributed to drafting the manuscript; and IC performed bioinformatics analysis and wrote the paper.
Kulminski AM, Shu L, Loika Y, et al. Genetic and regulatory architecture of Alzheimer's disease in the APOE region. Alzheimer's Dement. 2020;12:e12008 10.1002/dad2.12008
Funding information
This research was supported by the National Institute on Aging (grant no. P01 AG043352, R01 AG047310, and R01 AG061853). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
REFERENCES
- 1. Raichlen DA, Alexander GE. Exercise, APOE genotype, and the evolution of the human lifespan. Trends Neurosci. 2014;37(5):247‐255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Strittmatter WJ, Weisgraber KH, Huang DY, et al. Binding of human apolipoprotein E to synthetic amyloid beta peptide: isoform‐specific effects and implications for late‐onset Alzheimer disease. Proc Natl Acad Sci U S A. 1993;90(17):8098‐8102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Suri S, Heise V, Trachtenberg AJ, Mackay CE. The forgotten APOE allele: a review of the evidence and suggested mechanisms for the protective effect of APOE varepsilon2. Neurosci Biobehav Rev. 2013;37(10 Pt 2):2878‐2886. [DOI] [PubMed] [Google Scholar]
- 4. Knopman DS. Lowering of amyloid‐beta by beta‐secretase inhibitors–some informative failures. N Engl J Med. 2019;380(15):1476‐1478. [DOI] [PubMed] [Google Scholar]
- 5. Freudenberg‐Hua Y, Freudenberg J, Vacic V, et al. Disease variants in genomes of 44 centenarians. Mol Genet Genomic Med. 2014;2(5):438‐450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Cruchaga C, Haller G, Chakraverty S, et al. Rare variants in APP, PSEN1 and PSEN2 increase risk for AD in late‐onset Alzheimer's disease families. PLoS One. 2012;7(2):e31039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Nesse RM, Ganten D, Gregory TR, Omenn GS. Evolutionary molecular medicine. J Mol Med (Berl). 2012;90(5):509‐522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Oeppen J, Vaupel JW. Demography. Broken limits to life expectancy. Science. 2002;296(5570):1029‐1031. [DOI] [PubMed] [Google Scholar]
- 9. Corella D, Ordovas JM. Aging and cardiovascular diseases: the role of gene‐diet interactions. Ageing Res Rev. 2014;18:53‐73. [DOI] [PubMed] [Google Scholar]
- 10. Kulminski AM. Unraveling genetic origin of aging‐related traits: evolving concepts. Rejuvenation Res. 2013;16(4):304‐312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Vijg J, Suh Y. Genetics of longevity and aging. Annu Rev Med. 2005;56:193‐212. [DOI] [PubMed] [Google Scholar]
- 12. Crespi B, Stead P, Elliot M. Evolution in health and medicine Sackler colloquium: comparative genomics of autism and schizophrenia. Proc Natl Acad Sci U S A. 2010;107(Suppl 1):1736‐1741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Cupples LA, Heard‐Costa N, Lee M, Atwood LD. Genetics analysis workshop 16 problem 2: the Framingham Heart Study data. BMC Proc. 2009;3(Suppl 7):S3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Fried LP, Borhani NO, Enright P, et al. The cardiovascular health study: design and rationale. Ann Epidemiol. 1991;1(3):263‐276. [DOI] [PubMed] [Google Scholar]
- 15. Juster FT, Suzman R. An overview of the health and retirement study. J Hum Resour. 1995;30:S7‐S56. [Google Scholar]
- 16. Lee JH, Cheng R, Graff‐Radford N, Foroud T, Mayeux R, National Institute on Aging Late‐Onset Alzheimer's Disease Family Study Group . Analyses of the National Institute on Aging Late‐Onset Alzheimer's Disease Family Study: implication of additional loci. Arch Neurol. 2008;65(11):1518‐1526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Howie BN, Donnelly P, Marchini J. A flexible and accurate genotype imputation method for the next generation of genome‐wide association studies. PLoS Genet. 2009;5(6):e1000529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Willer CJ, Li Y, Abecasis GR. METAL: fast and efficient meta‐analysis of genomewide association scans. Bioinformatics. 2010;26(17):2190‐2191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kulminski AM, Huang J, Wang J, He L, Loika Y, Culminskaya I. Apolipoprotein E region molecular signatures of Alzheimer's disease. Aging Cell. 2018;17(4):e12779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Zaykin DV, Meng Z, Ehm MG. Contrasting linkage‐disequilibrium patterns between cases and controls as a novel association‐mapping method. Am J Hum Genet. 2006;78(5):737‐746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wellek S, Ziegler A. A genotype‐based approach to assessing the association between single nucleotide polymorphisms. Hum Hered. 2009;67(2):128‐139. [DOI] [PubMed] [Google Scholar]
