

Abstract
NMR-guided isolation (based on 1D 1H and 13C NMR resonances consistent with a chlorovinylidene moiety) resulted in the characterization of five new highly functionalized polyketides, trichophycins B-F (1-5) and one non-chlorinated metabolite tricholactone (6) from a collection of Trichodesmium bloom material from the Gulf of Mexico. The planar structures of 1-6 were determined using 1D and 2D NMR spectroscopy, mass spectrometry and complementary spectroscopic procedures. Absolute configuration analysis of 1 and 2 were carried out by 1H NMR analysis of diastereomeric Mosher esters in addition to ECD spectroscopy, J-based configuration analysis and DFT calculations. The absolute configurations of 3-6 were proposed based on comparative analysis of 13C NMR chemical shifts, relative configurations, and optical rotation values to compounds 1 and 2. Compounds 1-5 represent new additions to the trichophycin family and are hallmarked by a chlorovinylidene moiety. These new trichophycins and tricholactone (1-6) feature intriguing variations with respect to putative biosynthetic starting units, halogenation, and terminations and trichophycin E (4) features a rare alkynyl bromide functionality. The phenyl-containing trichophycins showed low cytotoxicity to neuro-2A cells, while the alkyne-containing trichophycins showed no toxicity.
Graphical Abstract
INTRODUCTION
Filamentous marine cyanobacteria continue to be an important source for the isolation of chemically diverse and biologically active secondary metabolites.1–4 Many of these molecules recognizably derive from polyketide synthase (PKS) biosynthetic pathways, non-ribosomal peptide synthetase (NRPS) pathways, or mixed PKS-NRPS pathways.5 Variations in the biosynthetic architecture that create these metabolites has led to remarkable chemical diversity and the evaluation of these cyanobacterial secondary metabolites in a broad range of biological assays has led to the identification of therapeutically-relevant biological activities including cytotoxicity,6,7 neuromodulation,8,9 anti-parasitism,10 and anti-inflammation.11 The availability of chloride, bromide and iodide in seawater and the action of halogenase enzymes in cyanobacterial biosynthetic pathways allows halide incorporation into marine natural products.12 Prior examples isolated from cyanobacteria include the lipoamides jamaicamide A,13 several vinyl chloride-containing malyngamides,14,15 and the polyketide kimbelactone.16
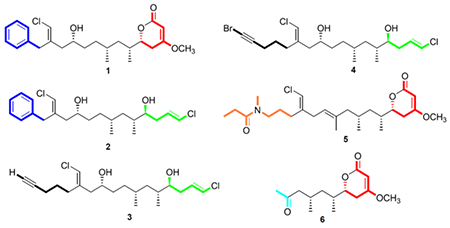
The vinyl chloride containing trichotoxins17 and the cytotoxic linear polyketide trichophycin A18 have been isolated from an environmental bloom of Trichodesmium by our laboratory. In an effort to gain greater insight into the secondary metabolite profile of these ecologically relevant bloom events and the chemical speciation of the chlorovinylidene-containing trichophycins, we examined lipophilic extracts and fractions of bloom material remaining in our laboratory from an event in the Gulf of Mexico in 2014. Six structurally similar compounds, five of which were halogenated and all presumably derived from a PKS system, were isolated and evaluated for cytotoxicity against the neuro-2A mouse neuroblastoma cell line. These new trichophycins and tricholactone (1-6) feature intriguing variations with respect to putative biosynthetic starting units, halogenation and terminations with trichophycin B (1), trichophycin F (5) and tricholactone (6) terminating in a functionalized lactone moiety and trichophycins C-E (2-4) featuring a terminal vinyl chloride group (Figure 1). A structure-activity relationship (SAR) suggests that phenyl-containing trichophycins and those with greater polyol character possess more significant cytotoxicity.
Figure 1.
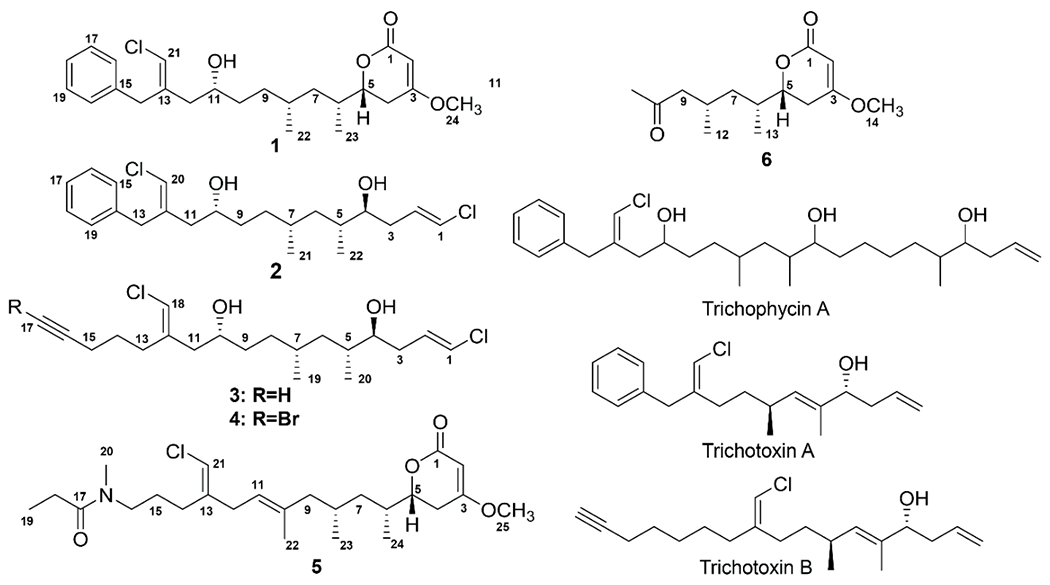
Structures of compound 1-6 and the previously reported trichophycin A18 and trichotoxins A and B.17
RESULTS AND DISCUSSION
Characterization of 1-6.
The extraction of Trichodesmium bloom material and fractionation of the crude extract guided by evaluation of 1D 1H NMR and 13C NMR spectra led to the isolation of six structurally similar compounds (1-6). Trichophycin B (1) was isolated as an optically active pale yellow oil. HRESIMS analysis of 1 gave an [M+H]+ of m/z 421.2153, suggesting a molecular formula of C24H33ClO4 and a requirement of 8 degrees of unsaturation. Examination and comparison of the 13C NMR, and 2D NMR spectra aided by a multiplicity-edited HSQC experiment showed the presence of 3 methyls, one of which was a methoxy group, 6 methylenes, 11 methines and 2 moderately deshielded olefinic carbons (δC 138.7 and 138.2), 1 deshielded olefinic carbon ((δC 173.3), and 1 carbonyl carbon with a chemical shift consistent with that of an ester group (δC 167.7) (Table 1). The 1H and 13C NMR spectra of 1 showed the characteristic signals of an aromatic ring for positions 15-20. Deshielded diastereotopic methylene protons (H2-14, δH 3.75, 3.51) showed HMBC correlations to the quaternary carbon of the aromatic ring (C-15, δC 138.2) and to a moderately polarized olefin comprised of a quaternary carbon (C-13, δC 138.7) and a carbon (C-21, δC 115.4) bearing a singlet methine proton at δH 6.07. These chemical shifts were consistent with the presence of a vinyl chloride functionality, and considering the aromatic moiety, accounted for 5 of the 8 required degrees of unsaturation. NOE correlations between H-21 (δH 6.07) and H2-12 (δH 2.17, 2.07) and H-11 (δH 3.60) supported a E geometry of the chlorovinylidene group. Moderately deshielded methylene protons H2-12 (H-2a, δH 2.17; H-2b, δH 2.07) showed HMBC correlations to the vinyl chloride-containing olefin, and examination of the 1H-1H COSY spectrum showed an extended spin system from H2-12 to H2-4 (Figure 2) and protons were correlated by interpretation of COSY NMR data. The H-5 oxymethine was considerably deshielded (δH 4.25) and showed correlations to H2-4 (δH 2.58, 2.17) and an HMBC correlation to C-1 (δC 167.7). The H2-4 methylene group showed HMBC correlations to an extremely polarized olefin (C-3, δC 173.3; C-2, δC 90.3), and H-4a showed an HMBC correlation to C-1. An HMBC correlation from the H3-24 methoxy group to C-3 completed the characterization of an α,β-unsaturated δ-lactone with a methoxy group at the β position. This functionalized lactone satisfied the 3 remaining degrees of unsaturation and completed the planar structure of 1.
Table 1.
