

Abstract
Several reports support the idea that μ- and δ-opioid receptors (ORs) may exist as heterodimers in brain regions involved in pain signaling. The unique pharmacology of these heteromers may present a novel analgesic target. However, the role of μ-δ heteromers in sensory neurons involved in pain and opioid analgesia remains unclear, particularly during neuropathic pain. We examined the effects of spinal nerve injury on μ-δ heteromer expression in dorsal root ganglion (DRG) neurons and the effects of a μ-δ heteromer–targeting agonist, CYM51010, on neuropathic pain behavior in rats and mice. An L5 spinal nerve ligation (SNL) in rats significantly decreased μ-δ heteromer expression in L5 DRG, but increased heteromer levels in uninjured L4 DRG. Importantly, in SNL rats, subcutaneous (s.c.) injection of CYM51010 inhibited mechanical hypersensitivity in a dose-related manner (EC50: 1.09 mg/kg) and also reversed heat hyperalgesia and attenuated ongoing pain (2 mg/kg, s.c.). HEK-293T cells surface-labeled with μ- and δ-ORs internalized both receptors after exposure to CYM51010. In contrast, in cells transfected with μ-OR alone, CYM51010 was significantly less effective at inducing receptor internalization. Electrophysiologic studies showed that CYM51010 inhibited the C-component and windup phenomenon in spinal wide-dynamic range neurons of SNL rats. The pain inhibitory effects of CYM51010 persisted in morphine-tolerant rats, but was markedly attenuated in μ-OR knockout mice. Our studies show that spinal nerve injury may increase μ-δ heterodimerization in uninjured DRG neurons and that μ-δ heteromers may be a potential therapeutic target for relieving neuropathic pain, even under conditions of morphine tolerance.
Keywords: Opioid receptor, heteromers, windup, neuropathic pain, dorsal root ganglion
1. Introduction
Opioid receptors (ORs) are G-protein-coupled receptors (GPCRs) that are widely distributed along the pain pathways. In sensory neurons, μ-, δ-, and κ-ORs play an important role in the modulation of pain signals. The analgesic effect of morphine is mediated primarily by μ-OR activation [41]. In the peripheral nervous system, μ- and δ-ORs are present in dorsal root ganglion (DRG) neurons [25,34]. Though μ-ORs are expressed primarily on small-diameter peptidergic neurons in DRG [60], δ-OR mRNA has been found in both small-diameter peptidergic and non-peptidergic (IB4+) cells and large-diameter neurons, in large dense-core vesicles [43,58]. Noxious stimulus-induced Ca2+ influx causes exocytosis of large dense-core vesicles and translocation of δ-ORs to the plasma membrane [17].
Whether μ- and δ-ORs colocalize in primary sensory neurons has been a subject of debate. Using δ-OR tagged with enhanced green fluorescent protein, Scherrer et al. [50] found that δ-OR and μ-OR were expressed in distinct subsets of mouse DRG neurons. However, evidence from the early immunohistochemical studies was challenged because the δ-OR antibody lacked specificity [50]. More recent studies that have used in situ hybridization, single-cell PCR, electrophysiologic, and immunostaining techniques provide evidence for the coexistence of functional δ-ORs and μ-ORs in a subpopulation of small-diameter peptidergic DRG neurons [25,59]. Importantly, both δ-OR- and μ-OR-specific agonists inhibited depolarization-induced Ca2+ currents in the same DRG neurons [59]. A pharmacologic study provided further evidence that the two receptors colocalize on a functionally important population of IB4-/TrkA+ nociceptors [35]. Intriguingly, δ-OR agonists can enhance μ-OR–mediated analgesia in opioid-naïve animals. However, antagonists of δ-OR decrease tolerance to the analgesic effects of morphine [1,51], and animals lacking δ-ORs or the gene encoding preprotachykinin do not exhibit morphine tolerance [22,61].
GPCRs can form homomers or heteromers. Heteromers exhibit novel pharmacology, such as altered ligand binding properties, G-protein coupling, and trafficking, that differs from that of homomers [5,11,18]. Studies suggest that μ-δ heteromers have distinct pharmacologic and functional properties [14,15,44,49]. For example, whereas activation of the μ-ORs induces G-protein–mediated signaling, the same agonist acting on the μ-δ heteromer may evoke β-arrestin–mediated signaling [15,49]. Chronic morphine exposure upregulates μ-δ heteromers in brain regions involved in pain signaling (e.g., rostroventral medulla, nucleus accumbens), suggesting that the central μ-δ heteromer may play a role in opioid tolerance [27]. The analgesic effects of opioids in inflammatory and neuropathic pain are mediated, in part, by μ-ORs expressed on primary sensory neurons. However, the role of μ-δ heteromers in pain and peripheral opioid tolerance has not been well examined, in part because specific histologic and pharmacologic tools have been lacking. The development of a heteromer-selective antibody has helped researchers to characterize the μ-δ heteromer distribution in DRG neurons and explore heteromer-mediated signaling [27]. Using this antibody, we examined the effects of spinal nerve injury and repeated μ-OR agonist administration on μ-δ heteromer expression in DRG neurons. We further tested whether a μ-δ heteromer–targeting agonist, CYM51010, inhibits neuropathic pain-related behavior and conducted molecular biologic and electrophysiologic experiments to study the mechanisms of drug action.
2. Materials and Methods
2.1. Animals
Male Sprague-Dawley rats (200–350 g; Harlan Bioproducts for Science, Indianapolis, IN), male wild-type (WT) C57BL/6J mice, μ-OR knockout (KO) mice (B6.129S2-Oprm1tm1Kff/J, 20–35 g, Jackson Laboratory, Bar Harbor, ME), and δ-OR KO mice were housed under optimal laboratory conditions with a 12-hour light/dark cycle and free access to food and water. The generation of δ-OR KO mice was described by Rozenfeld et al. [48]. Briefly, δ-OR+/− embryos (Oprd1tm1Dgen, which lack the first exon of δ-OR) [61] were purchased from the Mutant Mouse Resource and Research Center (MMRRC, Jackson Laboratory). δ-OR KO mice were generated at the Mount Sinai Mouse Genetics Research Facility by implanting the embryos into C57BL/6J mice and carrying out two successive breeding cycles with WT mice from the same background (C57BL/6J). Mice were housed in groups of five until testing.
Animals were acclimatized to laboratory conditions before the tests. All behavioral experiments were carried out between 9:00 a.m. and 5:00 p.m. by an investigator blinded to the drug assignment. The experimental protocols were approved by the Animal Care and Use Committees of Johns Hopkins University and the Mount Sinai School of Medicine and complied with the National Institutes of Health Guide for the Use of Experimental Animals to ensure minimal animal use and discomfort.
2.2. Drugs
Dermorphin [D-Arg2, Lys4] (1–4) amide (DALDA), a hydrophilic, peripherally acting μ-OR agonist, was purchased from US Biologicals (Salem, MA) and CYM51010, a μ-δ heteromer–targeting agonist, was purchased from ChemBridge (San Diego, CA). μ-δ heteromer antibody was purchased from Kerafast (Boston, MA). Morphine sulfate (Hospira, Inc., Lake Forest, IL) was procured from the Johns Hopkins Hospital pharmacy. Other drugs were purchased from Sigma-Aldrich (St. Louis, MO) or Tocris Bioscience (Bristol, UK). Stock solutions were freshly prepared as instructed by the manufacturer.
2.3. Neuropathic pain model in rodents
L5 spinal nerve ligation (L5-SNL) surgery was used for the induction of neuropathic pain-related behavior in rats. The procedure was a modification of that described in our previous studies [24,55]. Briefly, rats were anesthetized with isoflurane (2%; Abbott Laboratories, Chicago, IL, USA) delivered through a nose cone. Using aseptic conditions, we incised the skin at the midline over the lumbar spine to expose the L5, L6, and upper sacral vertebrae. The left transverse process of the L6 vertebra was removed, and the left L5 spinal nerve was exposed and dissected from the underlying tissue. The left L5 spinal nerve was tightly ligated with a 6–0 silk suture and cut distal to the ligature. The overlying muscle layer was approximated with 4–0 silk suture and the skin closed with metal clips. The rats were returned to their cages after surgery, kept warm under a heat lamp, and monitored during recovery. In control rats, the spinal nerve was exposed but not ligated. Since most fibers in the sciatic nerve arise from the L3 and L4 spinal nerves in mice, ligation and transection of L4 spinal nerve was performed to induce neuropathic pain in mice.
2.4. Animal behavioral tests
Animals were acclimatized and habituated to the test environment. All procedures have been described in our previous studies and brief descriptions are provided below [40,56].
2.4.1. Nociceptive assay in mice
Drug-induced antinociception was evaluated by using the tail-flick test [16]. The heat source intensity of a tail-flick apparatus (IITC Life Science, Woodland Hills, CA) was set at an intensity that resulted in a basal tail-flick latency of 5–7 seconds for most animals. Cutoff latency was set at 20 seconds to minimize tissue damage. The tail-flick latency was recorded at 0, 5, 10, 15, 30, 60, 90, and 120 minutes after drug administration. The percent maximum possible effect (%MPE) was calculated for each mouse at each time point according to the following formula: %MPE = [(latency after drug − baseline latency) / (20 − baseline latency)] x 100. We also calculated the area under the %MPE vs. time curves (AUCs) and percent of control (i.e., the anti-Flag Ab-treated group was set as 100%).
