

Abstract
In this study we investigate the ability of ethanol-inducible CYP2E1 to interact with other cytochrome P450 species and affect the metabolism of their substrates. As a model system we used CYP2E1-enriched human liver microsomes (HLM) obtained by incorporation of purified CYP2E1. Using a technique based on homo-FRET in oligomers of CYP2E1 labeled with BODIPY 577/618 maleimide we demonstrated that the interactions of CYP2E1 with HLM result in the formation of its mixed oligomers with other P450 species present in the microsomal membrane. Incorporation of CYP2E1 results in a multifold increase in the rate of metabolism of CYP2E1-specific substrates p-Nitrophenol and Chlorzaxozone. The rate of their oxidation remains proportional to the amount of incorporated CYP2E1 up to the content of 0.3–0.4 nmol/mg protein (or about 50% CYP2E1 in the P450 pool). The incorporated CYP2E1 becomes a fully-functional member of the P450 ensemble and do not exhibit any detectable functional differences with the endogenous CYP2E1. Enrichment of HLM with CYP2E1 results in pronounced changes in the metabolism of 7-ethoxy-4-cyanocoumarin (CEC), the substrate of CYP2C19 and CYP1A2 suggesting an increase in the involvement of the latter in its metabolism. This effect goes together with an augmentation of the rate of dealkylation of CYP1A2-specific substrate 7-ethoxyresorufin. Furthermore, probing the interactions of CYP2E1 with model microsomes containing individual P450 enzymes we found that CYP2E1 efficiently interacts with CYP1A2, but lacks any ability to form complexes with CYP2C19. This finding goes inline with CYP2E1-induced redirection of the main route of CEC metabolism from CYP2C19 to CYP1A2.
Keywords: cytochrome P450, CYP2E1, CYP1A2, protein-protein interactions, oligomerization, alcohol-drug interactions, fluorescence resonance energy transfer (FRET), BODIPY 577/618
The functional properties of the drug-metabolizing system of human liver are largely determined by the properties of the ensemble of cytochromes P450, which is responsible for metabolism of over 75% of all marketed drugs and new drug candidates [1–3]. It is commonly recognized that the inter-individual, age-dependent and temporal variations in drug metabolism are largely dictated by the changes in the composition of the ensemble of multiple drug-metabolizing cytochrome P450 species. However, the interconnection between the composition of this ensemble and its functional properties is far from being straightforward. Recent studies on a large selection of human liver microsomes (HLM) demonstrated the lack of a straight correlation between the composition of the cytochrome P450 pool in human liver microsomes and the profile of drug metabolism [4]. At the same time, there is an emerging recognition of the functional importance of physical interactions between multiple P450 species co-localized in the microsomal membrane [5–9]. Due to complex effects of these interactions any change in the content of a particular P450 enzyme may affect drug metabolism in a complex, hard-to-predict manner. Therefore, a systematic exploration of the network of P450-P450 interactions and identification of their functional consequences represents an issue of emerging importance.
Of particular practical significance are the functional effects of the changes in the composition of P450 ensemble induced by alcohol consumption. Although the multi-fold increase in the content of cytochrome P450 2E1 (CYP2E1) in liver observed in both alcoholics and moderate alcohol consumers is well documented [10–14], the role of CYP2E1 in the content of a particular P450 enzyme may affect drug metabolism in a complex, hard-to-predict manner, the instances of alcohol-drug interactions is commonly considered insignificant due to a minor role of this enzyme in drug metabolism. However, the involvement of CYP2E1 in alcohol-drug interactions may stretch far beyond its immediate effects on the metabolism of its substrates.
There is a limited amount of information on possible functional effects of CYP2E1 on other human P450 species. In an early study with model microsomes Tan and co-authors demonstrated mutual inhibitory effects of CYP2E1 and CYP2A6, which were attributed to a competition for NADPH-cytochrome P450 reductase (CPR) [15]. In the same study the authors also observed a potentially important effect of co-expression of CYP2E1 with CYP2A6 on H2O2 production by CYP2A6. While NADPH-dependent generation of H2O2 by CYP2A6 was drastically increased in the presence of coumarin, addition of this CYP2A6 substrate results in decreased H2O2 production by the system composed of CYP2A6, CYP2E1 and CPR [15]. In a latter study in a micellar reconstituted system Kelley and co-authors explored the reciprocal effects of rabbit CYP1A2 and CYP2E1 [16]. The authors demonstrate that, while the addition of CYP1A2 does not affect CYP2E1-dependent metabolism of N-nitrosodimethylamine, addition of CYP2E1 considerably intensifies CYP1A2-dependent dealkylation of 7-ethoxyresorufin (ERR). Based on the observation that this effect was most pronounced at sub-saturating concentrations of CPR, the authors concluded that the stimulating effect of CYP2E1 on CYP1A2 may be exerted through the formation of the CYP1A2-CYP2E1 complexes where the interactions of CYP1A2 with CPR are promoted [16].
In our study of interactions between CYP2E1 and CYP3A enzymes in model microsomes we demonstrated the formation of the complexes of CYP2E1 with either CYP3A4 or CYP3A5, where the activity of CYP2E1 is boosted, while the functionality of the CYP3A enzymes remains unaffected [17]. In our recent study we also explored the interactions between CYP2E1 with CYP2D6 and observed the formation of their heteromeric complexes in microsomal membrane. We demonstrated that these interactions inhibit the activity of CYP2E1, while exerting a time-dependent activating effect on CYP2D6 in the presence of its specific substrates, 3-[2-(N,N-diethyl-N-methyl ammonium)ethyl]-7-methoxy-4-methylcoumarin (AMMC) and bufuralol [9]. Of particular importance might be the effect of CYP2E1 on the stoichiometry of futile cycling and substrate oxidation by CYP2D6 revealed in a decrease in the electron leakage from CYP2D6 through the peroxide-generating pathways [9]. This effect is reminiscent of an impact of CYP2E1 on H2O2 production by CYP2A6 observed by Tan and co-workers [15].
All these results demonstrate a complex functional relationship between CYP2E1 and other drug-metabolizing P450 species which deserves detailed attention in view of its potential involvement in alcohol-drug interactions. Of particular interest is possible crosstalk between CYP2E1 and CYP1A2 evidenced in the study of Kelley and co-authors [16]. The effect of ethanol-induced increase in CYP2E1 concentration in liver on CYP1A2 may be involved in pronounced interactions of alcohol with such drug substrates of CYP1A2 as tricyclic antidepressnts (amitryptiline, clomipramine, imipramine, etc.) [18–19]. Most of the drugs of this type are also metabolized by CYP2C19 and possible rerouting of drug metabolism between the two enzymes may also play a role in their interactions with alcohol.
The aim of the present study is to probe the interactions of CYP2E1 with cytochromes P450 present in HLM and explore the effect of these interactions on the functional properties of CYP1A2 and CYP2C19. In a search for a model system suitable for these studies we recently introduced a method based on incorporation of purified cytochromes P450 into microsomal membrane [9, 17, 20]. An important advantage of this strategy is its applicability for incorporating genetically or chemically modified proteins. This includes the incorporation of proteins bearing fluorescent probes and light-activating crosslinkers. Therefore, this strategy may be used for monitoring protein-protein interactions in HLM with various biophysical techniques, as well as for identifying the P450 interaction partners with the technique of molecular fishing. Results of the present study demonstrate high utility of this approach in the studies of interactions between multiple cytochrome P450 species and their effect on drug metabolism. Using this strategy we evidenced the formation of the complexes of CYP2E1 with other P450 species present in HLM and demonstrated the effects of CYP2E1 on the activity of CYP1A2, the enzyme involved in metabolism of a wide variety of commonly used drugs known for their pharmacokinetic interactions with alcohol.
Experimental
Materials.
DL-dithiothreitol (DTT), p-Nitrophenol (pNP), p-Nitroanizol and L-α- phosphatidylcholine (PC) from egg yolk were the products of Sigma-Aldrich (St. Louis, MO). NADPH tetrasodium salt was from EMD Millipore (Billerica, MA). 7-ethoxyresorufm was from AnaSpec (San Jose, CA, USA). 7-ethoxy-4-cyanocoumarin (CEC), 7-hydroxy-4-cyanocoumarin (CHC) and BODIPY 577/618 maleimide were from Invitrogen/Molecular Probes Inc. (Eugene, CA).. Chlorzoxazone, 6-hydroxy chlorzoxazone and resorufin were the products of Cayman Chemical (Ann Arbor, MI). L-α-phosphatidylethanolamine (PE) from bovine liver and 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphate (phosphatidic acid, PA) were obtained from Avanti Polar Lipids, Inc. (Alabaster, AL). Octyl-β-D-glucopyranoside (octylglucoside) was the product of Fluka Honeywell Specialty Chemicals (Seelze, Germany). All other reagents were of ACS grade and used without additional purification.
Protein expression and purification.
N-terminally truncated (Δ3–20) and C-terminally His-tagged CYP2E1 [21] was expressed in E. coli TOPP3 cells as described for CYP2B4 in [22]. After spinning down the cells, the pellets were resuspended in ~10% of the initial volume of culture of 20mM K-phosphate buffer containing 20% glycerol, 10 mM β-mercaptoethanol (bME), 1mM phenylmethanesulfonyl fluoride (PMSF), and 10 μM leupeptin, pH 7.8. Lysozyme was added to the concentration of 0.3 mg/mL and the suspension was incubated at 4 °C for 30 min. at continuous stirring. After diluting with an equal volume of cold water and additional 10 min. of incubation, the suspension was centrifuged at 3800 r.p.m. for 20 min. The pellet was resuspended in 500 mM K-phosphate buffer containing 20% glycerol, 10 mM bME, 1 mM PMSF, 10 μM leupeptin, pH 7.4 (Buffer A) taken in the volume of approx. 2 ml per gram of cell pellet. The suspension was sonicated with an ultrasonic sonicator in 30–50 ml aliquots on ice. The aliquots were combined and a 10% stock solution of CHAPS in Buffer A was added to the concentration of 0.86% (14 mM). After 3 hours of incubation at continuous stirring at 4°C the suspension was centrifuged at 105,000g and 4 °C for 1 hour. The pellet was discarded and supernatant applied onto a column of HisPur™ Ni-NTA Resin (ThermoFisher Scientific, Waltham, MA) taken in the amount of 1 ml per ~100 nmols of cytochrome P450 found in supernatant pre-equilibrated with Buffer A containing 0.86% CHAPS (w/v) at the flow rate of 8–10 ml/hour. The column was subsequently washed with 20 volumes of buffer A containing 0.5% CHAPS., 5 volumes of buffer A containing 0.5% CHAPS and 7.5 mM imidazole and with 3 volumes of 100 mM K-P buf, containing 20% Glycerol, 2 mM TCEP and 0.5% CHAPS (buffer B)). CYP2E1 was eluted from the column with buffer B containing 200 mM histidine. The fractions containing CYP2E1 were combined and diluted with 3 volumes of water containg 20% glycerol and 0.5% CHAPS. Resulting solution was applied onto a column of Macro-Prep CM Resin (Bio-Rad Laboratories, Hercules, CA) taken in the amount of 1 ml per 60 nmol of CYP2E1. Before application the column was equilibrated with 37.5 mM K-phosphate buffer containing 20% glycerol, 3 mM dithiotreitol, 0.5 mM EDTA and 0.5% CHAPS. The column was washed with three volumes of 75 mM K-Phosphate buffer containing the same additions as above (Buffer C) and the protein was eluted with 20 column volumes of linear gradient of KC1 from 0 to 300 mM in Buffer C. The elution band of CYP2E1 was centered around KC1 concentration of 150 mM. The fractions containing CYP2E1 were combined and concentrated to ~10% of the initial volume on Vivaspin 20 100kDa MWCO centrifugal concentrators (Sartorius Stedim Lab. Ltd., Stonehouse, UK). After removal of CHAPS with DetergentOUT spin-out columns (G-Biosciences, St. Louis, MO) and further concentrating to CYP2E1 concentration of 100–200 μM, the protein solution was dialyzed against two changes of 500 volumes of 125 mM K-Phosphate buffer, pH 7.4 containing 20% glycerol (protein storage buffer) saturated with argon gas. The purified protein was stored at −80 °C before use.
Labeling of CYP2E1 with BODIPY-577/618 maleimide.
In order to prevent irreversible precipitation of the protein observed upon modification of CYP2E1 with BODIPY-577/618 maleimide in the protein storage buffer, we found it necessary to increase the ionic strength of the media. Buffer replacement was carried out by a passage through a spin-out column of Bio-Gel P6 (Bio-Rad, Hercules, CA, USA) equilibrated with 0.5 M K-phosphate buffer, pH 7.4, containing 20% glycerol and followed with a dilution with the same buffer to the final protein concentration of 10 μM. The resulting protein solution was placed into a fluorometer cell and thermostated at 4 °C with continuous siring. In order to incorporate the first molar equivalent of the probe a 3 mM solution of BODIPY 517/618 maleimide in acetone was added to the final concentration of 10.5 μM. The process of labeling was monitored by increase in fluorescence at 518 nm (excitation at 405 nm). The reaction was associated with a ~1.5 – 1.8 fold increase in fluorescence, which required around 60 min to be 95% complete. If desired, the second molar equivalent of the probe was introduced by a sequential addition of the second 10.5 μM aliquot of the label. The process of incorporation of the second molar equivalent of the probe was substantially slower and required around three hours for completion. The reaction was terminated by an addition of reduced gluthatione (GSH) to the concentration of 10 mM. No protein precipitation, heme depletion or conversion into the P420 state was observed in the protein modified at either 1:1 or 1:2 protein-to-label ratios. Modification with the second molar equivalent of the label resulted in some increase in the content of the high-spin state of the enzyme (see Fig. S1b in the Supplementary Material). Attempts to introduce the third molar equivalent of the label resulted in a massive irreversible precipitation of the protein. The protein modified at 1:1 or 1:2 protein-to-label ratios was concentrated to 25–30 μM with the use of a Centrisart I MWCO 100 kDa concentrator (Sartorius AG). After removal of any contaminating GSH-BODIPY adduct by a passage through a spin-out column of Bio-Gel P-6 equilibrated with the protein storage buffer the protein was stored at −80 °C before use.
Preparation of liposomes.
