

Abstract
Axonemal dynein is a microtubule-based molecular motor that drives ciliary/flagellar beating in eukaryotes. In axonemal dynein, the outer-arm dynein (OAD) complex, which comprises three heavy chains (α, β, and γ), produces the main driving force for ciliary/flagellar motility. It has recently been shown that axonemal dynein light chain-1 (LC1) binds to the microtubule-binding domain (MTBD) of OADγ, leading to a decrease in its microtubule-binding affinity. However, it remains unclear how LC1 interacts with the MTBD and controls the microtubule-binding affinity of OADγ. Here, we have used X-ray crystallography and pulldown assays to examine the interaction between LC1 and the MTBD, identifying two important sites of interaction in the MTBD. Solving the LC1-MTBD complex from Chlamydomonas reinhardtii at 1.7 Å resolution, we observed that one site is located in the H5 helix and that the other is located in the flap region that is unique to some axonemal dynein MTBDs. Mutational analysis of key residues in these sites indicated that the H5 helix is the main LC1-binding site. We modeled the ternary structure of the LC1-MTBD complex bound to microtubules based on the known dynein-microtubule complex. This enabled us to propose a structural basis for both formations of the ternary LC1-MTBD-microtubule complex and LC1-mediated tuning of MTBD binding to the microtubule, suggesting a molecular model for how axonemal dynein senses the curvature of the axoneme and tunes ciliary/flagellar beating.
Keywords: dynein, molecular motor, X-ray crystallography, structural biology, protein complex, axonemal dynein, light chain, cilium, flagellum, axoneme
Introduction
Dyneins, which consist of heavy, intermediate, light intermediate, and light chains, are microtubule-based molecular motors that move along microtubules toward the minus end (1). They are divided into two groups, cytoplasmic and axonemal dyneins, in terms of their physiological functions: cytoplasmic dynein is involved in intracellular transport and cell mitosis, whereas axonemal dynein produces the driving power for ciliary/flagellar beating. The axoneme of the cilia/flagella has the 9 + 2 structure, nine doublet microtubules surrounding two central microtubules (2, 3). Axonemal dyneins have a regular arrangement and are subdivided into two groups based on their location: dynein located on the outer side of the axoneme is called outer-arm dynein (OAD),3 and that on the inner side is inner-arm dynein (IAD). Both types of dynein are supramolecular motor complex coordinated to a number of subunits composing the dynein complex, as well as to various regulatory components.
The heavy chain, which is the largest subunit in a dynein motor, possesses a principal motor function with ATPase activity. Its structure comprises several functional domains, including the tail, linker, AAA+ ring, stalk, microtubule-binding domain (MTBD), strut/buttress, and C-sequence (see Fig. 1A) (4–8). To move along a microtubule, the communication between ATP hydrolysis in the AAA+ ring, and microtubule binding at the MTBD must be properly controlled. However, the MTBD are structurally separated about 15 nm from the AAA+ ring by the long coiled-coil structure of the stalk. This long-range intramolecular communication is currently explained by the helix-sliding model, whereby a registry change occurs in the packing between two helices in the coiled-coil region of the stalk (9–11). In this model, the MTBD is thought to switch between two structural conformations named the α- and β-registries. Indeed, microtubule co-sedimentation assays have confirmed that the α- and β-registries in both cytoplasmic and axonemal dyneins show high and low microtubule-binding affinity, respectively (9, 12). In several axonemal dyneins, there is a characteristic insertion in the MTBD, which is called the flap region. The structure of the flap has been determined in the MTBD structure of dynein-c (12), where it forms a β-hairpin structure extended from the globular domain. However, the biological function of the flap region remains unclear.
Figure 1.

Architecture of the OADγ heavy chain and overall structure of the LC1-MTBD complex. A, schematic diagram of OADγ. B, crystal structure of LC1-MTBD. LC1, MTBD, the H1 helix, the flap, and the H5 helix are shown in orange, cyan, blue, pink, and yellow, respectively. Crystal structure of LC1-MTBD shown in panel B corresponds to the dotted square in panel A. Left image of panel B corresponds to an image viewing from behind panel A.
In cilium/flagellum, axonemal dyneins need to respond to curvature of microtubules accompanied with beating to move precisely. There are many axonemal dynein heavy chains with and without the flap region (e.g. 14 in human) which are periodically aligned in the axoneme. A cryo-electron tomographic study had demonstrated that axonemal dyneins showed their different activity states in flagellum corresponding to the microtubule bending (13). However, it is not clear what the functional role of each specific axonemal dynein is, which displayed different motor properties in vitro observed in gliding assays (14, 15).
The other dynein chains are called accessory chains and play regulatory roles in the cargo-binding or ATPase activity of the heavy chain. Most of the dynein accessory chains are considered to interact with the N-terminal tail domain of the heavy chain. The axonemal dynein light chain-1 (LC1) from Chlamydomonas reinhardtii had been assumed to be bound to the AAA+ ring of the OAD γ-heavy chain (OADγ) (16). Recently, however, it was unveiled that LC1 is bound to the MTBD of OADγ (17) (see Fig. 1A). Mutational studies have shown that expression of an LC1 mutant leads to dominant-negative effects on swimming velocity and beat frequency in Chlamydomonas reinhardtii (18) and that knockdown of LC1 leads to low beat frequency in planaria (19). LC1 is also highly conserved in vertebrates (e.g. DNAL1 in human), and mutations of LC1 causes primary ciliary dyskinesia (20, 21).
