

Abstract
Hydroxylation of substituted phenols by flavin-dependent monooxygenases is the first step of their biotransformation in various microorganisms. The reaction is thought to proceed via electrophilic aromatic substitution, catalyzed by enzymatic deprotonation of substrate, in single-component hydroxylases that use flavin as a cofactor (group A). However, two-component hydroxylases (group D), which use reduced flavin as a co-substrate, are less amenable to spectroscopic investigation. Herein, we employed 19F NMR in conjunction with fluorinated substrate analogs to directly measure pKa values and to monitor protein events in hydroxylase active sites. We found that the single-component monooxygenase 3-hydroxybenzoate 6-hydroxylase (3HB6H) depresses the pKa of the bound substrate analog 4-fluoro-3-hydroxybenzoate (4F3HB) by 1.6 pH units, consistent with previously proposed mechanisms. 19F NMR was applied anaerobically to the two-component monooxygenase 4-hydroxyphenylacetate 3-hydroxylase (HPAH), revealing depression of the pKa of 3-fluoro-4-hydroxyphenylacetate by 2.5 pH units upon binding to the C2 component of HPAH. 19F NMR also revealed a pKa of 8.7 ± 0.05 that we attributed to an active-site residue involved in deprotonating bound substrate, and assigned to His-120 based on studies of protein variants. Thus, in both types of hydroxylases, we confirmed that binding favors the phenolate form of substrate. The 9 and 14 kJ/mol magnitudes of the effects for 3HB6H and HPAH-C2, respectively, are consistent with pKa tuning by one or more H-bonding interactions. Our implementation of 19F NMR in anaerobic samples is applicable to other two-component flavin-dependent hydroxylases and promises to expand our understanding of their catalytic mechanisms.
Keywords: flavin, flavoprotein, enzyme mechanism, enzyme kinetics, nuclear magnetic resonance (NMR), 19F NMR, flavin-dependent monooxygenase, pKa tuning
Introduction
Hydroxylation of aromatic compounds is important in catabolism and transformation of xenobiotics, for a wide variety of organisms. Such reactions are mostly catalyzed by metal-, pterin-, and flavin-dependent enzymes (1–5). These enzymes also metabolize drugs and thereby modify their therapeutic effects (4). Enzymatic hydroxylation reactions are important for synthesis of fine chemicals and have been employed in various biocatalytic applications (6–10). The enzymes are able to activate molecular oxygen (O2) and control the fate of the resulting reactive oxygen intermediates. Flavin-dependent monooxygenases catalyze aromatic hydroxylation by forming a reactive flavin–hydroperoxide adduct that can add a hydroxyl group to aromatic rings (11). Understanding how hydroxylases control the reactivity of activated O2 is both fundamentally important and potentially valuable for improving the utility of hydroxylases for practical applications.
There are two broad categories of flavin-dependent aromatic hydroxylases: single-component hydroxylases where flavin reduction and oxygenation occur on the same polypeptide chain, and two-component hydroxylases that require separate flavin reductase and substrate oxygenase components (11–13). Members of the first type utilize flavin as a cofactor, which remains bound throughout the catalytic cycle, and among the second type the oxygenase component binds reduced flavin as a substrate and releases oxidized flavin as a product. Both types of hydroxylases catalyze the reaction of reduced flavin with O2 to form a reactive C4a-hydroperoxyflavin (C4a-OOH)4 intermediate that reconciles the triplet nature of O2 with the singlet substrates and products (14, 15) and hydroxylates phenolic substrates (16).
For pyranose 2-oxidase and the oxygenase component (C2) of HPAH, density functional theory and transient kinetics indicate that reduced flavin reacts with O2 via proton-coupled electron transfer to form a radical pair consisting of flavin semiquinone and •OOH radicals, before recombination of the radical pair to form the C4a-OOH hydroperoxyflavin adduct (17–19). Subsequent hydroxylation of phenolic substrates is thought to occur via electrophilic aromatic substitution, based on theoretical calculations (20), and because the rates of hydroxylation are correlated with the favorability of flavin C4a-alkoxide formation, suggesting that this is a leaving group of the hydroxylation step (21, 22). However, recent quantum mechanical/molecular mechanical (QM/MM) calculations suggest that a hydroxyl radical–coupled electron transfer mechanism applies in 3-methylorcinolaldehyde monooxygenase from Talaromyces stipitatus (23).
Flavin-based hydroxylases are thought to activate their substrates for electrophilic attack by deprotonating them (24–30). Substrate binding to para-hydroxybenzoate hydroxylase appears to lower the substrate pKa by approximately two pH units (27, 29, 31). Similarly, substrate is deprotonated upon binding to 3-hydroxybenzoate (3HB) 6-hydroxylase (3HB6H), wherein a nearby His residue is proposed to serve as the base (32). NMR studies found that substrate is (partially) deprotonated to the phenolate when bound to reduced phenol hydroxylase (33). Similarly, 2-methyl-3-hydroxypyridine-5-carboxylate oxygenase appears to preferentially bind its substrate as the zwitterionic form in which the phenol group is deprotonated (34). Thus, stabilization of bound phenolates is well-established among single-component hydroxylases, and these therefore provide good systems in which to validate new methods for detecting and quantifying pKa depression applied to substrate upon binding. However, novel approaches are sorely needed to investigate two-component hydroxylases.
Much less is known about two-component hydroxylases because they bind substrates when their flavin is reduced and spectroscopically muted. Nevertheless, a low substrate pKa was found to correlate with rapid hydroxylation by HadA (a dehalogenating monooxygenase), implying that substrate deprotonation is a rate-contributing step (35). Similarly, the phenolic pKa of substrate bound to C2 may be unusually low (Fig. 1) because the hydroxylation rate constant and product yield are independent of pH from pH 6 to 10 (36). Active-site mutants that preserve positive charge at the position of His-120 retain activity consistent with a mechanism involving the phenolate form of substrate (37). However, quantitative studies of the effect have yet to be performed. There is a need for a method able to discern the protonation state of substrate when bound in the enzyme-active site, while maintaining an inert atmosphere.
Figure 1.

Proposed mechanism for electrophilic aromatic substitution as conducted by C2 on its substrate 4HPA (11, 36, 37, 74).
Ideally, one would like to know the pKa value of a substrate in the presence of the flavin C4a-hydroperoxide; however, the latter is very short-lived in the presence of substrate. As a proxy, we have studied the FMNH−-bound state of the C2 oxygenase, in which we can assume that the enzyme has adopted the conformation in which formation and reaction of the hydroperoxide occurs.
To directly observe the influence of the active-site environment on the pKa values of bound substrates, we used 19F NMR. 19F NMR provides high sensitivity comparable with 1H NMR, but with superior responsiveness to changes in the environment as well as effects of deprotonation of the fluorinated molecule itself (38). Moreover, with 19F incorporated only in the substrate analogs, the 19F NMR spectra are not complicated by contributions from the protein or the buffer, so we could replicate conditions under which turnover could occur if O2 were provided, without regard for the complexity of the 1H spectra (Fig. S1A). Although individual pKa values of the mono-fluorinated substrates are depressed somewhat by their fluorine substitutions, this should not greatly affect the change in pKa value associated with binding to enzyme. However, it is important to check that use of the analog does not result in mechanistic changes (39, 40).
We validated the method by applying it to the fluorinated substrate 4F3HB that binds to the single-component hydroxylase 3HB6H. We then used it to characterize the state of the substrate 3F4HPA when bound to the C2 component of HPAH. By using internal pH indicators monitored by 1H NMR, we eliminated use of a pH electrode. This, in turn, enabled the use of a septum-sealed anaerobic NMR tube and observation of the reduced (FMNH−-bound) state of C2, which is the state in which FMN binds to the protein (41). Because the NMR chemical shift indicators are small molecules, they can easily be identified by their sharp lines despite the presence of equal or greater concentrations of protons from the protein (Fig. S1A). This combination of 19F NMR to observe the bound substrate, with 1H NMR to measure the pH (Fig. S1B), permits monitoring of pH titrations of bound (and free) ligand to test the hypothesis that the pKa will be depressed upon binding to the enzyme.
The results indicate that the phenolate state of 4F3HB is stabilized by 1.6 pH units upon binding to 3HB6H, consistent with prior work (32). The method was then applied to a two-component hydroxylase, which proved more complicated. The dissociation constant describing 3F4HPA binding to C2 was found to be strongly pH-dependent consistent with preferential binding of the phenolate state to protonated protein and a pKa of 6.49 for bound 3F4HPA. However, contributions from an additional ionization event attributable to the protein were also detected via 19F chemical shifts and were tentatively assigned to an active-site histidine via specific amino acid substitutions.
Thus, a one- and a two-component hydroxylase are unified mechanistically in that both deprotonate substrate as part of binding, and we have quantified the extent to which the deprotonated form is stabilized. Although much is known about single-component hydroxylases in this regard, two-component hydroxylases are less well-documented. In this example, the exquisite sensitivity of the 19F NMR chemical shift enabled us to observe deprotonation of substrate analogs and also to infer the occurrence of additional deprotonation events in the active site. This approach can have great value in additional members of this fascinating and useful enzyme family.
Results
Single-component hydroxylase: pH dependence of 4F3HB alone
Before studying the fluorinated substrate (ligand) when bound to enzyme, we first characterized the free ligand to obtain a reference point. The 19F chemical shift of 4F3HB dissolved in water was measured at 0.3 pH unit intervals from pH 2.0 to 12.0 and plotted as a function of pH (Fig. 2). Two separate transitions could each be described by the Henderson-Hasselbalch equation (see Equation 3), yielding pKa values of 4.1 ± 0.01 and 8.7 ± 0.03 in excellent agreement with those obtained via titrations monitored by UV-visible spectrophotometry (4.1 ± 0.05 and 8.5 ± 0.01, see Fig. S2, uncertainties quoted are errors of the fits). The identities of the events responsible for the pKa values were determined by 13C NMR (Fig. 2B).
Figure 2.
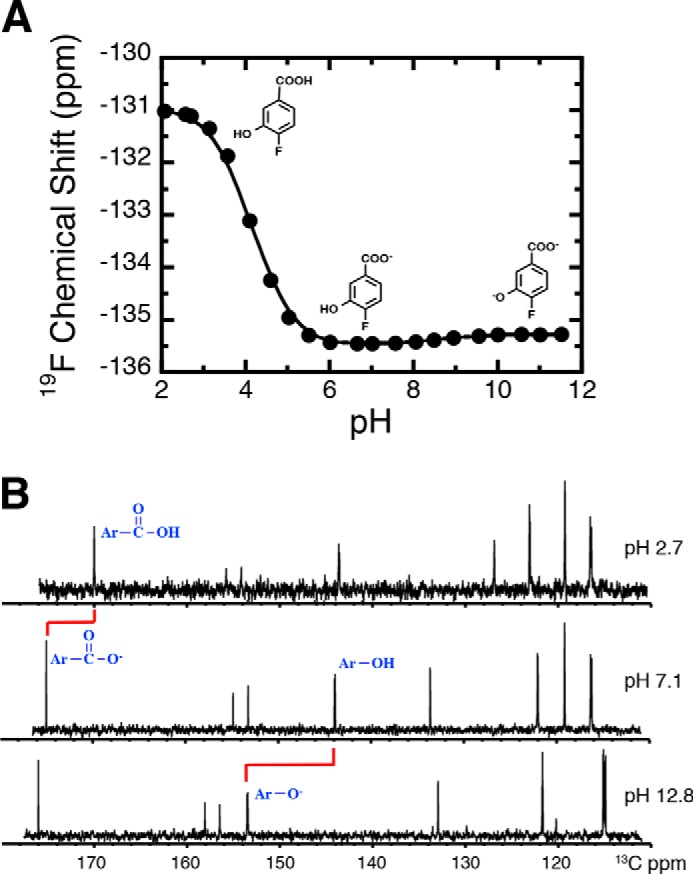
pH titration of the 19F NMR chemical shift of 4F3HB. A, fitting yielded pKa values of 4.1 ± 0.01 and 8.7 ± 0.03 for the carboxyl and phenol group, respectively. Structures of the ionization states of 4F3HB dominating in the three pH regions are shown, based on the data in B. B, 13C NMR was used to identify the functional group whose ionization state changed with each pKa. The carboxyl carbon near 180 ppm was the most affected by changing pH values from below to above the pKa of 4.2, whereas the phenolic carbon that moves from near 146 ppm to near 156 ppm was the most affected by changing the pH from below to above the pKa of 8.7. Thus, the data assign the pKa of 4.1 to deprotonation of the carboxyl and the pKa of 8.7 to deprotonation of the phenol. The signal near 135 ppm at pH 7.1 belongs to the carbon that subtends the carboxyl and the split signal near 158 ppm at pH 12.8 subtends the fluorine.