Giant liposomes used in this study were prepared by the octylglucoside dialysis/sorption technique [17]. To this end we prepared 15 mg of a mixture of phosphatidylcholine (PC), phosphatydilethanolamine (PE) and phosphatidic acid (PA) taken as chlorophorm solutions at 2:1:0.6 ratio (by weight). The chloroform was removed by evaporation under a stream of argon gas and subsequent drying under vacuum for 2 hours. The phospholipid pellet was suspended in 2.5 mL of 1.54% solution of octylglycoside in argon-saturated 100 mM HEPES buffer, pH 7.4, containing 150 mM KC1, 0.5 mM EDTA and 20% (v/v) glycerol (Buffer A) by vigorous stirring at a Vortex mixer under argon atmosphere. After 2 hours of incubation at room temperature under argon gas the suspension was diluted to the final volume of 9 ml by argon-saturated buffer A containing no octylglucoside. The resulting mixture was placed into a bag of Spectra/Por 6 dialysis membrane MWCO 25 kDa (Spectrum Chamical Mfg. Corp., New Brunswick, NJ) and dialyzed at 4 °C under argon atmosphere against three changes of 1L portions of buffer A, each containing 7 mL of Bio-Beads SM-2 Adsorbent (Bio-Rad, Hercules, CA, USA). The term of dialysis with each change of the buffer was 24 hours. The content of phospholipids in the resulting suspension of liposomes was quantified based on the determination of total phosphorus in a chloroform/methanol extract according to Bartlett [23]. The suspension was stored at −80 °C before use.
Microsomes from insect cells bearing recombinant human cytochromes P450 (Supersomes™) were the products of BD Gentest, now a part of Coming Life Sciences (Tewksbury, MA). In the present study we used the preparations containing CYP2E1 (SS2E1, lot 44748), CYP1A2 (SS1A2, lot 69375), CYP2B6 (SS2B6, lot 31487), CYP2C8 (SS2C8, lot 8239002), CYP2C9 (SS2C9, lot 41274), CYP2C19 (SS2C19, lot 73445), CYP2D6 (SS2D6, lot 7114001) and CYP3A4 (SS3A4, lot 35933). All those preparation contained human NADPH-cytochrome P450 reductase (CPR) and cytochrome b5, except for SS2C8 and SS1A2, which had no cytochrome b5 co-expressed. In the experiments on CYP2E1 interactions with microsomal membrane we also utilized Supersomes™ containing rat CPR (SS-CPR, lot 5274006) or human CPR and cytochrome b5 (SS-B5, lot 41774) as control membranes bearing no cytochromes P450.
Preparations of human liver microsomes (HLM).
In this study we used two different preparations of pulled human liver microsomes. The preparation obtained by differential centrifugation of pulled human liver S9 fraction (the product of BD Gentest, lot number 3212595) is reffered to as HLM-1. The HLM sample refered here as HLM-2 is an InVitroCYP™ M-class 50-donor mixed gender pooled HLM preparation (lot LFJ) obtained from BioIVT corporation (Baltimore, MD).
Incorporation of CYP2E1 into HLM was performed by incubation of undiluted suspensions of HLM (20–25 mg/ml protein, 10–13 mM phospholipid) in 125 mM K-Phosphate buffer containing 0,25M Sucrose with purified CYP2E1 for 16 – 20 hours at 4°C at continuous stirring. CYP2E1 was added in the amount ranging from 0.25 to 2 molar equivalents to the endogenous cytochrome P450 present in HLM. Following the incubation the suspension was diluted 4–8 times with 125 mM K-Phosphate buffer, pH 7.4 containing 0.25 M sucrose and centrifuged at 53,000 rpm (150,000 g) in an Optima TLX ultracentrifuge (Beckman Coulter Inc., Brea, CA, USA) with a TLA100.3 rotor for 90 min at 4 °C. The pellet was resuspended in the same buffer to the protein concentration of 15–20 mg/ml. The amount of incorporated cytochrome P450 was calculated from the difference between the heme protein added to the incubation media and the enzyme found in the supernatant. According to the results of this assay, our procedure resulted in incorporation of 96–98% of the added protein into the microsomal membrane.
Analysis of the content of microsomal preparations.
Determinations of protein and phospholipid concentrations in microsomal suspensions were performed with the bicinchoninic acid procedure [24] and through the determination of total phosphorus in a chloroform/methanol extract according to Bartlett [23], respectively.
The concentration of NADPH-eytochrome P450 reductase in microsomal membranes was determined based on the rate of NADPH-dependent reduction of cytochrome c at 25°C [25] monitored by an increase in the difference in absorbencies at 550 and 541 nm and calculated using the differential (“reduced-minus-oxidized”) extinction coefficient of cytocrome c of 18 mM−1cm−1 [26]. Effective molar concentration of CPR was estimated using the turnover number of 3750 min−1, which we determined with purified CPR quantified from its absorbance at 456 nm using the extinction coefficient of 21.4 mM−1 cm−1 [27].
The total concentration of cytochrome P450 in HLM was determined with a variant of “oxidized CO versus reduced CO difference spectrum” method [28]. In our assay we used Microsome Solubilization Buffer (MSB) composed of 100 mM potassium phosphate pH 7.3, 10% Glycerol, 0.5% Sodium Cholate and 0.4% Igepal CO-630. Prior to addition of the microsomal suspension the buffer was saturated with CO through bubbling the gas for 40–60 sec. 100 μl of CO- saturated MSB were placed into a quartz microcuvette. After recording a baseline in 380–600 nm region the cell was supplemented with 5μl of HLM sample (10–20 mg/ml) and the spectrum was recorded. Reduction was achieved by addition of a small amount of dithionite dust. The process of reduction was followed through monitoring the changes in the difference of optical densities (DOD) at 450 and 490 nm. The spectrum of absorbance of the reduced sample was taken when the DOD stabilizes. The difference between the spectra taken after and before reduction was calculated and used to determine the concentration of cytochromes P450 using the extinction coefficient of 106 mM-1 cm-1 for DOD between the maximum of the peak around 450 nm (448–451 nm) and the plateau at 490 nm. Determination of the content of cytochrome b5 in HLM was based on its NADH-dependent reduction and performed following the procedure described by Kennedy [29].
Mass-spectrometric analysis of the content of cytochromes P450 and NADPH-cytochrome P450 reductase in HLM was performed with a triple quadrupole mass spectrometer using the method of multiple reaction monitoring (MRM). In order to prepare samples of hydrolyzed microsomal proteins we mixed aliquots of microsomal preparations containing 100 μg of protein (~5μL) with 20 μL of the denaturation solution (100 mM K-Phosphate buffer pH 6.3 containing 5 M urea, 1% sodium deoxycholate, 15% acetonitrile, 300 mM NaCl and 20 mM TCEP). The samples were heated for 20 minutes at 50 °C, cooled to room temperature and mixed with 25 μL of 15 mM 2- iodoacetamide in 50 mM triethylammonium bicarbonate. The samples were incubated for 30 minutes at ambient temperature, diluted with 50 mM triethylammonium bicarbonate to the final volume of 120 μL and supplemented with 1 μg of trypsin added as 5 μL of 200 ng/mL solution. After 3 hours of incubation at 38 °C the second 1 μg aliquot of trypsin was added and the incubation continued for another 3 hours at the same temperature. At the end of digestion the samples were supplemented with 10 μL of 10% formic acid and centrifuged for 10 minutes at 12,000 g and 10 °C in order to remove deoxycholate, which is insoluble at these conditions. The supernatant was quantitatively (90 μL) transferred into a clean tube and dried under vacuum at 30°C for 70 minutes. Dried samples were dissolved in 40 μL of 15 mM ammonium acetate for further fractionation and supplemented with stable isotope labeled peptides. For each protein, one standard peptide with three transitions was used. The peptides were arranged into one Selected Reaction Monitoring (SRM) assay. The information on stable-isotope labeled internal standards (SIS) and MS platform used for the analysis (mass-to-charge (m/z) ratios of precursors and transition ions, collision energies, etc.) along with the sequences of the target peptides are provided in Table S1 found in the Supplementary Material.
Separation of peptides from digested HLM samples was carried out using the UPLC Agilent 1290 system including a pump and an autosampler. The sample was loaded into the analytical column Eclipse Plus SBC-18, 2.1 × 100 mm, 1.8 um, 100 A. Peptide elution was performed by applying a mixture of solvents A and B. Solvent A was HPLC grade water with 0.1% (v/v) formic acid, and solvent B was 80%(v/v) HPLC grade acetonitrile/water with 0.1% (v/v) formic acid. The separations were performed by applying a linear gradient from 3% to 32% solvent B over 50 min, then from 32% to 53% solvent B over 3 min at 300μl/min followed by a washing step (5 min at 90% solvent B) and an equilibration step (5 min at 3% solvent B). Ten microliters of each sample were applied on chromatographic column. The quantitative analysis was performed using Agilent 6495 Triple Quadrupole (Agilent, USA) equipped with a Jet Stream ionization source. The following parameters were used for the Agilent Jet Stream ionization source: the temperature of the drying gas of 280°C, 18 psi pressure in the nebulizer, 14 L/min flow rate of the drying gas, and 3000V voltage on the capillary.
The standard samples of target peptides were obtained using the solid-phase peptide synthesis on the Overture (Protein Technologies, USA) according to the published method [30]. The isotope-labeled lysine (13C6,15N2) or arginine (13C6,15N4) was used for isotope-labeled peptide synthesis instead of the unlabeled lysine or arginine, correspondingly. Concentration of the synthesized peptides was quantified through acidic hydrolysis followed by analysis of derived amino acids with fluorimetric detection [31].
Activity measurements with fluorogenic substrates.
The rate of O-dealkylation of CEC and ERR were measured with a real-time continuous fluorometric assay. A suspension of microsomes was added to 250 μl of 0.1 M Na-HEPES buffer, pH 7.4, containing 60 mM KC1 to a final CPR concentration of 0.003 – 0.01 μM. The mixture was placed into a 5 × 5 mm quartz cell with continuous stirring and thermostated at 30 °C. An aliquot of a 15–20 mM stock solution of CEC or 0.4–2 mM stock solution of ERR in acetone was added to attain the desired concentration in the range of 0.5 – 250 μM (CEC) or 0.03 – 10 μM (ERR). The reaction was initiated by addition of 20 mM solution of NADPH to the concentration of 200 μM. Increase in the concentration of products of dealkylation (CHC or resorufin) was monitored with a Cary Eclipse fluorometer (Agilent Technologies, Santa Clara, CA, USA) or a custom-modified PTI QM-1 fluorometer (Photon Technology International, New Brunswick, NJ) [9]. With both instruments the emission wavelength was set to 455 and 585 nm for the measurements with CEC and ERR, respectively. The excitation was performed with a monochromatic light centered at 405 or 560 nm (for CEC and ERR, respectively) with 5 nm bandwidth (Cary Eclipse) or with a light emitted by a 405 nm (CEC) or 532 nm (ERR) laser diode module (Thorlabs Inc, Newton, NJ). The rate of formation of the fluorescent products was estimated by determining the slope of the linear part of the kinetic curve recorded over a period of 3 – 5 min. Calibration of the assay was performed by measuring the intensity of fluorescence in a series of 5–10 samples of the same reaction mixture containing increasing concentrations of CHC or resorufin.
Measurements of the rate of hydroxylation of p-nitrophenol.
The rate of p-nitrophenol hydroxylation was determined with absorbance spectroscopy [32]. To measure the rate of formation of p-nitrocatechol (pNC) by HLM we mixed 3–5μl of the microsomal suspension with reaction buffer (0.1 M Na-Hepes buffer containing 60 mM KC1) and a desired amount of 10–20 mM stock solution of pNP in reaction buffer to the total volume of 80 μl. After a preincubation for 1 min in a water bath at 30 °C the reaction was initiated by addition of 0.8 μl of 20 mM NADPH solution. The probes were incubated at 30 °C for 30 min in a shaking water bath and the reaction was stopped by addition of 24 μl of 20% Trichloroacetic acid. The precipitated protein was removed by centrifugation at 15,000g for 2 min and the supernatant was supplemented with 16 μl of 5 M NaOH. The spectra of absorbance of the samples in 450 – 700 nm region were recorded in a 100μl microcuvette (optical path 1 cm). A spectrum of the sample treated as above and containing all ingredients except for pNP was used as a baseline. To quantify the amount of the formed pNC and obtain the dependencies of the reaction rate on substrate concentration we subjected a series of spectra obtained with pNP concentrations increasing from 0 to 250 μM to the principal component analysis (PCA) procedure. The spectra of the first two principal components were approximated with a combination of the standard spectra of absorbance of pNP and pNC supplemented with a first or second order polynomial, which was used to compensate for possible variation in turbidity of the samples. The maximum of absorbance of pNC was found at 514 nm and the value of the extinction coefficient of 10.3 mM−1 cm−1 at 546 nm [32] was used to normalize the spectral standard to 1μM concentration. The concentration factors found in this fitting were used to scale the first two vectors of loading factors and combine them into a dependence of the concentration of formed pNC on the concentration of pNP.
Activity measurements with chlorzoxazone were performed in 0.1 M Na-HEPES buffer, pH 7.4, containing 60 mM KCl. The reaction was started by addition of NADPH to the final concentration of 0.5 mM and carried on for 10 min at 30 °C. Chlorzoxazone was added as a 25 mM solution in 60 mM KOH. Reaction was quenched with an addition of 25% of the incubation volume of 1 M solution of formic acid in acetonitrile containing a known concentration 3,5-dibromo-4-hydroxybenzoic acid as internal standard, followed by centrifugation at 9,300 g for 10 min.
The amount of formed 6-hydroxychlorzoxazone was quantified by liquid chromatography-mass spectrometry. An LC-20AD series high-performance liquid chromatography system (Shimadzu, Columbia, MD) fitted with a HTC PAL autosampler (LEAP Technologies, Carrboro, NC) was used to perform chromatography on a Kinetex® reverse-phase column (50 × 2.1 mm, 2.6 μm; Phenomenex, Torrance, CA). Chromatographic separation was performed using an isocratic method at the flow rate of 0.18 ml/min (mobile phase A) and 0.22 ml/min (mobile phase B) for 5 min. The quantification of the metabolite was conducted using an API 4000 Q-Trap tandem mass spectrometry system manufactured by Applied Biosystems/MDS Sciex (Foster City, CA) using turbospray ESI operating in negative ion mode. The mass spectrometer parameters were set at curtain gas, 20; collision gas, medium; ion spray voltage, −4500; ion source gas 1, 70; ion source gas 2, 50; temperature, 500, declustering potential, −550; entrance potential, −5; collision energy, −25, collision cell exist potential, 10. The analyte (6-hydroxychlorzoxazone) and the internal standard (3,5-dibromo-4-hydroxybenzoic acid) were detected using multiple reaction monitoring (MRM) mode by monitoring the m/z transition from 184.0 to 120 and 294.8 to 251.0, respectively. Determination of the product amounts was achieved with a calibration curve ranging from 0 to 2000 nM concentration of 6-hydroxychlorzoxazone.