NMR data for trichophycin B (1)a
| position | δC, type | δH (J in Hz) | HMBC | COSY | NOESY |
|---|---|---|---|---|---|
| 1 | 167.7, C | ||||
| 2 | 90.3, CH | 5.13, d (1.6) | 1, 3, 4 | 24 | |
| 3 | 173.3, C | ||||
| 4a | 30.3, CH2 | 2.58, ddd (17.0, 12.9, 1.7) | 1, 2, 3, 5, 6 | 4b, 5 | 7a, 7b, 23 |
| 4b | 2.17, ovlpb | 2, 3, 5, 6 | 4a, 5 | 23 | |
| 5 | 79.0, CH | 4.25, dt (12.9, 3.9) | 1, 3, 4, 6, 7, 23 | 4a, 4b, 6 | 7b, 8, 23 |
| 6 | 33.9, CH | 1.83, m | 5, 7, 23 | 5, 7a, 7b, 23 | 4a, 22 |
| 7a | 39.7, CH2 | 1.46, ddd (13.5, 8.3, 5.7) | 5, 6, 8, 9, 22, 23 | 6, 7b | 4a |
| 7b | 1.08, ddd (13.5, 8.5, 5.5) | 5, 6, 8, 9, 22, 23 | 6, 7a, 8 | 4a, 5 | |
| 8 | 29.8, CH | 1.49, m | 7, 9, 10, 22 | 9b, 22 | 5, 11, 23 |
| 9a | 31.7, CH2 | 1.30, ovlp | 8, 10, 11, 22 | 9b, 10a | 12a |
| 9b | 1.14, m | 8, 10, 11, 22 | 8, 9a | 11 | |
| 10a | 34.1, CH2 | 1.42, m | 9, 11 | 10b, 11 | 12b |
| 10b | 1.30, ovlp | 9, 11 | 10a, 11 | ||
| 11 | 69.2, CH | 3.60, m | 9, 10, 12, 13 | 10a, 12b | 8, 9b, 21 |
| 12a | 42.7, CH2 | 2.17, ovlp | 10, 11, 13, 14, 21 | 11 | 9a, 21 |
| 12b | 2.07, dd (14.4, 9.0) | 10, 11, 13, 14, 21 | 11 | 10a, 21 | |
| 13 | 138.7, C | ||||
| 14a | 36.3, CH2 | 3.75, d (14.4) | 12, 13, 15, 16, 20, 21 | 14b | 16, 20 |
| 14b | 3.51, d (14.4) | 12, 13, 15, 16, 20, 21 | 14a | 16, 20 | |
| 15 | 138.2, C | ||||
| 16 | 128.7. CH | 7.22, ovlp | 14, 18 | 17 | 14a, 14b |
| 17 | 128.6, CH | 7.29, t (7.6) | 15 | 16 | |
| 18 | 126.5, CH | 7.22, ovlp | 16, 20 | ||
| 19 | 128.6, CH | 7.29, t (7.6) | 15 | 20 | |
| 20 | 128.7, CH | 7.22, ovlp | 14, 18 | 19 | 14a, 14b |
| 21 | 115.4, CH | 6.07, s | 12, 13, 14 | 11, 12a, 12b | |
| 22 | 20.3, CH3 | 0.85, d (6.4) | 7, 8, 9 | 8 | 6, 7b, 9a |
| 23 | 14.9, CH3 | 0.97, d (6.8) | 5, 6, 7 | 6 | 4a, 4b, 5, 8 |
| 24 | 56.0, CH3 | 3.74, s | 3 | 2 |
800 MHz for 1H NMR, 200 MHz for 13C NMR
overlapping signals
Figure 2.
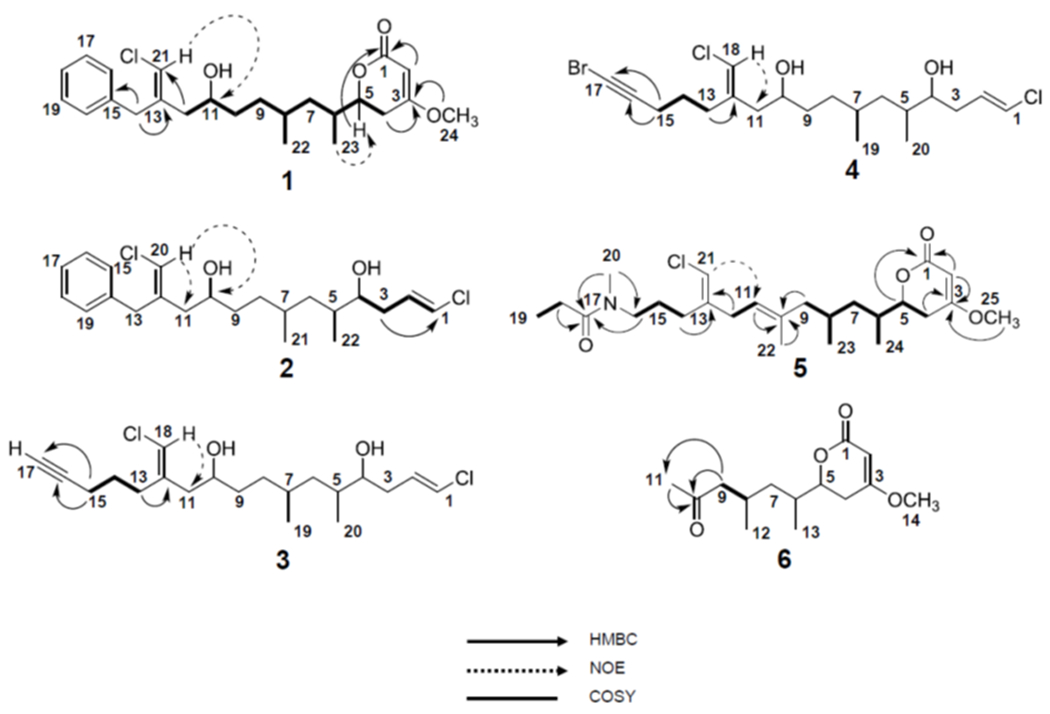
Selected 2D NMR correlations of 1-6.
HRESIMS analysis of 2 gave an [M+H]+ of m/z 399.1853, suggesting the molecular formula of C22H32Cl2O2 and a requirement of 6 degrees of unsaturation for this molecule. Using correlative information from 1D and 2D NMR experiments for 2, a polyketide chain was constructed from position 3 to position 22 in 2 nearly identical to that of position 4 to position 23 in trichophycin B (1) (cf. Tables 2 and 3). However, in 2 the oxygen-bearing carbon (C-4) is not part of a lactone ring functionality as it is in 1 as the more shielded oxymethine proton in 2 (H-4, δH 3.45) compared to that of 1 (H-5, δH 4.25) supported the free secondary alcohol. Whereas 1 is hallmarked by an extended polyketide chain which ultimately terminates in a lactone ring, the allylic methylene protons at position 3 in 2 (H2-3, δH 2.24, 2.13) showed HMBC correlations to a polarized olefin with chemical shifts consistent with that of a terminal vinyl chloride functionality. The C-1/C-2 olefin in 2 was determined to be E by virtue of a large vicinal 1H-1H coupling constant (13.2 Hz). NOE correlations established the chlorovinylidene group adjacent to the phenyl moiety as E in trichophycin C (2), identical to that of 1.
Table 2.
1H NMR for trichophycins C-E (2-4) (800 MHz, CDCl3)
| 2 | 3 | 4 | |
|---|---|---|---|
| pos | δH (J in Hz) | δH (J in Hz) | δH (J in Hz) |
| 1 | 6.04, d (13.2) | 6.05, d (13.2) | 6.05, d (13.2) |
| 2 | 5.94, m | 5.96, m | 5.95, m |
| 3a | 2.24, m | 2.25, m | 2.25, ovlp |
| 3b | 2.13, m | 2.14, m | 2.14, ovlp |
| 4 | 3.45, ddd (8.7, 5.3, 3.4) | 3.47, ddd (8.8, 5.2, 3.4) | 3.47, ddd (8.8, 5.2, 3.4) |
| 5 | 1.63, m | 1.68, ovlp | 1.67, ovlp |
| 6a | 1.35, ddd (13.3, 9.0, 4.5) | 1.40, ddd (13.3, 8.6, 4.5) | 1.40, ovlp |
| 6b | 0.96, ddd (13.5, 9.4, 5.5) | 1.00, ddd (13.5, 9.1, 5.4) | 1.00, ddd (14.0, 9.2, 5.5) |
| 7 | 1.46, m | 1.53, m | 1.53, m |
| 8a | 1.30, ovlp | 1.40, ovlp | 1.40, ovlp |
| 8b | 1.10, m | 1.20, m | 1.20, m |
| 9a | 1.41, m | 1.49, m | 1.50, m |
| 9b | 1.30, ovlp | 1.40, ovlp | 1.40, ovlp |
| 10 | 3.60, m | 3.68, m | 3.68, m |
| 11a | 2.17, dd (14.3, 3.6) | 2.27, m | 2.26, ovlp |
| 11b | 2.07, dd (14.3, 8.9) | 2.13, ovlp | 2.12, ovlp |
| 12 | |||
| 13a | 3.75, d (14.4) | 2.44, m | 2.42, m |
| 13b | 3.51, d (14.4) | 2.25, olvp | 2.24, ovlp |
| 14 | 1.69, ovlp | 1.69, ovlp | |
| 15 | 7.21, ovlp | 2.24, ovlp | 2.25, ovlp |
| 16 | 7.30, t (7.2) | ||
| 17 | 7.22, ovlp | 1.99, t (2.6) | |
| 18 | 7.30, t (7.2) | 5.94, s | 5.94, s |
| 19 | 7.21, ovlp | 0.91, ovlp | 0.91, d (6.6) |
| 20 | 6.07, s | 0.90, ovlp | 0.90, d (6.8) |
| 21 | 0.87, d (6.6) | ||
| 22 | 0.86, d (6.4) | ||
overlapping signals
Table 3.