Recently, it was also demonstrated that LC1 binding to the MTBD decreases the microtubule-binding affinity of the heavy chain (17). Because the ATPase activity of the heavy chain is increased in the presence of microtubules (10), the above-mentioned physiological and biochemical analyses imply that LC1 might change the ATPase activity of the AAA+ ring of OADγ from the remote MTBD region and thereby fine-tune ciliary/flagellar beating. However, the structural basis on how LC1 can tune ATPase activity through the MTBD and stalk regions remains unclear.
Here, we have performed mutational and structural studies to investigate the interaction between LC1 and the MTBD of OADγ, identifying two important sites in the MTBD for interaction with LC1. Our findings enable us to provide the first structural model of the LC1 accessory function, showing how LC1 is able to fine-tune the ATPase activity of the heavy chain without any direct interaction with the AAA+ ring or microtubule, because of its tight binding to the MTBD. Furthermore, our structural model addresses the possible functional role of the flap region that is directly bound to LC1, leading to a unique stepping model of OADγ on the microtubule based on tethering and release of the flap region by LC1.
Results
Overall structure of the LC1-MTBD complex
We solved the crystal structure of the LC1-MTBD complex from Chlamydomonas reinhardtii at 1.7 Å resolution by X-ray crystallography (Fig. 1B and Table 1). The asymmetric unit contains one molecule each of LC1 and the MTBD, suggesting that LC1 is bound to the MTBD with a stoichiometric ratio of 1:1, as predicted previously (17). The MTBD interacted with the hydrophobic core of LC1 mainly via two regions, the H5 helix and the flap region (Fig. 1B).
Table 1.
Crystallographic data and refinement statistics
Values in parentheses are for the highest resolution shell.
| Data collection | |
| X-ray source | SPring-8 BL44XU |
| Wavelength (Å) | 0.90000 |
| Space group | P212121 |
| Unit-cell parameters (Å) | a = 44.55, b = 73.03, c = 94.56 |
| Resolution (Å) | 50.00–1.70 (1.73–1.70) |
| Completeness (%) | 92.2 (89.4) |
| Rmerge (%)a | 7.3 (76.6) |
| 〈I/σ(I)〉 | 31.7 (2.7) |
| Refinement | |
| Resolution (Å) | 39.69–1.70 |
| Rworkb/Rfreec (%) | 17.25/21.39 |
| Overall mean B-factor (2) | 17.36 |
| Ramachandran plot (%) | |
| Favored (%) | 97.57 |
| Allowed (%) | 2.43 |
| Outliers (%) | 0.0 |
| r.m.s.d., bonds (Å) | 0.006 |
| r.m.s.d., angles (°) | 0.794 |
a Rmerge(I) = Σ|I(k) − 〈I〉|/ΣI(k), where I(k) is the value of the kth measurement of the intensity of a reflection, 〈I〉 is the mean value of the intensity of that reflection, and the summation is the overall measurement.
b Rwork = Σ‖Fobs(hkl)| − |Fcalc(hkl)‖/Σ Fobs(hkl) .
c Rfree is the R factor computed for the test set of reflections that were omitted from the refinement process.
Next, we compared published NMR and X-ray structures (PDB ID: 1M9L and 5YXM) of LC1 alone (22, 23) to the newly obtained MTBD-complexed LC1 structure (Fig. 2A). The r.m.s.d. of Cα atoms between the new structure and the NMR or X-ray structure was 2.92 Å (134 aa) or 0.48 Å (194 aa), respectively, suggesting that the structure of LC1 in the MTBD complex is more similar to the X-ray structure than to the NMR structure. This is understandable; the X-ray structure is likely to mimic the complexed conformation because of overlap between interaction sites with the next crystallographic molecule and interaction sites with the MTBD (76.9% residues of shared interaction sites, Fig. S1).
Figure 2.
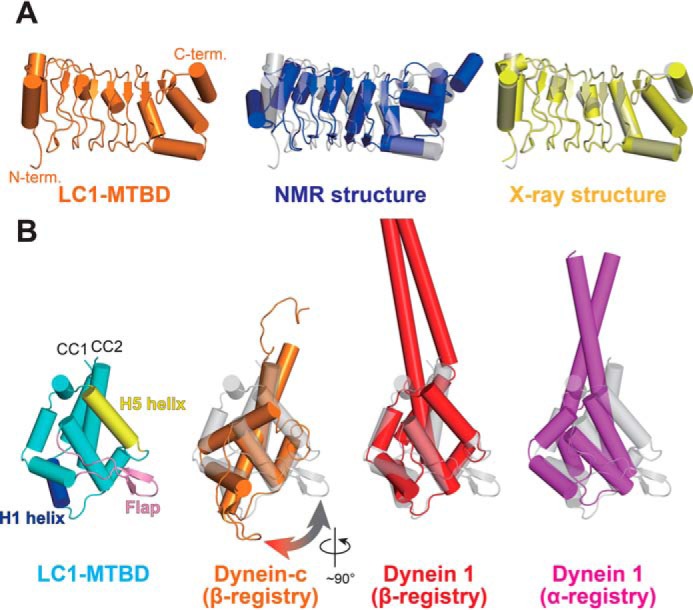
Structural comparison of LC1 and MTBD in the LC1-MTBD complex with known structures. A, structural comparison of LC1. The LC1 structure in LC-MTBD, the NMR structure (PDB ID: 1M9L), and the X-ray structure (PDB ID: 5YXM) are shown in orange, blue, and yellow, respectively. B, structural comparison of the MTBD. The MTBD structure in LC1-MTBD, that of dynein-c (PDB ID: 2RR7), that of dynein 1 in β-registry (PDB ID: 5AYH), and that of dynein 1 in α-registry (PDB ID: 3J1T) are shown in cyan, orange, red, and magenta, respectively. Please note that only the structure of dynein-c was determined by NMR spectroscopy and the cylindrical presentation of α-helices are slightly different from others because of the loosely determined main-chain torsion angles.
