

Abstract
Diabetic nephropathy (DN) is currently the most common complication of diabetes. It is considered to be one of the leading causes of end-stage renal disease (ESRD) and affects many diabetic patients. The pathogenesis of DN is extremely complex and has not yet been clarified; however, in recent years, increasing evidence has shown the important role of innate immunity in DN pathogenesis. Pattern recognition receptors (PRRs) are important components of the innate immune system and have a significant impact on the occurrence and development of DN. In this review, we classify PRRs into secretory, endocytic, and signal transduction PRRs according to the relationship between the PRRs and subcellular compartments. PRRs can recognize related pathogen-associated molecular patterns (PAMPs) and danger-associated molecular patterns (DAMPs), thus triggering a series of inflammatory responses, promoting renal fibrosis, and finally causing renal impairment. In this review, we describe the proposed role of each type of PRRs in the development and progression of DN.
Keywords: Diabetic nephropathy, Innate immunity, Pattern recognition receptor, Pathogenesis
1. Introduction
Diabetic nephropathy (DN) is a serious microvascular complication caused by long-term hyperglycemia and is considered to be a main cause of end-stage renal disease (ESRD) (Yang et al., 2013). ESRD is one of the leading causes of type 1 and type 2 diabetes mortality, with a prevalence of 25%–40% (D'addio et al., 2014; Huang et al., 2014). The main pathological changes in the early stages of DN are high intraglomerular pressure, hyper-perfusion and hyper-filtration, increasing glomerular filtration rate (GFR), widening of the mesangial area, and thickening of the glomerular basement membrane (GBM) (Guo et al., 2018). Late in the progression of the disease, the GFR gradually decreases, accompanied by glomerular sclerosis, tubulointerstitial fibrosis, tubular atrophy, and renal dysfunction, leading eventually to ESRD (Collins et al., 2012; Awad et al., 2015). There are many articles related to the pathogenesis of DN, but the specific mechanism of pathogenesis is yet to be fully elucidated (Lu et al., 2011). Research has revealed that the occurrence of DN is associated with a variety of factors, such as hemodynamic changes, oxidative stress, and the involvement of the renin angiotensin aldosterone system (RAAS), transforming growth factors (TGFs) and genetic factors (McKnight et al., 2015; Sifuentes-Franco et al., 2018; Rao et al., 2019). Chronic inflammation is also thought to be closely related to the development of ESRD. An extremely powerful kidney risk inflammatory signature (KRIS) consisting of 17 novel proteins rich in tumor necrosis factor (TNF) receptor superfamily members can mediate various inflammatory and immunoregulatory responses associated with DN (Niewczas et al., 2019). However, with the role of innate immunity in kidney diseases gradually becoming recognized, many scholars believe that innate immunity is involved in the occurrence and development of DN. As important components of the innate immune system, pattern recognition receptors (PRRs) play an important role in the pathogenesis of DN (Wada and Makino, 2016).
PRRs are a class of molecules expressed mainly on the surface of innate immune cells, especially some typical antigen-presenting cells such as macrophages and dendritic cells (DCs). PRRs can recognize one or more pathogen-associated molecular patterns (PAMPs) and danger-associated molecular patterns (DAMPs) (Wada and Makino, 2016). PAMPs are certain nonspecific, highly conserved molecules shared by a class or group of specific pathogenic microorganisms and their products, including lipopolysaccharide (LPS), teichoic acid peptidoglycan, and viral double-stranded RNA (Wilhelm et al., 2017). When tissue damage occurs due to various causes, such as inflammation, hypoxia, or stress, DAMPs can be released into the interstitial space or blood circulation. DAMPs are endogenous factors such as high-mobility group protein box 1 (HMGB1), heat shock protein (HSP), and uric acid crystals (Dowling and O'Neill, 2012). PRRs can exist in extracellular fluids such as the bloodstream and lymph, macrophage surfaces, and cellular and endosomal membranes, as well as in the cytosol (Medzhitov and Janeway, 1997). They can be divided into secretory, endocytic, and signal transduction PRRs, which include membrane type PRRs, endosomal membrane type PRRs, and cytoplasmic PRRs. Recent evidence has shown a close link between various types of PRR and DN. Various studies in vivo and in vitro have suggested that PRRs can mediate inflammatory responses and the process of fibrosis in the kidney, finally leading to renal dysfunction (Osborn and Olefsky, 2012).
The purpose of this review is to discuss the relationship between different types of PRRs and DN to provide new approaches and strategies for the treatment of DN.
2. Secretory PRRs and DN
Secretory PRRs exist mainly in the bloodstream, lymph, and interstitial fluids. Mannose-binding lectin (MBL), C-reactive protein (CRP), pentraxin-3 (PTX-3), and ficolin are all secretory PRRs, each of which has a close relationship with DN.
2.1. MBL
MBL is a complement-activating PRR of the innate immune system and is involved in the pathogenesis of DN (Axelgaard et al., 2017a). Humans have only one type of MBL, while mice have two subtypes of MBL, MBL-A and MBL-C. Østergaard et al. (2013) found that an increase in MBL-C was closely related to increasing plasma glucose levels, while the concentration of MBL-A did not differ significantly between DN patients and a control group. MBL is significantly elevated in the serum of DN patients, and is synthesized in the liver and secreted into the serum as an acute phase reaction component. There is increasing evidence suggesting an important link between genetic background and high serum MBL levels in patients with DN (Østergaard et al., 2012). In patients with the MBL2 gene polymorphism, circulating MBL levels are significantly higher than those in healthy controls, confirming that circulating MBL levels are elevated in an MBL2-genotype-dependent manner (Bijkerk et al., 2016). Further studies have revealed a significant correlation between single nucleotide polymorphisms (SNPs) of rs11003125 and rs1800450 of the MBL2 gene and DN. Serum MBL levels measured by enzyme-linked immunosorbent assay (ELISA; human MBL DuoSet, RD Systems) were significantly elevated in subjects with the CC genotype of rs11003125 (median serum MBL: 1553 ng/mL (CC) vs. 690 ng/mL (GG)) and the GG genotype of rs1800450 (median serum MBL: 1322 ng/mL (GG) vs. 621 ng/mL (GA) and 661 ng/mL (AA)) (Zhang et al., 2013). When circulating MBL is elevated, it can activate the lectin pathway and mediate the downstream inflammatory response via a series of mechanisms. In a long-term hyperglycemic state, some new epitopes, such as the Amadori-type new epitope, can be induced in several tissues. MBL can recognize these structures and activate the lectin pathway, thus inducing the inflammatory response (Hisano et al., 2007). There are many types of MBL-associated serine proteases (MASPs), including MASP-1, MASP-2, MASP-3, MBL-associated protein 19 (MAp-19), and MAp-44, of which only MASP-1 and MASP-2 are associated with the complement coagulation cascade (Dobó et al., 2009). After MBL recognizes carbohydrates, the initiator of the lectin pathway, MASP-1, is activated. The activated MASP-1 can cleave MASP-2, thereby initiating the complement cascade (Østergaard et al., 2017). MASP-2 is able to cleave complement proteins C4 and C2 to form C3 convertase. This in turn cleaves C3 to produce C5 convertase, which later enters the terminal pathway of complement activation to form a membrane attack complex (MAC) and lyse cells, resulting in impaired renal function, especially via tubulointerstitial damage (Henriksen et al., 2013; Zheng et al., 2018). After the cleavage of complement protein C3, the deposition of the released C3b fragment can also trigger a local inflammatory response, enhancing phagocytosis by activated macrophages and releasing various inflammatory cytokines and chemokines (C3a and C5a) (Axelgaard et al., 2017b). In addition, compared with a control group, the expression of MBL and nuclear factor-κB (NF-κB) was significantly increased in the glomeruli of DN rats, and the expression of MBL was positively correlated with the expression of NF-κB, which is involved in the development of DN (Yang et al., 2011). Activation of the MBL complement pathway promotes the expression of TNF-α and interleukin-6 (IL-6) in human renal glomerular endothelial cells (HRGECs). This can promote the proliferation of mesangial cells, increase the expression of fibronectin, and affect the dynamic stability of the extracellular matrix of mesangial cells and podocytes, leading to renal damage (Wu et al., 2011).