CYM51010 (30 nmol) was dissolved in 6% DMSO and 5% Tween 80 in saline. μ-δ mAb and control IgG (anti-Flag Ab; 1 μg) were diluted in saline. Corresponding vehicle was used for the control group. Mice were intrathecally (i.t.) injected with control IgG (anti-Flag IgG; 1 μg) or μ-δ mAb (1 μg) 30 minutes before i.t. administration of CYM51010 (30 nmol). For i.t. administration, we used the direct lumbar puncture method [19,33] in awake, conscious mice. We covered the head and upper body of the mice with a soft cloth and gripped them firmly by the pelvic girdle (iliac crest). We then administered 5 μL of drug with a 50 μL Hamilton syringe (Hamilton Co, Reno, NV) attached to a 30-gauge, 1-inch sterile disposable needle, which was inserted into the i.t. space at the cauda equina region according to the method described by Hylden and Wilcox [33]. Puncture of the dura was indicated by a flick of the tail.
2.4.2. von Frey hair test for mechanical pain sensitivity
To assess mechanical sensitivity to punctuate mechanical stimuli in rats, we measured paw withdrawal threshold (PWT) to von Frey filaments (Stoelting, Wood Dale, IL). Each filament (0.40–13 g) was applied to the test area on the plantar surface of the hind paw for 4–6 seconds according to the up-down method [10,55]. The 1.8 g stimulus was applied first. If a positive response occurred, the next smaller von Frey hair was used, and if a negative response was observed, the next higher force was used. The test was continued until (1) the responses to five stimuli were assessed after the first crossing of the withdrawal threshold or (2) the upper/lower end of the von Frey hair set was reached before a positive/negative response had been obtained. Abrupt paw withdrawal, licking, and shaking were considered positive responses. The pattern of positive and negative responses was converted to a 50% threshold value using the formula provided by Dixon [10]. As PWTs are often at the cutoff values in naïve animals, MPE values for inhibiting mechanical hypersensitivity were calculated with the equation: %MPE = [1 – (Cutoff PWT – Post-drug PWT)/(Cutoff PWT – Pre-drug PWT)] x 100, where the calculated cutoff PWT = 21.72 g.
In mice, we assessed sensitivity to punctuate mechanical stimuli as the frequency of paw withdrawal to calibrated von Frey monofilaments (Stoelting) applied to the hind paw. Animals were acclimated for 30 minutes before being tested with two calibrated von Frey monofilaments (low force = 0.07 g; high force = 0.4 g). Each von Frey filament was applied perpendicularly to the mid-plantar area of each hind paw for ∼1 second, and the stimulation was repeated 10 times at a rate of 0.5–1 Hz (1–2-second intervals). If a withdrawal response was evoked, the next stimulus was applied after the mouse resettled. The occurrence of paw withdrawal in each of these 10 trials was expressed as a percent response frequency: Paw withdrawal frequency (PWF) = (number of paw withdrawals/10 trials) × 100%. For each test force, the left hind paw was stimulated first, followed by the right hind paw, with no less than 5 minutes between the tests.
2.4.3. Hargreaves test for heat pain sensitivity
To test for signs of heat hypersensitivity, we used the Hargreaves test, which measures paw-withdrawal latency (PWL) to radiant heat stimuli. Animals were placed under individual plastic boxes on a heated glass floor (30°C) and allowed to habituate for at least 30 minutes before testing. Radiant heat was applied to the plantar surface of each hind paw three times (3–5-minute interval) with a plantar stimulator analgesia meter (IITC model 390). A cutoff time of 20 seconds was used to prevent tissue damage. The average paw withdrawal latency of the three trials was used for data analysis.
2.4.4. Rota-rod test
The rota-rod test was conducted as described in previous studies [29,56]. Rats were trained on a rotating rod (Ugo Basile, Italy) that accelerated from 0 to 30 rpm in 180 seconds. Their performance on the rod was examined before (pre-drug) and 30 minutes after drug administration. The time (in seconds) that each animal remained on the accelerating rod without falling was recorded.
2.4.5. Conditioned place preference (CPP) test
Animals were habituated for 30 minutes per day in a three-chamber box and were given free access to all chambers. In this apparatus, the two larger chambers contained distinct visual and tactile cues (vertical stripes vs. triangular shapes and smooth vs. grooved floor). The third, smaller chamber, interposed between the two larger chambers, was devoid of overt spatial or tactile cues. On the pre-conditioning day, the animal was again free to explore all three chambers, and the behavior was video recorded for 15 minutes. The results were used to quantify any basal chamber preference or aversion in individual rats. Consistent with our previous study [27], animals that spent more than 80% (>720 seconds) or less than 20% (<120 seconds) of the total time in any given chamber were excluded from further testing. The next day, animals received vehicle treatment in the morning session without anesthesia and, after 10 minutes, were placed in one of the conditioning chambers for 45 minutes. Four hours later, the same animals received drug treatment and, after another 10-minute interval, were restricted to the opposite conditioning chamber for 45 minutes. On the post-conditioning test day, the animals received no injection and were placed in the same three-chamber box with free access to all chambers. The animal’s behavior was recorded for 15 minutes and used to analyze chamber preference or aversion. Pairing of drug or vehicle with a given chamber was counterbalanced between groups. An increase in post-conditioning time spent in the drug-paired chamber, as compared with pre-conditioning time in the same chamber, was interpreted as indicating CPP. In addition, difference scores were calculated by subtracting pre-conditioning time from post-conditioning time in the vehicle- and drug-paired chambers.
2.5. DALDA tolerance-inducing paradigm
Based on previous studies and our pilot experiments, we developed a paradigm in which repeated drug injections were used to induce acute opioid tolerance in rats during days 6 to 9 post-SNL (n = 3–4 per group), a time when the neuropathic mechanical and heat hypersensitivity reaches a stable maximum [31]. Our goal was to determine how tolerance to a peripherally acting μ-OR–preferring agonist affects the expression of μ-δ heteromers in both L4 and L5 DRGs of L5-SNL rats. The dose of DALDA used to induce tolerance was twice the ED50 (dose estimated to produce 50% MPE). We carried out the pre-tolerance test in the morning on day 5 post-SNL. We first tested the rats to obtain baseline PWTs to von Frey filaments. L4 and L5 DRGs were collected in 3 animals before induction of tolerance. In other animals, on day 7 after SNL, we administered the first tolerance-inducing dose of DALDA (10 mg/kg, s.c.) in the back of rats. Tolerance-inducing doses were repeated on day 8 (morning and afternoon) and day 9 (morning only) post-SNL. On day 10 post-SNL, we carried out the post-tolerance test by measuring the PWT and PWL after administering DALDA at the same doses used in the pre-tolerance test. DRGs were collected from the rats after establishing the development of tolerance to DALDA. In SNL rats, levels of μ-δ heteromer in L4 and L5 DRGs were measured by ELISA as described in section 2.8.
2.6. Morphine tolerance-inducing paradigm in SNL rats
Before the SNL procedure in rats, we measured pre-morphine tolerance PWT baselines. After the SNL procedure, but before inducing morphine tolerance, we tested the effect of CYM51010 at 0.2 and 2 mg/kg (s.c.). Morphine tolerance was then induced by twice daily injections of morphine (10 mg/kg, s.c) for 8 consecutive days. We conducted post-morphine PWT testing daily to monitor the development of complete tolerance in rats. After the development of tolerance, we again tested the effect of CYM51010 at 0.2 and 2 mg/kg (s.c.).
2.7. Tissue immunofluorescence
After the behavior tests, all rats were anesthetized and perfused with 4% paraformaldehyde. L4 and L5 DRGs from sham control and nerve-injured rats were harvested, post-fixed, and dehydrated. The L5 DRG was identified based on its location in the lumbosacral (L5-S1) intervertebral foramen. All DRGs were cryosectioned at 20 μm and stored at −80°C until use. DRG sections were blocked for 1 hour in 10% goat serum and then incubated with mouse anti-μ-δ dimer and guinea pig anti-μ-OR (1:1000, EMD Millipore, Burlington, MA) overnight, followed by Cy3-conjugated goat anti-mouse IgG (1:200, Jackson ImmunoResearch, West Grove, PA) and Cy2-conjugated goat anti-guinea pig IgG (1:200, Jackson ImmunoResearch). All images were viewed and captured on an epifluorescence microscope (Nikon, Tokyo, Japan) under appropriate filter for Cy3 (excitation 550 nm; emission 570 nm) or Cy2 (excitation 492 nm; emission 510–520 nm).. For quantitative analysis, 2 sections from each DRG were chosen randomly for each rat and the data were collected from at least three animals in each group. To count the total number of DRG neurons in a section, cells were identified by morphology with a clearly defined, dark, condensed nucleolus. Positively stained cells were identified as neurons with a clear cell somata. The proportion of heterodimer-positive cells was determined as a percent of the total number of neurons counted in a section.
2.8. ELISA
Using a cryostat, we cut 10-μm-thick DRG sections, placed them on Superfrost Plus microscope slides (Thermo-Fisher Scientific, Waltham, MA), and circled them immediately with an ImmEdge PAP pen (Vector Laboratories, Inc., Burlingame, CA) to form a waterproof barrier. Tissue sections were incubated with 200 μL of 3% bovine serum albumin (BSA, Sigma) in phosphate-buffered saline (PBS) for 1 hour at 37°C. BSA solution was discarded, and 100 μL of μ-δ heteromer-selective monoclonal antibody (1:1000 dilution) was added to the wells. We added 100 μL of PBS to all wells to determine nonspecific binding and incubated the plates for 16–18 hours at 4°C. All wells were thoroughly washed four times (5 minutes each wash) at room temperature with 200 μL of 1% BSA in PBS. DRG sections were further incubated with 100 μL of peroxidase-conjugated secondary antibody (1:1000 in PBS containing 1% BSA) for 1 hour at 37°C. Wells were again washed four times (5 minutes each) at room temperature with 200 μL of 1% BSA in PBS and incubated with 100 μL of substrate (0.5 mg/10 mL orthophenylenediamine in 0.15 M citrate-phosphate buffer, pH 5, containing 10 μL of H2O2) for 10 minutes at room temperature. The reaction was terminated by adding 50 μL of 5 N H2SO4. The supernatant was transferred to another 96-well plate, and absorbance was read at 490 nm in an ELISA reader (Bio-Rad model 550).