To evaluate the extent to which the presence of a fluorine affects the pKa values, we also determined the pKa values of authentic substrate, obtaining values of 4.2 ± 0.08 and 10.2 ± 0.08 (Fig. S2) in agreement with literature (pKa, 1 = 4.0 and pKa, 2 = 9.7 (32)). The carboxyl pKa value is thus relatively unaffected; however, the phenolic pKa of the 4F3HB is 1.5 pH units lower than that of 3HB, consistent with electron withdrawal from the ring by the electronegative fluorine substituent (39). Our use of 4F3HB will therefore result in a lower phenolic pKa value relative to authentic substrate, but nonetheless it provides an informative probe of effects of the 3HB6H active site on its bound substrate.
Crucially, these controls demonstrate 19F NMR's ability to detect deprotonation of each of the compound's ionizable groups and to distinguish between the two events (Fig. 2A): deprotonation of the phenol group produced a small shift of the 19F resonance to a less-negative chemical shift (0.32 ppm), and deprotonating the carboxyl group resulted in a large change in chemical shift to more negative values (−4 ppm). Thus, the 19F chemical shift displays a nuanced response not just to accumulating negative charge via the inductive effect, but also to interactions through space and ortho effects (42, 43), producing an exquisitely responsive probe of local events (38).
pH dependence of the 19F chemical shift of 4F3HB in the presence of WT 3HB6H
For 4F3HB in the presence of a 2-fold excess of WT 3HB6H at pH 6.5 (Fig. 3A), the 19F resonance was shifted −0.20 ppm to a more negative chemical shift relative to its position in the absence of enzyme, and it was significantly broadened consistent with a larger correlation time for reorientation as part of a much larger entity (44).
Figure 3.
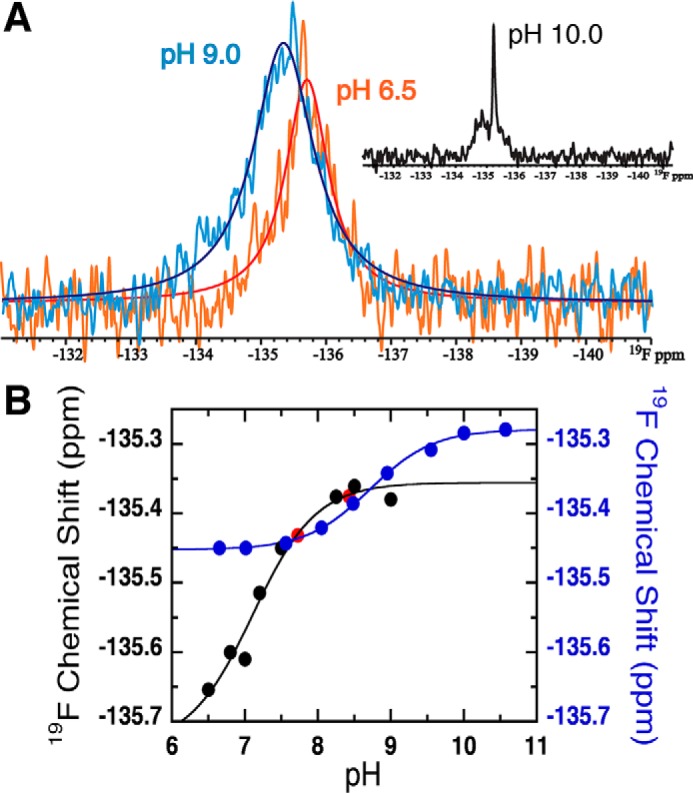
Effect of pH on the 19F signal of 4F3HB in the presence of WT 3HB6H at 4 °C. A, 19F signal of 4F3HB bound to 3HB6H at pH 6.5 (orange spectrum) and pH 9.0 (light blue spectrum), and the simulations of the NMR lines are shown in red and blue lines for the pH 6.5 and pH 9.0 spectra, respectively. Only bound ligand was observed from pH 6.0 to 9.0. However, the inset shows the 19F spectrum at pH 10.0 showing the signal of free 4F3HB at −135.24 ppm in addition to the broader signal of bound ligand (see text). B, overlay of the pH titration curves obtained for bound 4F3HB in the presence of 3HB6H (black circles) and free 4F3HB in the absence of 3HB6H (blue circles). Red dots correspond to points in the back-titration, demonstrating excellent reversibility. The solid lines show the fits of the Henderson-Hasselbalch equation (Equation 3) to points acquired in the ascending titrations, which yielded pKa values of 7.1 ± 0.2 (bound to 3HB6H) and 8.7 ± 0.03 (ligand alone).
The sample's pH was adjusted from 6.5 to 9.0 in small increments by repeated additions of 0.2 m KOH and from pH 10.0 back to 6.5 by additions of 1 m MES. At each point, the sample pH was determined from the 1H chemical shifts of the indicator molecules, and the 19F NMR spectrum of the ligand was collected. At pH values above pH 9.0, an additional sharper signal was observed (inset of Fig. 3A). This was determined to be free 4F3HB by addition of excess 4F3HB to the enzyme solution, resulting in a single stronger sharp signal at an unchanged chemical shift. Appearance of free ligand was accompanied by protein precipitation, indicating that liberation of the ligand was due to protein denaturation rather than elevation of the dissociation constant (Kd). No further measurements were made on such samples.
A plot of the chemical shift of bound 4F3HB versus pH and fitting with the Henderson-Hasselbalch equation (Equation 3) yielded a pKa of 7.1 ± 0.2 (Fig. 3B). We attribute this to ionization of the phenolic OH because it is associated with a 0.27 ppm change to a less negative chemical shift similar to the higher-pH ionization of free 4F3HB (Fig. 3B, blue curve). Thus, binding to the enzyme depressed the phenolic pKa of 4F3HB by 1.6 pH units relative to that of free 4F3HB (8.7 ± 0.03).
Binding of 4F3HB versus pH
A binding site that lowers the ligand pKa favors deprotonation of the ligand. Therefore, one predicts that the fluorinated analog with its intrinsically greater tendency to deprotonation should bind more readily than authentic substrate. Indeed, although 3HB's Kd of 0.05–0.16 mm for binding to 3HB6H (45, 46) predicts that some 60% of substrate would be protein-bound at the concentrations we used, the absence of any signal from free 4F3HB below pH 9.0 argues that this analog binds more tightly than does 3HB. This is consistent with 4F3HB's lower phenolic pKa and preferential binding of the deprotonated phenolate form.
Having confirmed the expected behavior of a single-component hydroxylase using 19F NMR, we applied the method to a two-component hydroxylase.
Two-component hydroxylase: 3F4HPA as a substrate for C2
As a model for the native substrate p-hydroxyphenylacetate (4HPA), we used 3F4HPA. This compound is itself a good substrate that reacts with the C2 component in ways that resemble those of authentic substrate. Pre-steady–state kinetics of the reaction of reduced C2 and 3F4HPA with a series of oxygen concentrations were examined by stopped-flow spectrophotometry. Absorbance changes at 380 and 450 nm (black and blue kinetic traces, respectively) revealed four phases for the reaction with 3F4HPA, similar to the reaction of C2 with 4HPA (compare Fig. 4 with Fig. 5 in Ref. 41). The first and second phases were dependent on the oxygen concentration, indicating that they reflect formation of a C4a-hydroperoxy–FMN intermediate.
Figure 4.

Reactions of C2·FMNH− complex with O2 in the presence of 3F4HPA. A, solution of C2 (100 μm), FMNH− (32 μm), and 2 mm 3F4HPA (before mixing) was mixed with buffers containing 2 mm 3F4HPA and various oxygen concentrations in a stopped-flow spectrophotometer. The reactions were monitored via the absorbance change at 380 and 450 nm (black and blue traces, respectively). Kinetic traces show four phases for A380. The first and second phases are dependent on the oxygen concentration. (1.03, 0.61, 0.31, or 0.13 mm oxygen, respectively, was present for traces shown from left to right.) The black traces highlight formation and then conversion of C4a-hydroperoxyflavin to C4a-hydroxyflavin and eventually to oxidized flavin. The third phase represents a slight increase in absorbance at 380 nm, and the fourth phase is flavin re-oxidation (highlighted in blue traces). Both these phases are independent of oxygen concentration (compare with WT data in Fig. 5 of Ref. 41). B, plots of kobs of the first and second phases versus oxygen concentrations. Second-order rate constants for the first and second phases were calculated from the slopes of the plots to be 1.16 × 106 m−1 s−1 (B, top) and 8.6 × 104 m−1 s−1 (B, bottom), respectively. C, reaction of C2/FMNH− with oxygen and 3F4HPA at various concentrations. A solution of the reduced enzyme (C2/FMNH−) was mixed with buffer containing oxygen and various concentrations of 3F4HPA in a stopped-flow spectrophotometer. The reactions were monitored via the absorbance at 380 and 450 nm, as shown with black and blue traces, respectively. After mixing, the reaction contained C2 (50 μm), FMNH− (16 μm), oxygen (0.13 mm), and various concentrations of 3F4HPA. The 450 nm kinetic traces reflect reaction with 2, 6, and 11 mm 3F4HPA, respectively, from left to right.
Figure 5.
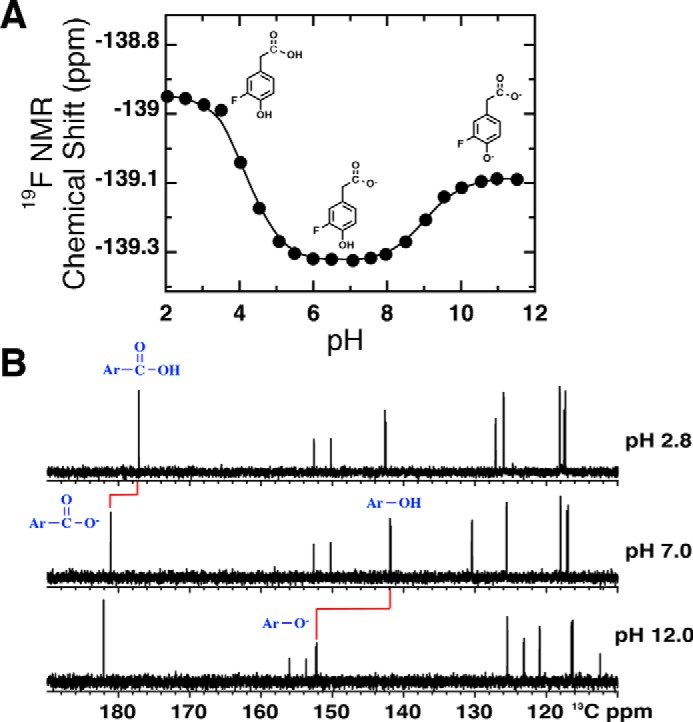
pH titration curves of 3F4HPA in the absence of WT C2 at 25 °C. A, pH dependence of the 19F chemical shift of 3F4HPA. Fitting to the Henderson-Hasselbalch equation (Equation 3) yielded pKa values of 4.2 ± 0.04 and 9.0 ± 0.02 for the carboxyl and hydroxyl group, respectively. Structures of the ionization states of 3F4HPA dominating in the three pH regions are shown, based on the data in B. B, 13C NMR determination of the events responsible for the different pKa values. The carboxyl carbon near 180 ppm is most sensitive to the pKa of 4.2, and the phenolic carbon near 140 ppm (at pH 7.0) is most sensitive to the pKa of 9.0.
The second-order rate constants of the first and second phases were calculated from the slopes of the plots of kobs versus oxygen concentration and found to be 1.16 × 106 and 8.6 × 104 m−1 s−1, respectively. These are similar to the rate constants measured for the reaction of authentic substrate with C2 (1.1 × 106 and 4.8 × 104 m−1 s−1 in the absence and presence of 4HPA, respectively (41)). Thus, the presence of the fluorinated analog decreased the rate constant of C4a-hydroperoxy–FMN formation as does the authentic substrate, and it appears that 3F4HPA, like 4HPA, may crowd the active site and decrease O2's access to reduced FMN (47). This further suggests that 3F4HPA binds in a position similar to that occupied by native substrate. Finally, at high concentrations, 3F4HPA inhibited dehydration of C4a-hydroxy-FMN, similar to the effect of 4HPA (Fig. 4C) (36). Thus, our pre-steady–state characterizations affirm that 3F4HPA reproduces the environment and perturbations associated with bound substrate.
pH dependence of free 3F4HPA
As a reference point for our 19F NMR studies of 3F4HPA bound to C2, we measured the 19F chemical shift of the free compound as a function of pH. Fig. 5A shows two discrete transitions that were each described by the Henderson-Hasselbalch equation (Equation 3) and yielded pKa values of 4.2 ± 0.04 and 9.0 ± 0.02, respectively, in excellent agreement with those obtained via UV-visible spectrophotometry (4.0 ± 0.05 and 8.9 ± 0.01, respectively, see Fig. S3). Similar to the case of 4F3HB, the phenolic pKa of 3F4HPA was 1.6 pH units lower than that of nonfluorinated substrate, although the carboxylic pKa was much less affected (Fig. S3A). As for 4F3HB, deprotonation of the p-hydroxyl group of 3F4HPA resulted in a small shift in the 19F resonance to less negative chemical shifts, in contrast with the effect of deprotonating the acetic acid group.
pH dependence of the 19F signal of 3F4HPA in the presence of WT HPAH-C2
To learn whether the pKa value of 3F4HPA is affected by interactions with the enzyme-active site, we compared the pH dependence of bound 3F4HPA with that of free 3F4HPA. The pH of an NMR sample of 3F4HPA with 2.5-fold excess WT-C2 and FMNH− was adjusted from 6.6 to 10.0 as described above, except that inert atmosphere was maintained. The highest pH investigated was pH 10.0 because C2 denatures above this pH. The 19F NMR spectrum exhibited a sharp signal from the unbound fraction at −139.312 ppm (pH 6.6) to −139.155 ppm (pH 10.0) in addition to a broad signal from the bound fraction at −137.959 (pH 6.6) to −139.692 ppm (pH 10.0) (Fig. 6). At pH 6.6 the bound ligand 19F chemical shift was 1.35 ppm less negative than that of the free ligand, but in the course of the titration to high pH it moved to −0.54 ppm more negative in 19F chemical shift relative to free ligand (pH 10.0). Thus, interaction with the protein had a significant effect on the ligand 19F chemical shift, and the effect had the opposite sign at high pH from at low pH, indicating that the nature of the interaction differs depending on the pH. This is in addition to, and different from, the effect of deprotonating the ligand, and therefore it suggests an additional pH dependence residing in the protein.
