

Abstract
Background
Since iPSC human develop into hepatic organoids through stages that resemble human embryonic liver development, they can be used to study developmental processes and disease pathology. Therefore, we examined the early stages of hepatic organoid formation to identify key pathways affecting early liver development.
Methods
Single cell RNA-sequencing and metabolomic analysis was performed on developing organoid cultures at the iPSC, hepatoblast (day 9) and mature organoid stage. The importance of the phosphatidyl-ethanolamine biosynthesis pathway to early liver development was examined in developing organoid cultures using iPSC with a CRISPR-mediated gene knockout and an over the counter medication (meclizine) that inhibits the rate-limiting enzyme in this pathway. Meclizine’s effect on the growth of a human hepatocarcinoma cell line in a xenotransplantation model and on the growth of acute myeloid leukemia cells in vitro was also examined.
Results
Transcriptomic and metabolomic analysis of organoid development indicated that the phosphatidyl-ethanolamine biosynthesis pathway is essential for early liver development. Unexpectedly, early hepatoblasts were selectively sensitive to the cytotoxic effect of meclizine. We demonstrate that meclizine could be repurposed for use in a new synergistic combination therapy for primary liver cancer: a glycolysis inhibitor reprograms cancer cell metabolism to make it susceptible to the cytotoxic effect of meclizine. This combination inhibited the growth of a human liver carcinoma cell line in vitro; and in a xenotransplantation model without causing significant side effets. This drug combination was also highly active against acute myeloid leukemic cells.
Conclusion
Our data indicates that the phosphatidyl-ethanolamine biosynthesis is a targetable pathway for cancer; and that meclizine may have clinical efficacy as a repurposed anti-cancer drug when used as part of a new combination therapy.
Keywords: hepatic organoid, cancer chemotherapy
Lay Summary
The early stages in human liver development were modeled using human hepatic organoids, and we identified a pathway that was essential for early liver development. Based upon this finding, a novel combination drug therapy was identified that could be used to treat primary liver cancer and possibly other types of cancers.
Graphical Abstract
Introduction
Organoids have provided a very powerful model system for analyzing organogenesis and disease pathophysiology (1, 2). We recently developed a novel method to direct the differentiation of induced pluripotent stem cells (iPSCs) into 3-dimensional human hepatic organoids (HOs), which consist of sheets of hepatocytes, and cholangiocytes that are organized into epithelia that surround the lumina of duct-like structures. The HOs have biosynthetic, and functional capabilities of human liver; and HOs with engineered mutations provide a unique model for studying human disease-causing genetic mutations (3). In response to changes in the growth factors added to the media, HOs develop through stages that resemble human liver during embryonic development. Because of these properties, HOs could be used to identify pathways regulating discrete stages in liver development and disease pathogenesis.
However, at each differentiation stage, HO cultures contain different types of cells, and subpopulations of each cell type can be in various stages of differentiation; which makes it very difficult to identify critical transcriptional changes through analyses performed on the bulk population (4). To deal with the cellular complexity within dynamically changing organoid cultures at different developmental stages, single cell RNA sequencing (scRNA-Seq) can be used to simultaneously analyze global changes in the transcriptome of many individual cells. This analytic methodology (5) has dramatically advanced our ability to analyze cellular fate determination (4, 6), cell ontogeny (7), and cell heterogeneity in various tissues (8–11), including liver (12). However, when any ‘omic’ methodology is used in isolation, it can be very difficult to determine which of many measured gene expression changes are critical for even a single differentiation step. To identify the key pathways for early liver development, we simultaneously characterized metabolomic and transcriptomic changes that occur during an early stage of HO formation. Our reasoning was that a change in the level of mRNA expression for a gene within a metabolic pathway with an altered metabolite is likely to be of importance. This integrative analysis identified a biosynthetic pathway that plays an important role in early liver development. Based upon this finding and an independently performed study of mitochondrial drug effect (13), we discovered that a commonly used over the counter medication targeting this pathway could be used as one component of a new combination treatment for primary liver cancer. Systemic therapies that are effective and well-tolerated are critically needed for primary liver cancer because it is one of the few cancers whose incidence has continued to rise in the US and worldwide (14, 15), it has a dismal 5-year survival with little improvement in recent times (12.2% in 2001–2003 and 14.8% in 2004–2009) (16); and limited treatment options are available (17).
Materials and Methods
Hepatic organoid (HO) preparation
The three control (C1, C2, C3) and two ALGS (ALGS1, ALGS2) iPSC lines used in this study, and the methods for directing their differentiation into HOs are fully described in (3).
scRNA-seq data generation
Single cells from iPSC (day 0), day 9 HB, and day 30 HO cultures, and PHHs were prepared using the C1 Single-Cell Auto Prep System (Fluidigm, South San Francisco, CA). The captured cells across the 96 wells were manually inspected as a quality control measure to remove empty wells, doublets or debris-containing wells. For quality control (normalization and estimation of technical noise), 0.05 pl of 1:200,000 dilution of External RNA Controls Consortium (ERCC) RNA spike-in Mix1 (Ambion, AM1780) was added to lysis buffer. High-quality cDNA libraries for single cell transcriptome analysis were prepared using the SMART-Seq v4 Ultra Low RNA Kit for the Fluidigm C1 System (Takara Bio USA, 635025) according to (18). The size distribution and the concentration of single-cell cDNA were assessed using a Agilent 2100 Bioanalyzer with the high sensitivity DNA Kit (Agilent Technologies, Santa Clara, CA, 5067). Sequencing libraries were constructed in 96-well plates using the Nextera XT DNA Sample Preparation kit according to the manufacturer’s protocol (Illumina, San Diego, CA, FC-131–1096) with 96 dual barcoded indices (Illumina; FC-131–1002). Library cleanup and pooling was performed using AMPure XP beads (Agencourt Biosciences; A63880). Up to 200 quantified single-cell libraries were pooled, and these were sequenced 150 bp paired-end on one lane of Illumina HiSeq 2000 to a depth of 2.1 million reads per cell. The methods used for scRNA-Seq data analysis are described in the supplemental methods. The raw and processed scRNA-Seq data are deposited in the GEO database (GSE139382).
Metabolomic Analysis
The semi-targeted metabolomic method (STMM) uses Dansyl [5-(dimethylamino)-1-napthalene sulfonamide] derivatization coupled with LC/MS analysis, which is the method that we have previously successfully used to analyze metabolomic changes in other systems (19–21). Dansylation increases metabolite detection sensitivity by 10–1000 fold, which enables changes in many metabolites with primary or secondary amino (and other) groups to be evaluated in an unbiased fashion (22). LCMS grade acetonitrile; methanol and water were purchased from Honeywell Burdick and Jackson (Muskegon). High purity formic acid was purchased from Thermo Scientific (Rockford, IL). A Folch extraction was performed on three independent samples of iPSC and hepatoblast cultures, each containing about 1 million cells. An aliquot of the aqueous layer containing the polar compounds were labeled with Dansyl [5-(dimethylamino)-1-napthalene sulfonamide], and the LCMS analysis was performed using our previously described methods (19). The identity of phosphoethanolamine was confirmed by its retention time and LC/MS/MS pattern relative to that of a chemical standard (Sigma). Of note, the abundance of phosphoethanolamine in iPSCs was below the minimal Signal to Noise ratio of 10; so its abundance was set to a baseline abundance of 1000 that was used for the fold abundance calculation.
Since we were searching for metabolic differences at an early stage of liver development, broad-spectrum metabolomic profiling was used to compare the levels of many different metabolites (>500) in iPSC and HBs. Because of this, the metabolomic results are reported as a ratio (i.e. fold change of the hepatoblast over the iPSC value), of what was actually measured. It is now is an accepted practice to report fold-changes and significance levels for this type of analysis (23).
PCYT2 gene knockout experiments
The sequences of three site-specific guide RNAs (sgRNAs) that target exons 8 and 12 of the PCYT2 gene, which were determined using the CRISPR Design Tool (24), are: (i) gccctggtgggcggaacccc, (ii) gatgactgtctcccctggct, and (iii) ggacgatgaggtctgtggtg. Oligonucleotides with these sequences were cloned into the lentiCRISPR v2 vector (Addgene #52961, Cambridge, MA), which uses the Puromycin selection to enrich for cells with a PCYT2 KO. Control iPSCs (C1, C2 and C3) were pelleted and mixed with 5~10 mg CRISPR sgRNA expression vectors in 100 ml of human ES cell solution 1 (Lonza), and nucleofection program A23 was applied. On day 3, puromycin was added to select for cells with a PCYT2 KO; and after 48 hrs, the iPSCs lines were split and placed into media to direct their differentiation into HB for 3 days, and the cells were transferred into another media to direct their differentiation into HBs (using previously described methods (3)). On day 4, we noted that the PCYT2 KO cells were dying, and the Trypan Blue dye exclusion assay was performed to assess cell viability. In brief, 100 ml of the cell suspension was mixed with 100 ml 0.4% Trypan Blue stock solution (T8154 Sigma), and cell counts were performed immediately under bright field microscopy.
Animal studies
The Stanford University Administrative Panel on Laboratory Animal Care (APLAC) approved the animal protocol and the experimental procedures. For these studies, highly immunocompromised NOG mice (25) were housed in a specific pathogen-free room, which was equipped with a HEPA filter system with 12 hours light/dark cycle. The methods used in performing the animal studies are described in the supplemental methods.
Patient samples and human ethics approval
The primary peripheral blood samples were obtained from subjects with acute myeloid leukemia prior to treatment, and after informed consents were obtained. The study was performed according to institutional guidelines that were approved by the Stanford University Institutional Review Board No. 6453. Patient details are provided in Supplemental Table 1.