Normalization of the rates of substrate metabolism determined in HLM samples was performed based on the concentration of CPR determined from the rate of NADPH-dependent reduction of cytochrome c as described above. The rates of substrate metabolism by Supersomes™ were normalized on the concentration of cytochrome P450 (due to excess of CPR over cytochrome P450 in these preparations).
Monitoring of the interactions of CYP2E1-BODIPY with microsomal membranes was performed with the use of a Cary Eclipse spectrofluorometer equipped with a Peltier 4-position cell holder. The excitation of donor phosphorescence was performed with monochromatic light centered at 405 or 505 nm with 20 nm bandwidth. Alternatively the measurements were done with the use a custom-modified PTI QM-1 fluorometer (Photon Technology International, New Brunswick, NJ) equipped with a thermostated cell holder and a refrigerated circulating bath. In this case the excitation was performed at 405 nm with a CPS405 collimated laser diode module (Thorlabs Inc, Newton, NJ). The spectra in the 570 – 800 nm wavelength region were recorded repetitively with the time interval varying from 0.5 to 15 min during the course of monitoring (5 – 16 hours). The experiments were performed at continuous stirring at 4 °C in 100 mM Na-Hepes buffer (pH 7.4) containing 150 mM KCl and 250 mM sucrose.
Fluorescence anisotropy measurements were performed with the use of a custom-modified PTI QM-1 fluorometer (Photon Technology International, New Brunswick, NJ) equipped with two broadband polarizing beamsplitter cubes PBSH-450-700-100 (CVI Laser, LLC, Albuquerque, NM), each for the exitation and the emission beam. Fluorescence excitation was performed at 405 nm with a CPS405 collimated laser diode module (Thorlabs Inc, Newton, NJ). The pairs of spectra of BODIPY fluorescence in 570–710 nm region were recorded sequentially for coplanar and orthogonal orientations of the polarizers. To determine the anisotropy of emission, the linear least square algorithm was used to approximate the spectrum taken with the coplanar orientation of polarizers with the one recorded at the orthogonal orientation. The resulting multiplication factor, RR/T, was then used to calculate the emission anisotropy (r) defined as r= (RP/T −1)/(RP/T +2).
Calculations of the surface density of cytochromes P450 in microsomal and proteoliposomal membranes were based on the molar ratio of membranous phospholipids to incorporated CYP2E1 (RL/P). For our calculations in the case of proteoliposomes we assumed the area of the membrane per one phospholipid molecule in a monolayer be of 0.72 nm2 (mean value for egg phosphatidylcholine membrane at 30 °C from Table 3 in [33]) and the footprint of one CYP3A4 molecule to be of 16 nm2. These assumptions result in the following relationship between the concentration of CYP2E1 in the membrane (CCYP2E1) and the RL/P:
| (Eq. 1) |
In the case of CYP2E1 incorporated into microsomal membranes we assumed the area of microsomal membrane corresponding to one molecule of phospholipid in a monolayer to be equal to 0.95 nm2 [34], similar to the approach used in our earlier reports [9, 17, 20]. In this case the relationship between CCYP2E1 and the RL/P has the following form:
| (Eq.2) |
Data analysis.
Analysis of series of spectra obtained in fluorescence spectroscopy experiments was done by principal component analysis (PCA) [35], which was used as a method of global analysis that minimizes the contribution of incidental variation in the shape of the spectra and makes the results more robust to a minor displacement of the emission band (a blue shift of approx. 2 nm) that takes place upon the interactions of CYP2E1-BODIPY with the membrane. To quantify the changes in the fluorescence of BODIPY we used a linear least-squares fitting of the spectra of the first and second principal components by a combination of the prototypical spectra of emission of CYP2E1-BODIPY in solution and in a membrane-bound state. These spectral standards were normalized to have the same spectral area.
The equation for the equilibrium of binary association (dimerization) used in the fitting of oligomerization isotherms (dependencies of FRET efficiency on the concentration of P450 in membranes) had the following form:
| (Eq. 3) |
where [E]0, [X], and KD are the total concentration of the associating compound (enzyme), the concentration of its dimers, and the apparent dissociation constant, respectively. In order to use this equation in fitting of the dependencies of the relative increase in fluorescence observed at enzyme concentration [E]0 (RE) equation (1) was complemented with the parameter Rmax. This parameter corresponds to the value of RE observed upon a thorough dissociation of completely oligomerized enzyme:
The parameter Rmax is determined by the efficiency of FRET (EFRET) according to the following relationship:
Fitting of the titration isotherms to the above equations was performed with non-linear regression using a combination of Nelder-Mead and Marquardt algorithms as implemented in our SpectraLab software [35].
Results
Design of the method for monitoring membrane incorporation of CYP2E1 with homo-FRET
In our previous studies we demonstrated that the incubation of either the microsomes from insect cells (Supersomes™ or Baculosomes®) or HLM with purified CYP3A4, CYP2D6 or CYP2E1 results in efficient incorporation of the heme proteins into the membrane [9, 17, 20]. In order to further refine and better characterize this model system we developed a new technique for monitoring the process of CYP2E1 incorporation into the membrane. Our method utilizes fluorescence resonance energy transfer (homo-FRET) between fluorophores attached to neighboring CYP2E1 molecules in the oligomer. Short Stockes shift characteristic to the dyes of the difluoro-boraindacene family (BODIPY-based dyes), where the distance between the maxima of the emission and the excitation bands (8 – 40 nm) is small relative to the widths of these bands (100 – 150 nm)[36], results in an efficient FRET between closely located fluorophores of the same kind (homo-FRET), Another important reason for choosing a BODIPY-based fluorophore for studying protein-protein interactions in phospholipid membranes is a remarkable resistance of fluorescence of most BODIPY dyes to changing polarity of the environment [37]. This insensitivity to environment provides for a significant prevalence of the FRET-based effects over the effects of membrane incorporation per se.
For our experiments we chose BODIPY-577/618 maleimide due to a minimal spectral overlap of its emission band (λmax=618 nm) with the absorbance bands of the P450 heme. With this florescent probe the effect of homo-FRET between two BODIPY moieties (R0=53 Å) is expected to predominate over the effect of FRET from BODIPY to the P450 heme groups of the neighboring heme proteins (R0=40 Å).
The fluorescence of BODIPY maleimides is known to exhibit a multifold increase upon their conjugation with SH-groups [36]. However, in the case of cytochromes P450 this increase is expected to be considerably diminished due to FRET from BODIPY to the heme (R0=40 Å). In an agreement with these expectations, the incorporation of the first molar equivalent of the label into CYP2E1 resulted in a quite modest (~1.8 fold) increase in the probe fluorescence. The process of this increase obeyed second-order reaction kinetics with the rate constant of 25–30 mM−1 min−1 (see Fig. S1 in the Supplementary Material). Importantly, the process of incorporation of the second molar equivalent of the probe was substantially slower (k≈7 mM−1 min−1) and the resulting increase in BODIPY fluorescence did not exceed 15% (Fig. S1). These results suggest that the two preferential points of attachment of BODIPY-maleimide in CYP2E1 (CYP2E1 possesses 7 potentially reactive cysteine residues – Cys174, Cys177, Cys261, Cys268, Cys452, Cys480 and Cys488) differ considerably in their accessibility to modification and their distance to the heme. The most accessible cysteine residue (presumably Cys488) appears to be located farther from the heme than the “second-choice” residue (presumably Cys261, Cys268 and/or Cys480), so that its fluorescence predominates in the emission of the labeled protein.
Similar to all purified membranous cytochromes P450, CYP2E1 appears to be heavily oligomerized in solution. As a result, the fluorescence of CYP2E1-BODIPY oligomers must be affected by homo-FRET between the labels located in the neighboring subunits of the oligomer. Dissociation of the oligomers caused by their incorporation into phospholipid membranes is expected to eliminate the effects of homo-FRET.
It is commonly accepted that, in contrast to FRET between dissimilar fluorophores, homo- FRET does not change the intensity and the lifetime of fluorescence [38]. The most distinctive effect of homo-FRET is a decrease in the degree of polarization (anisotropy) of fluorescence [38]. However, the conclusion on insensitivity of the fluorescence intensity to homo-FRET is valid only if the donor and the acceptor are located in the same environment, exhibit the same quantum yields and are not differentially quenched due to FRET to a third chromophore or other possible mechanisms. In the case of the labels incorporated into a protein with multiple possible points of attachment these conditions are not necessarily satisfied. In particular, in the case of cytochromes P450 the intensity of fluorescence of BODIPY probes attached at different positions is differentially affected by FRET to the heme. As a result, homo-FRET between the labels attached to different loci may result in a considerable change in the fluorescence intensity.
In order to probe the potential of homo-FRET between BODIPY-577/618 probes as a tool for monitoring dissociation of CYP2E1 oligomers we studied the effect of interactions of BODIPY- BODIPY with the membrane of phospholipid vesicles (liposomes) on the intensity and anisotropy of fluorescence. As shown in Fig. 1a, addition of liposomes to the labeled protein results in an increase in the anisotropy of BODIPY fluorescence that is indicative of a decrease in the efficiency of homo- FRET. However, the more pronounced effect is an increase in the intensity of BODIPY fluorescence (Fig. 1b,c). The kinetic curves of the changes in anisotropy and intensity of fluorescence may be approximated with a three-exponential equation with one and the same set of the rate constants (1.1, 0.06 and 0.008 min−1), although the relative amplitudes of the three phases differ considerably between the two modes of registration (Fig. 1a,c). Considerably higher contribution of the fast phase into the changes in anisotropy may suggest that at the initial stage of interactions the anisotropy may be affected by some factors other than homo-FRET. For instance, this initial increase in anisotropy is attributable to changes in rotational mobility of CYP2E1 aggregates due to breakdown of large multimeric units (e.g., hexamers and larger aggregates) into smaller modules (e.g. trimers) upon initial interactions with the phospholipid bilayer.
Fig, 1.
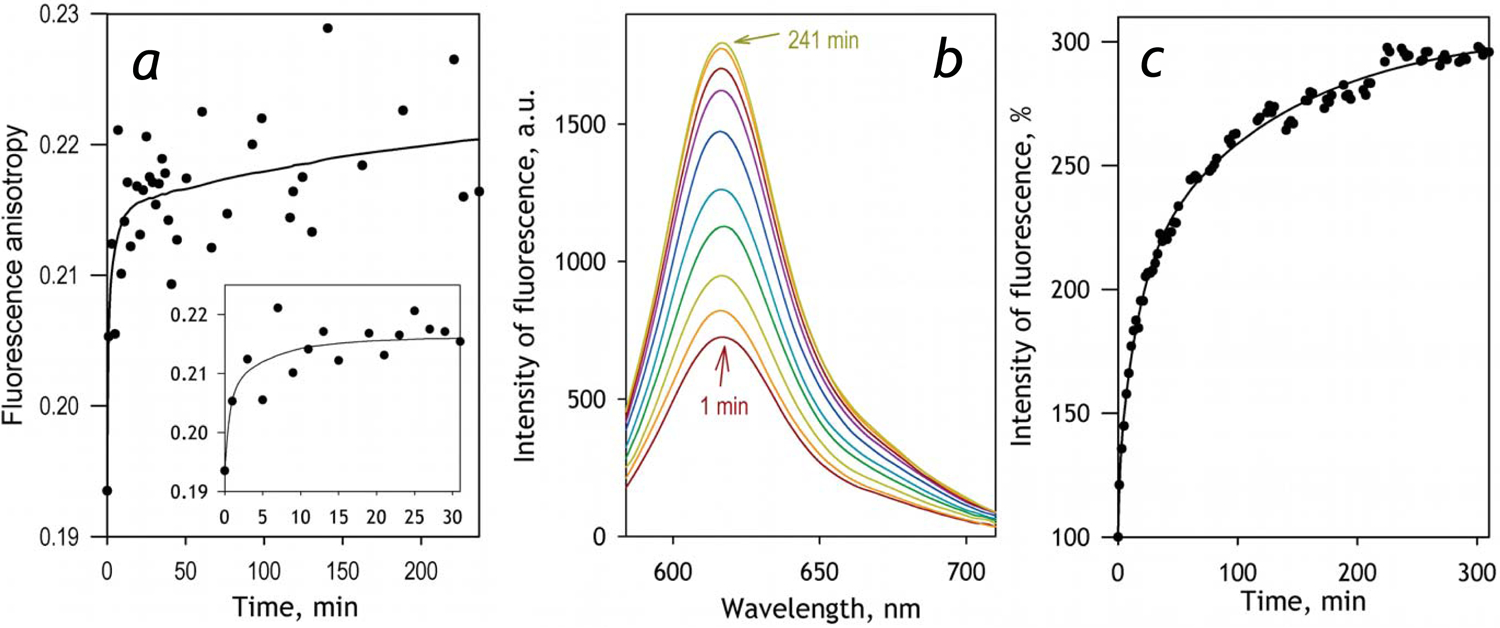
Effect of incorporation of CYP2E1-BODIPY into liposomes on the anisotropy and intensity of BODIPY fluorescence. The data shown in the figure were obtained during the incubation of 0.2 μM CYP2E1-BODIPY(1:1) with liposomes added to the concentration of 600 μM (per phospholipid). Panel a illustrates the changes in the anisotropy of fluorescence during the incubation. The solid line shown in the panel represents the approximation of the dataset with a three-exponential equation. The rate constants of the three phases found in this approximation are equal to of 1.1, 0.06 and 0.008 min−1 and the relative amplitudes amount to 66, 6 and 18% respectively. The inset zooms in the initial phase of the kinetics. Panel b exemplifies a series of spectra of fluorescence recorded during the incubation with a coplanar orientation of polarizers. The spectra shown here were recorded at 1, 3, 7, 15, 31, 61, 111, 191 and 241 min after addition of liposomes to CYP2E1-BODIPY. Panel c shows the changes in the relative intensity of fluorescence during the incubation. The solid line represents the approximation of the dataset with a sum of three exponential terms having the same rate constants as those specified for the kinetics shown in the panel a, and having the relative amplitudes of 12, 38, and 50% for the fast, mediun and the slow phases respectively.
