

SUMMARY
During cell division, the inheritance of a functional endoplasmic reticulum (ER) is ensured by the endoplasmic reticulum stress surveillance (ERSU) pathway. Activation of ERSU causes the septin ring to mislocalize, which blocks ER inheritance and cytokinesis. Here, we uncover that the septin ring in fact translocates to previously utilized cell division sites called cytokinetic remnants (CRMs). This unconventional translocation requires Nba1, a negative polarity regulator that normally prevents repolarization and re-budding at CRMs. Furthermore, septin ring translocation relies on the recruitment and activation of a key ERSU component Slt2 by Bem1, without activating Cdc42. Failure to transfer all septin subunits to CRMs delays the cell’s ability to re-enter the cell cycle when ER homeostasis is restored, and hinders cell growth after ER stress recovery. Thus, these deliberate but unprecedented rearrangements of cell polarity factors during ER stress safeguards cell survival and the timely cell cycle re-entry upon ER stress recovery.
Graphical Abstract
eTOC
How cells manage a sudden onset of stress is crucial for their survival. Chao et al. show that cells under ER stress can temporarily pause the cell cycle until the stress is mitigated. They accomplish this by transferring cell polarity components away from growth sites and onto cytokinetic remnants
INTRODUCTION
The functions of eukaryotic cells are organized and distributed into specific organelles. During the cell cycle, not only does the genome divide but organelles must be correctly distributed. Thus, one of the fundamental questions in cell biology is how specific organelles are inherited during the cell cycle. The endoplasmic reticulum (ER) is the major organelle responsible for the production and quality control of almost all secretory proteins. In addition, the ER is crucial for lipid biosynthesis and calcium homeostasis (Denic et al., 2006; Feige and Hendershot, 2011; Frakes and Dillin, 2017; Mori, 2000; Ron and Walter, 2007; Rutkowski and Kaufman, 2004). Importantly, the ER cannot be synthesized de novo and, instead, must be inherited from the mother cell. This suggests the presence of ER inheritance checkpoints.
The budding yeast, Saccharomyces cerevisiae, is an ideal model organism to study the division of the ER during the cell cycle, due to the asymmetric nature of yeast cell division. The ER in yeast is spatially segregated into the cortical ER (cER) that lies in the cell cortex, with some sections of the cER contacting the plasma membrane. The cER is connected by a few ER tubules with the peri-nuclear ER that is contiguous with the outer nuclear envelope (Barlowe, 2010; Bechmann et al., 2012; Du et al., 2004; Fehrenbacher et al., 2002; Hereford and Hartwell).
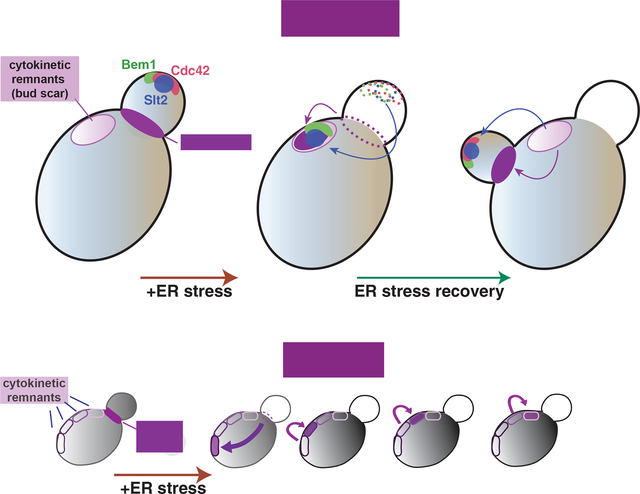
Previously, we discovered a cell cycle checkpoint for ensuring that functional ER is transferred to the daughter cell during the cell cycle, which we termed the ER stress surveillance (ERSU) pathway (Babour et al., 2010; Pina et al., 2018; Pina et al., 2016; Pina and Niwa, 2015). If the accumulation of unfolded or misfolded proteins exceeds ER functional capacity, ER homeostasis is disrupted, leading to a condition known as ER stress. In response to ER stress during the cell cycle, the ERSU pathway blocks the inheritance of the ‘stressed ER’ into the daughter cell and mobilizes the septin ring from the bud neck, ultimately leading to cell cycle arrest at cytokinesis.
Surprisingly, the ERSU is independent of the well-known unfolded protein response (UPR) signaling pathway that regulates ER functional homeostasis. In parallel to the ERSU pathway, ER stress activates the UPR pathway to up-regulate the transcription of genes coding for ER chaperones and protein folding components to re-establish ER functional homeostasis (Ron and Walter, 2007). When ER functional homeostasis is re-established, cells are released from cell cycle arrest, re-enter the cell cycle, and inherit a functional ER into the daughter cell (Babour et al., 2010). Thus, the ERSU is one of the checkpoint mechanisms, and works in concert with the UPR pathway to ensure proper organelle inheritance.
One of the hallmark events of the ERSU is septin ring mislocalization away from the bud neck. This process is a key mechanism leading to cytokinesis arrest in response to ER stress. The septin ring is composed of five septin subunits, Shs1, Cdc3, 10, 11, and 12, and its formation is dynamically regulated during the cell cycle (Field and Kellogg, 1999; Gladfelter et al., 2001; Mostowy and Cossart, 2012; Oh and Bi, 2011; Versele and Thorner, 2005a; Weirich et al., 2008). Septin ring formation occurs even prior to the emergence of the daughter cell (bud), marking the incipient bud site. The formation of the septin ring is tightly linked to the targeting of activated Cdc42, which allows emergence and polarized growth of the bud. Cdc42 activity is tightly regulated by its upstream components including Cdc24, a GTP exchange factor (GEF) of Cdc42, and Bud1/Rsr1. The septin ring stays at the bud neck between the mother and daughter cells throughout most of the cell cycle. At the end of the cell cycle, the dividing membrane between the mother and daughter cell (i.e., the septum) forms, followed by cell division. Finally, the septin ring disassembles into subunits, which then re-initiate the cycle of assembly/disassembly. Interestingly, septin ring assembly is normally inhibited at cytokinesis remnants (CRMs), which are previously utilized cell cycle division sites, through a block involving Nba1 (Meitinger et al., 2014). This ensures that budding and subsequent cytokinesis occurs only at naïve locations that have never been used as cell division sites. The number of CRMs increases as a yeast cell undergoes more rounds of cell cycle, and thus it provides a molecular clock for the age of the yeast cell (Caudron and Barral, 2009).
Septin ring mislocalization is important for the ERSU pathway. In slt2Δ cells that are unable to mount the ERSU response, the septin ring remains present at the bud neck during ER stress and cells subsequently die. However, we still do not know the details of how septin rings become mislocalized, or the functional significance of this process. Therefore, in this study we used molecular and cell biology approaches to investigate septin ring dynamics during ER stress.
RESULTS
The septin ring moves to the bud scar in response to ER stress
To characterize septin ring movement during ER stress, we performed time-lapse microscopy. We monitored the morphology and localization of the septin ring in a wild-type (WT) cell carrying green fluorescent protein (GFP)-tagged septin subunit Shs1 (Shs1-GFP). Previously, we found that the formation of septin rings occurred normally at the bud neck in small budded cells even under ER-stressed conditions (Babour et al., 2010). Thus, we reasoned that any mislocalization of the septin ring must occur at a later cell cycle stage, and monitored the behavior of Shs1-GFP in large budded G2 cells. Under normal growth, as cells entered mitosis, the Shs1-GFP ring split into two and its fluorescent levels started to decrease. Subsequently, Shs1-GFP began to accumulate at the incipient bud site (Figure 1A, Movie S1), consistent with previously reported septin ring dynamics (Oh and Bi, 2011; Versele and Thorner, 2005b).
Figure 1. Septin rings relocalize to CRMs during ER stress.

(A) Time-lapse analysis of septin dynamics (Shs1-GFP) in untreated WT cells. All scale bars, 2 μm. Later in the time course, a newly formed septin ring and a daughter cell are indicated by red arrows. Zoom up pictures of bud necks. Related to Movie S1.
(B) Time-lapse analysis of septin rings in WT cells that were treated with 1 μg/ ml Tm. Red arrows point to translocated septin rings without the emergence of a new bud that is indicated by red arrowheads. Related to Movie S2.
(C) Shs1-GFP cells were grown with (+) or without (−) 1μg/ ml Tm. CRMs were visualized by CW staining. Inserts contain zoomed images from the bud neck region.
(D) Quantifications for Shs1-GFP localization in +/− Tm conditions.
(E) Representative electron micrographs taken from the bud neck regions of WT cells. Cells were synchronized for 30 min with alpha factor, released and either treated with DMSO (no Tm) or incubated with 15 μg/ml Tm for 2 hr (+Tm), and then fixed. White arrows indicate septums; scale bar represents 0.5 μm. PS, primary septum; SS, secondary septum.
(F) Quantification of the experiment in (E). Upon ER stress induction by Tm treatment, the number of cells with primary septum (PS) (but not secondary septum (SS)) (shown in gray) significantly increased. In contrast, both PS and SS formed in unstressed cells (white column).
To monitor Shs1-GFP dynamics under ER stress, we switched to growth medium containing tunicamycin (Tm), a well-characterized ER stress inducer. Tm inhibits protein N-glycosylation in the ER, resulting in an accumulation of unfolded proteins (Kuo and Lampen, 1974). In marked contrast to cells grown in normal media, the septin ring moved to a site adjacent to the bud neck (Figure 1B, Movie S2). Importantly, we observed no apical growth of a new daughter cell at the site of septin translocation (Movie S2).
In the BY genetic background, haploid cells adopt an axial budding pattern in which new buds consistently form adjacent to the previous bud site or CRMs that include both bud and birth scars. We found that under ER stress, ~45% of Shs1-GFP co-localized with CRMs which could be visualized by calcofluor white (CW) staining (Figures 1C–D).
Septin translocation to CRMs was not specific to Tm, as we observed similar Shs1 behavior when we activated ER stress using ero1–1 temperature-sensitive mutant cells at the non-permissive temperature (Figure S1A) (ERO1 codes for the oxidoreductase that catalyzes disulfide bond formation in the ER. ero1–1 at 37°C reliably induces the ER stress response (Frand and Kaiser, 1998; Pollard et al., 1998; Tu et al., 2000)). Previously, we reported that septin subunits, Shs1, Cdc10, 12, and 11 were all mislocalized from the bud neck under ER stress (Babour et al., 2010). Consistently, we saw the co-localization other septin subunits such as Cdc11-GFP and Cdc10-GFP with CRMs (Figure S1B and 3D, respectively). In the W303 yeast strain, new buds emerge from distal positions with respect to the current site of division, presumably due to BUD4 mutations (Voth et al., 2005). Indeed, we found that Shs1-GFP in W303 background was also localized to CRMs in response to ER stress (Figures S1C–S1E, Movies S3 and S4), revealing that the septin ring translocation to CRMs occurs regardless of which budding pattern was being used.
Septin transfer correlates with cytokinesis block during ER stress
As the septin ring is coordinated with cytokinesis and the formation of primary and secondary septum, which are cell walls that form in between mother and daughter cells (Onishi et al., 2013; Schmidt et al., 2002; Weiss, 2012; Wloka and Bi, 2012), we hypothesized that the translocation of the septin ring might disrupt septum formation. Electron microscopy analyses of both primary and secondary septum revealed that many ER stressed cells had no secondary septum even though primary septum formed normally (Figures 1E–F). We also examined Myo1, which is a type II myosin that forms an actomyosin contractile ring targeted to the bud neck by septins to form the actomyosin contractile ring (Bi et al., 1998; Lippincott and Li, 1998; Tolliday et al., 2003). In unstressed WT cells carrying the genomic replacement of Myo1-GFP and Shs1-RFP, Myo1-GFP was localized at the bud neck and sandwiched between two septin rings (Figure 2A). Upon ER stress induction, Myo1-GFP co-localized with translocated Shs1-RFP, and thus was no longer in the bud neck (Figures 2A–B). The Myo1 recruiter, Bni5, also translocated to CRMs as well (Figure 2C). Taken together, these findings suggested that ER stress-induced septin movement occurred concurrently with the mislocalization of the actomyosin contractile ring, leading to a delay in cytokinesis. Curiously, MYO1 deletion results in multi-budded cells due to defects in cell division despite continued polarized growth (Bi et al., 1998; Lord et al., 2005; Watts et al., 1987). However, the growth phenotype of ER-stressed cells differs from that of myo1Δ cells, suggesting that polarized growth is terminated in ER-stressed cells.
Figure 2. The Bud1/Rsr1 GTPase complex is negatively regulated during ER stress.

(A) Myo1-GFP was co-localized with Shs1-RFP under both normal growth and ER stress. Cells were either grown without or with 1 μg/ml Tm. White arrows, RFP localization; yellow arrows, GFP. White and yellow arrowheads show altered localizations of Shs1 and Myo1, respectively. All scale bars, 2μm.
