

Abstract
The understanding of complex biological systems requires an ability to evaluate interacting networks of genes, proteins, and cellular reactions. Enabling technologies that support the rapid quantification of these networks will facilitate the development of biological models and help to identify treatment targets and to assess treatment plans. The biochemical process of protein phosphorylation, which underlies almost all aspects of cell signaling, is typically evaluated by immunoblotting procedures (Western blot) or more recently proteomics procedures, which provide qualitative estimates of the concentration of proteins and their modifications in cells. However, protein modifications are difficult to correlate with activity, and while immunoblotting and proteomics approaches have the potential to be quantitative, they require a complex series of steps that diminish reproducibility. Here, a complementary approach is presented that allows for the rapid quantification of a protein kinase activity in cell lysates and tumor samples. Using the activity of cellular ERK (extracellular signal-regulated kinase) as a test case, an array sensing approach that utilizes a library of differential peptide-based biosensors and chemometric tools was used to rapidly quantify nanograms of active ERK in micrograms of unfractionated cell lysates and tumor extracts. This approach has the potential both for high-throughput and for quantifying the activities of multiple protein kinases in a single biological sample. The critical advantages of this differential sensing approach over others are that it removes the need for the addition of exogenous inhibitors to suppress the activities of major off-target kinases and allows us to quantitate the amount of active kinase in tested samples rather than measuring the changes in its activity upon induction or inhibition.
Graphical Abstract
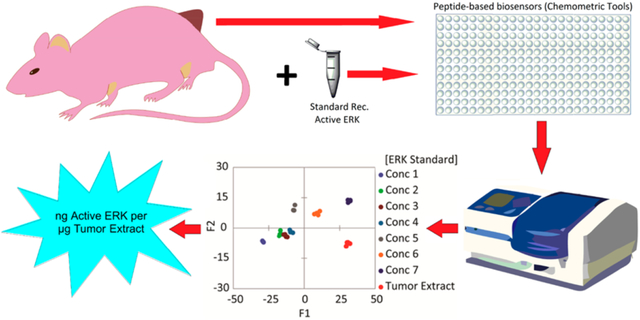
Protein kinases represent a set of attractive therapeutic targets due to their aberrant activity in human diseases, including numerous cancers.1,2 Understanding how to target kinases with small molecule inhibitors has significantly impacted drug discovery in oncology.3 However, despite often initial successful responses to kinase-targeted therapies, tumor cells eventually circumvent the action of such drugs, leading to resistance.
The protein kinases ERK1 and ERK2 (collectively termed ERK) are drug targets that modulate several regulatory mechanisms, including survival, proliferation, and metastatic potential of tumor cells.4 ERK1 and ERK2 are members of the mitogen-activated protein kinase (MAPK) family, which consists of three main groups of enzymes, the extracellular signal-regulated kinases (ERKs), the c-Jun-N-terminal kinases (JNKs), and the p38 MAP kinases. Specifically, ERK plays a pivotal role in the Ras-Raf-MEK-ERK pathway,5 components of which are frequently mutated in cancer, with more than 30% and ~8% of human tumors expressing RAS and RAF mutations, respectively.6–8 Somatic NRAS and BRAF mutations are extremely common in cutaneous melanoma, being present in up to 28% and 52% of cases, respectively (The Cancer Genome Atlas Network).9 Of the mutations reported, the constitutively active BRAF-V600E is highly prevalent.10 When this mutant is present, the ERK pathway downstream of Raf becomes refractory to regulatory signaling events.
A standard treatment for melanoma involves targeting the B-Raf and MEK kinases in the ERK pathway. For example, a combination of MEK and B-Raf inhibitors (dabrafenib and trametinib) yield objective response rates of 67%.11,12 However, such therapies are not always successful with resistance developing in patients that initially respond.13 As most of these resistance mechanisms drive the reactivation of ERK, there is a critical need for improving therapies targeting ERK and for anticipating and overcoming resistance mechanisms.14 Therefore, ERK activity represents a particularly informative biomarker for assessing ERK-driven cancers. A facile and direct method to evaluate ERK activity or inhibition in the context of cancer progression, treatment, and resistance15 would be of significant value to the scientific community.
The two most common approaches for studying ERK activity in cells or tissue samples are Western blot and immunohistochemistry.16 Some of the significant limitations of both assays are that (1) they do not measure kinase activity directly, (2) they are expensive and time-consuming, and (3) they are not amenable to high-throughput analysis (HTA) of many samples. These approaches contrast with fluorescence-based biosensors,15,17 such as the relatively selective SOX-based biosensors that have been used to detect changes in ERK activity in cell lysates18–20 directly.
The current fluorescence-based detection methods, as with the sensors introduced by Peterson et al.,20 report phosphorylation events with some selectivity; however, it is necessary to synthesize a specific receptor for each kinase target and with more than five hundred endogenous kinases, it is impossible to identify peptide biosensors that are uniquely specific for each. Furthermore, while these sensors are sufficient to estimate relative kinase activity in terms of percentage activity or fold changes, they are not generally amenable to absolute quantification of the target kinase due to their low specificity. To alleviate these difficulties, we developed a differential sensing approach to quantify the subtle changes in ERK activity in cancer cells. Differential sensing takes advantage of interactions between closely related biomolecules and cross-reactive biosensors, and a pattern-recognition protocol analyzes the responses from the semiselective biosensors.21,22
We21–25 and others have used cross-reactive arrays to fingerprint glycerides,23 proteins,24 and cell types.22,25–28 We recently reported a differential-based sensing approach to distinguish nine MAPKs from one another and developed a calibration regression model to measure ERK activation and inhibition in the presence of the closely related JNK and p38 MAPKs.29 However, the previous studies were carried out using purified MAPKs, which are not necessarily reflective of endogenous activity. Here, we report a differential-based sensing approach to rapidly quantify unknown endogenous levels of ERK activity in cancer cells. The ability to rapidly and simultaneously quantify ERK activity with high accuracy in multiple samples with high throughput provides an opportunity to comprehensively quantify the temporal changes in ERK signaling levels in response to modulators of the ERK pathway and relate these changes to observed cellular phenotypes.
The previously reported SOX-peptides Sub-D (1a), MEF2A (myocyte-specific enhancer factor 2A) (1b), NFAT4 (nuclear factor of activated T cells-4) (1c), and Sub-F (1d) were used to construct the cross-reactive array (Table S1 and Scheme 1). This differential sensing design exploits docking motifs found on MAPK substrates. In addition, the recruitment sites on MAPKs promote some level of binding specificity and affinity with peptide docking motifs within substrates.29 Achievement of cross-reactivity in the array results from the structural differences between the recruitment sites of MAPKs and their complementary substrate–peptide interactions.
RESULTS
Profiling MAPK Activity in MDA-MB-231 and A549 Cell Lysates.
As ERK is closely related to many other MAPKs and the purpose of these studies was to quantify ERK activity in cell lysates, it was first essential to demonstrate that an array of peptide sensors could distinguish between samples possessing varying levels of MAPK activity. Thus, we treated cell lines with activators, inhibitors, and a combination of both, which produce changes in the activity of not only ERK but also other MAPKs. Differences in the cell lines were generated by stimulation with either epidermal growth factor (EGF) (E), which induces ERK and other kinase activity,30 or anisomycin (A), which has been reported to activate both the JNK and p38 MAPK cascades.31 Addition of U0126, a MEK inhibitor (U),32 achieved inhibition of ERK. Addition of JNK-IN-8, a pan JNK inhibitor (J), achieved inhibition of JNK. Lastly, each cell line was treated with the MEK inhibitor then induced with EGF (E/U) or treated with JNK-IN-8 then induced with anisomycin (A/J). All the treatments were compared to control cells treated with serum-free media (n). Lysis of cells preceded assessment of MAPK activities by an array of SOX-peptides. Peptides were synthesized using standard Fmoc-SPPS protocol (supporting methods section), and their concentrations were determined following our previously published protocol.29 The arrays were prepared and the assays were conducted using 7 μg/well of the lysates following our general kinase assay protocol that is described in detail in the supporting methods section. ERK activity requires phosphorylation on both the threonine and tyrosine of a Thr-Pro-Tyr motif, and assessment of this phosphorylation in the lysates by Western blot analysis provides a useful comparison to the array activity assay data. Immune-complex protein kinase assays, which report on ERK activity, provide a further important layer of validation. As shown in Figure S1, the Western blots showed increased and decreased phosphorylation of ERK in cells treated with EGF and the MEK-inhibitor U0126, respectively. The phosphorylation of JNK, cJUN (JNK substrate), and p38 MAPK was enhanced upon induction by EGF or anisomycin in both cell lines tested and as expected, JNK-IN-8 inhibited cJUN phosphorylation but did not impact ERK phosphorylation in any of the cell lines treated.
We first evaluated the ability of our array to distinguish different levels of active MAPKs in MDA-MB-231 and A549 cell lines. The array primarily responds to all the kinase activities in the lysates and thus provides a “fingerprint” that relates to the activity status of the kinase signaling networks in the cells. Each combination of treatment and cell line generates a kinetic signature for each peptide sensor. The fluorescence output corresponding to the MEF2A-peptide, for example, is sensitive to the activity of both the ERK and JNK pathways and exhibits a response to modulators of these pathways (Figure 1). The array comprises four different sensors whose combined signatures represent a specific fingerprint for each treatment. As the individual peptides within the array are designed to be predominantly responsive to the MAPKs, the array is also expected to exhibit a bias toward their activity.