Culture of primary AML cells
The methods for AML blast culture were previously described (26). In brief, vials containing > 95% AML blasts were thawed in 20% fetal bovine serum + IMDM with DNAse; and plated at 1 × 105 - 1 × 106 cells /ml in IMDM with 0.5% serum and 20 ng/ml stem cell factor, Flt3L, thrombopoietin, interleukin-6, and interleukin-3. Various concentrations of meclizine and/or PFK148 were also added to the culture medium. After 96 hours in culture, the number of live cells was determined by flow cytometry using bead:cell ratios to count absolute numbers of propidium iodide-negative CD45 mid/side scatter blast gate cells as described (26).
Results
scRNA-Seq Analyses of developing HOs
To characterize the dynamic changes that occur as a control iPSC line (C1) differentiates into an organoid, scRNA-Seq was performed on the iPSC, day 9 hepatoblasts (HB), and day 21 HO cultures; and scRNA-Seq was also performed on primary human hepatocytes (PHHs) for comparison purposes. The t-Distributed Stochastic Neighbor Embedding (t-SNE) method was used to visualize this high dimensional data, which enables the relationships across each developmental stage to be evaluated. This analysis shows the developmental path as the iPSCs differentiate into hepatoblasts (HBs) and then into primary human organoids; and the transcriptome profile of many cells within the HO was similar to that of PHHs (Fig. 1A). Unexpectedly, this analysis identified two clusters of HBs: (i) early HBs whose expression profile was close to that of iPSC; and (ii) late HBs whose expression profile was similar to that of PHHs and HO. This finding is consistent with prior gene expression results, which indicated that there is a ‘resonant state’ between pluripotent cells and their progeny when the cells are in an early stage of differentiation (27). The early HBs express more pluripotency-related genes, which are lost as the cells are further differentiated.
Figure 1.

scRNA-Seq profiling of the transcriptome of cells during their in vitro differentiation from iPSC into primary HO. scRNA-Seq was performed on cells obtained from iPSC, hepatoblast (HB) (day 9), and primary HO cultures prepared from control donor 1; and from primary human hepatocytes (PHH). (A) t-SNE visualization of the scRNA-Seq data. The developmental path of the iPSC as they differentiate into hepatoblasts and then into primary human organoids, whose transcriptome profile is similar to that of PHHs, is evident. There were two clusters of HBs: early HBs are those at the bottom-left whose transcriptome was close to that of iPSC; and late HBs at the upper-right whose transcriptome was similar to that of PHHs and HO. The single cell clusters are visualized using t-SNE. (B-C) The fractional identity (FI) of the transcriptome of individual cells are superimposed upon the t-SNE plot shown in (A). The similarity of the transcriptome of each cell relative to iPSCs, PHHs (B) or cholangiocytes (C) was plotted as the FI. This calculation was performed so that the three FIs for each cell (each relative to iPSC, PHH or cholangiocytes) sums to 1. For example, the graph in (B) clearly shows a gradient in the relationship between the FI of the transcriptome of each cell type and PHH: iPSCs have zero FI, early HB have a low FI (indicative of proximity to iPSCs), while late HBs and HOs have a high FI (indicating they have greater similarity to PHH). Also, the FI for each of the PHHs are all virtually one, as expected. Of note, the gradient of FI values matches perfectly with the developmental path observed in the tSNE plot. In (C) a group of cells in the HO had a transcriptome profile that partially resembles that of cholangiocytes. The results in (B) and (C) indicate that the iPSC develop into cells resembling hepatocytes and cholangiocytes in a primary HO. (D,E) The relative similarities of the transcriptome profile of cultured cells to iPSCs and PHHs (D), or to that of adult or fetal hepatocytes (E). scRNA-Seq was performed on cells obtained from iPSC, hepatoblasts (HB), or hepatic organoid (HO) cultures prepared from 4 donors; or from PHHs. Quadratic programming was used to computationally infer their similarities, which was expressed as the FI relative to iPSC (FI = 0) and PHH (FI = 1) in (D), or to fetal (FI = 0) and adult (FI = 1) hepatocytes in (E). The cells were then ranked by their FI values relative to that of iPSC/PHH in (D), or relative to fetal/adult hepatocytes in (E). Each cell analyzed in this way is indicated by a colored dot that corresponds with the indicated cell type. Of note, most of the HBs had a FI relative to PHH that was <0.5, which was below the majority of the HO cells. However, a small percent of HB cells had a higher FI, which was closer to that of PHHs. This result is consistent with presence of early and late populations of HBs. A similar trend was observed for the FI relative to fetal or adult hepatocytes: most HBs had a FI relative to mature adult hepatocytes that was virtually zero, but the majority (~2/3) of the HO cells had a positive FI. However, a small percent of HBs had a higher inferred fractional identity, which was closer to that of PHHs.
Additional transcriptomic analyses revealed that HOs contain cells that are similar to hepatocytes, and others that are similar to cholangiocytes (Figs. 1B–E). The validity of the scRNA-Seq data is confirmed by the fact that mRNAs-which are characteristically expressed in iPSC (NANOG, OCT4), PHHs (A1AT, ALB), or cholangiocytes (CFTR)-were expressed in the appropriate cell type (Fig. S1). Analysis of this data (and from organoids formed from other iPSC lines) with another statistical method confirmed that cells within the different groupings were clustered, and the same developmental relationships were apparent (supplemental comments, Figs. S1–S2).
To better characterize the maturity of cells in HOs, we compared their transcriptome with previously obtained transcriptomic data for hepatocytes present in one fetal (gestation age 17.5 weeks) and three adult (ages 21–65) livers (28) using t-SNE visualization. As expected, the transcriptomes of hepatocytes in adult liver and PHHs were tightly clustered; while the transcriptomes of hepatocytes in fetal liver were separated from that of the adult liver hepatocytes/PHH cluster (Fig. 2A). Of importance, the transcriptome of HOs cells was more closely related to adult hepatocytes/PHH than to fetal hepatocytes. The SLICER method (29) was also used to computationally infer a developmental trajectory for the cells starting from iPSC. As with the t-SNE visualization, this method generated a developmental path from iPSC to HB, and then to HO, and then to adult hepatocytes/PHH (Fig. 2B). However, a small proportion of HB cells (~15 out of 149) diverged from the main path, which indicates that a subset of the HBs could have a different developmental fate. Of importance, most HO cells are located farther away from iPSCs, and are closer to adult hepatocytes/PHH than to fetal hepatocytes (Fig. 2B). Also, HBs were located closer to fetal hepatocytes, and were far away from the adult hepatocytes. These results indicate that the transcriptomic profile of HO cells resembled that of mature hepatocytes, while HBs had a closer resemblance to fetal hepatocytes. This analysis also identified many genes that were expressed at specific stages of HO development, and several were also associated with liver or liver cancer development (see supplemental comments, Table S1; Figs. S3–S4).
Figure 2.

(A) A comparison of the transcriptomes of iPSC, HB, HO and PHH cells with human fetal and adult liver cells. scRNA-Seq was performed on cells obtained from iPSC, day 9 hepatoblast (HB), and primary HO cultures prepared from four donors, and from primary human hepatocytes (PHH). Previously obtained transcriptomic data for hepatocytes present in one fetal (gestation age 17.5 weeks) and three adult (ages 21–65) livers was used for the comparison. The t-SNE visualization indicates that: adult liver hepatocytes and PHH were tightly clustered, and the hepatocytes in fetal liver were clearly separated from this cluster; and the transcriptome of HO cells was more closely related to adult hepatocytes/PHH than to fetal hepatocytes. (B) The SLICER method (29) was used to computationally infer a developmental trajectory (beginning from iPSC) for the cells shown in (A). Each cell type is indicated by a different color, and this method generated a developmental path from iPSC to HB, and then to HO, and then to adult hepatocytes/PHH. This pattern is the same as in the t-SNE visualization. Of note, a small proportion of HB cells diverged from the main path, indicating that they may have a different developmental fate. Most HO cells are located farther away from iPSCs, and are closer to adult hepatocytes/PHH than to fetal hepatocytes. In contrast, HBs were located closer to fetal hepatocytes, and were far away from the adult hepatocytes. Therefore, the transcriptomic profile of HO cells resembled that of mature hepatocytes, while that of HBs more closely resembled fetal hepatocytes.
A phosphatidylethanolamine (PE) biosynthesis pathway is essential for HB differentiation
To identify changes in mRNA levels that are essential for early liver development, a semi-targeted metabolomic method (22), which we have previously used to analyze other systems in an unbiased manner (19–21), was used to characterize metabolomic changes occurring when iPSC differentiate into HBs. Of the 494 metabolites evaluated, only eleven had a ≥3-fold abundance difference (P<0.05); and only three had a >10-fold difference in abundance between iPSC and HBs (Fig. 3A). Of these metabolites, phosphoethanolamine (P-Etn) had the greatest differential abundance; its abundance was ≥170-fold increased in HBs (p=0.018) relative to that in iPSCs. In contrast, there was no difference in ethanolamine abundance in iPSC vs. hepatoblasts (Fig. 3B). The marked increase in phosphoethanolamine abundance in HBs was of interest, since it is required for the biosynthesis of phosphatidylethanolamine (PE), which accounts for 25% of all cellular phospholipids and is critical for cellular membrane formation (30). Although it can be produced via four different biosynthetic pathways, the CDP-ethanolamine pathway is one of the major PE biosynthetic pathways (PEBP) in the liver (31–34). Analysis of bulk sample RNA-Seq data indicated that mRNAs encoding all three enzymes in this pathway (ENTK1, PCYT2 and SELENOI) (30) were increased in HBs (75, 58 and 41 fold, respectively) (Fig. 3B). PCYT2 encodes the rate-limiting second enzymatic step in the PEBP pathway (30, 35) (Fig. 3B). Consistent with our metabolomic results, it was previously observed that when physiologic ethanolamine concentrations are present, increased ENTK1 expression in cell lines produces a marked increase in cellular phosphoethanolamine, but not an increase in CDP-ethanolamine (36). Taken together, the transcriptomic and metabolomic changes indicate that the mRNAs for the enzymes and a key metabolite in the PEBP are coordinately increased at an early stage of liver development.
