

Abstract
Available assays for measuring cellular manganese (Mn) levels require cell lysis, restricting longitudinal experiments and multiplexed outcome measures. Conducting a screen of small molecules known to alter cellular Mn levels, we report here that one of these chemicals induces rapid Mn efflux. We describe this activity and the development and implementation of an assay centered on this small molecule, named manganese-extracting small molecule (MESM). Using inductively-coupled plasma–MS, we validated that this assay, termed here “manganese-extracting small molecule estimation route” (MESMER), can accurately assess Mn in mammalian cells. Furthermore, we found evidence that MESM acts as a Mn-selective ionophore, and we observed that it has increased rates of Mn membrane transport, reduced cytotoxicity, and increased selectivity for Mn over calcium compared with two established Mn ionophores, calcimycin (A23187) and ionomycin. Finally, we applied MESMER to test whether prior Mn exposures subsequently affect cellular Mn levels. We found that cells receiving continuous, elevated extracellular Mn accumulate less Mn than cells receiving equally-elevated Mn for the first time for 24 h, indicating a compensatory cellular homeostatic response. Use of the MESMER assay versus a comparable detergent lysis-based assay, cellular Fura-2 Mn extraction assay, reduced the number of cells and materials required for performing a similar but cell lethality-based experiment to 25% of the normally required sample size. We conclude that MESMER can accurately quantify cellular Mn levels in two independent cells lines through an ionophore-based mechanism, maintaining cell viability and enabling longitudinal assessment within the same cultures.
Keywords: manganese, metal homeostasis, transport metal, neurobiology, neurodegenerative disease, calcium transport, calcimycin, ionomycin, ionophore, MESM
Introduction
Manganese (Mn)2 is an essential metal required as a cofactor for numerous kinases and other enzymes such as glutamine synthetase, Mn superoxide dismutase, and arginase (1). Similar to other biological trace metals, sufficient Mn needs to be retained while avoiding excess toxic amounts. In the brain, this appears to be a narrow window: the estimated concentrations of physiological Mn in the brain are in the range of 20–55 μm Mn, whereas neurotoxic responses begin at concentrations corresponding to 60–160 μm Mn (2). However, mechanistic information on how cells maintain Mn homeostasis is exceedingly sparse.
Cellular Mn transport was initially thought to be only co-regulated by iron (Fe) transporters such as divalent metal transporter-1 (DMT1) or the transferrin receptor (1). A third transporter of Fe, ferroportin, is able to efflux both Fe and Mn from the plasma membrane (3). As mRNA expression of ferroportin is induced in a dose-dependent manner upon Mn exposure, it can act as a compensatory mechanism to prevent Mn toxicity (4, 5). However, a recent study noted that very low expression of ferroportin in mice did not impact Mn levels, suggesting that ferroportin may not play a role in Mn homeostasis at basal conditions (6). Although these Fe transporters are capable of trafficking Mn with some efficacy, without Mn specificity they alone are insufficient to explain how cells maintain a homeostatic balance of Mn (7).
Only recently have the first Mn-selective transporters been described (8). This cell-surface Mn-efflux transporter, SLC30A10, was first identified when patients suffering the symptoms of Mn toxicity (motor impairment, cirrhosis, and high blood Mn content) were matched with mutations in the gene expressing the SLC30A10 protein (9–11, 48). A set of cell-surface transporters, Zip8 and Zip14, coded by SLC39A8 and SLC39A14, have also been implicated in cellular Mn uptake based on clinical studies and recent animal models through a similar process (12–20, 49). Based on these clinical loss–of–function mutation studies and animal data, a model of how these three transporters work together to regulate whole-body Mn homeostasis and detoxification has been proposed (21). Mn is taken up from blood via ZIP14 in the liver and secreted into bile via SLC30A10. Hepatocytes are then able to take up Mn from the bile via ZIP8. The uptake of Mn by ZIP8 has been shown to be essential for the proper functioning of Mn-dependent enzymes throughout the whole body (14). This model is also consistent with the neuroprotective role of SLC30A10, as whole-body knockout mice have elevated brain Mn levels (22). The absorption of Mn from the blood is also neuroprotective, as mutations of ZIP14 lead to increased Mn accumulation in the brain, as well as increases in blood, bone, heart, and kidney Mn levels (23).
Mn has been shown to play a role in neurodevelopment. Epidemiological studies show that varying Mn exposures in children result in cognitive deficits (24–26). Patients suffering from Mn toxicity due to a genetic disorder (described above) present with similar symptoms to a syndrome known as manganism: a Parkinsonian-like condition that occurs in welders and other industrial workers chronically exposed to Mn. They share overlapping symptoms of the motor, cognitive, and emotional abnormalities seen in Parkinson's disease (27). However, instead of responding to common Parkinson's disease treatments, amelioration of the symptoms is only seen upon treatment with the calcium (Ca2+) disodium salt of the chelating agent EDTA (CaNa2EDTA) (28).
A cellular deficit of Mn has been implicated in the pathogenesis of Huntington's disease (HD), a fatal neurodegenerative disease characterized by the expression of mutant huntingtin protein and death of medium spiny neurons in the corpus striatum. The rescue of HD phenotypes by Mn has been seen both in vitro and in vivo (29–31). These findings support the idea that a Mn deficiency may be occurring and contributing to the pathology of HD. Understanding how cells maintain appropriate Mn concentrations in the brain under normal conditions is vital to understanding how a Mn deficit may occur in HD or how excess Mn might be corrected due to environmental exposure or other genetic mutations.
Although nonlethal, the use of the Ca2+ fluorescent indicator Fura-2 AM can measure intracellular Mn, but only in relative quantities (32–37). Fura-2 AM quantification works by loading cells full of membrane-soluble Fura-2, so that it directly interacts with intracellular Mn. Fura-2 fluorescence is quenched based on the Mn present in the cytosol, so that cells containing more Mn will have lesser fluorescence. However, the amount of Fura-2 that is loaded into the cells will vary between experiments and between cell types, making full quantification of Mn concentrations impossible. Furthermore, the amount of Mn that is quantified is based only where Fura-2 AM has access to it in the cytosol; Mn stored subcellularly is not considered. Beyond this, Fura-2 AM as well would need to be repeatedly loaded into cells to enable longitudinal studies. This loading would quite likely change cellular status and be toxic to the cells; thus, it has not allowed such analysis.
Therefore, the most practical standard (in terms of throughput and financial cost) for Mn quantification up until now has been the cellular Fura-2 Mn extraction assay (CFMEA), which relies on the use of a detergent to lyse open cells, and then the ability to quantify the now-extracellular Mn by a concentration-dependent quenching mechanism of the fluorophore Fura-2 (38). CFMEA is the cost-effective preferred method for measuring Mn in cells, which unlike Fura-2AM methods, allows for full quantification of Mn. The single disadvantage of the CFMEA method is one of the same disadvantages affecting the gold standards of spectrometry or spectroscopy—it requires the lysis and death of the cells being measured. In addition, alternative methods to measure Mn concentrations such as inductively-coupled plasma MS (ICP-MS) or atomic absorption spectroscopy (AAS) are prohibitively expensive for the number of samples, costing ∼$10 per/sample versus ∼$1 per 100 samples by CFMEA in 96-well plate format. Moreover, AAS and ICP-MS are hampered by detection limits precluding multititer plate cell culture–based experimental designs. With the exception using Fura-2 AM (for semi-quantitative Mn determination within an experiment), other techniques require the lysis and destruction of the biological sample, limiting longitudinal studies. This is important as the ability to look at Mn changes over time while limiting all other variables is not possible. The alternative is performing multiple experiments in parallel, greatly increasing the cells, resources, and financial costs required. Developing a nonlethal assay that bypasses these problems would facilitate the ability to explore new questions and mechanistic details of Mn transport, toxicity, and homeostasis. Here, we report the development of such a nonlethal multititer plate assay and the discovery of a novel small molecule to selectively and rapidly release total accumulated intracellular Mn for extracellular detection by a Fura-2–based method similar in concept and cost to CFMEA.
