

Abstract
Experimental Outer surface protein (Osp) C based subunit chimeritope vaccinogens for Lyme disease (LD) were assessed for immunogenicity, ability to elicit antibody (Ab) responses to divergent OspC proteins, and bactericidal activity. Chimeritopes are chimeric epitope based proteins that consist of linear epitopes derived from multiple proteins or multiple variants of a protein. An inherent advantage to chimeritope vaccinogens is that they can be constructed to trigger broadly protective Ab responses. Three OspC chimeritope proteins were comparatively assessed: Chv1, Chv2 and Chv3. The Chv proteins possess the same set of 18 linear epitopes derived from 9 OspC type proteins but differ in the physical ordering of epitopes or by the presence or absence of linkers. All Chv proteins were immunogenic in mice and rats eliciting high titer Ab. Immunoblot and enzyme linked immunosorbent assays demonstrated that the Chv proteins elicit IgG that recognizes a diverse array of OspC type proteins. The panel included OspC proteins produced by N. American and European strains of the LD spirochetes. Rat anti-Chv antisera also uniformly labeled intact, non-permeabilized Borreliella burgdorferi demonstrating that vaccinal Ab can bind to targets that are naturally presented on the spirochete cell surface. Vaccinal Ab also displayed potent complement dependent-Ab mediated killing activity. This study highlights the ability of OspC chimeritopes to serve as vaccinogens that trigger potentially broadly protective Ab responses. In addition to the current use of an OspC chimeritope in a canine LD vaccine, chimeritopes can serve as key components of human LD subunit vaccines.
Keywords: Borreliella burgdorferi, chimeritope, ticks, Lyme vaccine, Borrelia
INTRODUCTION
Lyme disease (LD) is the most common tick-borne disease in the United States. Borreliella burgdorferi (1), a spirochete formerly classified as Borrelia burgdorferi, is a causative agent of LD in North America and Europe (2,3). Since the discovery of B. burgdorferi, additional related species have been identified and determined to be pathogenic in humans (4,5). The LD spirochetes are transmitted to mammals by infected nymphal or female adult Ixodes ticks. I. scapularis and I. pacificus are the primary tick vectors in N. America. In Europe and Asia, I. ricinus and I. persulcatus are dominant (6,7). Ticks alone cannot maintain LD in nature since the spirochetes are not transovarially transmitted. Hence, Ixodes ticks can only become infected by feeding on an infected mammal. Since there is no animal-to-animal transmission, persistence of the LD spirochetes in nature is strictly dependent on its enzootic cycle (8). In the eastern half of N. America, Tamias striatus (eastern chipmunk), Sorex brevicauda (short-tailed shrew), Peromyscus leucopus (white-footed mouse) and Sorex cinereus (masked shrew) are the primary sources of infection for feeding larval stage ticks (9). In the western US, Neotoma fuscipes (dusky-footed wood rat) and Peromyscus maniculatus (deer mouse) are important reservoirs (10). In Europe, several species of mice, voles, squirrels, woodland birds, and pheasants serve as reservoirs (11).
Estimates from the Centers for Disease Control and Prevention indicate there are ~340,000 clinician diagnosed probable cases of LD each year in the United States (12). Due to the lack of uniform reporting across Europe, accurate estimates of LD incidence are not available. In contrast to current practices in human medicine, screening for LD and other tick-borne diseases is common in veterinary medicine, particularly in regions where LD is endemic or encroaching. The Companion Animal Parasite Council (CAPC) gathers canine LD antibody (Ab) test results from LD diagnostic assay manufacturers. CAPC reported 360,000 positive LD Ab tests in canines in 2019 in the US1. This is just a fraction of the total since <30% of testing data are captured. While a positive Ab test is not always indicative of active infection, screening for tick-borne infections in veterinary medicine has provided critical information for assessing disease risk in canine and human populations (13).
The diagnosis and clinical course of LD in canines and humans has been detailed in several reviews and thus is not covered here (14–16). Early diagnosis of LD is important, since untreated infections can result in serious multi-systemic clinical complications (16). In veterinary medicine, acaricides and LD bacterin and subunit vaccines are widely used for LD prevention (17). Bacterin and subunit vaccines fundamentally differ in terms of their composition. Bacterins, which consist of inactivated cell lysates of laboratory adapted LD strains, are complex as they contain all proteins and other cellular components produced during laboratory cultivation. A typical LD spirochete strain produces in excess of 1000 proteins in vitro (18,19) and the majority of these proteins are not presented on the cell surface and thus cannot serve as targets for vaccination induced Ab. Bacterins also lack or have low levels of several antigens that are selectively produced in the mammalian environment (20). In contrast, subunit vaccines consist of well characterized antigens that elicit protective or neutralizing Ab responses. At the present time, preventive strategies for LD in humans are largely ineffective (15) and there are no commercially available vaccines.
OspC derived proteins and OspA have been employed as vaccine antigens in canine LD subunit vaccines (21). OspA was also the sole vaccinogen in the only human LD vaccine to make it to market. After 3 years of use, LYMErix was voluntarily removed from the market by its manufacturer. Factors that contributed to the demise of LYMErix have been extensively reviewed in the literature (22–24) and hence, are only briefly discussed here. During the enzootic cycle, OspA is selectively produced in ticks with little to no expression in mammals (25,26). As a result, vaccinal OspA Ab can target spirochetes within ticks but not within mammals (27). In contrast, OspC is upregulated by the tick blood meal and is produced at high levels in infected mammals (25,26). Vaccinal Ab to OspC can target spirochetes both during transmission and upon entry into a host.
While the expression patterns of OspC are ideal for targeting with vaccinal Ab, its inherent sequence diversity and the localization of its immunodominant epitopes within variable domains of the protein have posed challenges in eliciting broadly protective Ab responses. It has been demonstrated that broad protection cannot be achieved with a single OspC protein (28–40). Variants of OspC, referred to as “OspC types”, have been delineated and are differentiated using letter, and other designations (type A, type B, etc.) (33,41,42). Sequence variation among OspC type proteins is clustered within the surface exposed helix 5 (H5) and loop5 (L5) epitopes (43) (40,41,44). Conserved domains of OspC do not contribute to protective Ab responses (17,21,45). In earlier studies, tetra- and octavalent OspC chimeric epitope based proteins (chimeritopes) consisting of L5 and H5 epitopes from different OspC types were developed to expand the protective range of OspC (41,44,46). Here, we extend and expand on that approach through the analysis of a series of OspC chimeritopes (Chv1, Chv2, and Chv3) that differ in their epitope organization. The Chv proteins were designed, produced and evaluated for structure using NMR. Immune responses were assessed by ELISA and immunoblot and the potential killing activity of vaccinal Ab measured using bactericidal assays. This study demonstrates that chimeritope vaccinogens can elicit broadly cross reactive Ab responses.
MATERIALS AND METHODS
B. burgdorferi strains, cultivation and ospC sequence analyses.
All LD spirochete isolates employed in this study (Table 1) were grown at 37 C in Barbour-Stoenner-Kelly (BSK)-H complete media supplemented with 6% rabbit serum (Sigma). Growth was monitored daily using simple wet-mounts and dark-field microscopy. Some isolates were subsurface plated (47) to obtain clonal populations (indicated in Table 1). Cultivation methods and media formulations have been previously described (48). The ospC genotype of each strain or clone was determined by PCR amplification and sequence analysis of the amplicons (44). Primers used in this study have been previously described (21).
Table 1.