13C NMR for trichophycins C-E (2-4) (200 MHz, CDCl3)
| 2 | 3 | 4 | Δδ 13C (2-3; 2-4) | |
|---|---|---|---|---|
| pos | δC, type | δC, type | δC, type | |
| 1 | 119.2, CH | 119.2, CH | 119.2, CH | 0.0; 0.0 |
| 2 | 130.7, CH | 130.7, CH | 130.7, CH | 0.0; 0.0 |
| 3 | 35.0, CH2 | 35.0, CH2 | 35.1, CH2 | 0.0; −0.1 |
| 4 | 74.8, CH | 74.8, CH | 74.8, CH | 0.0; 0.0 |
| 5 | 35.8, CH | 35.8, CH | 35.8, CH | 0.0; 0.0 |
| 6 | 39.8, CH2 | 39.7, CH2 | 39.8, CH2 | 0.1; 0.0 |
| 7 | 30.1, CH | 30.2, CH | 30.2, CH | −0.1; −0.1 |
| 8 | 31.5, CH2 | 31.6, CH2 | 31.6, CH2 | −0.1; −0.1 |
| 9 | 34.0, CH2 | 34.1, CH2 | 34.1, CH2 | −0.1; −0.1 |
| 10 | 69.2, CH | 69.2, CH | 69.3, CH | 0.0; −0.1 |
| 11 | 42.8, CH2 | 43.1, CH2 | 43.1, CH2 | −0.3; −0.3 |
| 12 | 138.6, C | 138.9, C | 138.8, C | −0.3; −0.2 |
| 13 | 36.3, CH2 | 29.4, CH2 | 29.4, CH2 | |
| 14 | 138.2, C | 26.1, CH2 | 25.9, CH2 | |
| 15 | 128.7, CH | 18.4, CH2 | 19.6, CH2 | |
| 16 | 128.6, CH | 83.9, C | 79.7, C | |
| 17 | 126.6, CH | 68.8, CH | 38.4, C | |
| 18 | 128.6, CH | 115.4, CH | 115.4, CH | |
| 19 | 128.7, CH | 20.7, CH3 | 20.7, CH3 | |
| 20 | 115.5, CH | 15.7, CH3 | 15.8, CH3 | |
| 21 | 20.6, CH3 | |||
| 22 | 15.7, CH3 | |||
HRESIMS analysis of 3 gave an [M+H]+ of m/z 375.1855, suggesting a molecular formula of C20H32Cl2O2 and a requirement of 4 degrees of unsaturation. Examination of the 13C NMR spectrum of 3 showed nearly identical signals to trichophyin C (2), with the exception of aromatic carbon signals, which were absent in 3 (Table 3). 1H-13C and 1H-1H correlations were made following interpretation of HMBC and COSY spectra, respectively, and established a partial structure of 3 identical to that of 2 from position 1 to position 12. Correlation analysis showed that the allylic methylene group H2-13 was coupled to a more shielded methylene group (H2-14, δH 1.69). Additionally, correlations showed that the H2-14 methylene group was coupled to a second deshielded methylene group (H2-15, δH 2.24). This methylene group showed HMBC correlations to the C-16 quaternary carbon (δC 83.9) and a carbon (C-17, δC 68.8) with an attached proton at δ1.99 establishing an alkyne functionality and completing the planar structure of trichophycin D (3). The NOE correlations and 1H-1H vicinal coupling constants showed that the chlorovinylidene and terminal vinyl chloride groups in 3 and had identical geometry to those in 2.
HRESIMS analysis of 4 gave an [M+H]+ of m/z 453.0961, suggesting a molecular formula of C20H32BrCl2O2 and a requirement of 4 degrees of unsaturation. Examination of 1H and 13C NMR signals showed that 4 was nearly identical in structure to 3 with the exception of the alkyne functionality (cf. Tables 2 and 3). The C-17 signal in 4 (δC 38.4) showed no proton attachment and was tremendously shifted upfield from that in 3 (δC 68.8). The upfield shift of C-16 (δC 83.9 in 3, δC 79.9 in 4) and the information gained from mass spectrometric analysis strongly supported an alkynyl bromide functionality in trichophycin E (4). The relative configurations of 4 were identical to that of 3. An alkynyl bromide functionality is also found in the mixed polyketide-peptide jamaicamide A and the alkyne signals in trichophycin E (δC 79.9 and 38.4) matched those of jamaicamide A quite well (δC 79.9 and 38.2).13
HRESIMS analysis of 5 gave an [M+H]+ of m/z 454.2739, suggesting a molecular formula of C25H40ClNO4, and a requirement of 6 degrees of unsaturation. Examination of the 1H NMR spectrum of 5 showed certain resonances with split signals in a 1:1 ratio. This phenomenon has been observed in several cyanobacteria metabolites with methylated tertiary amides such as smenamides A and B and kalkitoxin.19,20 After examining NOE correlations, these split signal effects were determined to be the result of conformers in the E and Z configuration at a tertiary amide functionality in 5. 1D and 2D NMR analysis of 5, led to the assignment of a partial structure that was identical to that of trichophycin B (1) from C-1 to C-9 including the methyl branches at C-6 and C-8 (Table 4). In a second partial structure, a moderately deshielded diastereotopic methylene group (H2-14, δH 2.19) was correlated by interpretation of COSY spectra to a second methylene group (H2-15, δH 1.63) which itself was correlated to a third methylene group (H2-16, δH 3.34). This deshielded methylene was correlated by examination of HMBC spectra to C-20 (δC 34.4) and the C-17 carbonyl (δC 173.4). A methylene (H2-18, δH 2.33) and methyl triplet (H3-19, δH 1.06) showed an HMBC correlation to C-17 and characterized the western half of 5 with an N-methyl propanamide functionality. H2-14 showed HMBC correlations to C-13 (δC 142.2) and C-21 (δC 112.6) establishing the chlorovinylidene moiety of the trichophycins. NOE correlations from H-21 to H-11 supported the chlorovinylidene configuration as E. The chemical shift value at C-22 (δC 15.0) and NOE correlations from H-11 to H2-9 and H3-22 to H2-12 supported an E configuration of C10/C11 olefin. H2-12 (δH 2.85) showed HMBC correlations to C-13 and C-21 as well as C-11 (δC 121.9) and the C-10 quaternary carbon (δC 136.8). A methyl correlated to C-10 by HMBC (H3-22 (δH 1.63) and an HMBC correlation from H2-9 to C-10 connected to two partial structures and completed the planar structure of 5.
Table 4.
NMR data for trichophycin F (5) Z-conformer (800 MHz for 1H NMR; 200 MHz for 13C NMR, CD3CN)
| position | δC, type | δH (J in Hz) | HMBC | COSY | NOESY |
|---|---|---|---|---|---|
| 1 | 167.0, C | ||||
| 2 | 89.6, CH | 5.11, s | 1, 3, 4 | 4a | 25 |
| 3 | 173.9, C | ||||
| 4a | 30.0, CH2 | 2.60, dd (17.0, 13.0) | 2, 3, 5, 6 | 2, 4b, 5 | 4b, 24 |
| 4b | 2.24, ddd (17.0, 3.6, 1.3) | 2, 3, 5 | 4a, 5 | 4a | |
| 5 | 78.7, CH | 4.30, dt (13.0, 3.9) | 3, 4, 6, 7, 24 | 4a, 4b, 6 | 4a, 4b, 7a, 8, 24 |
| 6 | 33.7, CH | 1.88, m | 5, 7, 24 | 24 | 23, 24 |
| 7a | 39.8, CH2 | 1.45, m | 5, 6, 8, 9, 22, 23 | 6, 7b, 8 | 7b |
| 7b | 1.07, m | 5, 6, 8, 9, 22, 23 | 6, 7a | 7a | |
| 8 | 27.5, CH | 1.75, m | 7, 9 | 9a, 23 | 5, 7a, 23, 24 |
| 9a | 46.9, CH2 | 2.10, m | 7, 8, 10, 11, 23 | 9b | 9a |
| 9b | 1.74, ovlpa | 7, 8, 10, 11, 23 | 9a | 9b | |
| 10 | 136.8, C | ||||
| 11 | 121.9, CH | 5.14, m | 9, 12, 22 | 12, 22 | 9a, 9b, 12 |
| 12 | 33.0, CH2 | 2.85, m | 10, 11, 13, 21 | 11, 21 | 22 |
| 13 | 142.2, C | ||||
| 14 | 27.8, CH2 | 2.19, ovlp | 12, 13, 15, 16 | 15 | |
| 15 | 24.6, CH2 | 1.63, m | 13, 14, 16 | 14, 16 | |
| 16 | 47.5, CH2 | 3.34, td, (7.1, 1.7) | 14, 15, 17, 20 | 15 | 15, 20 |
| 17 | 173.4, C | ||||
| 18 | 26.2, CH2 | 2.33, m | 17, 19 | 19 | 19, 20 |
| 19 | 9.0, CH3 | 1.06, t (7.4) | 17, 18 | 18 | 18 |
| 20 | 34.4, CH3 | 2.97, s | 16, 17 | 15, 16, 18 | |
| 21 | 112.6, CH | 5.92, s | 12, 13, 14 | 12 | 11, 12 |
| 22 | 15.0, CH3 | 1.63, s | 9, 10, 11 | 11 | 12 |
| 23 | 19.6, CH3 | 0.86, d (6.3) | 7, 8, 9 | 8 | 6, 7a, 7b, 8 |
| 24 | 14.3, CH3 | 0.99, d (6.8) | 5, 6, 7 | 6 | 4a, 5, 6, 8 |
| 25 | 56.0, CH3 | 3.75, s | 3 | 2 |
overlapping signals
HRESIMS analysis of 6 gave an [M+H]+ of m/z 255.1595, suggesting a molecular formula of C14H22O4 and a requirement of 4 degrees of unsaturation. NMR analysis determined that 6 contained the functionalized lactone moiety present in 1 and supported an identical planar structure from C-1 to C-8 in 6 as that from C-1 to C-8 in 1 including the position of the methyl groups (Table S2). However, in 6, a deshielded methylene group (H2-9, δH 2.44 and 2.18) showed HMBC correlations to C-8 and a carbonyl at C-10 (δC 208.9). A considerably deshielded methyl group at H3-11 (δH 2.08) showed HMBC correlations to C-10 and C-9 and completed the planar structure of tricholactone (6).