2.9. Cell culture and transfection
HEK-293T cells were cultured in growth medium that consisted of 90% DMEM, 10% fetal bovine serum, 100 U/mL penicillin, and 100 μg/mL streptomycin (Invitrogen) at 37°C in the presence of 95% O2 and 5% CO2. Lipofectamine 2000 (Thermo-Fisher Scientific) was used to transfect the cells with 2–4 μg plasmid/35 mm dish or 0.1–0.2 μg plasmid/96-well microplate well. The cells were cultured for 1–2 days before experiments. We routinely assessed the cells with DAPI staining to confirm the absence of mycoplasma.
2.10. Cell immunohistochemistry
HEK-293T cells co-transfected with HA-μ-OR and Myc-δ-OR expression plasmids were incubated with mouse anti-Myc antibody (M4439, Sigma-Aldrich, 1:100) and rabbit anti-hemagglutinin (HA) antibody (631207, Clontech [Takara Bio USA, Mountain View, CA] 1:100) for 30 minutes at 37°C. Cells were then treated with 5 μM DAMGO, DALDA, or CYM51010 for 30 or 90 minutes. Cells were fixed with 4% paraformaldehyde and 0.2% picric acid and then immunostained. For secondary antibodies, we used Alexa 568-conjugated goat antibody to rabbit (Thermo-Fisher Scientific) and Alexa 488-conjugated goat antibody to mouse (A10667, Molecular Probes, Eugene, OR). All secondary antibodies were diluted 1:100 in blocking solution.
2.11. Electrophysiologic studies
Animals were anesthetized with intraperitoneal (i.p.) pentobarbital (45–50 mg/kg) and mechanically ventilated via a tracheotomy tube. The lumbar enlargement of the spinal cord was exposed by a laminectomy between vertebral levels T12 and L1. Recordings from wide-dynamic range (WDR) neurons were obtained via parylene-coated tungsten microelectrodes (impedance 1–3 MΩ, Frederick Haer Company, Brunswick, ME, USA) placed at a depth of 500–1000 μm from the dorsal surface of L4 spinal cord level. Only WDR neurons with defined receptive fields in the plantar region of the hind paw were studied. The analog signals were amplified, filtered, displayed on an oscilloscope, and collected with a real-time computer-based data acquisition and processing system (CED 1401, Cambridge Electronic Design, Cambridge, UK). The WDR neuronal responses were evoked by graded peripheral intracutaneous electrical stimulation (0.1–10 mA, 2 ms). A train of 0.5-Hz electrical stimulation (16 pulses, 0.2 ms, 1.5 x C-component threshold) was used to induce a windup response.
2.12. Statistical analysis
The number of animals we used in each experiment was based on our experience with similar studies. We randomized animals to the different treatment groups and blinded the experimenter to drug treatment to reduce selection and observation bias. STATISTICA 6.0 software (StatSoft, Inc., Tulsa, OK) and GraphPad software Prism 6.0 (GrpahPad, San Diego, CA) were used to conduct all statistical analyses. For immunofluorescence, we compared the percentages of heteromer-positive cells in L4 and L5 DRGs between sham and SNL groups by one-way ANOVA followed by Tukey post hoc test. For behavioral tests, we determined PWT by converting the pattern of positive and negative responses to the von Frey filament stimulation to a 50% threshold value with the formula provided by Dixon [11]. For behavioral tests, we used unpaired t-test for comparison between two groups and one-way repeated-measures ANOVA to compare data between different time points in each group. Two-way ANOVA was used to compare data between different groups. When ANOVA showed a significant difference, pairwise comparisons between means were tested by the Tukey honestly significant difference post hoc test. Bonferroni correction was applied for multiple comparisons. In electrophysiology studies, we compared the number of action potentials evoked by test stimuli under pre-drug and post-drug conditions. For analysis of windup, we plotted the C-component of WDR neurons evoked by each stimulus against the stimulation number in a train of 16 stimuli. We compared the A- and C-components produced by graded electrical stimuli and the total C-component elicited in response to windup stimulation between pre- and post-drug conditions using two-way repeated measures ANOVA. All tests were two-tailed, and p<0.05 was considered statistically significant. The methods for statistical comparisons in each study are given in the figure legends. All values shown in the figures are mean ± SEM.
3. Results
3.1. L5-SNL induced an increase in μ-δ heteromers in uninjured L4 DRG neurons
More than 90% of the μ-OR–positive neurons were also μ-δ heteromer–positive, with only 7.7±1.2% of μ-OR–positive cells being heteromer-negative. By double labeling for μ-ORs and μ-δ heteromers, we found that L5-SNL induced a significant increase in μ-δ heteromer-positive neurons in L4 DRG, as compared to that in the control group (Fig. 1A, B). In contrast, and consistent with earlier reports of a reduction in μ-ORs at the level of spinal nerve injury, the number of μ-δ heteromer-positive neurons was decreased at L5 DRG (Fig. 1A, B) [39]. We used ELISA with a μ-δ heteromer-specific antibody to further confirm the immunohistochemical findings. Consistent with the histochemical results, we found that μ-δ heteromer levels increased in L4 DRG of L5-SNL rats (144.5 ± 14.5%), but decreased significantly in L5 DRG (31.7 ± 10.6%), as compared to that in the control group (Fig. 1C).
Figure 1. Changes to μ-δ heteromer expression in lumbar DRGs after nerve injury and tolerance to a peripherally restricted opioid.
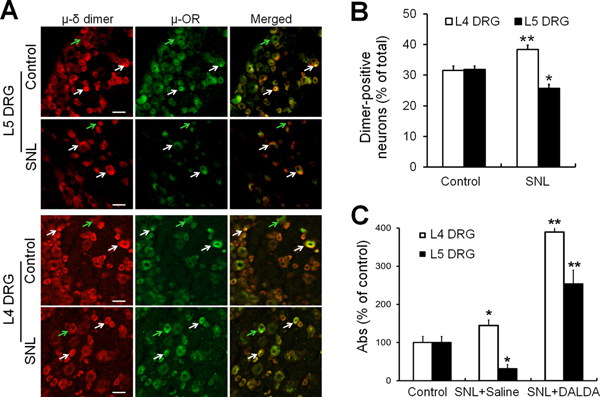
(A) Representative images of μ-OR and μ-δ heteromer immunoreactivity staining in L5 and L4 DRG of sham control and L5-SNL rats. White arrows indicate examples of double-labeled cells that are both μ-OR–positive and μ-δ heteromer–positive, whereas green arrows indicate single-labeled cells that are either primarily μ-OR or μ-δ heteromer–positive, but not both. Scale bar: 50 μm. (B) Quantified percentage of μ-δ heteromer–positive neurons in L4 and L5 DRGs of SNL rats and sham control rats (n = 3–4/group). The proportion of dimer-positive neurons is expressed as a percent of all neurons counted in sections of the DRG. The proportion of μ-δ heteromer–positive cells increased in L4 DRG of SNL rats and decreased in L5 DRG. (C) ELISA studies showed a similar increase in μ-δ heteromers in L4 DRG and decrease in L5 DRG after L5-SNL, as compared to that in the control group (n=3–4/group). In SNL rats that developed tolerance to the peripherally restricted opioid DALDA after repeated injections (10 mg/kg, b.i.d., s.c), levels of μ-δ heteromer increased in both uninjured (L4) and injured (L5) DRG, n=5–8/group. DRG, dorsal root ganglion; OR, opioid receptor. *p < 0.05, **p < 0.01 versus control, two-way ANOVA followed by Tukey’s post hoc test. Data are mean ± SEM.
3.2. Repeated treatment with a peripherally acting μ-OR agonist increased μ-δ heteromers in SNL rats
SNL rats that receive repeated administration of DALDA (10 mg/kg, s.c.), a peripherally acting μ-OR agonist, develop tolerance to inhibition of mechanical hypersensitivity [55]. We investigated the effect of tolerance to DALDA on μ-δ heteromers in both L4 and L5 DRGs of L5-SNL rats. ELISA results showed further increases in μ-δ heteromers in both uninjured (L4) and injured (L5) DRG neurons in these rats (Fig. 1C), suggesting possible involvement of heteromers in the development of tolerance to DALDA’s inhibition of mechanical hypersensitivity.
3.3. CYM51010 inhibited mechanical and heat hypersensitivities in SNL rats
At a dose of 2 mg/kg, the μ-δ heteromer agonist CYM51010 significantly attenuated SNL-induced mechanical and heat hypersensitivities (Fig. 2A, B). CYM51010–induced inhibition of mechanical hypersensitivity lasted for approximately 2 hours, whereas attenuation of heat hypersensitivity persisted for only 60 minutes (Fig. 2A, B).
Figure 2. Effects of μ-δ heteromer–targeting agonist CYM51010 on mechanical and heat hypersensitivities in SNL rats.