Description of bacterial isolates employed in this study.
| Species | Isolate or clone | OspC type* | Biological source |
|---|---|---|---|
| B. burgdorferi | B31–5A4 | A | Ixodes scapularis tick; NY, USA |
| LDP73 | B | Human blood; NY, USA | |
| LDP84 | C | Human blood; NY, USA | |
| LDP116 | D | Human blood; NY, USA | |
| N40 | E | I. scapularis tick; NY, USA; clonal | |
| DRI40h | F | Canine tissue (experimentally infected with ticks from Rl and CT); clonal | |
| B268 | G | Human skin; NY, USA | |
| DRI03a | H | Canine tissue (experimentally infected with ticks from Rl and CT); clonal | |
| B331 | I | Human blood; NY, USA | |
| B236 | J | Human skin; NY, USA | |
| LDP89 | K | Human blood; NY, USA | |
| SI-1 | L | Peromyscus gossypinus; GA, USA | |
| Veery | M | Veery bird; USA | |
| LDP63 | N | Human blood; NY, USA | |
| DRI40e | T | Canine tissue, experimentally infected with ticks from Rl and CT; clonal | |
| DRI85f | U | Canine tissue, experimentally infected with ticks from Rl and CT; clonal | |
| DRI85d | DRI85d | Canine tissue, experimentally infected with ticks from Rl and CT; clonal | |
| DRI85e | DRI85e | Canine tissue, experimentally infected with ticks from Rl and CT; clonal (note that the DRI85d, e and f clones all came from the same tissue biopsy). | |
| VCU1 | Not assigned | Human CSF; Germany | |
| B. garinii | Pwa | Pwa | Human cerebrospinal fluid (CSF); Germany |
| VSBP | Not assigned | Human CSF; Germany | |
| Jem5A | Not assigned | Human erythema migrans (EM) lesion; Japan; clonal; derived from isolate JEM5 | |
| Phi | Not assigned | Germany (biological origin not known) | |
| FRG | Not assigned | Germany | |
| B. afzelii | PKo | PKo | Human EM lesion; Germany |
| B5/92 | PKo | Human EM lesion, Germany | |
| SZ94 | Not assigned | I. ricinus; Europe (country of origin not known) | |
| VS461 | VS461 | I. ricinus; Germany | |
| B. bavariensis | PBi | PBi | Human CSF; Germany |
| B. mayonii | MN14–1420 | Mayoi | Human blood; USA |
| B. burgdorferi | B31-HE-OspC | A | Derived from parental strain B31–5A3; constitutively produces OspC type A; clonal |
| B31-ΔospC | N/A | ospC gene deletion mutant |
Note that some OspC proteins have not yet been classified as a specific OspC type and are thus indicated to be “not assigned”.
Cloning and production of recombinant proteins.
All cloning and protein purification methods were standard and have been previously described (49). In brief, genes encoding full length OspC type proteins minus the leader peptide were PCR amplified from the appropriate strain (Table 1) with primers (21) harboring tail sequences that allow for ligase independent cloning (LIC; pET46 Ek/LIC vector; Novagen) or ligation into pET45b(+) (Novagen). Phusion polymerase (New England Biolabs) was used in all PCR reactions under standard conditions. For cloning into the multi-cloning site of pET45b(+) (Novagen), ospC genes were amplified with primers harboring BamHI and EagI restriction sites. Some ospC genes and the Chv1, Chv2 and Chv3 chimeritopes were codon optimized and synthesized on a fee for service basis. The Chv genes were provided by the supplier in pJ201 (DNA2.0, Inc., Menlo Park, CA) and the synthesized ospC genes in pET45b(+) (Genscript). The sequence of all cloned genes was verified on a fee for service basis (Genewiz). The physical organization of each chimeritope is indicated in Figure 1. The Chv genes were PCR amplified from pJ201 using primers with LIC tails that allow for annealing with linearized pET100 vector (Novagen). After annealing or ligation of the amplicons into the appropriate plasmid, the plasmids were propagated in Escherichia coli NovaBlue DE3 cells (Novagen), purified and transformed into E. coli BL21/DE3 cells (Novagen).
Figure 1. Schematic depiction of the organization of the Chv series of chimeritopes.
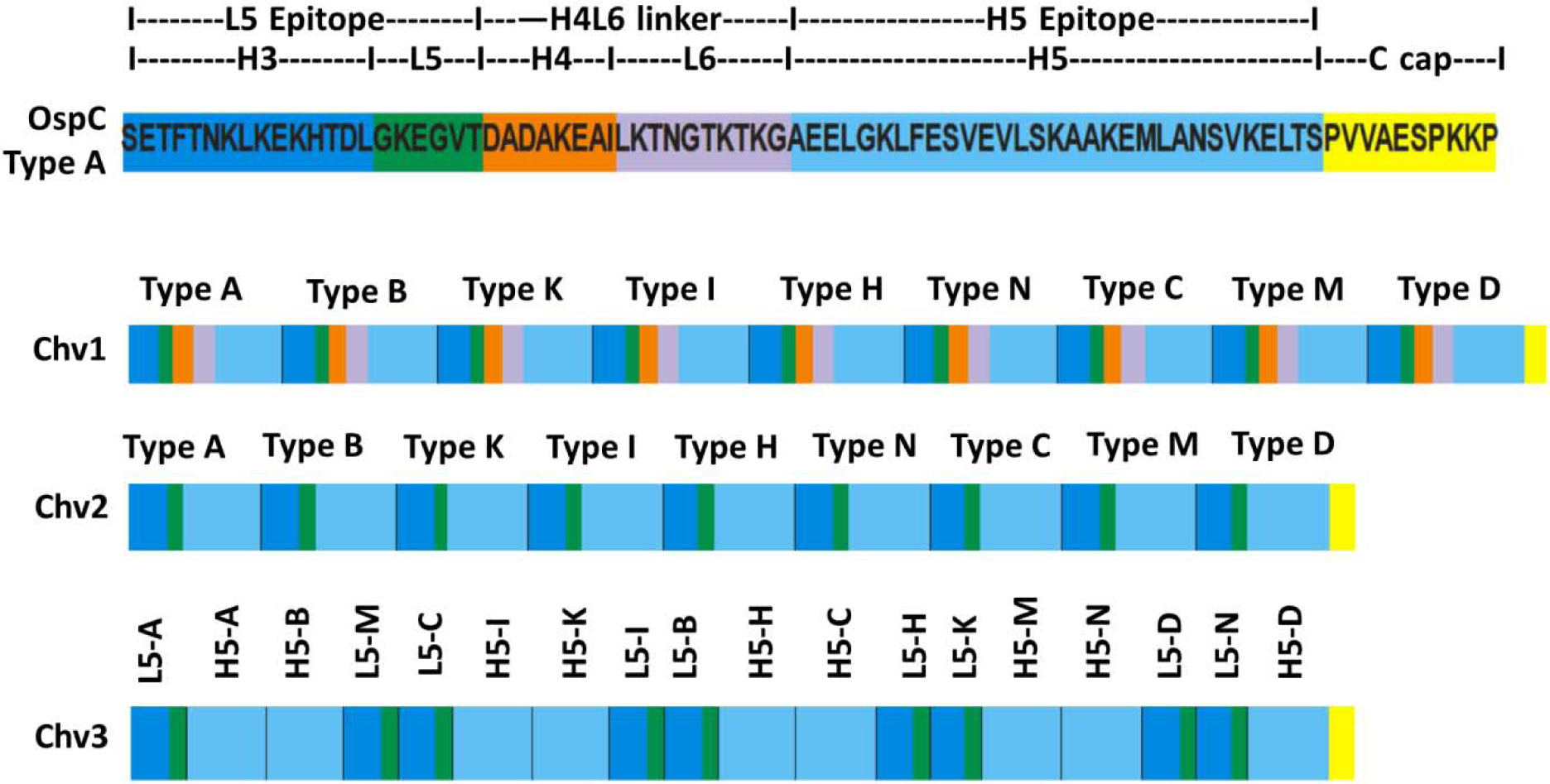
The top schematic is a linear representation of the Loop 5 (L5) and Helix 5 (H5) region of a type A OspC (B. burgdorferi B31) with the intervening sequence (Helix 4 Loop 6; H4L6) and structural elements shown. Coloring is used to highlight each structural domain. The lower schematics depict the organization of the Chv1, Chv2 and Chv3 chimeritopes. The color coding is the same as in the top panel and is included to highlight the organizational differences of each chimeritope.
Protein expression was induced with IPTG (final concentration of 1 mM), the induced cells were lysed and fractionated into soluble or insoluble phases using standard methods. Chv2 and all ospC genes separated with the soluble phase and were purified using non-denaturing conditions. Chv1 and Chv3 separated with the insoluble fraction and thus were purified under denaturing conditions. The soluble proteins were purified via their N-terminal hexahistidine tag using nickel affinity chromatography (ÄKTA purifier FPLC system, GE) as previously described (21). Imidazole was removed by dialysis into phosphate buffered saline (PBS; pH 7.4) across a 6 to 8 kDa molecular weight cut-off membrane (Spectrum Labs). To purify proteins from the insoluble phase, the insoluble fraction was resuspended in 8 M urea with 40 mM imidazole. Cellular debris was removed by centrifugation (15,500 × g; 30 m; 4 °C) and the supernatants were loaded onto a Poly-Prep Chromatography column (Biorad) pre-equilibrated in 50 mM Tris-HCl, 300 mM NaCl, pH 8, with 8 M urea. The protein bound resin was washed and the proteins eluted using 250mM imidazole. Urea and imidazole were removed by stepwise dialysis into phosphate buffered saline (PBS; pH 7.4) with decreasing concentrations of urea. Protein concentrations were determined using a BCA assay (Pierce).
Nuclear magnetic resonance spectroscopy.