Stereochemical assignment of 1.
The absolute configuration assignment of the trichophycins was challenging due to the occurrence of non-adjacent secondary alcohol groups in compounds 2-4, the presence of multiple stereocenters, and highly overlapped methylene regions in the 1D 1H NMR data.
Initial work to address this challenge began with assignment of the absolute configuration for the C-11 center of 1. This was determined by a modified Mosher’s esterification procedure21 using two equal portions of 1 that were acylated with R-(−)-and S-(+)-α-methoxy-α (trifluoromethyl)phenylacetyl chloride (α-MTPA-Cl). These reactions yielded the C-11 R ester from (S)-MTPA-Cl and the C-11 S ester from (R)-MTPA-Cl. Examination of the 1D 1H NMR and 1H-1H TOCSY spectra of the individual esters allowed calculation of Δ(δHS-δHR) values. These results showed negative Δ(δHS-δHR) values for H-21, H-14a, H-14b, H-12a and H-12b and positive Δ(δHS-δHR) values for H-10a, H-10b, H-9a, H-9b, H-8, H-7a, H-7b, H-4a, H-4b, and H-22 thus supporting an 11R configuration. The configuration of C-5 in 1, 5 and 6 was assigned as R following determination of the axial orientation of H-5 (large vicinal coupling constant between H-5 and H-4a, J = 13.0 Hz) and examination of ECD spectra of each compound which showed a positive n→π Cotton effect at 244 nm (Figures S43–S45). The stereochemistry at C-5 in these compounds was determined using the rules of Snatzke and Beecham22,23 coupled with a comparison of ECD curves of previously characterized similar molecules.24,25
The absolute configurations at C-6 and C-8 in 1 were addressed by application of the J-based configuration analysis method and supported by DFT calculations, which were performed for the four possible stereo-configurations, i.e. C11RC8SC6SC5R, C11RC8SC6RC5R, C11RC8RC6SC5R and C11RC8RC6RC5R. The configurations at C-11 and C-5 were kept fixed in each case. Briefly, 200 conformers generated through a previously described procedure26 were submitted to Gaussian 09 for geometry optimization at the B3LYP/6-31g(d,p) level of theory. Vibrational frequencies were calculated with the “freq” Gaussian keyword. After discarding redundant structures and structures with imaginary frequencies, the remaining conformers were ranked based on the sum of electronic and thermal free energies. For those conformers whose energies were within the 2.2 kcal/mol window of the lowest energy conformer, J-couplings were calculated in Gaussian 09 at the B3LYP/6-311+g(d,p) level of theory; the “mixed” Gaussian keyword was used to request a two-step coupling calculation.27 Finally, the J-couplings were averaged across the lowest energy conformers based on their calculated Boltzmann populations. The number of conformers used for J-coupling averaging and their respective Boltzmann distributions for each diastereomer are listed in Table 5.
Table 5.
Conformational distribution of different diastereomers of 1
| Stereochemistry | Number of lowest energy conformers (Boltzmann Populations, %) |
|---|---|
| C8SC6S | 12 (48.7, 15.3, 8.6, 5.9, 5.7, 3.8, 3.6, 2.1, 1.7, 1.7, 1.6, 1.4) |
| C8SC6R | 9 (57.5, 30.5, 2.8, 1.9, 1.7, 1.5, 1.5, 1.4, 1.2) |
| C8RC6S | 8 (43.3, 32.7, 7.1, 5.2, 4.2, 3.3, 2.7, 1.6) |
| C8RC6R | 12 (53.9, 9.2, 7.6, 6.9, 5.6, 4.0, 3.4, 2.9, 1.8, 1.7, 1.5, 1.4) |
Key calculated coupling constants obtained from the DFT calculations were compared to experimental J-coupling values measured using a variety of NMR experiments such as PSYCHEDELIC28 for the determination of 1H-1H coupling constants and HSQC-TOCSY IPAP,29 sel-HSQMBC IPAP,30 and HMBC IPAP31 for the measurement of long-range 1H-13C coupling constants. Unfortunately, the number of relevant experimental couplings that could be measured from all these techniques was still somewhat limited. Nonetheless, as shown in Table 6, a unique and optimal fit appeared for the C11RC8RC6RC5R configuration. This assignment was strengthened based on the key 2JH6-C5 coupling, which was measured to be a “small” value (< 2 Hz) that was only compatible with the proposed configuration (Table 6, shaded line).
Table 6.
DFT [B3LYP/6-311+g(d,p)//B3LYP/6-31g(d,p)] analysis of 1.
| Coupling | C8SC6S | C8SC6R | C8RC6S | C8RC6R | Experimental |
|---|---|---|---|---|---|
| H5 – H4a/H4b | 13.2/3.9 | 14.5/3.9 | 12.8/4.0 | 14.0/3.7 | 12.9/3.9 |
| H5-H6 | 6.3 | 3.6 | 8.6 | 3.0 | 4.2 |
| H6 – H7a/H7b | 11.8/2.5 | 11.7/4.3 | 11.2/2.7 | 10.7/5.5 | 8.5/5.7 |
| H8 – H7a/H7b | 12.1/3.2 | 11.9/4.1 | 11.9/3.1 | 10.7/4.8 | n.m.a/5.5 |
| H6 – C5 | −6.0 | −6.1 | −5.4 | 0.7 | <2.0 |
| H6 – C4 | 4.1 | 5.5 | 2.4 | 2.0 | n.m. |
| H6 – C8 | 2.7 | 1.5 | 2.3 | 1.5 | n.m. |
| H8 – C6 | 2.1 | 1.5 | 2.1 | 1.7 | n.m. |
| H4a/H4b – C6 | 2.9/0.5 | 2.7/0.3 | 3.1/0.5 | 2.4/0.6 | 3.5/1.2 |
| H7a/H7b – C5 | 4.2/1.4 | 8.2/2.4 | 3.2/2.2 | 6.2/2.2 | 4.2/4.0 |
not measured
To further bolster the preliminary stereochemical assignment, we carefully examined the most stable conformers for each of the four possible configurations obtained from the DFT output and carefully analyzed the 2D NOESY data. According to DFT, a strong NOE between H-5 and H-8 should be expected for the C8RC6R stereo-configuration, but not for the other three diastereomers.
The <R−6> averaged distance of H-5—H-8 for C8RC6R among the lowest energy conformers within a 2.2 kcal/mol cut-off window was 2.2, 4.2, 3.5 and 4.6 Å for C8RC6R, C8RC6S, C8SC6R, and C8SC6S, respectively. Acquisition of a high-resolution 2D NOESY spectrum was required to overcome some overlapping issues, which in combination with a 1D selective NOE experiment, shown in Figure 3, provided clear evidence of an NOE correlation between H-5 and H-8, thus further supporting the C8RC6RC5R stereochemistry.
Figure 3.
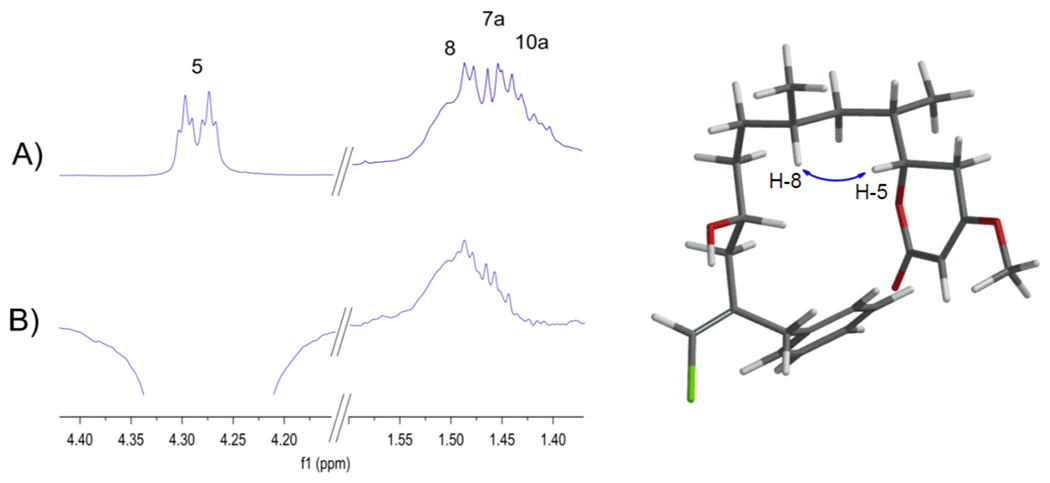
A) 1H NMR spectrum of 1. B) 1D sel-NOE after inversion of H-5.