(B) Quantification of Myo1-GFP in WT cells grown normally or under ER stress (1 μg/ml Tm) showed that Myo1-GFP moved with Shs1-RFP upon ER stress induction. Standard errors (SE) and statistic significances were calculated as described in materials and methods section unless otherwise stated.
(C) Bni5-GFP cells were grown without or with Tm. CRMs were visualized by CW staining.
(D, E) Spa2-GFP cells were grown without or with Tm. CW staining highlights the CRMs.
(F) Cells co-expressing an active Cdc42 biosensor (Gic2-PBD-RFP) and Shs1-GFP were grown with or without 1μg/ml Tm. Top, locations of active Cdc42 are indicated by white arrows; Shs1-GFP is shown by blue arrows. Bottem, quantification of the average fluorescence intensities of active Cdc42 (by Gic2-PBD-RFP) at the bud tip in cells grown with or without 1 μg/ml Tm. Mislocalized Gic2-RFP is indicated with yellow asterisks. Measurements of Gic2-RFP levels were made for cells in each frame of time-lapse experiments shown in Figures S2A and S2B (Related to Figures S2A and S2B). See also Movies S5 and S6.
(G) Schematics of the Cdc42-activating Bud1/Rsr1 GTPase module.
(H, I) Localizations of Cdc24-GFP in different stages of the WT cell cycle, grown either without (H) or with Tm (I). Cdc24 at the bud tip or bud neck is shown by white arrows; mislocalized Cdc24-GFP is indicated with yellow asterisks. CRMs were also visualized by staining with CW. Representative cells from each stage of the cell cycle are shown.
(J) Quantification of the Cdc24-GFP localizations shown in (H) and (I). The y-axis represents the ratios between the surface areas of buds and mothers; higher values indicate larger buds. The percentages of cells with the indicated localizations are shown above each column. Individual data points, as well as mean and SE are plotted, and the p value comparing localization of Cdc24-GFP to CRMs in untreated and treated samples was calculated. *, p < 0.0001.
Septin movement to CRMs occurs without induction of polarized growth
Our finding that the septin ring translocated to CRMs during ER stress could partially explain the differences in phenotypes between myo1Δ cells and ER-stressed cells. Under normal growth, septin ring formation at the presumptive bud site is directed by the master regulator of cell polarity, Cdc42 (Okada et al., 2013). However, at CRMs, a Cdc42-inhibitory circuit is present to prevent ‘refractory budding’ (Meitinger et al., 2014). Thus, translocation to CRMs during ER stress should allow septin ring to access the inactivation mechanism of Cdc42 and polarized growth at CRMs.
To confirm the status of polarized growth in ER stress, we first visualized the polarisome component Spa2, which acts to organize the actin cytoskeleton at sites of polarized growth (Sheu et al., 1998). Spa2 was dispersed and not enriched at CRMs in ER stressed cells, supporting the lack of the polarized growth (Figures 2D–E). A fluorescence biosensor, Gic2-PBD-RFP, visualizes only the active, GTP-bound form of Cdc42 (Okada et al., 2013). In unstressed cells, active Cdc42 was enriched in the bud cortex, whereas the septin ring was localized in the bud neck (Figure 2F), as reported previously (Okada et al., 2013). In contrast, during ER stress, septin translocation coincided with the dispersal of active Cdc42 (Figure 2F), indicating that active Cdc42 did not accumulate at CRMs. Using time-lapse microscopy, we found that during normal growth, active Cdc42 accumulated in the growing bud of small-budded cells (Figure S2A, Movie S5). In contrast, in time-lapse imaging of a stressed cell with a similarly sized bud, fluorescent signals of active Cdc42 dispersed within ~30 minutes and the bud did not grow (Figure S2B, Movie S6).
ER stress disperses Cdc42 from the site of polarized growth, disconnecting its upstream effectors
To investigate the mechanism of how ER stress leads to the inactivation of Cdc42 from the site of polarized growth, we examined an upstream component, Cdc24, the GEF for Cdc42 (Figure 2G) (Hereford and Hartwell, 1974). In normal growing cells, Cdc24-GFP localized to sites of polarized growth including the incipient bud site in G1, the bud cortex in S and G2, and the bud neck in M phase (Figure 2H), as previously reported (Bos et al., 2007). In contrast, during ER stress the majority (66%) of cells had Cdc24-GFP moved to CRMs (Figures 2I–J).
Bud1/Rsr1 interacts with and activates Cdc24 at sites of polarized growth. Thus, we examined the active (GTP-bound) form of Bud1 using a split-yellow fluorescent protein (YFP) protein complementation assay (PCA) in vivo. In this assay, the YFP protein is split into two fragments and each non-fluorescent fragment is attached to a protein of interest, a bait or a prey protein. The interaction of these two proteins brings together two YFP fragments and restores YFP fluorescence. Under normal growth, we observed YFP signals generated from Bud1-Cdc24 interactions at the bud tip and bud neck (Figure S2C, white arrowheads), consistent with previous reports (Park et al., 1997; Park et al., 2002). During ER stress, we did not detect significant YFP signal either at the bud tip or the bud neck (Figure S2D: yellow arrowheads for loss of or reduced YFP signals). Together, our results suggested that while some Cdc24 remained at the bud neck, the majority no longer interacted with Bud1.
Similarly, we examined the Bud1-activating components Bud5 (a GEF for Bud1) and Bud2 (a GTPase-activating protein, GAP, for Bud1) by split-YFP (Figures S2E–H) (Marston et al., 2001; Nelson et al., 2012). YFP generated from Bud1-Bud5 was localized at the bud tip (G1 and S) and the bud neck (G2/M and M) (Figure S2E). YFP generated from Bud1-Bud2 was also localized at the bud tip (G1 and S) and the bud neck (Figure S2G), consistent with previous report for localizations for Bud5 and Bud2 (Kang et al., 2001). In contrast, neither Bud1-Bud5 nor Bud1-Bud2 was localized at the site of polarized growth under ER stress (Figures S2F & 2H; yellow arrows indicated loss of interaction). Furthermore, we did not observe significant fluorescent signals at CRMs, indicating the absence of Bud1-Bud5 and Bud1-Bud2 interactions at CRMs. Taken together, these results showed that ER stress mislocalized Cdc24 to CRMs and attenuates the activity of the Bud1 GTPase.
Not all septin subunits are required to assemble at CRMs to inactivate Cdc42
We next examined whether septin ring translocation to CRMs is critical for inactivation of either Cdc42 or its activating components. To this end, we generated a septin mutant that moved away from the bud neck but did not reach to CRMs during ER stress: a C-terminal truncation of the Shs1 septin subunit, Shs1ΔCTE (CTE: C-terminal extension; Figure 3A). During normal growth, Shs1ΔCTE-GFP was localized at the bud neck like WT Shs1-GFP (Figure 3B). However, upon ER stress induction ~70% of shs1ΔCTE cells had no detectable Shs1ΔCTE localized at CRMs (Figure 3B). Shs1ΔCTE-GFP expression levels were similar to WT Shs1-GFP even after ER stress induction (Figure 3C), revealing that Shs1 were dispersed throughout the cytosol. In contrast to Shs1, ER stress induction caused other septin subunits including Cdc3, Cdc10, Cdc11 and Cdc12 to move from the bud neck to the CRMs (Figure 3E). Taken together, these results revealed that the CTE of Shs1 is an important element for its translocation, but not other septin subunits, to CRMs in response to ER stress.
Figure 3. The CTE of Shs1 diminishes Shs1 to move to CRMs in response to ER stress.

(A) Domain organization of Shs1 (Versele and Thorner, 2004), the G domain, and GTPase binding. Shs1ΔCTE lacks C-terminal end (CTE) of WT Shs1 (aa. 349–551).
(B) Shs1ΔCTE-GFP is dispersed in cells grown with 1 μg/ml Tm, while it is localized at the bud neck during normal growth. Arrowheads point absence of GFP localization. CFW staining shows CRMs (red). Quantification of Shs1ΔCTE-GFP in WT cells with or without 1 μg/ml Tm. * represents p<0.0001 comparing % of cells with dispersed Shs1ΔCTE-GFP.
(C) Western blot analysis of Shs1-GFP and Shs1ΔCTE-GFP expression in cells with or without 1 μg/ml Tm. Anti-Pgk1 was used as a loading control.
(D) CDC10-GFP in cells grown without or with Tm. All scale bars, 2 μm
(E) shs1ΔCTE cells expressing GFP tagged septin subunits at their genomic loci. Cells were grown without or with Tm, with CRMs visualized by CW staining.
(F) Localization of Cdc42 (Gic2) in shs1ΔCTE cells that were either untreated (no Tm) or treated with 1μg/ml Tm. White arrows show the polarized localization of Gic2-GFP in unstressed cells, which was lost in ER-stressed cells.
(G) Quantification of active Cdc42-GFP fluorescence levels in shs1ΔCTE cells grown with or without 1 μg/ml Tm. Mean and SE were calculated based on values from at least three independent experiments. * represents p<0.0001 comparing normalized Gic2-GFP levels between unstressed and ER stressed cells.
Localization of Cdc24-GFP in shs1ΔCTE cells that were either untreated or treated with 1μg/ml Tm. ER stress caused mislocalization of Cdc24-GFP (yellow arrow heads).
(I, J) Bni5-GFP (I) and Myo1-GFP (J) in shs1ΔCTE cells were grown without or with Tm. CRMs were visualized by CW staining.
Do shs1ΔCTE cells support ER stress-induced block of polarized growth? In ER-stressed shs1ΔCTE cells, active Cdc42 was no longer present at the bud tip or the bud neck or any other locations including CRMs; (Figures 3F–G). Cdc24 in shs1ΔCTE cells, on the other hand, was mislocalized to CRMs at the extent similar to ER stressed WT cells (Figure 3H). Thus, re-localizing the septin ring itself from the bud neck or molecular events associated with septin ring transfer from the bud neck contributes to the Cdc42 inactivation during ER stress. Furthermore, we tested the impact of ER stress on Bni5 in shs1ΔCTE cells (Figure 3I). We found that mislocalization of Bni5 also took place under ER stressed (Figure 3I). Similarly, Myo1 was also found at CRMs under ER stress (Figure 3J). Thus, the lack of Shs1 translocation to CRMs in shs1ΔCTE cells revealed that both polarized growth and cytokinesis was blocked under ER stress, at the extent similar to ER stressed WT cells. Together, these data suggest that not all septin subunits are required to stop polarized growth and cytokinesis in response to ER stress.
Translocation of Shs1 to CRMs is not required to induce the ERSU pathway
Does the lack of Shs1 from CRMs affect the ERSU pathway in shs1ΔCTE cells? To answer this question, we assessed the extent of ER inheritance block upon ER stress induction (Figure S3A). We classified cells as previously published to three groups: small-budded cells (< 2 μm; Group 1), medium-budded cells without nuclei (Group II), and large-budded cells with nuclei (Group III) (Babour et al., 2010; Pina et al., 2016; Pina and Niwa, 2015). As we found previously that ER stress has the most profound effect on Class I cells (Pina and Niwa, 2015), we focused on this class throughout this study. ER stress blocked ER inheritance in shs1ΔCTE cells at a level similar to that of WT cells (Figure S3A; lanes 1–2 for WT vs. lanes 7–8 for shs1ΔCTE cells). Thus, the functional significance of the transfer of Shs1 along with all other septin subunits to CRMs itself or events associated with septin ring transfer to CRMs may reside beyond the initial stages of the ERSU pathway.
Slt2 is required for ER stress-induced Cdc42 inactivation in both WT and Shs1 ΔCTE cells.
We reported previously that Slt2 is a component of the ERSU pathway and is important for cER inheritance block (Figure S3A) and septin ring translocation (Figure S3B) in ER stress (Babour et al., 2010; Pina et al., 2018). Here, we investigated the role of Slt2 in inhibiting polarized growth. In ER-stressed slt2Δ cells, Cdc42 (Figures S3C–D) and Cdc24 (Figures S3E–G) failed to move away from the site of polarized growth, demonstrating the requirement for Slt2 in blocking polarized growth. Cdc42 inactivation in shs1ΔCTE cells also depended on Slt2 (Figure S4A–C). In time-lapse experiments, Gic2-RFP remained localized at the bud tip (Movie S7; Figure S4D, blue arrowheads; Figure S4E) even in ER stressed slt2Δshs1ΔCTE cells. A second bud emerged (red arrowheads) without cytokinesis of the first daughter cell. Furthermore, Cdc24 was localized to the second bud site regardless of ER stress (Figure S4F–G). Interestingly, we did not see any multi-budded slt2Δ cells; thus, truncation of CTE in slt2Δ cells caused a failure to block cytokinesis in the absence of SLT2. This is consistent with the ability of cell polarity components to affect the cytokinesis machinery (Wu et al., 2013). Interestingly, the septin ring visualized by Cdc11-GFP in ER stressed slt2Δshs1ΔCTE cells was fragmented (Figures S4B), rather than an intact form remaining at the initial bud site as observed in slt2Δ cells.