Purified Chv1, Chv2 and Chv3 were exchanged into buffer (35 mM Tris-HCl; pH 7.4), 200 mM NaCl in 90% H2O/10% D2O) at final concentrations of 8.2 μM, 10.8 μM and 10.8 μM, respectively. The β1 immunoglobulin binding domain of protein G (GB1) was used as a folded protein control of known structure. GB1 (500 μM) was exchanged into NMR buffer (100 mM C2H3NaO2, pH 5.4, 0.02% NaN3, 50 μM EDTA, 1 mM DTT in 95% H20/5% D20). NMR spectra were collected on a Bruker Avance III 850 MHz spectrometer using a 5 mm1H (13C/15N) triple resonance probe equipped with cryogenically cooled preamplifiers and r.f. coils on1H and 13C channels. NMR experiments were conducted at 310K for the Chv proteins and at 298K for GB1. One-dimensional1H data were collected for each protein using 64K points (32K points for GB1), 1024 scans (32 scans for GB1), a relaxation delay of 1.5 seconds and excitation sculpting for solvent suppression. Data were processed using Bruker Biospin Topspin 3.5 software.
SDS-PAGE and immunoblot analyses.
Recombinant proteins were fractionated by SDS-PAGE using 12% Criterion precast gels or ANYkD gels (Biorad) and visualized by staining with Coomassie Brilliant Blue (CBB). The proteins were transferred to PVDF membranes (Pierce) by semi-dry blotting using a Trans-Blot Turbo apparatus per the manufacturer’s protocol (Biorad). As a control for protein loading, immunoblots were screened with α-His Tag Ab (Pierce) or with specific antisera (as indicated in each experiment). The appropriate anti-IgG horseradish peroxidase (HRP)-conjugated secondary Abs were added (1:40,000 dilution) and Ab binding detected by chemiluminescence using Clarity Max Western ECL blotting substrate (BioRad). The images presented in the immunoblot figures were cropped to center on the migration position of the recombinant proteins. All immunoblots for a given experiment were exposed together for an identical length of time (30 sec).
Generation of anti-Chv antisera.
Antisera were generated in C3H/HeJ mice and Sprague-Dawley rats. All animal studies were conducted in accordance with the Guide for the Care and Use of Laboratory Animals (Institute of Animal Research, National Research Council) with protocols approved by the Virginia Commonwealth University Institutional Animal Care and Use Committee. C3H/HeJ mice (Jackson Laboratories) were immunized by intraperitoneal (IP) inoculation with 3 doses of the individual Chv proteins in Rehydragel LV (Chemtrade Solutions) on days 0, 21, and 42 (25 μg per dose). Blood samples were harvested on days 0, 21, and 42 by tail snip and by cardiac puncture upon sacrifice on day 63. Sprague-Dawley rats (Charles River) were delivered a 4-dose series of recombinant protein by IP inoculation. The first dose consisted of 50 μg protein in Freund’s Complete Adjuvant. Booster vaccinations contained 25 μg protein in Freund’s Incomplete Adjuvant (Sigma) and each was delivered 2 weeks apart. One week after the last immunization, the rats were euthanized by CO2 asphyxiation and terminal bleeds were conducted by cardiac puncture. Serum was harvested using standard methods and vaccination induced IgG titers determined as above.
ELISAs and IgG titer determination.
Single dilution ELISA analyses were conducted using the appropriate immobilized vaccinogen or recombinant OspC protein (500 ng per well), a primary serum dilution of 1:100 and a HRP-conjugated secondary Ab dilution of 1:15000. IgG titers were determined using the vaccinogen as the immobilized antigen (500 ng per well) as previously described (50). Threefold serial dilutions of sera ranging from 1:50 to 1:109350 were tested and the titers (log transformed) calculated at 1/3 OD max (51).
Immunofluorescence assays (IFAs).
IFAs were performed using previously described methods (52). In brief, cells were collected from mid-log phase cultures, spotted on to Superfrost Plus slides (Fisher Scientific) and air-dried. After blocking with PBS-TB (3% BSA; PBS; 0.5% Tween 20), the samples were overlaid with the desired antiserum (diluted 1:1,000 in PBS-TB; 30 min), washed, and then overlaid with AlexaFluor 594-conjugated goat anti-rat IgG (1:500 in PBS-TB; 30 min) (Molecular Probes) in a dark chamber. The slides were washed, coverslips were mounted (ProLong Gold Antifade Mountant; Molecular Probes), and IgG binding visualized by fluorescence microscopy (BX51; Olympus). Images were digitally photographed (DP71; Olympus). Images of the cells in the identical fields of view were visualized using dark-field microscopy.
Assessment of bactericidal activity of vaccination induced Ab.
The killing activity of serum from hyperimmunized rats was measured using an in vitro bactericidal assay as previously described (41) with some modifications. In brief, 4 μl of mid-log phase B. burgdorferi B31-HE-OspC cells (20% v/v) were incubated with 4 μl complement preserved guinea pig serum (GPS; Complement Tech; 20% v/v) and 4 μl of heat-inactivated (HI; 56 °C, 30 min) hyperimmune sera (20% v/v). The final reaction volume was adjusted to 20 μl by the addition of BSK-H media and the mixtures were incubated at 37°C for 18 hr. Spirochete viability/motility was assessed using dark-field microscopy. Controls consisted of cells incubated in BSK-H media with: 1) Rat Serum (Innovative Research), 2) 20% hyperimmunized sera with 20% HI GPS, or 3) cells in BSK-H media with no additional serum added. All assays were performed in triplicate and multiple experimental replicates were performed. Calculations were performed by adding up the total number of live cells for five fields of view. Percent survival was obtained by dividing each group by control group 1. A One-Way ANOVA test was done using Graphpad Prism to evaluate statistical significance.
RESULTS
Chimeritope design rationale and production.
Schematics depicting the general organization of Chv1, Chv2 and Chv3 are provided in Figure 1. Each Chv protein harbors H5 and L5 epitopes (40,44) derived from the same set of 9 OspC types. Thus, each Chv protein is comprised of at least 18 well-characterized OspC epitopes (40,44). The Chv proteins differ from one another in their physical ordering of epitopes or by the presence or absence of intervening sequences between the L5 and H5 epitopes. The intervening sequence linker is comprised of helix 4 (H4) and loop 6 (L6) of OspC (53) and is henceforth referred to as the H4L6 linker. Chv1, the largest of the three proteins (60.6 kDa), harbors H4L6 linkers between each L5 and H5 epitope set. Chv2 (46.1 kDa) has the same epitope ordering as Chv1, but lacks the H4L6 linkers. The rationale for omitting the linkers was to reduce the size of the recombinant protein so as to allow for the potential inclusion of additional epitopes in downstream constructs. Relative to Chv1 and Chv2, the order of epitopes in Chv3 is distinctly different. For Chv3, the L5 and H5 epitopes from the same OspC type are not adjacent to one another, with the exception of OspC Type A. As in Chv2, the H4L6 linkers were omitted. The C-termini of Chv1, Chv2 and Chv3 are identical and consist of the C-terminal 10 amino acid residues of OspC. This C-terminal “cap” (C-cap) was added to protect against proteolytic degradation. The Chv proteins were efficiently expressed in E. coli. While all Chv proteins were predicted to be soluble upon expression in E. coli, Chv1 and Chv3 partitioned with the insoluble fraction and Chv2 with the soluble fraction. Chv1 and Chv3 were most likely packaged in inclusion vesicles. After purification and dialysis into PBS, all of the Chv proteins were soluble.
The Chv proteins are unstructured, extended random-coils.
To determine if the Chv proteins assume a defined structure, attempts were made to obtain crystals for X-ray crystallographic analysis. The determination of the structures could provide insight into epitope presentation. However, in spite of testing a wide range of conditions, crystals were not obtained. Hence, one-dimensional1H NMR was conducted to assess structural properties (Figure 2). The absence of1H signal dispersion in both the methyl (0.5 – 1 ppm) and amide (6.5 – 10 ppm) regions of the Chv proteins and overall line broadening throughout the spectra indicates an unstructured random-coil organization. Protons in the amide regions of the Chv proteins exhibited identical chemical environments, indicative of being solvent exposed. The NMR signal dispersion in the methyl region (−0.5–1) and separated proton resonances throughout the spectrum shows that GB1 control protein exists in a folded state. The lack of organized structure of the Chv proteins is consistent with the original goal of designing these proteins so that all of their individual epitopes are accessible and presented in a manner that will trigger Ab responses.
Figure 2. Nuclear magnetic resonance analyses of the Chv proteins.