Stereochemical assignment of 2.
In analogy to the work done to assign the absolute configuration of 1, we were able to generate and analyze bis-MTPA esters of 2. Examination of the 1D 1H NMR and 1H-1H COSY spectra of the individual bis-esters allowed calculation of Δ(δHS-δHR) values. These results revealed negative Δ(δHS-δHR) values for H-19, H-15, H-13a, H-20 and H-11 and positive Δ(δHS-δHR) values for H-9, H-8, H-7, H-6a, H-6b, H-5, H-4, H-21 and H-22. Additional negative Δ(δHS-δHR) values for H-3, H-2 and H-1 supported configurations of 10R and 4S.
Initial analysis of 3JHH couplings indicated the 2 was quite flexible so the stereochemical assignment of C-7 and C-5 was carried out by J-based configuration analysis supplemented with computational modeling by DFT, similarly adopted from the work done for 1. Four different stereo-configurations were considered, including C7RC5R, C7RC5S, C7SC5R, and C7SC5S, with C-10 and C-4 fixed to R and S, respectively. To better account for the flexibility of the molecule, geometry optimization was conducted at two different levels of theory in Gaussian 09, namely B3LYP/6-31g(d,p) and M062X/6-31g(d,p). These results are summarized in Table S3.
Calculation of J-couplings was performed as described previously for 1, and the averaged J-couplings are listed in Table 7 along with experimentally measured values. As is clearly shown in Table 7, the RMSD between theoretically calculated and experimentally measured J-couplings is the lowest for C7RC5R, using both B3LYP and MO62X functionals. The RMSDs of the other diastereomers are 2-3 fold larger. In Table S4 we compared the average J-couplings obtained from conformational distributions calculated at the B3LYP/6-31g(d,p) and MO62X/6-31g(d,p) levels of theory, in order to estimate the “error-bars” on the theoretical values. The relatively small RMSD between the theoretical couplings from B3LYP and MO62X, ranging from 0.7 to 1 Hz for the different stereoisomers, indicate good consistency between these two levels of theory. These results clearly favor C7RC5R as the correct stereoisomer for 2 and when taken together with the analysis of the Mosher esters support a 10R,7R,5R,4S absolute configuration assignment for 2 (Figure 4).
Table 7.
Comparison of experimentally measured J-couplings with theoretically calculated values for 2
| Coupling | B3LYP/6-311+g(d,p)//B3LYP/6-31g(d,p) |
B3LYP/6-311+g(d,p)//MO62X/6-31g(d,p) |
Exp (Hz) | ||||||
|---|---|---|---|---|---|---|---|---|---|
| C7SC5S | C7SC5R | C7RC5S | C7RC5R | C7SC5S | C7SC5R | C7RC5S | C7RC5R | ||
| H6a – C8 | 2.4 | 2.4 | 7.1 | 6.1 | 1.8 | 1.8 | 8.2 | 6.0 | 5.8 |
| H6b – C21 | 3.1 | 3 | 3.8 | 4.3 | 2.7 | 2.5 | 3.1 | 4.7 | 4.0 |
| H6b – C22 | 2.7 | 6.1 | 2.6 | 6.3 | 2.5 | 6.7 | 1.4 | 5.9 | 6.2 |
| H6a – H5 | 8.4 | 9.1 | 11.2 | 9.2 | 10.0 | 10.1 | 11.8 | 8.5 | 9.4 |
| H6b – C8 | 5.1 | 5.1 | 2.2 | 3.1 | 3.7 | 4.6 | 2.1 | 3.2 | 3.8 |
| H6a – H7 | 7.5 | 7.6 | 5.4 | 6.6 | 5.9 | 7.1 | 4.6 | 6.6 | 5.5 |
| H6b – H7 | 8.0 | 8.4 | 10.9 | 9.1 | 10.3 | 9.8 | 12.5 | 9.4 | 9.0 |
| H5 – H4 | 4.5 | 3.6 | 3.0 | 4.9 | 4.2 | 3.6 | 3.2 | 6.3 | 5.3 |
| H5 – C3 | 2 | 3.6 | 1.5 | 3.3 | 1.8 | 2.9 | 1.2 | 2.7 | 4.1 |
| H4 – C6 | 2.1 | 2.9 | 2.1 | 2.8 | 1.9 | 3.4 | 2.0 | 4.0 | 2.7 |
| H4 – C22 | 4.6 | 4.3 | 5.3 | 3.7 | 4.8 | 3.9 | 5.5 | 2.9 | 3.5 |
| H5 – C4 | 1.4a | 2.8a | 0.2 | 3.5a | 0.8a | 1.8a | 0.3 | 3.6a | 4.7 |
| RMSD (Hz) | 2.1 | 1.6 | 2.2 | 0.7 | 2.2 | 1.8 | 2.7 | 0.9 | |
The actual calculated value is negative.
Figure 4.
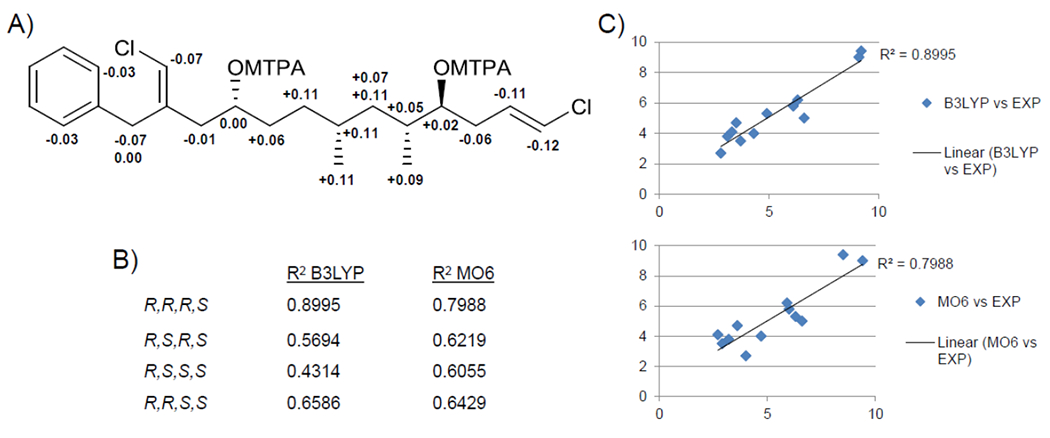
(A) ΔδS-R values of bis-Mosher esters of 2. B) R2 values for correlations between theoretical and experimental J-couplings for stereoconfigurations at C-7 and C-5 implementing both the B3LYP and MO6 levels of theory (configurations at C-10 and C-4 were held fixed as R and S, respectively. C) Plots of theoretical vs. experimental J-coupling values for C-7R and C-5R at the two levels of theory for 2.
The absolute configuration of 3 and 4 was proposed by analogy to 2 based on 13C NMR chemical shifts of the stereogenic carbons (Table 3, Δδ 13C 2-3; 2-4 values) and comparison of the relative configuration. The absolute configuration of C-5 in 5 and 6 was determined using ECD and remaining absolute configurations are proposed by relative configuration comparison to 1.
Cytotoxic Activity.
Trichophycin B (1) had an EC50 value against neuro-2A cells of 14.8 ± 2.4 μM. Trichophycin C (2) (EC50: 23.8 ± 4.2 μM) was less potent. The alkyne-containing trichophycins showed less potent activity than those containing phenyl groups. Trichophycin D (3) had an EC50 value of 39.8 ± 3.8 μM, while trichophycin E (4) showed low toxicity even at 100 μM and was essentially non-toxic. Trichophycin F (5) showed moderate toxicity against neuro-2A cells (EC50: 14.3 ± 2.3 μM) similar to that of 1. Tricholactone (6) degraded before it could be assessed for biological activity. Comparing the cytotoxicity of trichophycin C (2) in this study (EC50: neuro-2A = 23.8 ± 4.2 μM) to that previously reported for trichophycin A (EC50: neuro-2A = 6.5 ± 1.4 μM) and to that of trichotoxin A (EC50: neuro-2A ≥ 50 μM)18 provides increasing evidence of an association of increasing cytotoxicity with increasing polyol character. However, the trichophycins displaying greater polyol character have longer carbon chains. Determining the cytotoxicity of dehydrated trichophycin analogs would provide greater insight into this relationship.
Predicted Biosynthesis.