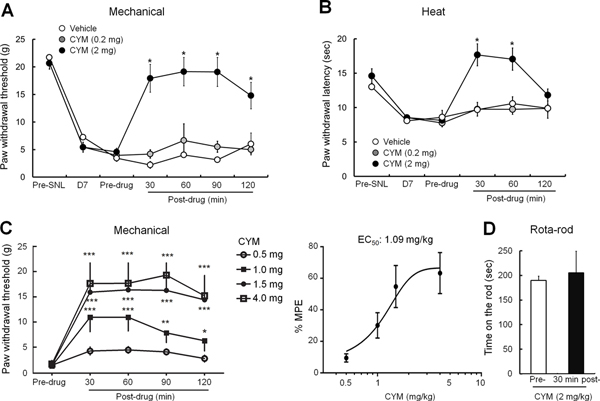
(A) Changes in ipsilateral paw withdrawal thresholds of SNL rats in response to mechanical stimuli after systemic administration of CYM51010 (0.2, 2 mg/kg, s.c.) or vehicle. (B) Effects of CYM51010 (0.2, 2 mg/kg, s.c.) or vehicle on ipsilateral paw withdrawal latency to heat stimulation in SNL rats. (C) Left: Time courses of changes in ipsilateral paw withdrawal thresholds to mechanical stimuli in SNL rats after systemic administration of CYM51010 (0.5, 1, 1.5, and 4 mg/kg, s.c.). Right: Dose-response function of CYM51010 was established based on the averaged %MPE at 30, 60, 90, and 120 minutes post-injection. EC50 (1.09 mg/kg; 95% CI, 0.71–1.46 mg/kg) was calculated by fitting a three-parameter logistic dose-response curve to estimated means resulting from the mixed linear model. n = 5–10/group. *p < 0.05, **p < 0.01, ***p < 0.001 versus pre-drug. (D) In the rota-rod test, the time that nerve-injured rats remained on the accelerating rod at 30 minutes after CYM51010 injection (2 mg/kg, s.c.) was unchanged from baseline. n = 5–6/group. *p < 0.05 versus vehicle, two-way mixed model ANOVA with Bonferroni correction for multiple comparisons. Data are mean ± SEM.
In a different group of rats at 2–3 weeks post-SNL, we examined the dose-response function for CYM51010 to inhibit mechanical hypersensitivity. At doses of 1, 1.5, and 4 mg/kg, CYM51010 significantly increased the ipsilateral PWT from pre-drug baseline from 30 to 120 minutes after injection (Fig. 2C). The EC50 value (1.09 mg/kg, 95% CI, 0.71–1.46 mg/kg) was calculated based on the averaged %MPEs from 30 to 120 minutes post-injection (Fig. 2C). In the rota-rod test, the fall time of rats treated with systemic CYM51010 (2 mg/kg, s.c.) was not significantly decreased at 30 minutes after injection, as compared to that at pre-drug baseline (Fig. 2D, n=5). Therefore, CYM51010 did not affect motor function.
3.4. Pain inhibitory effects of CYM51010 in SNL rats persisted after morphine tolerance
Rats with L5-SNL that were administered morphine (10 mg/kg, s.c.) twice a day for 8 days gradually developed tolerance to its analgesic effect. The %MPE in these morphine-tolerant rats was significantly decreased from the pre-tolerance value (Fig. 3A). Subcutaneous injection of CYM51010 (2 mg/kg) attenuated mechanical (Fig. 3B) and heat (Fig. 3C) hypersensitivities in SNL rats both before and after the development of morphine tolerance. In addition, the lower dose of CYM51010 (0.2 mg/kg, s.c.), which had not attenuated mechanical and heat hypersensitivities in SNL rats before the onset of morphine tolerance, became effective in attenuating thermal hypersensitivity after the development of morphine tolerance.
Figure 3. Effects of CYM51010 on mechanical and heat hypersensitivities in SNL rats before and after the induction of morphine tolerance.
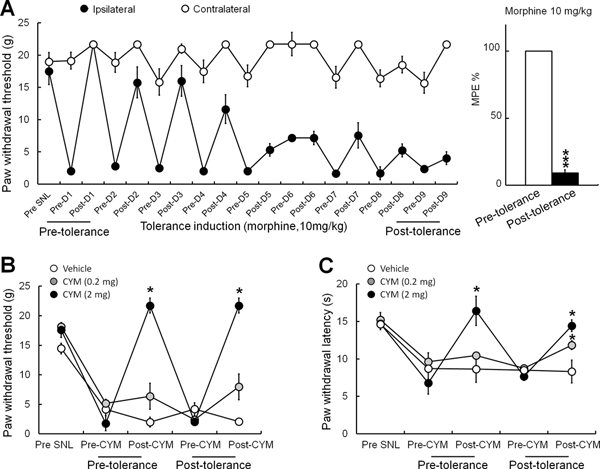
(A) Left: Time course of changes in ipsilateral and contralateral paw withdrawal thresholds of SNL rats in response to mechanical stimuli after morphine treatment. Morphine analgesic tolerance was gradually induced by twice-daily subcutaneous injections of morphine (10 mg/kg) for 8 consecutive days. Right: %MPEs of morphine (10 mg/kg, s.c.) at pre- and post-tolerance conditions. ***p < 0.001. (B, C) Subcutaneous injection of CYM51010 (2 mg/kg) significantly attenuated mechanical (B) and heat (C) hypersensitivities in SNL rats both before and after morphine tolerance. The lower dose of CYM51010 (0.2 mg/kg, s.c.) that did not attenuate the pain behavior in control rats did reduce thermal hyperalgesia in morphine-tolerant animals. n = 5–6/group. *p < 0.05 versus vehicle, two-way mixed model ANOVA. Data are mean ± SEM.
3.5. CYM51010 inhibited ongoing pain in SNL rats
We used the CPP test to assess drug-elicited relief from ongoing pain. SNL rats showed a preference for the chamber paired with CYM51010 (2 mg/kg, s.c.) over the vehicle-paired chamber, as evident from the significantly increased difference score in the CYM51010–paired chamber (Fig. 4A). Naïve animals did not show a preference for either chamber (Fig. 4B). These findings suggest that CYM51010 selectively inhibited ongoing pain in nerve-injured rats. We also tested the effect of 0.2 mg/kg CYM51010 on ongoing pain in SNL rats with and without morphine tolerance. At this lower dose, CYM51010 did not produce CPP in opioid-naïve SNL rats (Fig. 4C), but it did produce CPP in SNL rats that had developed morphine tolerance (Fig. 4D).
Figure 4. Systemic administration of CYM51010 induces conditioned place preference in SNL rats.
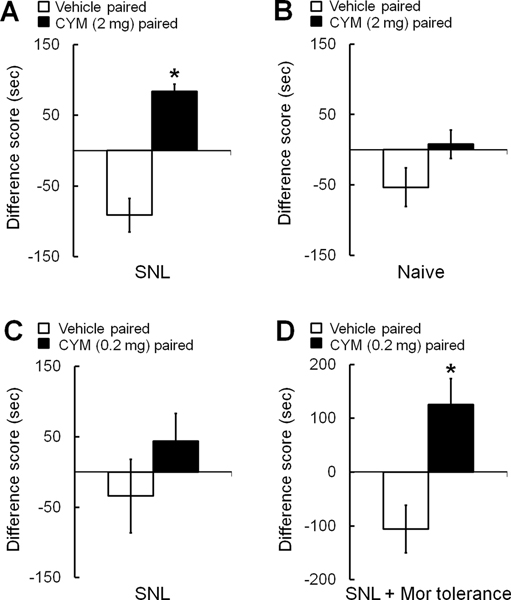
(A) The difference score of SNL rats for time spent in the chamber paired with CYM51010 (2 mg/kg, s.c.) was significantly higher than that for the vehicle-paired chamber during the post-conditioning period, indicating a preference for the chamber paired with CYM51010. (B) However, naïve rats did not exhibit a preference for the CYM51010–paired chamber. (C) A lower dose of the CYM51010 (0.2 mg/kg, s.c.) did not induce a significant preference for the drug-paired chamber in SNL rats. (D) However, 0.2 mg/kg s.c. CYM51010 did induce preference for the CYM51010–paired chamber in SNL rats with morphine tolerance. n = 6–8/group. Two-way repeated measures ANOVA with Bonferroni post hoc test, *p < 0.05 versus vehicle-paired chamber. Data are mean ± SEM.
3.6. Activation of μ-ORs led to co-internalization of surface δ-ORs
HEK-293T cells were co-transfected with HA-μ-OR and Myc-δ-OR and subsequently treated with 5 μM μ-OR agonist DAMGO, 5 μM μ-OR agonist DALDA, or 5 μM μ-δ heteromer–targeting agonist CYM51010. Immunohistochemistry was used to assess receptor internalization at 45 minutes after drug treatment (Fig. 5A). We observed significant co-internalization of HA-μ-OR and Myc-δ-OR in cells after exposure to DAMGO, DALDA, or CYM51010 (Fig. 5B). These findings suggest that both μ-δ heteromer agonist and μ-OR agonist can induce co-internalization of surface μ-ORs and δ-ORs.
Figure 5. Activation of μ-OR leads to co-internalization of surface δ-OR.
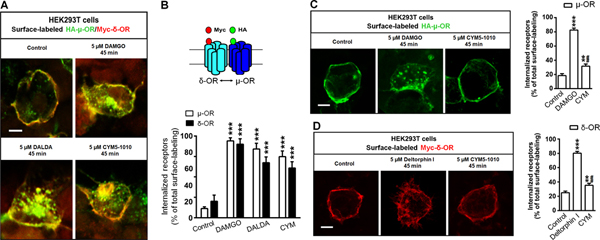
(A) Representative images showing co-internalization of HA-μ-OR and Myc-δ-OR in HEK-293T cells after 45 minutes of exposure to DAMGO (5 μM), DALDA (5 μM), or CYM51010 (5 μM). (B) Upper: Diagram depicts HA-μ-OR (green) and Myc-δ-OR (red) at the cell surface. Lower: Quantification of μ-OR and δ-OR internalization at 45 minutes after drug treatment. (C) Representative images and quantification of μ-OR internalization in HEK-293T cells transfected with HA-μ-OR alone and exposed for 45 minutes to DAMGO (5 μM) or CYM51010 (5 μM). (D) Representative images and quantification of δ-OR internalization in HEK-293T cells transfected with Myc-δ-OR alone and exposed for 45 minutes to deltorphin I (5 μM) or CYM51010 (5 μM). Scale bar: 10 μm. n = 15–17/group. One-way ANOVA and Bonferroni post hoc test. **p < 0.01, ***p < 0.001 versus control; ##p < 0.01 versus DAMGO and deltorphin I in C and D, respectively. Data are mean ± SEM.