Figure 1.
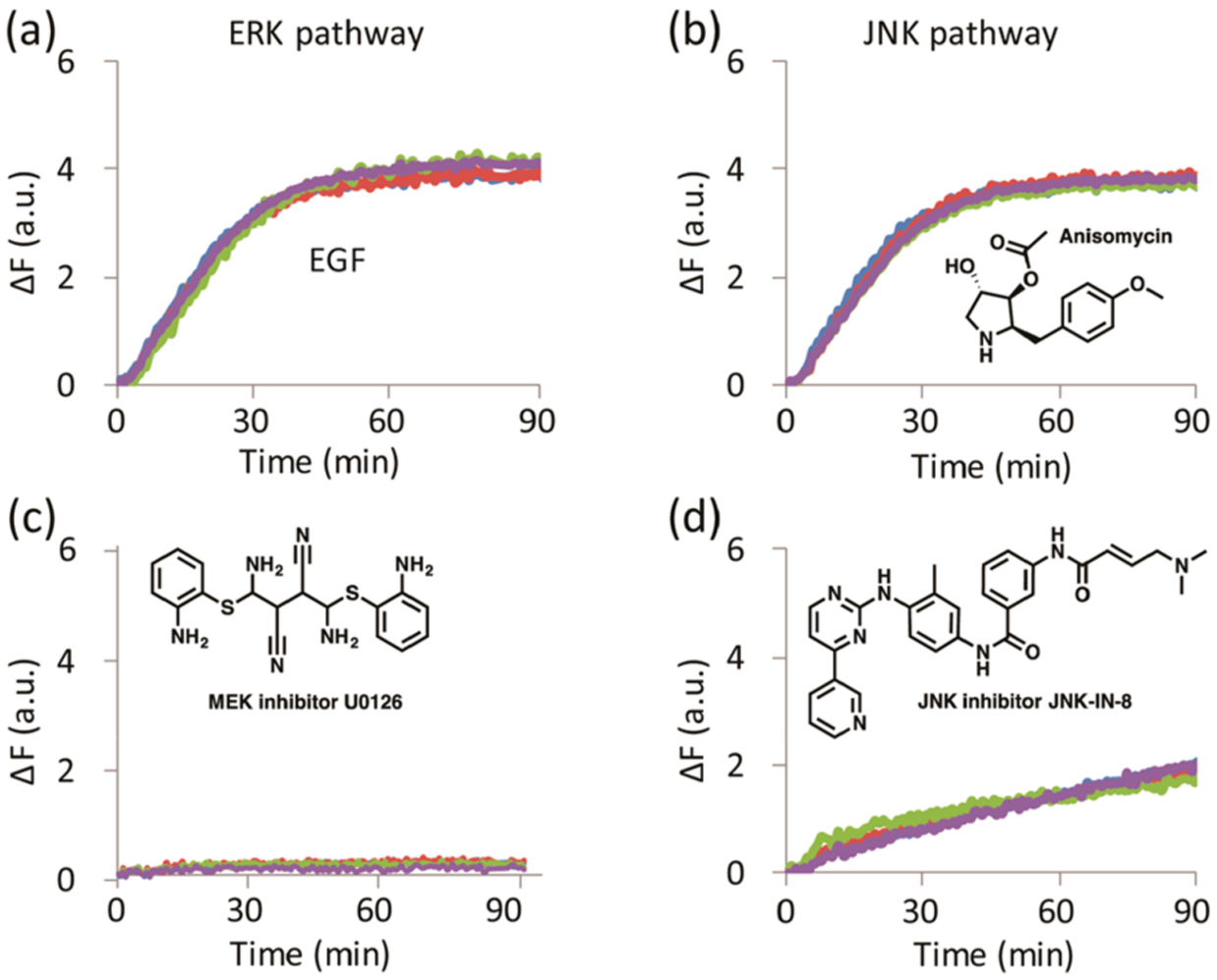
MAPK activity of MDA-MB-231 cell lysates towards SOX-MEF2A peptide. MDA-MB-231 cells treated with (a) EGF, (b) anisomycin, (c) MEK inhibitor U0126, and (d) JNK inhibitor JNK-IN-8, showing four experimental replicates per treatment.
The MDA-MB-231 and A549 cell lines that were incubated in serum-free media for 24 h and then treated with the MEK-inhibitor U0126 (M-U and A-U) were expected to display total suppression of ERK activity. Therefore, the M-U and A-U cell lysates were used to determine baseline references for each experiment in the corresponding cell line. The delta fluorescence (ΔF) values at 20 and 30 min, along with the calculated rate constants and errors, were used as multivariate data (4 peptides X 4 replicates X 4 inputs per each treatment) for chemometric analysis using linear discriminant analysis (LDA) in the program XLSTAT. LDA is a supervised method used to reduce the dimensionality of a data set and categorize the classes of analytes by maximizing the separation between preset classes while minimizing the distance within each class. The fluorescence and rate constant fingerprints of MDA-MB-231 and A549 cell lines generated by the array (Figure S2) gave the resulting LDA score plots in Figure 2a,b.
Figure 2.
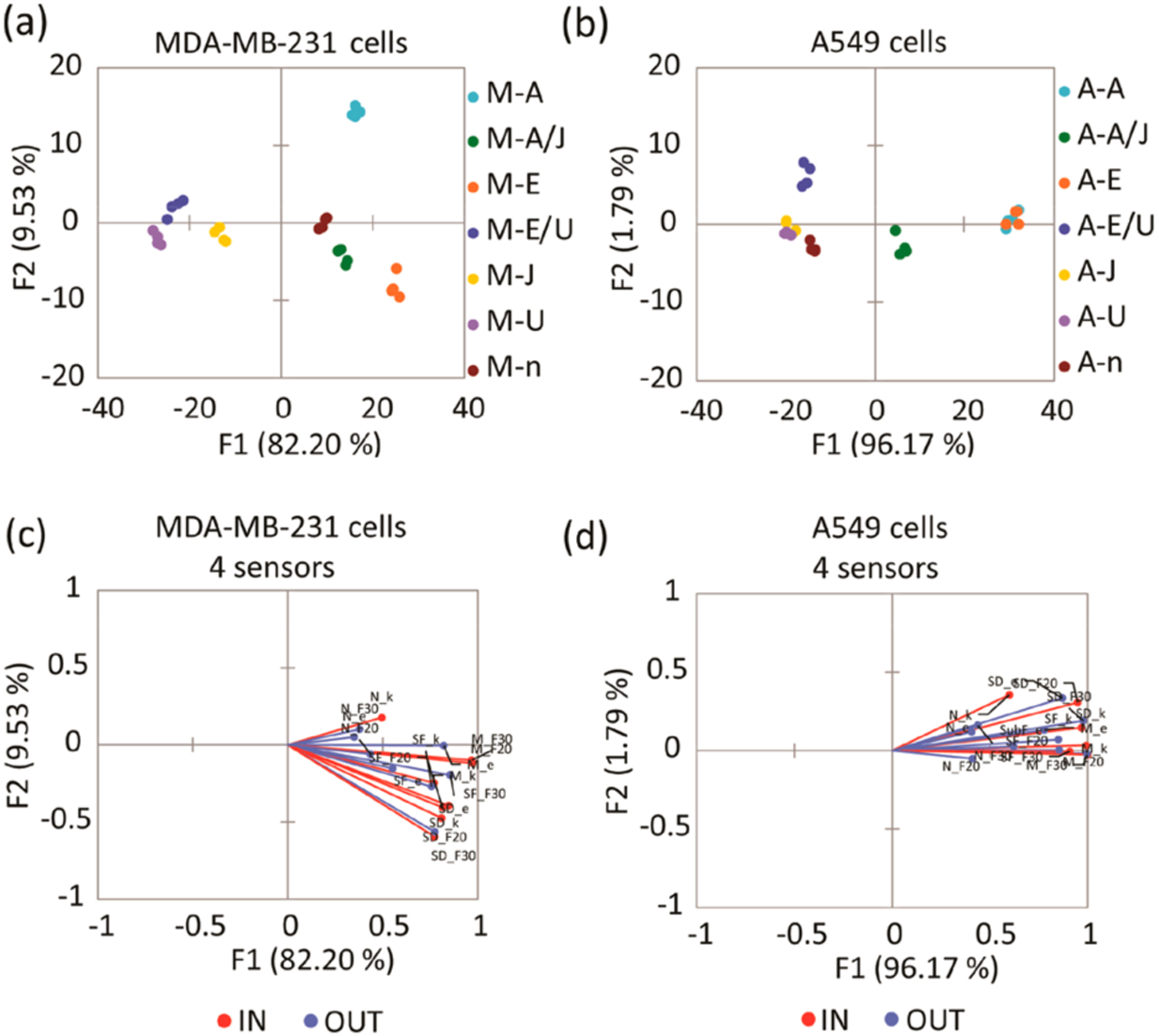
(a) LDA score plot of the fluorescence response pattern of MDA-MB-231 cell lines with different treatments, showing 100% cross-validation. (b) LDA score plot of the fluorescence response pattern of A549 cell lines with different treatments, showing 92.86% cross-validation. Serum-starved control (n), inhibition of ERK phosphorylation by U0126 (U), induction of ERK phosphorylation by EGF (E), cells treated with U0126 then induced by EGF (E/U), inhibition of JNK pathway by JNK-IN-8 (J), induction of JNK phosphorylation by anisomycin (A), cells treated with JNK-IN-8 then induced by anisomycin (A/J). (c, d) Loading plots of the response from the sensing array showing differentiation of sets of lysates from serum starved, inhibited, or stimulated MDA-MB-231 (c) and A549 (d) cell lines with 100% and 92.86% cross-validation using 4 sensors. Vectors corresponding to each sensor contribution to the differentiation of lysates, SOX-SubD (SD_F30,F20,k,e), SOX-MEF2A (M_F30,F20,k,e), SOX-SubF (SF_F30,F20,k,e), and SOX-NFAT4 (N_ F30,F20,k,e) are colored in blue and red. F30, F20, k, and e correspond to ΔF (30 min), ΔF (20 min), rate constant, and error, respectively.