We also compared the structure of the MTBD in the LC1-MTBD complex to structures of the MTBD from Chlamydomonas dynein-c (PDB ID: 2RR7), mouse cytoplasmic dynein 1 (PDB ID: 5AYH), and human cytoplasmic dynein 1 (PDB ID: 3J1T) (Fig. 2B). Surprisingly, the conformations of the axonemal dynein-specific flap region differed significantly between the LC1-complexed MTBD and the single MTBD of dynein-c. In the LC1-MTBD structure, the flap was kinked ∼90° around the axis parallel to the stalk as compared with that of dynein-c, indicating that the new conformation enables the flap to interact with LC1. We were also able to identify the structural state of the MTBD in terms of its affinity for microtubules, because the registry of the helical packing of stalk is exchangeable and linked to the microtubule-binding affinity. The r.m.s.d. of Cα atoms between the LC1-complexed MTBD and the MTBD of dynein-c or cytoplasmic dynein 1 (β-registry) was 2.13 Å (117 aa) or 1.74 Å (118 aa), respectively (11, 12). By contrast, between the complex and the MTBD of human cytoplasmic dynein 1 (α-registry) was 3.37 Å (111 aa) (24), suggesting that the LC1-complexed MTBD structure is in the β-registry, although the structure itself does not contain the coiled-coil region showing the actual registry of the helical packing.
The LC1 molecule alone possesses conformational flexibility as suggested by NMR, but it is clear from the complex structure that LC1 fixes the MTBD conformation to the β-registry if no structural or nucleotide-based restraints exist. This is compatible with the previous report that LC1 binding to the MTBD decreases the microtubule-binding affinity of the heavy chain (17).
Interactions between LC1 and MTBD
Next, we investigated the interaction between LC1 and the MTBD by pulldown assay. The MTBD binds to LC1 through the H5 helix and flap region with several hydrophobic and hydrophilic interactions (Fig. 3, A and B, and Table S1). Complex formation involves a total of 41 residues (21 in LC1 and 20 in the MTBD) using a wide range of molecular surfaces. We selected 10 residues of LC1 for mutational analyses to assess the effect of electrostatic interaction (His-31 and Arg-79), hydrogen bonding (Ser-56, Thr-57, Asn-59, and Tyr-102), and hydrophobic interaction (Trp-99, Tyr-121, Met-182, and Ile-34). Mutational analysis showed that most of the mutations decreased binding activity slightly as compared with WT LC1 (Fig. 3C). Eight mutational sites (colored yellow and red in Fig. 3C) are involved in the interaction with the H5 helix and five with the flap (pink and red). Three residues (Thr-57, Arg-79, and Tyr-102), which are located in adjacent β-strands of the leucine-rich repeat in LC1, are the only residues that interact with both the H5 helix and the flap (Fig. 4).
Figure 3.

Interactions between LC1 and the MTBD. A and B, open book representation of the structure of LC1-MTBD. The interacting residues with the H5 helix (yellow), the flap (pink), both in MTBD (red), and others (magenta) are shown as stick model in A. The electrostatic potential of the surface is shown in B. C, Pulldown assay of LC1 mutants. Residues that interact with the H5 helix, the flap, and both regions of the MTBD are shown in yellow, pink, and red, respectively. The values were normalized as the ratio of difference when the value of WT is 1, and averaged as the mean value ± S.E. from three independent experiments.
Figure 4.

Detailed view of the interactions between LC1 and the MTBD. A, close-up of the interaction surface between LC1 and MTBD. LC1, MTBD, the flap, and the H5 helix are shown in orange, cyan, pink, and yellow, respectively. The residues that interact with Arg-79 are shown as stick models. B, detailed view of interactions with the flap region. The interactions were calculated by using LigPlot+.
Among the mutational sites interacting with the H5 helix, T57A and R79Q mutations led to a substantial loss of activity (Fig. 3C), implying that proper positioning of the adjacent Thr-57 and Tyr-102 residues around Arg-79 places Arg-79 in an optimal position for binding to both the H5 helix and the flap. The fact that these three residues, Thr-57, Arg-79, and Tyr-102, are conserved from Chlamydomonas reinhardtii to Homo sapiens supports our interpretation.