The dependence of the amplitude of fluorescence increase on the amount of the added liposomal phospholipid is shown on Fig. 2. Here we compare the interactions of liposomes with the protein preparations labeled at 1:1 at 1:2 molar ratios referred hereinafter as CYP2E1-BODIPY(1:1) and CYP2E1-BODIPY(1:2) respectively. Increased degree of labeling must increase the probability of homo-FRET between the probes located at different positions. Consequently, the amplitude of fluorescence increase observed with CYP2E1-BODIPY(1:2) is expected to be considerably higher than that characteristic to the protein labeled at 1:1 ratio. As illustrated in Fig. 2, our results strongly comply with these expectations. In both cases the fluorescence increase enhances along with a decrease in the resulting concentration of CYP2E1 in the membrane (or with an increase in the lipid-to-CYP2E1 molar ratio, RL/P).
Fig. 2.
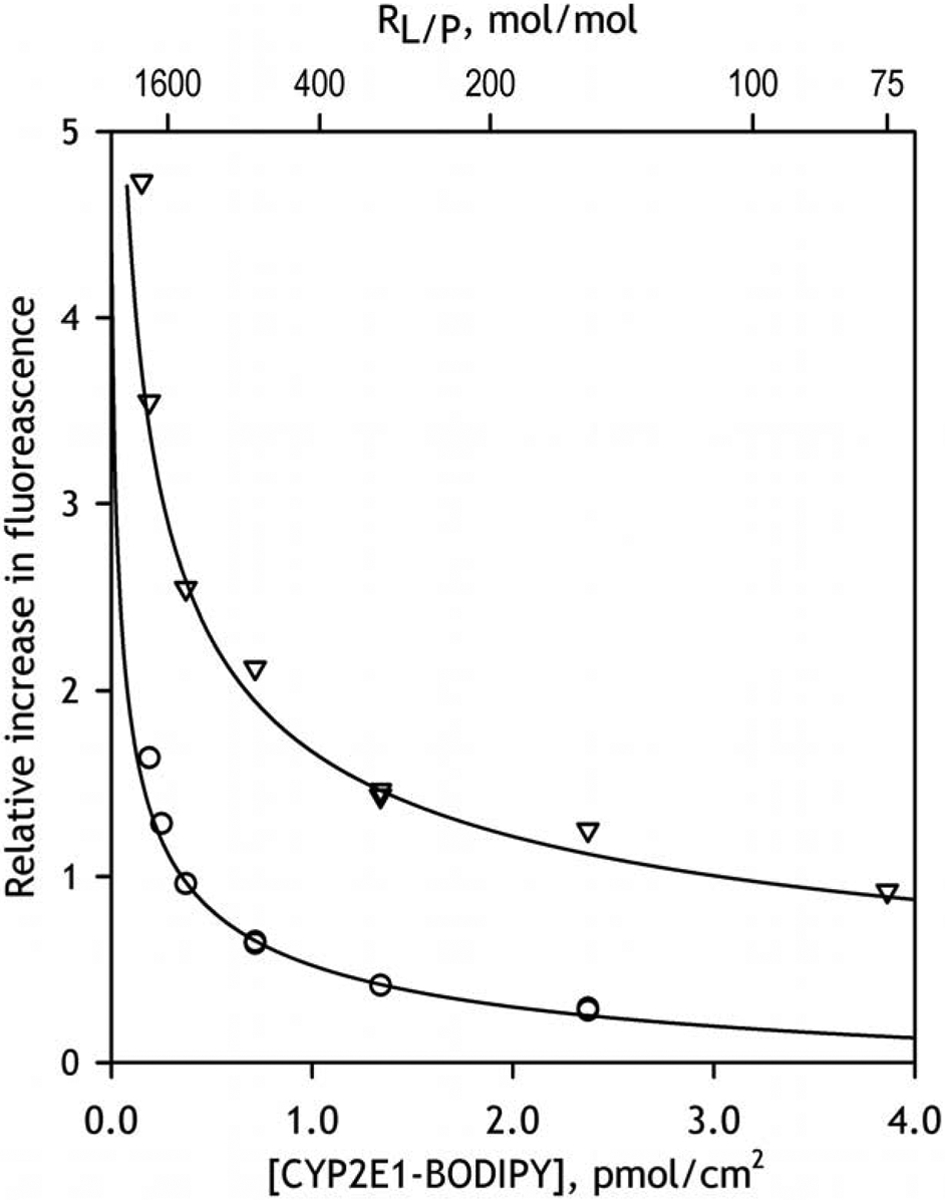
The dependencies of the relative increase in fluorescence intensity caused by incorporation of CYP2E1-BODIPY into the membranes of liposomes. The dataset shown in circles was obtained with CYP2E1-BODIPY(1:1), while the one shown in triangles represents the results obtained with μM CYP2E1-BODIPY(1:2). Solid lines show the approximations of the data sets with Eq. 3.
Similar to the approach used in our previous studies [9, 17, 20], we determined the value of the apparent dissociation constant of the homo-oligomers (KD) from the fitting of the dependencies of the amplitude of observed changes on the surface density of the protein to the equation for the equilibrium of binary association (Eq. 3). It should be noted however that the use of (Eq. 3) to describe the equilibrium of dissociation of the oligomers of unknown size in a two-dimensional space of the membrane is strictly provisional. The resulting values of apparent KD should be rather treated as rough estimates of the surface densities at which the degree of CYP2E1 oligomerization is equal to 50%. Correspondingly, we refer these estimates as [CYP2E1]50% values.
As shown in Fig 2, the dependencies obtained with the protein labeled at either 1:1 or 1:2 protein-to-label ratio closely obey this equation. The [CYP2E1]50% values determined from these approximations is equal to 0.036±0.003 pmol/cm2 and the efficiency of homo-FRET estimated for the homooligomers of CYP2E1-BODIPY(1:2) is as high as 90%. These results allow us to attribute a multifold increase in the intensity of fluorescence of CYP2E1-BODIPY observed in these experiments to a result of dissociation of the protein oligomers and a consequent elimination of homo-FRET between BODIPY fluorophores located in neighboring subunits. Studying the dependence of the amplitude of this increase on the amount of incorporated protein is therefore instrumental for assessing the degree of homo-oligomerization of CYP2E1 in microsomal membranes. Furthermore, comparison of the results obtained in HLM with those derived from the studies with model microsomes (Supersomes™) containing no P450 proteins or bearing a single recombinant P450 enzyme may be used to probe the interactions of CYP2E1 with other P450 species. Due to higher amplitude of signal and (presumably) more homogenous mode of protein modification in CYP2E1-BODIPY conjugates bearing two BODIPY fluorophores per one protein molecule, the double-labeled preparation was chosen for studying the interactions of CYP2E1 with HLM and model microsomes.
Characterization of HLM preparations used in this study
Both samples of HLM used in this study, namely HLM-1 and HLM-2, were characterized with measuring the total content of cytochromes P450 and the content of cytochrome b5 with UV-Vis spectroscopic assays. We also determined the content of CYP2E1, CYP1A2, CYP3A4 and NADPH-cytochrome P450 reductase in both samples with mass-spectrometry. The concentration of NADPH-cytochrome reductase (CPR) was also estimated on the basis of the rate of NADPH-dependent reduction of cytochrome c. Notably, the results of the two different methods of CPR quantification gave nearly identical results with both preparations of HLM.
Results of our analysis are summarized in Table 1. As seen from this table the two samples were significantly different in the composition of the P450 pool, as well as in the P450-to-phospholipids and P450-to-reductase molar ratios. HLM-2 preparation is characterized with higher P450 concentration (as judged from the phospholipids:P450 molar ratio, RL/P) and has a higher molar excess of P450 over the CPR. At the same time HLM-1 has higher content of both CYP1A2 and CYP2E1, while it is characterized with considerably lower abundance of CYP3A4 (Table 1).
Table 1.
Characterization of microsomal preparations used in this study.
| Parameter | HLM-1 | HLM-2 |
|---|---|---|
| Total P450, nmol/mg protein | 0.256± 0.020 | 0.398± 0.053 |
| CYP2E1, nmol/mg protein | 0.032 | 0.028 |
| CYP1A2, nmol/mg protein | 0.032 | 0.017 |
| CYP3A4, nmol/mg protein | 0.057 | 0.219 |
| CPR, nmol/mg (activity assay) | 0.058±0.010 | 0.051± 0.012 |
| CPR, nmol/mg (MS assay) | 0.059 | 0.051 |
| b5, nmol/mg protein | 0.375± 0.019 | 0.329± 0.002 |
| Phospholipid (PL), nmol/mg protein | 724± 136 | 650± 136 |
| PL/P450 molar ratio (RLP) | 2828 | 1633 |
| P450/CPR molar ratio | 4.4 | 7.7 |
| CYP2E1 molar fractiona, % | 12.5 | 7.1 |
| CYP1A2 molar fractiona,% | 12.7 | 4.3 |
| CYP3A4 molar fractiona,% | 22.4 | 55.1 |
The estimates obtained from spectroscopic and enzyme activity assays represent the averages of 2–8 individual measurements. The“±” values are the confidence intervals calculated for p=0.05. The values obtained with mass-spectroscopy are the results a single measurement.
The molar fraction was calculated from the ratio of the content of the individual P450 determined with mass spectrometry to the total P450 content determined with absorbance spectroscopy.
Incorporation of CYP2E1 into HLM and its effect on homo-oligomerization of the protein
In order to examine the kinetics of CYP2E1 incorporation into HLM and probe the interactions of the incorporated CYP2E1 with the endogenous cytochromes P450 we studied the process of interactions of CYP2E1-BODIPY(1:2) with HLM and compared it with the interactions with the model microsomes bearing no cytochromes P450 in their membranes (SS-CPR). In the experiments with HLM we used the HLM-1 preparation.
Incorporation of CYP2E1-BODIPY into HLM resulted in a multifold increase in the intensity of emission of BODIPY, similar to that observed in our experiments with liposomes. Fig. 3a exemplifies a series of kinetic curves of this increase recorded at various molar ratios of phospholipids of added HLM to CYP2E1-BODIPY. These time dependencies may be approximated with a three-exponential equation with the characteristic times of the three phases of 3 ± 0.5, 47 ± 20 and 600 ± 120 min. Analogous to that observed with liposomes, the maximal amplitude of the increase in fluorescence estimated from this approximation increases together with a decrease in the concentration of CYP2E1 in the membrane.
Fig 3.
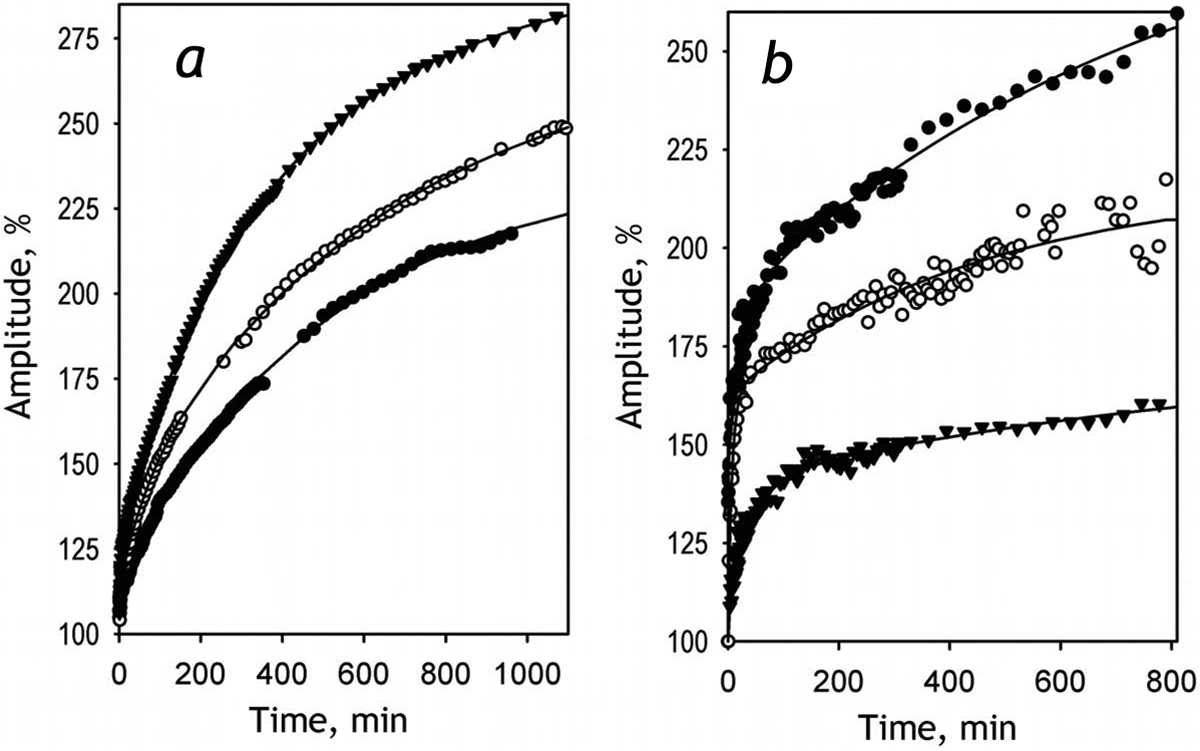
Effect of incorporation of CYP2E1-BODIPY into HLM-1 (a) and SS-CPR (b) on the intensity of BODIPY fluorescence, The plots show the kinetic curves of the increase in the relative intensity of fluorescence of the probe obtained upon incubation of CYP2E1-BODIPY(1:2) with HLM-1 (panel a) and SS-CPR (panel b). The kinetic curves shown in panel a were recorded at RL/P of 213:1 (filled circles), 560:1 (open circles) and 2240:1 (filled triangles). The data shown in panel b were recorded at RL/P of 600:1 (filled triangles), 1125:1 (open circles) and 3600:1 (filled circles). Solid lines represent the approximations of the datasets with a three-exponential equation.
The same behavior was also observed during the incorporation of CYP2E1-BODIPY into the membrane of control Supersomes™ (SS-CPR) containing recombinant CPR, but having no cytochromes P450 (Fig 3b). In this case the dependence of the amplitude on RL/P was considerably more rapid than that observed with HLM. This difference indicates that the interactions with cytochromes P450 present in HLM membrane promote the dissociation of the CYP2E1-BODIPY homo-oligomers.