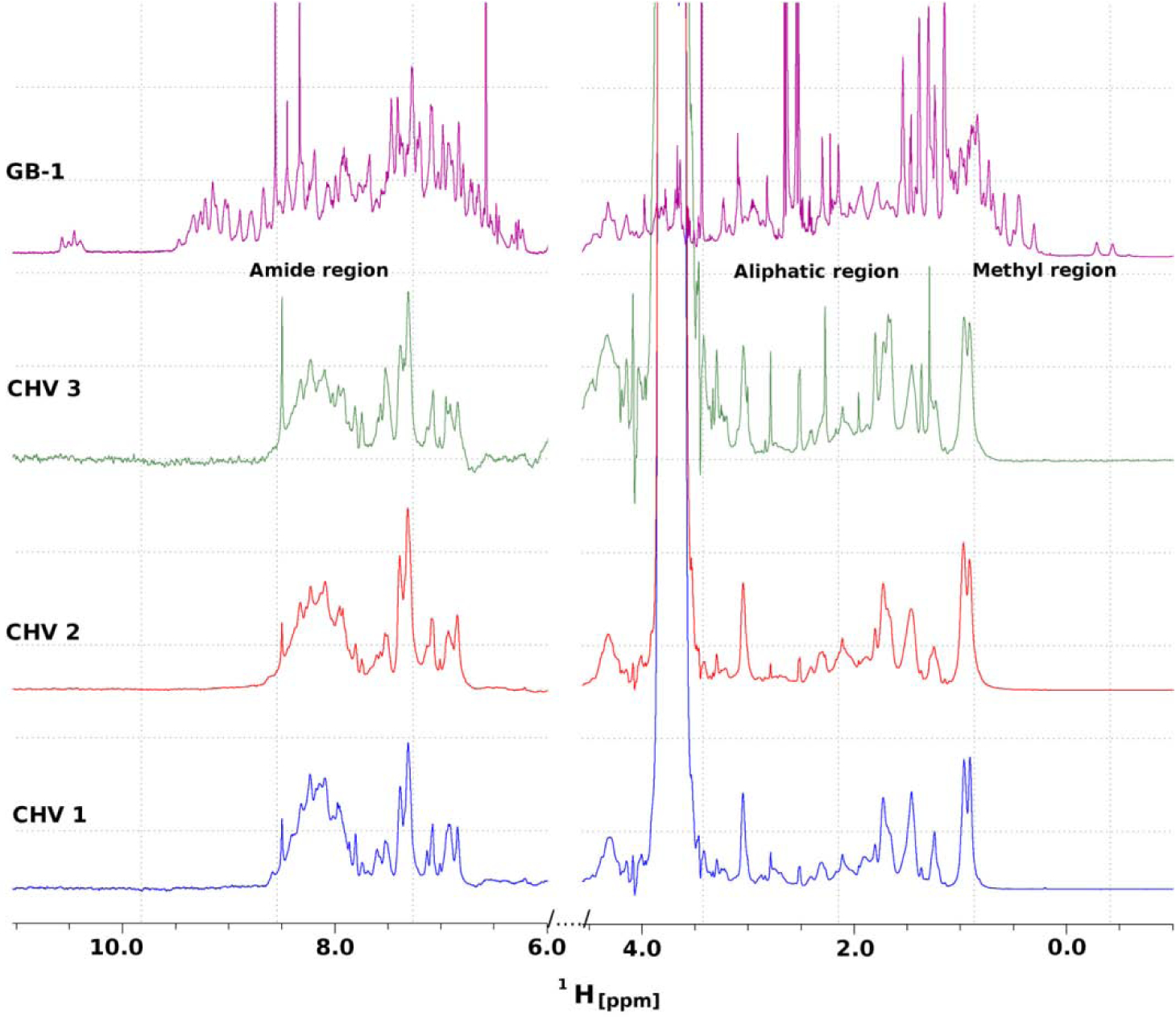
One-dimensional1H NMR spectra of Chv1, Chv2, and Chv3 and of the β1 immunoglobulin binding domain of protein G (GB1) are shown. The peak intensities of all spectra have been matched to each other and the strong peak near 3.5 ppm observed in the three Chv spectra is a result of 35mM Tris buffer. Methods were as detailed in the text.
The Chv proteins are immunogenic in mice and rats and elicit Ab that can bind to diverse OspC proteins.
The immunogenicity of the Chv proteins was assessed in mice and rats. Each Chv protein elicited similar high titer IgG responses (log transformed IgG titer values >4.0) suggesting that epitope ordering or the presence or absence of linkers did not significantly impact Ab titer to the vaccinogens (Figure 3). To determine if vaccinal Ab can recognize and bind to diverse OspC type proteins, a panel of OspC proteins (n=30; Table 1) were immunoblotted and screened with rat anti-Chv1, Chv2, and Chv3 antisera. The OspC proteins tested included those directly represented in the Chv constructs (A, B, C, D, H, I, K, M and N) and 21 that are not including European OspC types from B. garinii, B. bavariensis and B. afzelii and OspC from B. mayonii (OspC type Mayo1) (54). All OspC type proteins were bound by rat anti-Chv IgG. Figure 4 presents representative data for a subset of the OspC tested with complete ELISA based IgG binding results for all OspC types presented in Figure 5. The Chv1 and Chv2 proteins were more effective at eliciting broadly cross-reactive Ab responses that Chv3. The Factor H binding protein B (FhbB) from the periodontal disease pathogen, T. denticola, served as a negative control protein (55). As expected, FhbB was not bound by Ab (data not shown).
Figure 3. Determination of IgG titers to the Chv proteins in C3H-HeJ mice.
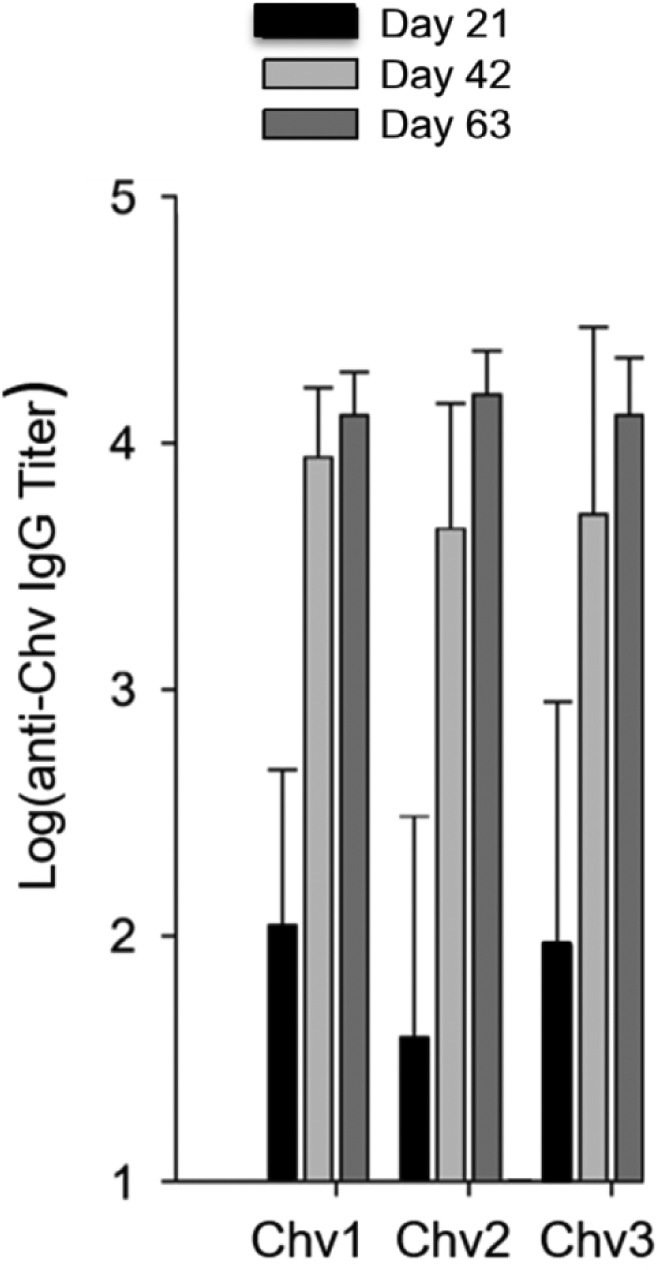
IgG titers to Chv1, Chv2 and Chv3 were determined by ELISA using serum harvested at the time points indicated. All methods were standard and are described in detail above.
Figure 4. The Chv chimeritopes elicit a broadly cross-reactive IgG responses.