Among the five mutational sites (Ile-34, Thr-57, Asn-59, Arg-79, and Tyr-102) tested for interaction with the flap, only T57A and R79Q led to decreased binding, whereas the other three showed similar binding affinity to that of WT LC1 (Fig. 3C), possibly because several other strong interactions remained. Overall, this suggests that the contribution of the flap to the LC1-MTBD interaction is relatively minor relative to that of the H5 helix, although Arg-79 is central to connecting the two types of interaction with the H5 helix and the flap.
Patel-King and co-workers have reported the phenotypic analysis of M182A mutant of Chlamydomonas reinhardtii (18). It is not surprising that mutation of Met-182 to Ala was least disruptive, because our mutational analysis in vitro showed almost no effect to the interaction between LC1(M182A) and MTBD as similarly as in vivo analysis.
Modeled structure of the microtubule-bound LC1-MTBD complex
Using a mid-resolution cryo-EM structure of the microtubule-cytoplasmic dynein MTBD complex (PDB ID: 3J1T) (24), we investigated how the LC1-complexed MTBD from OADγ (OADγ-MTBD) might bind to microtubules based on a hypothetically modeled structure of the ternary LC1-MTBD-microtubule complex. Using the MTBD core region of the OADγ-MTBD, the LC1-MTBD structure was superimposed onto the cryo-EM structure of the MTBD-microtubule complex (24) (Fig. 5) to predict the structure of the LC1-MTBD-microtubule complex.
Figure 5.
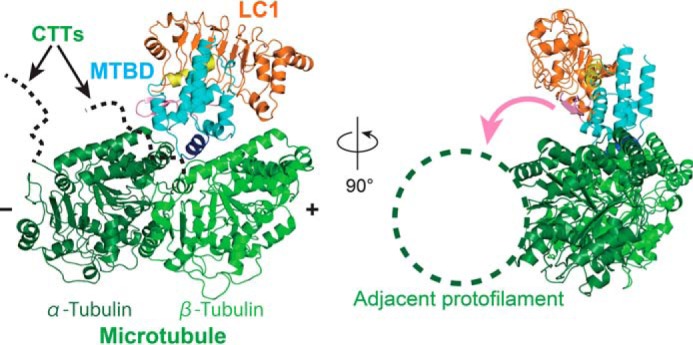
Modeled structure of the microtubule-binding LC1-MTBD complex. The crystal structure of LC1-MTBD was superimposed onto the cryo-EM structure of the MTBD-microtubule complex (PDB ID: 3J1T). Location of the CTTs invisible because of their flexibility were shown as the dotted lines in the left panel. The pink arrow shown in the right panel explain the possible path for the flap to reach the adjacent protofilament of microtubule.
In this modeled structure, the OADγ-MTBD was bound to α- and β-tubulins without any steric hindrance. Surprisingly, LC1 was located apart from the tubulins with no possibility of direct interaction with them. This is not consistent with the previous experimental results that LC1 can bind dynein-depleted axonemes with low affinity, which seemed to result from the direct binding of LC1 to microtubules (16). It should be noted that none of the reported structures contain the flexible C-terminal tail (CTT) region of tubulins with the posttranslational modification sites. Considering our structural model, it is possible that LC1 may bind to the invisible CTT regions of tubulins or to other accessory components contained in axonemes, which would position it near to the microtubules.
In the ternary model, the flap region of the OADγ-MTBD is positioned to connect LC1 and the MTBD, as mentioned above. Intriguingly, however, a relatively recent cryo-EM study reported that the coulomb map of the axonemal dynein (DNAH7) MTBD bound on the microtubule shows an additional contact with the adjacent protofilament (25). That study concluded that an extended flap region of the DNAH7-MTBD contacts the adjacent protofilament when the MTBD binds to the microtubule. Considering the cryo-EM structure together with our modeled structure of the ternary LC1-MTBD-microtubule complex, it is possible that the folded flap region in our modeled structure may be released upon binding to the microtubule and interact with the adjacent protofilament, as suggested by the cryo-EM structure of the DNAH7-MTBD bound to microtubules.
Discussion
In this study, we revealed the complex structure of LC1 bound to the MTBD of OADγ by X-ray crystallography and identified two interaction sites of MTBD for LC1, the H5 helix, and the flap region. In addition to the structural analysis, we performed a structure-based mutational analysis using a pulldown assay and 10 LC1 mutants to evaluate the importance of these two sites. The interaction surface and points between LC1 and the MTBD are so wide (buried surface area: 1021.1 Å2) and intense (Fig. 3A) that the effects of a single mutation were not marked; however, mutation of Arg-79 in LC1, which is positioned between the H5 and flap interaction sites of the MTBD (Fig. 4A), weakened the interaction between the two proteins. We assume that Arg-79 may help in coordinating interactions by the H5 helix and the flap in the MTBD. In fact, Arg-79 and its surrounding residues, such as Thr-57 and Tyr-102, are well-conserved from Chlamydomonas reinhardtii to Homo sapiens as mentioned above. The residues of MTBD interacting with LC1 were not entirely conserved in the other axonemal MTBDs (Fig. S2). This result might be a reason why LC1 binds only to OADγ.