2.2. CRP
CRP is an acute phase reaction protein and an inflammatory biomarker, which can be rapidly synthesized and secreted in the liver in response to inflammation and tissue damage. As a secretory PRR, CRP is closely related to the development of DN in diabetic patients (Overgaard et al., 2013). High-sensitivity CRP (hs-CRP) found in the plasma, has a higher sensitivity and is of more clinical significance. In patients with DN, serum hs-CRP significantly increases with the degree of albumin excretion and the severity of kidney damage (Shaheer et al., 2017). The hs-CRP concentration in DN patients ((5.15±1.27) mg/L) is significantly higher than that in healthy ((0.31±0.47) mg/L) and non-nephrotic diabetic controls ((2.53±0.71) mg/L). In addition, compared with a microalbuminuria group ((4.10±0.64) mg/L) and a non-albuminuria group ((2.54±0.86) mg/L), the hs-CRP concentration is higher ((5.90±2.10) mg/L) when substantial albuminuria occurs (Liu et al., 2015). When CRP binds to type II Fc receptor for immunoglobulin G (IgG) (FcγRII), it activates the NF-κB signal transduction pathway and induces the corresponding inflammatory response (Tang et al., 2017). CRP can also promote epithelial-mesenchymal transformation (EMT) and accelerate the process of renal fibrosis through the extracellular signal-regulated kinase 1/2 (ERK1/2) and Wnt/β-catenin signaling pathways (Zhang et al., 2019). In addition, CRP can be dependent on TGF-β1 or independently activate the FcγRII-Smad3-mTOR (mammalian target of rapamycin) signaling pathway, leading to renal fibrosis (Fig. 1) (You et al., 2016).
Fig. 1.
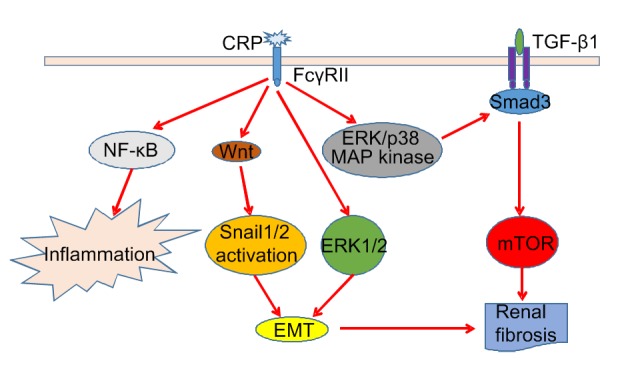
Pathogenic mechanisms of CRP causing inflammation and renal fibrosis in diabetic nephropathy
CRP binds to FcγRII and activates the NF-κB signal pathway to induce inflammation. CRP can also activate the ERK1/2, Wnt/β-catenin, TGF-β1/Smad3, and non-TGF-β1/Smad3 signaling pathways to induce renal fibrosis. CRP, C-reactive protein; FcγRII, type II Fc receptor for immunoglobulin G (IgG); NF-κB, nuclear factor-κB; ERK, extracellular signal-regulated kinase; TGF-β1, transforming growth factor-β1; MAP, mannose-binding lectin (MBL)-associated protein; mTOR, mammalian target of rapamycin; EMT, epithelial-mesenchymal transformation
2.3. PTX-3
PTX-3, a member of the conservative protein superfamily, is a multimeric inflammatory mediator and a better inflammatory marker than hs-CRP (Uzun et al., 2016). PTX-3 is the only long pentraxin protein detectable in kidney tissue and is closely associated with kidney damage in DN (Chen et al., 2018). There is a very close relationship between SNPs in PTX-3 and DN risk in diabetic patients. Genotyping of PTX-3 SNPs rs2305619 and rs2120243 shows that patients with GG variants of rs2305619 are significantly more sensitive to DN than patients with AA variants, whereas patients with AA variants of rs2120243 have a lower risk of DN (Zhu et al., 2017). Compared with a healthy control group ((0.81±0.25) ng/mL), plasma PTX-3 was significantly increased in patients with DN ((1.35±1.55) ng/mL), and was proportional to the severity of renal damage and increased albuminuria (Yilmaz et al., 2009). PTX-3 plays a key role in alleviating DN kidney damage and regulating the dynamic balance of macrophages M1/M2, promotes the differentiation of macrophages to the M2 type, and reduces inflammatory damage. In patients with DN, the levels of the anti-inflammatory cytokines IL-4 and IL-13 are significantly increased when treated with PTX-3, while IL-4 and IL-13 can induce the differentiation of macrophages to M2, thereby attenuating DN kidney damage (Sun et al., 2015). In addition, PTX-3 is closely related to endothelial injury in patients with DN. In diabetic patients with renal disease, plasma PTX-3 concentrations will be even more elevated if well-characterized diabetic microvascular complications exist (Yilmaz et al., 2010).
2.4. Ficolin
Ficolin is a soluble oligomeric defense protein with lectin-like activity. It is a newly discovered secretory PRR involved in inflammation and renal cell damage (Flyvbjerg, 2017). Ficolin is composed mainly of M-ficolin (ficolin-1), L-ficolin (ficolin-2), and H-ficolin (ficolin-3), of which H-ficolin is most closely associated with DN (Bidula et al., 2019). During a follow-up observation of albuminuria in patients with DN, Østergaard et al. (2014) found that H-ficolin levels were strongly associated with the risk of developing substantial albuminuria in microalbuminuria.
3. Endocytic PRRs and DN
Endocytic PRRs refer to a type of transmembrane receptor expressed on the surface of macrophages, which recognize and bind to the corresponding DAMP and are closely related to the pathogenesis of DN. Scavenger receptors are the most important endocytic PRRs, and include scavenger receptor A (SRA), scavenger receptor B (SRB), CXC chemokine ligand 16 (CXCL16), hemoglobin scavenger receptor CD163, and atypical chemokine receptor (ACKR2) (Pombinho et al., 2018).