We next examined whether CYM51010 specifically targets μ-δ heteromers to induce receptor internalization. In HEK-293T cells transfected with HA-μ-OR alone, CYM51010-induced HA-μ-OR internalization was significantly less than that resulting from 5 μM DAMGO (Fig. 5C). In HEK-293T cells transfected with Myc-δ-OR alone, 5 μM δ-OR agonist deltorphin I induced significant internalization of Myc-δ-OR (Fig. 5D). The level of Myc-δ-OR internalization induced by CYM51010 was significantly less than that by deltorphin I. Nevertheless, CYM51010 induced significant Myc-δ-OR internalization, as compared to the control. These findings suggest that CYM51010 may also activate some δ-ORs to induce receptor internalization.
3.7. CYM51010 inhibited C-component and windup responses of WDR neurons
Next we investigated whether CYM51010 affects the evoked response of spinal WDR neurons in SNL rats. Based on different response latencies produced by intracutaneous electrical stimulation of the hind paw, the Aα/β (0–25 ms)-, Aδ (25–100 ms)-, and C (100–500 ms)-components of WDR neuronal responses can be distinguished. The Aα/β-component is mediated by myelinated afferent fibers and represents the non-noxious peripheral signal. The Aδ- and C-components are induced in the thinly myelinated and unmyelinated afferent fibers and represent the response to noxious peripheral signals. At 30 minutes after injection of CYM51010 (2 mg/kg, s.c.), we observed no significant change in the stimulus-response function or the total number of action potentials in the Aα/β-component evoked by graded electrical stimuli with increasing amplitudes (Fig. 6A). However, the number of action potentials in the Aδ-component evoked by 4, 5, and 10 mA high-intensity electrical stimuli significantly decreased at 30 minutes after CYM51010 injection (Fig. 6B). Importantly, the C-component responses to 4, 5, and 10 mA electrical stimuli, as well as the total number of action potentials in the C-component, were also significantly decreased at 30 minutes after CYM51010 application (Fig. 6C). Administration of vehicle did not change the Aα/β-, Aδ-, or C-component (Supplemental Fig. 1).
Figure 6. Effect of systemic administration of CYM51010 on the excitability of spinal wide-dynamic range (WDR) neurons in SNL rats.
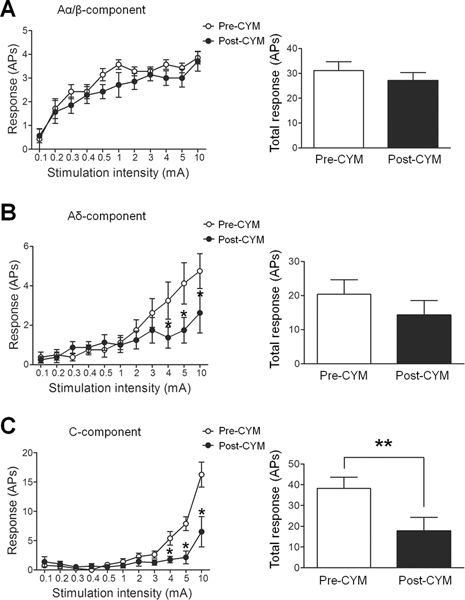
(A) Different components of the WDR neuronal responses to an intracutaneous electrical stimulation at the hind paw were distinguished based on the response latency (Aα/β: 0–25 ms; Aδ: 25–100 ms; C: 100–500 ms). Left: The stimulus-response functions of Aα/β-component to graded electrical stimulation before and 30 minutes after CYM51010 (2 mg/kg, s.c.) injection. Right: The total number of action potentials (APs) in the Aα/β-component produced in response to graded electrical stimuli. (B) Left: The stimulus-response functions of Aδ-component before and after CYM51010. Right: The total Aδ-component produced in response to graded electrical stimuli. (C) Left: The stimulus-response functions of C-component before and after CYM51010. Right: The total C-component produced in response to graded electrical stimuli. n=8/group; *p < 0.05, **p < 0.01 versus pre-CYM51010. Two-way repeated measures ANOVA with Bonferroni post-test was used to compare the responses to graded electrical stimuli between pre- and post-CYM51010 in each group. Student’s t-test was used to compare total responses between pre- and post-CYM51010. Data are mean ± SEM.
Repetitive electrical stimulation (0.5 Hz) with 1.5 times the C-fiber threshold induced windup in the C-component of the WDR neuronal response (Fig. 7), which represents the short-term sensitization of WDR neurons. Notably, at 30 minutes after systemic administration of CYM51010 (2 mg/kg, s.c.), both the windup function and total windup response were attenuated from those observed before drug treatment (Fig. 7A, B). Vehicle treatment did not affect the windup response (Fig. 7C).
Figure 7. Effect of systemic administration of CYM51010 on windup response of wide-dynamic range (WDR) neurons in SNL rats.
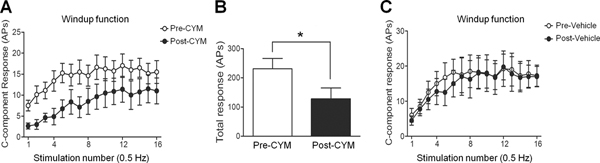
(A) Windup of the C-component response was elicited by applying 16 repetitive electrical stimuli to the hind paw (1.5x C-component activation threshold, 0.2 ms) at 0.5 Hz frequency. Windup function in WDR neurons was examined before and at 30 minutes after CYM51010 (2 mg/kg, s.c.) treatment. (B) The total number of C-components evoked by windup-inducing stimulation was decreased after CYM51010 treatment. (C) Windup functions were comparable before and after vehicle treatment. n=8/group; *p < 0.05 versus pre-CYM51010. Two-way repeated measures ANOVA with Bonferroni post-test was used to compare the responses to graded electrical stimuli between pre- and post-CYM51010 in each group. Student’s t-test was used to compare total responses between pre- and post-CYM51010. Data are mean ± SEM. APs, action potentials.
3.8. Effect of μ-δ heteromer–selective antibody on the antinociceptive effect of CYM51010 in WT and δ-OR KO mice
We examined the involvement of μ-δ heteromer in CYM51010-mediated antinociception by comparing the effects of a μ-δ heteromer–selective antibody in WT and δ-OR KO mice. Intrathecal injection of CYM51010 induced significant antinociception in WT mice (Fig. 8A). Importantly, CYM51010-induced antinociception was significantly decreased in WT mice, but not in δ-OR KO mice, that were pretreated with an i.t. injection of the μ-δ heteromer–selective antibody (1 μg, 30 min, Fig. 8A, B). This finding suggests that pain inhibition by CYM51010 is at least in part mediated by μ-δ heteromers. We observed a trend toward enhanced antinociceptive effect of CYM51010 in δ-OR knockout mice, when compared to WT mice, at 30 minutes in the tail-flick test, although this effect was not statistically significant (Tukey’s multiple comparison test). The antinociceptive effect of CYM51010 in the presence of μ-δ heteromer–selective antibody in WT and δ-OR KO mice may be mediated via μ-ORs, as we have demonstrated previously [19].
Figure 8. Antinociceptive effects of CYM51010 in wild-type (WT) and δ-OR knockout (KO) mice.
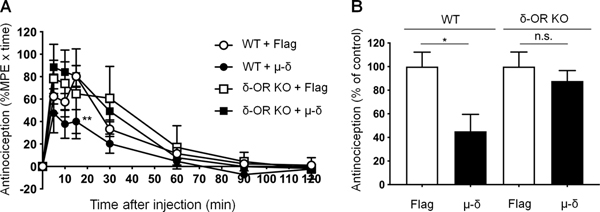
(A) Time course of CYM51010-induced antinociception in WT and δ-OR KO mice. Mice received an intrathecal (i.t.) injection of control IgG (anti-Flag IgG; 1 μg) or μ-δ heteromer–selective antibody (1 μg) 30 minutes before CYM51010 (30 nmol, i.t.) administration. Coadministration of μ-δ heteromer–selective antibody partially but significantly blocked i.t. CYM51010-mediated antinociception in WT mice but not in δ-OR KO mice. Antinociceptive activity was measured as %MPE x time, as described in Methods. (B) Quantification of CYM51010-induced antinociception. Data are expressed as mean ± SEM (n=6–9/group). *p < 0.05, **p < 0.01 versus Flag control as determined by ANOVA followed by multiple comparison test (Tukey’s multiple comparison test) or unpaired t test. n.s., not significant.
3.9. CYM51010 inhibited mechanical and heat hypersensitivities in WT mice but not μ-OR KO mice after SNL
The pain inhibitory effects of CYM51010 were further tested in WT and μ-OR KO mice at 2–3 weeks after SNL (n=6–8). The ipsilateral PWFs to both low force (0.07 g) and high force (0.4 g) punctuate mechanical stimulation were significantly decreased from pre-drug values at 60 minutes after CYM51010 (2 mg/kg, s.c.) injection in WT mice with SNL (Fig. 9A, B). The decrease in ipsilateral PWLs to radiant heat stimulation in SNL mice was significantly reversed from pre-drug values at 60 minutes after CYM51010 injection (Fig. 9C). These results suggest that CYM51010 significantly attenuates both mechanical (Fig. 9A, B) and heat (Fig. 9C) hypersensitivities in the ipsilateral hind paws of WT SNL mice, which is consistent with our findings in SNL rats. Importantly, the inhibitory effects of CYM51010 for both mechanical and heat hypersensitivity were markedly attenuated in μ-OR KO mice (Fig. 9A–C). CYM51010 also significantly increased PWLs of the contralateral hind paw from pre-drug baseline at 60 minutes in WT but not μ-OR KO mice.