Figure 6.

Effect of pH on the 19F signal of 3F4HPA free or bound to WT C2·FMNH−. A, 19F spectrum of 3F4HPA at a series of pH values shows both bound (B) and free (F) 3F4HPA in the presence of WT C2 and FMNH−. pH values reported are pH 6.6 (red spectrum), pH 7.2 (blue spectrum), pH 7.8 (green spectrum), pH 8.3 (magenta spectrum), pH 8.6 (cyan spectrum), pH 9.0 (black spectrum), pH 9.4 (light blue spectrum), and pH 10.0 (orange spectrum). All spectra are presented using the same vertical scale and were collected at 25 °C. Inset B shows the pH dependence of the 19F shifts of 3F4HPA bound to C2 (black circles) or free (blue circles). The green circles represent data from back-titration and demonstrate reversibility. The solid line provides the fit to the data of the Henderson-Hasselbalch equation (Equation 3) with pKa = 8.7 ± 0.03 for bound 3F4HPA. For free 3F4HPA, a pKa of 8.9 ± 0.09 was obtained (curve not shown). Inset C plots the observed dissociation constants versus the sample pH documenting tighter binding at higher pH, as predicted for favorable interaction between the phenolate form of substrate and a cationic protein site. All data were collected under inert atmosphere.
The 19F chemical shifts of bound and free 3F4HPA were plotted against pH (Fig. 6B, black and blue, respectively). The pKa values describing each were obtained from nonlinear least-square fits using the Henderson-Hasselbalch equation (Equation 3). Back-titration by addition of HCl gave the same results as those observed by addition of KOH indicating that the spectral changes were not due to increasing ionic strength in the course of the titrations nor an irreversible event such as protein denaturation. For free 3F4HPA, the chemical shift at each pH, the shape of the titration curve, and the pKLH value of 8.9 ± 0.09 were within error of those associated with the deprotonation of the p-hydroxyl group of 3F4HPA in the absence of C2 (9.0 ± 0.02) (Fig. 5A).
The bound form of 3F4HPA behaved differently. The observed pKa value of 8.7 ± 0.03 was similar to the pKa of phenol deprotonation for free 3F4HPA, but the 19F chemical shift change associated with the event was opposite in sign and much larger (−1.78 ppm versus +0.2 ppm for free 3F4HPA, δL − δLH). Indeed, the chemical shift change was more similar to the signature of deprotonating the carboxylic acid (Fig. 5A).
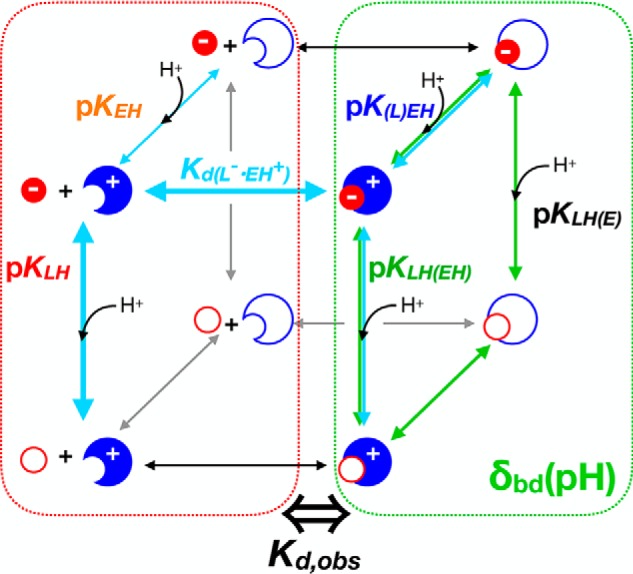
We considered the possibilities that the pKa of 8.7 represents the following: (a) deprotonation of the bound ligand's phenol (as in 3HB6H); (b) deprotonation of the bound ligand's carboxyl (based on the chemical shift signature); or (c) response of the ligand's 19F chemical shift to deprotonation of a protein residue nearby (“X”) (48). To arbitrate among the possibilities, we exploited the pH dependence of the apparent dissociation constant Kd, obs, as it provides an independent report on deprotonation events affecting the interaction between ligand and protein (Scheme 1). Because each of the pKa values of free ligand have been assigned, the pH dependence of Kd, obs can be used to test whether the pKa of 8.7 reflects either pKa of the bound ligand, where a given pKa of free ligand is pKLH, and the corresponding pKa of the bound ligand is pKLH(EH).
| (Eq. 1) |
Because binding of 3F4HPA to WT-C2 increased as the pH increased (Fig. 6A), we employed notation emphasizing binding of the deprotonated ligand, which in turn will interact more favorably with the positively-charged (protonated) enzyme active-site residue X. However, both protonation states of both groups are accounted for by the algebra employed below in Equations 4 and 5.
Scheme 1.
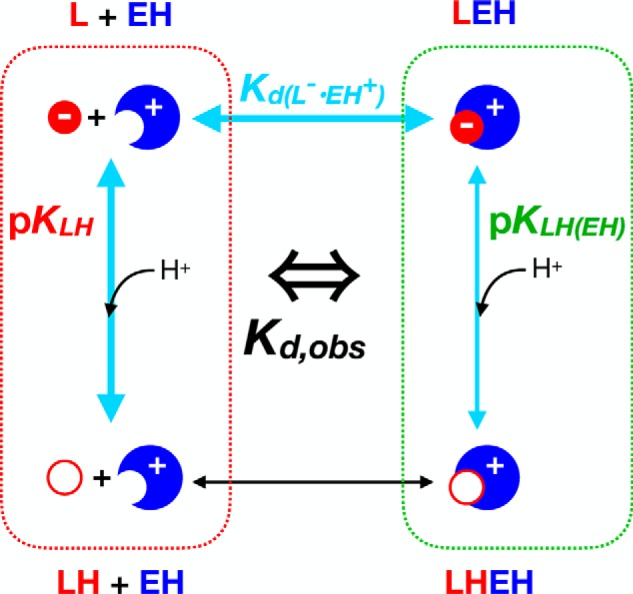
The predictions of scenarios a and b are plotted in Fig. S4 for comparison with the actual pH dependence of Kd, obs values. Neither are compatible with the data. Therefore, the simple model considering only the protonation state of the ligand is unable to account for the pKa of 8.7, and we must adopt the model that it reflects a protein residue. Thus, we now consider that there are two ionizable residues: the ligand phenol and X. This yields the expanded Scheme 2, which posits that the interaction will be favorable when the ligand is deprotonated and X is protonated (Fig. S5). X's pKa can then be extracted from the pH dependence of the chemical shift of bound ligand (pK(L)EH in Scheme 2), whereas the pKa of the bound ligand's phenol can be extracted from the pH dependence of Kd, obs (Equations 4 and 5).
Scheme 2.
Fits to the pH dependences of the chemical shift of bound ligand δobs(pH) and the Kd, obs(pH) indeed yielded two different pKa values. This is not unexpected; the chemical shift of bound 3F4HPA only reflects the states in which ligand is bound, and the Kd, obs reflects equilibria with states of free ligand and free enzyme as well (Schemes 1 and 2, and Equations 1 and 5).
A fit to the Kd, obs data assuming only one labile proton (e.g. pKa values for ligand, bound and free, Equation 1) and fixing the pKa free ligand at pKLH = 9.0 (above) display systematic deviations from the data between pH 7.0 and 10.0 (orange dashed curve in Fig. 7). Nevertheless, it replicates the observed tighter binding at higher pH values, with its convergence on a much lower pKLH(EH), and a lower pKa for bound than for free ligand (6.7 ± 0.1, versus 9.0 ± 0.02). Upon incorporating a second pKa associated with X (Equation 5), the fit to the data was excellent, replicating not only stronger binding at high pH, but also the biphasic behavior most evident near pH 8 (purple dotted curve in Fig. 7). This lends credibility to our two-residue model (i.e. our assumption of “EH” rather than “E” at pH 7 and below). Again the fit yielded a considerably depressed pKLH(EH) = 6.3 ± 0.1, versus pKLH = 9.0, which was again fixed. Regardless of whether or not a second protonation event is considered, the pKLH(EH) value that emerged was consistent with promotion of substrate deprotonation upon binding to the enzyme. However, the two-residue fit yielded a pKa of pK(L)EH = 8.1 ± 0.2 for the enzyme active-site residue, whereas the pKa obtained by fitting the 19F chemical shift of bound ligand using Equation 3 was 8.77 ± 0.03 (teal curve in Fig. 7).
Figure 7.
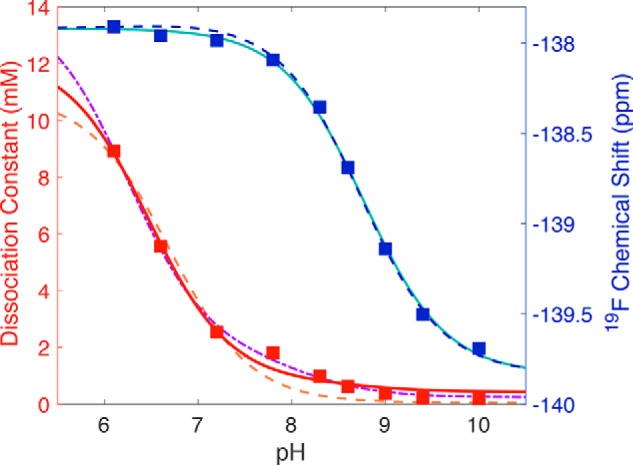
Application of model 2 to the pH dependence of the 19F chemical shift (blue squares) and the observed dissociation constant (red squares) of 3F4HPA binding to WT C2. Dissociation constants determined at different pH values using Equation 2 are plotted against pH values (red squares) and compared with the predictions of the different models. Model 1 (Scheme 1) produced the fit shown with the orange dashed line obtained using Equation 1, which yielded a high-pH limiting dissociation constant of 0.05 ± 0.01 mm for the L·EH complex and a pKLH(EH) of 6.7 ± 0.1, using the free ligand pKa of 9.0. This model provided an inadequate description of the data near pH 8.0 and so was replaced by the two-proton model (Scheme 2). The resulting fit to the data using Equation 5 better reproduced the intermediate-pH behavior (dot-dashed purple line) and yielded a limiting Kd, L·EH of 0.025 ± 0.005 mm, pKEH of free enzyme of 7.1 ± 0.2, pKLH(EH) for ligand bound to protonated enzyme of 6.2 ± 0.1, and pK(L)EH for enzyme with deprotonated ligand bound of 8.1 ± 0.2. The pH dependence of the chemical shift was well-described by the Henderson-Hasselbalch equation (Equation 3), yielding a pKa of 8.77 ± 0.03 (teal line). When the chemical shift and Kd, obs were fit simultaneously in Matlab, the intermediate pH behavior of Kd, obs was not as well-described (red solid line), but the parameters obtained from the simultaneous fit are comparable with those obtained from fits to each of chemical shift and Kd, obs alone, and the chemical shift's pH dependence was quite well-described (blue dashed line). Obtained parameters are provided in Table 1.
Simultaneous fitting to both δobs(pH) and Kd, obs(pH) provided good agreement with both (Fig. 7, blue and red curves, respectively), although the fit to Kd, obs(pH) is visibly inferior to the fit to δobs(pH). The simultaneous fit yielded a pKa for bound ligand of pKLH(EH) of 6.49 ± 0.09 (Table 1). The fact that similar values were obtained with different complexities of data sets and fits supports their validity. The 2.5 pH unit drop in ligand pKa is consistent with stabilization of phenolate upon binding. The pKa drop mirrors the 1.6 pH unit drop obtained for 4F3HB (above), although in the case of C2 it was inferred via the pH dependence of Kd, obs rather than that of δobs.