Inactivating Cdc42 by SLT2 MAP Kinase is essential for cell survival under ER stress
The above results revealed an unprecedented and essential role of the Slt2 MAP kinase in the inactivation of Cdc42 during ER stress. To further evaluate the functional significance of this role, we tested whether inactivating Cdc42 via its inhibitor ML141 could rescue the growth of slt2Δ cells. Indeed, ML141 rescued the growth of both slt2Δ and slt2Δ shs1ΔCTE cells under ER stress (Figure S4H; compare +Tm vs. +Tm+ML141). Significantly, ML141 treatment also rescued the ER inheritance block (Figure S5A; Figure S5B, compare lanes 4 and 6) and septin transfer to CRMs (Figure S5C; Figure S5D, compare lanes 4 and 6) in ER-stressed slt2Δ cells. These results are consistent with the idea that Slt2-induced Cdc42 inactivation is a hallmark of the ERSU pathway, coordinating with other ERSU events such as ER inheritance block and septin ring movement to CRMs to ultimately contribute to cell survival in response to ER stress.
Split-DHFR screen identifies Slt2 functional partners in the ERSU pathway
Under the normal growth, the importance of Slt2 is underscored by its involvement in a wide range of cellular functions, including genome silencing and cell wall responses (Chen and Thorner, 2007; Gustin et al., 1998). As a result, Slt2 is localized throughout the cell. This makes it challenging to delineate Slt2’s specific role in the ERSU pathway. To this end, we performed a split-DHFR screen to quantitatively identify Slt2 binding partners in vivo (Tarassov et al., 2008). Split-DHFR is a growth-based selection assay, in which bait and prey proteins are each tagged with complimentary fragments of a mutated DHFR (mDHFR); if bait and prey proteins interact, the two fragments of mDHFR reconstitute into a fully functioning enzyme that is not inhibited by methotrexate, an inhibitor of the endogenous and essential yeast DHFR (Tarassov et al., 2008). Therefore, growth levels on methotrexate are proportional to the amount of reconstituted mDHFR enzyme and enable quantification of protein-protein interactions. Using this assay, we screened ~6,000 genes (Table S1) and identified 100 that showed enhanced interactions with Slt2 specifically during ER stress (Table S2).
Bem1 is a unique binding partner for Slt2 during ER stress
We further conducted gene ontology (GO) analysis on the top 100 interactors and categorized them according to their GO molecular functions (Figure S6A, Table S3) (Robinson et al., 2002). We noticed that Bem1, Bnr1, and Vrp1 were the most connected genes (Figure S6A). Bem1 is an important polarity factor that functions as a scaffolding protein for Cdc24 and Cdc42, and recruits them to sites of polarized growth at both the bud tip and bud neck (Pruyne and Bretscher, 2000; Pruyne et al., 2004). Additionally, Bem1 is sequestered in CRMs by Nba1, which prevents it from binding to Cdc24, as a way of inhibiting Cdc42 recruitment to CRMs and of reducing the functional pool of Bem1 in cells (Meitinger et al., 2014). Thus, we focused on Bem1. Depending on which Bem1 pool Slt2 interacts with, we may be able to predict the functional significance of the Slt2-Bem1 interaction.
Using GFP-tagged Bem1, we found that Bem1 was localized at the bud tip in G1-S, and at the bud neck and CRMs in G2-M under normal growth (Figure 4A), in agreement with previous reports (Liu and Novick, 2014; Madden and Snyder, 1998; Smith et al., 2013; Toenjes et al., 2004). Upon ER stress induction, we observed Bem1-GFP primarily at the bud neck and CRMs, and only a small amount of Bem1-GFP remained at the site of polarized growth (Figure 4B). The loss of Bem1 localization at the bud tip is consistent with loss of activated Cdc42 (Gic2) from the bud tip and loss of polarized growth during ER stress (Figures 2F–J).
Figure 4. Bem1 interaction with Slt2 facilitates translocation of septin rings at CRMs during ER stress.

(A) Bem1-GFP in unstressed WT cells was localized to the bud tip, bud neck, and CRMs. All scale bars, 2 μm.
(B) Bem1-GFP localization was altered in Tm-treated WT cells. CRMs were visualized by staining with WGA-594. Zoomed-in views of Bem1-GFP at CRMs are shown.
(C) Split-YFP PCA between Slt2 and Bem1 in untreated WT cells (no Tm).
(D) Split-YFP PCA between Slt2 and Bem1 in cells treated with 1μg/ml Tm. Close-up views of Slt2 interacting with Bem1 at CRMs are also shown.
(E) co-IP of experiment with Bem1-GFP and Slt2-Myc (lane 6). Spa2 is a previously identified Slt2 binding protein (lane 5).
(F) The reconstituted YFP of the Slt2-Bem1 interaction (strain shown in C–D) was pulled down using anti-GFP beads. Cells were treated or untreated with Tm. Western blots for phosphorylated Slt2 (P-Slt2) and total Slt2 are shown.
(G) Split-YFP PCA between Shs1 and Bem1 in untreated (no Tm) and Tm-treated WT cells. Close-up views show Shs1 interacting with Bem1 localized at the bud neck in untreated cells and at CRMs in Tm-treated cells. Zoomed-in views showing PCA signals at the bud neck for unstressed and at CRMs for ER-stressed cells are shown.
(H) Shs1-GFP or Cdc10-GFP in unstressed cells was localized to the bud neck of bem1Δ cells, whereas Shs1-GFP or Cdc10-GFP was mislocalized away from the bud neck but was localized outside of CRMs in Tm-treated bem1Δ cells. Zoomed-in views show Shs1-GFP at the bud neck in unstressed cells and at a location distinct from CRMs in ER-stressed cells.
(I) Quantification of Shs1-GFP in bem1Δ cells shows that significant levels of Shs1-GFP become mislocalized upon ER stress induction. *, p < 0.01; **, p < 0.001; ns, not significant.
(J) Cartoon diagram summarizing the findings in this figure. Under normal conditions, Slt2 interacts with Bem1 at the bud tip while Shs1 is at the bud neck. None of these polarity components is seen at CRMs. When ER stress is triggered, Bem1 recruits Shs1 and phosphorylated Slt2 to CRMs.
These findings suggested that during ER stress, Slt2 might be recruited to CRMs by Bem1. We used split-YFP PCA in living cells to investigate. During normal growth, Slt2 interacted with Bem1 at sites of polarized growth, namely at the bud tip in small budded cells indicative of S phase, and the bud neck in large budded cells indicative of G2/M phases (Figure 4C). We further verified the interaction between Slt2 and Bem1 biochemically by coimmunoprecipitation (co-IP). The amount of Slt2 co-purified with Bem1 was similar to that with Spa2, a protein known to interact with Slt2 (Figure 4E, compare lanes 5 and 6) (van Drogen and Peter, 2002). During ER stress, Slt2-Bem1 PCA was localized at CRMs (Figure 4D), suggesting that Slt2 interacted with the pool of Bem1 at CRMs.
It is known that Slt2 becomes phosphorylated and activated under ER stress (Babour et al., 2010). Given the dynamic localization of Slt2, we tested the activation status of Slt2 which was localized to CRMs under ER stress. We pulled down the Slt2-Bem1 interacting pair by their reconstituted YFP, and probed for phosphorylated Slt2 (P-Slt2). Interestingly, we found that the Bem1 bound P-Slt2 increased in ER stress (Figure 4F), suggesting that binding to Bem1 could provide a mechanism for activating Slt2.
Bem1 recruits the Shs1 septin ring subunit to CRMs during ER stress
Next, we tested if Shs1 interacted with Bem1 at CRMs during ER stress using the Split-YFP PCA. In unstressed cells, we observed fluorescent signals at the bud neck, revealing that Shs1 interacted with Bem1 at this location; however, during ER stress the fluorescence signals representing the Shs1-Bem1 interaction were located at CRMs (Figure 4G). Shs1-GFP and Cdc10-GFP failed to accumulate on CRMs in ER-stressed bem1Δ cells (Figure 4H), but rather mislocalized at a location outside of CRMs (Figure 4I), revealing the importance of Bem1 for the full septin ring transfer to CRMs.
A negative polarity regulator, Nba1, remains at CRMs and is required for Shs1 localization to CRMs under ER stress
Our finding that Bem1 at CRMs binds to Slt2 and Shs1 was rather unexpected because Bem1 binding to Cdc24 at CRMs is normally prevented by Nba1, a recently identified CRM landmark (Meitinger et al., 2014). A genome-wide study revealed genetic interactions between Nba1 and Shs1 as well as components of the ERSU pathway, including Slt2, Bck1, and Pkc1 (Figure 5A) (Costanzo et al., 2011). This suggests that Nba1 is no loner found at CRMs in ER stressed cells or that Nba1 undergoes ER stress induced changes to become an integral part of the ERSU pathway (Figure 4J).
Figure 5. Nba1 recruits the septin ring to CRMs in ER stress.

(A) Genetic interactions between NBA1 and genes in the SLT2 pathway as well as SHS1 as identified by Costanzo et al. 2010. The strength of interactions are annotated on the edges.
(B) Nba1-RFP co-localizes with Shs1-GFP at the bud neck in untreated cells and at CRMs in Tm-treated cells.
(C) Split-YFP PCA between Shs1 and Nba1 in untreated (no Tm) and Tm-treated WT cells. Zoomed-in views show PCA signals at the bud neck in unstressed cells (no Tm) and at CRMs in ER-stressed cells (+Tm).
(D) We detected co-IP of Shs1-Myc and Nba1-GFP only after Tm treatment of WT cells (lane 5). In untreated cells, little physical interaction between Shs1-Myc and Nba1 was detected (lane 4).
(E) Nba1 is required for Shs1-GFP localization at CRMs during ER stress. Shs1-GFP in untreated (no Tm) or Tm-treated (+Tm) nba1Δ cells is shown.
(F) Quantification of Shs1-GFP in unstressed and ER-stressed nba1Δ cells. *, p < 0.05; **, p < 0.005.
(G) Cartoon diagram summarizing the results. Nba1 recruits Shs1 to CRMs during ER stress.
(H) Rax1 and Rax2 are important for localization of Nba1 to the CRMs at the plasma membrane (PM) (Meitinger et al. 2014).
(I) Nba1-GFP localization in WT or rax1Δ cells. The loss of RAX1 (rax1Δ) significantly diminished Nba1-GFP localization to CRMs and Nba1-GFP remained at the bud neck.
(J) Quantitation of Nba1-GFP localized at CRMs. T-test showed that Nba1-GFP localizations to CRMs was significantly reduced in rax1Δ compared to WT. (* represents p<0.0015)
(K) Shs1-GFP localization in untreated and Tm-treated rax1Δ cells. Shs1-GFP localizes to the bud neck during normal growth, and remained at the bud neck in ER-stressed rax1Δ cells.
(L) Quantification of Shs1-GFP localization in (K). P values from t-tests comparing each of the Shs1-GFP localizations between no Tm and +Tm conditions is shown (* represents p < 0.001).
Given our finding that Slt2, Shs1, and Cdc24 all found at CRMs under ER stress, we next tested if Nba1’s localization would also change. We found that Nba1-GFP was localized to the bud neck and CRMs as reported (Meitinger et al., 2014), and that its localization did not change during ER stress (Figure 5B), consistent with Nba1’s role as a landmark for CRMs. Using the split-YFP assay, we found that Shs1 and Nba1 interacted at CRMs only under ER stress (Figure 5C; +Tm). We also detected Shs1-Nba1 interaction using co-IP, but the interaction was only significant in the presence of Tm (Figure 5D, compare lanes 4 and 5). Importantly, in nba1Δ cells, only 23% of cells showed a transferred septin ring in CRMs (Figures 5E–F), in contrast to ~50% in ER stressed WT cells (Figures 1C–D). Thus, these results revealed a role for Nba1 in recruiting Shs1 to CRMs during ER stress (Figure 5G).
Two transmembrane proteins, Rax1 and Rax2, were reported to help localize Nba1 at CRMs (Figure 5H) (Meitinger et al., 2014). We confirmed that Nba1 localization at CRMs indeed depended on Rax1, but its localization to the bud neck did not (Figures 5I–J). If bud neck-localized Nba1 is sufficient to target the translocated septin ring to CRMs during ER stress, we would not expect RAX1 deletion to affect septin translocation. To differentiate which populations of Nba1 are responsible for mediating septin ring translocation during ER stress, we tested Shs1-GFP localization in ER-stressed rax1Δ cells. We found that Shs1-GFP remained at the bud neck, or mislocalized outside of CRMs in ER stressed rax1Δ cells (Figures 5K–L), indicating that Nba1 in CRMs is critical for septin ring translocation to CRMs.