The dependencies of the amplitude of fluorescence increase on the surface density of CYP2E1-BODIPY in the membrane are shown in Fig. 4. The parameters of CYP2E1 interactions with HLM and SS-CPR determined from these results may be found in Table 2. This table also contains the parameters obtained in the studies of interactions of CYP2E1-BODIPY with model microsomes containing recombinant CYP1A2 and CYP2C19 (see infra). The values of [CYP2E1]50% shown in this table correspond to a surface density at which the degree of CYP2E1 oligomerization is equal to 50%. This parameter is equal to the apparent KD of CYP2E1 homo-oligomers determined from the plots shown in Fig. 2 and Fig. 4 as described above. The values of (Lipid/CYP2E1)50% were calculated from the [GYP2E1]50% values and represent yet another representation of the same KD values. This way of representation may be more instrumental for judging the degree of homo-oligomerization at a given CYP2E1 content in the membrane as it does not require calculating the surface density of the protein.
Fig. 4.
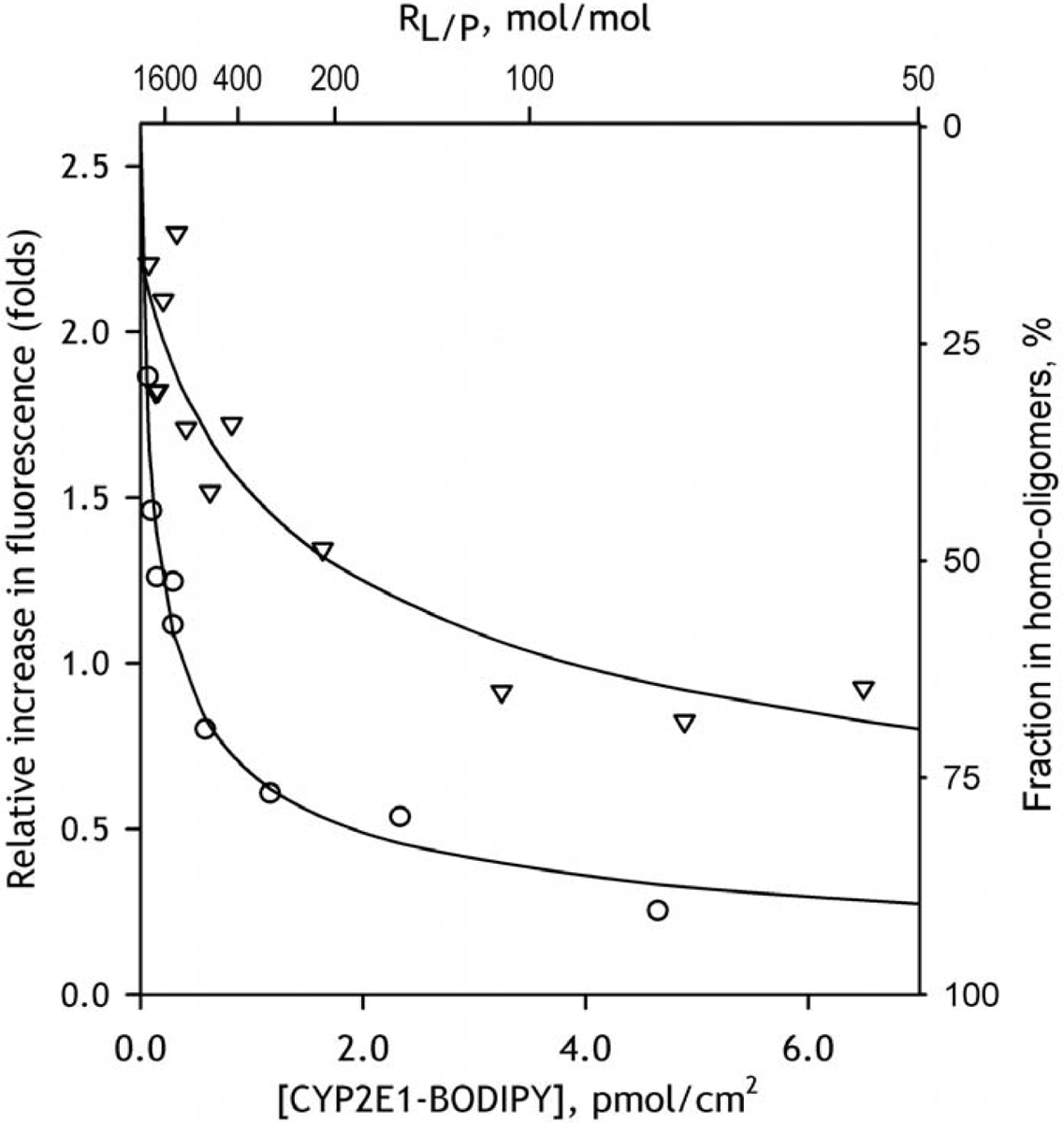
The dependencies of the relative increase in fluorescence intensity caused by incorporation of CYP2E1-BODIPY into the membranes of HLM-1 (triangles) and SS-CPR (circles) on the concentration (surface density) of CYP2E1-BODIPY in the membrane. Solid lines show the approximations of the data sets with Eq. 3. The scale shown on the right of the plot indicates the apparent fraction of CYP2E1 present in homo-oligomeric state estimated from the results of this fitting.
Table 2.
Effect of membrane incorporation of CYP2E1-BODIPY(1:2) on the degree of its homo-oligomerization explored with homo-FRET*
| Membranous preparation | [CYP2E1]50%a, pmol/cm2 | (Lipid/CYP2E1)50%b | FRET Efficiency, % |
|---|---|---|---|
| SS-CPR | 0.081 ± 0.020 | 4323 ±1113 | 72.9 |
| SS-b5 | 0.143 ± 0.038 | 2444 ± 699 | 71.8 |
| HLM | 0.914 ± 0.245 | 383 ± 111 | 79.0 |
| SS(1A2] | 0.890 ± 0.040 | 393 ± 18 | 78.6 |
| SS(2C19) | 0.059 ± 0.021 | 5891 ± 2441 | 80.0 |
The values given in the Table were obtained by fitting of the titration curves with the binary association equation (Eq. 3). The “±” values show the confidence interval calculated for p = 0.05.
Surface density of P450 in the membranes at which the amplitude of the titration curves reaches 50% of the maximal level. This value is equal to the value of KD determined from the fitting of the titration curves with Eq. 3. The surface density was calculated from the RL/P ratio as described in Experimental using equation (Eq. 2).
Lipid-to-P450 ratio at which the amplitude of the titration curves reaches 50% of the maximal level which was calculated from the respective [CYP2E1]50% values.
As seen from Table 2 the value of [CYP2E1]50% for CYP2E1 homo-oligomers is much lower than that observed with the control microsomes. Therefore, the decrease in the degree of CYP2E1 homo-oligomerization upon its incorporation into the membrane is much better pronounced in HLM as compared to that observed in the microsomes containing no P450 proteins. This observation evidence for the formation of mixed complexes of CYP2E1 with other P450 species that leads to the dissociation of CYP2E1 homo-oligomers.
Effect of enriching HLM with CYP2E1 on the metabolism of CYP2E1-specific substrates.
In order to probe if the incorporation of CYP2E1 into HLM makes the exogenous enzyme a fully functional member of the P450 ensemble we studied the effect of enriching HLM with CYP2E1 on the metabolism of CYP2E1-specific substrates, p-Nitrophenol (pNP) and Chlorzoxazone (CLZ). In our studies with pNP we compared two different preparations of HLM, HLM-1 and HLM-2, whereas the studies with CLZ were carried out with HLM-2 only due to limited amounts of HLM-1 preparation in hand. It should be noted that the rate of metabolism of all substrates used in this study were normalized on the concentration of CPR in the microsomal membrane (see Experimental). In view of a high excess of cytochromes P450 over CPR in HLM, this normalization was considered as the most appropriate approach for comparing the rates of metabolism in the HLM preparations with different composition of the cytochrome P450 ensemble.
The two preparations of HLM exhibited closely similar parameters of pNP metabolism and their dependencies on CYP2E1 concentration. With both preparations the dependencies of the reaction rate on pNP concentration obey the Hill equation more closely than they do with the Michaelis-Menten equation. The difference between the two ways of fitting is exemplified in Fig. 5a for the titration traces obtained with HLM-1. The fiiting with the Hill equation yields the values of the Hill coefficient equal to 1.5±0.5 and 1.3±0.2 in HLM-1 and HLM-2 respectively. This apparent homotropic cooperativity of CYP2E1-dependent metabolism of pNP has never been reported before and its relevance and possible mechanistic grounds require additional investigations.
Fig. 5.
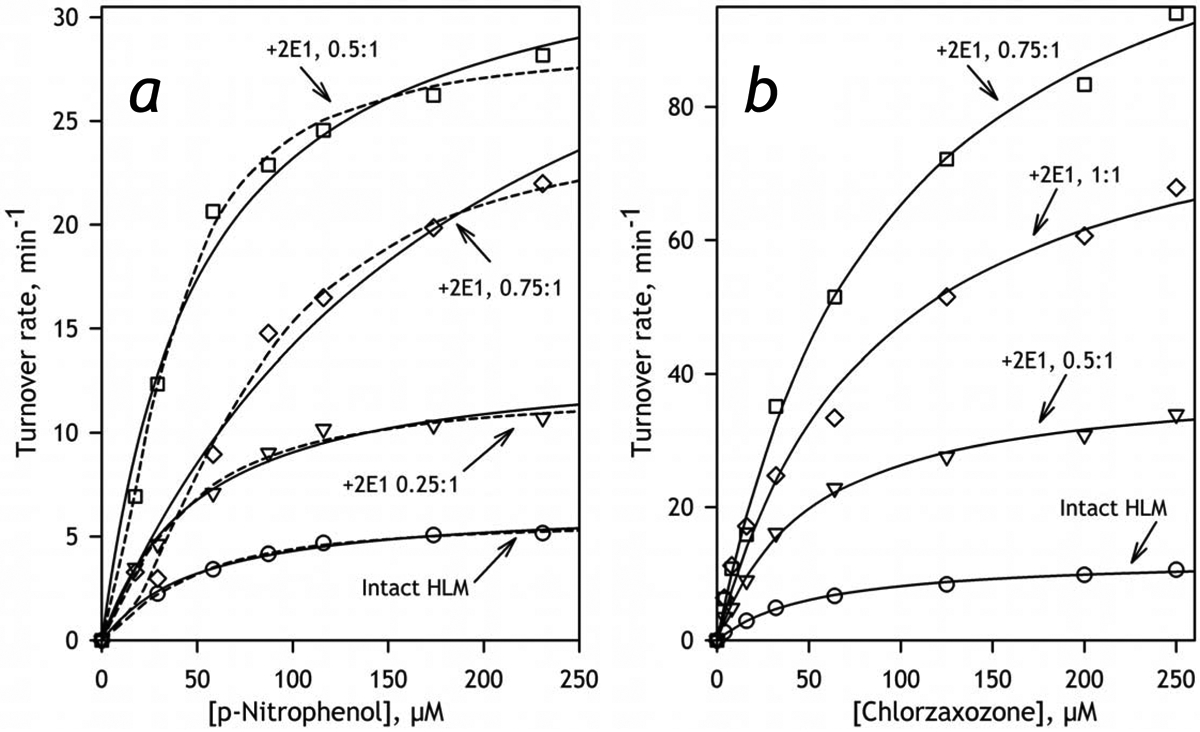
The effect of incorporation of CYP2E1 into HLM-2 on CYP2E1-dependent hydroxylation of pNP (a) and CLZ (b). The dependencies of the rate of reaction on the substrate concentration shown on this figure represent the averages of 3 – 6 individual experiments. The solid lines represent the results of the fitting of the experimental data to the Michaelis-Menten equation, while the dashed lines shown in panel a represent the approximations of the same data sets with the Hill equation. The ratios shown next to each curve indicate the molar ratio of added CYP2E1 to the internal cytochrome P450 content in HLM-2 (0.4 nmol/mg protein).
The parameters derived from the studies of pNP and CLZ metabolism are exemplified in Table 1 with the data obtained with HLM-2. The full set of parameters obtained in the experiments with pNP and HLM-1 may be found in Table S2 in the Supplementary Material. The values of Vmax derived from these experiments are also shown in Fig. 6a (see infra). As seen from these tables and illustrated in Fig. 5, where we compare the profiles of oxidation of pNP and CLZ obtained with HLM with different CYP2E1 concentrations, enrichment of HLM with CYP2E1 results in a multifold increase in the rates of metabolism of both substrates. As shown in Fig. 6, the apparent kcat derived from these titrations and calculated per concentration of CPR remains proportional to the amount of CYP2E1 in the membrane up to the content of 0.3–0.4 nmol/mg protein (or about 50% CYP2E1 in the P450 pool) and stabilizes (pNP) or decreases (CLZ) after that point. Other kinetic parameters (KM (CLZ) or S50 and the Hill coefficient (pNP)) do not show any statistically significant changes (Table 3). These results demonstrates that the incorporated CYP2E1 becomes a fully-functional member of the P450 ensemble and do not exhibit any detectable functional differences with the endogenous CYP2E1 in HLM.
Fig. 6.
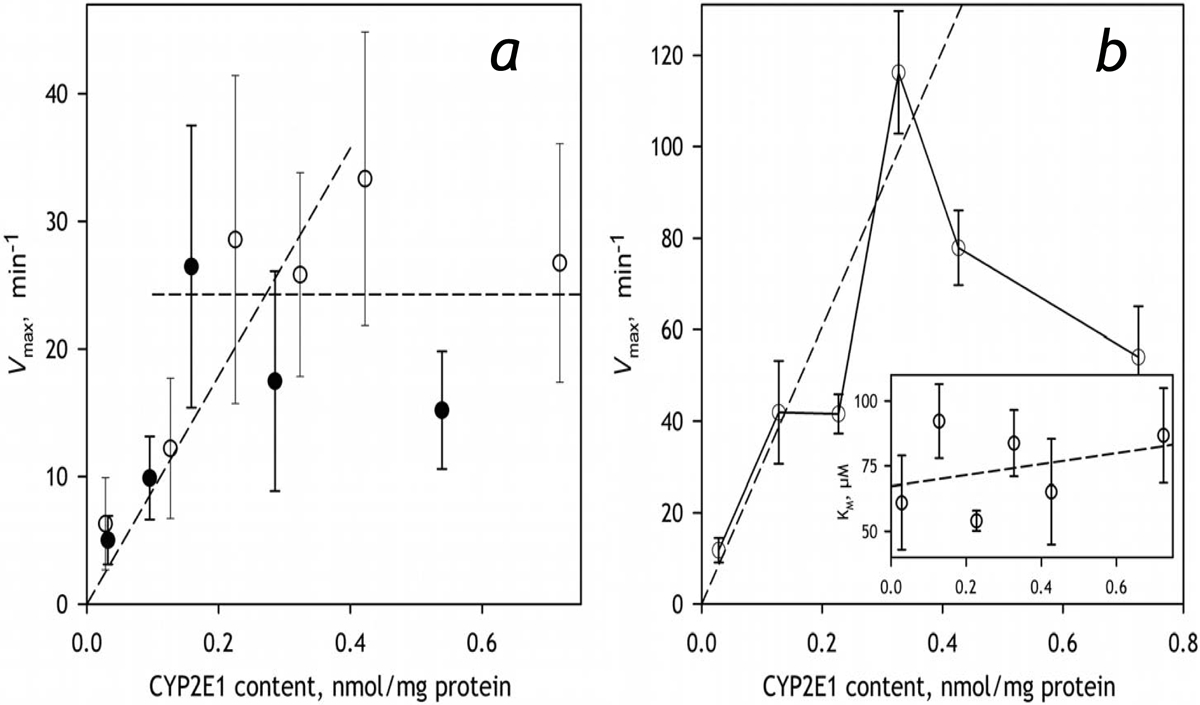
The effect of incorporation of CYP2E1 into HLM on the metabolism of CYP2E1-specific substrates pNP (a) and CLZ (b). The data shown in open and filled circles in panel a were obtained with HLM-1 and HLM-2 preparations respectively. The results presented in the panel b were obtained with HLM-2. Error bars represent the confidence intervals calculated for p=0.05 on the basis of 2–5 measurements.