Table 1.
Summary of pKa values observed and proposed chemical events associated with them

aCarboxyl of free 3F4HPA is shown.
b(EH) indicates that the proposed nearby enzyme active-site residue is protonated.
c(E) indicates that the proposed nearby enzyme active-site residue is not protonated.
d(L) indicates that the deprotonated state of ligand is bound, and imH indicates deprotonation of the imidazolium side chain of His.
eFixing pKa of 9.0 for free 3F4HPA is shown.
Because the simultaneous fit incorporates greater diversity and quantity of data, we adopt the parameters obtained from it as superior descriptors of the system. We emphasize that the model upon which they are based is not unique, and other solutions might be found that also fit our data. Nevertheless, the model in Scheme 2 has the virtues of being based on behavior also observed in 3HB6H, mechanistic studies of both enzymes, and residues known to be present in their active sites. In particular, the pKEH value of 7.7 that emerges from the model and the data is compatible with X being a His residue.
pH dependence of the 19F signal of 3F4HPA in the presence of C2 variants H120N, H120R, and S146A
We tested hypotheses for the identity of residue X by comparing our results on WT protein (above) with experiments on single-site variants. The crystal structure suggests that the conserved Ser-146 and His-120 are close to the ligand and could favor phenol deprotonation as indicated by the pH dependence of Kd, obs (Fig. 8B) (49). Analogs of C2's His and Ser pair are found in other monooxygenase components, including those of cholesterol monooxygenase from Mycobacterium tuberculosis and chlorophenol 4-monooxygenase from Burkholderia cepacia (50, 51). Moreover, these residues have been shown to affect the catalytic parameters and their dependence on pH (37).
Figure 8.
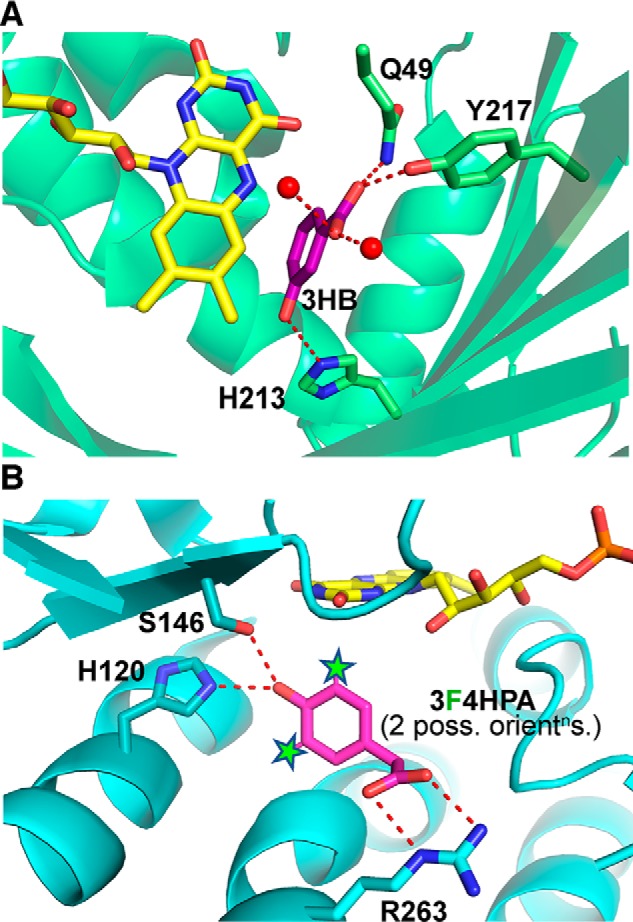
Ribbon structures of the active site of 3HB6H with 3HB bound (A) and the active site of WT-C2 with 4HPA bound and the two positions F can occupy indicated by stars (B). The 3HB6H structure was obtained by co-crystallization of the H213S variant of 3HB6H with 3HB (PDB code 4BK1) (53). UCSF's Chimera program (75) was used to model the WT His residue in place of Ser-213. The rotamer of His-213 that best overlays the Ser residue and approaches bound substrate without production of clashes was selected, and the energy of the resulting structure was minimized within Chimera. B, active site of WT-C2 co-crystallized with 4HPA is shown (PDB code 2JBT (49)). To show the locations that could be occupied by F atoms when 3F4HPA is bound, we designated both the 3 and 5 hydrogens of the 4HPA ligand as F atoms and identified them with stars to underscore that they are not experimentally determined F positions. Although the substrate analog we used has F in only one position, we do not know which of two possible orientations it might prefer when bound or whether both orientations are populated. Thus, the two “Fs” in the figure represent two possible positions of F, neglecting additional possible effects of enzyme dynamics. One of these positions is consistently ≈4 Å from the Nϵ of His-120 in our model active sites.
To test the possibility that His-120 is responsible for the pH-dependent environment sensed by bound 3F4HPA, we collected 19F spectra of 3F4HPA in the presence of H120N- and H120R-C2. These were chosen because the proteins are sufficiently stable for study by NMR, and because the H120R variant retains catalytic activity, albeit impaired (37). 19F NMR spectra collected as for WT revealed only the free form of 3F4HPA in the presence of the H120N or H120R variants. This is consistent with the Kd value for binding of 4HPA to H120N/C4a-hydroperoxyflavin of 2.2 mm versus 0.18 mm for the analogous WT-C2/C4a-hydroperoxyflavin complex (37). Thus, we confirm that His-120 aids in binding 3F4HPA. However, we could not explore the possibility that His-120 interacts directly with the F substituent of 3F4HPA.
As a control, we also studied S146A-C2, which eliminates an interaction with the phenol suggested by crystal structures (Fig. 8B) (49). 19F NMR spectroscopy revealed signals for both bound and free 3F4HPA, as for the WT-C2. The plot of the 19F chemical shift of bound 3F4HPA versus pH revealed a pKa of 8.70 ± 0.08 associated with a more negative chemical shift upon deprotonation, as for WT-C2 (Fig. S6). Thus, we can rule out Ser-146 as the source of the pKa that affects bound ligand's chemical shift. Moreover, because Ser-146 interacts with the ligand phenol, this strengthens our conclusion that the pKa of 8.7 is not due to the ligand phenol. This leaves His-120 as the remaining reasonable source of the pKa of 8.7, consistent with all the data in hand.
Single-turnover reaction of WT C2 with 3F4HPA at various pH values
To learn whether the protein residue with a pKa of 8.7 affects reactivity of the F, we turned to product analyses. The F substituent of 3F4HPA results in the molecule not being symmetric about the 1–4 axis and creates the possibility of forming two different products depending on whether hydroxylation displaces the F at position 3 or hydroxylates at position 5 (see Fig. 8B and Jadan et al. (40)). We hypothesized that the interaction affecting the ligand's 19F chemical shift could also bias the binding orientation of 3F4HPA and produce a pH-dependent ratio of products.
Product yields on the order of 25% were obtained, possibly because the electron-withdrawing F substituent diminishes 3F4HPA's reactivity as a target for electrophilic attack. Interestingly, the distribution of products obtained did not change with pH, revealing F− displacement in ∼53% of the reactions and hydroxylation at position 5 in ∼47%, over the pH range of 6.0 to 10.0 (Table S1). Thus, 3F4HPA appears able to bind in either orientation (Fig. 8B), and the absence of a change centered at pH 8.7 indicates that interaction between ionizable residue X and the F in the FMNH− state does not greatly bias the branching ratio between the two reactions that can occur. This agrees with Peelen et al. (33), who concluded that deprotonation of 3F-phenol in Trichosporon cutaneum phenol hydroxylase made the two ortho positions equally electron-rich and therefore equally subject to attack, although there was a 3-fold difference between them when the substrate was protonated (52). The pH-independent product ratio down to pH 6.0 agrees with the pH independence of the hydroxylation rate in indicating that bound substrate's pKa value is significantly lower than 6.0 in the state of the enzyme in which the reaction occurs.
The apparent discrepancy with our measured pKLH(EH) of 6.49 ± 0.09 can be because the reaction occurs in the presence of neutral C4a-hydroperoxyflavin, whereas the NMR studies were in the presence of anionic FNMH−, the charge of which would tend to raise the pKa values of nearby residues.
Discussion
Ionization state of ligand bound to 3HB6H and identities of residues affecting it
3HB6H served as a test system for validating our 19F NMR approach. The 19F chemical shift of bound 4F3HB changed as expected for deprotonation of the phenol and demonstrating a pKa depressed from 8.7 to 7.1 upon binding to enzyme. This contrasts with the earlier finding that the UV spectrum of 3HB did not appear to change significantly upon binding to enzyme at pH 8.0, arguing against deprotonation (32). However, 19F NMR provides a signal exclusive to the fluorinated ligand that is less subject to interference from other components of the sample (Fig. S1). Our pKa of 7.1 and 19F NMR spectra at pH values near 8.0 concur that bound ligand is mostly deprotonated near pH 8.0 (Fig. 3).
This study also demonstrated that replacement of His-213 with Ser eliminates active-site stabilization of the phenolate form of substrate. This is consistent with the larger Kd value reported for 3HB binding of 0.72 mm for H213S, versus the WT value of 0.15 mm (32), and it can explain the lowered (28%) efficiency with which H213S–3HB6H produces product (versus 86% for WT). Proton transfer from substrate to nearby His-213 was proposed to produce a His+·phenolate− pair in which substrate is activated for electrophilic attack (32). His-213's Nϵ2 is 3 Å from the phenolic OH of one of the substrate poses found in the H213S structure (Fig. 8A) (53). Moreover, His-213 Nδ1 is 3 Å from Nϵ2 of another His, His-363, which is itself polarized by an H-bond to the carbonyl O of Gly-359. Thus, His-213 appears to be supported by a relay of H-bonds that could prime it to abduct a proton from the substrate phenol (32, 53).
The crystal structures of 3HB6H also reveal a Cl− ion bound against the flavin re face, consistent with inhibition of 3HB6H's reactions with O2 and NADH by Cl− (53) and the ability of Cl− to act as a superoxide analog (54). Thus, we speculate that the Cl− may identify the pocket in which the hydroperoxide functional group forms. Such a location would enable the distal OH of C4a-hydroperoxide to attack the substrate carbon para to the hydroxyl group when the latter is interacting with His-213.
HPAH-C2: use of complementary observables to validate a model and extract pKa values for ligand and protein events
We exploited distinct 19F signals from bound and free ligand to determine Kd, obs as a function of pH. Kd, obs reflects states of free enzyme and ligand as well as bound states, so Kd, obs reports on ionization events additional to those affecting the ligand 19F chemical shift. The pH dependence of Kd, obs ruled out the ligand's own ionization events as sources of the pKa of 8.7 affecting the ligand chemical shift, so this pKa was assigned to a protein residue X. The resulting enlarged model (Scheme 2) explains the pH dependence of Kd, obs substantially in terms of a pKa for bound ligand of pKLH(EH) = 6.49, and it also yields a pKa value for X in the absence of ligand (pKEH) (Fig. S7). Thus, we learned the pKa values of X when bound and free, as well as the pKa values of ligand when bound and free.
The 2.5 pH unit drop in 3F4HPA's pKa upon binding to C2 demonstrates that the enzyme stabilizes the ligand's phenolate state, thereby activating it for electrophilic attack. The magnitude of the pKa shift corresponds to stabilization of the phenolate by 14 kJ/mol relative to the phenol. Similarly, the 1.6 pH unit shift in the pKa of 4F3HB upon binding to 3HB6H represents 9.1 kJ/mol stabilization of the deprotonated form relative to the protonated form.
The energies in question can derive from H-bonds, electrostatic interactions with active-site residues, and/or from effects of the polarity of the active site (34, 55). For example, the active-site Asp of α-lytic protease from Lysobacter enzymogenes has a pKa below 1.5 representing a shift of almost 3 pH units, attributed to H-bonds with backbone amides and side chains of a His and a Ser (56). Similarly, a salt bridge between two Asp residues in RNase H1 from Escherichia coli elevates the pKa of one Asp to 6.1 and depresses the pKa of the other to 2.6 (57), whereas in the β subunit of F1-ATPase from Bacillus PS-3, the active-site Glu has a pKa of 6.8, representing a 2.5 pH unit elevation (58). Thus, there are ample precedents for the sizes of pKa shifts we have measured (59), based on the same side chains as those found in C2's active site (60).
Identities of residues affecting the ionization state of substrate bound to HPAH-C2
Residue X has a pKa of 7.7 in our model, based on our data, making His-120 the most plausible candidate. Crystal structures indicate that the side chain Nϵ of His-120 is 3.0 Å from the hydroxyl group of 4HPA (Fig. 8B), where it is well-positioned to lower 4HPA's pKa via a NϵH+/phenolate− Coulomb interaction. Ser-146 is also in position to donate a hydrogen bond (Fig. 1). The flavin hydroquinone is nearby but is ruled out based on its pKa of 6 or lower, inferred from the pH-independence of the rate of reaction with O2 (Fig. 8B) (36, 61). Another His in the active site, His-396, was determined to aid formation of the C4a-hydroperoxy–FMN (19), suggesting that His-396's pKa is above 10 due to the pH independence of the reaction rate between pH 6.3 and 9.9 (61). Thus, the viable candidates are Ser-146 and His-120. Because the event and its pKa of 8.7 were retained when Ser-146 was replaced by Ala, but ligand binding was lost when His-120 was replaced, we identify X as His-120.