- 22. Kulminski AM, Huang J, Wang J, He L, Loika Y, Culminskaya I. Apolipoprotein E region molecular signatures of Alzheimer's disease. Aging Cell. 2018;17(4):e12779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Puri D, Gala H, Mishra R, Dhawan J. High‐wire act: the poised genome and cellular memory. FEBS J. 2015;282(9):1675‐1691. [DOI] [PubMed] [Google Scholar]
- 24. Creyghton MP, Cheng AW, Welstead GG, et al. Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc Natl Acad Sci U S A. 2010;107(50):21931‐21936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Yu CE, Seltman H, Peskind ER, et al. Comprehensive analysis of APOE and selected proximate markers for late‐onset Alzheimer's disease: patterns of linkage disequilibrium and disease/marker association. Genomics. 2007;89(6):655‐665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Takei N, Miyashita A, Tsukie T, et al. Genetic association study on in and around the APOE in late‐onset Alzheimer disease in Japanese. Genomics. 2009;93(5):441‐448. [DOI] [PubMed] [Google Scholar]
- 27. Lescai F, Chiamenti AM, Codemo A, et al. An APOE haplotype associated with decreased epsilon4 expression increases the risk of late onset Alzheimer's disease. J Alzheimers Dis. 2011;24(2):235‐245. [DOI] [PubMed] [Google Scholar]
- 28. Jazwinski SM, Kim S, Dai J, et al. HRAS1 and LASS1 with APOE are associated with human longevity and healthy aging. Aging Cell. 2010;9(5):698‐708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Fallin D, Cohen A, Essioux L, et al. Genetic analysis of case/control data using estimated haplotype frequencies: application to APOE locus variation and Alzheimer's disease. Genome Res. 2001;11(1):143‐151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Khachaturian AS, Hayden KM, Mielke MM, et al. Future prospects and challenges for Alzheimer's disease drug development in the era of the NIA‐AA Research Framework. Alzheimers Dement. 2018;14(4):532‐534. [DOI] [PubMed] [Google Scholar]
- 31. Schork NJ. Personalized medicine: time for one‐person trials. Nature. 2015;520(7549):609‐611. [DOI] [PubMed] [Google Scholar]
- 32. Finch CE. Evolution of the human lifespan, past, present, and future: phases in the evolution of human life expectancy in relation to the inflammatory load. Proc Am Philos Soc. 2012;156(1):9‐44. [PubMed] [Google Scholar]
- 33. Fiala M, Cribbs DH, Rosenthal M, Bernard G. Phagocytosis of amyloid‐beta and inflammation: two faces of innate immunity in Alzheimer's disease. J Alzheimers Dis. 2007;11(4):457‐463. [DOI] [PubMed] [Google Scholar]
- 34. Kigerl KA, Gensel JC, Ankeny DP, Alexander JK, Donnelly DJ, Popovich PG. Identification of two distinct macrophage subsets with divergent effects causing either neurotoxicity or regeneration in the injured mouse spinal cord. J Neurosci. 2009;29(43):13435‐13444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Theriault P, ElAli A, Rivest S. The dynamics of monocytes and microglia in Alzheimer's disease. Alzheimers Res Ther. 2015;7(1):41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Raj T, Rothamel K, Mostafavi S, et al. Polarization of the effects of autoimmune and neurodegenerative risk alleles in leukocytes. Science. 2014;344(6183):519‐523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Gjoneska E, Pfenning AR, Mathys H, et al. Conserved epigenomic signals in mice and humans reveal immune basis of Alzheimer's disease. Nature. 2015;518(7539):365‐369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Haan N, Zhu B, Wang J, Wei X, Song B. Crosstalk between macrophages and astrocytes affects proliferation, reactive phenotype and inflammatory response, suggesting a role during reactive gliosis following spinal cord injury. J Neuroinflammation. 2015;12:109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Li C, Zhao R, Gao K, et al. Astrocytes: implications for neuroinflammatory pathogenesis of Alzheimer's disease. Curr Alzheimer Res. 2011;8(1):67‐80. [DOI] [PubMed] [Google Scholar]