Trichophycins B-F (1-5) represent new analogs of trichophycin A and additions to the collection of structurally interesting halogenated cyanobacterial metabolites. The trichophycins recognizably derive from a standard PKS biosynthetic pathway(s). The most apparent structural differences among the trichophycins relate to the putative biosynthetic starter unit, the number of vinyl or alkynyl halide groups, and the mode of biosynthetic termination. While Trichodesmium theibautii was identified as the dominant species in the bloom from field characters, the bloom likely comprises multiple species of Trichodesmium.32 Thus, we cannot ascribe a single pathway to the origin of these metabolites. These metabolites may be generated by multiple pathways. This may explain the configuration differences at C-5 in 1 and C-4 in 2. Potential flexibility in the loading module in the putative biosynthetic pathway of trichophycins C, D, and E (2-4) may confer the ability to incorporate a probable phenylacetate unit (1 and 2) and a 5-hexynoate unit (3 and 4). The 5-hexynoate unit is the proposed starter group in the biosynthesis of jamaicamide A13 and terminal alkynes are present in a number of cyanobacterial lipopeptides.33–35 In jamaicamide A, the alkyne is proposed to arise from a 5-hexanoic acid precursor by the action of a fatty acid desaturase.13 Alkynyl bromide formation in jamaicamide A is proposed to occur through electrophilic addition by a haloperoxidase,36 which may also be the case with trichophycin E (4). The pendant vinyl group adjacent to the starter unit in the trichophycins would likely be generated by the action of an HMG-CoA synthase cassette, which has been shown to perform alkylation at the β position in a growing polyketide chain.5,37 Subsequent chlorination would give rise to the chlorovinylidene. The terminal vinyl chloride present in 2-4 could arise from the action of an FAD-dependent halogenase as is proposed in the formation of a terminal vinyl bromide functionality in the macrocyclic polyketide phormidolide.38 The formation of the terminal alkene in 2-4 is likely similar to that of curacin A, in which a conserved tridomain of an ACP, sulfotransferase and decarboxylating thioesterase participate to form a terminal double bond resulting from sulfate elimination from the beta position and decarboxylation.39,40
Trichophycin F (5) adds to the biosynthetic questions surrounding this group of molecules as the biosynthesis of 5 may incorporate a glycine unit or 4-aminobutyric acid. Tricholactone 6 may be an oxidative degradation product of 5, and is the only molecule in the group isolated that does not feature at least one vinyl chloride group. The trichophycins and tricholactone were stored neat. While trichophycins B-F have remained stable, tricholactone degraded.
Without information on the gene cluster or clusters encoding the trichophycins, we are limited in our ability to understand the biosynthetic processes involved. Genomic studies are planned to address these questions surrounding biosynthesis.
Experimental methods
General Experimental Procedures.
Optical rotations were measured using a Jasco P-2000 polarimeter. UV spectra were measured using a Beckman Coulter DU-800 spectrophotometer. CD spectra were recorded using a Jasco J-1100 CD spectrometer. NMR spectra were collected using Bruker 600 or 800 MHz NMR instruments equipped with a cryoprobe and a Varian 500 MHz instrument. HRESIMS analysis was performed using an AB SCIEX TripleTOF 4600 mass spectrometer with Analyst TF software. Semi-preparative HPLC was carried out using an Agilent 1100 series HPLC or a Dionex UltiMate 3000 HPLC system each equipped with a micro vacuum degasser, an autosampler and a photodiode-array detector.
Collection of Biological Material.
A localized bloom of Trichodesmium was collected from Padre Island, Corpus Christi, TX during 9-11 May 2014. Bloom material was collected from ca. 0.5-meter water depth by collecting surface bloom material in 5-gallon buckets. Approximately 300g wet weight cell mass was concentrated from this material by gentle filtration through 18 μm mesh. In the laboratory, a subsample of the cell mass was examined microscopically and identified according to Komarek (2005)41 by phycologist Paul V. Zimba at Texas A&M Corpus Christi.
Extraction and Isolation of Compounds 1-6.
Trichodesmium bloom material (14.4 g, dry wt) was repeatedly extracted with 2:1 CH2Cl2-CH3OH affording 3.95 g of crude lipophilic extract. The crude residue was reconstituted in hexanes and fractionated over silica gel using vacuum liquid chromatography (VLC) and a stepped gradient of hexanes, EtOAc and CH3OH. The VLC fraction eluting with 40% EtOAc in hexanes (Fraction D, 293.4 mg) was further fractionated over a 2 g Strata C18 SPE column eluting with 50% CH3CN in H2O, 100% CH3CN, 100% CH3OH and 100% EtOAc. The fraction eluting with 100% CH3CN (127.9 mg) was subjected to RP-HPLC using a YMC 5 μm ODS column (250 x 10 mm) with an elution solvent of 85% CH3CN in H2O with 0.1% formic acid added. Trichophycin D (3) (1.0 mg; tR 3.75 min) was isolated along with a second impure HPLC fraction. This fraction was separated using a YMC 5 μm ODS column (250 x 10 mm) with an elution solvent of 80% CH3OH in H2O with 0.1% formic acid added. Trichophycin C (2) (3.9 mg; tR 11.50 min) and trichophycin E (4) (0.1 mg; tR 12.50 min) were isolated from this fraction. The fractions eluting with 60% EtOAc in hexanes and 80% EtOAc in hexanes (Fractions E and F) were combined based on similarities in 1H NMR signals (306.4 mg) and subjected to RP-HPLC using a YMC 5 μm ODS column (250 × 10 mm) with an elution solvent of 80% CH3CN in H2O with 0.1% formic acid added and trichophycin B (1) (17 mg; tR 5.50 min) was isolated. The fractions eluting with 100% EtOAc and 25% CH3OH in EtOAc in hexanes (Fractions G and H) were combined based on similarities in 1H NMR signals and similar LC-MS profiles and the combined material (390.9 mg) was fractionated over a 2 g Strata C18 SPE column following an identical fractionation pattern as that for fraction D. The fraction eluting with 100% CH3CN (143.2 mg) was subjected to RP-HPLC using a YMC 5 μm ODS column (250 × 10 mm) with an elution solvent of 70% CH3CN in H2O with 0.1% formic acid and tricholactone (6) (0.5 mg; tR 16.00 min) was isolated along with a second impure fraction. A final purification of this impure material was carried out using a Kinetex 5 μm C18 column (250 × 10 mm); mobile phase: 75% CH3CN in water with 0.05% formic acid added to each solvent, flow 3 mL/min and 2.0 mg of 5 were isolated (tR, 9.0 min).
Trichophycin B (1):
Pale yellow oil; [α]22D +10.0 (c 0.20, MeOH); UV (MeOH) λmax (log ε) 211 (4.1), 231 (3.8) nm; 1H NMR (800 MHz, CDCl3) and 13C NMR (200 MHz, CDCl3), see Table 1; HRMS (ESI/QTOF) m/z: [M+H]+ (calcd for C24H34ClO4, 421.2146; found 421.2153).
Trichophycin C (2):
Colorless oil; [α]22D +26.8 (c 0.20, MeOH); UV (MeOH) λmax (log ε) 204 (4.0) nm; 1H NMR (800 MHz, CDCl3), see Table 2 and 13C NMR (200 MHz, CDCl3), see Table 3; HRMS (ESI/QTOF) m/z: [M+H]+ (calcd for C22H33Cl2O2, 399.1858; found 399.1853).
Trichophycin D (3):
Colorless oil; [α]22D +11.3 (c 0.08, MeOH); UV (MeOH) λmax (log ε) 202 (3.5) nm; 1H NMR (800 MHz, CDCl3), see Table 2 and 13C NMR (200 MHz, CDCl3), see Table 3; HRMS (ESI/QTOF) m/z: [M+H]+ (calcd for C20H33Cl2O2, 375.1858; found 375.1855).
Trichophycin E (4):
Colorless oil; UV (from UV scan during LC-MS analysis) 210 nm; 1H NMR (800 MHz, CDCl3), see Table 2 and 13C NMR (200 MHz, CDCl3), see Table 3; HRMS (ESI/QTOF) m/z: [M+H]+ (calcd for C20H32BrCl2O2, 453.0963; found 453.0961).
Trichophycin F (5):
Colorless oil; [α]22D +17.4 (c 0.2, MeOH); UV (MeOH) λmax (log ε) 202 (3.8), 229 (3.5) nm; 1H NMR (800 MHz, CD3CN) and 13C NMR (200 MHz, CD3CN), see Table 4; HRMS (ESI/QTOF) m/z: [M+H]+ (calcd for C25H41ClNO4, 454.2724; found 454.2739).
Tricholactone (6):
Colorless oil; UV (from UV scan during LC-MS analysis) 210, 228 nm; 1H NMR (800 MHz, DMSO) and 13C NMR (200 MHz, DMSO), see Table S2; HRMS (ESI/QTOF) m/z: [M+H]+ (calcd for C14H23O4, 255.1596; found 255.1595).
Preparation and Analysis of MTPA esters.