Figure 9. Effects of CYM51010 on mechanical and heat hypersensitivities in wild-type (WT) and μ-OR knockout (KO) mice after nerve injury.
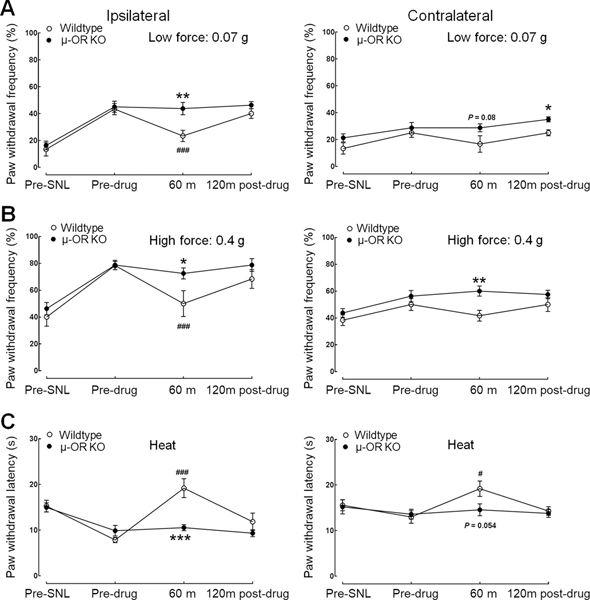
(A, B) Changes in ipsilateral (left panel) and contralateral (right panel) paw withdrawal frequencies to low force (0.07 g, A) and high force (0.4 g, B) mechanical stimuli after systemic administration of CYM51010 (2 mg/kg, s.c.) in WT and μ-OR KO mice with SNL. (C) Effects of CYM51010 (2 mg/kg) on the ipsilateral (left panel) and contralateral (right panel) paw withdrawal latency to heat stimulation in WT and μ-OR KO mice with SNL. n = 6–8/group. *p < 0.05, **p < 0.01, ***p < 0.001 versus WT; #p < 0.05, ###p < 0.001 versus pre-drug baseline. Two-way mixed model ANOVA. Data are mean ± SEM.
4. DISCUSSION
Opioids continue to have an important role in the management of severe acute pain and are currently recommended as third-line drugs for chronic neuropathic pain when anticonvulsant and antidepressant drugs do not provide adequate relief [8,13]. However, the use of opioids in patients with chronic pain is often limited by adverse effects, such as sedation, dizziness, cognitive dysfunction, and respiratory depression [42,47]. Physiologic and pharmacologic studies indicate that the μ-OR and δ-OR may modulate each other’s responses [1,7,32,35,47]. In addition, chronic morphine treatment has been reported to upregulate the δ-OR [7,44], which causes functional changes in the μ-OR and decreased responsiveness to morphine [22]. Reports that μ- and δ-ORs are co-expressed in subpopulations of DRG neurons [11,25,34,58] and the axonal terminals of the superficial dorsal horn [3] support the possibility of physical and functional interactions between the two receptors. Mounting evidence for the heterodimerization of GPCRs [18], particularly μ-OR and δ-OR [15,21], and for direct interaction between the two receptors, suggests that they may form heteromers in vivo [20]. Based on their unique pharmacologic and functional properties [36,49,52], μ-δ heteromers may be an attractive target for the development of new drugs to treat chronic pain [27].
In recent years, researchers have studied the roles of μ-δ heteromers expressed in the central nervous system during pain by using antibodies that selectively recognize the endogenous heteromers and by using cell-penetrating TAT peptides that disrupt μ-δ heteromers. However, the involvement of μ-δ heteromers in primary sensory neurons is not known, particularly in the context of nerve injury and opioid tolerance. We designed this study to investigate the effects of nerve injury on μ-δ heteromers in DRG and to examine the effects of a μ-δ heteromer–targeting agonist on neuropathic pain-related behavior and neurophysiology.
4.1. Peripheral nerve injury leads to an increase in μ-δ heteromer in DRG
Our immunohistochemical studies suggested that the number of heteromer-positive neurons increased in the uninjured L4 DRG after L5-SNL but decreased in axotomized L5 DRG. These findings are consistent with our earlier report of μ-OR expression undergoing similar dynamic regulation after nerve injury [39]. In line with our immunohistochemical findings, ELISA studies also showed an increase in μ-δ heteromers in L4 DRG after L5-SNL. In this context, it is interesting to consider the potential involvement of chaperone protein, such as receptor transport protein 4 (RTP4), in modulating levels of μ-δ heteromers in neuropathic pain. Previous studies have reported that RTP4 expression leads to an increase in cell surface expression of μ-δ heteromers [9] and that chronic morphine treatment results in significant upregulation of RTP4 and a concomitant increase of μ-δ heteromers in select brain regions [16]. It would be interesting to explore the regulation of RTP4 under conditions of neuropathic pain.
4.2. μ-δ heteromer–targeting agonist attenuates neuropathic pain-related behavior
Recent studies have suggested that μ-δ heteromers may be potential new targets for the treatment of chronic pain [4,46,49]. Gomes et al. [19] reported that systemic administration of CYM51010, a μ-δ heteromer–targeting agonist, induced an antinociceptive effect similar to that of morphine, but caused less analgesic tolerance. Based on our observations that μ-δ heteromers increased in uninjured L4 DRG neurons after L5-SNL, we used CYM51010 as a tool to investigate the effect of μ-δ heteromer activation on neuropathic pain-related behavior. Systemic administration of CYM51010 dose-dependently inhibited mechanical hypersensitivity in SNL rats. CYM51010 also reversed thermal hyperalgesia. Interestingly, this heteromer agonist remained effective even under conditions of morphine tolerance. In addition, CYM51010, at a dose that did not attenuate SNL-induced mechanical allodynia and thermal hyperalgesia in the absence of morphine tolerance, became effective after the development of morphine tolerance. Together, these findings suggest that neuropathic pain may be associated with an increase in μ-δ heteromers in DRG neurons and that this heteromer may be a potential new drug target for pain inhibition. Furthermore, the increased μ-δ heteromer availability for CYM51010 may result in enhanced pain inhibition under conditions of opioid tolerance.
We further examined the effects of CYM51010 on nerve injury-induced ongoing pain using a CPP behavioral test. CYM51010–treated rats with L5-SNL exhibited preference for the drug-paired chamber over the vehicle-paired chamber, suggesting an amelioration of ongoing pain. CYM51010 at the same dose did not induce CPP in uninjured rats, suggesting that CYM51010–induced CPP behavior is pain state-dependent and that CYM51010 may have a low potential to cause drug-seeking behavior (e.g., addiction). Additional studies are needed to determine the safety of drugs targeting the μ-δ heteromer. A lower dose of CYM51010 did not produce CPP in SNL rats. However, after development of morphine tolerance, when the levels of μ-δ heteromer were increased [16,27], nerve-injured rats did show CPP to even the low dose of CYM51010 (0.2 mg/kg, s.c). These observations indicate that CYM51010-induced amelioration of ongoing pain may be dependent on heteromer concentration.
We provide additional lines of evidence which indicate that the effects of CYM51010 are mediated predominantly through its actions on the μ-δ heteromer. Numerous studies have shown that μ-OR is internalized and recycled to the plasma membrane for re-sensitization after activation [28,38]. In studies of HEK-293T cells that co-expressed the μ-OR and δ-OR, we observed that selective μ- and δ-opioid agonists as well as CYM51010 caused internalization of both receptors. However, in HEK cells that were transfected with only the μ- or δ-OR, the selective agonists induced marked internalization of only their respective receptors. Comparatively, CYM51010 was significantly less effective at causing receptor internalization in these cells. These results support the earlier observations that CYM51010 is a μ-δ heteromer–targeting ligand. Furthermore, our observations that the inhibitory effects of CYM51010 on neuropathic pain-related behavior are reduced in μ-OR KO mice further support the premise that antihyperalgesic actions of the drug are not primarily mediated via the OR homomers, but likely through its effects on the heteromer.
Investigations into the actions of the μ-δ heteromer have been constrained by the limited availability of pharmacologic tools. A recent study evaluated the in vivo and in vitro effects of a novel bivalent selective antagonist for the μ-δ heteromer [45]. The investigators showed that the bivalent-linked peptide antagonist dose-dependently reversed the antinociceptive effects of CYM51010, but did not antagonize μ-OR and δ-OR monomer-selective agonists, DAMGO and DSLET, respectively. That study, in addition to current observations in HEK cells, provides further support for the relative selectivity of CYM51010 for the μ-δ heteromer.
4.3. Neurophysiologic effects of μ-δ heteromer–targeting agonist on neuronal responses
To identify the mechanism of action of CYM51010, we used in vivo electrophysiologic recordings of WDR neurons in nerve-injured rats. The results showed that CYM51010 did not affect A-fiber components of WDR neurons but significantly inhibited the C-fiber and windup responses. WDR neurons in the spinal dorsal horn display a windup phenomenon to repetitive C-fiber inputs that is considered to be a short-duration neuronal sensitization. We have shown in earlier studies that this windup is modulated by endogenous μ-OR mechanisms [23]. The effects of CYM51010 on dorsal horn windup suggest that the drug may suppress dorsal horn neuronal excitability and sensitization. Schuster et al. [52] previously reported synergistic analgesic interactions between spinally administered μ- and δ-OR agonists. The authors concluded that their ligand combination studies supported the existence of μ-δ heteromers at the spinal level.
4.4. Potential implications for neuropathic pain drug targets
In an attempt to improve analgesia with fewer adverse effects, researchers are examining new approaches such as biased agonists and non-selective mixed opioid agonists [4,12,26]. However, olecridine, the first in the class of μ-OR G-protein–biased agonists that was tested for its efficacy and adverse effects in clinical studies of acute moderate to severe post-surgical pain, failed to receive approval by the FDA [57]. The complexity of the molecular mechanisms involved in opioid-induced analgesia, tolerance, and side effects was highlighted in a recent report [37]. Knock-in mice with mutations at the carboxy-terminal phosphorylation site that render the μ-OR unable to recruit β-arrestins, exhibited enhanced analgesia and reduced tolerance, but unchanged or exacerbated respiratory depression, constipation, and opioid withdrawal signs. These observations suggest that the recruitment of β-arrestin alone does not contribute to the severity of opioid-associated adverse effects.