Figure 3.

(A) The relative abundance of 494 dansyl-labeled metabolites was measured in day 9 hepatoblasts and in iPSCs, which were generated from three different individuals. The fold difference (shown as the ratio of the hepatoblast/iPSC) in the measured relative abundance (Y-axis) of each metabolite (numbered along the X-axis) is graphed. The arrow indicates the metabolite with highest abundance difference, which was phosphoethanolamine. The identities of the two other metabolites with a fold change > 10 could not be determined. (B) The CDP (phosphoethanolamine cytidylyltransferase)-ethanolamine (Etn) pathway for phosphotidylethanolamine (PE) biosynthesis is activated when iPSC differentiate into hepatoblasts (HBs). In the first step, ethanolamine kinase (ETNK1) phosphorylates (Etn to produce P-Etn. The second step is rate limiting, and it is catalyzed by the product of the PCYT2 gene. This step generates CDP-Etn, which is then converted to PE by CDP-ethanolamine: 1,2-diacylglycerol ethanolaminephospho-transferase (SELENOI). The measured abundance of Etn and P-Etn in iPSC and HBs are shown below the pathway. Each measurement was the average of three independent measurements. The relative expression levels for ETNK1, PCYT2, and SELENOI mRNAs in iPSC and HBs, which were determined from bulk scRNA-Seq data, are shown above the pathway. There was a marked increase in P-Etn abundance (170-fold, p=0.018); and of ETNK1, PCYT2 and SELENOI mRNAs in HBs (75, 58 and 41-fold, respectively); which indicated that the CDP-Etn pathway for PE synthesis was activated when iPSC differentiated into HBs.
To determine if the PEBP is essential for early liver development, three different sgRNAs were used to prepare iPSCs with a CRISPR-mediated PCYT2 gene knockout from control iPSC lines (KO1–3). Two control iPSC (C1, C2) and three PCYT2 KO lines were differentiated into endoderm on day 3, and then transferred into the media that directs their differentiation into HBs. When the cultures were examined on day 4, the control iPSC lines could differentiate into endoderm, and remained viable in the HB media. While each of three PCYT2 KO cells could differentiate into endoderm, >95% of these cells were dead within 24 hrs after transfer to the HB media (Fig. 4A). This unexpected result is consistent with the fact that a homozygous Pcyt2 KO is embryonic lethal in mice (37). While mice with a hepatocyte-specific conditional Pcyt2 gene deletion had liver abnormalities, they were viable and fertile (38); which is probably due to the fact that the conditional knockout occurs after the liver has developed.
Figure 4.

(A) A PCYT2 gene knockout (KO) reduces cell viability. Two Control (C1, C2) and three iPSC lines with a CRISPR-mediated PCYT2 KO-which were prepared using three different sgRNAs (KO1, KO2, KO3)-were examined in these experiments. Each iPSC line was differentiated into endoderm (EN) through day 3; then transferred into media to direct their differentiation into HBs; and the developing HB cultures were examined on day 4. Bright field images of HB cultures prepared from the iPSC lines. While the cells generated from all of the iPSC lines were abundant and viable at the EN stage, the cell density and number of viable cells in all PCTY2 KO lines was dramatically reduced as they differentiated toward HBs. The scale bar is 50 μm. The bar graph shows the results of a trypan blue assay for cell viability (% viable cells) in day 4 differentiating HB cultures. Each bar is the mean (+ SD) of three independent determinations for each iPSC line. (B) Meclizine induces cell death in early hepatoblasts (HBs). Control iPSCs were differentiated for 3 days into endoderm. On day 4, the cells were placed in HB differentiation media containing: 50 uM Meclizine (M), 50 ng FGF10 (F10), 50 ng HGF (H), HGF and meclizine (HGF+M) or FGF10 and meclizine (F10+M) for 24 hrs. The bright field images showing the morphology of the cells indicate the extensive amount of cell death in occurring in meclizine-treated cells. Scale bar: 50 μm. The bar graph of the trypan blue assay for viable cells (% viability) in the day 4 cultures is shown. Each bar is the mean + SD of two independent determinations for each culture condition. (C) Meclizine impairs organoid regeneration. The cells in HOs were dissociated into single cells, and then placed in growth media to induce the formation of secondary organoids in the presence of meclizine at concentrations ranging from 0 to 80 uM. Top: Bright field images of cultures with 0, 4 or 8 uM meclizine examined on day 7 after staining with Calcein AM. Bottom: The efficiency of secondary organoid formation after 7 days in growth medium was quantitated by fluorescence measurement. Each data point is the mean of 2 independent determinations, and the results are normalized relative to the fluorescence of wells where organoids were re-formed in the absence of meclizine. (D,E) Meclizine selectively induces cell death in early hepatoblasts (HBs). (D) Cell Survival was measured 24 hour after the cells in iPSC (day 0); early (day 4), mid (day 7) or fully differentiated HB (day 10) cultures were exposed to 0, 1, 2, 4, 5, 8, 10, 15, 20, 40, 50, or 80 uM meclizine. Each point is the mean (+SD) of three independent trypan blue assays performed 24 hours after exposure to meclizine. (E) Meclizine survival curves were also examined in induced cardiomyocytes (CM) on day 5 or day 20; human fibroblasts (FB), primary human hepatocytes (PHH), and the HepG2 and Huh7 hepatocarcinoma lines (right). The red line indicates the meclizine concentration causing 50% cell death (LD50). (F) The culture media does not alter the susceptibility of iPSC to meclizine-induced cell death. iPSCs were incubated in either the iPSC or hepatoblast (HB) media containing the indicated concentrations of meclizine for 24 hours, and cell viability was then measured using the Prestoblue assay. Each bar is the mean + SD of two independent measurements performed for each condition.
Early HBs are selectively sensitive to PEBP inhibition
Meclizine is an over the counter antihistamine that is widely used for the treatment of motion sickness and vertigo (39, 40), and it was recently found to be a non-competitive inhibitor of PCYT2 enzyme activity (41). Therefore, we examined whether meclizine would have the same effect on early HBs as a CRISPR-mediated PCYT2 KO. Within 24 hrs after exposure to meclizine, virtually all cells in the day 4 cultures, which were in the early stage of differentiating toward HBs, were killed (Fig. 4B). Growth factor (FGF10 or HGF) addition did not reduce their sensitivity to meclizine-induced cell death. We previously demonstrated that HOs contained a population of SOX9+CK7+ cells that resemble liver progenitor cells; and that after HOs were formed, they could be dissociated into single cells, which could re-form hepatic organoids when placed in growth medium (3). Therefore, we examined whether meclizine could inhibit organoid re-formation. Meclizine was a potent inhibitor of organoid reformation; 4 μM meclizine inhibited organoid regeneration by 50% (Fig. 4C).
To better define the time period for sensitivity to meclizine-induced cell death, cell survival was examined after meclizine was added to iPSC cultures (day 0); or when it was added to early (day 4), mid (day 7) or fully differentiated HB (day 10) cultures. Only the early HBs (day 4) were sensitive to meclizine-induced cell death; 4 μM meclizine killed 50% of the cells at this stage. In contrast, the day 7 and day 10 HB cultures were relatively insensitive to meclizine-induced cell death (Fig. 4D). Human iPSCs were far less susceptible (LD50 ~60 μM), and human fibroblasts (LD50 >80 uM) and adult PHHs (LD50 >80 μM) were resistant to meclizine-induced cell death. Unlike the early HBs, human iPSC in the early stage of differentiating into cardiomyocytes (day 5) were no more sensitive to meclizine-induced cell death than were iPSCs; and fully differentiated induced cardiomyocytes (day 20) were not sensitive to meclizine-induced cell death (Fig. 4E). Moreover, HUH7 (42) and Hep G2 (43, 44) cell lines, which represent well differentiated hepatocellular carcinomas, were also relatively resistant to meclizine-induced cell death (Fig. 4E). It was possible that the hepatoblast differentiation media could increase the susceptibility of differentiating cells to meclizine-induced cytotoxicity. However, the susceptibility of iPSC, which were not differentiated into endoderm, to meclizine-induced cytotoxicity was not increased when they were cultured in the hepatoblast differentiation media (Fig. 4F). Thus, susceptibility to meclizine-induced cytotoxicity required that the iPSC were differentiated into endodermal cells. Taken together, these results indicate that there is a time window when cells in the early stage of organoid formation (which is reflective of early liver development) are extremely sensitive to PEBP inhibition.
PEBP in hepatocellular cancer
iPSCs express high levels of mRNAs encoding CD24, CD90, PROM1 (CD133) and EPCAM (Fig. S3). Based upon expression within a subset of cells within tumor samples that will form tumors in xenotransplantation models; CD133 (45, 46), EpCAM (47), CD24 and CD90 (48–51) have been identified as markers for cells contributing to the growth and maintenance of primary liver cancer. Moreover, DLK1, which is expressed in HBs (Fig. S3), has been identified as a potential therapeutic target liver cancer (52). To investigate the possibility that the PEBP could be a therapeutic target for liver cancer, two gene expression datasets in the Cancer Gene Atlas (53) were examined. One contained 374 primary (HCC1) or recurrent hepatocellular carcinomas (HCC2) and 50 normal liver samples (N1), and the other had 31 cholangiocarcinomas (CC) and 8 normal liver samples (N2). The SLICER method (29) was used to examine the relationship between the transcriptomes of HCC, normal liver tissue, and cells in developing HO cultures. This analysis revealed that the transcriptome of HCC clusters with that of HO and HB cells; and was distinct from that in normal liver tissue (Fig. 5A and supplemental comments,). We also compared the expression level for mRNAs encoding PEBP pathway genes (ETNK1, ETNK2, PCYT2, and SELENOI) in the HCC, CC and normal liver samples. Of importance, PCYT2 mRNA was significantly increased in primary HCC (1.7-fold, p value 1×10−7), and its expression was further increased in recurrent HCC (3.3-fold, p value 7×10−9) relative to normal liver (Fig. S5, Table S2).