SRA is a multifunctional PRR expressed on macrophages, whose expression can be regulated by a variety of cytokines (Sun et al., 2012). Platelet-derived growth factor and macrophage-colony-stimulating factor (M-CSF) can promote the expression of SRA, while IL-6, interferon-γ (IFN-γ), TNF-α, and peroxisome proliferator-activated receptor-γ (PPARγ) inhibit its expression. In diabetic patients, SRA can promote the migration of macrophages to the kidney and accelerate the adhesion of mesangial matrix to promote the process of fibrosis, leading to the occurrence of DN (Usui et al., 2007). SRA can also increase urinary albumin excretion and promote mesangial matrix expansion and glomerular hypertrophy in DN mice (Horiuchi et al., 2005). There are two isomers of SRB: SRB-I and CD36. SRB-I is expressed in human glomerular mesangial cells and proximal tubular epithelial cells and participates in the process of mediating cholesterol efflux to the serum as a receptor for high-density lipoprotein (HDL) (Tsun et al., 2014). In a long-term hyperglycemic state, the expression of SRB-I is decreased, resulting in significant impairment of its ability to regulate cholesterol efflux and excessive accumulation of lipids in the kidney, thus promoting the development of DN (Zhou et al., 2008; Tsun et al., 2013). CD36 plays a major role in renal tubular epithelial cells and podocytes. High glucose (HG)-induced CD36 expression is regulated by the AKT-PPARγ signaling pathway. HG stimulates AKT phosphorylation in HK-2 cells, which can significantly increase PPARγ expression, ultimately promoting CD36 expression. HG-induced CD36 overexpression can lead to excessive deposition of lipids in HK-2 cells, which in turn can reduce their viability (Feng et al., 2017). CD36 can also mediate reactive oxygen species (ROS) production in diabetic kidneys, which can induce EMT progression in renal tubular epithelial cells (Hou et al., 2015). It has also been reported that CD36 can interact with TGF-β1 and activate its fibrogenic activity, leading to renal fibrosis (Yang et al., 2007). In addition, CD36 can induce podocyte apoptosis and participate in the process of DN. CD36 can increase the uptake of fatty acids in podocytes, and then induce oxidative stress and an inflammatory response, leading to podocyte apoptosis (Yang et al., 2007). CXC motif chemokine ligand 16 (CXCL16) is a scavenger receptor for oxidized low-density lipoprotein (ox-LDL), which is expressed mainly in human podocytes and mediates ox-LDL uptake (Nosadini and Tonolo, 2011; Zhao et al., 2014). In addition to promoting the production of ROS in human podocytes, ox-LDL uptake by podocytes can downregulate α3-integrin expression and increase fibronectin production in human podocytes (Gutwein et al., 2009). Other studies have shown that CXCL16 may be involved in the AKT signal transduction pathway, which can infiltrate tubulointerstitial inflammatory factors and cytokines, accelerating the progression of DN (Ye et al., 2017; Hu et al., 2018). CD163 and ACKR2 are newly discovered scavenger receptors. CD163 can regulate the conversion of macrophages from the M1 to the M2 phase (Landis et al., 2018), while ACKR2 can significantly reduce kidney inflammation (Zheng et al., 2016). Both have a close relationship with DN.
4. Signal transduction PRRs and DN
Signal transduction PRRs are expressed mainly in the cell membrane, endosomal membrane, and cytoplasm. They can induce different gene expression by initiating specific signal transduction pathways, thus finely regulating the innate immune response and inflammatory response against different DAMPs and PAMPs. In this review, we consider mainly Toll-like receptors (TLRs) and nucleotide-binding oligomerization domain (NOD)-like receptors (NLRs) closely associated with DN.
4.1. Cell membrane and endosomal membrane PRRs
Both the cell membrane PRRs and endosomal membrane PRRs are essential TLRs. Cell membrane PRRs include mainly TLR2, TLR4, TLR5, and TLR11, while TLR3, TLR7, and TLR9 are all endosomal membrane PRRs.
TLRs are an important component of the innate immune system and an emerging family of receptors, which play a key role in the pathogenesis of DN (Lin and Tang, 2014). Under hyperglycemic conditions, DAMPs such as HMGB1 and HSP70 were found to be widely present in the kidneys of diabetic patients and experimental mice (Jheng et al., 2015; Thakur et al., 2017). In addition, compared with normal rats, the expression levels of TLR-2 (3.6-fold increase), TLR-4 (4-fold increase), myeloid differentiation factor 88 (MyD88; 1.8-fold increase), NF-κB (3.5-fold increase), and proinflammatory factors such as TNF-α ((23.22±2.35) ng/L vs. (10.23±1.26) ng/L), IL-6 ((7.67±0.89) ng/L vs. (2.48±0.53) ng/L), and monocyte chemoattractant protein 1 (MCP-1) were significantly increased in a DN group (Zhao and Han, 2018), indicating the essential role of TLRs in promoting the DN inflammatory response. The HMGB1 inhibitor glycyrrhizic acid significantly reduces TLR4-related ERK and p38 mitogen-activated protein kinase (MAPK)/NF-κB activation to reduce renal injury and the inflammatory response in diabetic rats (Zhang et al., 2017). In the treatment of diabetic patients, some drugs, such as umbelliferous drugs, inhibit TLR2 and TLR4 expression. The level of downstream inflammatory molecules is also significantly decreased, and finally renal function improves (Wang et al., 2019). All of these findings suggest that the activation of TLRs is an important mechanism for promoting DN immune-mediated injury. Recent studies have shown that genetic polymorphisms of TLRs and their related molecules play an indispensable role in the pathogenesis of DN. Sequence analysis of the TLR2 gene revealed that its complete sequence consists of 5'-untranslated region (UTR), coding domain sequence (CDS), and 3'-UTR, and its gene polymorphism can seriously affect TLR signaling (Subhash et al., 2018). Analysis of variants of rs5030717 and rs5030718 TLR4 in DN patients shows significant differences in genotype frequencies of TLR4 rs5030717 and rs5030718 GA and GG genotypes compared with the controls, suggesting that TLR4 gene polymorphism is closely related to the risk of DN (Abbas et al., 2018). MyD88, IL-1 receptor-associated kinase 4 (IRAK4), and TNF receptor-associated factor 6 (TRAF6) are all important molecules in the TLR signaling pathway, and there is a significant correlation between their gene polymorphism and DN. Some studies have found that the rs6853 AG genotype of the MyD88 gene, the rs4251532 CT genotype of the IRAK4 gene, and the rs16928973 gene polymorphism in the TRAF6 gene can significantly increase the risk of DN (Guo et al., 2016). Recently, there have been many studies on the pathogenesis of DN caused by TLRs; however, the specific mechanisms are not completely clear. TLR2, TLR4, TLR5, TLR7, TLR9, and TLR11 activate NF-κB in an MyD88-dependent manner, promoting the release of various inflammatory cytokines, including IL-6, macrophage inhibitory protein 2 (MIP-2), MCP-1, CC motif chemokine ligand 5 (CCL5), and vascular cell adhesion molecule 1 (VCAM-1) (Fig. 2). However, TLR3 and TLR4 can significantly upregulate their downstream signaling molecule phospho-IFN regulatory factor 3 (IRF3) in an MyD88-independent manner, which can promote the production of type 1 IFNs like IFN-α and IFN-β (Fig. 2) (Feng et al., 2015). Inflammatory cytokines and type 1 IFNs together promote the development of DN. In recent years, some other pathogenic mechanisms of DN have been discovered. Studies have shown that TLR4 mediates the upregulation of microRNA-146a-3p (miR-146a-3p), which in turn activates TLR8 to drive cells to secrete IL-8 (Gysler et al., 2016). In addition, Ding et al. (2017) found that HG can induce over-proliferation of mouse mesangial cells in a TLR4-dependent manner and excessive accumulation of mesangial matrix, thus accelerating the progression of DN.
Fig. 2.