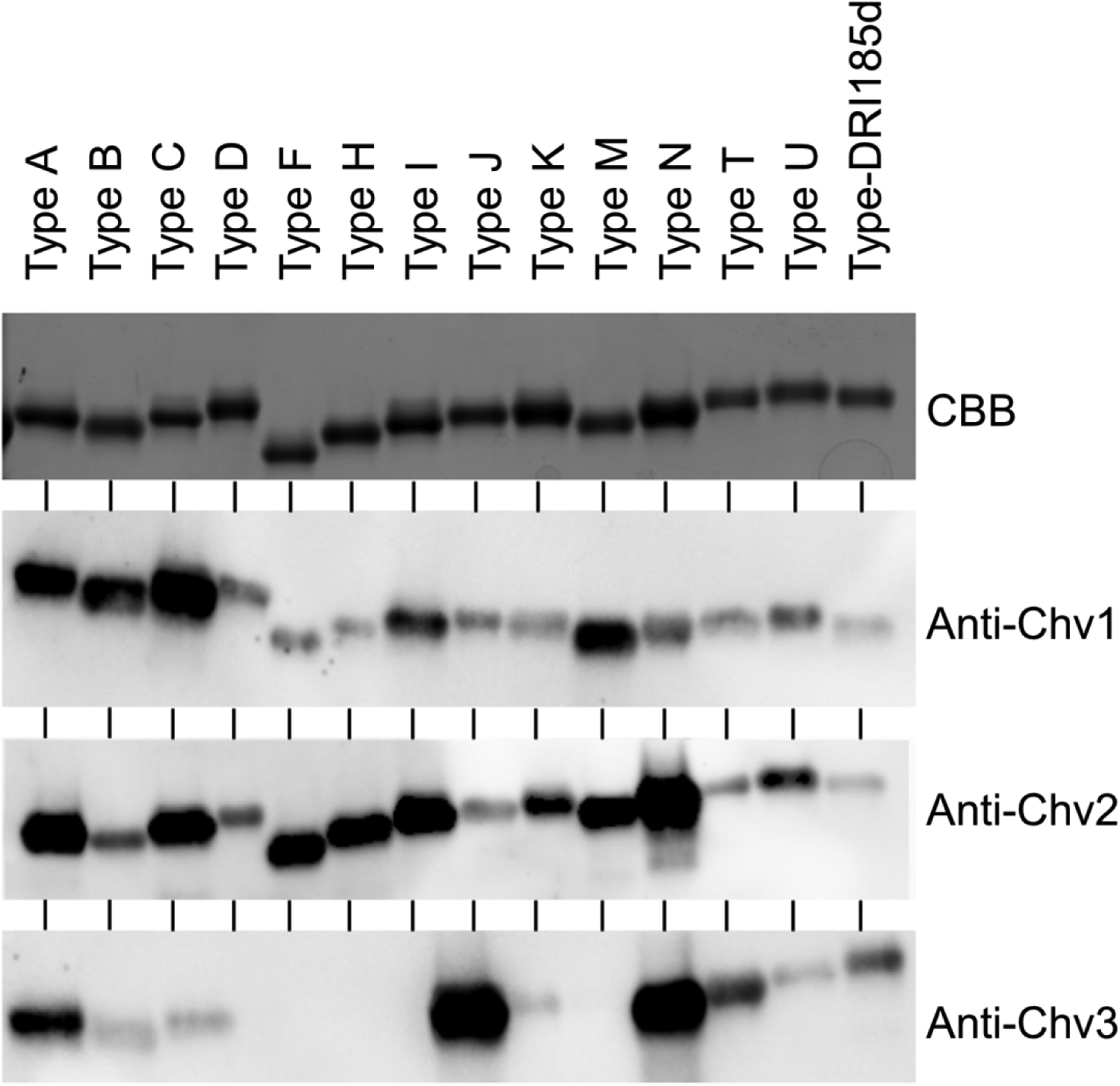
A panel of full-length recombinant OspC proteins (types indicated at the top of the figure) were subjected to SDS-PAGE, and then stained with CBB or transferred to PVDF membranes for immunoblot analyses. Blots were screened with the rat anti-Chv antiserum (1:1,000) as indicated to the right. All common reagents used in these immunoblots were derived from the same stocks and the immunoblots were exposed for the same amount of time. The images were cropped to remove blank space.
Figure 5. Vaccinal antibody binds to diverse full length recombinant OspC proteins.

An expanded set of OspC types were assessed for binding to vaccinal Ab using an ELISA based format. The OspC types and their species of origin are indicated along the Y axis. For OspC proteins that have not yet been assigned to an OspC type, the isolate of origin is indicated. The X axis indicates absorbance at 405nm. All methods were as detailed in the text.
Anti-Chv IgG binding was also assessed by ELISA. Thirty different OspC proteins were screened. As in the immunoblots, all OspC types were bound by IgG (Figure 5). The level of Ab binding to different OspC types was more consistent in the ELISAs than that observed in the immunoblots. This could result from a more favorable presentation of the L5 and H5 epitopes by non-denatured OspC proteins in the ELISA analyses. These analyses indicate that Ab elicited by the Chv chimeritopes can bind to a diverse OspC types originating from multiple LD spirochetes species and isolates for N. America and Europe.
Anti-Chv antisera surface labels B. burgdorferi strains.
To determine if Ab elicited by vaccination with the Chv proteins can recognize epitopes presented on the surface of B. burgdorferi, IFAs were performed using non-permeabilized intact cells. B. burgdorferi B31 wild type (wt) and B31-HE-OspC, an engineered strain that constitutively produces OspC during cultivation (56), were tested. The rationale for screening B31-HE-OspC, in addition to a wt laboratory cultivated strain, stems from the fact that N. American B. burgdorferi isolates (including strain B31) produce low and variable levels of OspC during cultivation (53). The percentage of cells in a B. burgdorferi B31 population that produce detectable levels of OspC is ~10% (45). Hence, the use of B31-HE-OspC facilitated efforts to assess anti-Chv IgG Ab binding to the cell surface. Consistent with the low level expression of OspC during in vitro cultivation, only a subset of B31 cells were surface-labeled by anti-Chv1, anti-Chv2 and anti-Chv3 antisera (Figure 6). In contrast, and as expected, all B31-HE-OspC cells were uniformly labeled with each Chv antisera. Surface labeling of cells was not observed when screened with preimmune rat serum (data not shown). The ability of Ab induced by vaccination with the Chv proteins to bind to intact B. burgdorferi cells indicates surface presentation of the L5 and H5 epitopes.
Figure 6. Immunofluorescence assays (IFAs) demonstrate surface exposure of the L5 and H5 OspC epitopes.
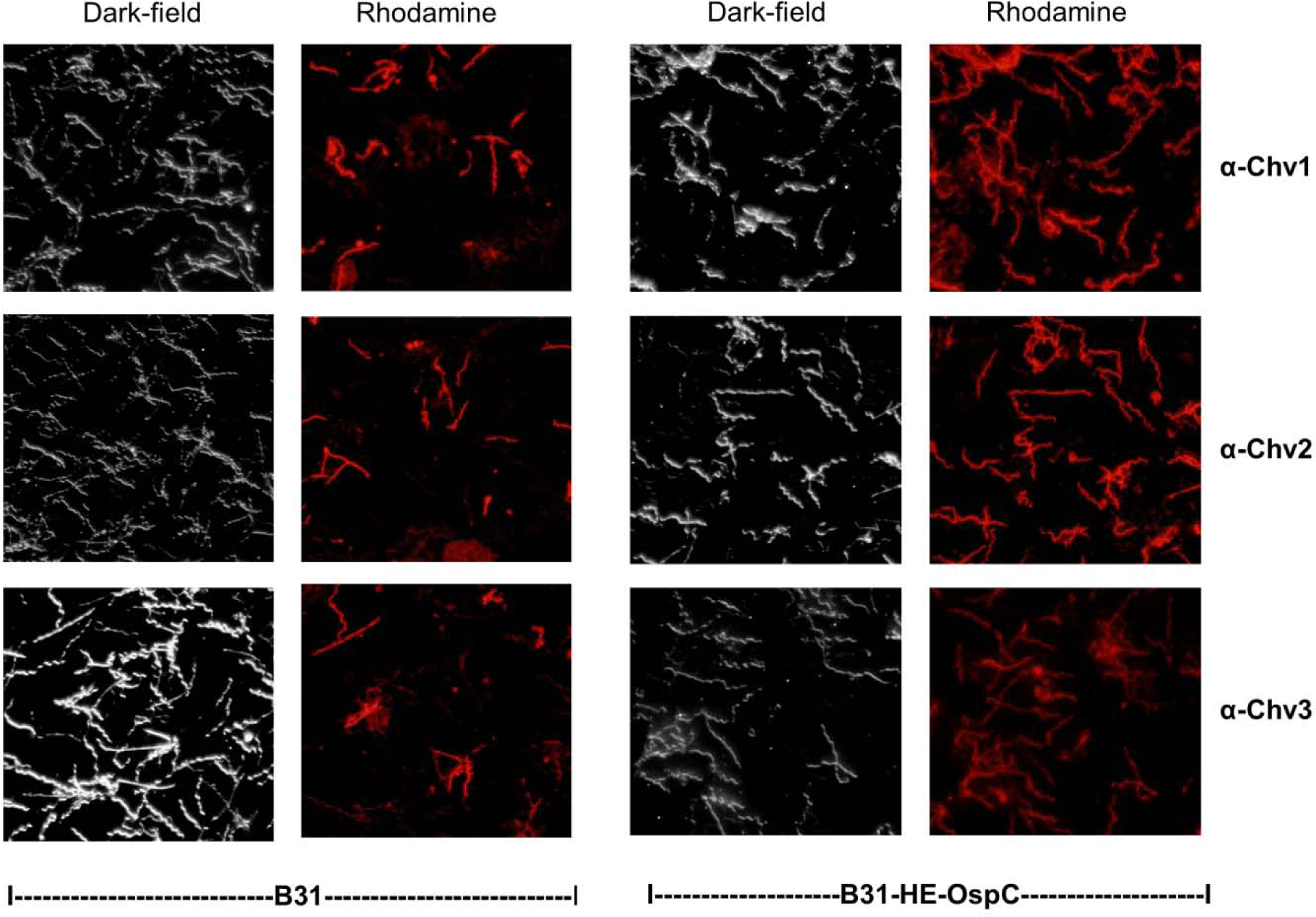
IFA analyses were performed using non-permeabilized intact cells as detailed in the text. The strains investigated and the antisera tested are indicated below and to the right of the images, respectively. Cells were visualized by dark-field microscopy or by fluorescence as indicated along the top.