The induction of heteromers represents a potential novel approach to creating unique pharmacologic conditions. Portoghese et al. [46] recently postulated that while some OR heteromers exist constitutively, colocalized receptors can also be induced to form heteromers by bivalent ligands. A series of bivalent ligands that contain μ-OR agonist and metabotropic glutamate receptor-5 (mGluR5) antagonist pharmacophores have been synthesized and studied. Bivalent ligands that induced μ-OR–mGluR5 heteromers produced potent antinociception in mouse models of inflammatory and cancer pain [2,54]. Recent studies suggest that oligomerization of μ-OR and other non-opioid GPCR complexes may also be a novel strategy to modulate μ-OR activity and enhance opioid analgesia. MrgC11, a primary sensory neuron-specific GPCR, may form heteromeric complexes with μ-OR in mouse DRG neurons, and a bivalent agonist acting at this site was shown to enhance opioid analgesia and decrease tolerance [30]. The physical interactions between different GPCRs have enormous implications for our understanding of how their actions are regulated. Our current findings suggest that heterodimerization of μ-OR and δ-OR in sensory neurons may represent a potential mechanism for regulating opioid analgesia. Furthermore, the μ-δ heteromer itself may represent a promising pharmacologic target for the development of specific and selective ligands that could alleviate neuropathic pain. In this context, it should be noted that previous studies have reported interactions between δ-OR and CB1 cannabinoid receptors in different models of neuropathic pain [6,48]. Furthermore, a recent study demonstrated increased δOR-CB1 receptor interacting complexes in the spinal cord of mice and humans after paclitaxel treatment, suggesting a role for these heteromers in chemotherapy-induced neuropathic pain [53]. Hence, targeting receptor interacting complexes with unique molecules or a combination of drugs could be a novel therapeutic strategy for the treatment of neuropathic pain.
Supplementary Material
Acknowledgements:
The authors thank Yuanxiang Tao, PhD, for technical consultation and assistance on the immunohistochemical studies and Dr. Achla Gupta for the purified μ-δ heteromer selective antibodies. We are grateful to Claire F. Levine, MS, ELS (scientific editor, Department of Anesthesiology/CCM, Johns Hopkins University), for her assistance in editing the manuscript.
Disclosure of funding: This study was conducted at the Johns Hopkins University and was supported mainly by grants from the National Institutes of Health (Bethesda, Maryland, USA): NS026363 (S.N.R.), NS070814 (Y.G.), NS110598 (Y.G.), DA08863 (L.A.D), and NS054791 (X.D.). This work was facilitated by the Pain Research Core funded by the Blaustein Fund and the Neurosurgery Pain Research Institute at the Johns Hopkins University. This study was also subsidized by a seed grant from the Johns Hopkins Blaustein Pain Research Fund (Y.G.). X.D. is also a faculty member of the Howard Hughes Medical Institute.
Footnotes
None of the authors have any conflicts of interest related to this work.
References
- [1].Abdelhamid EE, Sultana M, Portoghese PS, Takemori AE. Selective blockage of delta opioid receptors prevents the development of morphine tolerance and dependence in mice. J Pharmacol Exp Ther 1991;258:299–303. [PubMed] [Google Scholar]
- [2].Akgun E, Javed MI, Lunzer MM, Smeester BA, Beitz AJ, Portoghese PS. Ligands that interact with putative MOR-mGluR5 heteromer in mice with inflammatory pain produce potent antinociception. Proc Natl Acad Sci U S A 2013;110:11595–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Arvidsson U, Dado RJ, Riedl M, Lee JH, Law PY, Loh HH, Elde R, Wessendorf MW. delta-Opioid receptor immunoreactivity: distribution in brainstem and spinal cord, and relationship to biogenic amines and enkephalin. J Neurosci 1995;15:1215–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Azzam AAH, McDonald J, Lambert DG. Hot topics in opioid pharmacology: mixed and biased opioids. Br J Anaesth 2019;122:e136–e145. [DOI] [PubMed] [Google Scholar]
- [5].Belcheva MM, Clark AL, Haas PD, Serna JS, Hahn JW, Kiss A, Coscia CJ. Mu and kappa opioid receptors activate ERK/MAPK via different protein kinase C isoforms and secondary messengers in astrocytes. J Biol Chem 2005;280:27662–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Bushlin I, Gupta A, Stockton SD Jr., Miller LK, Devi LA. Dimerization with cannabinoid receptors allosterically modulates delta opioid receptor activity during neuropathic pain. PLoS One 2012;7:e49789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Cahill CM, Morinville A, Lee MC, Vincent JP, Collier B, Beaudet A. Prolonged morphine treatment targets delta opioid receptors to neuronal plasma membranes and enhances delta-mediated antinociception. J Neurosci 2001;21:7598–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Colloca L, Ludman T, Bouhassira D, Baron R, Dickenson AH, Yarnitsky D, Freeman R, Truini A, Attal N, Finnerup NB, Eccleston C, Kalso E, Bennett DL, Dworkin RH, Raja SN. Neuropathic pain. Nat Rev Dis Primers 2017;3:17002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Decaillot FM, Rozenfeld R, Gupta A, Devi LA. Cell surface targeting of μ-δ opioid receptor heterodimers by RTP4. Proc Natl Acad Sci U S A 2008;105:16045–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Dixon WJ. Efficient analysis of experimental observations. Annu Rev Pharmacol Toxicol 1980;20:441–62. [DOI] [PubMed] [Google Scholar]
- [11].Egan TM, North RA. Both mu and delta opiate receptors exist on the same neuron. Science 1981;214:923–4. [DOI] [PubMed] [Google Scholar]
- [12].Ehrlich AT, Kieffer BL, Darcq E. Current strategies toward safer mu opioid receptor drugs for pain management. Expert Opin Ther Targets 2019;23:315–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Finnerup NB, Attal N, Haroutounian S, McNicol E, Baron R, Dworkin RH, Gilron I, Haanpaa M, Hansson P, Jensen TS, et al. Pharmacotherapy for neuropathic pain in adults: a systematic review and meta-analysis. Lancet Neurol 2015;14:162–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Franco R, Casado V, Cortes A, Perez-Capote K, Mallol J, Canela E, Ferre S, Lluis C. Novel pharmacological targets based on receptor heteromers. Brain Res Rev 2008;58:475–82. [DOI] [PubMed] [Google Scholar]
- [15].Fujita W, Gomes I, Devi LA. Heteromers of μ-δ opioid receptors: new pharmacology and novel therapeutic possibilities. Br J Pharmacol 2015;172:375–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Fujita W, Yokote M, Gomes I, Gupta A, Ueda H, Devi LA. Regulation of an opioid receptor chaperone protein, RTP4, by morphine. Mol Pharmacol 2019;95:11–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Gendron L, Lucido AL, Mennicken F, O’Donnell D, Vincent JP, Stroh T, Beaudet A. Morphine and pain-related stimuli enhance cell surface availability of somatic delta-opioid receptors in rat dorsal root ganglia. J Neurosci 2006;26:953–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Gomes I, Ayoub MA, Fujita W, Jaeger WC, Pfleger KD, Devi LA. G protein-coupled receptor heteromers. Annu Rev Pharmacol Toxicol 2016;56:403–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Gomes I, Fujita W, Gupta A, Saldanha SA, Negri A, Pinello CE, Eberhart C, Roberts E, Filizola M, Hodder P, Devi LA. Identification of a mu-delta opioid receptor heteromer-biased agonist with antinociceptive activity. Proc Natl Acad Sci U S A 2013;110:12072–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Gomes I, Gupta A, Filipovska J, Szeto HH, Pintar JE, Devi LA. A role for heterodimerization of mu and delta opiate receptors in enhancing morphine analgesia. Proc Natl Acad Sci U S A 2004;101:5135–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Gomes I, Jordan BA, Gupta A, Trapaidze N, Nagy V, Devi LA. Heterodimerization of mu and delta opioid receptors: A role in opiate synergy. J Neurosci 2000;20:RC110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Guan JS, Xu ZZ, Gao H, He SQ, Ma GQ, Sun T, Wang LH, Zhang ZN, Lena I, Kitchen I, et al. Interaction with vesicle luminal protachykinin regulates surface expression of delta-opioid receptors and opioid analgesia. Cell 2005;122:619–31. [DOI] [PubMed] [Google Scholar]
- [23].Guan Y, Borzan J, Meyer RA, Raja SN. Windup in dorsal horn neurons is modulated by endogenous spinal mu-opioid mechanisms. J Neurosci 2006;26:4298–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Guan Y, Johanek LM, Hartke TV, Shim B, Tao YX, Ringkamp M, Meyer RA, Raja SN. Peripherally acting mu-opioid receptor agonist attenuates neuropathic pain in rats after L5 spinal nerve injury. Pain 2008;138:318–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Guerrero-Alba R, Valdez-Morales EE, Jimenez-Vargas NN, Bron R, Poole D, Reed D, Castro J, Campaniello M, Hughes PA, Brierley SM, Bunnett N, Lomax AE, Vanner S. Co-expression of mu and delta opioid receptors by mouse colonic nociceptors. Br J Pharmacol 2018;175:2622–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Gunther T, Dasgupta P, Mann A, Miess E, Kliewer A, Fritzwanker S, Steinborn R, Schulz S. Targeting multiple opioid receptors - improved analgesics with reduced side effects? Br J Pharmacol 2018;175:2857–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Gupta A, Mulder J, Gomes I, Rozenfeld R, Bushlin I, Ong E, Lim M, Maillet E, Junek M, Cahill CM, Harkany T, Devi LA. Increased abundance of opioid receptor heteromers after chronic morphine administration. Sci Signal 2010;3:ra54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].He L, Fong J, von ZM, Whistler JL. Regulation of opioid receptor trafficking and morphine tolerance by receptor oligomerization. Cell 2002;108:271–82. [DOI] [PubMed] [Google Scholar]