Our study indicates that LC1 is not able to bind directly to the microtubule track in its current binding geometry. Here it should be noted that LC1 is also able to interact with the MTBD stably even in α-registry. When we screened suitable constructs for crystallization, we prepared several variants with different coiled-coil lengths, and one of which fixed the α- or β-registry by the stable artificial coiled-coil at the base. The construct artificially fixed in the α-registry was designed as similarly as the case of previous cryo-EM analysis (26). Our preliminary results showed that LC1 could also bind to MTBD molecules that had been artificially fixed in the α-registry; this is not surprising because our documented interactions in this study between LC1 and the MTBD were broad and intense, as pointed out above. How, then, can LC1 alter the MTBD binding affinity and ultimately tune cilia/flagella activity? The clear structural difference in the MTBD flap region between our complex structure and the dynein-c structure (Fig. 2B) may be central to discussions of the basis for the regulatory function of LC1.
To examine whether the interactions between LC1 and the flap region observed in this study are conserved or not, the amino acid sequences of various MTBDs of OADγ were aligned and analyzed (Fig. S3). We found that the interaction sites in the flap region of OADγ are not conserved except for Asp-1681 near the root of the flap (Fig. 3A), suggesting that the interaction of LC1 with the H5 helix is physiologically dominant, whereas that with the flap region is relatively minor. This hypothesis is consistent with the ratio of residues involved in the two sites of interaction between LC1 and MTBD (19 residues for the H5 helix, and 14 residues for the flap region). These results may suggest that the interactions between the flap and LC1 are specific but also relatively weak and easily cancelled, consequently tethering or releasing the flap to and from the MTBD. These two structural states of the flap may be a key to discuss about the physiological functions of LC1. In addition, a recent cryo-EM study showed that the extended flap of the DNAH7-MTBD contacts the adjacent protofilament (25). Thus, in certain conditions, the flap released from the MTBD might be able to bind to the adjacent protofilament in the same manner of DNAH7 (Fig. 5).
To summarize newly obtained and already published information together, LC1 is not directly bound to the microtubule with enough space left between the microtubule track and LC1, and may interact remotely with the CTTs of tubulins (I). The binding of LC1 to MTBD is so tight that LC1 would bind to the MTBD permanently through the main interaction with the H5 helix (II), but the extended flap region of DNAH7 (human axonemal dynein which does not bind to LC1) can bind to the adjacent protofilament and strengthen the interaction with the microtubule (III). LC1 can alter the binding affinity of MTBD to the microtubule weaker (IV) and ultimately tune cilia/flagella activity (V). Based on these five pieces of information collectively, we propose a new structural mechanism in which LC1 may act as a regulatory switch that tethers or releases the flap to or from the MTBD in accordance with the local curvature of the microtubule for appropriate stepping of OADγ (Fig. 6). The regulatory stepping mechanism of the OADγ-MTBD would proceed as follows. (i) When the OADγ-MTBD is not bound to the microtubule, LC1 tethers the flap region as observed in the current X-ray structure. (ii) When the OADγ-MTBD binds to the microtubule in a nucleotide-dependent manner, LC1 is still tethering the flap from the MTBD to prevent it from reaching to the microtubule and making the interaction stronger. (iii) If the microtubule becomes closer to the flap tethered by LC1 depending on its local curvature (upper right panel of Fig. 6), the flap is released probably because of much stronger interaction against the adjacent protofilament of microtubule. (iv) After the power stroke accompanying the ATPase cycle in the AAA+ ring domain, the OADγ-MTBD detaches from the microtubule track and returns to the state in (i). In this model, LC1 is able to prevent the flap being involved in the additional interaction with the microtubule, depend on its local curvature, and eventually weaken the relative affinity to the microtubule compared with the case in absence of LC1.
Figure 6.
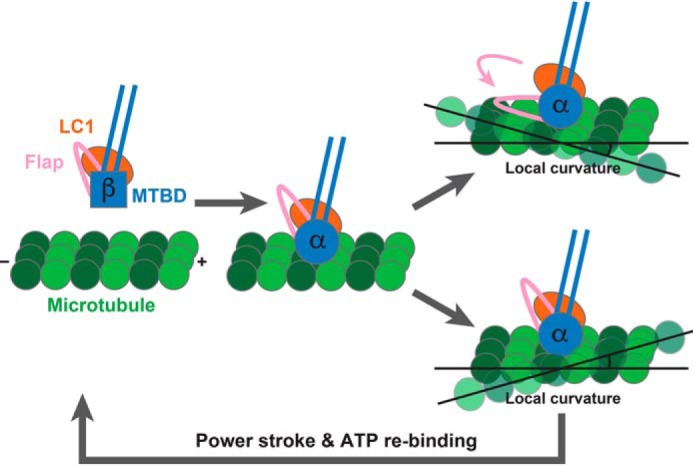
Proposed model of curvature sensing by LC1.
Our model may be partly supported by a previous in vitro 3D motility assay of microtubule sliding driven by OADα of Tetrahymena (OADα, β, and γ from Tetrahymena correspond to OADγ, β, and α from Chlamydomonas) (15). All OADs of Tetrahymena caused a clockwise rotation of each sliding microtubule around its longitudinal axis but only OADα (corresponding to Chlamydomonas OADγ) can tune the clockwise screwing pitch, whereas the OADβγ complex did not display any sensitivity. LC1 would be a regulatory protein to switch the additional microtubule-binding site of OADγ on or off, resulting in a rotational sliding of microtubule in gliding assay. To confirm our proposed mechanistic model of curvature sensing in vivo, functional studies will be undoubtedly needed.