The functional significance of the complete septin ring localization at CRMs
Our results so far revealed that polarity components undergo significant changes under ER stress. We next investigated the functional significance of the septin ring transfer to CRMs by comparing WT and shs1ΔCTE cells. ER inheritance block occurred normally in response to ER stress in both WT and shs1ΔCTE cells (Figure S3A). Thus, we tested a later event: the ability of cells to re-enter the cell cycle after ER functional homeostasis is re-established. In order to test whether shs1ΔCTE cells are able to re-enter the cell cycle in a manner similar to WT cells after recovery from ER stress, we devised an ER stress-recovery assay (Figure 6A). In this method, we first treated cells with Tm for 2 hrs, then washed Tm away before starting the recovery time course. At 4 min after washing out Tm and starting ER stress recovery, the septin ring re-appeared at a new location (Figures 6B and 6C, white arrow). This was followed by the emergence of a new bud after ~50 min of recovery time (Figure 6C, blue arrow, Movie S8). This result is in agreement with our previous report that the original daughter cell is never re-used when cells recover from stress (Babour et al., 2010). As the original daughter cell was not utilized, we observed two budded cells upon recovery, but the septin ring was only localized to the bud neck of the newly emerged daughter cell (Figure 6D).
Figure 6. The CTE of Shs1 dictates re-entry to the cell cycle when ER stress is recovered.

(A) Experimental design for the ER stress recovery assay. Cells expressing Shs1-GFP were treated with 1μg/ml Tm for 2 hrs. Then, Tm was removed by washing the cells with SC medium. Next, cells were monitored by live-cell imaging. In this assay, CRMs were visualized by staining with WGA-594.
(B, C) Recovery assay for WT cells expressing Shs1-GFP according to (A) to follow septin dynamics (Shs1-GFP) and bud emergence. The first frame of the time-lapse (time 0) is shown in (B) along with CRM staining. Subsequent frames are shown in (C). White arrow indicates Shs1-GFP. Blue arrow indicates newly emerging bud. Related to Movie S8.
(D) Location of Shs1-GFP and CRMs in WT cells upon ER stress recovery for 120 min. As cells re-enter the cell cycle, a new bud emerges and Shs1-GFP localizes to the new bud. The initial bud was not re-used even after cells recovered from ER stress.
(E) Recovery assay to monitor active Cdc42 (Gic2-PDB-RFP) in WT cells according to (A). Active Cdc42 (magenta arrow) started to appear within 4 min after Tm wash. A new bud started to appear at ~36–40 min (C) after ER stress recovery. Active Cdc42 appeared at the newly emerging bud cortex at ~64 min after Tm wash. Related to Movie S9.
(F, G) Recovery assay for Shs1-ΔCTE-GFP, which remained dispersed immediately after Tm recovery. The first frame of the time-lapse (time 0) is shown in (F) along with CRM staining. Subsequent frames are shown in (G). White arrow indicates Shs1-GFP and blue arrow shows a new bud. Related to Movie S10.
(H, I) Dynamics of ER stress recovery of bem1Δ cells. ER stress recovery assay for bem1Δ cells was done according to Figure 6A. The first frame of the time-lapse (time 0) is shown in (H) along with CRM staining. Subsequent frames are shown in (I). White arrow indicates Shs1-GFP. Blue arrow indicates bud emergence after recovery. Related to Movie S11.
(J) Growth of WT, shs1-ΔCTE, or bem1Δ cells after ER stress recovery. Mean and SE were calculated based on values from at least three independent experiments.
Furthermore, during recovery, activated Cdc42 was polarized to the new presumptive bud site (Figure 6E, Movie S9). These results are consistent with our observation that the previously translocated septin ring moved from CRMs to a new incipient bud site, allowing cells to re-enter the cell cycle. As such, we most frequently observed new daughters emerged from sites adjacent to CRMs in these recovery assays.
In contrast to WT cells, both the re-appearance of the septin ring and bud emergence in Shs1ΔCTE cells were significantly delayed after recovery from ER stress (Figures 6F and 6G, Movie S10). The slower kinetics of the re-appearance of the septin ring after Tm removal and the new bud emergence was further reflected in the slower growth during the recovery phase of 6 hrs (Figure 6J). Another striking difference with shs1ΔCTE cells was the specific location of the bud emergence: the new daughter cell emerged from within the existing daughter cell, instead of emerging from the mother cell as in WT (Figures 6C and 6G).
We further tested the importance of septin ring movement to CRMs in bem1Δ cells, in which Shs1-GFP moved to a discrete location outside of CRMs under ER stress (Figures 4H and 6H, Movie S11). Interestingly, during the recovery process, Shs1-GFP did not depart from the transferred location. Instead, the new bud emerged at this same location, although the kinetics of the bud emergence was significantly delayed at the extent similar to shs1ΔCTE cells (Figures 6H–I). Thus, these results unveiled that the transfer of the septin ring from the bud neck specifically to CRMs is important for timely recovery and re-entry into the cell cycle.
The impact of aging on ERSU cells
Establishment of a negative polarity cue (or disruption of a polarity cue) by ER stress-induced septin ring subunit translocation to CRMs raises an intriguing question regarding aged cells with multiple CRMs. After each cell division, CRMs accumulate on the cortex of the cell. Thus, aged cells have accumulated multiple CRMs (Powell et al., 2003; Sinclair et al., 1998). Taking this into account, how might aged cells respond to ER stress-induced septin ring translocation? Among asynchronously growing cells, over 50% are naive cells with only the birth scar (we refer to these as ‘young cells’). Here, we examined how ER stress impacts cells that have more than three CRMs (we refer to these as ‘aged cells’). First, we quantified the levels of Nba1 at each bud scar in aged cells (Figures 7A–B). Although Nba1-GFP was present at each bud scar, it was highest in the one most proximal to the bud neck (Figure 7B), consistent with a previous report (Meitinger et al., 2014). Based on our finding that Nba1 is a binding partner of Shs1 at CRMs, we tested if the septin ring might transfer to the most recent CRMs during ER stress. In aged cells grown in regular growth medium, the septin ring remained localized at the bud neck (Figure 7C). Upon ER stress induction, the septin ring transferred frequently (~60%) to the proximal bud scar, and less frequently (~20%) to distal ones (Figures 7C–E), indicating that septin ring transfer tends to occur in correlation with higher Nba1 levels.
Figure 7. Septin ring behaviors in aged ER stressed yeast cells with multiple bud scars.

(A, B) Nba1-GFP localization in Tm-treated (1 μg/ml) ‘aged’ cells (A) and quantification of Nba1-GFP signals at different CRMs (B). Note that ‘aged cells’ refers to cells two or more CRMs. “1st” “2nd” and “3rd” refer to the position of the CRM relative to the bud neck. p values comparing differences in GFP levels are indicated as: ns, not significant; *, p<0.001.
(C–E) Shs1-GFP localization in aged cells that were (C) untreated or (D) treated with Tm. Quantification of Shs1-GFP localization at each CRM is shown in (E). “Proximal” and “distal” refers to the location of the CRM relative to the bud neck. p values comparing differences in % of cells are indicated as *, p<0.001.
(F, G) ER stress recovery assay for ‘aged cells’ with Shs1-GFP transferred at the distal location. The first frame of the time-lapse (time 0) is shown in (F) along with CRM staining. Subsequent frames are shown in (G).
(H) Graphical representation of septin ring localization during the time lapse experiment shown in (F) and (G).
(I) Average time required for the reappearance of the septin ring at the new bud site, which was indicative of re-entry into the cell cycle, after ER stress recovery. p values were calculated to compare young cells and aged cells, and to compare aged cells with septin ring at either proximal or distal CRM prior to recovery in % of cells. *, p<0.001.
(J) In order for cells to effectively survive ‘ER stress’, a cellular strategy of hijacking the components that support cell growth and temporarily re-assembling them at a different location as an emergency complex allows proper handling of ER stress. Specifically, mobilizing components involved in the polarized growth at the bud tip, such as Bem1, Cdc24, and Slt2 and their re-assembly at CRMs along with association with the septin ring will achieve cell cycle halting while re-establishment of the ER functions is handled. Mobilization and re-formation of the polarized cell growth components may be a general emergency strategy for cells to cope with different types of stress or catastrophe. In response to a laser-induced wound, for example, components involved in polarized growth, such as Bni1 and Pkc1 (an upstream kinase of Slt2 MAP kinase), become mobilized from the bud tip, and their re-localization at the wound site allows for wound healing while cell growth is temporarily halted (Kono et al., 2012).
Age impacts the ability to undergo ER stress recovery
We next examined the consequence of translocating the septin ring to distal bud scars. Using the recovery assay (Figure 6A), we monitored ER-stressed and aged cells with septin ring translocated to the distal bud scar (Figures 7F–G). Upon removal of the ER stress-inducing drug, the translocated septin ring moved away from the distal bud scar and re-localized to the more recent (proximal) bud scar (Figures 7F–I, Movie S12). Ultimately, septin rings ended up at the most proximal site to the bud neck through stepwise re-localization of bud scars in the middle. The amount of time required for aged cells to translocate the septin ring from the distal CRM to the most recent (proximal) CRM was significantly delayed when compared to naïve cells (Figure 7I). Strikingly, despite the translocation of septin rings, we did not observe the emergence of a new daughter cell even after 120 min of ER stress recovery (Figure 7G). This is in stark contrast to naïve WT cells, where we detected re-entry into the cell cycle, scored by new bud emergence, within 90 min. Importantly, the recovery kinetics for aged WT cells was even slower than for shs1ΔCTE cells (Figure 6G). Thus, while ER stress-induced re-localization of septin ring could occur, the presence of multiple bud scars in aged cells diminished the ability of the cells to re-enter the cell cycle.
DISCUSSION
One of the most important elements of cell survival is the correct segregation of cellular contents during cell division. Previously, we reported that the ERSU pathway, a cell cycle checkpoint, ensures the inheritance of functional ER during the cell cycle (Babour et al., 2010; Pina et al., 2018; Pina et al., 2016; Pina and Niwa, 2015). When the ER is stressed, the ERSU pathway blocks the inheritance of stressed ER and cytokinesis, thereby preventing the generation of cells lacking sufficient levels of functional ER. Our previous work uncovered that the cytokinesis block occurs by mislocalization of the septin ring, a critical component of cytokinesis; however, the mechanism of this mislocalization and its potential role beyond cytokinesis block remained elusive. Here, we found that this movement of septin rings to CRMs occurs by partially overriding the Nba1-dependent negative polarity establishment component. In addition, we found that Nba1 allows Bem1 at CRMs to bind Cdc24 and Slt2; thus, CRMs serve as sites to gather key components in anticipation of cell cycle re-entry. Under normal growth, Nba1 at CRMs blocks both septin ring formation and Cdc24 association to CRM-localized Bem1, leading to the block of Cdc42 activation for polarized growth (Meitinger et al., 2014). Under ER stress, Nba1 is required for septin ring transfer to CRMs. Furthermore, Nba1 allows Cdc24 to associate with Bem1, while still preventing Cdc42 from being activated at CRMs. Bem1 localized at CRMs serves as the only binding site for activated Slt2, an ERSU MAP kinase. Taken together, our findings suggest that previous cell division sites, CRMs, serve as reservoir or an “ER stress recovery preparation” site, for gathering components that aid in ‘cell cycle re-entry’ upon ER stress recovery.
Our study here demonstrated that the septin ring transfer to CRMs in response to ER stress is important for the timely re-entry into the cell cycle. Furthermore, the ring must contain all five septin subunits. During the initial translocation steps upon ER stress induction, shs1ΔCTE cells could retain a partial septin complex at CRMs and missing only one subunit, Shs1 (Figure 3E). It is possible that the CTE of Cdc11 could replace the function of Shs1-CTE here (Finnigan, et al, 2015). Nonetheless, shs1ΔCTE cells are much delayed in the cell cycle re-entry, when compared to WT cells, suggesting that Cdc11-CTE could not completely cover for the loss of Shs1-CTE. The molecular basis of how the presence of the intact septin ring at CRMs ensures timely re-entry into the cell cycle will require further investigation. In this regard, recent studies have shown that septins are involved in other aspects of cell polarity, including 1) the formation of cytoskeletal scaffolding structures (Mostowy and Cossart, 2012); 2) the formation of a diffusion barrier in the plasma membrane and ER (Caudron and Barral, 2009); and 3) lending definition to daughter-cell differentiation during bud emergence (Okada et al., 2013), in addition to the well-known role of septin rings in polarized growth and cell division (Hartwell, 1974). All of these functions require septin rings to act as relatively static and passive landmarks during most of the cell cycle. The lack of septin ring formation at the bud neck prior to budding should trigger the Swe1-dependent morphogenesis checkpoint or mitotic delay (Barral et al., 1999). This highlights the importance of the timely placement and generation of the septin ring at the site of polarized growth for initiating a new round of the cell cycle. By contrast, an unprecedented translocation of septin rings to CRMs enables cells to retain cellular abilities to re-enter cell cycle during the ER stress.