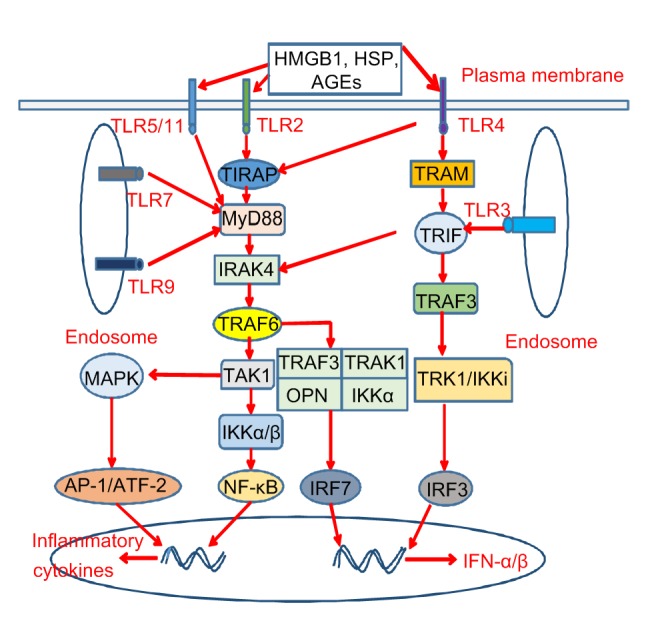
TLR signaling pathways in diabetic nephropathy
When activated by DAMPs (such as HMGB1, HSP, and AGEs), TLR2 and TLR4 directly activate MyD88 via TIRAP, TLR5, TLR7, TLR9, and TLR11, and finally activate NF-κB, IRF7, and AP-1/ATF-2. In addition, TLR4 can directly activate TRIF via TRAM and TLR3, which ultimately leads to the activation of IRF3 and IRAK4. Activation of NF-κB and AP-1/ATF-2 can induce the production of inflammatory factors, while activation of IRF3 and IRF7 can induce the production of type 1 IFNs, which together lead to the occurrence of DN. TLR, Toll-like receptor; DAMP, danger-associated molecular pattern; HMGB1, high-mobility group protein box 1; HSP, heat shock protein; AGEs, advanced glycation end products; MyD88, myeloid differentiation factor 88; TIRAP, Toll/interleukin-1 (IL-1) receptor domain-containing adaptor protein; NF-κB, nuclear factor-κB; IRF, interferon (IFN) regulatory factor; AP-1, activator protein 1; ATF-2, activating transcription factor-2; TRIF, Toll/IL-1-resistance domain-containing adaptor inducing IFN-β; TRAM, TRIF-related adaptor molecule; IRAK4, IL-1 receptor-associated kinase 4; DN, diabetic nephropathy; TRAF, tumor necrosis factor (TNF) receptor-associated factor; TAK1, transforming growth factor β-activated kinase-1; IKK, IκB kinase; MAPK, mitogen-activated protein kinase; TRAK, trafficking kinesin protein; OPN, osteopontin; TRK, tropomyosin-related kinase
4.2. Cytoplasmic PRRs
Cytoplasmic PRRs involved in the pathogenesis of DN are mainly NLRs, including NOD-like receptor protein 3 (NLRP3), NLRP1, NLR family caspase activation and recruitment domain (CARD)-containing protein 4 (NLRC4), NLRC5, and NOD1/2. They can all lead to the occurrence of DN in different ways (Fig. 3).
Fig. 3.
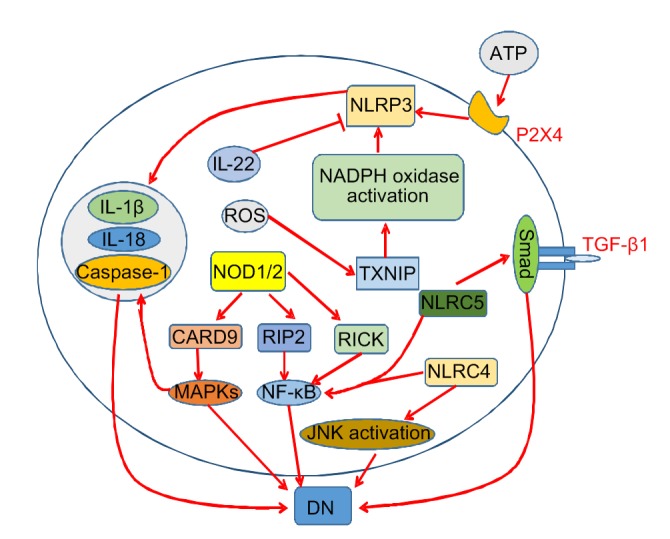
NLR signaling pathways in diabetic nephropathy
The NLRP3 inflammasome can be activated by the ROS/TXNIP/NADPH oxidase signaling pathway and extracellular ATP, and inhibited by IL-22. Activated NLRP3 can cause excessive amounts of inflammatory cytokines such as IL-1β and IL-18. NOD1/2 can activate MAPKs and NF-κB, and NLRC4 and NLRC5 can also activate NF-κB. In addition, NLPC4 and NLRC5 affect the JNK and TGF-β/Smad pathways, respectively. NLR, nucleotide-binding oligomerization domain (NOD)-like receptor; NLRP3, NLR protein 3; ROS, reactive oxygen species; TXNIP, thioredoxin-interacting protein; NADPH, nicotinamide adenine dinucleotide phosphate; ATP, adenosine triphosphate; IL, interleukin; MAPK, mitogen-activated protein kinase; NF-κB, nuclear factor-κB; NLRC, NLR family caspase recruitment domain (CARD)-containing protein; JNK, c-Jun N-terminal kinase; TGF, transforming growth factor; RICK, receptor interacting protein kinase (RIP)-like-interacting caspase-like apoptosis-regulatory protein (CLARP) kinase; DN, diabetic nephropathy
Examination of renal tissue of mice with DN has demonstrated that NLRP3, apoptosis-associated speck-like protein containing a CARD (ASC), caspase-1, IL-1β, and IL-18 expression levels were significantly upregulated (Wang et al., 2018). The expression levels of the NLRP3 inflammasome and its related molecules were also upregulated in in vitro experiments (Qiao et al., 2018), indicating that this inflammasome is responsible for the pathogenesis of DN. When mice lack NLRP3, or are administered NLRP3 antagonists or drugs that inhibit NLRP3 inflammasome activation, kidney damage in diabetic mice is significantly alleviated, confirming that NLRP3 is an important cause of diabetes-induced kidney damage (Liu et al., 2018; Zhu et al., 2018). Numerous studies have shown that ROS is an important promoter of NLRP3 inflammasome activation. ROS can activate the NLRP3 inflammasome in podocytes by ROS/thioredoxin-interacting protein (TXNIP)/nicotinamide adenine dinucleotide phosphate (NADPH) oxidase signaling and then induce podocyte injury (Han YH et al., 2018). Some studies suggest that sweet taste receptors (STRs) are involved in the activation of ROS-NLRP3 inflammasome signaling (Zhou et al., 2018). In addition, extracellular adenosine triphosphate (ATP) can bind to the P2X4 receptor, which in turn activates the NLRP3 inflammasome and causes IL-1β and IL-18 to mature and release (Chen et al., 2013). However, recent studies have shown that IL-22 can significantly reverse the renal activation of NLRP3 and downregulate the NLRP3/caspase-1/IL-1β pathway to protect the kidney (Wang et al., 2017). In contrast to the role of NLRP3, NLRP1 plays a protective role in DN, and NLRP1 rs11651270 and rs2670660 polymorphisms are significantly associated with a low risk of developing DN (Soares et al., 2018). NLRP6 and NLRP12 play an important role in the development of many diseases in an inflammasome-dependent manner. Inflammasomes nucleated by NLRP6, NLRP12, and NLRP3 integrate signals from metabolic systems and contribute to diabetes, but its role in the occurrence and development of DN remains to be studied (Janowski et al., 2013; Masters, 2013). In kidney specimens of patients with DN, a significant increase in the expression of NLRC4 and NLRC5 was also observed. NLRC4 can induce the production of IL-1β and promote the infiltration of F4/80+ macrophages in the kidney, while NLRC5 can promote fibronectin and collagen IV expression and macrophage infiltration (Yuan et al., 2016; Luan et al., 2018). In addition, under HG conditions, the NLRC4 inflammasome is able to induce NF-κB and c-Jun N-terminal kinase (JNK) activation in renal tissues of DN mice (Yuan et al., 2016). NLRC5 can promote NF-κB and TGF-β/Smad signaling pathways and inhibit the PIK3/Akt signaling pathway, ultimately leading to inflammation, fibrosis, and damage to HK-2 cells (Han F et al., 2018; Luan et al., 2018). NOD1 and NOD2 are also closely associated with the production and persistence of inflammation in DN patients and mice. When NOD1 is activated by HG, it can promote the production of various inflammatory cytokines in renal tissues through the NOD1-receptor interacting protein kinase (RIP)-like-interacting caspase-like apoptosis-regulatory protein (CLARP) kinase (RICK)-NF-κB signaling pathway. Under HG stimulation, NOD1, RICK, NF-κB, and IL-1β expression significantly increased, and renal function decreased in rat mesangial cells (Huang et al., 2016). In renal biopsy samples from patients with DN, NOD2 was overexpressed and positively correlated with the severity of renal injury. Overexpression of NOD2 can activate the MAPK/ERK kinase (MEK)/ERK signaling pathway to promote EMT in glomerular endothelial cells (GEnCs) and accelerate the process of renal fibrosis (Shang et al., 2017). One of the causes of proteinuria in patients with DN is an increase in human antigen R (HuR) in the kidney, whereas under the action of HG, HuR can bind to the 3'-UTR of NOD2 mRNA, thus increasing its stability, leading to overexpression of NOD2 and impairment of renal function (Shang et al., 2015).