Figure 5.

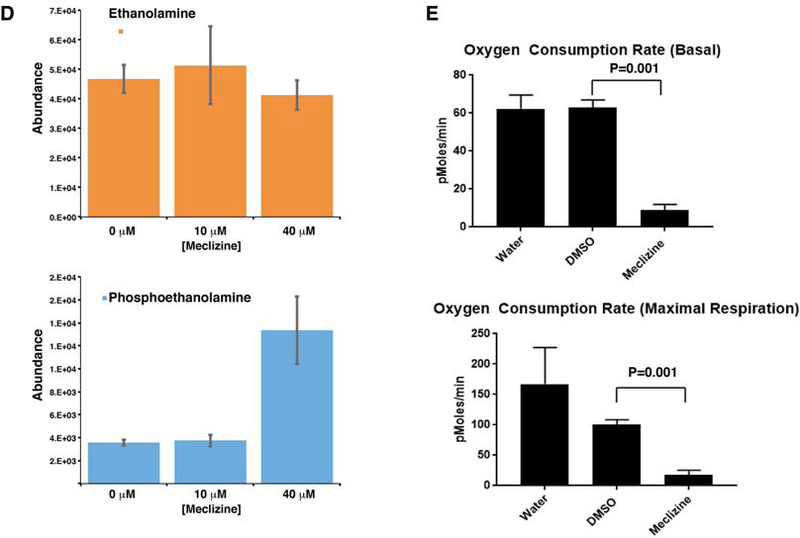
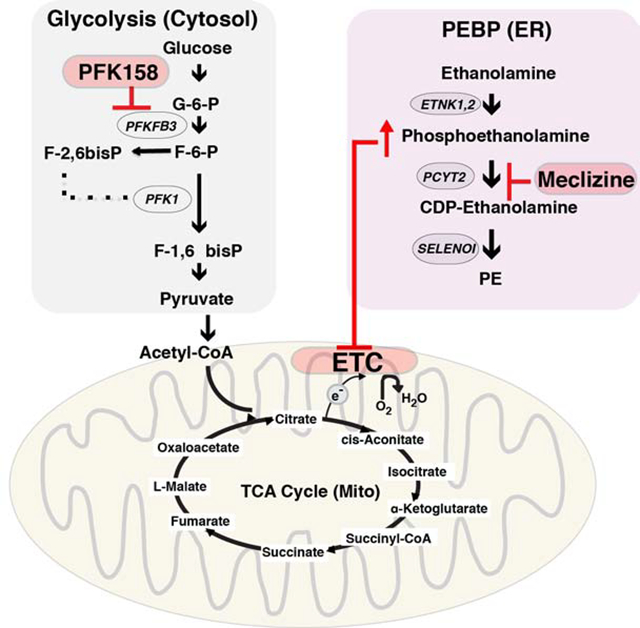
(A) The SLICER method 21 was used to examine the relationships between the transcriptomes of cells in developing organoid cultures (iPSC, HB and HO) cells and PHH, 271 HCCs in the TCGA dataset, and normal liver. Each cell type is indicated by a different color. The previously identified developmental pathway from iPSC ->HB-> HO was preserved in this analysis. Normal liver clustered with HO, and is distinct from the other cells types. HCC transcriptomes overlapped with HO and HB cells, and were distinct from iPSCs and normal liver. These results indicate that the transcriptomic profile of HB and HO cells resembles that of liver cancer cells. (B) Diagram of the mechanism for the synergistic inhibitory effect of meclizine and PFK158 on cell growth. Within the endoplasmic reticulum (ER), meclizine inhibits the rate-limiting enzyme (PCYT2) in the PEBP, which increases cellular phosphoethanolamine. ATP is generated by glycolysis occurring in the cytoplasm. PFK158 inhibits an enzyme (PFKFB3) whose reaction product activates the rate-limiting step in glycolysis. By inhibiting glycolysis, PFK158 increases cellular dependence upon energy production by mitochondria. Phosphoethanolamine inhibits the mitochondrial electron transport chain (ETC). By this mechanism, treatment with the combination of PFK158 and meclizine synergistically inhibits cell growth. (C) Meclizine and PFK158 inhibit the growth of the HepG2 (top) and HuH7 (bottom) hepatocarcinoma cell lines. The cells were grown for 7 days in a standard high glucose medium, or in media where galactose and glutamine replaced the glucose. While meclizine (40 uM) did not significantly affect the proliferation of either cell line in the glucose medium, it significantly reduced their proliferation in the galactose medium. Addition of a low concentration (2 or 5 uM) of PFK158 had no effect on cell proliferation in either media. However, addition of a high PFK158 (5 or 10 uM) concentration did impair cell growth. When meclizine was combined with a low concentration of PFK158, cell viability was inhibited. C: vehicle treated cells; M: Meclizine (40 uM); P2, 2 uM PFK158; P5, 5 uM PFK158; P10, 10 uM PFK; M+P*, 40 uM Meclizine plus the indicated uM concentration of PFK158. Each data point is the average of 8 independent measurements obtained from 2 different experiments. (D) Meclizine increases cellular phosphoethanolamine. HepG2 cells (2×106 cells per 100 mm dish) were grown for 2 days to ~70% confluence in a high glucose medium (DMEM with 1% FBS); and then treated with vehicle (0 μM), 10 μM meclizine, or 40 μM meclizine for 24 hours. Then, the ethanolamine and phosphoethanolamine levels were measured by MS analysis. Each bar shows the average ± SEM of 3 independent measurements for each group. Phosphoethanolamine abundance was significantly increased (*p=0.004) by 40 (but not 10) uM meclizine, while there was no significant change in ethanolamine abundance. (E) Meclizine decreases the basal and maximal oxygen consumption rate of a hepatocarcinoma cell line. HepG2 cells were cultured in the presence of water, vehicle (DMSO), or 50 uM meclizine. The oxygen consumption rate was measured using the Mito Stress Test on a XFe96 Seahorse Analyser at 2 hours after compound or vehicle addition. The upper graph shows the measured basal oxygen consumption rate, and the lower graph shows the maximal respiration rate measured after addition of uncoupling agent carbonyl cyanide-4 trifluoromethoxy phenylhydrazone (FCCP). Each bar is the average + SD of 16 independent measurements; and 50 uM meclizine induced a significant decrease in basal (p<0.001) and maximal (p<0.001) oxygen consumption.
Many types of cancer cells have very high rates of glycolysis, which results from oncogene-induced increases in glucose uptake and in the enzymes involved in glucose metabolism (54). Rapidly growing hepatoma cell lines are metabolically adapted to grow in high glucose media, and they derive the bulk of their energy from glycolysis (55) (Fig. 5B). Although this pathway produces only 5% of the potential energy that could be generated from a glucose substrate by mitochondrial oxidative phosphorylation (OXPHOS), the flux through this pathway can be greatly accelerated when glucose is abundant. Since conversion of galactose to pyruvate generates no net ATP, when mammalian cells are switched to a media where galactose is the sole sugar source, they use OXPHOS as their energy source. Also, cancer cells use glutamine metabolism via the tricarboxylic acid cycle as the major pathway for producing the molecules required for proliferation (56, 57). Since HCC may exist within a metabolically constrained environment in the liver, we examined the effect of meclizine on the growth of two hepatoma cell lines (Hep G2 and HuH7) in media containing galactose and glutamine. In this media, 40 μM meclizine inhibited the growth of both hepatoma lines at 24 and 48 hrs (Fig. 5C). These results indicate that meclizine could inhibit the hepatocarcinoma cell line growth when they are dependent upon mitochondrial OXPHOS for energy production. Studies were then performed to characterize the mechanism of action of meclizine-induced cytotoxicity. After HepG2 cells were incubated with 40 uM meclizine for 24 hrs, cellular phosphoethanolamine was significantly increased (3.7-fold, p=0.004), while the ethanolamine level was not changed (Fig. 5D). Meclizine also induced a decrease in both the basal (p=0.001) and maximal (p=0.001) oxygen consumption rates in the hepatocellular carcinoma line (Fig. 5E). These effects are all consistent with the previously proposed mechanism of action for meclizine (41).