Table 3.
Effect of incorporation of CYP2E1 into HLM-2 on the kinetic parameters of oxidation of p-Nitrophenol and Chlorzoxazone*
| Molar ratio of added CYP2E1 to P450 present in HLM | CYP2E1 content, nmol/mg protein | p-Nitrophenol | Chlorzoxazone | |||
|---|---|---|---|---|---|---|
| S50, μMa | Na | Vmax, min−1 | KM, μM | Vmax, min−1 | ||
| 0 | 0.028 | 39 ± 17 | 1.3 ±0.2 | 6.3 ± 3.6 | 54 ± 16 | 12 ± 3 |
| 0.25 | 0.13 | 43 ± 7 | 1.3 ±0.4 | 12 ±6 | 79 ±29 | 42 ± 11 |
| 0.5 | 0.23 | 37 ± 11 | 1.6 ±0.3 | 29 ± 13 | 54 ± 4.4 | 42 ±4 |
| 0.75 | 0.32 | 81 ± 11 | 1.7 ±0.2 | 26 ±8 | 84 ± 13 | 105 ±25 |
| 1 | 0.42 | 81 ± 5 | 1.2 ±0.3 | 33 ± 12 | 65 ±20 | 78 ±8 |
| 1.75 | 0.72 | 48 ±2 | 1.5 ±0.4 | 27 ±9 | 87 ± 18 | 54 ± 11 |
The estimates given in the table correspond to the averages of 2–4 individual measurements. The“±” values are the confidence intervals calculated for p=0.05.
S50 are N are the concentration of half-saturation with substrate and the Hill coefficient obtained from the fitting of the titration curves with the Hill equation.
Probing the ability of recombinant human P450 species to metabolize CEC and ERR.
In our further studies of the effect of added CYP2E1 on the metabolism of substrates of other P450 species we used fluorogenic substrates 7-ethoxy-4-cyanocoumarin (CEC) and 7- ethoxyresorufin (ERR), which are primarily metabolized in HLM by CYP1A2 and CYP2C19 [39–40]. In order to better characterize the ability of human cytochrome P450 species to metabolize these substrates we investigated their metabolism by recombinant human CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, CYP2E1, and CYP3A4 using commercial preparations of Supersomes™ containing the respective enzymes. Results of these assays are summarized in Table 4.
Table 4.
Parameters of metabolism of 7-CEC and 7-ERR by recombinant human cytochromes P450*.
| P450 species | CEC | ERR | ||
|---|---|---|---|---|
| KM, μM | Vmax, min−1 | KM, μM | Vmax, min−1 | |
| CYP1A2 | 2.3 ± 0.10 | 7.9 ± 2.2 | 0.40 ± 0.14 | 1.1 ± 0.3 |
| CYP2B6 | 8.2 ± 2.4 | 1.0 ± 0.2 | No activity detected | |
| CYP2C8 | No activity detected | 0.26 ± 0.14 | 0.02 ± 0.01 | |
| CYP2C9 | No activity detected | No activity detected | ||
| CYP2C19 | 24 ± 5 | 2.3 ± 0.5 | 0.33 ± 0.13 | 0.06 ± 0.02 |
| CYP2D6 | No activity detected | No activity detected | ||
| CYP2E1 | No activity detected | 33.0 ± 9.2 | 0.05 ± 0.02 | |
| CYP3A4 | No activity detected | No activity detected | ||
The estimates given in the table represent the averages of 2–4 individual measurements. The“±” values are the confidence intervals calculated for p=0.05.
In a good agreement with the previous reports, the highest activity in dealkylation of ERR was exhibited by CYP1A2. Some activity with this substrate was also detected with CYP2C8 and CYP2C19. However, although affinity of these CYP2C enzymes to ERR was quite close to that specific to CYP1A2, the rates of reaction observed with them were considerably lower. Although CYP2E1 also reveals some activity in ERR dealkylation, the observed values of Vmax and KM (Table 4) leave out a possibility of any sizeable participation of CYP2E1 in ERR metabolism by HLM, at least at the substrate concentrations below 5 μM used in our studies with CYP2E1-enriched HLM.
Although the highest rate of metabolism of CEC was also exhibited by CYP1A2, the activity rendered by CYP2C19 is high enough to consider this enzyme as an important contributor to CEC metabolism, especially taking into account its relatively high abundance in HLM [4]. Another P450 enzyme capable of metabolizing CEC is CYP2B6. However, this enzyme is unlikely to make any important contribution to CEC metabolism in HLM due to low rate of turnover and low concentration of CYP2B6.
Therefore, probing the impact of CYP2E1 incorporation on the parameters of metabolism of CEC and ERR by HLM is expected to provide information on the activities of CYP1A2 and CYP2C19 and discriminate between the CYP2E1 effects on the two enzymes.
Impact of CYP2E1 incorporation on the metabolism of CEC and ERR by HLM.
The dependencies of the rate of CEC dealkylation on substrate concentration obtained with intact and CYP2E1-enriched HLM samples of HLM-1 and HLM-2 are shown in Fig. 7. The parameters obtained from the approximation of these dependencies with the Michaelis-Menten equation are summarized in Table 5. The rate of metabolism of CEC by HLM-1 was noticeably higher than that observed with HLM-2, consistent with higher concentration of CYP1A2 in HLM-1 as compared to HLM-2 (Table 1). As suggested by the titration traces obtained with HLM-1 and HLM-2 shown in Fig. 7, CYP2E1 enrichment may result in some increase in the rate of CEC metabolism in both preparations of HLM. However, this increase cannot be considered statistically significant (Table 5). More important is a statistically significant decrease in the value of KM observed with both HLM preparations (Table 5). While in the intact microsomes this value is close to that characteristic of CYP2C19-dependent metabolism of CEC (Table 4), incorporation of CYP2E1 results in its decrease and makes it closer to that expected for CYP1A2-catalyzed reaction (Table 5).
Fig. 7.

The effect of incorporation of CYP2E1 into HLM on the dependencies of the rate of CEC metabolism on substrate concentration. The results obtained with HLM-1 and HLM-2 are shown in the panels a and b respectively and represent the averages of 3 – 5 individual experiments. The data obtained with intact HLM are shown in circles while the triangles designate the results obtained with the samples containing CYP2E1 incorporated at 1:1 ratio to the microsomal P450 content (0.26 and 0.4 nmol/mg protein for HLM-1 and HLM-2, respectively). The solid limes represent the results of fitting of the datasets to the Michaelis-Menten equation. The dashed lines in the panel b shows the approximations with the equation of the sum of two Michaelis-Menten functions.
Table 5.
Parameters of dealkylation of CEC and ERR in intact and CYP2E1-enriched HLM*.
| HLM sample | CEC | ERR | ||
|---|---|---|---|---|
| KM, μM | Vmax, min−1 | KM, μM | Vmax, min−1 | |
| HLM-1 | 14.8 ± 1.8 | 3.8 ± 1.0 | 0.35 ± 0.01 | 0.19 ± 0.015 |
| HLM-1 + CYP2Ela | 9.7 ± 1.7 (0.01) | 3.9 ± 0.3 (0.80) | 0.43 ± 0.23 (0.59) | 0.31 ± 0.01 (0.004) |
| HLM-2 | 32.2 ±14.7 | 2.1 ± 0.5 | 0.46 ± 0.16 | 0.17 ± 0.04 |
| HLM-2 + CYP2E1a | 7.4 ±2.1(0.01) | 2.2 ± 0.7 (0.32) | 0.46 ± 0.11 (0.96) | 0.64 ± 0.30 (0.05) |
The estimates given in the table represent the averages of 2–4 individual measurements. The“±” values are the confidence intervals calculated for p=0.05. The values in parentheses represent the p-values of Student’s t-test for the hypothesis of equality of the respective values with the value presented in the previous line (i.e., at no CYP2E1 incorporated). The values of KM and Vmax characterized with p-values ≤0.05 are shown in bold to emphasize the effects with high statistical significance.
The results obtained with the samples containing CYP2E1 incorporated at 1:1 molar ratio to the microsomal P450 content (0.26 and 0.4 nmol/mg protein for HLM-1 and HLM-2, respectively).
The change in the main route of CEC metabolism suggested by this initial analysis may be better revealed through fitting the titration curves to the equation of the sum of two Michaelis- Menten functions. In the case of HLM-1, however, this fitting was incapable to resolve two components with different KM values and inefficient in improving the quality of approximation. However, in the case of HLM-2 the fitting of the datasets with a single Michaelis-Menten function results in systematic deviations suggesting a noticeable biphasicity of the titration curves (Fig. 7b). The use of the equation of the sum of two Michaelis-Menten functions for fitting the datasets obtained with HLM-2 (either with or without incorporated CYP2E1) suggests the values of KM for the two phases to be in the range of 2–4 and 30–70 μM.
Global fitting of the entire dataset obtained with a series of HLM-2 preparations containing various amounts of incorporated CYP2E1 gives the best results with the estimates of KM of 2.5±0.9 and 58±13 μM for the high- and low-affinity components respectively. These estimates are in a reasonable agreement with the values of 2.3±0.1 and 24±5 μM obtained with CYP1A2- and CYP2C19-containing Supersomes respectively (Table 4). As illustrated in Fig. 8a, the fraction of the high-affinity phase derived from the global fitting increases proportionally to the concentration of CYP2E1 in HLM up to the content of ~0.2 nmol/mg protein and tends to stabilize after that point. This change is likely to be also associated with some increase in the rate of CEC metabolism, although the latter effect cannot be considered statistically significant (Fig. 8b).
Fig. 8,
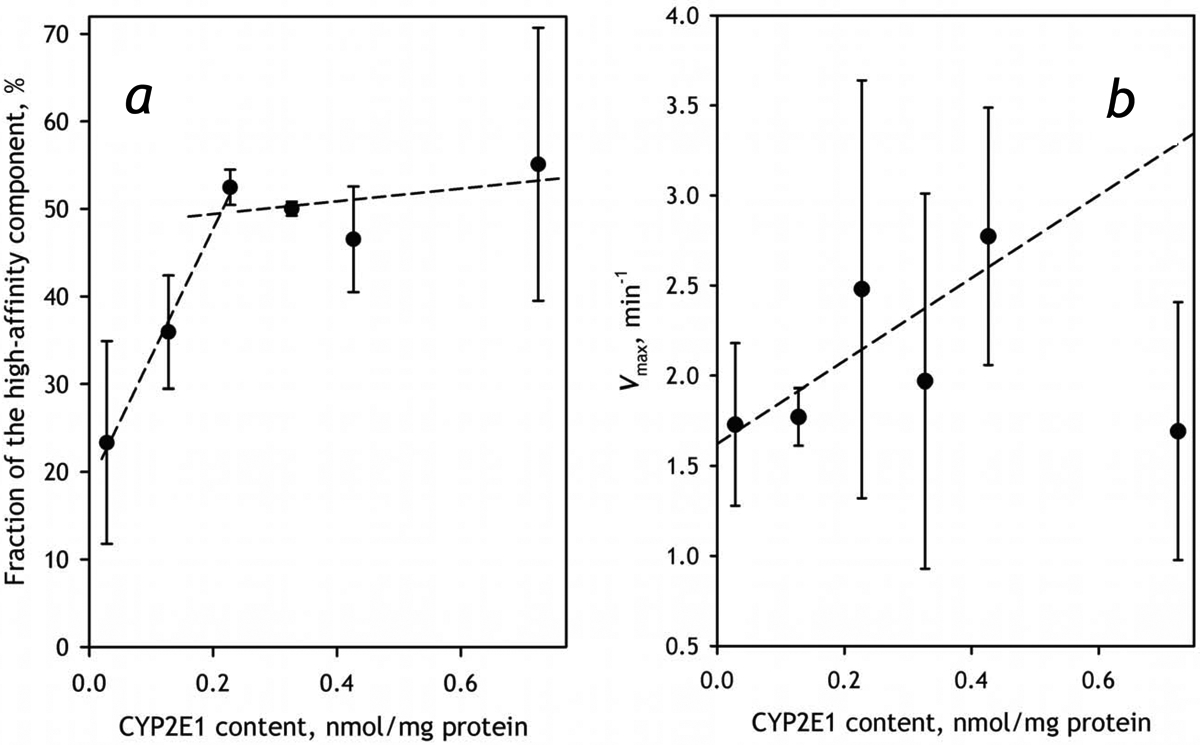
The effect of incorporation of CYP2E1 into HLM-2 on the parameters of metabolism of CEC. Panel a shows the dependence of the fraction of the high-affinity component on CYP2E1 content. The dependence of the total maximal rate of metabolism is shown in panel b. Error bars represent the confidence intervals calculated for p=0.05 on the basis of 2–4 measurements.
The effect of CYP2E1 enrichment on the contribution of the high affinity component in the biphasic profile of CEC dealkylation suggests an important augmentation of the involvement of CYP1A2. In order to probe this possible activation further we investigated the effect of incorporation of CYP2E1 on the metabolism of ERR, the substrate almost exclusively metabolized by CYP1A2. As seen from the data presented in Table 5, an increase in the concentration of CYP2E1 causes a remarkable increase in the rate of ERR dealkylation in both preparations of HLM, although this effect is better pronounced in HLM-2. In order to study this effect further we investigated the dependence of the parameters of ERR metabolism in HLM-2 preparation. As shown in Fig. 9, the rate of ERR metabolism increases along with increasing concentration of CYP2E1 in the membrane. This nearly-linear increase is observed up to the CYP2E1 content of 0.4 nmol/mg protein (or about 50% CYP2E1 in the P450 pool). The fact that this multifold increase in Vmax is not associated with any substantial changes in the value of KM (Fig. 9b, inset) rules out a possibility of direct involvement of CYP2E1-dependent metabolism of CEC and substantiates our conclusion that incorporation of CYP2E1 results in an ample activation of CYP1A2 present in HLM.