Results
Small molecule VU0028386 (MESM) rapidly evokes cellular efflux of Mn
A set of small molecules that were found to alter net Mn levels (39) were screened to determine whether their mechanism of action was due to an influence in Mn efflux rates. We found that the majority of these small molecules could in fact influence Mn efflux rates (Fig. 1, A–C, and Fig. S1). Most notably, the small molecule VU0028386 (which henceforth we will call MESM) greatly facilitated Mn efflux from cells almost immediately and after 15 min released nearly all of the intracellular Mn (Fig. 1C). In comparison, an analogous amount of effluxed Mn was not reached under vehicle conditions even after 6 h. The time for 50% of the total Mn to be released under vehicle conditions was 79.1 min (t½) with 95% CI (confidence interval) falling between 70.73 and 89.87 min.
Figure 1.

Small molecule VU0028386 (MESM) rapidly induces cellular efflux of Mn. Cells of the murine striatal neuron lineage, STHdhQ7/Q7 (Q7), were pre-exposed to 125 μm MnCl2 for 2 h in HBSS before washing away the extracellular Mn and allowing the cells to efflux their intracellular Mn to the extracellular space in PBS (lacking Ca2+ and Mg) with co-incubation of a small molecule (10 μm). Fluorescent dye (Fura-2; 500 nm) was added to the PBS so its fluorescence could be quenched by effluxed Mn. Fluorescence of Fura-2 was measured at 360/535 (excitation/emission) every 15 min for 6 h at 37 °C, and Mn was calculated based on percent fluorescence quenching. The maximum quantity of Mn effluxed after 6 h was normalized to 100%. A, representative sample from a partial screen of small molecules performed in duplicate was performed (all 22 molecules can be seen in Fig. S1). B, following the screen, small molecule VU0026921 was assayed again (total n = 3) as another “Mn-increaser” to compare with VU0028386 (MESM), which was also assayed again (total n = 6) for confirmation of its efflux kinetics. C, sum of squares F-test comparison of MESM and vehicle data sets, using a nonlinear regression model of one-phase exponential decay, determined both data sets fit significantly different curves (p < 0.001). The biological half-life (t½) of Mn efflux in vehicle was calculated at 79.11 min. In comparison, the efflux of Mn with MESM is so fast a half-life value cannot be accurately calculated as the 50% mark occurs before measurement begins. Considering the lag time between assay and measurement, we can estimate the half-life value is less than 2 min. Error bars are the standard deviation of the biological replicates from each experiment. D, STHdhQ7/Q7 (Q7) was co-incubated with a known “Mn-increaser” VU0028386 or VU0026921 at 10 μm or 0.1% DMSO (vehicle) with 125 μm MnCl2 for 2 h in HBSS. Cells were washed five times in PBS to remove extracellular Mn. Cells were lysed open with Triton, and intracellular Mn was quantified using the CMFEA method, as described previously (38). Mn levels were normalized to vehicle levels, and their fold-change is plotted (n = 3). Error bars shown represent standard deviation (A–D). An ordinary one-way ANOVA with Dunnett's multiple comparisons test to vehicle to show statistical significance for intracellular Mn accumulation increases (*, p < 0.05).
In contrast, the rate at which Mn effluxes in the presence of MESM is extremely fast to the point that a t½ cannot be accurately calculated. Considering the lag period between assay and first measurement, we can estimate that the t½ value is less than 2 min. This is based on recordings of the length of this period from adding a small molecule to the first plate reader data point, which yielded an average of 94.4 ± 4.72 S.D. seconds (n = 5; data not shown). Taking this into consideration, because a true zero cannot be recorded, we imputed a 0-s time, 0% Mn efflux point in all of the graphs 94 s before the first measured point. We compare MESM's effects on efflux with another small molecule (VU0026921), which was also screened and previously referred to as a “Mn-increaser” (Fig. 1B).
Because MESM was originally identified as a “Mn increaser” in terms of net cellular uptake (39), we sought to confirm that MESM does in fact increase Mn levels in cells when incubated with extracellular Mn at the same time (Fig. 1D). The difference between the two experiments is the cellular concentration gradient of Mn during MESM exposure. When cells are pre-exposed to Mn to increase intracellular Mn levels, and extracellular Mn concentration is low during the MESM treatment, MESM facilitates Mn to move to the extracellular space (Fig. 1C). When MESM is co-incubated with high extracellular Mn levels, intracellular Mn levels are increased compared with Mn co-incubation with vehicle alone (Fig. 1D). In summary, MESM can induce Mn influx in conditions where extracellular Mn concentrations are higher than its intracellular concentration. MESM can also induce Mn efflux when extracellular Mn concentration is lower than intracellular Mn.
To establish that the effect of MESM on apparent Mn levels was not an artifact of interfering with the quantification of Mn (which is a function of the quenching of the fluorophore Fura-2), cell-free concentration curves of Mn to quench Fura-2 were generated in the presence or absence of MESM (Fig. 2). The data showed no evidence that MESM alone impacted Fura-2 fluorescence at the concentrations being used for extraction. This supports the conclusion that MESM was yielding the true quantities of intracellular Mn from cells via efflux rather than non-Mn–dependent changes in Fura-2 fluorescence. Although MESM was used at 3–10 μm concentrations for Mn extractions throughout these studies, at concentrations 30 μm and higher there was evidence of an interaction that is consistent with a competition for binding of Mn to both MESM and Fura-2 (Fig. S2).
Figure 2.
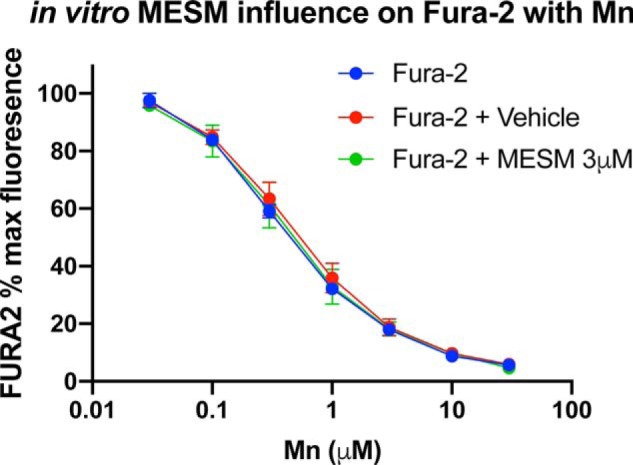
MESMER does not interfere with Fura-2–based Mn quantification. Various concentrations of Mn were added to empty wells (no cells) in PBS (without Ca2+ and Mg) in the presence of Fura-2 (0.5 μm), Fura-2 with 3 μm MESM, or Fura-2 with an equivalent DMSO vehicle. The plates were read at 360/535 nm excitation/emission as described. n = 3 of the individual replicates performed on different days is shown, with four technical replicate wells for each condition each day. Error bars show standard deviation of the three independent replicate averages. As expected, a two-way ANOVA yielded a significant main effect for Mn concentration (p < 0.0001), but no main effect of MESM or interaction. Sidak's multiple comparisons test yielded no significant differences in Fura-2% fluorescence between groups at each Mn concentration (p > 0.05).