- 40. Zhong N, Weisgraber KH. Understanding the basis for the association of apoE4 with Alzheimer's disease: opening the door for therapeutic approaches. Curr Alzheimer Res. 2009;6(5):415‐418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Dodd JW. Lung disease as a determinant of cognitive decline and dementia. Alzheimers Res Ther. 2015;7(1):32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Lahiri DK. Apolipoprotein E as a target for developing new therapeutics for Alzheimer's disease based on studies from protein, RNA, and regulatory region of the gene. J Mol Neurosci. 2004;23(3):225‐233. [DOI] [PubMed] [Google Scholar]
- 43. Subramanian S, Gottschalk WK, Kim SY, Roses AD, Chiba‐Falek O. The effects of PPARgamma on the regulation of the TOMM40‐APOE‐C1 genes cluster. Biochim Biophys Acta. 2017;1863(3):810‐816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Bouhlel MA, Derudas B, Rigamonti E, et al. PPARgamma activation primes human monocytes into alternative M2 macrophages with anti‐inflammatory properties. Cell Metab. 2007;6(2):137‐143. [DOI] [PubMed] [Google Scholar]
- 45. Iglesias J, Morales L, Barreto GE. Metabolic and inflammatory adaptation of reactive astrocytes: role of PPARs. Mol Neurobiol. 2017;54(4):2518‐2538. [DOI] [PubMed] [Google Scholar]
- 46. Banno A, Reddy AT, Lakshmi SP, Reddy RC. PPARs: key regulators of airway inflammation and potential therapeutic targets in asthma. Nucl Receptor Res. 2018;5 10.11131/2018/101306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Albright JM, Dunn RC, Shults JA, Boe DM, Afshar M, Kovacs EJ. Advanced age alters monocyte and macrophage responses. Antioxid Redox Signal. 2016;25(15):805‐815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Koellhoffer EC, McCullough LD, Ritzel RM. Old maids: aging and its impact on microglia function. Int J Mol Sci. 2017;18(4):pii: E769 10.3390/ijms18040769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Franceschi C, Valensin S, Lescai F, et al. Neuroinflammation and the genetics of Alzheimer's disease: the search for a pro‐inflammatory phenotype. Aging (Milano). 2001;13(3):163‐170. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Basic information on SNPs and the associations of these SNPs with AD in the combined sample and each cohort separately.
Table S2. Linkage disequilibrium estimates using haplotype‐based method in the pooled sample of all cohorts.
Table S3. Linkage disequilibrium estimates using genotype‐based method in the pooled sample of all cohorts.
Table S4. Linkage disequilibrium estimates using haplotype‐based method in the pooled sample of all cohorts and each cohort separately for rs7412 and rs429358.
Table S5A. Linkage disequilibrium estimates using haplotype‐based method in the pooled sample of all cohorts for women.
Table S5B. Linkage disequilibrium estimates using haplotype‐based method in the pooled sample of all cohorts for men.
Table S6. Linkage disequilibrium estimates using haplotype‐based method in younger and older individuals.
Table S7. Functional annotation of potential regulatory SNPs in the APOE region.
Data Availability Statement
This article was prepared using limited access data sets obtained though dbGaP (accession numbers phs000007.v28.p10, phs000287.v5.p1, phs000428.v1.p1, and phs000168.v2.p2) and the University of Michigan. Phenotypic Health and Retirement Study (HRS) data are available publicly and through restricted access from http://hrsonline.isr.umich.edu/index.php?p=data.
RESEARCH IN CONTEXT
Systematic review: Recently, we reported significant molecular signatures of Alzheimer's disease (AD) in the apolipoprotein E (APOE) region, which excluded the ε2 and ε4 alleles. A literature review (PubMed and Google Scholar) identified few other publications, which reported significant associations of linkage disequilibrium (LD) structures with AD. These relevant publications are appropriately cited.
Interpretation: Susceptibility to AD is the result of a complex interplay of the ε2 and ε4 alleles with other alleles from different genes in the APOE region, which is not driven by common evolutionary forces. Accordingly, this interplay is the result of AD‐specific exposures, which, therefore, can be amendable to AD preventive interventions even with natural, for example, lifestyle, factors.
Future directions: This work suggests an approach to examine the potential role of complex genotypes/haplotypes in the AD etiology in loci with complex LD structures. Further work should be focused on elucidating personalized, that is, more homogeneous, group‐specific, polygenic profiles of AD risk and protection.