Only C2 variants with a positive charge at position 120 produced product (37), consistent with electrostatic stabilization of a phenolate substrate transition state. Finally, our pKa values of pKEH = 7.7 and pK(L)EH = 8.7 are comparable with pKa values measured for other His residues. For example, the active site His in α-chymotrypsin from bovine pancreas has a pKa of 7.5 that rises to ∼10.3–12.1 upon binding of substrate analogs (62).
From an energetic standpoint, a protein/ligand interaction that stabilizes the anionic state of the ligand should conversely stabilize the cationic form of the binding site. Indeed, His-120's pKa is elevated when ligand is bound, from pKEH = 7.7 to pK(LH)EH = 8.7. The magnitude of this change is only 1.0 pH unit, indicating that additional residues(s) contribute to the 2.5 pH unit depression of the ligand's pKa. Based on its position (49) and conservation (50, 51), Ser-146 is an obvious candidate, and it has been credited with positioning the substrate (37).
For S146A-C2, the pH dependence of the 19F chemical shift of bound ligand as well as its Kd, obs yielded mostly similar parameters to those obtained for WT protein (compare Fig. S6 with Fig. 7). However, pKEH of His-120 is 0.4 pH units lower in S146A-C2 than in WT, compatible with a modest hydrogen bond between His-120 and Ser-146, and the pKLH(EH) of bound ligand is also lowered to 4.8 versus 6.49 for WT. Thus, it appears that Ser-146 raises the pKa values of both His-120 and bound substrate. The latter effect may be inconsequential because pKLH(EH) is quite low in WT and S146A, but elevation of His-120's pKEH to 7.7 can improve enzyme proficiency by extending the pH range over which the active site contains a cation. The effects of Ser-146 are nevertheless small compared with the effect of the interaction between His-120 and ligand, as pK(L)EH is unchanged at 8.7 in S146A-C2.
Here, too, our results concur with the literature, as replacement of Ser-146 by Ala barely changed the Kd (to 0.32 mm at pH 8.0 versus the WT Kd of 0.35 mm (41)), and S146A-C2 can perform hydroxylation reasonably well (37), affirming a correlation between activity (36) and stabilization of the phenolate form of substrate demonstrated here.
Mechanistic implications of the residue ionization states that emerge
Our data support a model in which ligand binding to C2 causes His-120 to gain a proton at pH values between 7.7 and 8.7, while the ligand loses one. In effect, a proton is transferred from ligand to His-120 converting the two neutral functionalities to a counterion pair that stabilizes the anionic substrate (Fig. 9). However, the hydroxylation rate is pH-independent over a larger pH range, from 6.2 to 9.9 (36). This was thought to require that the pKLH(EH) of bound substrate be below 6.0 and the pK(L)EH of His-120 with substrate bound be above 10. The current analysis finds that a His-120 pK(L)EH higher than 8.7 is not necessary, because the ligand deprotonates without assistance from His-120 at higher pH values, due to its pKLH(E) of 7.5 (Fig. 9). Thus, the active site has the effect of stabilizing >25% of substrate in its deprotonated state from pH 6.0 up, whereas free substrate is >25% deprotonated only above pH 8.5.
Figure 9.
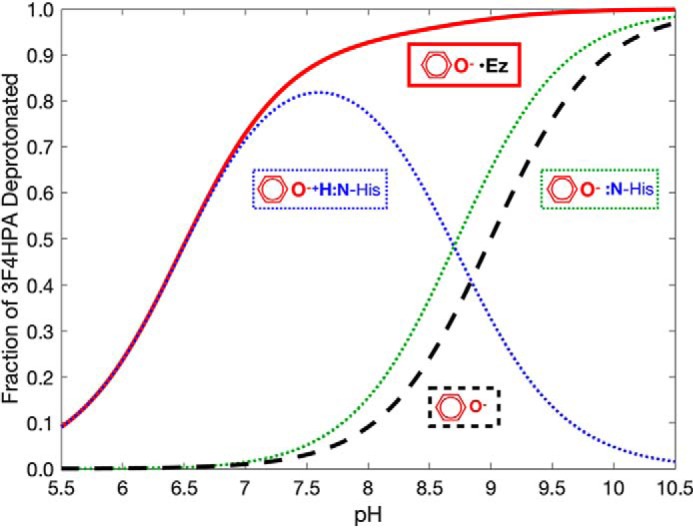
Extent of deprotonation of 3F4HPA free or bound to WT C2·FMNH− as a function of pH. The extent to which the two different states containing deprotonated 3F4HPA are populated (dotted lines), along with the total extent to which 3F4HPA is deprotonated (solid red line), based on our model and the pKa values that emerged from our fits to the data. At high pH values above 8.7, the active-site His-120 and bound 3F4HPA are both predominantly deprotonated (dotted green). At lower pH values where free 3F4HPA would normally be protonated (dashed black), the protonated state of His-120 stabilizes deprotonated 3F4HPA (dotted blue). Thus, our model indicates that enzyme-bound substrate is deprotonated to a much lower pH than free substrate (red curve versus dashed black) and that the enlarged pH span for the substrate phenolate is due to cationic His-120 (dotted blue). Our data thereby predict a substantially pH-independent catalytic rate over the pH range of 6.5 to 10.
At pH values near 6, we expect that the extent of substrate deprotonation would be greater in the state of the enzyme that actually undergoes reaction. We were constrained to study FMNH−-containing enzyme, although it is neutral C4a-hydroperoxy–flavin that participates in the rate-limiting step. The absence of anionic flavin in that case would tend to lower the pKa value of a nearby ligand, explaining why our value for pKLH(EH) is higher than the pKa <6.0 determined from kinetics (36), despite our use of a fluorinated substrate.
Broader impact of the methodology
19F-containing substrate analogs in combination with the excellent responsiveness of 19F's NMR chemical shift have afforded unique insight into the identities and energies of proton dissociation equilibria that contribute to the catalytic mechanisms of aromatic hydroxylases. There are previous examples of 19F NMR of fluorinated ligands bound to enzymes (63); however, ours stands out in having used both 19F and 1H spectra to determine the ligand protonation state and pH values in sealed samples that preserve the enzyme's redox state. Moreover, we combined the NMR with kinetic measurements to demonstrate that our 19F probe is in fact a substrate. Our approach has wide applicability to diamagnetic aromatic monooxygenases, flavin-based or not, and indeed to any enzymes for which fluorinated substrate analogs can be found and shown to mimic the behavior of authentic substrate. Because of 19F's high gyromagnetic ratio, the NMR spectra are comparable in strength to 1H spectra, so sub-millimolar concentrations of protein can be studied (38, 64, 65). However, because 19F is naturally absent from proteins, the spectra are uncomplicated by protein resonances or signals from most common buffers (Fig. S1). Because the 19F chemical shift is highly responsive to the environment (66) and there are very few distinct F atoms in the samples, the signals tend not to overlap. Thus, simple one-dimensional (1D) NMR spectra suffice, and the experiments can be applied to proteins too large for study by 2D and 3D NMR. Moreover, the large chemical shift change associated with ligand binding means that faster on- and off-rates nonetheless remain in the slow-exchange limit providing separate bound and free signals.
In the case of C2, an unexpected chemical shift signature and a large effect on the dissociation constant could both be explained on the basis of deprotonation of ligand in concert with a protein residue whose protonated form favors substrate binding. Thus, 19F NMR afforded information on a protein residue as well as the fluorinated ligand itself. It would be very interesting to apply this method to the reduced state of para-hydroxybenzoate hydroxylase where a pKa of 7.1–7.4 has been assigned to bound substrate when the enzyme is oxidized based on UV spectra (27, 29–31) but to His-72 when the enzyme is reduced, despite a comparative dearth of data (67).
Conclusions
Our 19F NMR studies demonstrate that the pKa of substrate is depressed in both a single-component and a two-component flavoprotein hydroxylase, supporting deprotonation of the phenol group as a general mechanism of substrate activation for electrophilic attack among these enzymes. Several one-component oxygenases were known to bind substrate in phenolate form (30, 31, 33, 34), but we now place this on quantitative footing, demonstrating 9.1 and 14 kJ/mol stabilization of substrate phenolate consistent with electrostatic or hydrogen bonding mechanisms.
The case of C2 demonstrates the capacity of 19F NMR to distinguish ligand deprotonation from other events, based on companion 13C NMR experiments that identify specific deprotonation events (Figs. 2 and 5). Thus, our data and model indicate that the protonation state of a nearby amino acid is coupled to that of the ligand. For both enzymes, comparisons with protein variants supported an active site His as substantially responsible for stabilizing bound ligand as the phenolate. In C2, our experimentally-validated model supports formation of a His+·phenolate− pair.
Experimental procedures
Materials
3F4HPA, 4F3HB, maleic acid, and 4-methylimidazole (4-MI) were from Sigma. 2,4-Dimethylimidazole (2,4-DMI) was from Lancaster Synthesis. Acetic acid was from EMD Millipore.
Enzymes
The genes for wildtype (WT) and variants of C2 from Acinetobacter baumanii and 3HB6H from Rhodococcus jostii RHA1 were constructed and expressed, and proteins were purified as described previously (46, 68, 69). Immediately before NMR data collection, enzymes were exchanged into 100 mm potassium chloride, pH 6.0, by passage through a SephadexTM G-25 gel-filtration chromatographic column equilibrated with 100 mm potassium chloride, pH 6.0. The compounds used in the experiment (3F4HPA, 4F3HB, maleic acid, acetic acid, 4-MI, and 2,4-DMI) were dissolved in 100 mm potassium chloride to final concentrations of 10 mm, and the resulting solutions were adjusted to pH ∼6.0. The concentrations of the following compounds were determined using the known extinction coefficients: FMN, ϵ446 = 12.2 mm−1 cm−1; FAD, ϵ450 = 11.3 mm−1 cm−1; WT and all variants of C2, ϵ280 = 56.7 mm−1 cm−1 (41); WT and all variants of 3HB6H, ϵ452 = 11.0 mm−1 cm−1 (46).
NMR samples
NMR samples of WT and variants of C2 were prepared under inert atmosphere in an anaerobic glove box (M-BRAUN UNIlab). The reduced holoenzyme solution (C2/FMNH−) was prepared by equilibration of 0.5 mm C2 and 0.5 mm oxidized FMN with O2-free atmosphere followed by reduction with a stoichiometric amount of sodium dithionite. Anaerobic solution of 3F4HPA was then added to yield a final concentration of 0.2 mm followed by addition of an aliquot of stock solution of NMR pH indicators (acetic acid, maleic acid, 4-MI, and 2,4-DMI) to yield final concentrations of 1 mm, and sodium trimethylsilylpropanesulfonate (DSS) to yield a final concentration of 0.5 mm (internal chemical shift standard for 1H). This solution was augmented with 25% (v/v) 2H2O for field locking, and then 700 μl were transferred to a 5-mm NMR tube with a septum-lined screw cap (Wilmad catalog no. 535-TR-7). A 1-mm capillary tube was filled with 0.1 mm potassium fluoride in 100 mm potassium chloride, pH 6.0, to serve as a chemical shift reference for 19F, and flame-sealed. This was placed in the NMR tube, which was then sealed before removal from the glove box.
For 3HB6H experiments, the NMR samples (aerobic condition) contained 0.8 mm 3HB6H enzyme (in the oxidized state, WT, or variants), 0.4 mm 4F3HB substrate analog, 1 mm indicators, 0.5 mm DSS, 0.1 mm KF (in a sealed capillary), and 25% 2H2O in 100 mm potassium chloride, pH 6.0. The provision of excess enzyme favored complete binding of the substrate analog.
NMR spectroscopy
19F NMR spectra were acquired on a Varian INOVA 600 MHz NMR spectrometer using a 600 MHz 1H-19F/15N-31P 5-mm PFG switchable probe with Z gradients, at 25 °C (for WT and variants of C2) or 4 °C (for WT 3HB6H to accommodate the enzyme's moderate stability). The sample was equilibrated at each temperature for at least 30 min, and the probe was tuned for detection of 19F. After inserting the sample into the magnet bore, N2 gas was used to maintain an inert atmosphere, control the temperature, maintain probe cooling, and spin NMR tubes at 20 Hz. 19F spectra were obtained as 2.323-s acquisitions following 90° excitation pulses (25 μs at 25 °C and 21.1 μs at 4 °C) separated by 3.9-s relaxation delays. A total of 6400 transients were averaged per 28,000 Hz–wide 1D spectrum (total time ≈12 h). Data were processed using 20 Hz Lorentz line-broadening prior to Fourier transformation. Chemical shift values were referenced to potassium fluoride in a capillary at −121 ppm. 19F spectra of 4F3HB bound to 3HB6H were analyzed by Gaussian deconvolution using the software provided by the spectrometer manufacturer (VnmrJ 3.2) to yield the positions, widths, and integrated areas of individual resonances (see Fig. 3A).