Using MESM as a tool to extract and quantify Mn
As noted above, current quantitative methods of assessing intracellular Mn are cell-lethal, as the cells ultimately need to be lysed open for Mn to be quantified. The most high-throughput and inexpensive way this can be presently carried out is using CFMEA (Fig. 3). CFMEA relies on the lysis of cells with detergent to release Mn and quantify it by the quenching of Fura-2 fluorescence (38, 40). The lysis of cells every time Mn levels are quantified severely limits the type of experiments that are possible. Examples of such limited experiments are longitudinal or large multiplexed experiments. There are also time and financial burdens of cell-lethal assays, especially in human-induced pluripotent stem cell (hiPSC) model systems that are expensive to maintain and take weeks to differentiate. Thus, there is a strong appeal for an assay that could extract Mn in a nonlethal approach. We reasoned that a nontoxic small molecule that leads to the rapid efflux of total cellular Mn could be used to develop such an assay. Therefore, we sought to develop a new assay, manganese-extracting small molecule estimation route (MESMER) using the MESM molecule to rapidly extract and quantify cellular Mn content by a nonlethal alternative method based upon the CFMEA (Fig. 3). The only difference between the two methods is the extraction step, in which MESMER uses the small molecule MESM instead of detergent lysis. Considering the rate of efflux seen in Fig. 1C, we selected 15 min as the time for incubating MESM with the cells, as this time was sufficient to release >93.0 ± 5.1% of total cellular Mn.
Figure 3.
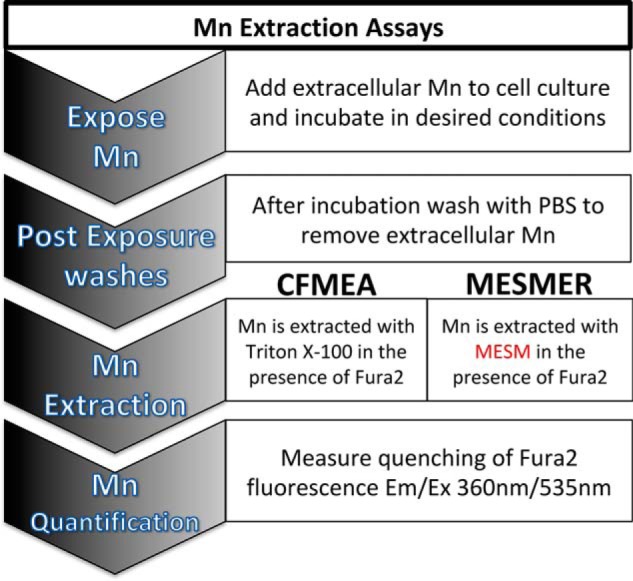
Already-validated CFMEA method and the proposed MESMER method differ only at their extraction step. In both assays, cells are “loaded” with Mn by an extracellular Mn concentration. After this incubation, this extracellular Mn is washed away so that only intracellular Mn remains. In the CFMEA method, PBS with 0.1% Triton is used to lyse open the cells and release intracellular Mn, and 0.5 μm Fura-2 is also present to act as a fluorescent Mn indicator. In contrast, the MESMER assay extracts Mn using a small molecule, VU0028386, or MESM, while also in the presence of Fura-2. The last step remains the same, as Mn is quantified by measuring the quenching of the Fura-2 signal at excitation/emission 360/535 nm.
MESMER extractions are specific to Mn and are comparable with the CFMEA and ICP-MS quantification
To test whether MESMER could be used to quantify Mn accurately, the levels of Mn in cells were compared between the new assay and the AAS-validated CFMEA (38). In both cell types tested (STHdhQ7/Q7 and HEK293), MESMER demonstrated a Mn concentration–dependent effect similar to CFMEA, with comparable % maximum values of Fura-2 in each condition (Fig. 4, A and B). The % maximum values in all cases was defined as the fluorescence measured divided by maximum fluorescence of Fura-2 (excitation/emission 360/535 nm) seen in the absence of additional extracellular Mn added to the base culture media. In the STHdh and HEK293 cells, the % maximum value of Fura-2 quenching at each Mn exposure was statistically indistinguishable between the MESMER and CFMEA extractions (Fig. 4, A and B). To ensure that the MESM molecule was responsible for this effect, extraction was attempted with efflux buffer using just the DMSO vehicle that MESM is dissolved in instead at the same dilution. This method did not yield a Mn concentration–dependent effect, and it was statistically significantly different from both CFMEA and MESMER extractions in all conditions (Fig. 4, A and B).
Figure 4.
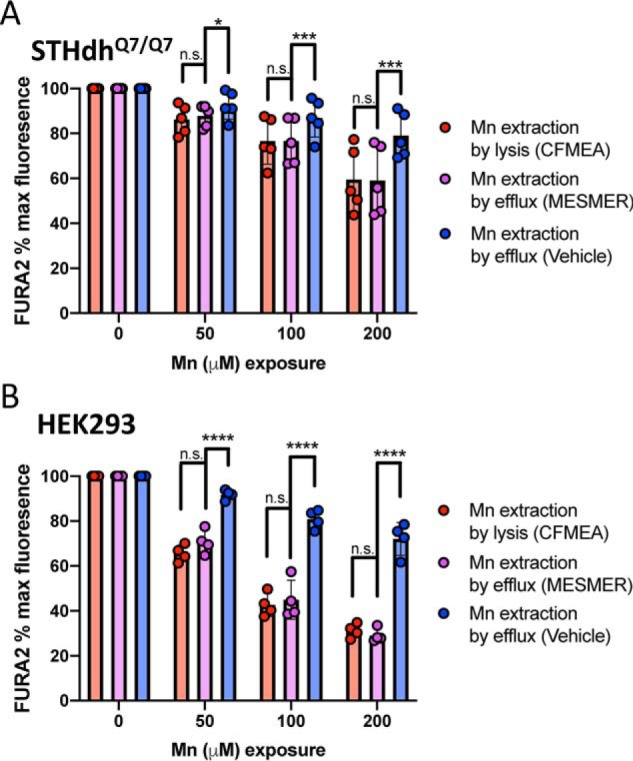
MESMER assay yields comparable readouts as current CFMEA method standard. Murine striatal neuron lineage STHdh Q7 cells (A) or HEK cells (B) were exposed to 0, 50, 100, or 200 μm Mn for 2 h at 37 °C. Q7 cells (n = 5) were exposed to Mn in HBSS and HEK cells (n = 4) in DMEM, respectively. The cells were then washed in PBS (lacking Ca2+ and Mg2+) and then exposed to 3 μm MESM (MESMER), a DMSO vehicle equivalent (vehicle), or 0.1% Triton in PBS (CFMEA). All solutions contained 0.5 μm Fura-2. After 15 min at 37 °C, the plates were read at 360/535 nm (see Fig. 3). Error bars shown represent standard deviation. Statistical significance was determined by two-way ANOVA and is denoted by the following: n.s., not significant; *, p < 0.05; ***, p < 0.001; ****, p < 0.0001.
To fully confirm that MESMER is in fact quantifying Mn accurately and specifically, STHdh cells were exposed to 50 μm MnCl2 for 2 h in media. After washing with PBS, intracellular Mn content was measured by MESMER or ICP-MS. ICP-MS was also run on samples that had already been quantified by MESMER as a control (Fig. 5A). A one-way ANOVA showed no significant difference in Mn content as measured by MESMER or ICP-MS. In contrast, samples that had Mn extracted by MESMER and then assayed by ICP-MS had significantly less Mn than either MESMER or ICP-MS alone. When cells were not exposed to extracellular Mn, but were exposed to MESM prior to measurement by ICP-MS, the intracellular Mn content of those cells was significantly lower (>95%) than those exposed to vehicle (Fig. 5B). This indicated that MESM could quantitatively release intracellular concentrations of Mn into the extracellular environment.
Figure 5.