The six treatments in the MDA-MB-231 cell line, along with functional clustering of the experimental replicates, were differentiated well by the array on the F1 and F2 axes (Figure 2a). Jack-knife analysis was performed, and the estimated percentage of well-classified classes was reported, showing high classification accuracy by our LDA model (100% cross-validation, Table S2). The cells treated with the activators (M-E and M-A) were classified on the right quadrants, whereas the cells treated with the inhibitors (M-U and M-J) locate to the opposite side. Finally, the control (M-n) along with the combined treatments (M-E/U and M-A/J) locate to the center of the score plot. Overall, while F1 carries 80% of the variance, F2 and F3 (not shown) pick up the cross-reactivity of the peptides to the various cellular treatments. In contrast, the A549 cell line with the same treatments (shown in Figure 2b) shows some overlap of both activators and inhibitors along the F2 axes, located on the right and the left axes, respectively. This means that these treatments cannot be differentiated, and the LDA model only shows 92.86% cross-validation (Table S3). Further, the data tightly clustered according to analyte identity along the F1 axis, which carries most of the overall variance (around 96%). Thus, the peptides give highly similar response trends to different treatments in A549 cell lines, which explains their lower analyte differentiation than obtained for MDA-MB-231 cell lines.33
Assessing Peptide Sensor Contribution toward MAPK Profiling.
Loading plots (Figure 2c,d) of the LDA score plots (Figure 2a,b) were generated to assess the contributions of each peptide to the output of the array. The red and blue vectors shown in both loading plots correspond to the contribution of each peptide parameter (ΔF at 20 and 30 min, rate constant, and error) to the overall array response. The peptides mainly responded toward cell lines treated with EGF (E), anisomycin (A), and anisomycin/JNK-IN-8 inhibitor (A/J), which would present higher ERK activity compared to the cell lines treated with MEK inhibitors. Thus, the vectors were expected to actively respond to the cell lines located on the right quadrants of the LDA score plots. Importantly, the length of the vectors is indicative of the contribution to the overall response of the array. Thus, the SOX-SubD, SOX-MEF2A, and SOX-SubF are sensors that respond primarily to ERK activity. These vectors have the most significant length and contribution to the response of the array. Further, the fact that the A549 loading vectors primarily all point in the same direction and have less scatter along F2 compared to those derived from the MDA-MB-231 line suggests that cross-reactivity among the peptides is lower with the A549 treatments.
Furthermore, the data suggest that the array is primarily influenced by the activity of ERK, with the other MAPKs contributing to a lesser extent to the overall signal. Thus, the corresponding SOX-NFAT4 vectors did not significantly contribute to the array response. As a result, the input data derived from SOX-SubD, SOX-MEF2A, and SOX-SubF was used to obtain new LDA score plots without SOX-NFAT4 (Figure S3a,c), along with the corresponding loading plots (Figure S3b,d), without significant deterioration of the kinase classification. Overall, we replicated the previously described differentiation and distribution of the treated lysates using the four sensors with the three sensors along the F1 and F2 axes. Thus, SOX-SubD, SOX-MEF2A, and SOX-SubF peptide sensors were the sensors used in the chemometric analysis for the following experiments.
Assessing the Array at Lower MAPK Concentrations.
An additional experiment was performed to test the sensitivity of the array toward a lower amount of the lysates. In this experiment, we used only 2 μg/well of the lysates with the same concentrations of the SOX-peptides (5 μM) and ATP. This approach allowed for the assessment of whether the same pattern of activation/inhibition by each treatment could be reproduced in both cell lines using a minimal amount of lysates. The LDA score plots (Figure S4) showed the same general pattern according to the positions of each treatment. Thereby, the array was found to be reproducible and independent of the amount of lysates used in the assay.
Construction of a Calibration Regression Model for Cancer Cell Lines.
Our goal for this study was to accurately quantify, for the first time, the activated kinase content in both cancer cell lysates and tumor extracts. It was essential to build calibration regression models to achieve this goal. Thus, we explored the possibility of using the SOX-peptide array to develop a quantitative regression method, specifically with a focus on quantifying the concentration of active ERK in the cell lines tested. A parallel Western blot analysis served as a means to corroborate the amount of phosphorylated ERK present in each sample (Figure S1). As the array demonstrated a strong bias toward ERK activity, a regression model was derived using the M-U and A-U cell lysates to calibrate ERK activity in each cell line. Each lysate was enriched using known concentrations of recombinant, activated ERK1 (0–6.4 nM). The fluorescence and rate responses of each sample were estimated using the sensor array and 7 μg/well from each lysate. The resulting fingerprints showed a dependence of the fluorescence and rate responses on the concentrations of ERK in both cell lines (Figure S5). The corresponding LDA score plot for both cell lines revealed a curved dependence of the concentration variance along the F1 and F2 axes (Figure 3a,b).
Figure 3.
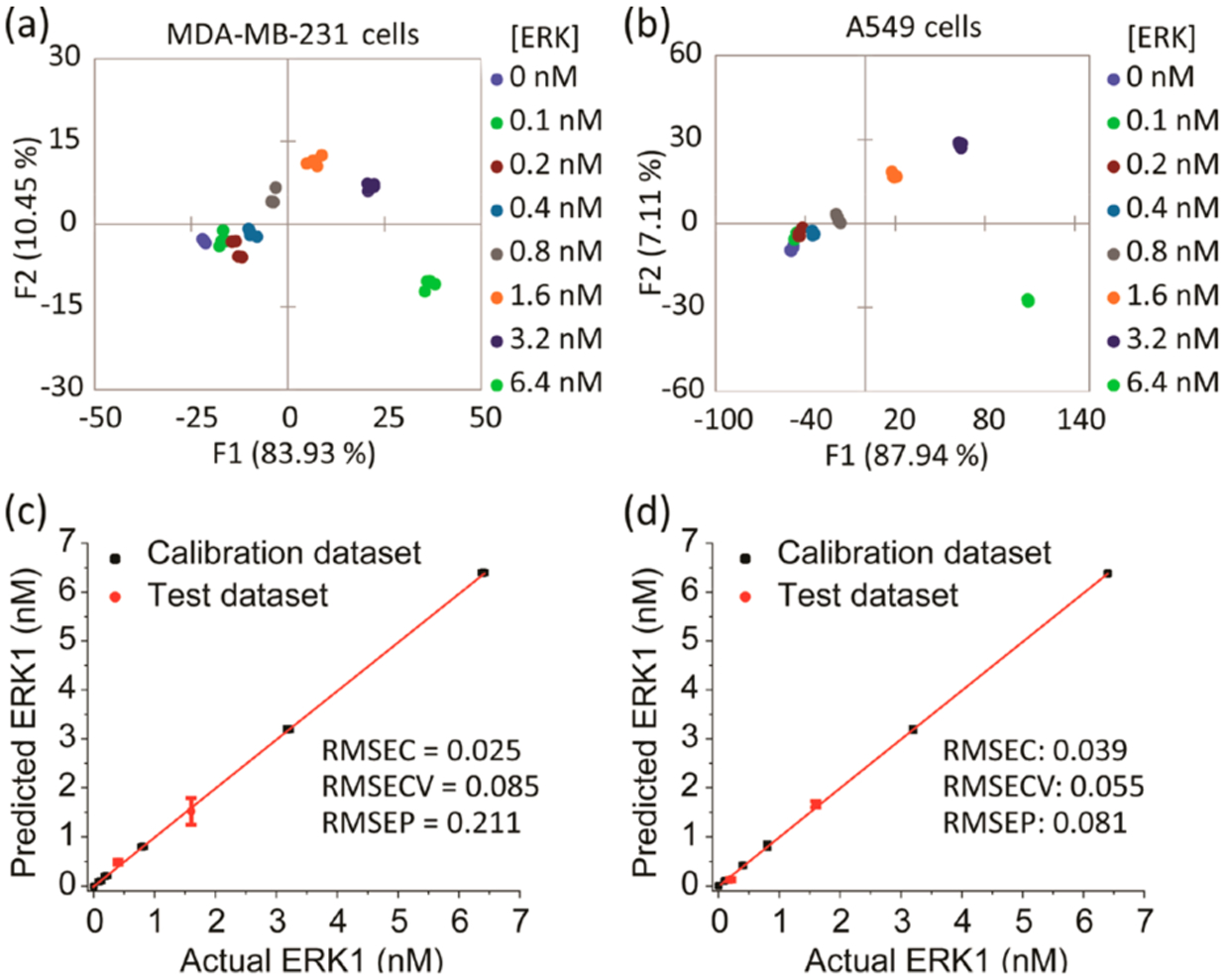
Results of the quantitative analysis of cell lysates enriched by different amount of recombinant, fully phosphorylated ERK1. (a, b) LDA score plots showing the response of the biosensors to cell lysates enriched by increasing amounts of recombinant ERK1. MDA-MB-231 (a) or A549 (b) cells were serum starved for 24 h then treated by U0126 MEK inhibitor for 2 h before lysis. (c, d) Results of the SVM regression method of MDA-MB-231 (c) and A549 (d) cell lysates enriched by different concentrations of recombinant ERK1. The unknown samples (red circle dots) represent correct predictions.