Bactericidal activity of anti-Chv antisera.
The killing activity of sera was measured using in vitro bactericidal assays; a standard and well-accepted approach for assessing correlates of protection. After incubation of cells with the anti-Chv antisera, bacterial viability was assessed by dark-field microscopy. Antisera raised against each Chv protein displayed “high level” bactericidal activity which is defined as >75% killing of the cell population. No killing was observed when HI-GPS was used as the exogenous complement source indicating that killing occurs through a strictly complement-dependent manner (Figure 7). Anti-OspA antisera served as a positive control, and as expected, displayed high-level killing activity. This is consistent with the high-level expression of OspA during cultivation which provides ample target for Ab binding on the cell surface.
Figure 7. The Chv proteins elicit production of potent bactericidal Ab.
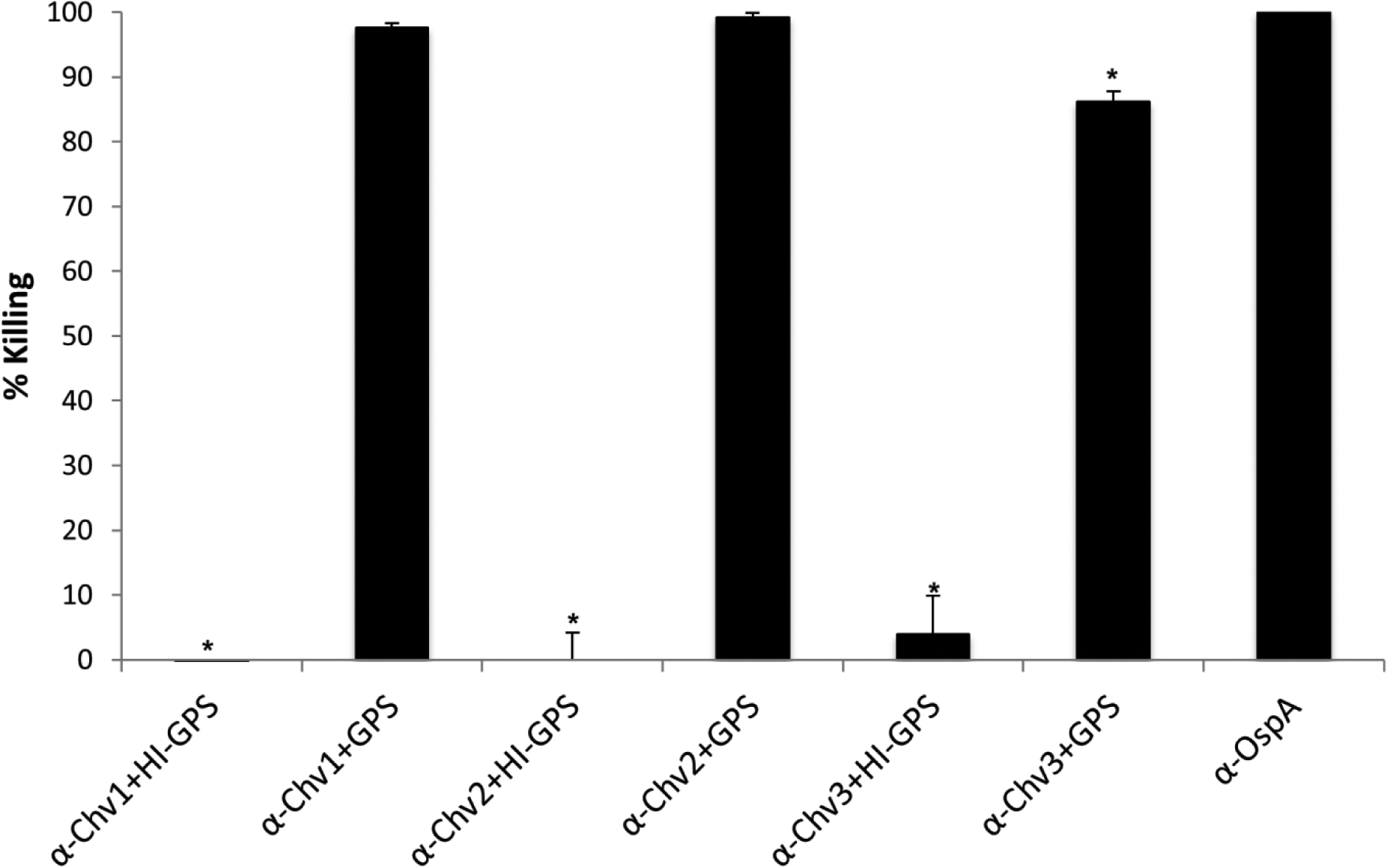
B. burgdorferi B31-HE-OspC was incubated with Chv hyperimmune serum as indicated in the figure with GPS serving as the exogenous complement source. To determine if Ab mediated killing is complement dependent, cells were incubated with hyperimmune serum and HI-GPS. Antisera generated against OspA served as a positive control for bactericidal activity. The One-Way ANOVA with post-hoc Tukey’s Multiple Comparison test was used to assess statistical significance (p<0.05). α-OspA served as the statistical reference.
Discussion
To inform efforts to generate optimized OspC based chimeritopes for use in LD vaccines, the Chv1, Chv2, and Chv3 proteins were designed, produced and purified. The Chv proteins are comprised of the immunodominant H5 and L5 linear epitopes derived from 9 different OspC type proteins (Figure 1). The epitopes are joined within a single contiguous polypeptide (41,57). The advantage of chimeritope vaccinogens is that through careful selection of epitopes, they can be constructed to specifically elicit productive Ab responses (i.e., Ab with protective potential) (41,51,57). Earlier studies demonstrated that the physical ordering of epitopes in a chimeritope can influence its molecular and immunological properties (51). The key differences between the Chv proteins investigated here are described in detail above and are illustrated in Figure 1. This study highlights the need to consider epitope ordering and the presence or absence of linkers. The data demonstrate that OspC based chimeritopes are immunogenic in multiple animal models and elicit broadly cross-reactive IgG with potent bactericidal activity.
The first step in these analyses was to determine if the organizational differences of the Chv proteins influences production in E. coli. While the Chv proteins were expressed at similar levels, their soluble-insoluble partitioning patterns differed. Chv1 and Chv3 localized with the insoluble fraction (presumably due to inclusion body formation), while Chv2 localized to the soluble fraction. To determine if structural differences influence partitioning properties, NMR analyses were conducted (Figure 2). Chv1, Chv2 and Chv3 were all found to exist as unstructured, extended random coils. Hence, the basis for differential partitioning upon induction remains unclear. Importantly, after purification and dialysis into PBS, all of the Chv proteins were soluble.