MESMER assay yields comparable readouts to ICP-MS and is Mn-specific. Murine striatal neuron lineage STHdh Q7 cells were exposed to 50 μm Mn in DMEM for 2 h before being washed in PBS. Samples were subjected to MESMER, ICP-MS, or both (A). For comparison, some cells were not exposed to any extracellular Mn in DMEM, and they received MESM or vehicle prior to sample preparation for ICP-MS (B). Data were normalized to protein via a BCA assay of the samples. A one-way ANOVA with Tukey's multiple comparison tests revealed a significant difference between the samples that were extracted with MESMER prior to ICP-MS compared with those samples that were just extracted with ICP-MS (*, p < 0.05). These samples extracted with MESMER prior to ICP-MS were also significantly different from samples that were just extracted with MESMER alone (**, p < 0.01). Importantly, there was no significant difference between the samples that were measured by MESMER alone or ICP-MS alone (n.s.). For the cells that received no Mn treatment in DMEM (B), there was a significant decrease of extracted Mn in those treated with MESM prior to ICP-MS, compared with those treated with vehicle as measured by a two-tailed t test (*, p < 0.05). ICP-MS for other divalents was analyzed in the samples that had Mn pre-exposed prior to extraction (C) or with Mn pre-exposure (D). There was a significant difference in Ca and Cu in C as measured by Student's two-tailed t test (*, p < 0.05; **, p < 0.01).
To understand the specificity of the effect that MESM might have on other essential metals, the content of magnesium, calcium, iron, cobalt, copper, zinc, and molybdenum were also measured by ICP-MS in the samples that were ± MESM and ± Mn. Because MESM is able to facilitate the movement of Mn out of the cell to the extracellular space, it was reasoned that it might do the same to other divalent metals. There was no statistical decrease in the levels of any other metal tested by ICP-MS when treated with 50 μm extracellular Mn (Fig. 5C) or no Mn treatment (Fig. 5D). As of result of MESM, the only statistically significant difference seen besides a decrease in Mn levels was a significant but small increase in calcium and copper levels limited to cells that have been previously exposed to Mn (Fig. 5, C and D). The ionophore activity of MESM therefore appears to be Mn-specific.
MESMER and CFMEA extractions in hiPSC-derived cells
There exists a great value in developing a nonlethal method in hiPSCs given their utility for longitudinal studies across developmental trajectories, e.g. being able to follow cells as they proceed from the pluripotent state to post-mitotic neuroprogenitors and then to immature and mature neurons. Considering the model that stem cells can provide in terms of neurodevelopmental toxicity (41), we sought to confirm that MESMER could provide quantification of Mn in hiPSC-derived Islet-1–positive striatal-like neuroprogenitors (NPCs). Islet-1–positive striatal-like NPCs were exposed to varying concentrations of Mn for 1, 3, or 6 h. After 6 h, cells were returned back to Mn-free media for 1 or 6 more hours before being assayed. MESMER was able to detect expected concentration-dependent effects of Mn exposure (Fig. S3A). Consistent and predictable time-dependent differences in Mn levels were also evident in these cells (Fig. S3B). The cells that were returned to Mn-free medium before being assayed showed time-dependent effects, where those allowed to incubate longer in Mn-free media had less cellular Mn measured.
MESM extracts Mn more efficiently than known Mn ionophores without cellular toxicity
Despite its name, calcimycin (also known as A23187) is an ionophore with a higher affinity toward Mn2+ than Ca2+ (42, 43). Of similar relation and function, ionomycin is another ionophore often paired with calcimycin (44, 45). We tested whether known Mn ionophores such as ionomycin, calcimycin, or a commercially-available molecule named manganese ionophore II (Mi2) could extract Mn in a similar way as MESM. Effectively, the MESMER assay was performed except, instead of MESM, ionophores (calcimycin, ionomycin, or Mi2) were used at varying concentrations. MESM at varying concentrations was used as a control, and a CFMEA was performed at the same time for comparison (Fig. 6, A and B). Although there was an effect of concentration of calcimycin in the STHdh cells that approached the amount of Fura-2 quenching seen in the CFMEA, neither ionomycin, calcimycin, nor Mi2 yielded quenching of Fura-2 to the same degree as measured by CFMEA detergent lysis of the cells, at any concentration between 1 and 30 μm. In contrast, Mn extraction by MESM was not significantly different from CFMEA at 3 and 10 μm in both cell types tested (Fig. 6, A and B). Calcimycin continued to extract more Mn over time (as indicated by Fura-2 quenching), but even after 45 min it failed to reach the CFMEA range (Fig. S4).
Figure 6.
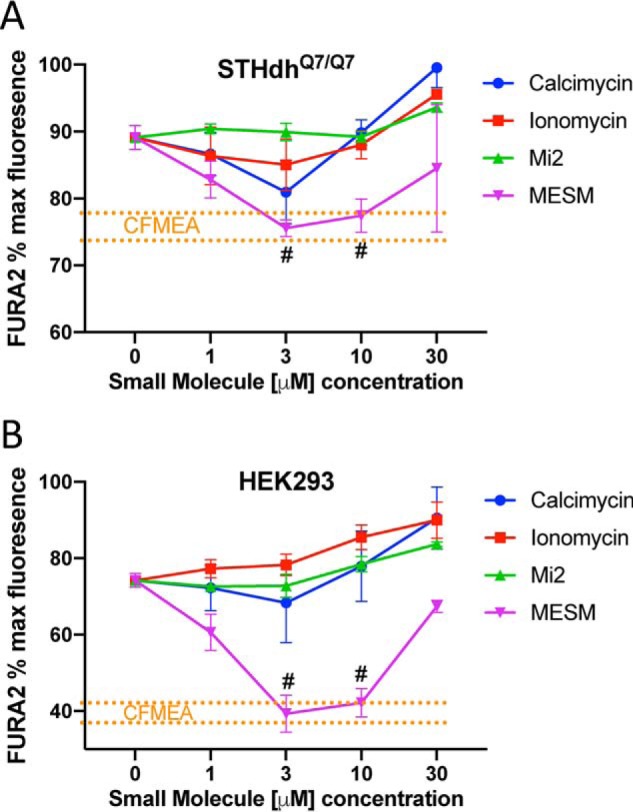
Extraction with MESM is comparable with CFMEA output from 3 to 10 μm and outperforms other known Mn ionophores. Murine striatal neuron lineage of Q7 cells (A) or HEK cells (B) was exposed to 100 μm Mn for 2 h at 37 °C. Q7 cells (n = 3–6) were exposed to Mn in HBSS and HEK cells (n = 3) in DMEM, respectively. The cells were then washed five times in PBS (lacking Ca2+ and Mg2+) and then exposed to 0.5 μm Fura-2 and varying concentrations of calcimycin, ionomycin, manganese ionophore II (Mi2), or MESM for 15 min before reading at 360/535 nm excitation/emission. A separate group of cells had Mn extracted by traditional CFMEA means after Mn exposure, for comparison. CFMEA dotted lines (in orange) denote range of 1 S.D. from CFMEA average. Each point outside the orange lines is significantly different from CFMEA, as determined by a two-way ANOVA with Sidak's multiple comparisons. The points within the orange lines are not significantly different from CFMEA levels and are labeled with #. Error bars represent standard deviation of all biological replicates.
Following the exposure to ionophores of different concentrations, the STHdh and HEK cells were tested for viability using the Cell-Titer Blue (CTB) assay. Viability is significantly reduced in STHdh and HEK293 cells with concentrations of calcimycin 3 μm and higher (Fig. 7, A and B). Ionomycin also reduces cell viability at concentrations at or above 10 μm in HEK293 cells and above 30 μm in STHdh cells. As demonstrated previously (39), MESM does not significantly alter viability in STHdh cells or in HEK293 cells below 10 μm (Fig. 7, A and B). In an assay of membrane integrity, neither MESM nor the other ionophores were significantly different from vehicle. As a positive control, Triton was used to disrupt membrane integrity (Fig. 7C).
Figure 7.