Experimental procedures
Construct
The GST-tagged LC1 gene (UniProtKB: Q9XHH2, 1–198 aa) from Chlamydomonas reinhardtii and the His-tagged MTBD region (1614–1757 aa) of the OADγ heavy chain gene (UniProtKB: A8JFP1) from Chlamydomonas reinhardtii were inserted into a modified pET-17b plasmid with a linker sequence (agaaataattttgtttaactttaagaaggagatatacat) between the stop codon of LC1 and MTBD for bi-cistronic expression. For the pulldown assay, stalk region (1542–1832 aa) of the OADγ was used instead of the MTBD in the same construct, and the mutations were introduced by overlap extension PCR.
Expression and purification
The plasmid was transformed into BL21(DE3) cells. Cells were precultured at 37 °C overnight, transferred to 1.5 liters of modified LB medium (27) and cultured at 37 °C overnight. The cells were harvested by centrifugation and stored at −80 °C. Frozen cells obtained from 4.5 liters of culture were resuspended in 150 ml of buffer A (50 mm Tris-HCl, pH 8.0, 50 mm NaCl) supplemented with 1 mm benzamidine hydrochloride, 1 mm 6-aminocaproic acid, and 0.1 mm PMSF. The cells were lysed by sonication and the lysate was clarified by ultracentrifugation at 200,000 × g for 30 min. The supernatant was mixed with Ni-IMAC agarose (Bio-Rad) and then incubated at 4 °C for 30 min. The agarose was washed with 5 column volumes of buffer B (50 mm Tris-HCl, pH 8.0, 50 mm NaCl, 10 mm imidazole-HCl) and eluted by buffer C (50 mm Tris-HCl, pH 8.0, 50 mm NaCl, 300 mm imidazole-HCl). The eluted sample was mixed with 1/70 (w/w) TEV protease to remove affinity tags and the buffer was exchanged to buffer D (10 mm Tris-HCl, pH 8.0, 50 mm NaCl) by dialysis at 20 °C overnight. The sample was passed through Ni-IMAC agarose (Bio-Rad) and Glutathione Sepharose 4B (Bio-Rad), and then loaded onto a HiLoad 16/600 Superdex 200 pg column (GE Healthcare) equilibrated in buffer D. Fractions containing LC1-MTBD were pooled and concentrated to 70 mg/ml using an Amicon Ultra-4 Centrifugal Filter Unit (10 kDa). The sample was stored at −80 °C until use.
Crystallization and data collection
LC1-MTBD crystals were grown at 4 °C by the sitting drop vapor-diffusion method, in which 200 nanoliters of LC1-MTBD (70 mg/ml in buffer D) was mixed with an equal volume of reservoir solution (20% PEG3350, 0.2 m sodium phosphate monobasic). The crystals were soaked in a cryo-protectant solution (PEG3350 and sodium phosphate monobasic added to buffer D to give 35% and 0.2 m, respectively) for several seconds, and then flash-cooled in liquid nitrogen. The X-ray diffraction experiment was performed at the beamline of BL44XU, SPring-8.
Structure determination
The diffraction images were processed by using HKL2000 software (28). The structure was solved by molecular replacement with Phaser-MR in the PHENIX program suite (29) using the LC1 structure (PDB ID: 5YXM) as a starting model (23). Structure refinement was performed by using phenix.refine in the PHENIX program suite (29). The final structure was validated using MolProbity (30). The crystallographic data and refinement statistics are summarized in Table 1.
Structural analysis
For structural comparisons of LC1s and MTBDs, the structures were superimposed using the secondary structure matching function by the program LSQKAB in the CCP4 package (31) calculating the r.m.s.d. values. For the ternary modeled structure of the LC1-MTBD bound to the microtubule, the MTBD molecule of the LC1-MTBD complex structure (this study) and the MTBD-microtubule complex structure (PDB ID: 3J1T) were superimposed using the secondary structure matching function by the program LSQKAB (31). The protein-protein interactions were quantitatively analyzed by LigPlot+ (32).
Pulldown assay
The transformed cells were cultured in 2 ml of modified LB medium at 37 °C for overnight, and then harvested by centrifugation. Cells from 1 ml of culture were resuspended in 0.5 ml of buffer A and lysed by sonication. The lysate was clarified by centrifugation at 200,000 × g for 10 min. The supernatant was mixed with 10 μl of Ni-IMAC agarose (Bio-Rad) and then incubated at 4 °C for 20 min. The agarose was washed three times with 400 μl of buffer A. Samples were eluted by adding 20 μl of buffer C. The eluted samples were mixed with 4× SDS sample buffer and subjected to SDS-PAGE. The gels were stained by Coomassie Brilliant Blue Stain One Super (nakalai tesque). Band intensities were measured by Quantity One software (Bio-Rad), and analyzed by SigmaPlot 13 (Systat Software, Inc.).
Accession number
The coordinates and structure factors for LC1-MTBD have been deposited in the worldwide Protein Data Bank under accession number 6L4P.