The absence in melanoma 2 (AIM2) inflammasome is a non-NLR cytoplasmic PRR and one of the DNA recognition receptor families. It can bind to double-stranded DNA (dsDNA) and play an important role in the pathogenesis of DN (Fernandes-Alnemri et al., 2009). In the kidneys of patients with DN, immunofluorescence showed that AIM2 was highly expressed in glomeruli and renal tubules. When AIM2 was deficient, inflammation, fibrosis and kidney damage were significantly reduced in a mice model, indicating that the AIM2 inflammasome contributes to kidney inflammation and fibrosis in mice. During the death of necrotic cells in damaged kidneys, dsDNA is released and phagocytosed by proinflammatory macrophages, thereby activating the AIM2 inflammasome. The activation of the AIM2 inflammasome can promote the expression of caspase-1 and IL-1β, thus contributing to chronic damage to the kidney and accelerating the progression of DN (Komada et al., 2018).
5. Conclusions
DN is a microvascular disease that is common in patients with type 1 or type 2 diabetes and seriously affects health. The specific mechanisms are not fully understood. However, in recent years the important role of PRRs in the pathogenesis and progression of DN has received increasing attention. Research on PRRs has become a hot topic in nephrology research. Renal inflammation and fibrosis are the two most important processes of renal tissue damage in DN. This review describes how various types of PRRs affect the inflammatory response and fibrosis process ultimately leading to DN. Most previous reviews have shown that activation of TLR and NLR signaling is involved in the initiation of the innate immune response to cause DN. This review also highlights the roles of secretory PRRs and endosomal PRRs. The mechanism of action of PRRs has not yet been fully clarified and is extremely complex. More research is needed to clarify DN pathogenesis to provide new ideas and clues for the prevention and treatment of DN, thus developing safer and more effective treatment strategies.
Footnotes
Project supported by the National Natural Science Foundation of China (Nos. 81060063 and 81660129)
Contributors: Zhi-feng ZHOU and Lei JIANG set up the theme and the frame of this review, wrote and edited the manuscript. Qing ZHAO, Yu WANG, and Jing ZHOU performed the references collection and selection. Jin-lei LV and Qin-kai CHEN contributed the design and revision of the manuscript. All authors have read and approved the final manuscript.
Compliance with ethics guidelines: Zhi-feng ZHOU, Lei JIANG, Qing ZHAO, Yu WANG, Jing ZHOU, Qin-kai CHEN, and Jin-lei LV declare that they have no conflict of interest.
This article does not contain any studies with human or animal subjects performed by any of the authors.
References
- 1.Abbas SA, Raza ST, Mir SS, et al. Role of variants rs5030717 and rs5030718 of TLR4 in the risk prediction of nephropathy, hypertension and dyslipidaemia in type 2 diabetes mellitus. Br J Biomed Sci. 2018;75(4):163–168. doi: 10.1080/09674845.2018.1477033. [DOI] [PubMed] [Google Scholar]
- 2.Awad AS, You HN, Gao T, et al. Macrophage-derived tumor necrosis factor-α mediates diabetic renal injury. Kidney Int. 2015;88(4):722–733. doi: 10.1038/ki.2015.162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Axelgaard E, Østergaard JA, Thiel S, et al. Diabetes is associated with increased autoreactivity of mannan-binding lectin. J Diabetes Res, 2017:6368780. 2017 doi: 10.1155/2017/6368780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Axelgaard E, Østergaard JA, Haxha S, et al. Global autorecognition and activation of complement by mannan-binding lectin in a mouse model of type 1 diabetes. Mediators Inflamm, 2017:9403754. 2017 doi: 10.1155/2017/9403754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bidula S, Sexton DW, Schelenz S. Ficolins and the recognition of pathogenic microorganisms: an overview of the innate immune response and contribution of single nucleotide polymorphisms. J Immunol Res, 2019: 3205072. 2019 doi: 10.1155/2019/3205072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bijkerk R, van der Pol P, Khairoun M, et al. Simultaneous pancreas-kidney transplantation in patients with type 1 diabetes reverses elevated MBL levels in association with MBL2 genotype and VEGF expression. Diabetologia. 2016;59(4):853–858. doi: 10.1007/s00125-015-3858-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen KH, Zhang JG, Zhang WW, et al. ATP-P2X4 signaling mediates NLRP3 inflammasome activation: a novel pathway of diabetic nephropathy. Int J Biochem Cell Biol. 2013;45(5):932–943. doi: 10.1016/j.biocel.2013.02.009. [DOI] [PubMed] [Google Scholar]
- 8.Chen XH, Luo J, Wu MM, et al. Study on association of pentraxin 3 and diabetic nephropathy in a rat model. J Diabetes Res, 2018:8968573. 2018 doi: 10.1155/2018/8968573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Collins AJ, Foley RN, Chavers B, et al. United States Renal Data System 2011 Annual Data Report: atlas of chronic kidney disease & end-stage renal disease in the United States. Am J Kidney Dis. 2012;59(1 Suppl 1):A7. doi: 10.1053/j.ajkd.2011.11.015. [DOI] [PubMed] [Google Scholar]
- 10.D'addio F, Trevisani A, Nasr MB, et al. Harnessing the immunological properties of stem cells as a therapeutic option for diabetic nephropathy. Acta Diabetol. 2014;51(6):897–904. doi: 10.1007/s00592-014-0603-1. [DOI] [PubMed] [Google Scholar]
- 11.Ding T, Chen W, Li J, et al. High glucose induces mouse mesangial cell overproliferation via inhibition of hydrogen sulfide synthesis in a TLR-4-dependent manner. Cell Physiol Biochem. 2017;41(3):1035–1043. doi: 10.1159/000461483. [DOI] [PubMed] [Google Scholar]
- 12.Dobó J, Harmat V, Beinrohr L, et al. MASP-1, a promiscuous complement protease: structure of its catalytic region reveals the basis of its broad specificity. J Immunol. 2009;183(2):1207–1214. doi: 10.4049/jimmunol.0901141. [DOI] [PubMed] [Google Scholar]
- 13.Dowling JK, O'Neill LA. Biochemical regulation of the inflammasome. Crit Rev Biochem Mol Biol. 2012;47(5):424–443. doi: 10.3109/10409238.2012.694844. [DOI] [PubMed] [Google Scholar]