MESM exposure during the assay does not affect viability as measured by the CTB assay of membrane integrity as measured by CytoTox Green assay. Following Mn treatment and ionophore extraction/treatment, the ionophore/PBS mixture was removed from Q7 cells (A) or HEK cells (B) and then put back into their regular DMEM for 24 h at 37 °C. An injection of 20 μl of CTB reagent was added to each well and returned to the incubator for another 2 h before measuring the absorbance of each well at 590 nm. n = 3–6 biological replicates for Q7 cells (A) and n = 5 for HEK cells (B). The CTB assay relies on viable cells to reduce the reagent resazurin to a fluorescent product, see under “Experimental procedures.” Signals of vehicle conditions were normalized to 100%, and a two-way ANOVA with Sidak's multiple comparisons test to vehicle for every condition was used. Significance from vehicle is denoted by the following: ***, p < 0.001; ****, p < 0.0001. C, separate measure of toxicity was used as validation at the most effective extraction and the least toxic by CTB conditions (3 μm compound) in Q7 cells called CytoTox Green. It is based on membrane integrity, so that a dye will fluoresce if it binds DNA; see under “Experimental procedures.” n = 3 of independent biological replicates for all. Error bars shown represent standard deviation of the average across biological replicates. One-way ANOVA with Dunnett's multiple comparisons only shows statistically significant difference from vehicle in the positive control, which was 0.1% Triton X-100.
MESM facilitates the transport of Mn independent of cellular metabolism or cell-surface proteins
The ability of MESM to transport Mn in or out of a cell based on a concentration gradient suggests an activity consistent with an ionophore. An ionophore would not be expected to be dependent on cellular metabolism for its mechanism. To test the hypothesis that MESM does not depend on cellular metabolism to move Mn across cell membranes, STHdh cells were co-exposed with MESM and Mn at 37 °C or on ice to limit cellular energetics and ATP metabolism. Compared with cells co-incubated with MESM and Mn at 37 °C, cells co-incubated with MESM and Mn while on ice accumulated Mn by the same quantity (Fig. 8A). This is in contrast to other known “Mn-increasers” such as VU0026921 or VU003765, which have significant decreases in net Mn uptake when co-incubated on ice.
Figure 8.

MESM transports Mn independent of cellular metabolism or transmembrane proteins. A, Q7 cells were co-incubated with 100 μm Mn and 10 μm MESM, two other known “Mn-increasers” (VU0026921 and VU0003765), or equivalent vehicle for 2 h at 37 °C or on ice (n = 4). Cells were then washed five times in PBS, and then CFMEA extraction was performed. In contrast to the other “Mn-increasers,” MESM does not lose any ability to move Mn into the cell while on ice, and ATP is limited. Significance from a two-way ANOVA with multiple Sidak's comparisons test is denoted by the following: *, p < 0.05; **, p < 0.01. B, PAMPA shows the results of an incubation with 2 μm Mn on one side of the artificial membrane for 30 min when paired with DMSO (vehicle), 3 μm MESM, 3 μm calcimycin (Mn-ionophore; positive control), or 3 μm VU0026921 (another “Mn-increaser” capable of binding Mn). Fura-2 quenching is measured on the receiver side (opposite end) of the artificial membrane, showing that Mn can cross this lipid layer only in the presence of a known ionophore (calcimycin) and purported ionophore (MESM). Statistical significance was determined by a one-way ANOVA (n = 3) and denoted by the following: **, p < 0.01; or n.s., not significant). n = 3 independent plates performed on different days.
Regarding a mechanism of action, we sought to distinguish the possibility between MESM modulating cell-surface ion transport proteins to become Mn-permeable and from MESM itself forming a Mn-permeable pore or directly facilitating the passage of Mn across cell membranes. To test whether MESM needs cell-surface proteins to transport Mn, we used an artificial membrane to see whether Mn transport across it was possible with MESM. We found that although the artificial membrane was impermeable to Mn, co-incubation with MESM facilitates Mn transport (Fig. 8B). This is similar behavior to known Mn-ionophore calcimycin, but in contrast to a second small molecule increaser of net Mn uptake known to bind Mn, VU0026921, which is unable to transport Mn across the artificial membrane (Fig. 8B). Finally, we tested whether MESM requires co-exposure with Mn to promote subsequent Mn transport across membranes. When STHdh cells were pretreated with MESM, and then washed off prior to a Mn exposure, the cells accumulated more Mn compared with a vehicle pretreatment (Fig. S5A). This suggests that MESM may partially permeate the cell during the pre-exposure (and Mn is not required to be present for this action). However, pretreatment alone to MESM led to lower overall Mn accumulation than co-treatment of MESM with Mn exposure. This suggested that the presence of Mn promotes the process by which MESM alters membranes to be Mn-permeable. This links MESM with Mn, implying a physical interaction between them.
MESM does not transport calcium across the plasma membrane
While considering that other Mn ionophores such as ionomycin and calcimycin have a significant affinity for Ca2+, we tested the hypothesis that MESM could transport Ca2+ across the cellular membrane as well as Mn. HEK293 cells were loaded with the Ca-sensitive fluorescent dye, Fluo-4 (46), prior to adding varying concentrations of calcimycin, ionomycin, Mi2, and MESM. Cells were imaged in real time upon adding the ionophores. As expected, the fluorescence of the cells (indicating increased calcium) following 30 μm ionomycin or calcimycin began to spike after just 15 s and peaked around 40 s. In contrast, a change in fluorescence was not detected following 30 μm MESM or Mi2 even after 5 min, indicating that both MESM and Mi2 do not alter intracellular Ca2+ concentrations (Fig. 9).
Figure 9.
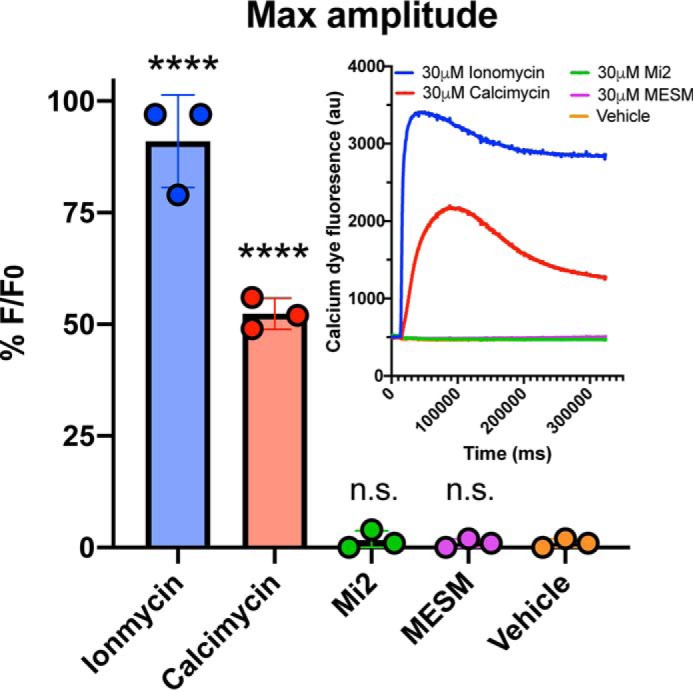
MESM does not influence intracellular calcium. HEK293 cells (n = 3, each experiment was performed in triplicate wells) were plated the day before the experiment at 15,000 cells per well and grown until 100% confluency. Cells were soaked with Fluo-4 dye for 1 h at room temperature prior to adding varying concentrations of MESM, ionomycin, calcimycin, and manganese ionophore II (Mi2) ranging from 3 nm to 30 μm. Shown here is the maximum concentration (30 μm). Using PanOptic to measure fluorescence every second for 5 min, an ionophore from a donor plate was added to the acceptor plate containing cells. Fluorescence was measured at 482 ± 16 nm excitation and 536 ± 20 nm emission. Inset shows a representative trace of each ionophore at maximum concentration (30 μm). The maximum amplitude was calculated after normalizing for light variations with a static ratio and subtracting background. Percent fluorescence was normalized to the maximum fluoresced wells in each plate (30 μm ionomycin). An ordinary one-way ANOVA was performed with Dunnett's multiple comparisons test. Statistical significance from vehicle is indicated as not significant (n.s.) or **** (p < 0.0001).