1H 1D NMR spectra were acquired on a Varian INOVA 400 MHz spectrometer using the “Wet1D” pre-sequence with 300 Hz-wide on-resonance eburp1 waveforms to selectively excite the signal of bulk water in preparation for pulse-field gradient suppression, followed by a 90° excitation pulse to excite the full spectrum and a 1-s acquisition time, with a 1-s relaxation delay time between scans. The pH indicators' chemical shifts were tabulated relative to internal DSS at 0 ppm in 10,000 Hz–wide spectra produced by averaging 64 scans. Data were zero-filled once, weighted with a Gaussian or squared sine bell apodization function, and Fourier was transformed.
13C NMR spectra were obtained using a Varian INOVA 400 MHz spectrometer (100 MHz for 13C) at 25 °C. The sample solutions for 13C NMR measurement were 0.1 m 3F4HPA, 10% 2H2O, with CDCl3 in a sealed capillary serving as an internal standard for 13C, 77.23 ppm. The 13C NMR spectra were obtained using a 45° excitation pulse, a 12-s delay between scans, and proton decoupling throughout.
Calculation of the apparent dissociation constant, Kd, obs
For the case of 3F4HPA binding to C2, we calculated Kd, obs at each pH based on the integrated peak areas of the bound and free forms of 3F4HPA. In the absence of chemical exchange, the 6.2-s interval we employed between 90° pulses would allow recovery of >95 and >93% of bound and free ligand magnetization, respectively, based on the T1 values of 2.25 ± 0.1 s for free 3F4HPA and T1 ≤2 s for C2 (calculated based on measurements for two other proteins and scaling for τc with the assumption that bound ligand tumbles with the protein (70)). The almost-complete recovery and similar degrees of saturation indicate that the two signals will similarly report on the concentration of the species they represent. Moreover, taking into account chemical exchange employing the on-rate of 10 s−1 reported (41) or adjusted to the concentrations employed here (30 s−1), simulations as per Ref. 71 show that the degree of saturation of the two ligands differs by less than 0.1% with the 6.2 s recycle time we used.
The peak areas were used with knowledge that the total concentration of ligand was 0.2 mm to calculate bound and free ligand concentrations. Then, the total enzyme concentration of 0.5 mm and the concentration of bound ligand (=ligand·enzyme complex, [LigEz]) was used to calculate the concentration of free enzyme, and thereby the dissociation constant in effect (Equation 2):
| (Eq. 2) |
In Equation 2, the protonation states of the ligand and enzyme are not specified (this feature is incorporated below).
pH titrations
The internal NMR pH indicators with pKa values spanning the range of 4.8 to 8.7 were chosen based on the simplicity of their spectra and noninteraction with the enzymes and their substrate binding. The pKa value of each of the indicators used was determined under our conditions (in 100 mm potassium chloride with 10% 2H2O and 0.5 mm DSS at 25 °C), and the NMR response was documented by withdrawing NMR samples at 0.3 pH unit intervals in a conventional pH electrode-monitored titration extending from pH 1 to 12. 1H NMR spectra were acquired on a Varian INOVA 400 MHz as described above. The pKa of each indicator was determined from plots of the indicators' chemical shifts versus the pH values provided by the electrode (Table 2). Thereafter, the known chemical shifts versus pH were used to infer internal pH from the set of indicator chemical shifts without opening the sample to air or inserting a pH probe, because the indicators' chemical shift(s) vary with the pH near their pKas (Fig. S1).
Table 2.
List of pH indicators used in the experiment
| Indicator compound | Resonance observed | Resonancea | Measured pKab | Literature pKac |
|---|---|---|---|---|
| ppm | ||||
| Acetic acid | Methyl proton | 1.90–2.09 | 4.78 ± 0.08 | 4.76 |
| Maleic acid | C(2) proton | 6.00–6.34 | 6.08 ± 0.03 | 6.26d |
| 4-Methylimidazole | C(2) proton | 7.63–8.51 | 7.87 ± 0.0 | 7.52 |
| C(5) proton | 6.82–7.15 | 7.89 ± 0.03 | 7.52 | |
| C(4) methyl protons | 2.19–2.33 | 7.86 ± 0.07 | 7.52 | |
| 2,4-Dimethylimidazole | C(5) proton | 6.65–6.95 | 8.70 ± 0.06 | 8.64e |
| C(2) methyl protons | 2.30–2.55 | 8.67 ± 0.08 | 8.64e | |
| C(4) methyl protons | 2.13–2.25 | 8.68 ± 0.08 | 8.64 |
a Resonance versus internal 0.5 mm DSS (chemical shift reference for 1H) is shown.
b pKa values were determined in 10% 2H2O, 0.5 mm DSS, with 100 mm potassium chloride. Values are the averages of two independent measurements. An additional error may be contributed by the electrode.
c Data are from Perrin and Dempsey (72), except where noted.
d The higher of maleic acid's two pKa values is named pKa2 by Perrin and Dempsey (72).
e Data were determined in 100% 2H2O with 100 mm potassium chloride at 25 °C (73).
For experiments on C2, an anaerobic solution of potassium hydroxide (KOH) was titrated into 700 μl of anaerobic NMR samples as described above. Each addition of KOH was made in an anaerobic glove box so the sample was never opened to air. Each 2 μl addition of 0.2 m KOH produced an ∼0.3 pH unit change. After each KOH addition, a 1H NMR spectrum was acquired at 400 MHz to determine the new pH, and a 19F NMR spectrum was acquired at 596 MHz (600 MHz for 1H) to determine the 3F4HPA 19F chemical shift. KCl (100 mm) was also present in the enzyme solution to minimize the change in overall ionic strength, and the reversibility of the titration was confirmed by addition of 0.1 m HCl to return the pH to its original range. Fewer points were taken from this back-titration because the enzyme is less amenable to addition of HCl. The 19F chemical shifts versus pH from the back-titration were compared with those obtained from the upward titration and confirmed that titrations were reversible (Figs. 3 and 6).
For 3HB6H experiments, titrations from pH 6.5 to 10.0 were accomplished by additions of aerobic 0.2 m KOH, and the return to low pH was accomplished via additions of aerobic 1.0 m MES, see above. All titrations were performed at least twice.
Analysis of the pH dependence
The dissociation constants and chemical shifts measured at a series of pH values were modeled as functions of pH, with fitting to different models performed using KaleidaGraph (Synergy Software Version 4.5.3) and MatLab (Mathworks R2019a).
The pH dependence of the observed 19F chemical shift δobs was modeled in terms of the Henderson-Hasselbalch equation (Equation 3),
| (Eq. 3) |
in which δLH and δL are the 19F chemical shifts of the acid and base forms of the ligand (LH and L) and are the asymptotes obtained from the fit. Equation 3 was adapted to analyze absorbance as a function of pH.
Observed dissociation constants, Kd, obs, are understood to encompass multiple microscopic binding possibilities differing with respect to the protonation states of participating species. To shed light on these, the pH dependence of Kd, obs was analyzed in terms of two different models.
1) In the first model, binding of ligand depends only on the protonation state of the ligand itself, and it reflects pKa values of one proton in the enzyme-bound as well as the free state of the ligand pKLH(EH) and pKLH, respectively (Scheme 1, EH denotes enzyme in its protonated state). In our enzymes, the tightest binding is attained for deprotonated ligand (L), so Kd, obs(pH) is described relative to binding of L to EH: L + EH ⇔ L·EH.
2) In the second model, we additionally considered that the protonation state of an enzyme residue might also change in the pH range of interest (Scheme 2, the two-proton model, and Equations 4 and 5). The model incorporates interaction between the protonation states of the ligand (LH or L) and the enzyme (EH or E), where the species responsible for a pKa is indicated in the subscript with the state of the other species providing context in parentheses. Thus pKLH(EH) is the pKa of ligand bound to the protonated state of the enzyme, whereas pK(L)EH indicates the pKa of the enzyme residue when deprotonated ligand is bound. Favorable interaction between a protonated (cationic) enzyme residue and deprotonated (anionic) ligand will tend to raise the enzyme's pKa and lower that of the ligand. Thus, we expect that the ligand's pKa will decrease more upon binding to protonated enzyme than to deprotonated enzyme: pKLH(EH) < pKLH(E) and that bound ligand will display a pKa of pKLH(EH) when the enzyme is protonated, i.e. when pH < pK(L)EH,
| (Eq. 4) |
and δLHE = δLE − 0.15 and δLEH = δLHEH + 0.15, where 0.15 is the chemical shift change produced by ligand protonation, obtained from fits to data for free ligand.
| (Eq. 5) |
In the slow-exchange binding regime that applies to our systems, chemical shifts of bound ligand describe only the environment sensed by the bound population of ligand and reflect the protonation state of enzyme residues that interact with ligand as well as the protonation state of ligand itself (portion of Scheme 2 boxed in green). The Kd, obs describes the equilibrium between all bound and all free ligand, and thus additionally reports on the pKa of free ligand (pKLH, known from titrations of free ligand) and free enzyme (pKEH), in the portion of Scheme 2 boxed in red.
Details of fits
The pH dependence of the apparent dissociation constant Kd, obs was fit with Equations 1 (model 1) and 5 (model 2), and the chemical shift of bound ligand was fit with Equations 3 or 4, respectively, to assess the merits of the different models and to extract parameters able to simultaneously describe both pH dependences. Equations 4 and 5 together were used to fit a merged data set of δobs and Kd, obs (Scheme 2).
Global fits to δobs and Kd, obs were implemented in MatLab, wherein scaled chemical shifts (CSsc = (δobs + 137.7)·5) were employed so that the magnitude of the total change would be 8.9, comparable with the total change of 9 for the Kd, obs. In fitting model 2 to the δobs of bound ligand versus pH, we made the simplifying assumption that the chemical shift would respond similarly to the ligand's own protonation state when bound as when free, regardless of the protonation state of the protein residue. This allowed us to treat the difference δL(EH) − δLH(EH) as equal to δL(E) − δLH(E) and δL − δLH, of which the latter is known from titrations of free ligand. This decreased the number of unknown chemical shifts from 4 to 2 in the global fits. When pKa values obtained from these simplified fits were fixed and all four chemical shifts were optimized independently, the results did not change significantly.
Rapid kinetics experiments
Rapid kinetics measurements were performed with a Hi-Tech Scientific Model SF-61DX stopped-flow spectrophotometer in single-mixing and double-mixing modes. The optical path-length of the observation cell was 1 cm. The reactions were conducted at 4 °C. The stopped-flow instrument was made anaerobic by flushing with an oxygen-scrubbing solution consisting of 400 μm glucose, 1 mg/ml glucose oxidase (15.5 unit/ml), and 4.8 μg/ml catalase in 50 mm sodium phosphate buffer, pH 7.0. The oxygen-scrubbing solution was allowed to stand in the flow system overnight, and the system was thoroughly rinsed with anaerobic buffer before experiments were performed. To study the reaction of C2 with oxygen, the reduced enzyme in the presence of substrate was prepared by rendering a solution of C2 (100 μm) plus FMN (32 μm) anaerobic by equilibration in an anaerobic glove box, followed by stoichiometric reduction with sodium dithionite. The resulting solution was placed in a tonometer and transferred to the stopped-flow instrument. The reduced enzyme was mixed with buffers containing various oxygen concentrations (0.13, 0.31, 0.61, and 1.03 mm) and 2 mm 3F4HPA (41). All quoted concentrations are those obtained after mixing. Apparent rate constants (kobs) were calculated from exponential fits to the kinetic traces, performed using KineAsyst3 software (Hi-Tech Scientific, Salisbury, UK) or Program A (written at the University of Michigan by Rong Chang, Jung-yen Chiu, Joel Dinverno, and David P. Ballou). Rate constants were obtained by fitting plots of kobs versus concentrations of oxygen with a Marquardt-Levenberg nonlinear fit algorithm that is included in the KaleidaGraph software.
Single-turnover reactions of WT C2 with 3F4HPA at various pH values
500 μl of an anaerobic solution of 200 μm C2 with 100 μm FMNH− in 10 mm sodium phosphate buffer at pH 7.0 was mixed with an equal volume of 4.0 mm 3F4HPA in 100 mm buffer at 25 °C. After 15 min, the reaction was quenched with 500 μl of 0.15 m HCl. The experiment was repeated at a series of pH values by employing various pH values for the buffer of the 3F4HPA solution, including 100 mm sodium phosphate for pH 6.0 or 7.0, 100 mm Tris-HCl for pH 8.0, and 100 mm Gly-NaOH for pH 9.0 or 10.0. The quenched solutions were separated from the enzyme using Microcon® ultracentrifugal filters (10 kDa cutoff). The filtrates were analyzed using HPLC with diode array detection and MS detection.