In differential sensing protocols, support vector machine (SVM) is often used to analyze multivariate data with nonlinear behavior.34 SVM algorithms can be applied to both classification and regression. For regression purposes, it can compute a linear regression function in a high dimensional feature space where the input data are mapped via a nonlinear function. Hence, we used SVM regression to build calibration models, which are validated within the analysis to predict the properties (concentration of active ERK in our study) of unknown samples. Division of the numerical data into two sets allowed for the development of the calibration model and validation. For each cell line, two concentrations out of the eight recombinant ERK1 concentrations (25% of the data set) were used to test the model, and the calibration model was used to predict and calculate the corresponding concentrations of these two test lysates. The array yielded an accurate quantitative regression analysis of active ERK in the test lysates of MDA-MB-231 and A549 cells, where the test concentrations were correctly predicted (red dots) as shown in Figure 3c,d, respectively. The validation of the model produces a root-mean-square error of prediction (RMSEP) value, which gives a measure of the predictive accuracy of the model when presented with test data.
Quantifying Unknown ERK in Cells with Differing Kinase Expression.
Given the SVM results introduced above, we anticipated that various treatments in the MDA-MB-231 and A549 cells would result in distinct MAPK activity profiles, and thus each treatment group would display its own unique MAPK activity signature, allowing us to perform quantitation of ERK activity. Unlike in the current study, sensors were previously employed to evaluate relative changes in the activities of the targeted kinase in different cell lysates when exposed to different stress situations, without giving an absolute quantification of the active kinase in these lysates.20 Thus, using SVM algorithms guided by the resulting calibration models, we challenged our array to predict definite concentrations of active ERK in MDA-MB-231 and A549 cell lysates that had been subject to the six treatments described earlier (Figure S3a,c) and that produced six groups of cell lysates expressing variable levels of active ERK. Importantly, the active ERK measured in MDA-MB-231 and A549 cells corresponds to total ERK1 and ERK2.
We estimated the amount of active ERK with antibodies that specifically recognize the doubly phosphorylated form of ERK in each lysate by determining the intensity of the corresponding immuno-active band obtained by Western blot. Comparison of these results occurred through a calibration curve built using different known amounts of recombinant activated ERK1 run in the same gel (Figure S1). Prediction by our models allowed a comparison of the amount of active ERK (ng μg−1 lysate) in each sample. The values obtained by our model used the average of four experimental replicates, whereas the Western blot values were the result of two experimental replicates (Table 1 and Figure 4). Initially, assessment of lysates derived from MDA-MB-231 cells using the array (Figure 4a) suggested that cells induced with EGF (M-E) or anisomycin (M-A) or treated with JNK-IN-8 then induced with anisomycin (M-A/J) exhibited strong ERK activation when compared to the basal level in the nontreated cells (M-n).
Table 1.
Comparison of Predicted Active ERK Contents Obtained Using the Sensor Array with the Calibration Regression Model, Western Blot, and Immunoprecipitation–Kinase Assay Protocolsa
| pp-ERK (ng μg−1 Lysate) | ||||||||
|---|---|---|---|---|---|---|---|---|
| sensorsb | Western blot | kinase assay | ||||||
| cells | treatment | mean (n = 4) | ±SD | mean (n = 2) | ±SD | p-value (two-tailed)c p ≤ 0.05 | value (n = 1) | ±SEd |
| MDA-MB-231 | M-n | 0.374 | 0.029 | 0.450 | 0.125 | 0.264 | 0.352 | 0.107 |
| M-U | 0.027 | 0.005 | 0.035 | 0.011 | 0.248 | 0.034 | 0.017 | |
| M-E | 0.898 | 0.031 | 0.656 | 0.071 | 0.003 | 0.643 | 0.103 | |
| M-E/U | 0.007 | 0.016 | 0.062 | 0.037 | 0.051 | 0.010 | 0.017 | |
| M-J | 0.067 | 0.013 | 0.110 | 0.055 | 0.168 | 0.073 | 0.012 | |
| M-A | 0.351 | 0.032 | 0.407 | 0.008 | 0.082 | 0.383 | 0.086 | |
| M-A/J | 0.412 | 0.014 | 0.402 | 0.005 | 0.422 | 0.421 | 0.057 | |
| A549 | A-n | 0.175 | 0.002 | 0.243 | 0.082 | 0.128 | 0.161 | 0.016 |
| A-U | 0.155 | 0.004 | 0.202 | 0.041 | 0.059 | 0.141 | 0.051 | |
| A-E | 0.508 | 0.011 | 0.504 | 0.102 | 0.933 | 0.480 | 0.135 | |
| A-E/U | 0.211 | 0.009 | 0.302 | 0.086 | 0.074 | 0.236 | 0.225 | |
| A-J | 0.149 | 0.005 | 0.053 | 0.094 | 0.079 | 0.162 | 0.113 | |
| A-A | 0.495 | 0.011 | 0.244 | 0.087 | 0.003 | 0.504 | 0.431 | |
| A-A/J | 0.32 | 0.005 | 0.213 | 0.106 | 0.081 | 0.338 | 0.059 | |
Recombinant ERK1 calibration curves were employed to generate the data in this table. Non-treated cells in serum-free media (n), inhibition of ERK phosphorylation with U0126 (U), induction of ERK phosphorylation with EGF (E), cells treated with U0126 then induced with EGF (E/U), inhibition of JNK pathway with JNK-IN-8 (J), induction of JNK phosphorylation with anisomycin (A), and cells treated with JNK-IN-8 then induced with anisomycin (A/J).
Cell lysates were used at a concentration of 7 μg/well.
Unpaired t-test and p-value calculations for the sensors and the Western blot means were done using GraphPad Prism 7 software.
Standard error of regression slope.
Figure 4.
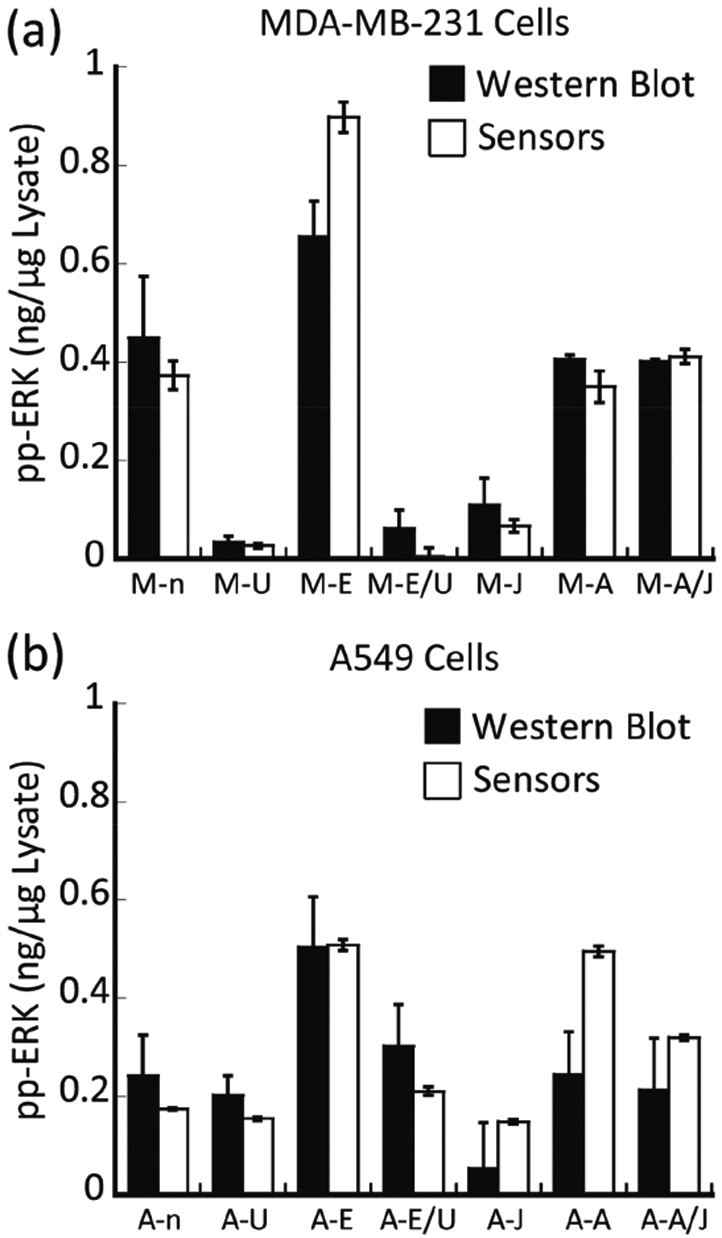
Comparing the estimated amount of active ERK in the tested lysates using two different protocols. The amount of active ERK in different MDA-MB-231 (a) and A549 (b) cell lysates was calculated using both the SVM calibration models (the sensors) and the Western blot. Serum-starved control (n), cells treated with MEK inhibitor U0126 (U), cells induced with EGF (E), cells treated with U0126 then induced with EGF (E/U), cells treated with JNK inhibitor JNK-IN-8 (J), cells induced with anisomycin (A), and cells treated with JNK-IN-8 then induced with anisomycin (A/J).