Recently, 6-phosphofructo-2-kinase/fructose-2,6-biphosphatase 3 (PFKFB3) inhibitors have been produced (58–60), and a third generation inhibitor (PFK158) is now in clinical testing as an anti-cancer agent (61–64). PFKFB3 stimulates glycolysis because the product of its enzymatic activity (fructose-2,6-bisphosphate F2,6P2) is an allosteric activator of the rate-limiting enzyme (phosphofructokinase-1, PFK-1) in the glycolysis pathway (Fig. 5B). PFKFB3 expression is induced by hypoxia (65), and it is overexpressed (66) and highly phosphorylated (67) in many different types of cancer. Therefore, it was possible that PFKFB3 inhibition, which would increase cellular dependence upon mitochondrial OXPHOS, could inhibit the growth of HCC when used in combination with meclizine. Consistent with this possibility, the combination of meclizine and PFK158 inhibited the growth of both hepatoma cell lines (Fig. 5C). Although meclizine alone did not inhibit their growth in the glucose media, the mitochondrial membrane potential of the HepG2 hepatocellular carcinoma line in the glucose media (and also in the galactose/glutamine media) was altered by meclizine (Fig. S6). This effect is consistent with meclizine’s known mechanism of action (discussed below). To more fully characterize the effect of this drug combination, we examined the effect of exposure to 24 different combinations of meclizine and PFK158 concentrations for 24 and 48 hrs on HepG2 viability (Fig. S7). This data indicates that there is a very significant interaction between the two drugs, which appeared at both time points (p < 2.2 ×10−16 for both exposure periods). Specifically, in the presence of a high dose of Meclizine (40 μm) and PFK158 (2 or 5 μm), a significant synergistic effect on cell growth appeared at both time points: this drug combination reduced cell viability by 1.4-fold more than expected by simply adding the effect of each individual drug at the same dose. A lesser, but consistent effect on cell viability, was also observed with the combination of 20 uM Meclizine and 2 (or 5) μm PFK158. The synergistic effect of this drug combination was also observed at lower drug concentrations; this effect was especially apparent at 48 hr time point: 5 μM concentrations of both drugs induced an additional 1.5-fold decrease in cell viability relative to total additive effect of each of the two drugs (Fig. S7). Additional studies were also performed to characterize the mechanism of action of meclizine-induced cytotoxicity. Low concentrations of meclizine (2 or 5 μM) altered the mitochondrial membrane potential of HepG2 cells (Fig. S8). The effect on the mitochondrial membrane potential, which is a very sensitive assay for cellular perturbation, is consistent with the synergistic effect that low meclizine concentrations had when combined with PFK158. Also, after HepG2 cells were incubated with 40 uM meclizine for 24 hrs, cellular phosphoethanolamine was significantly increased (3.7-fold, p=0.004), while the ethanolamine level was not changed (Fig. 5D). Meclizine also induced a decrease in both the basal and the maximal oxygen consumption rate in the hepatocellular carcinoma line (Fig. 5E). These effects on mitochondrial respiration are consistent with the previously characterized mechanism for the inhibitory effect of meclizine on cell growth (41).
Because of these results, we investigated whether mRNA levels for PEBP pathway mRNAs or PFKFBK3 could predict survival risk after HCC diagnosis. Kaplan-Meier survival curves showing the survival time after primary HCC diagnosis in The Cancer Gene Atlas (TCGA) (50) (n=371) cohort were prepared based upon the level of PCYT2, ETNK2 or PFKFB3 mRNA expression in each HCC sample (Fig. 6). The expression level for each mRNA was classified as high (or low) if it was above (or below) the median level in the respective cohort. To confirm this finding, the same analysis was performed using the data obtained from 242 HCC subjects in the Fudan (61) cohort. Subjects whose HCCs had a higher level of ETNK2 mRNA expression had a significantly longer median survival time than those with a lower level in both cohorts (TCGA p=0.04, Fudan p=3×10−5) (Fig. 6, S9). Also, HCC with a lower level of PFKFB3 mRNA expression had a significantly longer survival time (TCGA p=0.01, Fudan p=9×10−4). In contrast, the level of PCYT2 mRNA expression was not associated with a significant survival difference in either cohort (Fig 6). The fact the ETNK2 and PFKFB3 mRNA expression level predicted survival in both cohorts was somewhat unexpected, especially since prior genome-wide mRNA profile clustering failed to identify TCGA subgroups with different survival periods (53). However, the fact that subjects with a lower PFKFB3 mRNA expression level had a longer survival is not surprising. Since PFKB3 enzyme activity generates a metabolite that increases the rate of glycolysis, cancers with a lower level of PFKFB3 enzyme activity will have a lower rate of glycolysis, i.e. they will have less Warburg Effect type of metabolic alterations that are associated with uncontrolled proliferation of cancer cells (68). Although the basis for the relationship between ETNK2 mRNA levels and survival is less clear, it is possible that HCC with higher ETNK2 mRNA levels retain properties of hepatic cells and are more differentiated than HCC with a low level of ETNK2 mRNA. The decrease in ENTK2 mRNA and increase in PFKFB3 mRNA levels as the HCC stage increases is consistent with these possibilities (Fig. S9). Also, HCC with lower PFKFB3 activity will be more dependent upon mitochondrial OXPHOS as an energy source, and thus would be more sensitive to the mitochondrial effect of the elevated P-Etn concentration caused by meclizine-mediated inhibition of PCYT2.
Figure 6.

Survival risk prediction in hepatocellular carcinoma based upon mRNA expression levels. Kaplan-Meier survival curves showing the survival time after hepatocellular cancer diagnosis in The Cancer Gene Atlas (TCGA) (n=371) (53) and Fudan (n=242) (88) cohorts based upon the level of PCYT2, ETNK2 or PFKFB3 mRNA expression in each HCC sample. The expression level for each mRNA was classified as high (or low) if it was above (or below) the median level in each cohort. The p values of a Cox proportional hazard model, which compared the survival times between the two groups within each cohort, are also displayed. A high level of ETNK2 mRNA or a low level of PFKFB3 mRNA expression was associated with significantly increased survival in both cohorts.
Drug efficacy in a human hepatocellular carcinoma liver xenotransplantation model
To assess the efficacy of meclizine and PFK158 treatment in vivo, we developed a novel xenotransplantation model in which a well differentiated human hepatocarcinoma cell line (Hep G2) (43, 44) was transplanted into the liver of immunocompromised NOG mice. The transplanted Hep G2 cells stably expressed a GFP-luciferase fusion protein, which enabled a bioluminescence-based assessment of the human tumor burden within the liver (Fig 7A). One day after transplantation of 1×106 human cells, mice with similar signal intensities in their livers (p=0.98) were randomly separated into two groups (Fig. S10A). Each group was then treated for a two-week period (orally) each day with either vehicle, 25-mg/kg meclizine, 25 mg/kg PFK158 or with both drugs. After two weeks of treatment with either meclizine (p=0.92) or PFK158 alone (p=0.54), the liver tumor burden was not reduced relative to vehicle treated mice (Fig. S10B). However, the liver tumor burden was substantially reduced in mice that received both meclizine and PFK158 relative to vehicle treated mice (P=0.02) (Fig. 7B). Of note, five drug-treated mice had no detectable human tumor cells within their livers, while all of the vehicle treated mice had significant levels of human tumor cells in their livers. Drug combination efficacy in reducing the liver tumor burden was confirmed in an independent experiment (p=0.037) (Fig. 7C). In the vehicle treated mice, the transplanted human hepatocarcinoma cells formed macroscopic tumors that were visible to the naked eye when the liver tissue was harvested, which infiltrated the livers (Fig. 7D). Also, the drug-treated mice had a normal appearance, and no liver toxicity was apparent by serologic testing (P=0.8) (Fig. 7E). Moreover, mice treated with meclizine (25 mg/kg PO qd) and PFK158 (25 mg/kg PO qd) maintained their body weight after 26 days of treatment (p= 0.24) (Fig. S11). Even when drug treatment was delayed for 3 weeks after human hepatocarcinoma cell transplantation, treatment with the combination of meclizine and PFK158 for one (p=0.14), two (p=0.037) or three weeks (p=0.021) caused a significant reduction in liver tumor burden (Fig. 7F). These results demonstrate that treatment with meclizine and PFK158 can synergistically reduce the growth of a human hepatocarcinoma cell line in vivo.
Figure 7.

Treatment with meclizine and PFK158 inhibits the growth of a human hepatocellular carcinoma line in vivo. (A) Diagram of the model. Hep G2 cells expressing a GFP-luciferase fusion protein were transplanted into the livers of NOG mice. One day later, the mice were treated with either vehicle (Veh) or 25 mg/kg Meclizine and 25 mg/kg PFK158 by oral gavage (OG) each day for 14 days (n=7 mice per group). Bioluminescent imaging was then used to measure the amount of human hepatocarcinoma cells within the liver of these mice. (B) Bioluminescent images show the tumor area within the livers of the vehicle and drug treated NOG mice on day 14. The luminescence scale is shown on the right. Of note, the 1st and 4th mouse shown have a very small area of bioluminescence within the splenic area; which represents residual cells present at the site of transplantation. The bioluminescence in the 6th mouse is also not in the liver and is within the colon (or bladder). The graph compares the tumor area within the livers of vehicle and drug treated mice. Each dot shows the measured bioluminescent signal (total flux in photons/second) within the liver region for each mouse; and the horizontal lines are the mean ± SEM for each group. The results of a t-test comparing the measured human tumor areas of the vehicle and P+M treated groups is shown. (C) The day 14 bioluminescent images and graph of the measurements obtained in an independently performed second experiment is shown. (D) Macroscopic tumors are formed within the livers of mice after xenotransplantation of human hepatocarcinoma cells. Left and Center Panels: Representative H/E stained livers section obtained 4 weeks after human hepatocarcinoma cells were transplanted into NOG mice that were treated with either vehicle (Veh) or meclizine and PFK-158 (M+P). While tumor is not present within the M+P treated liver a macroscopic tumor is seen in the vehicle treated liver. The scale bar is 50 uM. Right Panel: Immunofluorescence image of a liver section obtained from a vehicle-treated mouse one month after transplantation of a human hepatocarcinoma cell line expressing GFP. To visualize the areas with the human hepatocarcinoma, the slide was stained with an anti-GFP antibody. The scale bar is 250 uM. In both the H/E and immunostained images, there is an extensive amount of human carcinoma within the liver of vehicle treated mice. (E) PFK158 and Meclizine treatment does not cause liver toxicity. The serum ALT and AST levels were measured on day 14 in the vehicle or drug (P+M) treated (n=7 mice/ group) mice shown in B. The measurement for each mouse is indicated by a dot; and the line indicates the average and SEM for each group. The average serum ALT and AST levels in the P+M group were not different from that of the vehicle treated mice (ALT: 30.9 ± 2.3 vs 38.6 ± 11.1 U/L, p=0.1; AST: 74.1 ± 26.5 vs 78 ± 29.7 U/L, p=0.8). (F) Three weeks after Hep G2 cells expressing a GFP-luciferase fusion protein were transplanted into the livers of NOG mice, the mice were treated with either vehicle (Veh) or 25 mg/kg Meclizine and 25 mg/kg PFK158 by oral gavage (OG) each day for 21 days (n=10 mice per group). The graph compares the tumor area within the livers of vehicle and drug treated mice, which was measured by bioluminescent imaging after 0 (day 21), 1,2, or 3 weeks of drug treatment. Each dot shows the measured bioluminescent signal (total flux in photons/second) within the liver region for each mouse; and the horizontal lines are the mean ± SEM for each group. The results of a t-test comparing the measured human tumor areas of the vehicle and P+M treated groups is shown.