Author contributions
W. P., P. Chaiyen, and A.-F. M. conceptualization; W. P. data collection; W. P., P. Chaiyen, and A.-F. M. formal analysis; W. P. and A.-F. M. validation; W. P., P. Chenprakhon, T. D., D. M., J. S., C. T., W. J. v. B., and P. Chaiyen investigation; W. P. visualization; W. P., P. Chaiyen, and A.-F. M. methodology; W. P. writing-original draft; W. J. v. B. resources; W. J. v. B., P. Chaiyen, and A.-F. M. supervision; W. J. v. B., P. Chaiyen, and A.-F. M. writing-review and editing; P. Chaiyen and A.-F. M. funding acquisition; P. Chaiyen and A.-F. M. project administration; A.-F. M. software.
Supplementary Material
Acknowledgment
We thank A. Sebesta for repair of the anaerobic glove box.
This work was supported by the Vice President for Research of the University of Kentucky, the Center for Pharmaceutical Development, National Science Foundation Grant I/UCRC IIP-1540011 (to A.-F. M.), a Research Challenge Trust Fund Fellowship (to W. P.), the National Science Foundation, Chemistry of Life Processes Grant CHE-1808433 (to A.-F. M.), the Thailand Research Fund grant RTA5980001, Thailand Science Research and Innovation Global Partnership Program and Vidyasirimedhi Institute of Science and Technology (VISTEC) (to P. Chaiyen), and Thailand Research Fund Grants MRG6180156 (to P. Chenprakhon) and RSA5980062 (to J. S.). The authors declare that they have no conflicts of interest with the contents of this article.
This article contains Figs. S1–S7 and Table S1.
- C4a-OOH
- C4a-hydroperoxyflavin
- 3F4HPA
- 3-fluoro-4-hydroxyphenylacetate
- 3HB
- 3-hydroxybenzoate
- 3HB6H
- 3-hydroxybenzoate 6-hydroxylase
- 4F3HB
- 4-fluoro-3-hydroxybenzoate
- C2
- oxygenase component of p-hydroxyphenylacetate 3-hydroxylase
- DSS
- 2,2-dimethyl-2-silapentane-5-sulfonic acid
- HPAH
- p-hydroxyphenylacetate 3-hydroxylase
- 4HPA
- 4-hydroxyphenylacetate = p-hydroxyphenylacetate
- MES
- 2-(N-morpholino)ethanesulfonic acid
- 4-MI
- 4-methylimidazole
- 2,4-DMI
- 2,4-dimethylimidazole
- DSS
- sodium trimethylsilylpropanesulfonate
- PDB
- Protein Data Bank.
References
- 1. van Berkel W. J., Kamerbeek N. M., and Fraaije M. W. (2006) Flavoprotein monooxygenases, a diverse class of oxidative biocatalysts. J. Biotechnol. 124, 670–689 10.1016/j.jbiotec.2006.03.044 [DOI] [PubMed] [Google Scholar]
- 2. Huijbers M. M., Montersino S., Westphal A. H., Tischler D., and van Berkel W. J. (2014) Flavin dependent monooxygenases. Arch. Biochem. Biophys. 544, 2–17 10.1016/j.abb.2013.12.005 [DOI] [PubMed] [Google Scholar]
- 3. Torres Pazmiño D. E., Winkler M., Glieder A., and Fraaije M. W. (2010) Monooxygenases as biocatalysts: classification, mechanistic aspects and biotechnological applications. J. Biotechnol. 146, 9–24 10.1016/j.jbiotec.2010.01.021 [DOI] [PubMed] [Google Scholar]
- 4. Fagan R. L., and Palfey B. A. (2010) in Comprehensive Natural Products Chemistry II (Begley T., ed) pp. 37–114, Elsevier, Oxford, UK [Google Scholar]
- 5. Costas M., Mehn M. P., Jensen M. P., and Que L. Jr. (2004) Dioxygen activation at mononuclear nonheme iron active sites: enzymes, models, and intermediates. Chem. Rev. 104, 939–986 10.1021/cr020628n [DOI] [PubMed] [Google Scholar]
- 6. Dhammaraj T., Phintha A., Pinthong C., Medhanavyn D., Tinikul R., Chenprakhon P., Sucharitakul J., Vardhanabhuti N., Jiarpinitnun C., and Chaiyen P. (2015) p-Hydroxyphenylacetate 3-hydroxylase as a biocatalyst for the synthesis of trihydroxyphenolic acids. ACS Catal. 5, 4492–4502 10.1021/acscatal.5b00439 [DOI] [Google Scholar]
- 7. Pinthong C., Phoopraintra P., Chantiwas R., Pongtharangkul T., Chenprakhon P., and Chaiyen P. (2017) Green and sustainable biocatalytic production of 3,4,5-trihydroxycinnamic acid from palm oil mill effluent. Process Chem. 63, 122–129 10.1016/j.procbio.2017.08.006 [DOI] [Google Scholar]
- 8. Tinikul R., Chenprakhon P., Maenpuen S., and Chaiyen P. (2018) Biotransformation of plant-derived phenolic acids. Biotechnol. J. 13, e1700632 10.1002/biot.201700632 [DOI] [PubMed] [Google Scholar]
- 9. Zhu Y., Zhang Q., Li S., Lin Q., Fu P., Zhang G., Zhang H., Shi R., Zhu W., and Zhang C. (2013) Insights into caerulomycin A biosynthesis: a two-component monooxygenase CrmH-catalyzed oxime formation. J. Am. Chem. Soc. 135, 18750–18753 10.1021/ja410513g [DOI] [PubMed] [Google Scholar]
- 10. Baker Dockrey S. A., Lukowski A. L., Becker M. R., and Narayan A. R. (2018) Biocatalytic site- and enantioselective oxidative dearomatization of phenols. Nat. Chem. 10, 119–125 10.1038/nchem.2879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Chenprakhon P., Wongnate T., and Chaiyen P. (2019) Monooxygenation of aromatic compounds by flavin-dependent monooxygenases. Protein Sci. 28, 8–29 10.1002/pro.3525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Sucharitakul J., Tinikul R., and Chaiyen P. (2014) Mechanisms of reduced flavin transfer in the two-component flavin-dependent monooxygenases. Arch. Biochem. Biophys. 555, 33–46 10.1016/j.abb.2014.05.009 [DOI] [PubMed] [Google Scholar]
- 13. Holtmann D., and Hollmann F. (2016) The oxygen dilemma: a severe challenge for the application of monooxygenases? Chembiochem 17, 1391–1398 10.1002/cbic.201600176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Massey V. (1994) Activation of molecular oxygen by flavins and flavoproteins. J. Biol. Chem. 269, 22459–22462 [PubMed] [Google Scholar]
- 15. Eberlein G., and Bruice T. C. (1983) The chemistry of a 1,5-diblocked flavin. 2. Proton and electron transfer steps in the reaction of dihydroflavins with oxygen. J. Am. Chem. Soc. 105, 6685–6697 10.1021/ja00360a024 [DOI] [Google Scholar]
- 16. Mattevi A. (2006) To be or not to be an oxidase: challenging the oxygen reactivity of flavoenzymes. Trends Biochem. Sci. 31, 276–283 10.1016/j.tibs.2006.03.003 [DOI] [PubMed] [Google Scholar]
- 17. Wongnate T., Surawatanawong P., Visitsatthawong S., Sucharitakul J., Scrutton N. S., and Chaiyen P. (2014) Proton-coupled electron transfer and adduct configuration are important for C4a-hydroperoxyflavin formation and stabilization in a flavoenzyme. J. Am. Chem. Soc. 136, 241–253 10.1021/ja4088055 [DOI] [PubMed] [Google Scholar]
- 18. Chenprakhon P., Trisrivirat D., Thotsaporn K., Sucharitakul J., and Chaiyen P. (2014) Control of C4a-hydroperoxyflavin protonation in the oxygenase component of p-hydroxyphenylacetate-3-hydroxylase. Biochemistry 53, 4084–4086 10.1021/bi500480n [DOI] [PubMed] [Google Scholar]
- 19. Visitsatthawong S., Chenprakhon P., Chaiyen P., and Surawatanawong P. (2015) Mechanism of oxygen activation in a flavin-dependent monooxygenase: a nearly barrierless formation of C4a-hydroperoxyflavin via proton-coupled electron transfer. J. Am. Chem. Soc. 137, 9363–9374 10.1021/jacs.5b04328 [DOI] [PubMed] [Google Scholar]
- 20. Ridder L., Harvey J. N., Rietjens I. M. C. M., Vervoort J., and Mulholland A. J. (2003) Ab initio QM/MM modeling of the hydroxylation step in p-hydroxybenzoate hydroxylase. J. Phys. Chem. B 107, 2118–2126 10.1021/jp026213n [DOI] [Google Scholar]
- 21. Ortiz-Maldonado M., Ballou D. P., and Massey V. (1999) Use of free energy relationships to probe the individual steps of hydroxylation of p-hydroxybenzoate hydroxylase: studies with a series of 8-substituted flavins. Biochemistry 38, 8124–8137 10.1021/bi990560e [DOI] [PubMed] [Google Scholar]
- 22. Chaiyen P., Sucharitakul J., Svasti J., Entsch B., Massey V., and Ballou D. P. (2004) Use of 8-substituted-FAD analogues to investigate the hydroxylation mechanism of the flavoprotein 2-methyl-3-hydroxypyridine-5-carboxylic acid oxygenase. Biochemistry 43, 3933–3943 10.1021/bi035734d [DOI] [PubMed] [Google Scholar]
- 23. Tweedy S. E., Rodríguez Benítez A., Narayan A. R. H., Zimmerman P. M., Brooks C. L. 3rd., and Wymore T. (2019) Hydroxyl radical-coupled electron-transfer mechanism of flavin-dependent hydroxylases. J. Phys. Chem. B 123, 8065–8073 10.1021/acs.jpcb.9b08178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wang X., Hou Q., and Liu Y. (2018) Insights into the decarboxylative hydroxylation of salicylate catalyzed by the flavin-dependent monooxygenase salicylate hydroxylase. Theor. Chem. Acc. 137, 89 10.1007/s00214-018-2278-0 [DOI] [Google Scholar]
- 25. Ridder L., Mulholland A. J., Rietjens I. M., and Vervoort J. (2000) A quantum mechanical/molecular mechanical study of the hydroxylation of phenol and halogenated derivatives by phenol hydroxylase. J. Am. Chem. Soc. 122, 8728–8738 10.1021/ja0007814 [DOI] [Google Scholar]
- 26. Van Berkel W. J., and Müller F. (1989) The temperature and pH dependence of some properties of p-hydroxybenzoate hydroxylase from Pseudomonas fluorescens. Eur. J. Biochem. 179, 307–314 10.1111/j.1432-1033.1989.tb14556.x [DOI] [PubMed] [Google Scholar]
- 27. Entsch B., Palfey B. A., Ballou D. P., and Massey V. (1991) Catalytic function of tyrosine residues in para-hydroxybenzoate hydroxylase as determined by the study of site-directed mutants. J. Biol. Chem. 266, 17341–17349 [PubMed] [Google Scholar]
- 28. Entsch B., and van Berkel W. J. (1995) Structure and mechanism of para-hydroxybenzoate hydroxylase. FASEB J. 9, 476–483 10.1096/fasebj.9.7.7737455 [DOI] [PubMed] [Google Scholar]
- 29. Eschrich K., van der Bolt F. J., de Kok A., and van Berkel W. J. (1993) Role of Tyr201 and Tyr385 in substrate activation by p-hydroxybenzoate hydroxylase from Pseudomonas fluorescens. Eur. J. Biochem. 216, 137–146 10.1111/j.1432-1033.1993.tb18125.x [DOI] [PubMed] [Google Scholar]
- 30. Benítez A. R., Tweedy S., Baker Dockrey S. A., Lukowski A. L., Wymore T., Khare D., Brooks C. L. 3rd., Palfey B. A., Smith J. L., and Narayan A. R. H. (2019) Structural basis for selectivity in flavin-dependent monooxygenase-catalyzed oxidative dearomatization. ACS Catal. 9, 3633–3640 10.1021/acscatal.8b04575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Shoun H., Beppu T., and Arima K. (1979) On the stable enzyme·substrate complex of p-hydroxybenzoate hydroxylase. J. Biol. Chem. 254, 899–904 [PubMed] [Google Scholar]
- 32. Sucharitakul J., Medhanavyn D., Pakotiprapha D., van Berkel W. J., and Chaiyen P. (2016) Tyr217 and His213 are important for substrate binding and hydroxylation of 3-hydroxybenzoate 6-hydroxylase from Rhodococcus jostii RHA1. FEBS J. 283, 860–881 10.1111/febs.13636 [DOI] [PubMed] [Google Scholar]
- 33. Peelen S., Rietjens I. M., van Berkel W. J., van Workum W. A., and Vervoort J. (1993) 19F NMR study on the pH-dependent regioselectivity and rate of the ortho-hydroxylation of 3-fluorophenol by phenol hydroxylase from Trichosporon cutaneum. Eur. J. Biochem. 218, 345–353 10.1111/j.1432-1033.1993.tb18383.x [DOI] [PubMed] [Google Scholar]