Novel longitudinal Mn exposure assay to determine whether Mn transport is altered by prior Mn exposures in cultured cells
It is currently unknown whether cells alter Mn transport properties after prior exposure to elevated levels of extracellular Mn. With a validated MESMER assay capable of longitudinal studies, we set out to test the effect of continuous Mn exposures in cell cultures over the course of 8 days. We designed a parallel experiment in which individual wells are assayed by MESMER without prior Mn exposures and matched for the duration in culture. Cells were continuously exposed to 1, 10, or 50 μm Mn concentrations over the course of 8 days and were compared with control cells that received Mn only for the first time, for 24 h, matched for days in culture post-plating. These were controlled with multiple wells seeded the same day (day −1), so the plate being MESMERized on any day has control wells for comparison that is the same age and is receiving Mn for the first time. In addition, we had previously verified that multiple MESM exposures did not affect Mn uptake (Fig. S6). To minimize cell growth over time, the cells were kept at 37 °C starting at day 0. These cells are conditionally immortal at 33 °C, as higher temperatures lead to the destabilization of the temperature-sensitive SV40 large T antigen and the cells substantially reduce proliferation.
Because MESMER allows for repeated Mn assessment of the same cells, we were able to measure the net uptake of Mn after each exposure in the same cell cultures. To our knowledge, this is the first report of longitudinal quantification of Mn in the same cell cultures. We hypothesized that cells that were continuously exposed to elevated Mn would accumulate less Mn over a 24-h period than cells receiving Mn for the first time. We reasoned that this would occur due to compensatory homeostatic responses to decrease net Mn uptake (perhaps via down-regulation of uptake transporters), although this might depend on the concentration of Mn exposed.
STHdh cells were plated on day −1, brought to 37 °C on day 0, and treated with 1, 10, or 50 μm Mn beginning on day 1. Every 24 h, cells were assayed by MESMER and then returned to media containing the same Mn concentration as before. Every 24 h, new cells received Mn for the first time, and MESMER would assay their uptake 24 h later. The first direct comparison between treatment type (continuous or first exposure) occurred at 48 h post-initial Mn exposure of the controls. The results were analyzed by a repeated measures two-way ANOVA for each exposure concentration. There was a significant main effect of day (hours post-initial Mn) seen at each concentration (####; p < 0.0001; Fig. 10, A–C). There was a significant main effect of day (hours post-initial Mn) seen at each concentration (p < 0.0001; Fig. 10, A–C). There was also a main effect of treatment type (Continuous or First Mn Exposure, Fig. 10, A and B, p < 0.01; C, p < 0.0001) in all three concentrations exposed. Furthermore, Sidak's multiple comparisons noted significant differences between treatment-type groups at specific days (Fig. 10, *, p < 0.05; **, p < 0.01; *** p < 0.001).
Figure 10.
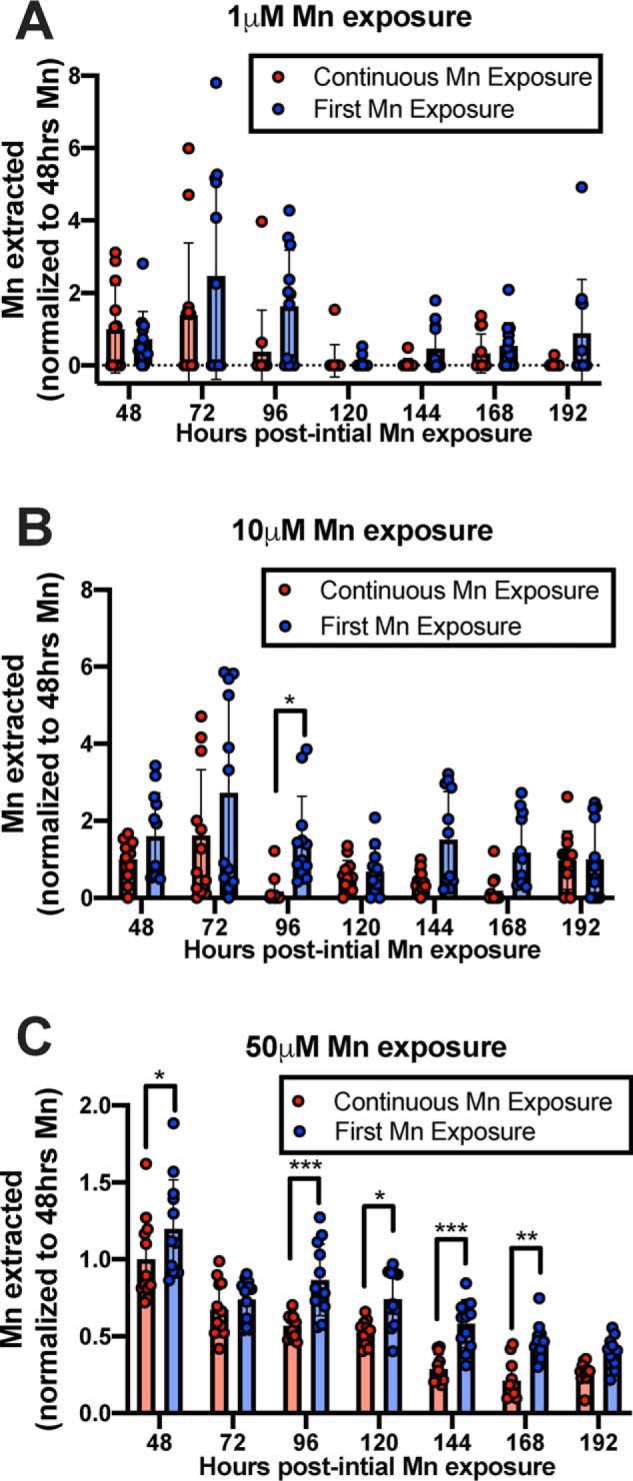
Continuous Mn exposure to neuronal cultures leads to decreased Mn accumulation relative to cells exposed to Mn for the first time. STHdh Q7 cells were exposed to 1, 10, or 50 μm MnCl2 in cell medium at 37 °C. Every 24 h, control cells (Continuous Mn Exposure) were washed with PBS and assayed with MESMER so that intracellular Mn could be quantified and then returned to media with Mn (red). Every 24 h, new cells that were previously unexposed to Mn, but the same age as the control, were exposed to Mn for the first time and MESMERized 24 h later (blue). n = 12 for each group, across two independent trials. Each point represents one well of cells. A repeated-measures two-way ANOVA showed significant main effects for the number of hours at all Mn concentrations (p < 0.0001; A–C). There was also a main effect of treatment type (A and B, p < 0.01; C, p < 0.0001) but no significant interaction between the two at any concentration exposed. Sidak's multiple comparisons confirmed significant differences in Mn extracted from cells between treatment groups (*, p < 0.05; **, p < 0.01; and *** p < 0.001).
It is noteworthy that by using MESMER to perform this experiment, compared with multiple CFMEA plates for each time point, the experiment took (one) 96-well plate × (three) biological replicates = (three) 96-well plates total. In contrast, multiple CFMEAs would require (four) 96-well plates just for one replicate and 12 plates total for three replicates. This MESMER experiment was four times smaller, saving the cost of 864 wells worth of cells and reagents.
Discussion
Here, we report on the identification of a small molecule in an efflux screen of Mn influencers, and we demonstrate it shows functional properties consistent with being a selective ionophore for Mn (47). We have adapted this small molecule as a tool and modified an assay around it to assess intracellular Mn accumulation. We then used this assay to examine a basic scientific question about cellular Mn homeostasis and compensatory changes due to prior exposures. Advancements in technology and innovation drive research science discoveries. Thus, in this case, we were lacking the ability to quantitatively measure Mn concentrations in living cells without killing them, a “necessity” for data concerning biological Mn research. Altered Mn biology has been implicated in neurodevelopment and multiple neurological motor disorders, such as Huntington's disease and a parkinsonian-like condition known as manganism (1). Patients with genetic mutations encoding proteins implicated in Mn homeostasis can suffer from manganese toxicity with symptoms ranging from motor impairment to liver cirrhosis (8–18, 41, 42).