Efficacy for Acute Myeloid Leukemia (AML)
We wanted to determine if PEBP pathway activity could play a role in the progression of a broader range of cancers than was previously known, and if the combination therapy would be effective in other types of cancers. To investigate this, we first examined the effect of meclizine on primary cultures of AML blasts generated from 3 AML patients at the time of their diagnosis (Table S3). Interestingly, after 96 hrs in culture, AML blasts obtained from 1 subject were somewhat sensitive to meclizine alone (IC50=17.7 μM). However, addition of 0.9 μM PFK-158 resulted in the elimination of >95% of the blasts in these cultures in the presence of 10 μM meclizine, and the effect of the drug combination was significantly increased relative to that of 10 μM meclizine alone (p=0.005) (Fig. 8A). The synergistic effect of this drug combination was also observed in AML blast cultures prepared from the two other subjects. When 0.9 μM PFK158 was combined with 0.5 μM meclizine, >95% of the AML blasts from both subjects were eliminated; and the drug combination effect was significantly increased relative to that of meclizine alone (AML SU582, p=0.0001; AML SU839, p=0.0001) (Fig. 8B). These results indicate that the combination of meclizine and PFK158 had a synergistic effect on AML blasts.
Figure 8.

(A) Primary AML blasts obtained from an AML patient at the time of diagnosis were cultured in media containing the indicated concentrations of meclizine in absence (left) or presence of 0.9 uM PKF158 (middle). After 96 hours in culture, the number of live cells was determined by flow cytometry. In the right panel, the numbers of live cells after treatment with the various drug combinations at the indicated concentrations are shown; and the p-values (where significant) for assessing the effect of the different drug concentrations are indicated. The corresponding IC50 for the effect of meclizine on their growth is also indicated. (B) Primary AML blasts obtained from two AML patients were cultured in media containing 0 or 0.5 uM meclizine in absence or presence of 0.9 uM PKF158. After 96 hours in culture, the number of live cells was determined by flow cytometry. The p-values (where significant) for assessing the effect of the meclizine and PFK158 relative to meclizine alone are indicated. In all figures, each bar is the average + SE of 2 independent measurements; and an unpaired t-test was performed on the replicate analyses.
Discussion
The ability to form HOs from human iPSC enables the pathways regulating the various stages in liver development to be analyzed in detail. The early stages are of particular interest, since they are more difficult to analyze in vivo. The developmental stages for liver lineage cells have been well demarcated, which provides a useful map of the route followed during liver development. However, we often lack detailed information about the underlying control mechanisms, which limits our ability to develop therapies for human liver diseases that alter (as in genetic diseases) or usurp (as in liver cancer) the early developmental pathways. Our transcriptomic and metabolomic analysis, along with the results of our PCYT2 gene knockout and meclizine effect studies, indicated that the de novo PEBP is essential for early hepatoblast development. At this stage, there is a coordinate up-regulation of the enzymes required for hepatic PE biosynthesis, and this is accompanied by massive increase in the abundance of the metabolite consumed by the rate-limiting step of this pathway. Similarly, after hepatectomy, there is a marked increase in PE synthesis (69), and ethanolamine availability has been shown to be a primary determinant of the rate of PE synthesis in regenerating liver (70).
Why is meclizine selectively cytotoxic for early hepatoblasts?
The inhibitory effect of meclizine on the growth of hepatocellular carcinoma cells (or AML blasts) cannot be mediated through histamine 1 receptor binding. Analysis of multiple gene expression datasets indicated that the mRNA encoding this receptor is not expressed in Hep G2 cells [(71) and (72)], nor is it expressed in AML blasts (http://firebrowse.org/viewGene.html?gene=hrh1# and (72)). However, meclizine has been shown to significantly alter cellular energy metabolism in a variety of different types of cells in vitro; meclizine treatment causes the cells to utilize a larger percentage of their mitochondrial respiration for ATP production (13). Meclizine’s effect on energy metabolism is rapid (it occurs within minutes), and is mediated via an indirect effect on mitochondria. When isolated mitochondria were treated with meclizine, their respiration, membrane potential and redox potential were not altered (13). The mechanism for this metabolic effect became clear after it was discovered meclizine inhibits PCYT2, which markedly increased cellular phosphoethanolamine levels. Moreover, meclizine increased cellular phosphoethanolamine to a level that inhibited mitochondrial respiration (41), and meclizine-induced effects on cellular respiration were potentiated by addition of ethanolamine (13). The marked increase in the cellular phosphoethanolamine during early HB differentiation renders these cells particularly susceptible to meclizine-induced cell death due to PCYT2 inhibition. However, it is important to note that the mechanism by which phosphoethanolamine alters mitochondrial respiration has not been fully characterized.
Could a commonly used over the counter medication be used to treat primary liver cancer?
The marked increase in the metabolite consumed by the rate-limiting step and of the enzymes required for PE biosynthesis indicates that early hepatoblasts in HO cultures have a need for PE biosynthesis. The similarities in the transcriptome profiles of cells in HO cultures and HCC tissue enabled us to identify an ‘Achilles heel’ for treating certain types of cancers. Of importance, cancer cell susceptibility to meclizine is not directly due to inhibition of PE biosynthesis. In contrast, it results from interrupting the pathway at the level of the rate-limiting enzyme with an inhibitor; this increases the cellular level of phosphoethanolamine, which has damaging effects on mitochondrial function, albeit with an as-yet undefined mechanism.
Alterations in cell metabolism are critical for cancer cell growth (54, 73–76). In addition, fatty acid biosynthesis, which provides the precursors required for PE biosynthesis, is notably the pathway that was identified as most consistently up regulated in primary liver cancer (77). It has also been shown that PCYT2 mRNA and protein levels are increased in a breast cancer cell line in vitro in response to metabolic stress (78). The effect of the combination of meclizine and PFK158 is consistent with the known mechanism of action for each of these drugs. PFK158 reduces the amount of energy produced by glycolysis; this renders cancer cells more dependent upon the energy generated by OXPHOS, which makes them more susceptible to the toxicity caused by the meclizine-induced increase in phosphoethanolamine. A direct inhibitory effect on the cancer cell growth represents a new way in which PFKFB3 inhibitors could be used for cancer treatment. Because glycolysis is the predominant bioenergitic pathway in endothelial cells (where it generates 85% of cellular ATP), it regulates blood vessel sprouting (56). Although PKFB3 inhibitors were originally developed to inhibit metastasis by altering metabolism within the blood vessels supplying the tumor (79), our data demonstrates that PKFB3 inhibitors can be combined with meclizine to synergistically inhibit the growth of the cancer itself. In this novel approach, a glycolysis inhibitor is used to reprogram cancer cell metabolism to make it susceptible to the cytotoxic effect of meclizine. Combining anti-cancer therapies with metabolic alterations is a recently discovered method for increasing their effectiveness. As examples, cancer cell sensitivity to treatment with methotrexate or phosphotidylinositol-3 kinase inhibitors is enhanced by dietary interventions that increase histidine metabolism (80) or reduce hyperglyemia-induced insulin sectretion (81), respectively. Similarly, administration of tetrahydrobioperin (BH4), a metabolite that is an enzyme cofactor, augments anti-cancer immunity (82).
A major factor affecting survival after treatment of HCC is tumor recurrence; the 5-year recurrence rate ranges from 60–70% (83). Most recurrences are limited to the liver and develop within 1-year after resection (84). Even if an HCC is resected at an early stage (size < 2 cm and no evidence of vascular invasion), the 5-year recurrence rate is ~60% (85). Of note, the peak plasma concentration (~0.2 μM) measured after a standard 25 mg oral dose of meclizine in humans (86) is 20-fold below that required for meclizine-induced cell death in early HB cultures. The intrinsic potency of meclizine as an inhibitor of PCYT2 enzyme activity is relatively weak, since it is an off target effect of a drug that was developed as a histamine receptor antagonist. It is likely that a drug with increased potency for PCYT2 inhibition could be quickly identified. Nevertheless, our in vivo xenotransplantation model results demonstrate that meclizine can have a therapeutic effect within the liver. After an oral dose, the liver is exposed to a higher level of meclizine than is found in plasma. Also, post-marketing surveillance data on the use of nonprescription strength meclizine in humans, and published animal studies indicate that higher doses of meclizine can be tolerated (50, 87). Since the meclizine and a glycolysis inhibitor combination was also highly active against acute myeloid leukemic cells, our data indicates that phosphatidyl-ethanolamine biosynthesis is a targetable pathway for cancer treatment, and that meclizine may have clinical efficacy as a repurposed drug for the treatment of HCC and other cancers.