In contrast, cells treated with U0126 (either accompanied by EGF induction or not) (M-U and M-E/U) and cells treated with JNK-IN-8 (M-J) without anisomycin induction showed a consistently low concentration of active ERK. Studies in A549 lysates furnished similar results (Figure 4b). These results demonstrate the ability of the array to predict the relative activity of ERK in different cell lysates. Significantly, and as a unique property of our approach, the amount of active ERK (ng μg−1 lysate) determined by the array compared well to the Western blot analysis (Table 1 and Figure 4). The calculated p-values (Table 1) showed no significant difference between the values estimated using the two methods, except for MDA-MB-231 cells that were induced by EGF and A549 cells that were induced by anisomycin. For these two experiments, the array measured slightly higher activity of ERK when compared to the phosphorylated ERK levels that were estimated by Western blot. As a final comparison, ERK was immunoprecipitated from cell lysates using an antibody that recognizes both ERK1 and ERK2, and its kinase activity toward a specific protein substrate, Ets-1, was determined directly using a radioactive-based immune-complex protein kinase assay (supporting methods section). The agreement between the quantification using the array and the immune kinase assay was excellent, providing further validation of the results (Table 1).
Quantifying ERK Inhibition in MDA-MB-231 Cells.
We next explored whether the array could be used to monitor the extent of inhibition of ERK activity in cancer cells. The goal was to assess whether active ERK could be quantified accurately in cells treated with therapeutic levels of an inhibitor targeting the ERK pathway. First, we evaluated the ability of the array to fingerprint the inhibition of ERK in MDA-MB-231 cell lysates. The resulting LDA score plot (Figure 5a) revealed a curved progression of the data along the F1 and F2 axes. A second replicate was performed to assess the reproducibility of the inhibition assay. The second LDA plot (Figure 5b) revealed the same trend, indicating a dependence of the response on increasing inhibitor concentrations. Both experiments demonstrated high classification accuracy (100% cross-validation, Table S4 and S5, respectively), and also, the inhibition assay was found to be reproducible. As with the treatment studies, we used our calibration model to estimate concentrations of active ERK in the MDA-MB-231 cell lysates treated with different doses of U0126 (0–15.65 μM) for 2 h followed by ERK pathway stimulation using 100 nM EGF for 30 more minutes (Figure S6). Once again, Western blot was used as a comparison to provide a value of double-phosphorylated ERK concentrations in the same lysates (Figure S7). The amount of suppressed ERK activity (ng μg−1 lysate) against increasing concentrations of U0126 was plotted to compare the results of both methods (Figure 5c). Our array was able to provide a similar measurement of the concentration of active ERK in the lysate at each inhibitor concentration when compared to the values of the band intensities from the Western blot analysis (Table S6). The value of the IC50 determined from the array (0.33 ± 0.09 μM) was found to be quite comparable to that obtained by the Western blot analysis (0.31 ± 0.043 μM). In general, the values from the two methods overlapped well and displayed a linear correlation when plotted as active ERK (ng μg−1 lysate) determined by the array versus Western blot (Figure 5d). In both methods, systematic errors, as well as the intrinsic variability of the Western blot as a multistep process, may contribute to minor deviations. Immune-complex kinase assays, which were also in agreement, provided further validation of the results (Table S6). Taken together, this experiment revealed the power of our array to monitor inhibition of ERK in MDA-MB-231 lysates, even at high inhibitor concentrations in the presence of other kinases.
Figure 5.
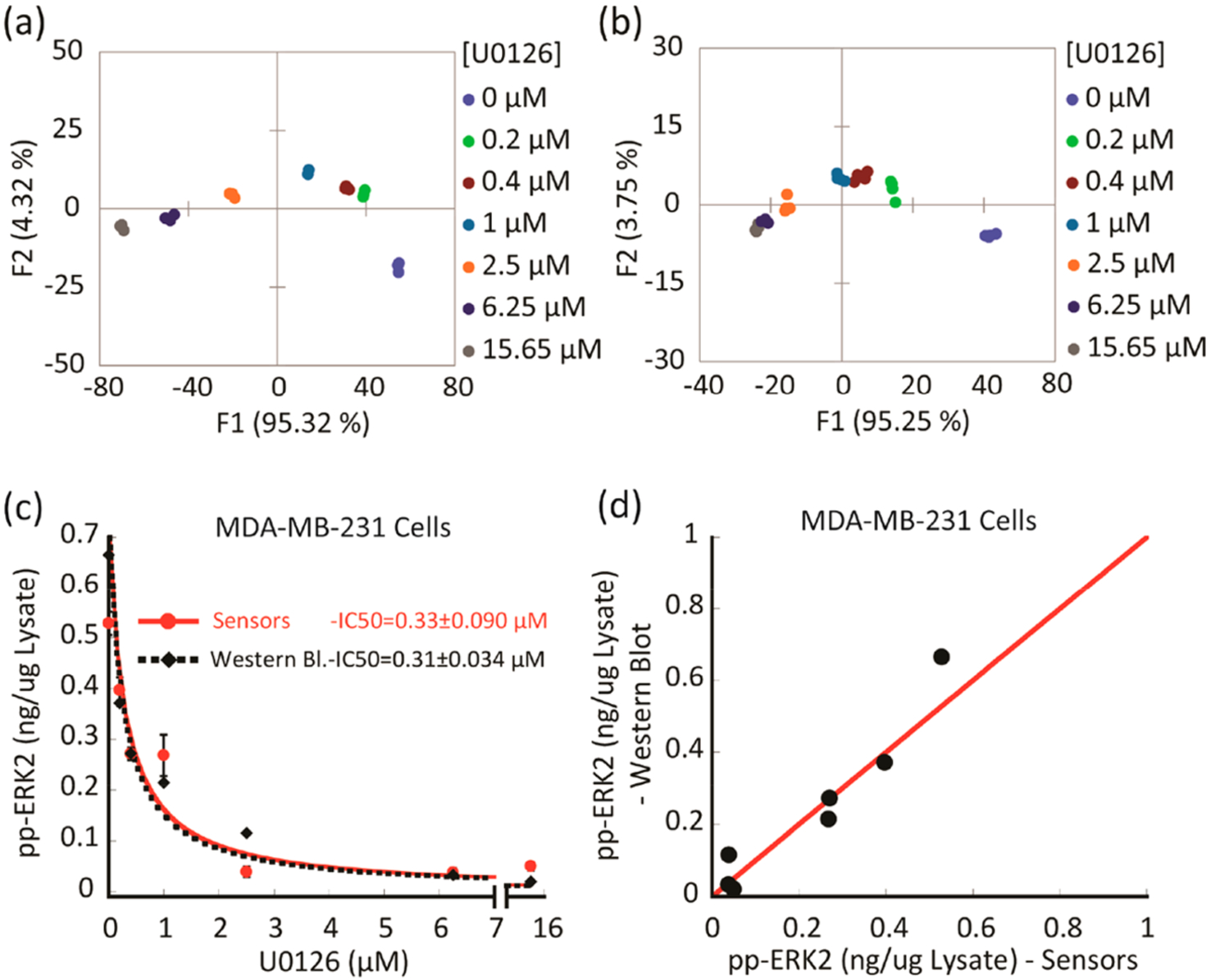
Following ERK phosphorylation suppression in MDA-MB-231 cells treated with different doses of MEK inhibitor (U0126).(a, b) LDA score plots showing two experimental replicates of the response from the sensing array showing differentiation of lysates of MDA-MB-231 cells that were treated with increasing concentrations of MEK inhibitor (U0126) before induction with EGF and lysis. (c, d) Comparison between the amount of phosphorylated ERK, which was estimated using either the array sensor protocol or Western blot in the MDA-MB-231 cell lysates. (c) Plot of suppressed ERK phosphorylation versus increasing concentration of MEK inhibitor (U0126) determined by the array sensors (red dots) and Western blot analysis (black diamonds). Data were fitted to equation 1 (Table S6 footnote). (d) Scatter plot of suppressed ERK activity, showing a correlation of the values obtained with both methods. The red line corresponds to a linear correlation of 1.0.
Quantifying Unknown ERK Activity in Tumor Samples.