- 14.Feng L, Gu CW, Li YX, et al. High glucose promotes CD36 expression by upregulating peroxisome proliferator-activated receptor γ levels to exacerbate lipid deposition in renal tubular cells. BioMed Res Int, 2017:1414070. 2017 doi: 10.1155/2017/1414070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Feng YY, Yang SL, Ma YX, et al. Role of Toll-like receptors in diabetic renal lesions in a miniature pig model. Sci Adv. 2015;1(5):e1400183. doi: 10.1126/sciadv.1400183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fernandes-Alnemri T, Yu JW, Datta P, et al. AIM2 activates the inflammasome and cell death in response to cytoplasmic DNA. Nature. 2009;458(7237):509–513. doi: 10.1038/nature07710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Flyvbjerg A. The role of the complement system in diabetic nephropathy. Nat Rev Nephrol. 2017;13(5):311–318. doi: 10.1038/nrneph.2017.31. [DOI] [PubMed] [Google Scholar]
- 18.Guo CC, Zhang LJ, Nie LH, et al. Association of polymorphisms in the MyD88, IRAK4 and TRAF6 genes and susceptibility to type 2 diabetes mellitus and diabetic nephropathy in a southern Han Chinese population. Mol Cell Endocrinol. 2016;429:114–119. doi: 10.1016/j.mce.2016.04.003. [DOI] [PubMed] [Google Scholar]
- 19.Guo XX, Wang Y, Wang K, et al. Stability of a type 2 diabetes rat model induced by high-fat diet feeding with low-dose streptozotocin injection. J Zhejiang Univ Sci-B (Biomed & Biotechnol) 2018;19(7):559–569. doi: 10.1631/jzus.B1700254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gutwein P, Abdel-Bakky MS, Doberstein K, et al. CXCL16 and oxLDL are induced in the onset of diabetic nephropathy. J Cell Mol Med. 2009;13(9b):3809–3825. doi: 10.1111/j.1582-4934.2009.00761.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gysler SM, Mulla MJ, Guerra M, et al. Antiphospholipid antibody-induced miR-146a-3p drives trophoblast interleukin-8 secretion through activation of Toll-like receptor 8. Mol Hum Reprod. 2016;22(7):465–474. doi: 10.1093/molehr/gaw027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Han F, Gao Y, Ding CG, et al. Knockdown of NLRC5 attenuates renal I/R injury in vitro through the activation of PI3K/Akt signaling pathway. Biomed Pharmacother. 2018;103:222–227. doi: 10.1016/j.biopha.2018.04.040. [DOI] [PubMed] [Google Scholar]
- 23.Han YH, Xu XX, Tang CY, et al. Reactive oxygen species promote tubular injury in diabetic nephropathy: the role of the mitochondrial ROS-TXNIP-NLRP3 biological axis. Redox Biol. 2018;16:32–46. doi: 10.1016/j.redox.2018.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Henriksen ML, Brandt J, Andrieu JP, et al. Heteromeric complexes of native collectin kidney 1 and collectin liver 1 are found in the circulation with MASPs and activate the complement system. J Immunol. 2013;191(12):6117–6127. doi: 10.4049/jimmunol.1302121. [DOI] [PubMed] [Google Scholar]
- 25.Hisano S, Matsushita M, Fujita T, et al. Activation of the lectin complement pathway in post-streptococcal acute glomerulonephritis. Pathol Int. 2007;57(6):351–357. doi: 10.1111/j.1440-1827.2007.02107.x. [DOI] [PubMed] [Google Scholar]
- 26.Horiuchi S, Unno Y, Usui H, et al. Pathological roles of advanced glycation end product receptors SR-A and CD36. Ann N Y Acad Sci. 2005;1043(1):671–675. doi: 10.1196/annals.1333.076. [DOI] [PubMed] [Google Scholar]
- 27.Hou YJ, Wu M, Wei JY, et al. CD36 is involved in high glucose-induced epithelial to mesenchymal transition in renal tubular epithelial cells. Biochem Biophys Res Commun. 2015;468(1-2):281–286. doi: 10.1016/j.bbrc.2015.10.112. [DOI] [PubMed] [Google Scholar]
- 28.Hu ZB, Ma KL, Zhang Y, et al. Inflammation-activated CXCL16 pathway contributes to tubulointerstitial injury in mouse diabetic nephropathy. Acta Pharmacol Sin. 2018;39(6):1022–1033. doi: 10.1038/aps.2017.177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Huang W, Gou F, Long Y, et al. High glucose and lipopolysaccharide activate NOD1-RICK-NF-κB inflammatory signaling in mesangial cells. Exp Clin Endocrinol Diabetes. 2016;124(8):512–517. doi: 10.1055/s-0042-105641. [DOI] [PubMed] [Google Scholar]
- 30.Huang YQ, Gou R, Diao YS, et al. Charlson comorbidity index helps predict the risk of mortality for patients with type 2 diabetic nephropathy. J Zhejiang Univ-Sci B (Biomed & Biotechnol) 2014;15(1):58–66. doi: 10.1631/jzus.B1300109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Janowski AM, Kolb R, Zhang WZ, et al. Beneficial and detrimental roles of NLRs in carcinogenesis. Front Immunol, 4:370. 2013 doi: 10.3389/fimmu.2013.00370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jheng HF, Tsai PJ, Chuang YL, et al. Albumin stimulates renal tubular inflammation through an HSP70-TLR4 axis in mice with early diabetic nephropathy. Dis Model Mech. 2015;8(10):1311–1321. doi: 10.1242/dmm.019398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Komada T, Chung H, Lau A, et al. Macrophage uptake of necrotic cell DNA activates the AIM2 inflammasome to regulate a proinflammatory phenotype in CKD. J Am Soc Nephrol. 2018;29(4):1165–1181. doi: 10.1681/asn.2017080863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Landis RC, Quimby KR, Greenidge AR. M1/M2 macrophages in diabetic nephropathy: Nrf2/HO-1 as therapeutic targets. Curr Pharm Des. 2018;24(20):2241–2249. doi: 10.2174/1381612824666180716163845. [DOI] [PubMed] [Google Scholar]
- 35.Lin M, Tang SCW. Toll-like receptors: sensing and reacting to diabetic injury in the kidney. Nephrol Dial Transplant. 2014;29(4):746–754. doi: 10.1093/ndt/gft446. [DOI] [PubMed] [Google Scholar]
- 36.Liu Q, Jiang CY, Chen BX, et al. The association between high-sensitivity C-reactive protein concentration and diabetic nephropathy: a meta-analysis. Eur Rev Med Pharmacol Sci. 2015;19(23):4558–4568. [PubMed] [Google Scholar]
- 37.Liu YW, Hao YC, Chen YJ, et al. Protective effects of sarsasapogenin against early stage of diabetic nephropathy in rats. Phytother Res. 2018;32(8):1574–1582. doi: 10.1002/ptr.6088. [DOI] [PubMed] [Google Scholar]
- 38.Lu L, Peng WH, Wang W, et al. Effects of atorvastatin on progression of diabetic nephropathy and local RAGE and soluble RAGE expressions in rats. J Zhejiang Univ-Sci B (Biomed & Biotechnol) 2011;12(8):652–659. doi: 10.1631/jzus.B1101004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Luan PP, Zhuang JH, Zou J, et al. NLRC5 deficiency ameliorates diabetic nephropathy through alleviating inflammation. FASEB J. 2018;32(2):1070–1084. doi: 10.1096/fj.201700511RR. [DOI] [PubMed] [Google Scholar]
- 40.Masters SL. Specific inflammasomes in complex diseases. Clin Immunol. 2013;147(3):223–228. doi: 10.1016/j.clim.2012.12.006. [DOI] [PubMed] [Google Scholar]
- 41.McKnight AJ, Duffy S, Maxwell AP. Genetics of diabetic nephropathy: a long road of discovery. Curr Diab Rep. 2015;15(7):41. doi: 10.1007/s11892-015-0610-9. [DOI] [PubMed] [Google Scholar]
- 42.Medzhitov R, Janeway CA., Jr Innate immunity: impact on the adaptive immune response. Curr Opin Immunol. 1997;9(1):4–9. doi: 10.1016/S0952-7915(97)80152-5. [DOI] [PubMed] [Google Scholar]