- 34. Chaiyen P., Brissette P., Ballou D. P., and Massey V. (1997) Reaction of 2-methyl-3-hydroxypyridine-5-carboxylic acid (MHCP) oxygenase with N-methyl-5-hydroxynicotinic acid: studies on the mode of binding, and protonation status of substrate. Biochemistry 36, 13856–13864 10.1021/bi9715122 [DOI] [PubMed] [Google Scholar]
- 35. Pimviriyakul P., Surawatanawong P., and Chaiyen P. (2018) Oxidative dehalogenation and denitration by a flavin-dependent monooxygenase is controlled by substrate deprotonation. Chem. Sci. 9, 7468–7482 10.1039/C8SC01482E [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ruangchan N., Tongsook C., Sucharitakul J., and Chaiyen P. (2011) pH-dependent studies reveal an efficient hydroxylation mechanism of the oxygenase component of p-hydroxyphenylacetate 3-hydroxylase. J. Biol. Chem. 286, 223–233 10.1074/jbc.M110.163881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Tongsook C., Sucharitakul J., Thotsaporn K., and Chaiyen P. (2011) Interactions with the substrate phenolic group are essential for hydroxylation by the oxygenase component of p-hydroxyphenylacetate 3-hydroxylase. J. Biol. Chem. 286, 44491–44502 10.1074/jbc.M111.284463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kitevski-LeBlanc J. L., and Prosser R. S. (2012) Current applications of 19F NMR to studies of protein structure and dynamics. Prog. Nucl. Magn. Reson. Spectrosc. 62, 1–33 10.1016/j.pnmrs.2011.06.003 [DOI] [PubMed] [Google Scholar]
- 39. Husain M., Entsch B., Ballou D. P., Massey V., and Chapman P. J. (1980) Fluoride elimination from substrates in hydroxylation reactions catalyzed by p-hydroxybenzoate hydroxylase. J. Biol. Chem. 255, 4189–4197 [PubMed] [Google Scholar]
- 40. Jadan A. P., Moonen M. J., Boeren S., Golovleva L. A., Rietjens I. M., and van Berkel W. J. (2004) Biocatalytic potential of p-hydroxybenzoate hydroxylase from Rhodococcus rhodnii 135 and Rhodococcus opacus 557. Adv. Synth. Catal. 346, 367–375 10.1002/adsc.200303146 [DOI] [Google Scholar]
- 41. Sucharitakul J., Chaiyen P., Entsch B., and Ballou D. P. (2006) Kinetic mechanisms of the oxygenase from a two-component enzyme, p-hydroxyphenylacetate 3-hydroxylase from Acinetobacter baumannii. J. Biol. Chem. 281, 17044–17053 10.1074/jbc.M512385200 [DOI] [PubMed] [Google Scholar]
- 42. Sanders L. K., and Oldfield E. (2001) Theoretical investigation of 19F NMR chemical shielding tensors in fluorobenzenes. J. Phys. Chem. A 105, 8098–8104 10.1021/jp011114f [DOI] [Google Scholar]
- 43. Arunima, and Kurur N. D. (2005) Ortho effect in fluorobenzenes: cross-correlated relaxation and quantum chemical studies. Magn. Reson. Chem. 43, 132–138 10.1002/mrc.1520 [DOI] [PubMed] [Google Scholar]
- 44. Gerig J. T. (2003) in Biophysics Textbook Online (Society B., ed) pp. 1–35, Biophysical Society, Bethesda, MD [Google Scholar]
- 45. Montersino S., and van Berkel W. J. (2012) Functional annotation and characterization of 3-hydroxybenzoate 6-hydroxylase from Rhodococcus jostii RHA1. Biochim. Biophys. Acta 1824, 433–442 10.1016/j.bbapap.2011.12.003 [DOI] [PubMed] [Google Scholar]
- 46. Sucharitakul J., Wongnate T., Montersino S., van Berkel W. J., and Chaiyen P. (2012) Reduction kinetics of 3-hydroxybenzoate 6-hydroxylase from Rhodococcus jostii RHA1. Biochemistry 51, 4309–4321 10.1021/bi201823c [DOI] [PubMed] [Google Scholar]
- 47. Baron R., Riley C., Chenprakhon P., Thotsaporn K., Winter R. T., Alfieri A., Forneris F., van Berkel W. J., Chaiyen P., Fraaije M. W., Mattevi A., and McCammon J. A. (2009) Multiple pathways guide oxygen diffusion into flavoenzyme-active sites. Proc. Natl. Acad. Sci. U.S.A. 106, 10603–10608 10.1073/pnas.0903809106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Kahyaoglu A., and Jordan F. (2002) Direct proton magnetic resonance determination of the pKa of the active center histidine in thiolsubtilisin. Protein Sci. 11, 965–973 10.1110/ps.3890102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Alfieri A., Fersini F., Ruangchan N., Prongjit M., Chaiyen P., and Mattevi A. (2007) Structure of the monooxygenase component of a two-component flavoprotein monooxygenase. Proc. Natl. Acad. Sci. U.S.A. 104, 1177–1182 10.1073/pnas.0608381104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Dresen C., Lin L. Y., D'Angelo I., Tocheva E. I., Strynadka N., and Eltis L. D. (2010) A flavin-dependent monooxygenase from Mycobacterium tuberculosis involved in cholesterol catabolism. J. Biol. Chem. 285, 22264–22275 10.1074/jbc.M109.099028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Webb B. N., Ballinger J. W., Kim E., Belchik S. M., Lam K. S., Youn B., Nissen M. S., Xun L., and Kang C. (2010) Characterization of chlorophenol 4-monooxygenase (TftD) and NADH:FAD oxidoreductase (TftC) of Burkholderia cepacia AC1100. J. Biol. Chem. 285, 2014–2027 10.1074/jbc.M109.056135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Vervoort J., Rietjens I. M., van Berkel W. J., and Veeger C. (1992) Frontier orbital study on the 4-hydroxybenzoate-3-hydroxylase-dependent activity with benzoate derivatives. Eur. J. Biochem. 206, 479–484 10.1111/j.1432-1033.1992.tb16950.x [DOI] [PubMed] [Google Scholar]
- 53. Montersino S., Orru R., Barendregt A., Westphal A. H., van Duijn E., Mattevi A., and van Berkel W. J. (2013) Crystal structure of 3-hydroxybenzoate 6-hydroxylase uncovers lipid-assisted flavoprotein strategy for regioselective aromatic hydroxylation. J. Biol. Chem. 288, 26235–26245 10.1074/jbc.M113.479303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Kommoju P.-R., Chen Z. W., Bruckner R. C., Mathews F. S., and Jorns M. S. (2011) Probing oxygen activation sites in two flavoprotein oxidases using chloride as an oxygen surrogate. Biochemistry 50, 5521–5534 10.1021/bi200388g [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Isom D. G., Castañeda C. A., Cannon B. R., and Garcia-Moreno B. (2011) Large shifts in pKa values of lysine residues buried inside a protein. Proc. Natl. Acad. Sci. U.S.A. 108, 5260–5265 10.1073/pnas.1010750108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Everill P., Sudmeier J. L., and Bachovchin W. W. (2012) Direct NMR observation and pKa determination of the Asp(102) side chain in a serine protease. J. Am. Chem. Soc. 134, 2348–2354 10.1021/ja210091q [DOI] [PubMed] [Google Scholar]
- 57. Oda Y., Yamazaki T., Nagayama K., Kanaya S., Kuroda Y., and Nakamura H. (1994) Individual ionization constants of all the carboxyl groups in ribonuclease HI from Escherichia coli determined by NMR. Biochemistry 33, 5275–5284 10.1021/bi00183a034 [DOI] [PubMed] [Google Scholar]
- 58. Tozawa K., Ohbuchi H., Yagi H., Amano T., Matsui T., Yoshida M., and Akutsu H. (1996) Unusual pKa of the carboxylate at the putative catalytic position of thermophilic F-1-ATPase β subunit determined by C-13-NMR. FEBS Lett. 397, 122–126 10.1016/S0014-5793(96)01155-6 [DOI] [PubMed] [Google Scholar]
- 59. Fersht A. (1985) Enzyme Structure and Mechanism, 2nd Ed., p. 174, W. H. Freeman & Co., London [Google Scholar]
- 60. Anderson D. E., Becktel W. J., and Dahlquist F. W. (1990) pH-induced denaturation of proteins: a single salt bridge contributes 3–5 kcal/mol to the free energy of folding of T4 lysozyme. Biochemistry 29, 2403–2408 10.1021/bi00461a025 [DOI] [PubMed] [Google Scholar]
- 61. Thotsaporn K., Chenprakhon P., Sucharitakul J., Mattevi A., and Chaiyen P. (2011) Stabilization of C4a-hydroperoxyflavin in a two-component flavin-dependent monooxygenase is achieved through interactions at flavin N5 and C4a atoms. J. Biol. Chem. 286, 28170–28180 10.1074/jbc.M111.241836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Lin J., Cassidy C. S., and Frey P. A. (1998) Correlations of the basicity of His 57 with transition state analogue binding, substrate reactivity, and the strength of the low-barrier hydrogen bond in chymotrypsin. Biochemistry 37, 11940–11948 10.1021/bi980278s [DOI] [PubMed] [Google Scholar]
- 63. Dugad L. B., and Gerig J. T. (1988) NMR studies of carbonic anhydrase-4-fluorobenzenesulfonamide complexes. Biochemistry 27, 4310–4316 10.1021/bi00412a018 [DOI] [PubMed] [Google Scholar]
- 64. Salopek-Sondi B., and Luck L. A. (2002) 19F NMR study of the leucine-specific binding protein of Escherichia coli: mutagenesis and assignment of the 5-fluorotryptophan-labeled residues. Protein Eng. 15, 855–859 10.1093/protein/15.11.855 [DOI] [PubMed] [Google Scholar]
- 65. Stockman B. J. (2008) 2-Fluoro-ATP as a versatile tool for 19F NMR-based activity screening. J. Am. Chem. Soc. 130, 5870–5871 10.1021/ja801588u [DOI] [PubMed] [Google Scholar]
- 66. Gerig J. T. (1994) Fluorine NMR of proteins. Prog. Nucl. Magn. Reson. Spec. 26, 293–370 10.1016/0079-6565(94)80009-X [DOI] [Google Scholar]
- 67. Palfey B. A., Moran G. R., Entsch B., Ballou D. P., and Massey V. (1999) Substrate recognition by “password” in p-hydroxybenzoate hydroxylase. Biochemistry 38, 1153–1158 10.1021/bi9826613 [DOI] [PubMed] [Google Scholar]
- 68. Chaiyen P., Suadee C., and Wilairat P. (2001) A novel two-protein component flavoprotein hydroxylase. Eur. J. Biochem. 268, 5550–5561 10.1046/j.1432-1033.2001.02490.x [DOI] [PubMed] [Google Scholar]
- 69. Thotsaporn K., Sucharitakul J., Wongratana J., Suadee C., and Chaiyen P. (2004) Cloning and expression of p-hydroxyphenylacetate 3-hydroxylase from Acinetobacter baumannii: evidence of the divergence of enzymes in the class of two-protein component aromatic hydroxylases. Biochim. Biophys. Acta 1680, 60–66 10.1016/j.bbaexp.2004.08.003 [DOI] [PubMed] [Google Scholar]
- 70. Ernst R. W., and Anderson W. A. (1966) Application of Fourier transform spectroscopy to magnetic resonance. Rev. Sci. Instrum. 37, 93–102 10.1063/1.1719961 [DOI] [Google Scholar]
- 71. Spencer R. G., and Fishbein K. W. (2000) Measurement of spin-lattice relaxation times and concentrations in systems with chemical exchange using the one-pulse sequence: breakdown of the Ernst mode for partial saturation in nuclear magnetic resonance. J. Magn. Reson. 142, 120–135 10.1006/jmre.1999.1925 [DOI] [PubMed] [Google Scholar]
- 72. Perrin D. D., and Dempsey B. (1983) Buffers for pH and Metal Ion Control, p. 159, Chapman and Hall, London [Google Scholar]
- 73. Valcour A. A., and Woodworth R. C. (1986) Internal proton magnetic resonance probes for pH titration of proteins. J. Magn. Reson. 66, 536–541 10.1016/0022-2364(86)90197-6 [DOI] [Google Scholar]
- 74. Dhammaraj T., Pinthong C., Visitsatthwong S., Tongsook C., Surawatanawong P., and Chaiyen P. (2016) A single-site mutation at Ser-146 expands the reactivity of the oxygenase component of p-hydroxyphenylacetate 3-hydroxylase. ACS Chem. Biol. 11, 2889–2896 10.1021/acschembio.6b00402 [DOI] [PubMed] [Google Scholar]
- 75. Pettersen E. F., Goddard T. D., Huang C. C., Couch G. S., Greenblatt D. M., Meng E. C., and Ferrin T. E. (2004) UCSF Chimera–a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 10.1002/jcc.20084 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.