Author contributions
A. T., Y. N., H. T., T. Y., and G. K. conceptualization; A. T. and H. T. data curation; A. T. and H. T. formal analysis; A. T. and G. K. supervision; A. T. and G. K. funding acquisition; A. T. validation; A. T. investigation; A. T. visualization; A. T. methodology; A. T. writing-original draft; A. T., Y. N., H. T., and G. K. project administration; A. T., T. Y., and G. K. writing-review and editing; T. Y. resources.
Supplementary Material
Acknowledgments
We thank the staffs of beamline BL44XU at SPring-8 for helping with data collection.
This work was supported by a Grant-in-Aid for JSPS Fellows Number 17J07553 (to A. T.) and a Grant-in-Aid for Scientific Research B number 18H02390 (to G. K.). The authors declare that they have no conflicts of interest with the contents of this article.
This article contains Figs. S1–S3 and Table S1.
The atomic coordinates and structure factors (code 6L4P) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- OAD
- outer-arm dynein
- IAD
- inner-arm dynein
- MTBD
- microtubule-binding domain
- LC
- light chain
- aa
- amino acids
- CTT
- C-terminal tail
- buffer A
- 50 mm Tris-HCl, pH 8.0, 50 mm NaCl
- buffer C
- 50 mm Tris-HCl, pH 8.0, 50 mm NaCl, 300 mm imidazole-HCl
- buffer D
- 10 mm Tris-HCl, pH 8.0, 50 mm NaCl
- r.m.s.d.
- root mean square deviation.
References
- 1. Roberts A. J., Kon T., Knight P. J., Sutoh K., and Burgess S. A. (2013) Functions and mechanics of dynein motor proteins. Nat. Rev. Mol. Cell Biol. 14, 713–726 10.1038/nrm3667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bui K. H., Sakakibara H., Movassagh T., Oiwa K., and Ishikawa T. (2008) Molecular architecture of inner dynein arms in situ in Chlamydomonas reinhardtii flagella. J. Cell Biol. 183, 923–932 10.1083/jcb.200808050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Pigino G., Maheshwari A., Bui K. H., Shingyoji C., Kamimura S., and Ishikawa T. (2012) Comparative structural analysis of eukaryotic flagella and cilia from Chlamydomonas, Tetrahymena, and sea urchins. J. Struct. Biol. 178, 199–206 10.1016/j.jsb.2012.02.012 [DOI] [PubMed] [Google Scholar]
- 4. Carter A. P., Cho C., Jin L., and Vale R. D. (2011) Crystal structure of the dynein motor domain. Science 331, 1159–1165 10.1126/science.1202393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kon T., Sutoh K., and Kurisu G. (2011) X-ray structure of a functional full-length dynein motor domain. Nat. Struct. Mol. Biol. 18, 638–642 10.1038/nsmb.2074 [DOI] [PubMed] [Google Scholar]
- 6. Kon T., Oyama T., Shimo-Kon R., Imamula K., Shima T., Sutoh K., and Kurisu G. (2012) The 2.8 Å crystal structure of the dynein motor domain. Nature 484, 345–350 10.1038/nature10955 [DOI] [PubMed] [Google Scholar]
- 7. Schmidt H., Zalyte R., Urnavicius L., and Carter A. P. (2015) Structure of human cytoplasmic dynein-2 primed for its power stroke. Nature 518, 435–438 10.1038/nature14023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Schmidt H., Gleave E. S., and Carter A. P. (2012) Insights into dynein motor domain function from a 3.3-Å crystal structure. Nat. Struct. Mol. Biol. 19, 492–497 10.1038/nsmb.2272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Gibbons I. R., Garbarino J. E., Tan C. E., Reck-Peterson S. L., Vale R. D., and Carter A. P. (2005) The affinity of the dynein microtubule-binding domain is modulated by the conformation of its coiled-coil stalk. J. Biol. Chem. 280, 23960–23965 10.1074/jbc.M501636200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kon T., Imamula K., Roberts A. J., Ohkura R., Knight P. J., Gibbons I. R., Burgess S. A., and Sutoh K. (2009) Helix sliding in the stalk coiled coil of dynein couples ATPase and microtubule binding. Nat. Struct. Mol. Biol. 16, 325–333 10.1038/nsmb.1555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Nishikawa Y., Inatomi M., Iwasaki H., and Kurisu G. (2016) Structural change in the dynein stalk region associated with two different affinities for the microtubule. J. Mol. Biol. 428, 1886–1896 10.1016/j.jmb.2015.11.008 [DOI] [PubMed] [Google Scholar]
- 12. Kato Y. S., Yagi T., Harris S. A., Ohki S. Y., Yura K., Shimizu Y., Honda S., Kamiya R., Burgess S. A., and Tanokura M. (2014) Structure of the microtubule-binding domain of flagellar dynein. Structure 22, 1628–1638 10.1016/j.str.2014.08.021 [DOI] [PubMed] [Google Scholar]