Based on time-lapse experiments, septin ring translocation to CRMs appears to occur via a transfer of a septin ring complex that is initially formed at the bud neck. In response to ER stress, we noticed that the diameter of the septin ring complex appeared to become smaller than the septin ring at the bud neck prior to ER stress. This suggests that ER stress causes a septin ring to take a more compact structure, allowing for increased mobility. Alternatively, some of the septin subunits may disassemble from the bud neck to generate a different septin ring at CRMs. At this point, we cannot rule out the possibility that some of the septin subunits disassemble at the bud neck and reassemble to a ring at CRMs. Supporting such a mechanism, a previous study showed that septin ring subunits gradually transfer from the previous bud neck to the incipient bud site (Chen et al., 2011). Regardless of the mechanism, an important factor that distinguishes ER stress-induced septin ring is its transfer to CRMs rather than to the incipient bud site.
The translocation of septin rings at CRMs also accompanied other unexpected changes. Under normal growth, septin ring formation is coordinated with polarized cell growth. Under ER stress, the presence of septin rings at CRMs was not associated with bud emergence or polarized bud growth. Furthermore, under normal growth conditions, yeast cells have an elaborate mechanism involving a Cdc42 antagonist, Nba1, that prevents polarized growth from CRMs by interfering with the Cdc24 association with Bem1 localized at CRMs. Surprisingly, we found that Nba1 remained localized at CRMs under ER stress, but might have undergone certain character changes while retaining other features. For example, septin ring formation is normally coordinated with Cdc42 activation and polarized bud growth, but these events did not take place in septin rings transferred at CRMs under ER stress. Further, both Cdc24 and Slt2, but not Cdc42, localize to CRMs upon binding to Bem1. These results reveal that ER stress incapacitates a part of Nba1 functions, such that septin ring formation and Cdc24 localization can take place while Cdc42 activation or initiating polarized growth are continued to be blocked.
How can Bem1-Cdc24 be localized at CRMs in the presence of Nba1? Previous studies have shown that Bem1 serves two main scaffolding functions: 1) for the Cdc42 GEF, Cdc24; and 2) for Cdc42 effectors such as Cla4 or Ste20, which are p21-activated kinases (PAK) (Atkins et al., 2008; Kozubowski et al., 2008). PAK phosphorylates Cdc42 during polarized growth in yeast cells. This process helps ensure that the temporal and spatial regulation of Cdc42 activity determines the site and timing of symmetry breaking on the cell surface. Even after bud emergence, Bem1 remains at the bud tip until the direction of bud growth switches from an orthogonal to a bilateral direction. Our finding that Bem1 interacts with activated Slt2 during ER stress suggests that Slt2 may mediate one of the molecular switches that disassembles the Bem1-Cdc24 complex from the bud tip and assembles it at CRMs. Interestingly, Slt2 appears to be temporally activated during the cell cycle at around the time when the polarized growth switches directions even in the absence of ER stress (Li et al., 2010). Thus, under normal growth, the Slt2-Bem1 association may ultimately dictate Cdc42 activity in a spatially and temporally regulated manner to establish a switch for polarized growth. Upon ER stress activation, the Slt2-Bem1 interaction occurred at CRMs, which, as a consequence, may generate “hyper-activated” Bem1 or partially weaken Nba1 function. Thus, this process somehow facilitates Bem1-Cdc24 to localize to CRMs even in the presence of Nba1. The continued presence of Nba1 may contribute to disconnecting the activation of Cdc42. Furthermore, the interaction of Bem1, Cdc24, and activated Slt2 might allow septin ring subunits to translocate to CRMs even in the presence of a negative regulator such as Nba1. These observations are consistent with the idea that septin ring translocation to CRMs also requires Bem1.
Importantly, the lack of a “ER-stress recovery complex” at CRMs in ER-stressed bem1Δ or shs1-ΔCTE cells underscores the functional significance of concentrating Slt2, Cdc24, and all five septin subunits at CRMs under ER stress. In both cases, the absence of translocated Shs1 at CRMs significantly delayed the cell’s ability to re-enter the cell cycle even when ER function is re-established. Thus, ER stress-induced Shs1 transfer to CRMs represents a key event in anticipation of re-established cellular competence of resuming cell cycle division following ER stress recovery. Interestingly, upon re-entry into the cell cycle, we found that the original daughter cell was never utilized. Instead, a new second bud emerged from the original mother cell. Under normal growth, establishing polarized growth requires an intrinsic competition between different foci of activated Cdc42 and its upstream GEFs and GAPs, which are aided by positive feedback to establish the “winning” foci for polarized growth that leads to the bud’s emergence from that foci (Wu et al., 2015). Once ER functional homeostasis is re-established, polarized growth can be re-established by re-mobilizing these components to the outside of the inhibitory zone that is defined by CRMs. Indeed, we found that upon recovery from ER stress new buds emerged from the sites directly adjacent to CRMs, retaining the usual axial budding pattern seen in haploid yeasts. The kinetics of the cell cycle recovery was significantly diminished in both shs1-ΔCTE and bem1Δ cells, in which septin ring, Bem1, Cdc24, and Slt2 failed to gather at CRMs upon ER stress. Thus, these findings underscore the functional significance of strategically localizing key components at CRMs for resuming polarized growth in order to re-enter the cell cycle.
An interesting implication associated with CRMs as the site of septin ring translocation is aging, as cells accumulate CRMs as they undergo replication cycles. Therefore, unlike young cells, aging mother cells with multiple CRMs provide more choices for septin rings and other key components to congregate. Although young and aged cells appear to be equally effective at translocating septin rings, older cells with multiple CRMs struggle at re-localizing septin rings during recovery. Nba1 is present in several CRMs in such older cells. In addition, as cells age further, some of the CRMs may not have Nba1, which could alter the nature of septin ring transfer to occur at CRMs. One of the challenges of septin ring transfer to CRMs with decreased levels of Nba1 or little Nba1 includes strategies to block polarized growth. Interestingly, UPR activation is also significantly slower in older cells (data not shown). Whatever the mechanisms might be, the prolonged kinetics of ER stress and UPR induction may provide additional time to gather components key to cell cycle re-entry. We found that septin rings transferred to older CRMs and then kept moving between CRMs until they reached the most recent CRMs before re-initiating the cell cycle. This might reveal that Nba1 concentration dictates the establishment towards recovery state. Furthermore, strong evidence suggests that during replicative aging, aging factors such as extra-chromosomal DNA circles and damaged-protein aggregates that decrease fitness of aged cells accumulate asymmetrically in the aging mothers (Erjavec et al., 2007; Higuchi-Sanabria et al., 2014; Shcheprova et al., 2008; Singh et al., 2017; Spokoini et al., 2012); this may also contribute to the delay of re-entry into the cell cycle during ER stress.
There is a precedent for a molecular strategy to respond to cellular emergencies by re-directing polarized cell growth components. For example, the mobilization/re-organization of Bni1—which regulates polarized cytoskeleton and Pkc1, an upstream kinase of Slt2—plays a major role in recovery from a laser-induced wound on the cell surface of yeast (Kono et al., 2012) (Figure 7J). The driving force behind halting polarized growth and mobilizing the necessary components for cell membrane surface repair is generated from rapidly degrading Bni1, a formin that nucleates actin filaments, via proteasomes. Concomitantly, Bnr1, another formin is mobilized to reach to the wound site to generate a ‘wound recovery complex’ along with Pkc1. A conceptual parallel can be found in ER-stressed cells: in response to ER stress, re-localization of Bem1, Slt2 kinase, and Cdc24 from the bud tip to CRMs is coordinated with the timely induction of the ERSU pathway. Re-establishment of the polarized growth and a platform for inheritance of the ER in the emerging daughter cell can be facilitated by the presence of this ‘ER stress recovery complex’ at a reservoir of regulators of polarized growth at CRMs. Thus, our ER stress studies and those examining laser-induced wounds may have revealed an underlying principle and cellular strategy of handling catastrophes: by linking cell growth with the handling of a specific cellular stress, cells mobilizing components involved in polarized cell growth and re-functions them to take care of a specific cellular stress. Such a strategy ensures a break on the continued growth and provides effective means to handle stress recovery. One important element of such a stress-handling strategy is to retain the ability to resume polarized growth. CRMs may provide an ideal location for transferring ‘ER stress recovery complex’ under ER stress in the absence of specific targets such as wounded sites in the cell.
Finally, as ER stress is conserved among eukaryotic cells, it is tempting to speculate that a similar mechanism might exist for effectively handling ER stress in mammalian cells. While the molecular basis of the CRM and its constituents are unique to yeast cells, recent studies have suggested that the mid body (MB) formed at the cleavage furrow during cytokinesis may be a functional equivalent of CRMs (Chen et al., 2013; Ettinger et al., 2011; Kuo et al., 2011; Pohl and Jentsch, 2009; Thieleke-Matos et al., 2017). Structurally, the MB takes a ring-like shape that resembles CRMs. As CRMs represent prior cytokinesis sites, the MB is also formed during cytokinesis and plays a role in cell division. While the exact MB constituents differ from those of CRMs, recent studies revealed that the MB also retains the post-mitotic structure. Specifically, upon division of mammalian two daughter cells, the MB ends up in one of the daughter cells. Ultimately, many MBs can be removed from the cell by a few mechanisms including autophagy, although its half-life appears to differ depending on the cell type. The half-life of an MB appears to dictate the pluripotency of stem cells. For example, stem cells with a long-lived or persistent MB normally retain pluripotency. By contrast, during asymmetric division of stem cells, in which one cell retains pluripotency and the other differentiates into a specific cell type, the extent of potency is correlated with the half-life of the MB. Given our findings on CRMs, it will be interesting to test if pluripotency of MBs change in response to ER stress.
STAR METHODS
Detailed methods are provided in the online version of this paper and include the following:
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Maho Niwa (mniwarosen@ucsd.edu)
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Unless otherwise indicated, all yeast strains used in this study is of the S288C background. See Table S5 for a full list of strains. All yeast was grown at 30°C in synthetic defined medium containing yeast nitrogenous base, 2% dextrose and the appropriate amino acids for the genotype unless otherwise stated.
METHODS DETAILS
Plasmid construction
Epitope-tagging of endogenous proteins was done by homologous recombination of PCR-generated fragments from templates in haploids at the C-terminus of the specified genes at their endogenous locus in either BY7043 (Tong and Boone, 2006) or BY4741. The templates include pKT128 (GFP::SpHIS5)(Sheff and Thorn, 2004), pFA6A-pmRFP-KanMX6, pFA6A-13Myc-KanMX6 (Bahler et al., 1998), pHVF1CT and pUVF2CT (Chao et al., 2014). KanMX deletion strains were obtained from freezer stocks of the haploid yeast deletion collection (BY4741, MAT a, KanMX; Thermo Fischer). NatR deletion or truncation strains were constructed in BY7092 using p4339 (Tong and Boone, 2006). All deletion and truncation strains were confirmed by PCR. Double epitope tagged (including, GFP/RFP) and double deletion strains were generated by standard yeast genetic techniques of sporulation and tetrad dissection. Split-YFP PCA strain construction was done as described in (Chao et al., 2014). For the expression of BUD2, BUD5 and SHS1, full length ORFs were cloned from BY4741 genomic DNA and inserted into p416-TEF at XbaI/SalI. To create p316-Gic2-PBD-RFP, Gic2prom>Gic2-PBD-RFP was PCR-cloned from YIp211-GIC2PBD(W23A)-RFP into pRS316 plasmid at Xba1I/ KpnI.
Light Microscopy techniques
Log phase live yeast cells were imaged using the Zeiss Axiovert 200M with a 100×1.3 NA objective, or the DeltaVision system (Applied Precision) consisting of an inverted epifluorescence microscope (IX71, Olympus).
All time-lapse imaging was done using in-house fabricated microfluidics devices except for ER stress recovery experiments, in which we immobilized cells 1.6% agarose pads containing SC medium. For microfluidics, cells were immobilized in a microfluidic chamber using concanavalin A prepared at 2mg/ml with 50mM CaCl2 and 50mM MnCl2. Microfluidics devices were fabricated exactly as described in (Hansen et al., 2015).
To visualize CRMs, we used either 0.1μg/ml calcofluor white (Sigma) or Wheat Germ Agglutinin-555 conjugate as indicated. ER stress was induced by treating log-phase cultures with 1μg/ml Tunicamycin for at least 1.5hr.