Supplementary Material
Highlights.
Transcriptomic and metabolomic analysis of human hepatic organoids identified the phosphoethanolamine biosynthesis pathway as essential for early liver development.
Meclizine, an over the counter medication that inhibits the rate limiting enzyme in the phosphoethanolamine biosynthesis pathway, could be repurposed for use in a new synergistic combination therapy for treatment of primary liver cancer. A glycolysis inhibitor reprograms cancer cell metabolism to render it susceptible to the cytotoxic effect of meclizine.
This drug combination inhibited the growth of a human liver carcinoma cell line in vitro; and in a xenotransplantation model without causing significant side effets. This drug combination was also highly active against acute myeloid leukemia cells.
Acknowledgements
We also thank Dr. Julien Sage (Stanford University Medical School) for providing the human fibroblasts used in these studies; and Drs. Bob Lewis and Julien Sage for thoughtful review of this manuscript.
Financial Support: This study was supported by an award (1R01DK10218201A1) made to GP from the NIH/NIDDK; and by an award from the Emmerson Collective to GP. The primary AML samples were obtained from the Stanford Hematology Division Tissue Bank.
Footnotes
Conflict of Interest: Stanford University Medical School has filed (on 11/6/18) a U.S. provisional patent application (Serial No. 62/756,230; Combination Therapy for Cancer; G. Peltz and Y. Guan are the named inventors) covering the combined use of PFKFB3 and PCYT2 inhibitors for the treatment of cancer. There are no other conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Kretzschmar K, Clevers H, Organoids: Modeling Development and the Stem Cell Niche in a Dish. Dev Cell 38, 590–600 (2016). [DOI] [PubMed] [Google Scholar]
- 2.Lou YR, Leung AW, Next generation organoids for biomedical research and applications. Biotechnology advances 36, 132–149 (2018). [DOI] [PubMed] [Google Scholar]
- 3.Guan Y et al. , Human Hepatic Organoids for the Analysis of Human Genetic Diseases. JCI Insight 2, pii: 94954 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Trapnell C, Defining cell types and states with single-cell genomics. Genome Res 25, 1491–1498 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Stegle O, Teichmann SA, Marioni JC, Computational and analytical challenges in single-cell transcriptomics. Nat Rev Genet 16, 133–145 (2015). [DOI] [PubMed] [Google Scholar]
- 6.Shapiro E, Biezuner T, Linnarsson S, Single-cell sequencing-based technologies will revolutionize whole-organism science. Nat Rev Genet 14, 618–630 (2013). [DOI] [PubMed] [Google Scholar]
- 7.Tanay A, Regev A, Scaling single-cell genomics from phenomenology to mechanism. Nature 541, 331–338 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Villani AC et al. , Single-cell RNA-seq reveals new types of human blood dendritic cells, monocytes, and progenitors. Science 356, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zeisel A et al. , Brain structure. Cell types in the mouse cortex and hippocampus revealed by single-cell RNA-seq. Science 347, 1138–1142 (2015). [DOI] [PubMed] [Google Scholar]
- 10.Montoro DT et al. , A revised airway epithelial hierarchy includes CFTR-expressing ionocytes. Nature 560, 319–324 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Plasschaert LW et al. , A single-cell atlas of the airway epithelium reveals the CFTR-rich pulmonary ionocyte. Nature 560, 377–381 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Halpern KB et al. , Single-cell spatial reconstruction reveals global division of labour in the mammalian liver. Nature 542, 352–356 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gohil VM et al. , Nutrient-sensitized screening for drugs that shift energy metabolism from mitochondrial respiration to glycolysis. Nat Biotechnol 28, 249–255 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Global Burden of Disease Cancer Collaboration et al. , Global, Regional, and National Cancer Incidence, Mortality, Years of Life Lost, Years Lived With Disability, and Disability-Adjusted Life-years for 32 Cancer Groups, 1990 to 2015: A Systematic Analysis for the Global Burden of Disease Study. JAMA Oncol 3, 524–548 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Petrick JL, Kelly SP, Altekruse SF, McGlynn KA, Rosenberg PS, Future of Hepatocellular Carcinoma Incidence in the United States Forecast Through 2030. J Clin Oncol 34, 1787–1794 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Momin BR, Pinheiro PS, Carreira H, Li C, Weir HK, Liver cancer survival in the United States by race and stage (2001–2009): Findings from the CONCORD-2 study. Cancer 123 Suppl 24, 5059–5078 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Heimbach JK et al. , AASLD guidelines for the treatment of hepatocellular carcinoma. Hepatology 67, 358–380 (2018). [DOI] [PubMed] [Google Scholar]
- 18.Ramskold D et al. , Full-length mRNA-Seq from single-cell levels of RNA and individual circulating tumor cells. Nat Biotechnol 30, 777–782 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wu M et al. , Opiate-induced Changes in Brain Adenosine Levels and Narcotic Drug Responses. Neuroscience 228, 235–242 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Park W et al. , Metabolomic Markers Differentiate Mucinous and Non-Mucinous Pancreatic Cysts. Gastrointestinal Endoscopy 78, 295–302 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wu Manhong et al. , Increased Dipeptide Abundance in Non-Small Cell Lung Cancer Rapid Commun. Mass Spectrom. 27, 2091–2098 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Guo K, Li L, Differential 12C-/13C-isotope dansylation labeling and fast liquid chromatography/mass spectrometry for absolute and relative quantification of the metabolome. Anal Chem 81, 3919–3932 (2009). [DOI] [PubMed] [Google Scholar]
- 23.Guijas C, Montenegro-Burke JR, Warth B, Spilker ME, Siuzdak G, Metabolomics activity screening for identifying metabolites that modulate phenotype. Nat Biotechnol 36, 316–320 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hsu PD et al. , DNA targeting specificity of RNA-guided Cas9 nucleases. Nat Biotechnol 31, 827–832 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ito M et al. , NOD/SCID/gamma(c)(null) mouse: an excellent recipient mouse model for engraftment of human cells. Blood 100, 3175–3182 (2002). [DOI] [PubMed] [Google Scholar]
- 26.Sinha S et al. , Mutant WT1 is associated with DNA hypermethylation of PRC2 targets in AML and responds to EZH2 inhibition. Blood 125, 316–326 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Islam S et al. , Quantitative single-cell RNA-seq with unique molecular identifiers. Nat Methods 11, 163–166 (2014). [DOI] [PubMed] [Google Scholar]
- 28.Camp JG et al. , Multilineage communication regulates human liver bud development from pluripotency. Nature 546, 533–538 (2017). [DOI] [PubMed] [Google Scholar]
- 29.Welch JD, Hartemink AJ, Prins JF, SLICER: inferring branched, nonlinear cellular trajectories from single cell RNA-seq data. Genome Biol 17, 106 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vance JE, Tasseva G, Formation and function of phosphatidylserine and phosphatidylethanolamine in mammalian cells. Biochim Biophys Acta 1831, 543–554 (2013). [DOI] [PubMed] [Google Scholar]
- 31.Sundler R, Akesson B, Regulation of phospholipid biosynthesis in isolated rat hepatocytes. Effect of different substrates. J Biol Chem 250, 3359–3367 (1975). [PubMed] [Google Scholar]
- 32.Tijburg LB, Geelen MJ, Van Golde LM, Biosynthesis of phosphatidylethanolamine via the CDP-ethanolamine route is an important pathway in isolated rat hepatocytes. Biochem Biophys Res Commun 160, 1275–1280 (1989). [DOI] [PubMed] [Google Scholar]
- 33.Tijburg LB, Houweling M, Geelen MJ, van Golde LM, Effects of dietary conditions on the pool sizes of precursors of phosphatidylcholine and phosphatidylethanolamine synthesis in rat liver. Biochim Biophys Acta 959, 1–8 (1988). [DOI] [PubMed] [Google Scholar]
- 34.Bleijerveld OB, Brouwers JF, Vaandrager AB, Helms JB, Houweling M, The CDP-ethanolamine pathway and phosphatidylserine decarboxylation generate different phosphatidylethanolamine molecular species. J Biol Chem 282, 28362–28372 (2007). [DOI] [PubMed] [Google Scholar]
- 35.Patel D, Witt SN, Ethanolamine and Phosphatidylethanolamine: Partners in Health and Disease. Oxid Med Cell Longev 2017, 4829180 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lykidis A, Wang J, Karim MA, Jackowski S, Overexpression of a mammalian ethanolamine-specific kinase accelerates the CDP-ethanolamine pathway. J Biol Chem 276, 2174–2179 (2001). [DOI] [PubMed] [Google Scholar]
- 37.Fullerton MD, Hakimuddin F, Bakovic M, Developmental and metabolic effects of disruption of the mouse CTP:phosphoethanolamine cytidylyltransferase gene (Pcyt2). Mol Cell Biol 27, 3327–3336 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Leonardi R, Frank MW, Jackson PD, Rock CO, Jackowski S, Elimination of the CDP-ethanolamine pathway disrupts hepatic lipid homeostasis. J Biol Chem 284, 27077–27089 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.(Pfizer, New York, NY, 1999). [Google Scholar]
- 40.Paul MA, MacLellan M, Gray G, Motion-sickness medications for aircrew: impact on psychomotor performance. Aviat Space Environ Med 76, 560–565 (2005). [PubMed] [Google Scholar]