- 13. Lin J., and Nicastro D. (2018) Asymmetric distribution and spatial switching of dynein activity generates ciliary motility. Science 360, eaar1968 10.1126/science.aar1968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kikushima K., and Kamiya R. (2008) Clockwise translocation of microtubules by flagellar inner-arm dyneins in vitro. Biophys. J. 94, 4014–4019 10.1529/biophysj.107.123083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Yamaguchi S., Saito K., Sutoh M., Nishizaka T., Toyoshima Y. Y., and Yajima J. (2015) Torque generation by axonemal outer-arm dynein. Biophys. J. 108, 872–879 10.1016/j.bpj.2014.12.038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. King S. M., and Patel-King R. S. (2012) Functional architecture of the outer arm dynein conformational switch. J. Biol. Chem. 287, 3108–3122 10.1074/jbc.M111.286211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ichikawa M., Saito K., Yanagisawa H.-A., Yagi T., Kamiya R., Yamaguchi S., Yajima J., Kushida Y., Nakano K., Numata O., and Toyoshima Y. Y. (2015) Axonemal dynein light chain-1 locates at the microtubule-binding domain of the γ heavy chain. Mol. Biol. Cell. 26, 4236–4247 10.1091/mbc.e15-05-0289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Patel-King R. S., and King S. M. (2009) An outer arm dynein light chain acts in a conformational switch for flagellar motility. J. Cell Biol. 186, 283–295 10.1083/jcb.200905083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Rompolas P., Patel-King R. S., and King S. M. (2010) An outer arm dynein conformational switch is required for metachronal synchrony of motile cilia in planaria. Mol. Biol. Cell. 21, 3669–3679 10.1091/mbc.e10-04-0373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Mazor M., Alkrinawi S., Chalifa-Caspi V., Manor E., Sheffield V. C., Aviram M., and Parvari R. (2011) Primary ciliary dyskinesia caused by homozygous mutation in DNAL1, encoding dynein light chain 1. Am. J. Hum. Genet. 88, 599–607 10.1016/j.ajhg.2011.03.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Horváth J., Fliegauf M., Olbrich H., Kispert A., King S. M., Mitchison H., Zariwala M. A., Knowles M. R., Sudbrak R., Fekete G., Neesen J., Reinhardt R., and Omran H. (2005) Identification and analysis of axonemal dynein light chain 1 in primary ciliary dyskinesia patients. Am. J. Respir. Cell Mol. Biol. 33, 41–47 10.1165/rcmb.2004-0335OC [DOI] [PubMed] [Google Scholar]
- 22. Wu H., Blackledge M., Maciejewski M. W., Mullen G. P., and King S. M. (2003) Relaxation-based structure refinement and backbone molecular dynamics of the dynein motor domain-associated light chain. Biochemistry 42, 57–71 10.1021/bi026762j [DOI] [PubMed] [Google Scholar]
- 23. Toda A., Tanaka H., and Kurisu G. (2018) Structural atlas of dynein motors at atomic resolution. Biophys. Rev. 10, 677–686 10.1007/s12551-018-0402-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Redwine W. B., Hernandez-Lopez R., Zou S., Huang J., Reck-Peterson S. L., and Leschziner A. E. (2012) Structural basis for microtubule binding and release by dynein. Science 337, 1532–1536 10.1126/science.1224151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lacey S. E., He S., Scheres S. H., and Carter A. P. (2019) Cryo-EM of dynein microtubule-binding domains shows how an axonemal dynein distorts the microtubule. Elife 8, e47145 10.7554/eLife.47145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Uchimura S., Fujii T., Takazaki H., Ayukawa R., Nishikawa Y., Minoura I., Hachikubo Y., Kurisu G., Sutoh K., Kon T., Namba K., and Muto E. (2015) A flipped ion pair at the dynein-microtubule interface is critical for dynein motility and ATPase activation. J. Cell Biol. 208, 211–222 10.1083/jcb.201407039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Nair R., Salvi P., Banerjee S., Raiker V. A., Bandyopadhyay S., Soorapaneni S., Kotwal P., and Padmanabhan S. (2009) Yeast extract mediated autoinduction of lacUV5 promoter: An insight. N. Biotechnol. 26, 282–288 10.1016/j.nbt.2009.08.002 [DOI] [PubMed] [Google Scholar]
- 28. Otwinowski Z., and Minor W. (1997) Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 276, 307–326 10.1016/S0076-6879(97)76066-X [DOI] [PubMed] [Google Scholar]
- 29. Adams P. D., Grosse-Kunstleve R. W., Hung L.-W., Ioerger T. R., McCoy A. J., Moriarty N. W., Read R. J., Sacchettini J. C., Sauter N. K., and Terwilliger T. C. (2002) PHENIX: Building new software for automated crystallographic structure determination. Acta Crystallogr. D Biol. Crystallogr. 58, 1948–1954 10.1107/S0907444902016657 [DOI] [PubMed] [Google Scholar]
- 30. Lovell S. C., Davis I. W., Arendall W. B. 3rd, de Bakker P. I. W., Word J. M., Prisant M. G., Richardson J. S., and Richardson D. C. (2003) Structure validation by Cα geometry: φ,ψ and Cβ deviation. Proteins 50, 437–450 10.1002/prot.10286 [DOI] [PubMed] [Google Scholar]
- 31. Collaborative Computational Project, Number 4. (1994) The CCP4 suite: Programs for protein crystallography. Acta Crystallogr. D Biol. Crystallogr. 50, 760–763 10.1107/S0907444994003112 [DOI] [PubMed] [Google Scholar]
- 32. Laskowski R. A., and Swindells M. B. (2011) LigPlot+: Multiple ligand–protein interaction diagrams for drug discovery. J. Chem. Inf. Model. 51, 2778–2786 10.1021/ci200227u [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.