Electron Microscopy
Transmission electron microscopy was performed as previously described (Onishi et al., 2013). Briefly, WT (W303) yeast cells were untreated or treated with 1μg/ml Tunicamycin for 2 hrs at 30 °C. The cells were harvested by filtration, fixed with 3% glutaraldehyde in 0.1 M sodium cacodylate buffer, pH 6.8, first for 1 h at RT, then at 4°C overnight. The fixed cells were washed three times with the same buffer, post-fixed with 4% potassium permanganate at 4°C for 2h, washed three times with H2O, incubated in 2% uranyl acetate at RT for 1h, washed twice with H2O, and dehydrated through a graded series of increasing ethanol concentrations. The cells were then embedded in LR white resin (Fluka; Sigma-Aldrich), and sections of ~70 nm were cut using a Leica Ultracut S microtome, collected on formvar-coated 100-mesh copper grids (Electron Microscopy Sciences), and post-stained for 30 sec in 1:1 3% uranyl acetate and 50% acetone, followed by 0.2% lead citrate for 3 min. Sections were then imaged at 120 kV using a JEM-1400 transmission electron micro- scope (JEOL) equipped with a Gatan Orius 4k X 4k digital camera.
Split-DHFR screen
Two libraries were employed for the DHFR PCA screen: a library with C-terminal DHFR F[1,2] tag and a library with C-terminal DHFR F[3] tag (Tarassov et al., 2008). A query strain was taken out from each library with Slt2 tagged with either fragment and mated against the opposite libraries by overnight incubation on YPD. After mating, diploid cells were selected for by incubation for 2 days on YPD medium with 100 μg/ml nourseothricin (Werner Bioagents, Jena, Germany) and 250 μg/ml hygromycin B (Wisent Bioproducts, Saint-Jean-Baptiste, Canada). This step was repeated once. Next, the strains were transferred to synthetic complete medium (4% (w/v) Noble agar) with 200 μg/ml methotrexate (Bioshop Canada, Montréal, Canada) and without adenine or ammonium sulfate. Pictures of the strains were taken after 4 days incubation at 30°C. Colony size was analyzed using the Balony software (Young and Loewen, 2013).
Co-purification Assays
Cells were harvested by centrifugation and resuspended with lysis buffer (20mM Tris-HCl, pH 7.4, 150mM NaCl, 1mM EDTA and 0.5% NP-40) containing protease inhibitor cocktail (Sigma) and bashed with acid-washed beads for 10min at 4°C. Cell lysates were homogenized by sonication. To purify GFP-tagged proteins, we used GFP-nAb magnetic agarose beads (Allele Biotech) and followed their recommended procedures. Purified proteins were confirmed by western blotting using anti-GFP antibody (Roche) at 1:1000, and co-purified binding partners by using anti-Myc antibody (Sigma) at 1:2000.
Yeast Spot Assays
10- fold serial dilutions of log phase cells were spotted using a pin-frogger onto agar plates containing synthetic complete (SC) media with 2% glucose and grown for 48hr at 30°C.
QUANTIFICATION AND STATISTICAL ANALYSIS
Quantification of microscopy images
The ImageJ software (National Institute of Health) was used for all quantifications, and a minimum of 100 cells was measured for each experiment. Progression through the cell cycle was arbitrarily classified as follows: G1, cells with no buds; S, bud area less than 1/3 of the mother; G2, bud area greater than 1/3, but less than 2/3 of the mother; and M phase, bud area greater than 2/3 of the mother. Bud to mother size ratios were determined by tracing bud and mother cell perimeters on the corresponding transmission images and measuring the area.
Statistical analysis
Statistical testing was performed using Graph Pad Prism (GraphPad Software, La Jolla, CA). Experiments used for statistics were repeated 3 times. Quantitative data are expressed as mean ± SEM. Student’s T test was used to generate p values.
Gene-set enrichment analysis for split-DHFR screen
The split-DHFR screen was analyzed by using FunSpec (Robinson et al., 2002). The resulting network was visualized using Cytoscape 3.2 using force-directed layout.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-GFP mouse monoclonal (no longer available) | Roche | 11814460001 |
| Anti-Myc mouse monoclonal | Sigma | M4439 |
| Anti-PGK1 mouse monoclonal | Invitrogen | 459250 |
| Bacterial and Virus Strains | ||
| N/A | ||
| Biological Samples | ||
| N/A | ||
| Chemicals, Peptides, and Recombinant Proteins | ||
| GFP-nAb magnetic agarose beads | Allele Biotech | ABP-NAB-GFPXK20 |
| Protease inhibitor cocktail | Sgima | P8215 |
| concanavalin A | Sigma | C2010 |
| calcofluor white | Sigma | 18909 |
| Wheat Germ Agglutinin, Alexa Fluor 555 Conjugate | Invitrogen | W32464 |
| ML 141 | Sigma | SML0407 |
| Tunicamycin | Calbiochem | CAS 11089-65-9 |
| Critical Commercial Assays | ||
| N/A | ||
| Deposited Data | ||
| N/A | ||
| Experimental Models: Cell Lines | ||
| N/A | ||
| Experimental Models: Organisms/Strains | ||
| See Supplementary Table S5 | ||
| Oligonucleotides | ||
| See Supplementary Table S4 | ||
| Recombinant DNA | ||
| P4339 | Boone lab, U of Toronto | N/A |
| pHVF1CT | Loewen lab, UBC | Chao Cell 2014 |
| pUVF2CT | Loewen lab, UBC | Chao Cell 2014 |
| YIp211-GIC2PBD(W23A)-RFP | Bi lab, U of Pennsylvania | |
| 316>GIC2-PBD-RFP | This paper | N/A |
| 416-TEF>BUD2 | This paper | N/A |
| 416-TEF>BUD5 | This paper | N/A |
| 416-TEF>SHS1 | This paper | N/A |
| pKT128 | Sheff Yeast 2004 | N/A |
| pFA6A-pmRFP-KanMX6 | Bahler Yeast 1998 | N/A |
| pFA6A-13Myc-KanMX6 | ||
| Software and Algorithms | ||
| ImageJ | NIH | N/A |
| Prism 5.0 | GraphPad | N/A |
| Cytoscape 3.2 | N/A | |
| Other | ||
Strains
| Name | Genotype | Source |
|---|---|---|
| BY4741 | MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 | This lab |
| BY7092 | MATalpha can1delta::STE2pr-Sp_his5 lyp1delta his3delta1 leu2delta0 ura3delta0 met15delta0 | Boone lab |
| BY7043 | MATalpha can1Δ::STE2pr-lue2 lyp1Δ his3Δ 1 leu2Δ0 ura3Δ0 met15Δ0 | Boone lab |
| MNY1032 | SHS1-GFP::KanMX W303 background | This lab |
| MNY2434 | UPRE-GFP::URA in ABY100 | This lab |
| MNY2659 | Nba1-GFP::HIS in BY 7043 | This lab |
| MNY2660 | Pho88-GFP::HIS in BY 7043 | This lab |
| MNY2660 | Pho88-GFP::HIS in BY 7043 | This lab |
| MNY2662 | Shs1-GFP::HIS in BY 7043 | This lab |
| MNY2663 | Shs1-cte-GFP::HIS in BY 7043 | This lab |
| MNY2664 | Cdc11-GFP::HIS in BY 4741 | This lab |
| MNY2669 | shs1-cte::NAT in BY7092 | This lab |
| MNY2671 | Δslt2::NAT in BY7092 | This lab |
| MNY2672 | Δnba1::NAT in BY7092 | This lab |
| MNY2685 | Δslt2::KanMX Shs1-GFP::HIS | This lab |
| MNY2687 | Δnba1::KanMX Shs1-GFP::HIS | This lab |
| MNY2689 | Δnba1::NAT Δice2::KanMX | This lab |
| MNY2690 | Δslt2::NAT Δnba1::KanMX | This lab |
| MNY2691 | Δslt2::KanMX shs1-cte::NAT by tetrad (MAT a) | This lab |
| MNY2700 | Shs1-RFP::KanMX X Nba1-GFP::HIS | This lab |
| MNY2718 | Shs1-RFP::KanMX Myo1-GFP::HIS | This lab |
| MNY2724 | ero1–1::KanMX Shs1-GFP::HIS | This lab |
| MNY2785 | Bud2-GFP::HIS BY7043 | This lab |
| MNY2788 | Bud5-GFP::HIS BY7043 | This lab |
| MNY2789 | Shs1-VF1::HIS in 4741 | This lab |
| MNY2790 | VF1-Slt2::HIS in 4741 | This lab |
| MNY2794 | Nba1-VF2::URA BY 7043 | This lab |
| MNY2815 | Δslt2::KanMX shs1-cte::NAT Pho88-GFP::HIS | This lab |
| MNY2816 | Δslt2::KanMX shs1-cte::NAT Cdc11-GFP::HIS | This lab |
| MNY2817 | Δslt2::NAT Pho88-GFP::HIS | This lab |
| MNY2818 | shs1-cte::NAT Pho88-GFP::HIS | This lab |
| MNY2830 | Bud1-VF1::HIS Cdc24-VF2::URA | This lab |
| MNY2832 | Bud1-VF1::HIS in 4741 | This lab |
| MNY2833 | Bud2-VF2::URA in 7043 | This lab |
| MNY2834 | Bud5-VF2::URA in 7043 | This lab |
| MNY2835 | Cdc24-VF2::URA in 7043 | This lab |
| MNY2836 | Bem1-VF2::URA in 7043 | This lab |
| MNY2840 | Bud5-GFP::HIS Δslt2::KanMX shs1-cte::NAT | This lab |
| MNY2841 | Cdc24-GFP::HIS Δslt2::G418 shs1-cte::NAT | This lab |
| MNY2850 | Bud2-GFP::HIS shs1-cte::NAT | This lab |
| MNY2851 | Bud5-GFP::HIS shs1-cte::NAT | This lab |
| MNY2852 | Cdc24-GFP::HIS shs1-cte::NAT | This lab |
| MNY2853 | Cdc24-GFP::HIS Δslt2::KanMX | This lab |
| MNY2854 | Bud5-GFP::HIS Δslt2::G418 | This lab |
| MNY2856 | Slt2–13myc:: KanMX 4741 | This lab |
| MNY2857 | Shs1–13myc::KanMX 4741 | This lab |
| MNY2858 | Bud2-GFP::HIS Δslt2::KanMX | This lab |
| MNY2859 | Bem1-GFP::HIS 7043 | This lab |
| MNY2879 | Shs1-GFP::HIS Δbem1::KanMX | This lab |
| MNY2881 | Shs1-GFP::HIS Δrax1::KanMX | This lab |
| MNY2890 | 316>Gic2-PBD-RFP in 4741 | This lab |
| MNY2891 | Cdc24-GFP::HIS 7043 | This lab |
| MNY2828 | Bud1-VF1::HIS x Bud2-VF2::URA | This lab |
| MNY2829 | Bud1-VF1::HIS x Bud5-VF2::URA | This lab |
| MNY2831 | VF1-Slt2::HIS x Bem1-VF2::URA | This lab |
Primers
| oligo name | sequence 5′−3′ |
|---|---|
| Δslt2_KO_F′ | tagaaataattgaagggcgtgtataacaattctgggagACATGGAGGCCC |
| Δslt2_KO_R′ | ggtgattctatacttccccggttacttatagttttttgCAGTATAGCGACCAGCATTCAC |
| Δslt2_Ko_Chk_F′ | CCTGTGTGTAGTGAAAAATTCGAAT |
| Δslt2_Ko_Chk_R′ | ctatggtgattctatacttccccgg |
| Nba1_KO_F | atattcgactaacaagaagaccattatcaaaaccagatACATGGAGGCCCAGAATACCCT |
| Nbal_KO_R | ACCGGAAGAGAAAGAAACTTATATATTACCACTATACTcagtatagcgaccagcattcac |
| Nba1_Chk_F | CCACAGTTAGTGAACAAAAA |
| Nbal_Chk_R | GCTTTGTCTAATCTTTTCAG |
| Sec63-GFP_KI_F | ATCGATACGGATACAGAAGCTGAAGATGATGAATCACCAGAAGGTgacgg |
| Sec63-GFP_KI_R | cgtctaagagctaaaatgaaaaactatactaatcacttatatTCGatgaattcgagctcg |
| Shs1-CTE_KO_F | CACCACGCAAAATTTGCTTTACGAGAATTACCGTTCCGACATGGAGGCCCAGAATACCCT |
| Shs1-CTE_KO_R | gctttggattttgtacagatacaacTCAATCTCTACCCCAGTATAGCGACCAGCATTCAC |
| Shs1_KO_Chk_F | CCACGCAAAATTTGCTTTACG |
| Shs1_KO_Chk_R | CGATGCAATAGAGGCTAAATC |
| Shs1-GFP_KI_F | GACACGTATACTGATTTAGCCTCTATTGCATCGGGTAGAGATGGTgacggtgctggttta |
| Shs1-GFP_KI_R | tatttatttatttgctcagctttggattttgtacagatacaaTCGatgaattcgagctcg |
| ShslΔCTE_GFP_F | AAACTATCGTCCGTGGCCAACGCTGAAGAAATTGGTCCTAATGGTgacggtgctggttta |
| Pho88-GFP_KI_F | GAAGAAGCTGAAAGAGCCGGTAACGCTGGTGTTAAGGCTGAAGGTgacgg |
| Pho88-GFP_KI_R | gcagcaactgcgtagagaaaaaaatgaatatatttttacataTCGatgaattcgagctcg |
| Pho88-RFP-KI-F | AGAAGCTGAAAGAGCCGGTAACGCTGGTGTTAAGGCTGAACGGATCCCCGGGTTAATTAA |
| Pho88-RFP-KI-R | gcagcaactgcgtagagaaaaaaatgaatatatttttacaGAATTCGAGCTCGTTTAAAC |
| Shsl-RFP-KI-F | CACGTATACTGATTTAGCCTCTATTGCATCGGGTAGAGATCGGATCCCCGGGTTAATTAA |
| Shsl-RFP-KI-R | tatttatttatttgctcagctttggattttgtacagatacGAATTCGAGCTCGTTTAAAC |
| Cdc11-GFP_KI_F | GAAGCCAGGTTGGAAAAAGAGGCGAAAATCAAACAGGAAGAAGGTgacggtgctggttta |
| Cdcll-GFP_KI_R | atatagagaaagaagaaataagtgaggaagccaaaagcggacTCGatgaattcgagctcg |
| VF1_R_Chk | CATTAACATCACCATCTAATTCAACC |
| VF2_R_Chk | ACCACCATCTTCAATGTTGTGTC |
| Nbal-VF-KI_F | GATTAGACAAAGCTACAAAGGCTCTTGAAGGGTTTtatgtatcatacacatacgatttag |