- [29].He SQ, Li Z, Chu YX, Han L, Xu Q, Li M, Yang F, Liu Q, Tang Z, Wang Y, et al. MrgC agonism at central terminals of primary sensory neurons inhibits neuropathic pain. Pain 2014;155:534–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].He SQ, Xu Q, Tiwari V, Yang F, Anderson M, Chen Z, Grenald SA, Raja SN, Dong X, Guan Y. Oligomerization of MrgC11 and mu-opioid receptors in sensory neurons enhances morphine analgesia. Sci Signal 2018;11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].He SQ, Yang F, Perez FM, Xu Q, Shechter R, Cheong YK, Carteret AF, Dong X, Sweitzer SM, Raja SN, Guan Y. Tolerance develops to the antiallodynic effects of the peripherally acting opioid loperamide hydrochloride in nerve-injured rats. Pain 2013;154:2477–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].He SQ, Zhang ZN, Guan JS, Liu HR, Zhao B, Wang HB, Li Q, Yang H, Luo J, Li ZY, Wang Q, Lu YJ, Bao L, Zhang X. Facilitation of mu-opioid receptor activity by preventing delta-opioid receptor-mediated codegradation. Neuron 2011;69:120–31. [DOI] [PubMed] [Google Scholar]
- [33].Hylden JL, Wilcox GL. Intrathecal morphine in mice: a new technique. Eur J Pharmacol 1980;67:313–6. [DOI] [PubMed] [Google Scholar]
- [34].Ji RR, Zhang Q, Law PY, Low HH, Elde R, Hokfelt T. Expression of mu-, delta-, and kappa-opioid receptor-like immunoreactivities in rat dorsal root ganglia after carrageenan-induced inflammation. J Neurosci 1995;15:8156–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Joseph EK, Levine JD. Mu and delta opioid receptors on nociceptors attenuate mechanical hyperalgesia in rat. Neuroscience 2010;171:344–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Kabli N, Fan T, O’Dowd BF, George SR. μ-δ opioid receptor heteromer-specific signaling in the striatum and hippocampus. Biochem Biophys Res Commun 2014;450:906–11. [DOI] [PubMed] [Google Scholar]
- [37].Kliewer A, Schmiedel F, Sianati S, Bailey A, Bateman JT, Levitt ES, Williams JT, Christie MJ, Schulz S. Phosphorylation-deficient G-protein-biased mu-opioid receptors improve analgesia and diminish tolerance but worsen opioid side effects. Nat Commun 2019;10:367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Koch T, Widera A, Bartzsch K, Schulz S, Brandenburg LO, Wundrack N, Beyer A, Grecksch G, Hollt V. Receptor endocytosis counteracts the development of opioid tolerance. Mol Pharmacol 2005;67:280–7. [DOI] [PubMed] [Google Scholar]
- [39].Lee CY, Perez FM, Wang W, Guan X, Zhao X, Fisher JL, Guan Y, Sweitzer SM, Raja SN, Tao YX. Dynamic temporal and spatial regulation of mu opioid receptor expression in primary afferent neurons following spinal nerve injury. Eur J Pain 2011;15:669–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Li Z, Tseng PY, Tiwari V, Xu Q, He SQ, Wang Y, Zheng Q, Han L, Wu Z, Blobaum AL, et al. Targeting human Mas-related G protein-coupled receptor X1 to inhibit persistent pain. Proc Natl Acad Sci U S A 2017;114:E1996-E2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Matthes HWD, Maldonado R, Simonin F, Valverde O, Slowe S, Kitchen I, Befort K, Dierich A, Le MM, Dolle P, Tzavara E, Hanoune J, Roques BP, Kieffer BL. Loss of morphine-induced analgesia, reward effect and withdrawal symptoms in mice lacking the mu-opioid-receptor gene. Nature 1996;383:819–23. [DOI] [PubMed] [Google Scholar]
- [42].Meert TF, Vermeirsch HA. A preclinical comparison between different opioids: antinociceptive versus adverse effects. Pharmacol Biochem Behav 2005;80:309–26. [DOI] [PubMed] [Google Scholar]
- [43].Minami M, Maekawa K, Yabuuchi K, Satoh M. Double in situ hybridization study on coexistence of mu-, delta- and kappa-opioid receptor mRNAs with preprotachykinin A mRNA in the rat dorsal root ganglia. Brain Res Mol Brain Res 1995;30:203–10. [DOI] [PubMed] [Google Scholar]
- [44].Morinville A, Cahill CM, Esdaile MJ, Aibak H, Collier B, Kieffer BL, Beaudet A. Regulation of delta-opioid receptor trafficking via mu-opioid receptor stimulation: evidence from mu-opioid receptor knock-out mice. J Neurosci 2003;23:4888–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Olson KM, Keresztes A, Tashiro JK, Daconta LV, Hruby VJ, Streicher JM. Synthesis and evaluation of a novel bivalent selective antagonist for the mu-delta opioid receptor heterodimer that reduces morphine withdrawal in mice. J Med Chem 2018;61:6075–86. [DOI] [PubMed] [Google Scholar]
- [46].Portoghese PS, Akgun E, Lunzer MM. Heteromer Induction: An Approach to Unique Pharmacology? ACS Chem Neurosci 2017;8:426–8. [DOI] [PubMed] [Google Scholar]
- [47].Rozenfeld R, Abul-Husn NS, Gomez I, Devi LA. An emerging role for the delta opioid receptor in the regulation of mu opioid receptor function. ScientificWorldJournal 2007;7:64–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Rozenfeld R, Bushlin I, Gomes I, Tzavaras N, Gupta A, Neves S, Battini L, Gusella GL, Lachmann A, Ma’ayan A, Blitzer RD, Devi LA. Receptor heteromerization expands the repertoire of cannabinoid signaling in rodent neurons. PLoS One 2012;7:e29239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Rozenfeld R, Devi LA. Receptor heterodimerization leads to a switch in signaling: β-arrestin2-mediated ERK activation by μ-δ opioid receptor heterodimers. FASEB J 2007;21:2455–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Scherrer G, Imamachi N, Cao YQ, Contet C, Mennicken F, O’Donnell D, Kieffer BL, Basbaum AI. Dissociation of the opioid receptor mechanisms that control mechanical and heat pain. Cell 2009;137:1148–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Schiller PW. Bi- or multifunctional opioid peptide drugs. Life Sci 2010;86:598–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Schuster DJ, Metcalf MD, Kitto KF, Messing RO, Fairbanks CA, Wilcox GL. Ligand requirements for involvement of PKCε in synergistic analgesic interactions between spinal μ and δ opioid receptors. Br J Pharmacol 2015;172:642–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Sierra S, Gupta A, Gomes I, Fowkes M, Ram A, Bobeck EN, Devi LA. Targeting cannabinoid 1 and delta opioid receptor heteromers alleviates chemotherapy-induced neuropathic pain. ACS Pharmacol Transl Sci 2019;2:219–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Smeester BA, Lunzer MM, Akgun E, Beitz AJ, Portoghese PS. Targeting putative mu opioid/metabotropic glutamate receptor-5 heteromers produces potent antinociception in a chronic murine bone cancer model. Eur J Pharmacol 2014;743:48–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Tiwari V, Anderson M, Yang F, Tiwari V, Zheng Q, He SQ, Zhang T, Shu B, Chen X, Grenald SA, Stephens KE, Chen Z, Dong X, Raja SN, Guan Y. Peripherally acting μ-opioid receptor agonists attenuate ongoing pain-associated behavior and spontaneous neuronal activity after nerve injury in rats. Anesthesiology 2018;128:1220–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Tiwari V, Yang F, He SQ, Shechter R, Zhang C, Shu B, Zhang T, Tiwari V, Wang Y, Dong X, Guan Y, Raja SN. Activation of peripheral μ-opioid receptors by dermorphin [D-Arg2, Lys4] (1–4) amide leads to modality-preferred inhibition of neuropathic pain. Anesthesiology 2016;124:706–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Trevena. FDA Advisory Committee Briefing Document: Oliceridine. 2018. Available at https://www.fda.gov/media/121230/download. Accessed November 9, 2019.
- [58].Wang H, Wessendorf MW. Equal proportions of small and large DRG neurons express opioid receptor mRNAs. J Comp Neurol 2001;429:590–600. [DOI] [PubMed] [Google Scholar]
- [59].Wang HB, Zhao B, Zhong YQ, Li KC, Li ZY, Wang Q, Lu YJ, Zhang ZN, He SQ, Zheng HC, Wu SX, Hokfelt TG, Bao L, Zhang X. Coexpression of δ- and μ-opioid receptors in nociceptive sensory neurons. Proc Natl Acad Sci U S A 2010;107:13117–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Zhang X, Bao L, Shi TJ, Ju G, Elde R, Hokfelt T. Down-regulation of mu-opioid receptors in rat and monkey dorsal root ganglion neurons and spinal cord after peripheral axotomy. Neuroscience 1998;82:223–40. [DOI] [PubMed] [Google Scholar]
- [61].Zhu Y, King MA, Schuller AGP, Nitsche JF, Reidl M, Elde RP, Unterwald E, Pasternak GW, Pintar JE. Retention of supraspinal delta-like analgesia and loss of morphine tolerance in delta opioid receptor knockout mice. Neuron 1999;24:243–52. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.