- 41.Gohil VM et al. , Meclizine inhibits mitochondrial respiration through direct targeting of cytosolic phosphoethanolamine metabolism. J Biol Chem 288, 35387–35395 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nakabayashi H, Taketa K, Miyano K, Yamane T, Sato J, Growth of human hepatoma cells lines with differentiated functions in chemically defined medium. Cancer Res 42, 3858–3863 (1982). [PubMed] [Google Scholar]
- 43.Pang RW, Poon RT, Cancer stem cell as a potential therapeutic target in hepatocellular carcinoma. Curr Cancer Drug Targets 12, 1081–1094 (2012). [DOI] [PubMed] [Google Scholar]
- 44.Knowles BB, Howe CC, Aden DP, Human hepatocellular carcinoma cell lines secrete the major plasma proteins and hepatitis B surface antigen. Science 209, 497–499 (1980). [DOI] [PubMed] [Google Scholar]
- 45.Ma S et al. , Identification and characterization of tumorigenic liver cancer stem/progenitor cells. Gastroenterology 132, 2542–2556 (2007). [DOI] [PubMed] [Google Scholar]
- 46.Ma S et al. , miR-130b Promotes CD133(+) liver tumor-initiating cell growth and self-renewal via tumor protein 53-induced nuclear protein 1. Cell Stem Cell 7, 694–707 (2010). [DOI] [PubMed] [Google Scholar]
- 47.Yamashita T et al. , EpCAM-positive hepatocellular carcinoma cells are tumor-initiating cells with stem/progenitor cell features. Gastroenterology 136, 1012–1024 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ho CM et al. , Histopathological evidence for the existence of primary liver progenitor cell cancer: insight from cancer stem cell pathobiology. Discov Med 23, 41–50 (2017). [PubMed] [Google Scholar]
- 49.Yang ZF et al. , Identification of local and circulating cancer stem cells in human liver cancer. Hepatology 47, 919–928 (2008). [DOI] [PubMed] [Google Scholar]
- 50.Wan X et al. , CD24 promotes HCC progression via triggering Notch-related EMT and modulation of tumor microenvironment. Tumour Biol 37, 6073–6084 (2016). [DOI] [PubMed] [Google Scholar]
- 51.Lee TK et al. , CD24(+) liver tumor-initiating cells drive self-renewal and tumor initiation through STAT3-mediated NANOG regulation. Cell Stem Cell 9, 50–63 (2011). [DOI] [PubMed] [Google Scholar]
- 52.Xu X et al. , DLK1 as a potential target against cancer stem/progenitor cells of hepatocellular carcinoma. Mol Cancer Ther 11, 629–638 (2012). [DOI] [PubMed] [Google Scholar]
- 53.w. b. e. Cancer Genome Atlas Research Network. Electronic address, N. Cancer Genome Atlas Research, Comprehensive and Integrative Genomic Characterization of Hepatocellular Carcinoma. Cell 169, 1327–1341 e1323 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Pavlova NN, Thompson CB, The Emerging Hallmarks of Cancer Metabolism. Cell metabolism 23, 27–47 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rodriguez-Enriquez S, Juarez O, Rodriguez-Zavala JS, Moreno-Sanchez R, Multisite control of the Crabtree effect in ascites hepatoma cells. Eur J Biochem 268, 2512–2519 (2001). [DOI] [PubMed] [Google Scholar]
- 56.DeBerardinis RJ et al. , Beyond aerobic glycolysis: transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proc Natl Acad Sci U S A 104, 19345–19350 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Le A et al. , Glucose-independent glutamine metabolism via TCA cycling for proliferation and survival in B cells. Cell metabolism 15, 110–121 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Clem BF et al. , Targeting 6-phosphofructo-2-kinase (PFKFB3) as a therapeutic strategy against cancer. Mol Cancer Ther 12, 1461–1470 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Clem B et al. , Small-molecule inhibition of 6-phosphofructo-2-kinase activity suppresses glycolytic flux and tumor growth. Mol Cancer Ther 7, 110–120 (2008). [DOI] [PubMed] [Google Scholar]
- 60.Conradi LC et al. , Tumor vessel disintegration by maximum tolerable PFKFB3 blockade. Angiogenesis 20, 599–613 (2017). [DOI] [PubMed] [Google Scholar]
- 61.Boyd S et al. , Structure-Based Design of Potent and Selective Inhibitors of the Metabolic Kinase PFKFB3. J Med Chem 58, 3611–3625 (2015). [DOI] [PubMed] [Google Scholar]
- 62.Telang S et al. , Discovery of a PFKFB3 inhibitor for phase I trial testing that synergizes with the B-Raf inhibitor vemurafenib. Cancer & Metabolism 2(Suppl 1), 14–15 (2014).25215185 [Google Scholar]
- 63.Redman R, Pohlmann P, Kurman M, Tapolsky G, Chesney J, PFK-158, first-in-man and first-in-class inhibitor of PFKFB3/ glycolysis: A phase I, dose escalation, multi-center study in patients with advanced solid malignancies. Cancer Research 75 (Suppl 15), Abstract nr CT206 (2015). [Google Scholar]
- 64.Clem BF, O’Neal J, Klarer AC, Telang S, Chesney J, Clinical development of cancer therapeutics that target metabolism. QjM 109, 367–372 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Obach M et al. , 6-Phosphofructo-2-kinase (pfkfb3) gene promoter contains hypoxia-inducible factor-1 binding sites necessary for transactivation in response to hypoxia. J Biol Chem 279, 53562–53570 (2004). [DOI] [PubMed] [Google Scholar]
- 66.Atsumi T et al. , High expression of inducible 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase (iPFK-2; PFKFB3) in human cancers. Cancer Res 62, 5881–5887 (2002). [PubMed] [Google Scholar]
- 67.Atsumi T et al. , Expression of inducible 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase/PFKFB3 isoforms in adipocytes and their potential role in glycolytic regulation. Diabetes 54, 3349–3357 (2005). [DOI] [PubMed] [Google Scholar]
- 68.Liberti MV, Locasale JW, The Warburg Effect: How Does it Benefit Cancer Cells? Trends Biochem Sci 41, 211–218 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Abel S, Smuts CM, de Villiers C, Gelderblom WC, Changes in essential fatty acid patterns associated with normal liver regeneration and the progression of hepatocyte nodules in rat hepatocarcinogenesis. Carcinogenesis 22, 795–804 (2001). [DOI] [PubMed] [Google Scholar]
- 70.Houweling M, Tijburg LB, Vaartjes WJ, van Golde LM, Phosphatidylethanolamine metabolism in rat liver after partial hepatectomy. Control of biosynthesis of phosphatidylethanolamine by the availability of ethanolamine. Biochem J 283 ( Pt 1), 55–61 (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Liguori MJ, Blomme EA, Waring JF, Trovafloxacin-induced gene expression changes in liver-derived in vitro systems: comparison of primary human hepatocytes to HepG2 cells. Drug Metab Dispos 36, 223–233 (2008). [DOI] [PubMed] [Google Scholar]
- 72.Cancer Cell Line Encyclopedia Consortium, Genomics of Drug Sensitivity in Cancer Consortium, Pharmacogenomic agreement between two cancer cell line data sets. Nature 528, 84–87 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Boroughs LK, DeBerardinis RJ, Metabolic pathways promoting cancer cell survival and growth. Nat Cell Biol 17, 351–359 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Zhang J, Pavlova NN, Thompson CB, Cancer cell metabolism: the essential role of the nonessential amino acid, glutamine. EMBO J 36, 1302–1315 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Nwosu ZC et al. , Identification of the Consistently Altered Metabolic Targets in Human Hepatocellular Carcinoma. Cell Mol Gastroenterol Hepatol 4, 303–323 e301 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.DelNero P, Hopkins BD, Cantley LC, Fischbach C, Cancer metabolism gets physical. Sci Transl Med 10, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Bjornson E et al. , Stratification of Hepatocellular Carcinoma Patients Based on Acetate Utilization. Cell reports 13, 2014–2026 (2015). [DOI] [PubMed] [Google Scholar]
- 78.Zhu L, Bakovic M, Breast cancer cells adapt to metabolic stress by increasing ethanolamine phospholipid synthesis and CTP:ethanolaminephosphate cytidylyltransferase-Pcyt2 activity. Biochem Cell Biol 90, 188–199 (2012). [DOI] [PubMed] [Google Scholar]
- 79.Cantelmo AR et al. , Inhibition of the Glycolytic Activator PFKFB3 in Endothelium Induces Tumor Vessel Normalization, Impairs Metastasis, and Improves Chemotherapy. Cancer Cell 30, 968–985 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kanarek N et al. , Histidine catabolism is a major determinant of methotrexate sensitivity. Nature 559, 632–636 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Hopkins BD et al. , Suppression of insulin feedback enhances the efficacy of PI3K inhibitors. Nature 560, 499–503 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Cronin SJF et al. , The metabolite BH4 controls T cell proliferation in autoimmunity and cancer. Nature 563, 564–568 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Doganay L, Hepatocellular Cancer: Is Recurrence Inevitable? J Gastrointest Cancer, (2017). [DOI] [PubMed] [Google Scholar]
- 84.Wang Y et al. , Radiofrequency ablation versus hepatic resection for small hepatocellular carcinomas: a meta-analysis of randomized and nonrandomized controlled trials. PLoS One 9, e84484 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Roayaie S et al. , Resection of hepatocellular cancer </=2 cm: results from two Western centers. Hepatology 57, 1426–1435 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Wang Z et al. , Meclizine metabolism and pharmacokinetics: formulation on its absorption. J Clin Pharmacol 52, 1343–1349 (2012). [DOI] [PubMed] [Google Scholar]
- 87.Lo RC, Ng IO, Hepatic progenitor cells: their role and functional significance in the new classification of primary liver cancers. Liver Cancer 2, 84–92 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Roessler S et al. , A unique metastasis gene signature enables prediction of tumor relapse in early-stage hepatocellular carcinoma patients. Cancer Res 70, 10202–10212 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.