| Nba1-VF-KI_R | CGGACTTGTCCAAGTATCAATGAATACAAGCCATTGAATTACtcgatgaattcgagctcg |
| Myo1-GFP_F | AAAAATATTGATAGTAACAATGCACAGAGTAAAATTTTCAGTGGTgacggtgctggttta |
| Myo1-GFP_R | cgtgtcgtctttttctgttaataatgcatattctcattctgtTCGatgaattcgagctcg |
| Shsl-VFl/2_KI_F | TAGCCTCTATTGCATCGGGTAGAGATggtgctggttatgtatcatacacatacgatttag |
| Shsl-VFl/2_KI_R | atttatttatttgctcagctttggattttgtacagatacaactcgatgaattcgagctcg |
| Bud2-GFP_KI_F | CTGACAAGATGGTTCAAAAAGAAAAAAGAAACAGGGGGATCTGGTgacggtgctggttta |
| Bud2-GFP_KI_R | ctttcaaaggaaagaatatgaagtgaacatttttttctacgaTCGatgaattcgagctcg |
| Bud5-GFP_KI_F | AGGGCGTATCAAGTCAGTATAGCTAAGGTTCCAAGGCTTACCGGTgacggtgctggttta |
| Bud5-GFP_KI_R | aagaagcaaaaggaagtcatctttctttgaacagttctgtttTCGatgaattcgagctcg |
| Cdc24-GFP_F | TTGGCGGAAAACAATGAGAAATTCTTGAACATTCGTCTGTATGGTgacggtgctggttta |
| Cdc24-GFP_R | ttcttgaattatttagtatttgctgtatactagttttatttaTCGatgaattcgagctcg |
| Bud5_KO_F | gacctcttgagcggtgagcctctggcaaagaagaaagaACATGGAGGCCCAGAATACCCT |
| Bud5_KO_R | agcaaaaggaagtcatctttctttgaacagttctgtttCAGTATAGCGACCAGCATTCAC |
| Bud5_KO_Chk_F | ACTGACCTCAGTGATTTACTTTTCC |
| Bud5_KO_Chk_R | GCAGTGATGTAAAAGGTACACAAGG |
| Bud2_KO_F | gcatacgtcgtggtgtttatctttgattgtatcatattACATGGAGGCCCAGAATACCCT |
| Bud2_KO_R | tcaaaggaaagaatatgaagtgaacatttttttctacgCAGTATAGCGACCAGCATTCAC |
| Bud2_KO_Chk_R | tatttccacattctggatcgc |
| Bud5-VF_KI_F | ATCAAGTCAGTATAGCTAAGGTTCCAAGGCTTACCtatgtatcatacacatacgatttag |
| Bud2-VF_KI_F | GATGGTTCAAAAAGAAAAAAGAAACAGGGGGATCTtatgtatcatacacatacgatttag |
| Cdc24-VF_KI_F | AAAACAATGAGAAATTCTTGAACATTCGTCTGTATtatgtatcatacacatacgatttag |
| Budl-VF_KI_F | AAAGAAGAAAAACGCTTCCACTTGCACTATTCTAtatgtatcatacacatacgatttag |
| Budl-VF_KI_R | ttttatctgatatcttgattcatttataataaaattaagtgatcgatgaattcgagctcg |
| Beml-GFP_F | AACATAATCCAAGCCAAACTGAAAATTTCCGTTCACGATATTGGTgacggtgctggttta |
| new Bem1-VF_R | aaagaagaaaaatgcttcgtcttctaacactagatactagattcgatgaattcgagctcg |
| Bud3_KO_Chk_F | GACAAAGAGAACGATGAAACC |
| Bud3_KO_Chk_R | CCTGATGTAAAGAAGCGCTTC |
| Bud4_KO_Chk_F | AGGAGATAGACAATGAAATGG |
| Bud4_KO_Chk_R | GCATCTTCTTCCTCTTCATCT |
| new_Bud2_cloning_F | cccggggtcgacATGAGCTCCAACAATGAACCGGCCC |
| new_Bud2_cloning_R | cccggggaattccgaTTAAGATCCCCCTGTTTCTTTT |
| XbaI-Bud2_F | gcgctctagaATGAGCTCCAACAATGAACC |
| Bud2-SalI_R | ggccgtcgaccgaTTAAGATCCCCCTGTTT |
| XbaI-Shs1_F | gcgctctagaATGAGCACTGCTTCAACACC |
| Shsl-SalI_R | ggccgtcgacTCAATCTCTACCCGATGCAA |
| Gic2PBD-RFP_F | ccccgagctcgatctagatgttgcc |
| Gic2PBD-RFP_R | ccagtgaattcgagctcGGTACC |
| Slt2-13Myc-KI_F | TGAAAAAGAGCTGGAGTTTGGATTAGATAGAAAATATTTTCGGATCCCCGGGTTAATTAA |
| Slt2-13Myc-KI_R | ggtgattctatacttccccggttacttatagttttttgtcGAATTCGAGCTCGTTTAAAC |
Highlights.
The septin ring moves from the bud neck to the bud scar in response to ER stress.
Transfer of the septin ring is required for timely re-entry into the cell cycle.
Slt2 inactivates Cdc42 and is essential for cell survival under ER stress.
Age impacts cell cycle re-entry upon ER stress recovery.
ACKNOWLEDGMENTS
We thank Dr. Stephan Michnick for the DHFR libraries, The work was supported by grants from the NIH (R01GM087415), and UCSD Chancellor Excellent Scholarship (formally FISP), and the UCSD Academic Senate fund to M.N.; and by grants from VolksWagen foundation “Life” Grant and a DFG grant (SFB1190) to M. S. J. T. C. and F. P. were supported by a UCSD Molecular Biology Cancer postdoctoral fellowship and an NIH 5T32AI007469-20UCSD/LIAI allergy postdoctoral training grant, respectively. M. S. is an Incumbent of the Dr. Gilbert Omenn and Martha Darling Professorial Chair in Molecular Genetics.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
The authors declare no competing interests.
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Maho Niwa (mniwarosen@ucsd.edu).
References
- Atkins BD, Yoshida S, and Pellman D (2008). Symmetry breaking: scaffold plays matchmaker for polarity signaling proteins. Curr Biol 18, R1130–1132. [DOI] [PubMed] [Google Scholar]
- Babour A, Bicknell AA, Tourtellotte J, and Niwa M (2010). A surveillance pathway monitors the fitness of the endoplasmic reticulum to control its inheritance. Cell 142, 256–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bahler J, Wu JQ, Longtine MS, Shah NG, McKenzie A 3rd, Steever AB, Wach A, Philippsen P, and Pringle JR (1998). Heterologous modules for efficient and versatile PCR-based gene targeting in Schizosaccharomyces pombe. Yeast 14, 943–951. [DOI] [PubMed] [Google Scholar]
- Barlowe C (2010). ER sheets get roughed up. Cell 143, 665–666. [DOI] [PubMed] [Google Scholar]
- Barral Y, Parra M, Bidlingmaier S, and Snyder M (1999). Nim1-related kinases coordinate cell cycle progression with the organization of the peripheral cytoskeleton in yeast. Genes Dev 13, 176–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bechmann LP, Hannivoort RA, Gerken G, Hotamisligil GS, Trauner M, and Canbay A (2012). The interaction of hepatic lipid and glucose metabolism in liver diseases. J Hepatol 56, 952–964. [DOI] [PubMed] [Google Scholar]
- Bi E, Maddox P, Lew DJ, Salmon ED, McMillan JN, Yeh E, and Pringle JR (1998). Involvement of an actomyosin contractile ring in Saccharomyces cerevisiae cytokinesis. J Cell Biol 142, 1301–1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bos JL, Rehmann H, and Wittinghofer A (2007). GEFs and GAPs: critical elements in the control of small G proteins. Cell 129, 865–877. [DOI] [PubMed] [Google Scholar]
- Caudron F, and Barral Y (2009). Septins and the lateral compartmentalization of eukaryotic membranes. Dev Cell 16, 493–506. [DOI] [PubMed] [Google Scholar]
- Chao JT, Wong AK, Tavassoli S, Young BP, Chruscicki A, Fang NN, Howe LJ, Mayor T, Foster LJ, and Loewen CJ (2014). Polarization of the endoplasmic reticulum by ER-septin tethering. Cell 158, 620–632. [DOI] [PubMed] [Google Scholar]
- Chen CT, Ettinger AW, Huttner WB, and Doxsey SJ (2013). Resurrecting remnants: the lives of post-mitotic midbodies. Trends Cell Biol 23, 118–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Howell AS, Robeson A, and Lew DJ (2011). Dynamics of septin ring and collar formation in Saccharomyces cerevisiae. Biol Chem 392, 689–697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen RE, and Thorner J (2007). Function and regulation in MAPK signaling pathways: lessons learned from the yeast Saccharomyces cerevisiae. Biochim Biophys Acta 1773, 1311–1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costanzo M, Baryshnikova A, Myers CL, Andrews B, and Boone C (2011). Charting the genetic interaction map of a cell. Curr Opin Biotechnol 22, 66–74. [DOI] [PubMed] [Google Scholar]
- Denic V, Quan EM, and Weissman JS (2006). A luminal surveillance complex that selects misfolded glycoproteins for ER-associated degradation. Cell 126, 349–359. [DOI] [PubMed] [Google Scholar]
- Du Y, Ferro-Novick S, and Novick P (2004). Dynamics and inheritance of the endoplasmic reticulum. Journal of Cell Science 117, 2871–2878. [DOI] [PubMed] [Google Scholar]
- Erjavec N, Larsson L, Grantham J, and Nystrom T (2007). Accelerated aging and failure to segregate damaged proteins in Sir2 mutants can be suppressed by overproducing the protein aggregation-remodeling factor Hsp104p. Genes Dev 21, 2410–2421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ettinger AW, Wilsch-Brauninger M, Marzesco AM, Bickle M, Lohmann A, Maliga Z, Karbanova J, Corbeil D, Hyman AA, and Huttner WB (2011). Proliferating versus differentiating stem and cancer cells exhibit distinct midbody-release behaviour. Nat Commun 2, 503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fehrenbacher KL, Davis D, Wu M, Boldogh I, and Pon LA (2002). Endoplasmic reticulum dynamics, inheritance, and cytoskeletal interactions in budding yeast. Molecular Biology of the Cell 13, 854–865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feige MJ, and Hendershot LM (2011). Disulfide bonds in ER protein folding and homeostasis. Curr Opin Cell Biol 23, 167–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Field CM, and Kellogg D (1999). Septins: cytoskeletal polymers or signalling GTPases? Trends Cell Biol 9, 387–394. [DOI] [PubMed] [Google Scholar]
- Frakes AE, and Dillin A (2017). The UPRER: Sensor and Coordinator of Organismal Homeostasis. Mol Cell 66, 761–771. [DOI] [PubMed] [Google Scholar]
- Frand AR, and Kaiser CA (1998). The ERO1 gene of yeast is required for oxidation of protein dithiols in the endoplasmic reticulum. Molecular Cell 1, 161–170. [DOI] [PubMed] [Google Scholar]
- Gladfelter AS, Pringle JR, and Lew DJ (2001). The septin cortex at the yeast mother-bud neck. Curr Opin Microbiol 4, 681–689. [DOI] [PubMed] [Google Scholar]
- Gustin MC, Albertyn J, Alexander M, and Davenport K (1998). MAP kinase pathways in the yeast Saccharomyces cerevisiae. Microbiol Mol Biol Rev 62, 1264–1300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen AS, Hao N, and O’Shea EK (2015). High-throughput microfluidics to control and measure signaling dynamics in single yeast cells. Nat Protoc 10, 1181–1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartwell LH (1974). Saccharomyces cerevisiae cell cycle. Bacteriol Rev 38, 164–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hereford LM, and Hartwell LH (1974). Sequential gene function in the initiation of Saccharomyces cerevisiae DNA synthesis. J Mol Biol 84, 445–461. [DOI] [PubMed] [Google Scholar]
- Higuchi-Sanabria R, Pernice WM, Vevea JD, Alessi Wolken DM, Boldogh IR, and Pon LA (2014). Role of asymmetric cell division in lifespan control in Saccharomyces cerevisiae. FEMS Yeast Res 14, 1133–1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang PJ, Sanson A, Lee B, and Park HO (2001). A GDP/GTP exchange factor involved in linking a spatial landmark to cell polarity. Science 292, 1376–1378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kono K, Saeki Y, Yoshida S, Tanaka K, and Pellman D (2012). Proteasomal degradation resolves competition between cell polarization and cellular wound healing. Cell 150, 151–164. [DOI] [PubMed] [Google Scholar]
- Kozubowski L, Saito K, Johnson JM, Howell AS, Zyla TR, and Lew DJ (2008). Symmetry-breaking polarization driven by a Cdc42p GEF-PAK complex. Curr Biol 18, 1719–1726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuo SC, and Lampen JO (1974). Tunicamycin--an inhibitor of yeast glycoprotein synthesis. Biochem Biophys Res Commun 58, 287–295. [DOI] [PubMed] [Google Scholar]