The immunogenicity of the Chv proteins was comparatively assessed in mice and rats. High-titer IgG responses developed against each protein (Figure 3). Anti-Chv1, 2 and 3 Ab bound to OspC types derived from a diverse panel of N. American and European LD isolates (Figures 4 and 6). This was anticipated as some of the L5 and H5 epitopes selected for inclusion were chosen because they share amino acid identity with L5 and H5 epitopes from other OspC types (45). It is noteworthy that OspC proteins of several B. burgdorferi, B. afzelii, B. garinii, B. bavariensis, and B. mayonii isolates were bound by anti-Chv Ab. B. mayonii is a recently identified LD spirochete species that was detected in I. scapularis ticks and cultured from the blood of two human LD patients from the upper Midwest (54). It is clear from these analyses that the Chv chimeritopes to are immunogenic and induce production of IgG that recognizes diverse OspC types.
The presentation of epitopes incorporated into a chimeritope may differ versus that of the structured native protein. As noted above, the Chv proteins are monomeric extended random coils. In contrast, native OspC is largely alpha-helical and its biologically active form is a homodimer (53). In addition to the linear epitopes identified in OspC (40,41,58), there is also evidence that some epitopes are conformationally defined or dependent on the formation of an OspC dimer (34,59). To determine if Ab elicited by vaccination with the Chv proteins can recognize the L5 and H5 epitopes as presented on the spirochetal cell surface, IFA analyses were conducted (Figure 5). Ab against all Chv proteins surface-labeled B. burgdorferi B31. As expected, due to the low expression levels of OspC during cultivation, labeling was weak and not all cells were bound by Ab. To mirror in vivo expression levels, the B31-HE-OspC strain, which constitutively produces OspC, was screened by IFA. Ab raised against each Chv protein strongly labeled this strain. This is consistent with the known OspC structures, which indicate surface exposure of the L5 and H5 epitopes on the membrane distal and membrane proximal regions of OspC, respectively (53,60). The recognition of the Ab binding targets by anti-Chv Ab in the context of an intact cell is a necessary property for vaccinal Ab to have potential bactericidal activity.
The demonstration of bactericidal activity of vaccinal Ab is an important in vitro correlate of protective efficacy (61–65). However, as alluded to above an inherent complication in measuring the bactericidal activity of Ab directed at OspC is the low and variable expression level of OspC in laboratory cultivated wild type strains (45,66). To address this, B. burgdorferi B31-HE-OspC served as the test strain in the bactericidal assays. High-level killing (defined as >75%) was observed with anti-Chv1, 2 and 3 antisera (Figure 7). Killing activity was determined to occur through an Ab mediated-complement dependent mechanism as occurs with recombinant full length OspC (41,52,67). It is important to note that some studies that have sought to quantify the bactericidal activity of OspC or OspC derived proteins did not determine OspC expression levels in the test strains (36,68–71). Lack of attention to this critical variable most certainly resulted in a significant underestimate of bactericidal activity.
An OspC chimeritope is one of two antigens in the commercially available, USDA approved canine LD vaccine, VANGUARD® crLyme (Zoetis) (72). While the information derived from this study informed the rationale design of the OspC chimeritope in VANGUARD® crLyme, it is important to note that the chimeritopes studied in this report are distinct from that used in the canine LD vaccine, which was specifically tailored for veterinary applications. The Chv proteins differ in that they consist of L5 and H5 epitopes from OspC types that are associated with invasive infections in humans (44,73,74). While there is overlap, some OspC types identified that have been identified in dogs have never been recovered from humans (42). In summary, in this study we demonstrate that the Chv chimeritopes induce high titer Ab that recognizes a highly diverse group of OspC type proteins and is bactericidal. The data support the hypothesis that chimeritopes can provide protection against diverse LD spirochete species and strains. The goal of producing OspC based chimeritopes and vaccine formulations for use as a human LD vaccine is now within reach.
Highlights.
Chv chimeritopes are recombinant proteins that contain linear epitopes from multiple variants of outer surface protein C.
Epitope ordering and the presence or absence of inter-epitope linkers influences antibody responses to OspC chimeritopes.
Immunization with the Chv proteins elicits the production of broadly cross-reactive antibody with potent bactericidal activity.
Funding:
This study was supported in part by awards, grants or donations from the NIH, NIAID (R01AI141801 and R56AI127801), Steven and Alexandra Cohen Foundation, The Christian Foundation, Zoetis, and the VCU VETAR (Value and Efficiency Teaching and Research) to RTM and by a grant to JAC and RTM from NIH, NIAID (R01 AI072683)
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflicts of interest:
RTM and CGE are inventors and receive royalties from a commercially available canine Lyme disease vaccine (VANGUARD®crLyme; produced and distributed by Zoetis) through the VCU Intellectual Property Foundation. RTM is a paid key opinion leader and speaker for ZoetisUS and Zoetis Canada. Virginia Commonwealth University receives royalties from sale of VANGUARD® crLyme.
Literature cited
- 1.Mead PS (2015) Infect Dis Clin North Am 29, 187–210 [DOI] [PubMed] [Google Scholar]
- 2.Benach JL, Bosler EM, Hanrahan JP, Coleman JL, Habicht GS, Bast TF, Cameron DJ, Ziegler JL, Barbour AG, Burgdorfer W, Edelman R, and Kaslow RA (1983) N Engl J Med 308, 740–742 [DOI] [PubMed] [Google Scholar]
- 3.Burgdorfer W, Barbour AG, Hayes SF, Benach JL, Grunwaldt E, and Davis JP (1982) Science 216, 1317–1319 [DOI] [PubMed] [Google Scholar]
- 4.Marconi RT, and Garon CF (1992) J Gen Microbiol 138, 533–536 [DOI] [PubMed] [Google Scholar]
- 5.Marconi RT, and Garon CF (1992) J Bacteriol 174, 241–244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bissett ML, and Hill W (1987) Journal of Clinical Microbiology 25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Oliver JH Jr., Owsley MR, Hutcheson HJ, James AM, Chen C, Irby WS, Dotson EM, and McLain DK (1993) J Med Entomol 30, 54–63 [DOI] [PubMed] [Google Scholar]
- 8.Radolf JD, Caimano MJ, Stevenson B, and Hu LT (2012) Nat Rev Microbiol 10, 87–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brisson D, Dykhuizen DE, and Ostfeld RS (2008) Proc Biol Sci 275, 227–235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Swei A, Briggs CJ, Lane RS, and Ostfeld RS (2012) Vector Borne Zoonotic Dis 12, 623–632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Heyman P, Cochez C, Hofhuis A, van der Giessen J, Sprong H, Porter SR, Losson B, Saegerman C, Donoso-Mantke O, Niedrig M, and Papa A (2018) Expert Rev Anti Infect Ther 8, 33–50 [DOI] [PubMed] [Google Scholar]
- 12.Nelson CA, Saha S, Kugeler KJ, Delorey MJ, Shankar MB, Hinckley AF, and Mead PS (2015) Emerg Infect Dis 21, 1625–1631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bowser NH, and Anderson NE (2018) Vet Sci 5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Little SE, Heise SR, Blagburn BL, Callister SM, and Mead PS (2010) Trends Parasitol 26, 213–218 [DOI] [PubMed] [Google Scholar]
- 15.Littman MP, Gerber B, Goldstein RE, Labato MA, Lappin MR, and Moore GE (2018) J Vet Intern Med 32, 887–903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Steere AC, Strle F, Wormser GP, Hu LT, Branda JA, Hovius JW, Li X, and Mead PS (2017) Nat Rev Dis Primers 2, 16090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Izac JR, and Marconi RT (2019) Vet Clin North Am Small Anim Pract [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Iyer R, Caimano MJ, Luthra A, Axline D Jr., Corona A, Iacobas DA, Radolf JD, and Schwartz I (2015) Mol Microbiol 95, 509–538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fraser CM, Casjens S, Huang WM, Sutton GG, Clayton R, Lathigra R, White O, Ketchum KA, Dodson R, Hickey EK, Gwinn M, Dougherty B, Tomb JF, Fleischmann RD, Richardson D, Peterson J, Kerlavage AR, Quackenbush J, Salzberg S, Hanson M, van Vugt R, Palmer N, Adams MD, Gocayne J, Weidman J, Utterback T, Watthey L, McDonald L, Artiach P, Bowman C, Garland S, Fuji C, Cotton MD, Horst K, Roberts K, Hatch B, Smith HO, and Venter JC (1997) Nature 390, 580–586 [DOI] [PubMed] [Google Scholar]