Finally we explored the utility of the array in examining more complex biological systems by quantifying unknown ERK activity in tumor samples derived from nude mice bearing melanoma A375 xenografts. In this experiment, we suppressed ERK activity in tumor-bearing mice using BI-78D3, a covalent inhibitor of ERK.35 In addition to covalently binding ERK and directly inhibiting its ability to phosphorylate substrates, BI-78D3 also induces the dephosphorylation of ERK in cells. As a correlation between the activity of ERK in the extracts and its phosphorylation cannot be assumed following BI-78D3 treatment, we also utilized an immune complex kinases assay to measure the activity of ERK.
Thus, 20 nude athymic mice received one million A375 melanoma cells injected sc into the right flank. Once the tumors became palpable, the mice were assigned randomly into two groups; one group was injected intraperitoneally with 15 (mg/kg)/day of BI-78D3, and the other group was injected with vehicle control. Following euthanasia after 10 days of a daily treatment, tumors were extracted from five mice of each group, lysed, and pooled into one lysate sample. To quantify the amount of phosphorylated ERK present in each tumor sample, we used recombinant activated ERK1 and ERK2 for calibration purposes (Western blots, Figure S8). The SOX-peptide assays were conducted in 96-well plates (four replicates) with 7 μg aliquots of tumor extract per well. In these experiments, the sensors displayed a lower signal intensity compared to experiments using the cell lysates (Figure S9a,b). Accordingly, delta fluorescence (ΔF) values at 20, 50, and 90 min, along with the calculated rate constants and errors, were used as the multivariate data (3 peptides X 4 replicates X 5 inputs per each treatment) for chemometric analysis. A new calibration regression model was constructed using the tumor sample extracts from the treated mice as the background (Figure S9). Similar to the previous lysate calibration experiment, the tumor extract was enriched by different known amounts of recombinant, activated ERK1 (0–6.4 nM) to generate a calibration. The fingerprints of these samples revealed a dependence of the fluorescence and rate responses on the concentrations of active ERK in these enriched tumor samples (Figure S9a,b). As before, the corresponding LDA score plot also presented a curved-shape dependence of the concentration variance along the F1 and F2 axes (Figure S9c). Thus, an SVM algorithm was used to build and validate the calibration regression model. Two concentrations out of the six recombinant ERK1 concentrations (33% of the data set) were used to test the model. The array produced an accurate quantitative regression analysis of active ERK in the two test samples, predicting the correct concentration in those samples, as shown in Figure 6a (circled red dots).
Figure 6.
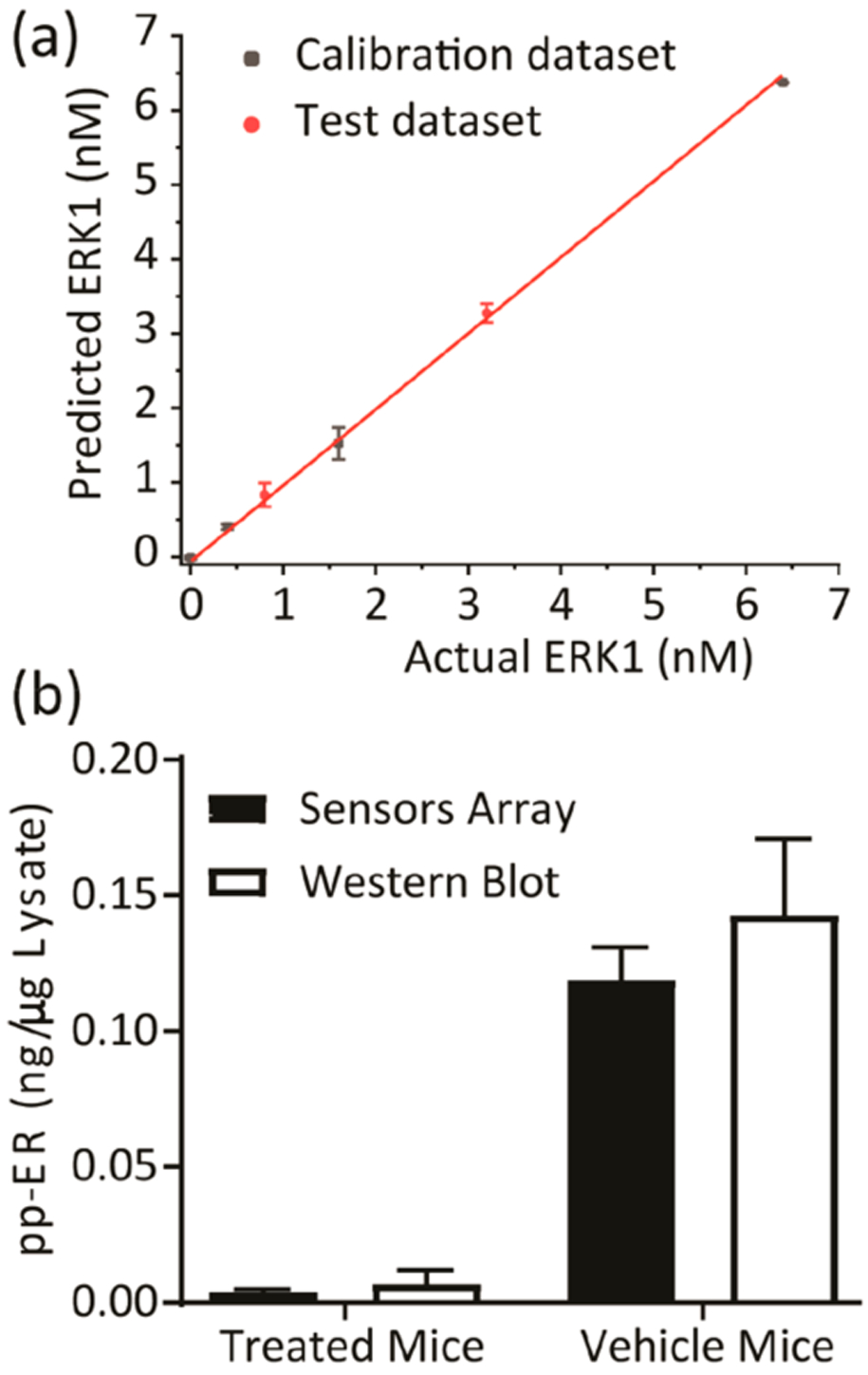
Quantification of phosphorylated ERK in tumor samples. (a) Results of the SVM regression method for treated tumor samples enriched by increasing concentration of recombinant, fully phosphorylated ERK1. (b) The content of active ERK in tumor samples was estimated using the SVM calibration model and western Blot. Tumor samples extracted from two groups of nude mice (5 mice per group) bearing the melanoma A375 xenografts, either treated by 15 mg kg−1 ERK covalent inhibitor (BI-78D3) or by control vehicle (supporting methods section). The pooling of lysates from each group provided one sample (treated or vehicle mice samples).
The calibration model was then used to predict and calculate the actual concentration of active ERK in each tumor extract. The values predicted by our model (average of four experimental replicates) were comparable to those calculated using both the Western blot protocol (average of two experimental replicates) (Figure 6b and Table S7) and the immune kinase assay (Table S7). Assessment of the tumor extracts using the array (Figure 6b) suggested that the mice that were treated by the vehicle exhibited elevated ERK activity in the extracted tumor when compared to the lower level quantified in tumors extracted from the BI-78D3 treated mice.
Finally, we tested the possibility of building a general regression model with the data from both cell and tumor samples. Regarding the different features that we applied to the chemometric analysis, here we selected a new combination of five features (ΔF at 20, 30, and 90 min, rate constant, and error) for three peptides (SOX-Sub-D, SOX-MEF2A, and SOX-Sub-F). With both the data from cell lysates (U0126 treated) and tumor samples (BI-78D3 treated) that were enriched by 0–6.4 nM recombinant active ERK1, the LDA model shows 92.86% cross-validation in total (Table S27). To test the general applicability, we generated the SVM regression model using the LDA score factors (F1–F3) as the input data set (Table S28). At first, tumor samples were selected as calibration data and cell samples as test data. The regression model built on tumor samples produced an accurate ERK quantification for cell samples, showing both low root-mean-square error of calibration and prediction (RMSEC 0.1903, RMSEP 0.3138). Then we used the cell samples as calibration data and tumor samples as test data; the general model also works in reverse (RMSEC 0.2385, RMSEP 0.4324). Detailed model performance and predictions are shown in Tables S29–32. These results demonstrate that our general model from tumor samples could predict ERK activity in cell samples and vice versa.
DISCUSSION
In this study, we sought to demonstrate the utility of using a SOX peptide-based biosensor array in conjunction with chemometric modeling to directly profile and quantify varied levels of active ERK in cancer cells. First, we applied a differential sensing protocol to MDA-MB-231 triple-negative breast cancer (TNBC) and A549 non-small-cell lung cancer (NSCLC) cell lines. This protocol utilized chemometric (machine learning) modeling methods to enable the interpretation of complex multivariate data obtained by high-throughput analysis (HTA)38 to facilitate the profiling of ERK pathway activity in cancer cells. Second, we developed a multivariate regression model to quantify unknown levels of active ERK in these cells accurately. The semiselective biosensors exploit the cross-reactivity associated with other kinases in the cells, allowing the extraction of signals that are only relevant to ERK activity. We envisage this approach being useful in directly monitoring ERK dynamics and its suppression by clinical inhibitors of upstream components of the pathway.