- 43.Niewczas MA, Pavkov ME, Skupien J, et al. A signature of circulating inflammatory proteins and development of end-stage renal disease in diabetes. Nat Med. 2019;25(5):805–813. doi: 10.1038/s41591-019-0415-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nosadini R, Tonolo G. Role of oxidized low density lipoproteins and free fatty acids in the pathogenesis of glomerulopathy and tubulointerstitial lesions in type 2 diabetes. Nutr Metab Cardiovasc Dis. 2011;21(2):79–85. doi: 10.1016/j.numecd.2010.10.002. [DOI] [PubMed] [Google Scholar]
- 45.Osborn O, Olefsky JM. The cellular and signaling networks linking the immune system and metabolism in disease. Nat Med. 2012;18(3):363–374. doi: 10.1038/nm.2627. [DOI] [PubMed] [Google Scholar]
- 46.Østergaard JA, Bjerre M, Ramachandrarao SP, et al. Mannan-binding lectin in diabetic kidney disease: the impact of mouse genetics in a type 1 diabetes model. Exp Diabetes Res, 2012:678381. 2012 doi: 10.1155/2012/678381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Østergaard JA, Bjerre M, Dagnaes-Hansen F, et al. Diabetes-induced changes in mannan-binding lectin levels and complement activation in a mouse model of type 1 diabetes. Scand J Immunol. 2013;77(3):187–194. doi: 10.1111/sji.12027. [DOI] [PubMed] [Google Scholar]
- 48.Østergaard JA, Thiel S, Hovind P, et al. Association of the pattern recognition molecule H-ficolin with incident microalbuminuria in an inception cohort of newly diagnosed type 1 diabetic patients: an 18 year follow-up study. Diabetologia. 2014;57(10):2201–2207. doi: 10.1007/s00125-014-3332-7. [DOI] [PubMed] [Google Scholar]
- 49.Østergaard JA, Thiel S, Hoffmann-Petersen IT, et al. Incident microalbuminuria and complement factor mannan-binding lectin-associated protein 19 in people with newly diagnosed type 1 diabetes. Diabetes Metab Res Rev. 2017;33(5):e2895. doi: 10.1002/dmrr.2895. [DOI] [PubMed] [Google Scholar]
- 50.Overgaard AJ, McGuire JN, Hovind P, et al. Serum amyloid A and C-reactive protein levels may predict microalbuminuria and macroalbuminuria in newly diagnosed type 1 diabetic patients. J Diabetes Complications. 2013;27(1):59–63. doi: 10.1016/j.jdiacomp.2012.06.016. [DOI] [PubMed] [Google Scholar]
- 51.Pombinho R, Sousa S, Cabanes D. Scavenger receptors: promiscuous players during microbial pathogenesis. Crit Rev Microbiol. 2018;44(6):685–700. doi: 10.1080/1040841x.2018.1493716. [DOI] [PubMed] [Google Scholar]
- 52.Qiao YC, Tian XX, Men L, et al. Spleen tyrosine kinase promotes NLR family pyrin domain containing 3 inflammasome-mediated IL-1β secretion via c-Jun N-terminal kinase activation and cell apoptosis during diabetic nephropathy. Mol Med Rep. 2018;18(2):1995–2008. doi: 10.3892/mmr.2018.9164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rao V, Tan SH, Candasamy M, et al. Diabetic nephropathy: an update on pathogenesis and drug development. Diabetes Metab Syndr. 2019;13(1):754–762. doi: 10.1016/j.dsx.2018.11.054. [DOI] [PubMed] [Google Scholar]
- 54.Shaheer AK, Tharayil JK, Krishna PW. A comparative study of high sensitivity C-reactive protein and metabolic variables in type 2 diabetes mellitus with and without nephropathy. J Clin Diagn Res. 2017;11(9):BC01–BC04. doi: 10.7860/jcdr/2017/30272.10528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Shang J, Wan Q, Wang XJ, et al. Identification of NOD2 as a novel target of RNA-binding protein HuR: evidence from NADPH oxidase-mediated HuR signaling in diabetic nephropathy. Free Radic Biol Med. 2015;79:217–227. doi: 10.1016/j.freeradbiomed.2014.12.013. [DOI] [PubMed] [Google Scholar]
- 56.Shang J, Zhang Y, Jiang YM, et al. NOD2 promotes endothelial-to-mesenchymal transition of glomerular endothelial cells via MEK/ERK signaling pathway in diabetic nephropathy. Biochem Biophys Res Commun. 2017;484(2):435–441. doi: 10.1016/j.bbrc.2017.01.155. [DOI] [PubMed] [Google Scholar]
- 57.Sifuentes-Franco S, Padilla-Tejeda DE, Carrillo-Ibarra S, et al. Oxidative stress, apoptosis, and mitochondrial function in diabetic nephropathy. Int J Endocrinol, 2018: 1875870. 2018 doi: 10.1155/2018/1875870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Soares JLS, Fernandes FP, Patente TA, et al. Gain-of-function variants in NLRP1 protect against the development of diabetic kidney disease: NLRP1 inflammasome role in metabolic stress sensing? Clin Immunol. 2018;187:46–49. doi: 10.1016/j.clim.2017.10.003. [DOI] [PubMed] [Google Scholar]
- 59.Subhash V, Monika S, Richa S, et al. Distribution of single nucleotide polymorphisms and protein domain architecture of Toll-like receptor-2 in Pahari cattle (Indian non-descript indigenous breed) Res Vet Sci. 2018;117:144–149. doi: 10.1016/j.rvsc.2017.12.003. [DOI] [PubMed] [Google Scholar]
- 60.Sun HB, Tian J, Xian WH, et al. Pentraxin-3 attenuates renal damage in diabetic nephropathy by promoting M2 macrophage differentiation. Inflammation. 2015;38(5):1739–1747. doi: 10.1007/s10753-015-0151-z. [DOI] [PubMed] [Google Scholar]
- 61.Sun L, Wen JH, Sun HL, et al. Perindopril attenuates renal tubulointerstitium injury by inhibiting scavenger receptor A over-expression in diabetic rats. J Endocrinol Invest. 2012;35(5):511–515. doi: 10.3275/7867. [DOI] [PubMed] [Google Scholar]
- 62.Tang Y, Fung E, Xu AP, et al. C-reactive protein and ageing. Clin Exp Pharmacol Physiol. 2017;44(S1):9–14. doi: 10.1111/1440-1681.12758. [DOI] [PubMed] [Google Scholar]
- 63.Thakur V, Nargis S, Gonzalez M, et al. Role of glycyrrhizin in the reduction of inflammation in diabetic kidney disease. Nephron. 2017;137(2):137–147. doi: 10.1159/000477820. [DOI] [PubMed] [Google Scholar]
- 64.Tsun JGS, Shiu SWM, Wong Y, et al. Impact of serum amyloid A on cellular cholesterol efflux to serum in type 2 diabetes mellitus. Atherosclerosis. 2013;231(2):405–410. doi: 10.1016/j.atherosclerosis.2013.10.008. [DOI] [PubMed] [Google Scholar]
- 65.Tsun JGS, Yung S, Chau MKM, et al. Cellular cholesterol transport proteins in diabetic nephropathy. PLoS ONE. 2014;9(9):e105787. doi: 10.1371/journal.pone.0105787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Usui HK, Shikata K, Sasaki M, et al. Macrophage scavenger receptor-A-deficient mice are resistant against diabetic nephropathy through amelioration of microinflammation. Diabetes. 2007;56(2):363–372. doi: 10.2337/db06-0359. [DOI] [PubMed] [Google Scholar]
- 67.Uzun S, Ozari M, Gursu M, et al. Changes in the inflammatory markers with advancing stages of diabetic nephropathy and the role of pentraxin-3. Ren Fail. 2016;38(8):1193–1198. doi: 10.1080/0886022x.2016.1209031. [DOI] [PubMed] [Google Scholar]