- 20.Ramamoorthy R, and Philipp MT (1998) Infect Immun 66, 5119–5124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Izac JR, Camire AC, Earnhart CG, Embers ME, Funk RA, Breitschwerdt EB, and Marconi RT (2019) Vaccine 37, 2401–2407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Marconi RT, and Earnhart C (2010) in Borrelia: Molecular biology, host interaction and pathogenesis (Samuels, D. S., and Radolf, J., eds), pp. 467–486, Caister Academic Press, Norfolk [Google Scholar]
- 23.Parenti D (1999) Conn Med 63, 570. [PubMed] [Google Scholar]
- 24.Zundorf I, and Dingermann T (2008) Pharm Unserer Zeit 37, 38–39 [DOI] [PubMed] [Google Scholar]
- 25.Schwan TG (2003) Biochem Soc Trans 31, 108–112 [DOI] [PubMed] [Google Scholar]
- 26.Schwan TG, and Piesman J (2000) J Clin Microbiol 38, 382–388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fikrig E, Telford SR 3rd, Barthold SW, Kantor FS, Spielman A, and Flavell RA (1992) Proc Natl Acad Sci U S A 89, 5418–5421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Brown EL, Kim JH, Reisenbichler ES, and Hook M (2005) Vaccine 23, 3687–3696 [DOI] [PubMed] [Google Scholar]
- 29.Scheiblhofer S, Weiss R, Durnberger H, Mostbock S, Breitenbach M, Livey I, and Thalhamer J (2003) Microbes and Infection 5, 939–946 [DOI] [PubMed] [Google Scholar]
- 30.Bauer Y, Hofmann H, Jahraus O, Mytilineos J, Simon MM, and Wallich R (2001) Eur J Immunol 31, 767–776 [DOI] [PubMed] [Google Scholar]
- 31.Probert WS, Crawford M, Cadiz RB, and LeFebvre RB (1997) J Infect Dis 175, 400–405 [DOI] [PubMed] [Google Scholar]
- 32.Probert WS, and LeFebvre RB (1994) Infection and Immunity 62, 1920–1926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Seinost G, Dykhuizen DE, Dattwyler RJ, Golde WT, Dunn JJ, Wang IN, Wormser GP, Schriefer ME, and Luft BJ (1999) Infect Immun 67, 3518–3524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gilmore RD Jr., and Mbow ML (1999) Infect Immun 67, 5463–5469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rousselle JC, Callister SM, Schell RF, Lovrich SD, Jobe DA, Marks JA, and Wieneke CA (1998) J Infect Dis 178, 733–741 [DOI] [PubMed] [Google Scholar]
- 36.Jobe DA, Lovrich SD, Schell RF, and Callister SM (2003) Clinical and Diagnostic Laboratory Immunology 10, 573–578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mathiesen MJ, Christiansen M, Hansen K, Holm A, Asbrink E, and Theisen M (1998) J Clin Microbiol 36, 3474–3479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Barthold SW, Feng S, Bockenstedt LK, Fikrig E, and Feen K (1997) Clin Infect Dis 25 Suppl 1, S9–17 [DOI] [PubMed] [Google Scholar]
- 39.Gilmore RD Jr., Kappel KJ, Dolan MC, Burkot TR, and Johnson BJ (1996) Infect Immun 64, 2234–2239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Buckles EL, Earnhart CG, and Marconi RT (2006) Clin Vaccine Immunol 13, 1162–1165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Earnhart CG, Buckles EL, and Marconi RT (2007) Vaccine 25, 466–480 [DOI] [PubMed] [Google Scholar]
- 42.Rhodes DV, Earnhart CG, Mather TN, Meeus PF, and Marconi RT (2013) Vet J 198, 412–418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Earnhart CG, and Marconi RT (2007) Clin Vaccine Immunol 14, 628–634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Earnhart CG, Buckles EL, Dumler JS, and Marconi RT (2005) Infect Immun 73, 7869–7877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Oliver LD Jr, Earnhart CG, Virgina-Rhodes D, Theisen M, and Marconi R (2016) The Veterinary Journal 218, 27–33 [DOI] [PubMed] [Google Scholar]
- 46.Yang CK, Kim JH, and Stallcup MR (2006) Mol Endocrinol 20, 3251–3262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sung SY, McDowell JV, and Marconi RT (2001) J Bacteriol 183, 5855–5861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sung SY, McDowell JV, Carlyon JA, and Marconi RT (2000) Infect Immun 68, 1319–1327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mallory KL, Miller DP, Oliver LD Jr., Freedman JC, Kostick-Dunn JL, Carlyon JA, Marion JD, Bell JK, and Marconi RT (2016) Pathog Dis 74 [DOI] [PubMed] [Google Scholar]
- 50.Earnhart CG, Leblanc DV, Alix KE, Desrosiers DC, Radolf JD, and Marconi RT (2010) Mol Microbiol 76, 393–408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Earnhart CG, and Marconi RT (2007) Vaccine 25, 3419–3427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Izac JR, Oliver LD Jr., Earnhart CG, and Marconi RT (2017) Vaccine 35, 3178–3185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Eicken C, Sharma V, Klabunde T, Owens RT, Pikas DS, Hook M, and Sacchettini JC (2001) J Biol Chem 276, 10010–10015 [DOI] [PubMed] [Google Scholar]
- 54.Pritt BS, Mead PS, Johnson DK, Neitzel DF, Respicio-Kingry LB, Davis JP, Schiffman E, Sloan LM, Schriefer ME, Replogle AJ, Paskewitz SM, Ray JA, Bjork J, Steward CR, Deedon A, Lee X, Kingry LC, Miller TK, Feist MA, Theel ES, Patel R, Irish CL, and Petersen JM (2016) Lancet Infect Dis [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Miller DP, Bell JK, McDowell JV, Conrad DH, Burgner JW, Heroux A, and Marconi RT (2012) J Biol Chem 287, 12715–12722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tilly K, Bestor A, Jewett MW, and Rosa P (2007) Infect. Immun 75, 1517–1519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Earnhart CG, and Marconi RT (2007) Hum Vaccin 3, 281–289 [DOI] [PubMed] [Google Scholar]
- 58.Baum E, Randall AZ, Zeller M, and Barbour AG (2016) PLoS One 8, e67445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Edmondson DG, Prabhakaran S, Norris SJ, Ullmann AJ, Piesman J, Dolan M, Probst C, Radzimski C, Stocker W, and Komorowski L (2017) Clin Vaccine Immunol 24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kumaran D, Eswaramoorthy S, Luft BJ, Koide S, Dunn JJ, Lawson CL, and Swaminathan S (2001) Embo J 20, 971–978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Cevenini R, Sambri V, Massaria F, La Placa M, Brocchi E, and De Simone F (1992) FEMS Microbiol Lett 69, 147–152 [DOI] [PubMed] [Google Scholar]
- 62.Neary JM, Kyungcheol Y, Karalus RJ, and Murphy TF (2001) Infection and Immunity 69, 773–778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Welsch JA, Moe GR, Rossi R, Adu-Bobie J, Rappuoli R, and Granoff DM (2003) J Infect Dis 188, 1730–1740 [DOI] [PubMed] [Google Scholar]
- 64.Welsch JA, Ram S, Koeberling O, and Granoff DM (2008) J Infect Dis 197, 1053–1061 [DOI] [PubMed] [Google Scholar]
- 65.Hsieh CL, Ptak CP, Tseng A, Suguiura IMS, McDonough SP, Sritrakul T, Li T, Lin YP, Gillilan RE, Oswald RE, and Chang YF (2017) Elife 6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Xiang X, Yang Y, Du J, Lin T, Chen T, Yang XF, and Lou Y (2017) Front Cell Infect Microbiol 7, 131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Nowling JM, and Philipp MT (1999) Infect Immun 67, 443–445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lovrich SD, Jobe DA, Schell RF, and Callister SM (2005) Clinical and diagnostic laboratory immunology 12, 746–751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lovrich SD, La Fleur RL, Jobe DA, Johnson JC, Asp KE, Schell RF, and Callister SM (2007) Clin Vaccine Immunol 14, 635–637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.LaFleur RL, Dant JC, Wasmoen TL, Callister SM, Jobe DA, Lovrich SD, Warner TF, Abdelmagid O, and Schell RF (2009) Clin Vaccine Immunol 16, 253–259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Jobe DA, Kowalski TJ, Bloemke M, Lovrich SD, and Callister SM (2011) Clin Vaccine Immunol 18, 1034–1037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ball EC (2016) Zoetis SAB-00193 [Google Scholar]
- 73.Golovchenko M, Sima R, Hajdusek O, Grubhoffer L, Oliver JH Jr., and Rudenko N (2014) Parasit Vectors 7, 538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Strle K, Jones KL, Drouin EE, Li X, and Steere AC (2011) Am J Pathol 178, 2726–2739 [DOI] [PMC free article] [PubMed] [Google Scholar]