Current assays of intracellular Mn measurements (except using Fura-2 AM, which is responsive to cytosolic (50) Mn specifically as the dye does not typically penetrate intracellular organelles and provides relative quantification or semi-quantitative measures only (27, 46)) require lysis of the cells being studied, which restricts longitudinal experiments and multiplexed outcome measures. This includes the CFMEA, which has been previously validated by AAS (38). In addition to this limitation, it's a time and financial burden to kill cells that may have required substantial funds and time to grow. For example, the differentiation of hiPSCs down the floor plate lineage to make mature (150 day) dopamine neurons currently costs approximately $1000 per multititer plate (calculations not shown). For this reason, it is notable that we are able to quantify concentration- and time-dependent effects by MESMER in NPCs (Fig. S3). This opens the door for any future experiments with hiPSC-derived cultures, and it fully takes advantage of repeatability/nontoxicity of the assay that would normally be a financial and time burden in lethal assays.
We postulate that MESM is acting as an ionophore at the cell surface, so that Mn ions may move in either direction, and whether there is net efflux or accumulation would depend on the relative concentration of Mn in the cytosol versus extracellular space. Thus under different contexts (e.g. high extracellular Mn or previous Mn exposure followed by low extracellular Mn), it may induce a net increase or decrease in cellular Mn levels following the direction of the Mn concentration gradient. The differences between the net Mn efflux and net Mn accumulation experiments (e.g. Fig. 1, C versus D) are due to differences in the direction of the Mn concentration gradient. For the efflux experiments, cells are first exposed to high extracellular Mn to “load” more Mn into the cells. Then, the extracellular Mn concentration is brought back to low again as the extracellular Mn is washed away. This creates a concentration gradient of Mn to move outside (efflux) from the cell, and this is greatly facilitated when MESM is then added. In contrast, if MESM is instead simply co-exposed with micromolar Mn, and extracellular Mn is kept high, Mn will accumulate more with MESM than vehicle alone. These data suggest that cytosolic levels of Mn are lower than the extracellular concentrations we are exposing them to, thus leading to a net influx of Mn.
MESMER allows total intracellular Mn to be quantified by equilibrating total cellular Mn with the extracellular buffer where it is quantified by Fura-2 quenching. Given the small total intracellular volume of the cultured cells in one well (∼100–1000 nl), compared with the 100 μl of extracellular volume Mn is extracted in, the dilution of intracellular to extracellular Mn is at least 1:100. Thus, provided MESM enables a Mn equilibrium where intracellular and extracellular Mn concentrations are the same, the majority of total Mn (>99%) would be extracellular and quantifiable by Fura-2 quenching. This is supported by the ICP-MS data (Fig. 5A), where the Mn remaining after MESMER, as measured by ICP-MS, is not zero but ∼1% of the total Mn effluxed by MESMER.
The results of the ICP-MS analysis (Fig. 5) independently confirm that MESMER is accurately and precisely measuring Mn content. Indeed, the ionophore appears to be Mn-specific; other divalent metals are not depleted from the cell by MESM exposure, whether extracellular Mn is added or not. Notably, there is a small but statistically significant increase of Ca2+ and Cu2+ from vehicle to MESM treated as measured by ICP-MS; however, several aspects make this statistic misleading. First, the cells are washed with PBS prior to extraction with MESM, and this efflux buffer contains no added Ca2+ or Cu2+. In other words, there aren't any extracellular sources that Ca2+ or Cu2+ could be coming from to increase Ca2+ or Cu2+ in the cell. Second, under these conditions, the concentration gradient is facing outward, so an ionophore would only move Ca2+ or Cu2+ out of the cell, rather than in the cell. Thus, we postulate that Ca2+ and Cu2+ are normally lost from the cell during the wash process, but MESM is decreasing this loss only in cells that had previously been exposed to Mn. Third, calcium transport experiments following ionophore exposures (Fig. 9) previously confirmed the null affinity MESM has for Ca2+.
Finally, we point out that the Ca2+ measured by ICP-MS is 44Ca, rather than 40Ca, which makes up only 2.9% of all Ca (40Ca is not readable as it coincides with argon, which is an important component used in the ICP-MS methodology). The amount of 44Ca may not necessarily be representative of all cellular Ca2+, as isotope fractionation variability exists biologically (51, 52).
Regarding the efflux screen, one could hypothesize that the small molecules that increase net Mn uptake when co-incubated with Mn might do so by blocking efflux or those that decrease net Mn levels do so by facilitating efflux. Combined with the data shown in Fig. S1, it is clear that whether a molecule was identified to increase or decrease net Mn levels did not predict an increase or decrease in efflux rates. However, the only molecule that was seen, based upon the n = 2 screen, to substantially slow Mn efflux was in fact a “Mn decreaser,” VU00243195 (Fig. 1A and Fig. S1D). This was the only one of six decreasers screened that had a significant impact with an altered t½ value. Out of the 22 total, nine classified “increasers” had significantly different t½ values than vehicle.
The two cell lines primarily used in this study were chosen because the STHdh cells are neuronally-derived from the striatum, an area of the brain that is particularly affected by Mn accumulation and toxicity. Furthermore, this cell line is among the most sensitive cell lines to Mn cytotoxicity with an LC50 value of ∼1 mm after 24 h (31). These WT cells also have an HD model counterpart that demonstrates a clear deficit in Mn accumulation. HEK293 cells were chosen as they are a common human cell line and are quite dissimilar from a mouse neuronal lineage, with an LC50 value of 2.1 mm after 16 h (53). As MESMER was also able to replicate concentration- and time-dependent effects of Mn on hiPSC-derived human striatal precursors, we would suspect that MESMER would work on most mammalian cell types (Fig. S3). This is further supported by our data showing that MESM does not appear to require energy or cellular transporters for its actions (Fig. 8A), so cell type differences in Mn transporters should not influence the ability of the assay to work. Of course, this does not mean MESMER will be effective in all types of cells. Of particular interest, specialized polarized cells such as enterocytes or hepatocytes may not be quantifiable with MESMER. This remains to be seen. We should also note that due to the Ca/Mg-free conditions required for MESMER to detect Mn, this technique could not be used in animal tissue. MESM cannot influence intracellular Mn while in the presence of media (Fig. S8), presumably due to the majority of the molecule being bound by protein in the serum.
There are two broadly-defined types of ionophores, namely channel-forming and carrier- mediated (54). Our data suggest that MESM is the latter. The observation that co-treatment is significantly more effective than sequential treatment of MESM and Mn (Fig. S5A) is more consistent with a mechanism of a carrier-mediated ionophore than one that forms a pore. Channel formers also tend to have a longer effect than carrier-mediated ionophores (54). When STHdh cells are pre-exposed to MESM for 1 h, washed, and then incubated an additional hour before Mn exposure, no effect of MESM is seen (Fig. S5B). This relatively fast degradation time is consistent with MESM not being a channel former.