- 68.Wada J, Makino H. Innate immunity in diabetes and diabetic nephropathy. Nat Rev Nephrol. 2016;12(1):13–26. doi: 10.1038/nrneph.2015.175. [DOI] [PubMed] [Google Scholar]
- 69.Wang C, Hou XX, Rui HL, et al. Artificially cultivated Ophiocordyceps sinensis alleviates diabetic nephropathy and its podocyte injury via inhibiting P2X7R expression and NLRP3 inflammasome activation. J Diabetes Res, 2018:1390418. 2018 doi: 10.1155/2018/1390418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wang HQ, Wang SS, Chiufai K, et al. Umbelliferone ameliorates renal function in diabetic nephropathy rats through regulating inflammation and TLR/NF-κB pathway. Chin J Nat Med. 2019;17(5):346–354. doi: 10.1016/s1875-5364(19)30040-8. [DOI] [PubMed] [Google Scholar]
- 71.Wang SF, Li YB, Fan JJ, et al. Interleukin-22 ameliorated renal injury and fibrosis in diabetic nephropathy through inhibition of NLRP3 inflammasome activation. Cell Death Dis. 2017;8(7):e2937. doi: 10.1038/cddis.2017.292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Wilhelm I, Nyúl-Tóth A, Kozma M, et al. Role of pattern recognition receptors of the neurovascular unit in inflamm-aging. Am J Physiol Heart Circ Physiol. 2017;313(5):H1000–H1012. doi: 10.1152/ajpheart.00106.2017. [DOI] [PubMed] [Google Scholar]
- 73.Wu XH, Huang SM, Fan WX, et al. Influence of high glucose and mannose binding lectin complement pathway activation to IL-6 and TNF-alpha’s expression by human renal glomerular endothelial cells. J Sichuan Univ (Med Sci Ed) 2011;42(1):90–94. (in Chinese) [PubMed] [Google Scholar]
- 74.Yang WF, Han F, Zhang XH, et al. Extra-pulmonary tuberculosis infection in the dialysis patients with end stage renal diseases: case reports and literature review. J Zhejiang Univ-Sci B (Biomed & Biotechnol) 2013;14(1):76–82. doi: 10.1631/jzus.B1200244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Yang YL, Lin SH, Chuang LY, et al. CD36 is a novel and potential anti-fibrogenic target in albumin-induced renal proximal tubule fibrosis. J Cell Biochem. 2007;101(3):735–744. doi: 10.1002/jcb.21236. [DOI] [PubMed] [Google Scholar]
- 76.Yang YX, Huang SM, Yan XY, et al. Relationship between activation of mannan-binding lectin complement and NF-κB in diabetic nephropathy. J Sichuan Univ (Med Sci Ed) 2011;42(4):490–493. (in Chinese) [PubMed] [Google Scholar]
- 77.Ye YN, Chen QZ, Li JM, et al. CXCL16 deficiency attenuates diabetic nephropathy through decreasing oxidative stress and inflammation. Biochem Biophys Res Commun. 2017;491(3):848–854. doi: 10.1016/j.bbrc.2017.05.013. [DOI] [PubMed] [Google Scholar]
- 78.Yilmaz MI, Axelsson J, Sonmez A, et al. Effect of renin angiotensin system blockade on pentraxin 3 levels in type-2 diabetic patients with proteinuria. Clin J Am Soc Nephrol. 2009;4(3):535–541. doi: 10.2215/cjn.04330808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Yilmaz MI, Carrero JJ, Martin-Ventura JL, et al. Combined therapy with renin-angiotensin system and calcium channel blockers in type 2 diabetic hypertensive patients with proteinuria: effects on soluble TWEAK, PTX3, and flow-mediated dilation. Clin J Am Soc Nephrol. 2010;5(7):1174–1181. doi: 10.2215/cjn.01110210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.You YK, Huang XR, Chen HY, et al. C-reactive protein promotes diabetic kidney disease in db/db mice via the CD32b-Smad3-mTOR signaling pathway. Sci Rep. 2016;6(1):26740. doi: 10.1038/srep26740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Yuan F, Kolb R, Pandey G, et al. Involvement of the NLRC4-inflammasome in diabetic nephropathy. PLoS ONE. 2016;11(10):e0164135. doi: 10.1371/journal.pone.0164135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Zhang H, Zhang R, Chen J, et al. High mobility group box1 inhibitor glycyrrhizic acid attenuates kidney injury in streptozotocin-induced diabetic rats. Kidney Blood Press Res. 2017;42(5):894–904. doi: 10.1159/000485045. [DOI] [PubMed] [Google Scholar]
- 83.Zhang L, Shen ZY, Wang K, et al. C-reactive protein exacerbates epithelial-mesenchymal transition through Wnt/β-catenin and ERK signaling in streptozocin-induced diabetic nephropathy. FASEB J. 2019;33(5):6551–6563. doi: 10.1096/fj.201801865RR. [DOI] [PubMed] [Google Scholar]
- 84.Zhang NN, Zhuang MQ, Ma AX, et al. Association of levels of mannose-binding lectin and the MBL2 gene with type 2 diabetes and diabetic nephropathy. PLoS ONE. 2013;8(12):e83059. doi: 10.1371/journal.pone.0083059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Zhao LP, Wu F, Jin LG, et al. Serum CXCL16 as a novel marker of renal injury in type 2 diabetes mellitus. PLoS ONE. 2014;9(1):e87786. doi: 10.1371/journal.pone.0087786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Zhao M, Han JG. Dendrobium officinale Kimura et Migo ameliorates insulin resistance in rats with diabetic nephropathy. Med Sci Monit Basic Res. 2018;24:84–92. doi: 10.12659/MSMBR.909242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Zheng JM, Ren XG, Jiang ZH, et al. Lectin-induced renal local complement activation is involved in tubular interstitial injury in diabetic nephropathy. Clin Chim Acta. 2018;482:65–73. doi: 10.1016/j.cca.2018.03.033. [DOI] [PubMed] [Google Scholar]
- 88.Zheng SR, Coventry S, Cai L, et al. Renal protection by genetic deletion of the atypical chemokine receptor ACKR2 in diabetic OVE mice. J Diabetes Res, 2016: 5362506. 2016 doi: 10.1155/2016/5362506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Zhou HL, Tan KCB, Shiu SWM, et al. Cellular cholesterol efflux to serum is impaired in diabetic nephropathy. Diabetes Metab Res Rev. 2008;24(8):617–623. doi: 10.1002/dmrr.895. [DOI] [PubMed] [Google Scholar]
- 90.Zhou LP, Huang W, Xu YH, et al. Sweet taste receptors mediated ROS-NLRP3 inflammasome signaling activation: implications for diabetic nephropathy. J Diabetes Res, 2018:7078214. 2018 doi: 10.1155/2018/7078214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Zhu H, Yu WH, Xie YY, et al. Association of pentraxin 3 gene polymorphisms with susceptibility to diabetic nephropathy. Med Sci Monit. 2017;23:428–436. doi: 10.12659/MSM.902783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Zhu XG, Shi J, Li HC. Liquiritigenin attenuates high glucose-induced mesangial matrix accumulation, oxidative stress, and inflammation by suppression of the NF-κB and NLRP3 inflammasome pathways. Biomed Pharmacother. 2018;106:976–982. doi: 10.1016/j.biopha.2018.07.045. [DOI] [PubMed] [Google Scholar]