- Kuo TC, Chen CT, Baron D, Onder TT, Loewer S, Almeida S, Weismann CM, Xu P, Houghton JM, Gao FB, et al. (2011). Midbody accumulation through evasion of autophagy contributes to cellular reprogramming and tumorigenicity. Nat Cell Biol 13, 1214–1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Du Y, Siegel S, Ferro-Novick S, and Novick P (2010). Activation of the mitogen-activated protein kinase, Slt2p, at bud tips blocks a late stage of endoplasmic reticulum inheritance in Saccharomyces cerevisiae. Mol Biol Cell 21, 1772–1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lippincott J, and Li R (1998). Dual function of Cyk2, a cdc15/PSTPIP family protein, in regulating actomyosin ring dynamics and septin distribution. J Cell Biol 143, 1947–1960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu D, and Novick P (2014). Bem1p contributes to secretory pathway polarization through a direct interaction with Exo70p. J Cell Biol 207, 59–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lord M, Laves E, and Pollard TD (2005). Cytokinesis depends on the motor domains of myosin-II in fission yeast but not in budding yeast. Mol Biol Cell 16, 5346–5355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madden K, and Snyder M (1998). Cell polarity and morphogenesis in budding yeast. Annu Rev Microbiol 52, 687–744. [DOI] [PubMed] [Google Scholar]
- Marston AL, Chen T, Yang MC, Belhumeur P, and Chant J (2001). A localized GTPase exchange factor, Bud5, determines the orientation of division axes in yeast. Curr Biol 11, 803–807. [DOI] [PubMed] [Google Scholar]
- Meitinger F, Khmelinskii A, Morlot S, Kurtulmus B, Palani S, Andres-Pons A, Hub B, Knop M, Charvin G, and Pereira G (2014). A memory system of negative polarity cues prevents replicative aging. Cell 159, 1056–1069. [DOI] [PubMed] [Google Scholar]
- Mori K (2000). Tripartite management of unfolded proteins in the endoplasmic reticulum. Cell 101, 451–454. [DOI] [PubMed] [Google Scholar]
- Mostowy S, and Cossart P (2012). Septins: the fourth component of the cytoskeleton. Nat Rev Mol Cell Biol 13, 183–194. [DOI] [PubMed] [Google Scholar]
- Nelson SA, Sanson AM, Park HO, and Cooper JA (2012). A novel role for the GTPase-activating protein Bud2 in the spindle position checkpoint. PLoS One 7, e36127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh Y, and Bi E (2011). Septin structure and function in yeast and beyond. Trends Cell Biol 21, 141–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okada S, Leda M, Hanna J, Savage NS, Bi E, and Goryachev AB (2013). Daughter cell identity emerges from the interplay of Cdc42, septins, and exocytosis. Dev Cell 26, 148–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onishi M, Ko N, Nishihama R, and Pringle JR (2013). Distinct roles of Rho1, Cdc42, and Cyk3 in septum formation and abscission during yeast cytokinesis. J Cell Biol 202, 311–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park HO, Bi E, Pringle JR, and Herskowitz I (1997). Two active states of the Ras-related Bud1/Rsr1 protein bind to different effectors to determine yeast cell polarity. Proc Natl Acad Sci U S A 94, 4463–4468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park HO, Kang PJ, and Rachfal AW (2002). Localization of the Rsr1/Bud1 GTPase involved in selection of a proper growth site in yeast. J Biol Chem 277, 26721–26724. [DOI] [PubMed] [Google Scholar]
- Pina F, Yagisawa F, Obara K, Gregerson JD, Kihara A, and Niwa M (2018). Sphingolipids activate the endoplasmic reticulum stress surveillance pathway. J Cell Biol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pina FJ, Fleming T, Pogliano K, and Niwa M (2016). Reticulons Regulate the ER Inheritance Block during ER Stress. Dev Cell 37, 279–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pina FJ, and Niwa M (2015). The ER Stress Surveillance (ERSU) pathway regulates daughter cell ER protein aggregate inheritance. Elife 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pohl C, and Jentsch S (2009). Midbody ring disposal by autophagy is a post-abscission event of cytokinesis. Nat Cell Biol 11, 65–70. [DOI] [PubMed] [Google Scholar]
- Pollard MG, Travers KJ, and Weissman JS (1998). Ero1p: a novel and ubiquitous protein with an essential role in oxidative protein folding in the endoplasmic reticulum. Molecular Cell 1, 171–182. [DOI] [PubMed] [Google Scholar]
- Powell CD, Quain DE, and Smart KA (2003). Chitin scar breaks in aged Saccharomyces cerevisiae. Microbiology 149, 3129–3137. [DOI] [PubMed] [Google Scholar]
- Pruyne D, and Bretscher A (2000). Polarization of cell growth in yeast. I. Establishment and maintenance of polarity states. J Cell Sci 113 (Pt 3), 365–375. [DOI] [PubMed] [Google Scholar]
- Pruyne D, Legesse-Miller A, Gao L, Dong Y, and Bretscher A (2004). Mechanisms of polarized growth and organelle segregation in yeast. Annual Review of Cell and Developmental Biology 20, 559–591. [DOI] [PubMed] [Google Scholar]
- Robinson MD, Grigull J, Mohammad N, and Hughes TR (2002). FunSpec: a web-based cluster interpreter for yeast. BMC Bioinformatics 3, 35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ron D, and Walter P (2007). Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol 8, 519–529. [DOI] [PubMed] [Google Scholar]
- Rutkowski DT, and Kaufman RJ (2004). A trip to the ER: coping with stress. Trends Cell Biol 14, 20–28. [DOI] [PubMed] [Google Scholar]
- Schmidt M, Bowers B, Varma A, Roh DH, and Cabib E (2002). In budding yeast, contraction of the actomyosin ring and formation of the primary septum at cytokinesis depend on each other. J Cell Sci 115, 293–302. [DOI] [PubMed] [Google Scholar]
- Shcheprova Z, Baldi S, Frei SB, Gonnet G, and Barral Y (2008). A mechanism for asymmetric segregation of age during yeast budding. Nature 454, 728–734. [DOI] [PubMed] [Google Scholar]
- Sheff MA, and Thorn KS (2004). Optimized cassettes for fluorescent protein tagging in Saccharomyces cerevisiae. Yeast 21, 661–670. [DOI] [PubMed] [Google Scholar]
- Sheu YJ, Santos B, Fortin N, Costigan C, and Snyder M (1998). Spa2p interacts with cell polarity proteins and signaling components involved in yeast cell morphogenesis. Mol Cell Biol 18, 4053–4069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinclair DA, Mills K, and Guarente L (1998). Molecular mechanisms of yeast aging. Trends Biochem Sci 23, 131–134. [DOI] [PubMed] [Google Scholar]
- Singh P, Ramachandran SK, Zhu J, Kim BC, Biswas D, Ha T, Iglesias PA, and Li R (2017). Sphingolipids facilitate age asymmetry of membrane proteins in dividing yeast cells. Mol Biol Cell 28, 2712–2722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith SE, Rubinstein B, Mendes Pinto I, Slaughter BD, Unruh JR, and Li R (2013). Independence of symmetry breaking on Bem1-mediated autocatalytic activation of Cdc42. J Cell Biol 202, 1091–1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spokoini R, Moldavski O, Nahmias Y, England JL, Schuldiner M, and Kaganovich D (2012). Confinement to organelle-associated inclusion structures mediates asymmetric inheritance of aggregated protein in budding yeast. Cell Rep 2, 738–747. [DOI] [PubMed] [Google Scholar]
- Tarassov K, Messier V, Landry CR, Radinovic S, Serna Molina MM, Shames I, Malitskaya Y, Vogel J, Bussey H, and Michnick SW (2008). An in vivo map of the yeast protein interactome. Science 320, 1465–1470. [DOI] [PubMed] [Google Scholar]
- Thieleke-Matos C, Osorio DS, Carvalho AX, and Morais-de-Sa E (2017). Emerging Mechanisms and Roles for Asymmetric Cytokinesis. Int Rev Cell Mol Biol 332, 297–345. [DOI] [PubMed] [Google Scholar]
- Toenjes KA, Simpson D, and Johnson DI (2004). Separate membrane targeting and anchoring domains function in the localization of the S. cerevisiae Cdc24p guanine nucleotide exchange factor. Curr Genet 45, 257–264. [DOI] [PubMed] [Google Scholar]
- Tolliday N, Pitcher M, and Li R (2003). Direct evidence for a critical role of myosin II in budding yeast cytokinesis and the evolvability of new cytokinetic mechanisms in the absence of myosin II. Mol Biol Cell 14, 798–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong AH, and Boone C (2006). Synthetic genetic array analysis in Saccharomyces cerevisiae. Methods Mol Biol 313, 171–192. [DOI] [PubMed] [Google Scholar]
- Tu BP, Ho-Schleyer SC, Travers KJ, and Weissman JS (2000). Biochemical basis of oxidative protein folding in the endoplasmic reticulum. Science 290, 1571–1574. [DOI] [PubMed] [Google Scholar]
- van Drogen F, and Peter M (2002). Spa2p functions as a scaffold-like protein to recruit the Mpk1p MAP kinase module to sites of polarized growth. Curr Biol 12, 1698–1703. [DOI] [PubMed] [Google Scholar]
- Versele M, and Thorner J (2004). Septin collar formation in budding yeast requires GTP binding and direct phosphorylation by the PAK, Cla4. Journal of Cell Biology 164, 701–715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Versele M, and Thorner J (2005a). Some assembly required: yeast septins provide the instruction manual. Trends Cell Biol 15, 414–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Versele M, and Thorner J (2005b). Some assembly required: yeast septins provide the instruction manual. Trends in Cell Biology 15, 414–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voth WP, Olsen AE, Sbia M, Freedman KH, and Stillman DJ (2005). ACE2, CBK1, and BUD4 in budding and cell separation. Eukaryot Cell 4, 1018–1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watts FZ, Shiels G, and Orr E (1987). The yeast MYO1 gene encoding a myosin-like protein required for cell division. EMBO J 6, 3499–3505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weirich CS, Erzberger JP, and Barral Y (2008). The septin family of GTPases: architecture and dynamics. Nat Rev Mol Cell Biol 9, 478–489. [DOI] [PubMed] [Google Scholar]
- Weiss EL (2012). Mitotic exit and separation of mother and daughter cells. Genetics 192, 1165–1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wloka C, and Bi E (2012). Mechanisms of cytokinesis in budding yeast. Cytoskeleton (Hoboken) 69, 710–726. [DOI] [PubMed] [Google Scholar]
- Wu CF, Chiou JG, Minakova M, Woods B, Tsygankov D, Zyla TR, Savage NS, Elston TC, and Lew DJ (2015). Role of competition between polarity sites in establishing a unique front. Elife 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu N, Sarna LK, Hwang SY, Zhu Q, Wang P, Siow YL, and O K (2013). Activation of 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase during high fat diet feeding. Biochim Biophys Acta 1832, 1560–1568. [DOI] [PubMed] [Google Scholar]
- Young BP, and Loewen CJ (2013). Balony: a software package for analysis of data generated by synthetic genetic array experiments. BMC Bioinformatics 14, 354. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.