1 mg of trichophycin B (1) was dissolved in dry CDCl3 and separated into two equal portions in 4 mL vials. Dry pyridine (10 μL) and (S)-(+)-α-methoxy-α-(trifluoromethyl)phenylacetyl chloride (15 μL) were added to the first vial. The vial was capped and the reaction mixture was stirred for 24 h. The identical procedure was repeated with an equal amount of 1 and (R)-(−)-a-methoxy-a-(trifluoromethyl)phenylacetyl chloride. After 24 h, the contents of the vials were immediately transferred to NMR tubes for further analysis. S-ester 1H NMR (800 MHz, CDCl3) δ 7.28 (2H, t, J = 7.4 Hz, H-17, H-19), 7.22 (1H, d, J = 7.4 Hz, H-18), 7.19 (2H, d, J = 7.4 Hz, H-16, H-20), 5.89 (1H, s, H-21), 5.19 (1H, s, H-2), 5.17 (1H, m, H-11), 4.24 (1H, dt, J = 13.0, 3.7 Hz, H-5), 3.73 (3H, s, H-24), 3.72 (1H, d, J = 14.8 Hz, H-14a), 3.46 (1H, d, J = 14.8 Hz, H-14b), 2.57 (1H, m, H-4a), 2.26 (1H, dd, J = 14.6, 8.3 Hz, H-12a), 2.17 (1H, m, H-4b), 2.16 (1H, m, H-12b), 1.74 (1H, m, H-6), 1.63 (1H, m, H-8), 1.46, m, H-10a), 1.37 (1H, ovlp, H-7a), 1.36 (1H, ovlp, H-9a), 1.23 (1H, m, H-10b), 1.12 (1H, m, H-9b), 1.07 (1H, m, H-7b), 0.94 (3H, d, J = 6.8 Hz, H-23), 0.81 (3H, d, J = 6.6 Hz, H-22); R-ester 1H NMR (800 MHz, CDCl3) δ 7.28 (2H, m, H-17, H-19), 7.22 (1H, m, H-18), 7.21 (2H, d, J = 7.6 Hz, H-16, H-20), 5.97 (1H, s, H-21), 5.19 (1H, s, H-2), 5.17 (1H, m, H-11), 4.24 (1H, dt, J = 13.0, 3.6 Hz, H-5), 3.73 (3H, s, H-24), 3.78 (1H, d, J = 14.4 Hz, H-14a), 3.47 (1H, d, J = 18.0 Hz, H-14b), 2.71 (1H, dd, J = 17.0, 12.8 Hz, H-4a), 2.55 (1H, dd, J = 17.0, 3.5 Hz, H-4b), 2.32 (1H, dd, J = 14.6, 8.5 Hz, H-12a), 2.20 (1H, dd, J = 14.4, 4.5 Hz, H-12b), 1.74 (1H, m, H-6), 1.53 (1H, m, H-8), 1.41 (1H, m, H-10a), 1.33 (1H, m, H-7a), 1.29 (1H, m, H-9a), 1.15 (1H, m, H-10b), 1.06 (1H, m, H-7b), 0.90 (1H, m, H-9b), 0.94 (3H, d, J = 6.8 Hz, H-23), 0.77 (3H, d, J = 6.6 Hz, H-22).
The procedure above was performed on 1.8 mg of trichophycin C (2).
Trichophycin C MTPA: S-ester 1H NMR (500 MHz, CDCl3) δ 7.27 (2H, t, J = 7.4 Hz, H-16, H-18), 7.22 (1H, d, J = 7.4 Hz, H-17), 7.17 (2H, d, J = 7.4 Hz, H-15, H-19), 5.89 (1H, s, H-20), 5.87 (1H, s, H-1), 5.68 (1H, m, H-2), 5.16 (1H, m, H-10), 4.99 (1H, m, H-4), 3.70 (1H, d, J = 14.5, H-13a), 3.49 (1H, m, H-13b), 2.25 (2H, m, H-3), 2.15 (2H, dd, J = 14.5, 4.5 Hz, H-11), 1.83 (1H, m, H-5), 1.55 (2H, m, H-9), 1.42 (1H, m, H-7), 1.19 (2H, ovlp, H-8), 1.18 (1H, ovlp, H-6a), 0.95 (1H, m, H-6b), 0.86 (3H, d, J = 6.8 Hz, H-21), 0.82 (3H, d, J = 6.4 Hz, H-22); R-ester 1H NMR (500 MHz, CDCl3) δ 7.27 (2H, m, H-16, H-18), 7.22 (1H, m, H-17), 7.20 (2H, d, J = 7.5 Hz, H-15, H-19), 5.96 (1H, s, H-20), 5.99 (1H, s, H-1), 5.79 (1H, m, H-2), 5.16 (1H, m, H-10), 4.97 (1H, m, H-4), 3.77 (1H, d, J = 14.5, H-13a), 3.49 (1H, m, H-13b), 2.31 (2H, m, H-3), 2.16 (2H, m, H-11), 1.78 (1H, m, H-5), 1.49 (2H, m, H-9), 1.31 (1H, m, H-7), 1.08 (2H, ovlp, H-8), 1.11 (1H, ovlp, H-6a), 0.84 (1H, m, H-6b), 0.75 (3H, d, J = 6.7 Hz, H-21), 0.73 (3H, d, J = 6.8 Hz, H-22).
Cytotoxicity Assay.
Neuro-2A cells were added to assay plates in 100 μl of Eagle’s Minimum Essential Media (EMEM) supplemented with 10% FBS at a density of 5,000 cells/well. Cells were incubated overnight (37 °C, 5% CO2) and examined microscopically to confirm confluence and adherence. Test substances were dissolved in DMSO (1% v/v) and added to the cells in the range of 100, 10, 1, 0.1 and 0.01 μM in order to construct a dose response curve. Three technical replicates were prepared for each concentration and each assay was performed in triplicate. Doxorubicin was used as a positive control (EC50: 0.112 ± 0.021 μM) and DMSO was used as a negative control. Plates were incubated for 72 h after which 15 μl of MTT dye were added each assay well. The dye was allowed to incubate with the cells for 4 h after which media was aspirated and the remaining formazan crystals were solubilized in 100 μl of DMSO. Absorbance at 540 nm was measured using a Molecular Devices SpectraMax plate reader and EC50 curves were generated and statistical procedures were performed using Graphpad Prism software.
Supplementary Material
ACKNOWLEDGMENT
We thank Paul V. Zimba and I-Shuo Huang at Texas A&M Corpus Christi for field collection assistance and cyanobacteria identification. We thank Peter D. R. Moller of NOAA for assistance with NMR experiments. Research reported in this publication was made possible by the use of spectrometric and spectroscopic equipment and services available through the RI-INBRE Centralized Research Core Facility, which is supported by the Institutional Development Award (IDeA) Network for Biomedical Research Excellence from the National Institute of General Medical Sciences of the National Institutes of Health under grant number P20GM103430. Certain NMR experiments were conducted at a research facility at the University of Rhode Island supported in part by the National Science Foundation EPSCoR Cooperative Agreement #EPS-1004057. This work was supported in part by an American Society of Pharmacognosy Starter Grant awarded to M. Bertin.
Footnotes
Publisher's Disclaimer: This document is the Accepted Manuscript version of a Published Work that appeared in final form in The Journal of Organic Chemistry, copyright © American Chemical Society after peer review and technical editing by the publisher. To access the final edited and published work see https://pubs.acs.org/articlesonrequest/AOR-vRKxX4NKsdfyWkM93BEb
Supporting Information
Tables of NMR spectroscopic data, DFT and J-coupling analysis, 1D and 2D NMR data, CD data, 1H NMR of MTPA derivatives and dose-response curves for compounds 1-6 are available free of charge at http://pubs.acs.org.