Fig. 9,
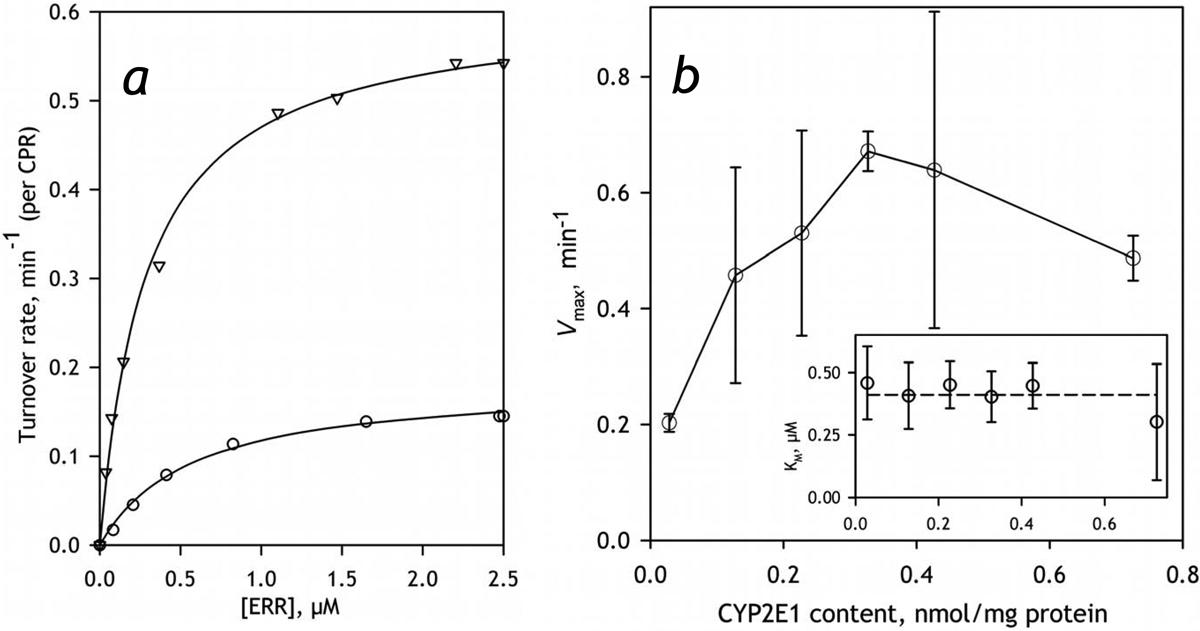
The effect of incorporation of CYP2E1 into HLM-2 on the parameters of metabolism of ERR. Panel a exemplified the dependence of the rate of the turnover on substrate concentration obtained with intact HLM (circles) and the sample containing CYP2E1 incorporated in the amount of 0.3 nmol per mg protein that corresponds to 0.75:1 ratio to the microsomal P450 content (triangles). These traces represent the averages of 3 and 2 individual experiments respectively. Panel b shows the dependencies of Vmax (main panel) and KM on CYP2E1 content. Error bars represent the confidence intervals calculated for p=0.05 on the basis of 2–5 measurements.
Probing the association of CYP2E1 with CYP1A2 and CYP2C19 in Supersomes.
In order to probe if the observed activation of CYP1A2 may be attributed to a direct consequence of the formation of mixed oligomers of CYP1A2 and CYP2E1 we probed the association of these two proteins in the microsomal membrane using CYP1A2-containing Supersomes™ and our FRET-based technique that utilizes CYP2E1 labeled with BODIPY 577/618. The same approach was also used to probe possible interactions of CYP2E1 with CYP2C19, another enzyme metabolizing CEC. In order to have an appropriate control dataset we also monitored the interactions of CYP2E1 with Supersomes™ containing human CPR and cytochrome b5 but bearing no P450 enzymes.
Similar to what was observed upon incorporation of CYP2E1-BODIPY into liposomes and HLM (Fig. 1 and Fig. 3), addition of CYP2E1-BODIPY to the control Supersomes™ as well as to those containing CYP1A2 or CYP2C19 resulted in a stepwise increase in the intensity of fluorescence that obeyed three-exponential kinetics and took 18 – 20 hours for completion. The dependencies of the maximal amplitude of this increase on CYP2E1 concentration in the membrane (Fig. 10) demonstrate a significant difference between the CYP1A2-containing microsomes with the microsomes bearing no P450 protein or having CYP2C19 in their membranes. In the latter two cases a substantial degree of dissociation of the CYP2E1-BODIPY homo-oligomers was observed only at low surface densities of the incorporated enzyme, at RL/P ratio in the range of several thousands. In contrast, in the case of CYP1A2 containing microsomes, 50% dissociation of homooligomers was observed at RL/P ratio around 400:1, similar to what was seen in HLM (Table 2). These results suggest that CYP2E1 efficiently forms mixed oligomers with CYP1A2 but lacks any ability to interact with CYP2C19, which stands in one-to-one correspondence with the observed effect of CYP2E1 on CYP1A2-specific activities in HLM.
Fig. 10.

Effect of increasing concentrations of CYP2E1-BODIPY on the relative increase in fluorescence intensity caused by incorporation of CYP2E1-BODIPY into the membranes of Supersomes™ containing recombinant CYP1A2 and CPR (circles, solid line), CYP2C19, CPR and cytochrome b5 (triangles, solid line) or control Supersomes™ containing human CPR and cytochrome b5 but bearing no cytochromes P450 (squares, dashed line). The lines show the approximations of the data sets with Eq. 3.
Discussion
In this study we utilized the approach based on homo-FRET (fluorescent resonance energy transfer where donor and acceptor are represented by the fluorophores with one and the same structure) to monitor the incorporation of CYP2E1 into HLM and probe its interactions with other cytochrome P450 species in the microsomal membrane. Studying the effect of this incorporation on CYP2E1-specific activities of HLM we demonstrate that the incorporated enzyme becomes a fully-functional member of the P450 ensemble and do not exhibit any detectable functional differences with the intrinsic CYP2E1 enzyme present in HLM. Exploring the functional effect of increase in CYP2E1 concentrations we evidenced an important activation of CYP1A2 that results in rerouting of metabolism of CEC, the substrate jointly metabolized by CYP1A2 and CYP2C19, toward preferential dealkylation by CYP1A2. Furthermore, probing the interactions of CYP2E1 with model microsomes containing individual P450 enzymes we found that CYP2E1 efficiently interacts with CYP1A2, but lacks any ability to form complexes with CYP2C19. This observation stands in one-to-one correspondence with the observed effect of CYP2E1 on CYP1A2-specific activities in HLM.
What though may be the mechanism of activation of one P450 species by its interactions with another, dissimilar P450 enzyme observed in this study and also evidenced in earlier reports from our group and by others [9, 16–17, 41–42]? Our understanding of these instances is based on a concept of “positional heterogeneity”, which we introduced to explain a persistent distribution of P450 pool between the fractions with different functional properties [5, 8, 17]. This concept is based on an assumption that the P450 subunits forming homo- and heterooligomers are not identical in their conformation and orientation, but are characterized by different abilities to be reduced, bind substrates and interact with redox partners. In effect, oligomerizartion of cytochromes P450 results in abstracting an important part of the P450 pool from the catalytic activity. A discussion of possible structural basis for positional heterogeneity in P450 oligomers may be found in a recent comprehensive review by Reed and Backes [43].
From the standpoint of the above concept, the most obvious explanation for non-additive effects of changing the content of cytochrome P450 ensemble might be a difference in the propensities of different P450 species for occupying the positions of two different types in the oligomer. In particular, it might be hypothesized that, when CYP1A2 interacts with CYP2E1, the former preferentially occupies the “active” positions, whereas CYP2E1 fills the “restrained” ones. This re-distribution of P450 species between the positions of two different types result in increase in the active fraction of CYP1A2.
This hypothesis is illustrated in Fig. 11, which schematically represents the sequence of events taking place upon incorporation of CYP2E1 (orange subunits) into the microsomal membrane. In this figure we arbitrary depicted P450 oligomer in solution as a hexamer composed of two types of subunits – “active” (triangle-shaped) and “restrained” (diamond-shaped). The same types of subunits also present in the P450 oligomers in the membrane, which are shown in Fig. 11 as trimers. Prior to incorporation of CYP2E1, the P450 species present in HLM form heterooligomers, where certain P450 species including CYP1A2, which is shown as green subunits, preferentially occupy the “restrained” locations, while the active positions are occupied by other P450 species (blue subunits). Upon incorporation of CYP2E1 (event 1) and after a slow dissociation and reassembly of the oligomers (event 2) the “restrained” positions become preferentially occupied by CYP2E1 due to some inherent structural features of the latter. This reorganization displaces CYP1A2 to “active” positions (triangle-shaped subunits) and thereby activates the enzyme.
Fig. 11.
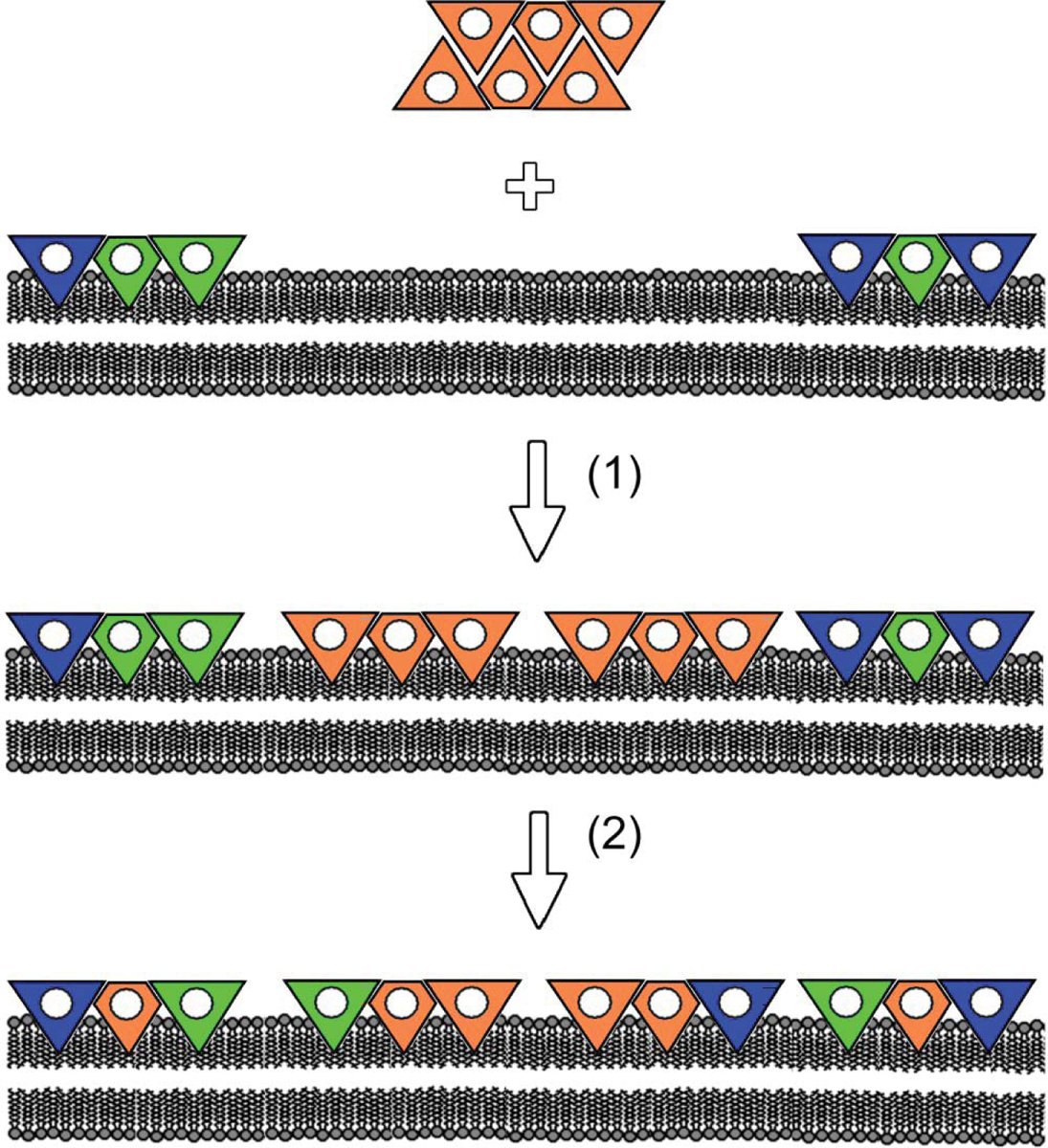
A schematic representation of the hypothesis explaining the mechanism of activation of CYP1A2 (green subunits) by CYP2E1 (orange subunits). The mechanism involves incorporation of added CYP2E1 into the membrane of HLM (event 1), where important part of CYP1A2 is rendered inactive due to a preferential occupation of the “restrained” positions (diamond-shaped subunits) in heterooligomers with other P450 species (blue subunits). After a slow dissociation and reassembly of the oligomers (event 2) the “restrained” positions become preferentially occupied by CYP2E1 due to some inherent structural features of the latter. This reorganization displaces CYP1A2 to the “active” positions (triangle-shaped subunits) and thereby activates the enzyme.
Obviously, the scheme shown in Fig. 11 is extremely simplified. It limits the number of interacting P450 species to three, does not show reversible dissociation of the oligomers and does not take into account modulation of P450 oligomerization by isoform-specific substrates discussed in our previous publication [9]. In reality the difference in the structural properties between multiple microsomal P450 species may result in very complex dependence of the degree of position-determined “restraining” of any particular P450 on the composition of the P450 pool and its exposure to P450 substrates and allosteric modulators.