The biosensors Sub-D (1a), MEF2A (1b), NFAT4 (1c), and Sub-F (1d) were used to construct the cross-reactive array (Supplementary Scheme 1). The array is particularly sensitive to ERK activity changes over other MAPKs, due to the differential responsiveness of each of the biosensors. Thus, using this array our goals were to fingerprint ERK activity and profile its activation and inhibition in different cancer cell lines. We developed a calibration model to identify the changes in ERK activity as well as to quantify the active ERK content in the two tested cell lines (MDA-MB-231 and A549) exposed to various treatments. To achieve these goals, we sought to ensure the array exhibited the following characteristics: (1) the array must exhibit high sensitivity to changes in ERK activity levels; (2) the array must be compatible with the complex nature of cell extracts, (3) the method should be applicable to HTA; (4) the biosensor library must exhibit substantial fluorescence changes upon phosphorylation and have robust signals; (5) chemometric analysis must be applied to the deconvolution of the fingerprints with high validation and reproducibility.
We found that the array was suitable for quantifying unknown levels of ERK activity in cell lysates when employed with a multivariate regression model. The predicted values provided by our model were found to be comparable to those obtained by traditional Western blot and immune-complex protein kinase assay protocols (Tables 1, S6, and S7). Additionally, the differential sensing approach, along with chemometric analysis, provided evidence for the notion that a SOX-peptide array can be used to detect and quantify ERK activity in tissue samples. This conclusion is supported by an agreement between Western blot and immune-complex protein kinase analyses and the array analysis for tumor extracts isolated from mice treated with or without BI-78D3, a covalent inhibitor of ERK that impedes both ERK dephosphorylation and activity.35
Western blot and other immune complex based assays (e.g., ELISA or AlphaLISA) require highly specific antibodies to detect either the active or inactive form of ERK. These can involve time-consuming steps that are not amenable to HTA (See Table 2 for a comparison between our differential sensing assay and Western blot). Our approach circumvents the high specificity requirement and takes advantage of the cross-reactive interactions within the cellular environment to display representative signals associated with ERK activity. Further, our approach does not require the addition of inhibitors to suppress the activities of major off-target kinases,19 and thereby baseline corrections for other kinases are not necessary to remove nonspecific kinase activity.
Table 2.
Comparison of Traditional Western Blot and Differential Sensing Array
| time (h) | differential sensing array | time (h) | Western blot analysis36,37 |
|---|---|---|---|
| 0.5 | Solutions preparation: (l) 2× assay buffer (50 mM HEPES buffer (pH = 7.4), 100 mM KC1, 0.2 mM EDTA, 0.2 mM EGTA, 4 mM DTT and 20 mM MgCl2); (2) cell lysate; (3) 2× substrate solution (10 μM of each sensor and 1 mM ATP) | 2.5 | (l) mix 60—70 μg of each lysate or different amounts of recombinant activated target kinase with 5× SDS-PAGE loading dye and 1× lysis buffer, boil for 5 min then load into the wells of the gel; (2) gel electrophoresis at 120 V for 1—1.5 h |
| 1 | plate preparation: (l) training set preparation, addition of increasing concentrations of recombinant activated target kinase and the cell lysate (7 μg/well) in the assay buffer; (2) test set preparation, cell lysates (7 μg/well) diluted in the assay buffer and loaded into the same plate to quantify the target kinase | 2.5–15.5 | transfer at 90 V for 2 h or at 30 V for overnight at 4 °C then wash 3 × 5 min |
| 1.5 | readout: (l) plate reader injector is used to add the substrates solution into the wells that contain the lysates diluted in the assay buffer. (2) recording of changes in the emission (ex.360 nm; em.485 nm) over time | 2.5–16.5 | membrane block for 1 h at room temp then incubation with primary Ab for 1 h at room temp or overnight at 4 °C; wash 5 × 5 min |
| 0.5 | data analysis: (l) curve fitting (calculation of rate constants); (2) chemometrics (LDA); (3) developing a calibration model or training set; (4) prediction of unknown kinase concentrations in the test set | 1.5 | incubation with secondary Abs for 1 h at room temp, wash 5 × 5 min |
| 1 | ECL reagent treatment and film development or Licor System analysis | ||
| 1 | data analysis | ||
| ~3.5 | total | 11–38 | total |
Significantly, our approach allows for the quantification of the actual amount of active ERK, which is different from measuring phosphorylated ERK using Western blot, which is a proxy for activity. This is easily appreciated if one considers a situation where a covalent inhibitor could bind to the active site of ERK to inhibit it and influence feedback mechanisms that affect its phosphorylation status. Previous to our array assay, the only general approach to quantify actual enzymatic activity was by isolating a specific kinase by immunoprecipitation, followed by a kinase assay. Thus, our differential sensing approach has the power to provide a significant alternative to detect and quantify the enzymatic activity of ERK in cell extracts in real-time.
If performed manually, the array assay, including sample preparation, readouts, and chemometric analysis, can be applied to a new sample in only a few hours (3.5 h) compared to Western blot, which can vary between 11 and 38 h36,37 (Table 2). We have demonstrated that this time can be even shorter if all solutions are distributed in the plate using an Echo 550 Acoustic liquid handler (San Jose, CA), making the technique a HTS-compatible approach.
Conclusions.
In summary, this study showed that a library of cross-reactive SOX-peptide-based biosensors along with chemometric analysis could be used to profile ERK activity, even in the presence of other MAPKs, in cancer cell lines and tumor samples presenting different levels of ERK activation and inhibition. The approach can be used to measure multiple samples quickly, allowing for a faster analysis than other methods of the response to actual ERK activity in different treatments in cell line models. This approach might also provide an alternative to assessing ERK activity in human tumor biopsies, which would be valuable for evaluating treatments and inhibitor resistance. By utilizing arrays comprised of different substrates, we believe this approach can be expanded to many other protein kinases. Currently, we are developing a larger array that includes more peptides designed to target members of the entire MAP kinase family. We believe that such an array will facilitate the quantification of more than one kinase simultaneously, and by further expanding the array to include other peptides as well as protein substrates, we expect to be able to expand our analysis to other subfamilies of the protein kinase family. Furthermore, with appropriate assays, we believe this general approach will be applicable to the quantification of other enzyme families that modulate biological molecules, such as proteases and protein phosphatases. Thus, we believe this general approach has the potential to be applied to the challenges of modern medicine where the quantification of specific cellular activities can be used to guide treatments.
METHODS
The Supporting Information (methods section) describes detailed procedures for peptide synthesis and characterization, the assay protocols, cell culture and maintenance, cell lysate preparation, animal study, and tumor extraction. The Supporting Information Table S1 and Figures S10–S13 provide the exact sequences and full characterizations of the synthesized peptides. Tables S8–S32 in the Supporting Information show detailed data of the SVM analysis.
Supplementary Material
Funding
The following support is acknowledged: GM123252 (KND), CPRIT RP140648 (KND), CPRIT RP180880 (KND), Welch F-1390 (KND), the Freshman Research Initiative (FRi) at UT Austin (DZO), and the Welch Regents Chair F-0046 (EVA).
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acschembio.9b00580.
Methods, sequences and characterization of the SOX-peptide substrates, Western blots of the cell lysates and tumor samples used in this study, the fluorescence and rate constant fingerprints of MDA-MB-231, A549 cell lysates and tumor samples generated by the array, LDA scores plots and/or loading plots for the lysates at high and low protein concentrations and for the tumor samples enriched with different concentrations of active recombinant ERK1, jack-knife analysis of different treatments for each tested cell line, predicted ERK concentrations in cell lysates with inhibitor treatment, and in the tumor samples, the detailed data of the SVM analysis and the general model performance and predictions (PDF)
The authors declare no competing financial interest.