References
- (1).Gerwick WH; Moore BS Lessons from the past and charting the future of marine natural products drug discovery and chemical biology. Chem. Biol 2012, 19, 85–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Nunnery JK; Mevers E; Gerwick WG Biologically active secondary metabolites from marine cyanobacteria. Curr. Opin. Biotechnol 2010, 21, 787–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Tan LT Bioactive natural products from marine cyanobacteria for drug discovery. Phytochemistry 2007, 68, 954–979. [DOI] [PubMed] [Google Scholar]
- (4).Tan LT Pharmaceutical agents from filamentous marine cyanobacteria. Drug. Discov. Today 2013, 18, 863–871. [DOI] [PubMed] [Google Scholar]
- (5).Kehr JC; Picchi DG; Dittmann E Natural product biosyntheses in cyanobacteria: a treasure trove of unique enzymes. Beilstein J. Org. Chem 2011, 7, 1622–1635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Pereira AR; Kale AJ; Fenley AT; Byrum T; Debonsi HM; Gilson MK; Valeriote FA; Moore BS; Gerwick WH The carmaphycins: new proteasome inhibitors exhibiting an α,β-epoxyketone warhead form a marine cyanobacterium. Chembiochem. 2012, 13, 810–817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Luesch H; Yoshida WY; Moore RE; Paul VJ; Corbett TH Total structure determination of apratoxin A, a potent novel cytotoxin from the marine cyanobacterium Lyngbya majuscula. J. Am. Chem. Soc 2001, 123, 5418–5423. [DOI] [PubMed] [Google Scholar]
- (8).Aráoz R; Molgó J; Tandeau de Marsac N Neurotoxic cyanobacterial toxins. Toxicon. 2010, 56, 813–828. [DOI] [PubMed] [Google Scholar]
- (9).Berman FW; Gerwick WH; Murray TF Antillatoxin and kalkitoxin, ichthyotoxins from the tropical cyanobacterium Lyngbya majuscula, induce distinct temporal patterns of NMDA receptor-mediated neurotoxicity. Toxicon. 1999, 37, 1645–1648. [DOI] [PubMed] [Google Scholar]
- (10).Vining OB; Medina RA; Mitchell EA; Videau P; Li D; Serrill D; Kelly JX; Gerwick WH; Proteau PJ; Ishmael JE; McPhail KL Depsipeptide companeramides from a Panamanian marine cyanobacterium associated with the coibamide producer. J. Nat. Prod 2015, 78, 413–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Choi H; Mascuch SJ; Villa FA; Byrum T; Teasdale ME; Smith JE; Preskitt LB; Rowley DC; Gerwick L; Gerwick WH Honaucins A-C, potent inhibitors of inflammation and bacterial quorum sensing: synthetic derivatives and structure-activity relationships. Chem. Biol 2012, 19, 589–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Neumann CS; Fujimori DG; Walsh CT Halogenation strategies in natural product biosynthesis. Chem. Biol 2008, 15, 99–109. [DOI] [PubMed] [Google Scholar]
- (13).Edwards DJ; Marquez BL; Nogle LM; McPhail K; Goeger DE; Roberts MA; Gerwick WH Structure and biosynthesis of the jamaicamides, new polyketide-peptide neurotoxins from the marine cyanobacterium Lyngbya majuscula. Chem. Biol 2004, 11, 817–833. [DOI] [PubMed] [Google Scholar]
- (14).Cardellina JH; Marner FJ; Moore RE Malyngamide A, a novel chlorinated metabolite of the marine cyanophyte Lyngbya majuscula. J. Am. Chem. Soc 1979, 101, 240–242. [Google Scholar]
- (15).Gerwick WH; Reyes S; Alvarado B Two malyngamides from the Caribbean cyanobacterium Lyngbya majuscula. Phytochemistry 1987, 26, 1701–1704. [Google Scholar]
- (16).Nunnery JK; Engene N; Byrum T; Cao Z; Jabba SV; Pereira AR; Teatulohi M; Murray TF; Gerwick WH Biosynthetically intriguing chlorinated lipophilic metabolites from geographically distant tropical marine cyanobacteria. J. Org. Chem 2012, 77, 4198–4208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Schock TB; Huncik K; Beauchesne KR; Villareal TA; Moeller PDR Identification of trichotoxin, a novel chlorinated compound associated with the bloom forming cyanobacterium, Trichodesmium thiebautii. Environ. Sci. Technol 2011, 45, 7503, 7509. [DOI] [PubMed] [Google Scholar]
- (18).Bertin MJ; Wahome PG; Zimba PV; He H; Moeller PDR Trichophycin A, a cytotoxic linear polyketide isolated from a Trichodesmium thiebautii bloom. Mar. Drugs 2017, 15, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Teta R; Irollo E; Sala GD; Pirozzi G; Mangoni A; Costantino V Smenamides A and B, chlorinated peptide/polyketide hybrids containing a dolapyrrolidinone unit from the Caribbean sponge Smenospongia aurea. Evaluation of their role as leads in antitumor drug research. Mar Drugs 2013, 11, 4451–4463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Wu M; Okino T; Nogle LM; Marquez BL; Williamson RT; Sitachitta N; Berman FW; Murray TF; McGough K; Jacobs R; Colsen K; Asano T; Yokokawa F; Shioiri T; Gerwick WH Structure, synthesis, and biological properties of kalkitoxin, a novel neurotoxin from the marine cyanobacterium Lyngbya majuscula. J. Am. Chem. Soc 2000, 122, 12041–12042. [Google Scholar]
- (21).Hoye TR; Jeffrey CS; Shao F Mosher ester analysis for the determination of absolute configuration of stereogenic (chiral) carbinol carbons. Nat. Protoc 2007, 2, 2451–2458. [DOI] [PubMed] [Google Scholar]
- (22).Snatzke G Circular dichroism and optical rotary dispersion - principles and application to the investigation of the stereochemistry of natural products. Angew. Chem. Int. Ed 1968, 7, 14–25. [Google Scholar]
- (23).Beecham AF The CD of αβ-unsaturated lactones. Tetrahedron 1972, 28, 5543–5554. [Google Scholar]
- (24).Ellestad GA; McGahren WJ; Kunstmann MP Structure of a new fungal lactone, LL-P880, from an unidentified Penicillium sp. J. Org. Chem 1972, 37, 2045–2047. [DOI] [PubMed] [Google Scholar]
- (25).Tian JF; Yu RJ; Li XX; Gao H; Guo LD; Tang JS; Yao XS 1H and 13C NMR spectral assignments of 2-pyrone derivatives from an endophytic fungus of sarcosoataceae. Magn. Reson. Chem 2015, 53, 866–871. [DOI] [PubMed] [Google Scholar]
- (26).Sherer EC; Lee CH; Shpungin J; Cuff JF; Da C; Ball R; Bach R; Crespo A; Gong X; Welch CJ Systematic approach to conformational sampling for assigning absolute configuration using vibrational circular dichroism. J. Med. Chem 2014, 57, 477–494. [DOI] [PubMed] [Google Scholar]
- (27).Deng W; Cheeseman JR; Frisch MJ Calculation of nuclear spin-spin coupling constants of molecules with first and second row atoms in study of basis set dependence. J. Chem. Theory and Comput 2006, 2, 1028–1037. [DOI] [PubMed] [Google Scholar]
- (28).Sinnaeve D; Foroozandeh M; Nilsson M; Morris GA A general method for extracting individual coupling constants from crowded (1)H NMR spectra. Angew. Chem. Int. Ed. Engl 2016, 55, 1090–1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Nolis P; Espinosa JF; Parella T Optimum spin-state selection for all multiplicities in the acquisition dimension of the HSQC experiment. J. Magn. Reson 2006, 180, 39–50. [DOI] [PubMed] [Google Scholar]
- (30).Gil S; Espinosa JF; Parella T Accurate measurement of small heteronuclear coupling constants from pure-phase α/β HSQMBC cross-peaks. J. Magn. Reson 2011, 213, 145–150. [DOI] [PubMed] [Google Scholar]
- (31).Saurí J; Parella T On the interference of J(HH) modulation in HSQMBC-IPAP and HMBC-IPAP experiments. Magn. Reson. Chem 2013, 51, 509–516. [DOI] [PubMed] [Google Scholar]
- (32).Hynes AM; Webb EA; Doney SC; Waterbury. Comparison of cultured Trichodesmium (Cyanophyceae) with species characterized from the field. J. Phycol 2012, 48, 196–210. [DOI] [PubMed] [Google Scholar]
- (33).Mevers E; Liu WT; Engene N; Mohimani H; Byrum T; Pevzner PA; Dorrestein PC; Spadafora C; Gerwick WH Cytotoxic veraguamides, alkynyl bromide-containing cyclic depsipeptides from the marine cyanobacterium cf. Oscillatoria margaritifera. J. Nat. Prod 2011, 74, 928–936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Jiménez JI; Scheuer PJ New lipopeptides from the Caribbean cyanobacterium Lyngbya majuscula. J. Nat. Prod 2001, 64, 200–203. [DOI] [PubMed] [Google Scholar]
- (35).Hooper GJ; Orjala J; Schatzman RC; Gerwick WH Carmabins A and B, new lipopeptides from the Caribbean cyanobacterium Lyngbya majuscula. J. Nat. Prod 1998. 61, 529–533. [DOI] [PubMed] [Google Scholar]
- (36).Boudreau PD; Monroe EA; Mehrotra S; Desfor S; Korobeynikov A; Sherman DH; Murrary TF; Gerwick L; Dorrestein PC; Gerwick WH Expanding the described metabolome of the marine cyanobacterium Moorea producens JHB through orthogonal natural products workflows. PLoS One. 2015, 10, e0133297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Buchholz TJ; Rath CM; Lopanik NB; Gardner NP; Håkansson K; Sherman DH Polyketide β-branching in bryostatin biosynthesis: identification of surrogate acetyl-ACP donors for BryR, and HMG-ACP synthase. Chem. Biol 2010, 17, 1092–1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Bertin MJ; Vulpanovici A; Monroe EA; Korobeynikov A; Sherman DH; Gerwick L; Gerwick WH The phormidolide biosynthetic gene cluster: a trans-AT PKS pathway encoding a toxic macrocyclic polyketide. ChemBioChem. 2016, 17, 164–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Chang Z; Sitachitta N; Rossi JV; Roberts MA; Flatt PM; Jia J; Sherman DH; Gerwick WH Biosynthetic pathway and gene cluster analysis of curacin A, an antitubulin natural product from the tropical marine cyanobacterium Lyngbya majuscula. J. Nat. Prod 2004, 67, 1356–1367. [DOI] [PubMed] [Google Scholar]
- (40).Gehret JJ; Gu L; Gerwick WH; Wipf P; Sherman DH; Smith JL Terminal alkene formation by the thioesterase of curacin A biosynthesis: structure of a decarboxylating thioesterase. J. Biol. Chem 2011, 286, 14445–14454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Komárek J; Anagnostidis K Cyanoprokarota Part 2: Oscillatoriales; Elsevier: München, Germany, 2005; pp. 1–759. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.