Of particular interest is the nature of saturation-like effects observed at high concentrations of CYP2E1 in the microsomal membrane. Stabilization or inhibition of CYP2E1-dependent activities at the CYP2E1 content higher than 0.3–0.5 nmol/mg protein are clearly seen from our results obtained with CYP2E1 substrates pNP and CLZ (Fig. 6). Importantly, the same kind of behavior is also revealed in the studies with CEC (Fig. 8) and ERR (Fig. 9), the substrates metabolized by CYP2C19 and/or CYP1A2. The most obvious explanation for stabilization of the rate of pNP metabolism at high CYP2E1 concentration (Fig. 6a) may be based on a hypothesis of saturation of CPR with CYP2E1 at these concentrations. However, the observation that this stabilization comes together with a pronounced inhibition of the metabolism of CLZ, another CYP2E1 substrate (Fig. 6b), and is not associated with a complete suppression of the metabolism of CEC and ERR (the substrates of CYP1A2) makes this explanation quite unlikely. Another explanation for these effects may be provided by a hypothesis of increased probability of formation of inactive large aggregates of P450 enzymes or their non-productive complexes with CPR at very high non-physiological levels of P450 content (≥0.8 nmol/mg protein). In this case the effect must be substrate-unspecific and observable with the activities of all cytochromes P450 present in HLM. This possibility will be probed in our further studies with the substrates of other P450 species.
The present study reveals one potentially important aspect of the complex inter-enzyme crosstalk in the P450 ensemble – activation of CYP1A2 caused by increased content of CYP2E1 in the heterooligomers of multiple P450 species. From the practical standpoint this observation suggests that the ethanol-dependent induction of CYP2E1 in human liver may have a pronounced effect on pharmacokinetics of drugs metabolized by CYP1A2. Of special interest are those jointly metabolized by CYP1A2 and CYP2C19, such as tricyclic antidepressants (amitriptylline, imipramine). CYP2E1-dependent activation of CYP1A2 may reroute the metabolism of mutual drug substrates of CYP2C19 and CYP1A2 towards the latter, similar to what was observed here for the metabolism of CEC. This rerouting may have important practical consequences from the standpoint of pharmacogenomics, as the activity of the human CYP2C19 enzyme is known to be largely affected by its widespread polymorphism [44].
A series of investigations with HLM preparations varying in the composition of the P450 ensemble is needed to reveal other principal elements of the entangled system of interrelations between multiple P450 species. The present study demonstrates that the approach based on incorporation of any particular purified P450 enzyme into HLM may provide a potent means for untangling this complex system and obtaining the information necessary for unambiguous prediction of the profile of drug metabolism specific to any given composition of the P450 ensemble in human liver.
Supplementary Material
Acknowledgments:
The authors are grateful to Jeffrey P. Jones (WSU) for research support, assistance in obtaining and analyzing LC-MS/MS data and continuous interest to this study. We also gratefully acknowledge the assistance of the “Human Proteome” Core Facility of the Institute of Biomedical Chemistry (IBMC, Moscow, Russia) in generating mass-spectrometry data used in characterization of HLM preparations.
Funding. This research was supported by the National Institute On Alcohol Abuse And Alcoholism of NlH under Award Number R21AA024548.
Abbreviations:
- CEC
7-ethoxy-4-cyanocoumarin
- CHC
7-hydroxy-4-cyanocoumarin
- CLZ
chlorzoxazone
- CPR
NADPH-cytochrome P450 reductase
- ERR
7-ethocyresorufin
- HLM
human liver microsomes
- pNP
p-nitrophenol
- RL/P
the ratio of the molar content of phospholipids to that of cytochromes P450 in microsomal membranes
- SS2E1, SS1A2, SS2B6, SS2C8, SS2C9, SS2C19, SS2D6 and SS3A4, insect cell microsomes (Supersomes™) containing recombinant human CYP2E1, CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6 and CYP3A4, respectively
Footnotes
Declaration of interest: The authors declare that they have no conflicts of interest with the contents of this article.
References
- 1.Rendre S (2002) Summary of information on human CYP enzymes: Human P450 metabolism data. Drug Metab. Rev 34, 83–448 [DOI] [PubMed] [Google Scholar]
- 2.Guengerich FP and Rendre S (2010) Update Information on Drug Metabolism Systems- 2009, Part I. Cur. Drug Metab 11,1–3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wienkers LC and Heath TG (2005) Predicting in vivo drug interactions from in vitro drug discovery data. Nature Reviews Drug Discovery. 4, 825–833 [DOI] [PubMed] [Google Scholar]
- 4.Zhang HF, Wang HH, Gao N, Wei JY, Tian X, Zhao Y, Fang Y, Zhou J, Wen Q, Gao J, Zhang YJ, Qian XH and Qiao HL (2016) Physiological Content and Intrinsic Activities of 10 Cytochrome P450 Isoforms in Human Normal Liver Microsomes. J. Pharm. Exp. Ther. 358, 83–93 [DOI] [PubMed] [Google Scholar]
- 5.Davydov DR (2011) Microsomal monooxygenase as a multienzyme system: the role of P450- P450 interactions. Expert Opin. Drug Metab. Toxicol 7, 543–558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Reed JR and Backes WL (2012) Formation of P450·P450 complexes and their effect on P450 function. Pharm. Ther 133, 299–310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Reed J and Backes W (2016) The functional effects of physical interactions involving cytochromes P450: putative mechanisms of action and the extent of these effects in biological membranes. Drug Metab Rev.. 48,453–469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Davydov DR (2016) Molecular Organization of the Microsomal Oxidative System: a New Connotation for an Old Term. Biochemistry Mo scow-Supplement Series B-Biomedical Chemistry. 10,10–21 [Google Scholar]
- 9.Davydov DR, Davydova NY, Rodgers JT, Rushmore TH and Jones JP (2017) Toward a systems approach to the human cytochrome P450 ensemble: interactions between CYP2D6 and CYP2E1 and their functional consequences. Biochem. J 474, 3523–3542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lieber CS and Abittan CS (1999) Pharmacology and metabolism of alcohol, including its metabolic effects and interactions with other drugs. Clinics in Dermatology. 17, 365–379 [DOI] [PubMed] [Google Scholar]
- 11.Cederbaum AI (1998) Ethanol-related cytotoxicity catalyzed by CYP2E1-dependent generation of reactive oxygen intermediates in transduced HepG2 cells. Biofactors. 8, 93–96 [DOI] [PubMed] [Google Scholar]
- 12.Dupont I, Lucas D, Clot P, Menez C and Albano E (1998) Cytochrome P4502E1 inducibility and hydroxyethyl radical formation among alcoholics. J. Hepatol 28, 564–571 [DOI] [PubMed] [Google Scholar]
- 13.Cederbaum AI (2006) CYP2E1 - Biochemical and toxicological aspects and role in alcohol- induced liver injury. Mount Sinai J. Med 73, 657–672 [PubMed] [Google Scholar]
- 14.Meskar A, Plee-Gautier E, Amet Y, Berthou F and Lucas D (2001) Alcohol-xenobiotics interactions. Role of cytochrome P450 2E1. Pathologie Biologie. 49, 696–702 [DOI] [PubMed] [Google Scholar]
- 15.Tan Y, Patten CJ, Smith T and Yang CS (1997) Competitive interactions between cytochromes P450 2A6 and 2E1 for NADPH-cytochrome P450 oxidoreductase in the microsomal membranes produced by a baculovirus expression system. Arch Biochem Biophys. 342, 82–91 [DOI] [PubMed] [Google Scholar]
- 16.Kelley RW, Cheng DM and Backes WL (2006) Heteromeric complex formation between CYP2E1 and CYP1A2: Evidence for the involvement of electrostatic interactions. Biochemistry. 45,15807–15816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Davydov DR, Davydova NY, Sineva EV and Halpert JR (2015) Interactions among Cytochromes P450 in Microsomal Membranes: Oligomerization of Cytochromes P450 3A4, 3A5 and 2E1 and its Functional Consequences. J. Biol.Chem 453, 219–230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dorian P, Sellers EM, Reed KL, Warsh JJ, Hamilton C, Kaplan HL and Fan T (1983) Amitriptyline and ethanol pharmaco kinetic and pharmacodynamic interaction. European J. Clin. Pharm 25, 325–332 [DOI] [PubMed] [Google Scholar]
- 19.Tanaka E (2003) Toxicological interactions involving psychiatric drugs and alcohol: An update. J. Clin. Pharm. Ther 28, 81–95 [DOI] [PubMed] [Google Scholar]
- 20.Davydov DR, Davydova N,Y, Sineva EV, Kufareva I and Halpert JR (2013) Pivotal role of P450-P450 interactions in CYP3A4 allostery: the case of alpha-naphthoflavone. Biochem. J 453,219–230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Spatzenegger M, Liu H, Wang QM, Debarber A, Koop DR and Halpert JR (2003) Analysis of differential substrate selectivities of CYP2B6 and CYP2E1 by site-directed mutagenesis and molecular modeling. J. Pharm. Exp. Ther 304,477–487 [DOI] [PubMed] [Google Scholar]
- 22.Jang H-H, Dayydov DR, Lee G-Y, Yun C-H and Halpert JR (2014) The role of cytochrome P450 2B6 and 2B4 substrate access channel residues predicted based on crystal structures of the amlodipine complexes. Arch. Biochem. Biophys 545,100–107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bartlett GR (1959) Phosphorus assay in column chromatography. J. Biol. Chem 234,466–468 [PubMed] [Google Scholar]
- 24.Smith PK, Krohn RI, Hermanson GT, Mallia AK, Gartner FH, Provenzano MD, Fujimoto EK, Goeke NM, Olson BJ and Klenk DC (1985) Measurement of protein using bicinchoninic acid. Anal. Biochem 150, 76–85 [DOI] [PubMed] [Google Scholar]
- 25.Shen AL, Porter TD, Wilson TE and Kasper CB (1989) Structural analysis of the FMN binding domain of NADPH-cytochrome P-450 oxidoreductase by site-directed mutagenesis. J. Biol. Chem 264, 7584–7589 [PubMed] [Google Scholar]
- 26.Massey V (1959) The microestimation of succinate and the extinction coefficient of cytochrome c. Biochim. Biophys. Acta 34, 225–256 [DOI] [PubMed] [Google Scholar]
- 27.French JS and Coon MJ (1979) Properties of NADPH-cytochrome P-450 reductase purified from rabbit liver microsomes. Arch. Biochem. Biophys 195, 565–577 [DOI] [PubMed] [Google Scholar]
- 28.Guengerich FP, Martin MV, Sohl CD and Cheng Q (2009) Measurement of cytochrome P450 and NADPH-cytochrome P450 reductase. Nat. Protoc 4, 1245–1251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kennedy CJ (1994) CHAPTER 35 - Xenobiotics: designing an in vitro system to study enzymes and metabolism In Biochemistry and Molecular Biology of Fishes (Hochachka PW and Mommsen TP, eds.). pp. 417–430, Elsevier [Google Scholar]
- 30.Hood CA, Fuentes G, Patel H, Page K, Menakuru M and Park JH (2008) Fast conventional Fmoc solid-phase peptide synthesis with HCTU. J. Pept. Sci 14, 97–101 [DOI] [PubMed] [Google Scholar]
- 31.Fekkes D (1996) State-of-the-art of high-performance liquid chromatographic analysis of amino acids in physiological samples. J. Chromatogr. B-Biomed. Appl 682, 3–22 [DOI] [PubMed] [Google Scholar]
- 32.Reinke LA and Moyer MJ (1985) Para-nitrophenol hydroxylation - a microsomal oxidation which is highly inducible by ethanol. Drug Metab. Dispos 13, 548–552 [PubMed] [Google Scholar]
- 33.Nagle JF and Tristram-Nagle S (2000) Structure of lipid bilayers. Biochim. Biophys. Acta 1469, 159–195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wibo M, Amar-Costesec A, Berthet J and Beayfay H (1971) Electron microscope examination of subcellular fractions III. Quantitative analysis of the microsomal fraction isolated from rat liver. J Cell Biol. 51, 52–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Davydov DR, Deprez E, Hui Bon Hoa G, Knyushko TV, Kuznetsova GP, Koen YM and Archakov AI (1995) High-pressure-induced transitions in microsomal cytochrome P450 2B4 in solution - evidence for conformational inhomogeneity in the oligomers. Arch. Biochem. Biophys 320, 330–344 [DOI] [PubMed] [Google Scholar]
- 36.Haugland RP (2005) The Molecular Probes® handbook, a guide to fluorescent probes and labeling technologies Invitrogen Corp., Carlsbad, CA [Google Scholar]
- 37.Karolin J, Johansson LBA, Strandberg L and Ny T (1994) Fluorescence and absorption spectroscopic properties of dipyrrometheneboron difluoride (BODIPY) derivatives in liquids, lipid-membranes, and proteins. J. Am. Chem. Soc 116, 7801–7806 [Google Scholar]
- 38.Tramier M, Piolot T, Gautier I, Mignotte V, Coppey J, Kemnitz K, Durieux C and Coppey-Moisan M (2003) Homo-FRET versus hetero-FRET to probe homodimers in living cells. Methods Enzymol. 360, 580–597 [DOI] [PubMed] [Google Scholar]
- 39.Donato MT, Jimenez N, Castell JV and Gomez-Lechon MJ (2004) Fluorescence-based assays for screening nine cytochrome P450 (P450) activities in intact cells expressing individual human P450 enzymes. Drug Metab. Dispos 32, 699–706 [DOI] [PubMed] [Google Scholar]
- 40.Ghosal A, Hapangama N, Yuan Y, Lu XW, Horne D, Patrick JE and Zbaida S (2003) Rapid determination of enzyme activities of recombinant human cytochromes P450, human liver microsomes and hepatocytes. Biopharm. Drug Dispos 24, 375–384 [DOI] [PubMed] [Google Scholar]
- 41.Backes WL, Batie CJ and Cawley GF (1998) Interactions among P450 enzymes when combined in reconstituted systems: formation of a 2B4–1A2 complex with a high affinity for NADPH- cytochrome P450 reductase. Biochemistry. 37,12852–12859 [DOI] [PubMed] [Google Scholar]
- 42.Reed JR, Eyer M and Backes WL (2010) Functional interactions between cytochromes P450 1A2 and 2B4 require both enzymes to reside in the same phospholipid vesicle. Evidence for physical complex formation. J. Biol. Chem 285, 8942–8952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Reed JR and Backes WL (2017) Physical Studies of P450-P450 Interactions: Predicting Quaternary Structures of P450 Complexes in Membranes from Their X-ray Crystal Structures. Front. Pharmacol 8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sienkiewicz-Oleszkiewicz B and Wiela-Hojenska A (2018) CYP2C19 polymorphism in relation to the pharmacotherapy optimization of commonly used drugs. Pharmazie. 73, 619–624 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.