REFERENCES
- (1).Zhang C, Habets G, and Bollag G (2011) Interrogating the kinome. Nat. Biotechnol 29, 981–983. [DOI] [PubMed] [Google Scholar]
- (2).Chaikuad A, M C Tacconi E, Zimmer J, Liang Y, Gray NS, Tarsounas M, and Knapp S (2014) A unique inhibitor binding site in ERK1/2 is associated with slow binding kinetics. Nat. Chem. Biol 10, 853–860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Zhang J, Yang PL, and Gray NS (2009) Targeting cancer with small molecule kinase inhibitors. Nat. Rev. Cancer 9, 28–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Toulany M, Minjgee M, Saki M, Holler M, Meier F, Eicheler W, and Rodemann HP (2014) ERK2-dependent reactivation of Akt mediates the limited response of tumor cells with constitutive K-RAS activity to PI3K inhibition. Cancer Biol. Ther 15, 317–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Roskoski R Jr (2012) ERK1/2 MAP kinases: Structure, function, and regulation. Pharmacol. Res 66, 105–143. [DOI] [PubMed] [Google Scholar]
- (6).Holderfield M, Deuker MM, McCormick F, and McMahon M (2014) Targeting RAF kinases for cancer therapy: BRAF-mutated melanoma and beyond. Nat. Rev. Cancer 14, 455–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Stephen AG, Esposito D, Bagni RK, and McCormick F (2014) Dragging ras back in the ring. Cancer Cell 25, 272–281. [DOI] [PubMed] [Google Scholar]
- (8).Goetz EM, Ghandi M, Treacy DJ, Wagle N, and Garraway LA (2014) ERK mutations confer resistance to mitogen-activated protein kinase pathway inhibitors. Cancer Res. 74, 7079–7089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Cancer Genome Atlas Network (2015) Genomic Classification of Cutaneous Melanoma. Cell 161, 1681–1696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Smalley I, and Smalley KSM (2018) ERK Inhibition: A New Front in the War against MAPK Pathway-Driven Cancers? Cancer Discovery 8, 140–142. [DOI] [PubMed] [Google Scholar]
- (11).Gibney GT, and Atkins MB (2015) Immunotherapy or molecularly targeted therapy: what is the best initial treatment for stage IV BRAF-mutant melanoma? Clin Adv. Hematol Oncol 13, 451–458. [PubMed] [Google Scholar]
- (12).Long GV, Stroyakovskiy D, Gogas H, Levchenko E, de Braud F, Larkin J, Garbe C, Jouary T, Hauschild A, Grob JJ, Chiarion Sileni V, Lebbe C, Mandala M, Millward M, Arance A, Bondarenko I, Haanen JB, Hansson J, Utikal J, Ferraresi V, Kovalenko N, Mohr P, Probachai V, Schadendorf D, Nathan P, Robert C, Ribas A, DeMarini DJ, Irani JG, Casey M, Ouellet D, Martin AM, Le N, Patel K, and Flaherty K (2014) Combined BRAF and MEK inhibition versus BRAF inhibition alone in melanoma. N. Engl. J. Med 371, 1877–1888. [DOI] [PubMed] [Google Scholar]
- (13).Konieczkowski DJ, Johannessen CM, Abudayyeh O, Kim JW, Cooper ZA, Piris A, Frederick DT, Barzily-Rokni M, Straussman R, Haq R, Fisher DE, Mesirov JP, Hahn WC, Flaherty KT, Wargo JA, Tamayo P, and Garraway LA (2014) A melanoma cell state distinction influences sensitivity to MAPK pathway inhibitors. Cancer Discovery 4, 816–827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Sammons RM, Ghose R, Tsai KY, and Dalby KN (2019) Targeting ERK beyond the boundaries of the kinase active site in melanoma. Mol. Carcinog 58, 1551–1570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Warthaka M, Adelmann CH, Kaoud TS, Edupuganti R, Yan C, Johnson WH Jr., Ferguson S, Tavares CD, Pence LJ, Anslyn EV, Ren P, Tsai KY, and Dalby KN (2015) Quantification of a Pharmacodynamic ERK End Point in Melanoma Cell Lysates: Toward Personalized Precision Medicine. ACS Med. Chem. Lett 6, 47–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Garai A, Zeke A, Gogl G, Toro I, Fordos F, Blankenburg H, Barkai T, Varga J, Alexa A, Emig D, Albrecht M, and Remenyi A (2012) Specificity of linear motifs that bind to a common mitogen-activated protein kinase docking groove. Sci. Signaling 5, No. ra74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Martić S, and Kraatz H-B (2013) Chemical biology toolkit for exploring protein kinase catalyzed phosphorylation reactions. Chem. Sci 4, 42–59. [Google Scholar]
- (18).Stains CI, Lukovic E, and Imperiali B (2011) A p38alpha-selective chemosensor for use in unfractionated cell lysates. ACS Chem. Biol 6, 101–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Stains CI, Tedford NC, Walkup TC, Lukovic E, Goguen BN, Griffith LG, Lauffenburger DA, and Imperiali B (2012) Interrogating signaling nodes involved in cellular transformations using kinase activity probes. Chem. Biol 19, 210–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Peterson LB, Yaffe MB, and Imperiali B (2014) Selective mitogen activated protein kinase activity sensors through the application of directionally programmable D domain motifs. Biochemistry 53, 5771–5778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Umali AP, and Anslyn EV (2010) A general approach to differential sensing using synthetic molecular receptors. Curr. Opin. Chem. Biol 14, 685–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Zamora-Olivares D, Kaoud TS, Dalby KN, and Anslyn EV (2013) In-situ generation of differential sensors that fingerprint kinases and the cellular response to their expression. J. Am. Chem. Soc 135, 14814–14820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Diehl KL, Ivy MA, Rabidoux S, Petry SM, Muller G, and Anslyn EV (2015) Differential sensing for the regio- and stereoselective identification and quantitation of glycerides. Proc. Natl. Acad. Sci. U. S. A 112, E3977–3986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Motiei L, Pode Z, Koganitsky A, and Margulies D (2014) Targeted Protein Surface Sensors as a Tool for Analyzing Small Populations of Proteins in Biological Mixtures. Angew. Chem 126, 9443–9447. [DOI] [PubMed] [Google Scholar]
- (25).Xu Q, Zhang Y, and Zhang CY (2015) A triple-color fluorescent probe for multiple nuclease assays. Chem. Commun. (Cambridge, U. K.) 51, 9121–9124. [DOI] [PubMed] [Google Scholar]
- (26).Tomita S, Sakao M, Kurita R, Niwa O, and Yoshimoto K (2015) A polyion complex sensor array for markerless and noninvasive identification of differentiated mesenchymal stem cells from human adipose tissue. Chem. Sci 6, 5831–5836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Bajaj A, Rana S, Miranda OR, Yawe JC, Jerry DJ, Bunz UHF, and Rotello VM (2010) Cell surface-based differentiation of cell types and cancer states using a gold nanoparticle-GFP based sensing array. Chemical Science 1, 134–138. [Google Scholar]
- (28).Bajaj A, Miranda OR, Kim IB, Phillips RL, Jerry DJ, Bunz UH, and Rotello VM (2009) Detection and differentiation of normal, cancerous, and metastatic cells using nanoparticle-polymer sensor arrays. Proc. Natl. Acad. Sci. U. S. A 106, 10912–10916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Zamora-Olivares D, Kaoud TS, Jose J, Ellington A, Dalby KN, and Anslyn EV (2014) Differential sensing of MAP kinases using SOX-peptides. Angew. Chem., Int. Ed 53, 14064–14068. [DOI] [PubMed] [Google Scholar]
- (30).zu Schwabedissen HEM, Grube M, Dreisbach A, Jedlitschky G, Meissner K, Linnemann K, Fusch C, Ritter CA, Volker U, and Kroemer HK (2006) Epidermal growth factor-mediated activation of the map kinase cascade results in altered expression and function of ABCG2 (BCRP). Drug Metab. Dispos 34, 524–533. [DOI] [PubMed] [Google Scholar]
- (31).Rosser EM, Morton S, Ashton KS, Cohen P, and Hulme AN (2004) Synthetic anisomycin analogues activating the JNK/SAPK1 and p38/SAPK2 pathways. Org. Biomol Chem 2, 142–149. [DOI] [PubMed] [Google Scholar]
- (32).Duncia JV, Santella JB 3rd, Higley CA, Pitts WJ, Wityak J, Frietze WE, Rankin FW, Sun JH, Earl RA, Tabaka AC, Teleha CA, Blom KF, Favata MF, Manos EJ, Daulerio AJ, Stradley DA, Horiuchi K, Copeland RA, Scherle PA, Trzaskos JM, Magolda RL, Trainor GL, Wexler RR, Hobbs FW, and Olson RE (1998) MEK inhibitors: the chemistry and biological activity of U0126, its analogs, and cyclization products. Bioorg. Med. Chem. Lett 8, 2839–2844. [DOI] [PubMed] [Google Scholar]
- (33).Stewart S, Ivy MA, and Anslyn EV (2014) The use of principal component analysis and discriminant analysis in differential sensing routines. Chem. Soc. Rev 43, 70–84. [DOI] [PubMed] [Google Scholar]
- (34).Minami T, Esipenko NA, Zhang B, Kozelkova ME, Isaacs L, Nishiyabu R, Kubo Y, and Anzenbacher P Jr. (2012) Supramolecular sensor for cancer-associated nitrosamines. J. Am. Chem. Soc 134, 20021–20024. [DOI] [PubMed] [Google Scholar]
- (35).Kaoud TS, Johnson WH, Ebelt ND, Piserchio A, Zamora-Olivares D, Van Ravenstein SX, Pridgen JR, Edupuganti R, Sammons R, Cano M, Warthaka M, Harger M, Tavares CDJ, Park J, Radwan MF, Ren P, Anslyn EV, Tsai KY, Ghose R, and Dalby KN (2019) Modulating multi-functional ERK complexes by covalent targeting of a recruitment site in vivo. Nat. Commun 10, 5232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Eslami A, and Lujan J (2010) Western blotting: sample preparation to detection. J. Visualized Exp, 2359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Mahmood T, and Yang PC (2012) Western blot: technique, theory, and trouble shooting. N Am. J. Med. Sci 4, 429–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Duncan JS, Whittle MC, Nakamura K, Abell AN, Midland AA, Zawistowski JS, Johnson NL, Granger DA, Jordan NV, Darr DB, Usary J, Kuan PF, Smalley DM, Major B, He X, Hoadley KA, Zhou B, Sharpless NE, Perou CM, Kim WY, Gomez SM, Chen X, Jin J, Frye SV, Earp HS, Graves LM, and Johnson GL (2012) Dynamic reprogramming of the kinome in response to targeted MEK inhibition in triple-negative breast cancer. Cell 149, 307–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.