Future experiments should explore the structure–activity relationship of MESM and Mn by testing similarly-structured small molecules. It is noteworthy that MESM lacks a polyether motif common among ionophores, and thus how it binds Mn is currently unknown. We suspect that at concentrations above 10 μm, MESM competes with Fura-2 for Mn binding, which is why Fig. 5 is parabolic. This is not seen in Fig. 2 because it does not go above 3 μm, but this idea is supported in Fig. S2. Two structural analogs have already been tested in the screen: VU0028040 and VU0027832. They demonstrate a clear increase in the rate of efflux compared with vehicle, with t½ values of 22.3 and 22.6 min, respectively (compared with vehicle of 79.1 min). However, they still require an hour to release nearly 100% of Mn compared with MESM's 15 min Fig. S1, P and Q (39). Radiolabeled or fluorescent analogs could help tighten the kinetics of the molecule and help identify its localization within the cell, as it is currently uncertain where MESM is located at a subcellular level, although evidence supporting Mn uptake into the nucleus, mitochondria, and Golgi apparatus has been reported (55–59). The use of a selective and cell-permeable Mn sensor in combination with MESM would identify where MESM affects Mn within the cell (55, 60). Considering how much Mn is accumulated within the cell during Mn/MESM co-exposure (an amount much higher than predicted if only equilibrium is being reached), we hypothesize that MESM is only impacting transport of Mn across the plasma membrane or an extracellular cytosol transporter, but not where Mn is being stored in a subcellular compartment. Most literature suggests that the majority of subcellular Mn is stored in the mitochondria (56–59). In this model, MESM would bring Mn from the extracellular space across the plasma membrane to the cytosol, where Mn is then taken up by a compartment not accessible to MESM-selective Mn permeabilization. MESM is thus able to promote an increase in total cellular Mn levels over time, while only maintaining equilibrium between the cytosol and extracellular space. Because the location of where MESM is binding at a subcellular level is unknown, likewise we do not know which proteins might be “de-metalled” following MESMER. This may have metabolic or homeostatic consequences not detected by our studies.
The experiments in Fig. 10 are the first longitudinal studies of cellular Mn levels performed in cultured cells of the same wells following continued Mn exposures. The results provide evidence of a compensatory homeostatic response to continuous elevated Mn exposures. Although the overall decline in Mn uptake over time seen in Fig. 10C is unprecedented, the magnitude in effect of treatment does not appear to change across time ranges between 48 and 168 h. This effect (main effect of treatment type) can be detected with just a 1 μm addition of Mn, well-below the physiological concentration range of Mn in humans (20–55 μm). The effect size varies at the 1 and 10 μm levels up to 8-fold, which is at least in part because the amount of Mn quantified was hovering around the detection threshold of the assay (∼100 nm or 10 pmol of Mn), and the average amount of Mn detected in cells exposed to 1 μm Mn is at or close to zero. It is noteworthy that when using higher concentrations of Mn (50 μm), the maximum fold-change is only ∼1.5-fold. In conditions where extracellular Mn is elevated above physiological intracellular levels (e.g. >100 μm), no differences in Mn uptake were detected with prior Mn exposure (Fig. S9). This suggests that cells are incapable of adjusting to extracellular Mn concentrations above their total intracellular concentration levels. Unfortunately, detection limits by the assay prevent us from investigating sub-physiological Mn homeostasis, although the importance of staying within physiological levels of Mn while studying Mn homeostasis should inform future experiments.
Physiological intracellular concentrations of Mn are in the low micromolar range, and 50 μm represents the high end of that range (2). However, pathophysiological levels could be 3-fold higher, bringing us to ∼150 μm. We used exposure levels comparable with what is utilized in the literature to examine the biological effects of Mn toxicity in the absence of cell death. These concentrations though are total tissue or cellular levels, with the majority believed to be intracellular. Extracellular levels of Mn are typically in the low 10–100 nm range in serum and cerebrospinal fluid. When we use MESM to equilibrate Mn between intracellular and extracellular, the high extracellular volume substantially dilutes the intracellular concentration, thus making detection of normal physiological levels not possible by Fura-2, which has a limit of detection near 100 nm. The dynamic range for the assay is 10 pmol to 3 nmol per sample in 100 μl of efflux buffer (96-well format).
Other exposure paradigms might yield stronger effects than seen here. We expect future studies to pursue these instances, so that subsequent experiments can identify protein and expression level changes of putative transporters or accompanying players. One may expect to see changes in the activation of Mn-responsive proteins such as AKT, p53, and GLT-1 if Mn homeostasis is altered. If there are decreases in Mn accumulation, this might be correlated to changes in expression of Mn transporters like ZIP8, ZIP14, and SLC30A10. Most importantly, we would want to know whether these changes in Mn accumulation/protein expression are meaningful. Are there changes in Mn-dependent enzyme activity, such as arginase or glutamine synthetase? Do these changes have pathological consequences? Now that we are able to monitor Mn accumulation longitudinally with MESMER, if we can induce a model of altered Mn homeostasis, then we can ask questions about what it really means to have altered Mn homeostasis on a cellular level.
Experimental procedures
Cell culture
STHdhQ7/Q7 WT immortalized murine striatal derived cells (STHdh) were obtained from the Coriell Cell Repository (Camden, NJ). They were plated at a density of 10,000 cells per well of a 96-well plate and incubated at 33 °C with 5% CO2 in Dulbecco's modified Eagle's medium (DMEM) (high-glucose, Sigma; D6546) with 10% fetal bovine serum (FBS) (Atlanta Biologicals; Flowery Branch, GA), 1% penicillin/streptomycin (15140-122; Life Technologies, Inc.), 2 mm GlutaMAX (Life Technologies, Inc.), 0.5 mg/ml G418 sulfate (Life Technologies, Inc.), 1× nonessential amino acid solution (Life Technologies, Inc.), and 14 mm HEPES (Life Technologies, Inc.) for 24 h prior to the assay. Cells were dissociated using 0.05% trypsin/EDTA solution (Life Technologies, Inc.).
HEK293 (ATCC) cells were plated at a density of 60,000 cells per well of a 96-well plate and incubated at 37 °C with 5% CO2 in DMEM (Corning, 15-013-CV) with 10% FBS and 1% penicillin/streptomycin for 24 h prior to the assay. Cells were dissociated using 0.05% trypsin/EDTA solution (Life Technologies, Inc.).
hiPSCs cell culture
hiPSCs line was derived from a healthy male control subject, CD12, at the age of 53 with no family history of neurodegenerative disease (61). hiPSCs were maintained in mTeSR1 medium (StemCell Technologies, Vancouver, British Columbia, Canada) on Matrigel (BD Biosciences)-coated plates. hiPSCs were dissociated by incubating for 10 min in Accutase at 37 °C (Innovative Cell Technologies, San Diego, CA), then centrifuged and resuspended in mTeSR1 with 10 μm ROCK inhibitor Y-27632 (Tocris), and replated at 100,000 cells/ml. Striatal neural differentiation was started after the hiPSCs cultures reached 100% confluency. Islet1 striatal differentiation was conducted for 10 days via dual SMAD neural induction protocol using LDN (4 μm) (Stemgent catalog no. 04-0074) and SB431542 (10 μm) (Stemgent catalog no. 04-0010), but in addition purmorphamine (0.65 μm) (Stemgent, Cambridge, MA) was added to pattern striatal NPCs. NPCs were replated at 300,000 cells/ml. Additional details and validation of this differentiation method have been previously reported (29, 31, 62).
Mn efflux with small molecules
Cells (excluding NPCs) were washed twice with HBSS (with Ca2+ and Mg2+; Gibco,+ 14025-092) and exposed to varying MnCl2 concentrations (AC205891000; Thermo Fisher Scientific, Waltham, MA) for 2 h in HBSS and incubated at 37 °C unless otherwise indicated. After incubation, the cells were washed three times with PBS (lacking Ca2+ and Mg2+) to remove the extracellular Mn. The cells were then incubated in efflux buffer (PBS containing 500 nm Fura-2; Enzo Life Sciences) to allow for intracellular Mn to efflux into the extracellular space. Small molecules (or corresponding vehicle 0.1% DMSO, D8418, Sigma) were often included in this efflux buffer to assess their impact on Mn efflux rates, as in Fig. 1A. VU0026921 and VU0028386 (MESM) were synthesized at the Vanderbilt Synthesis Core. All other small molecules were obtained from the ChemBridge Library. The fluorescence of the Fura-2 in the efflux buffer was then measured at 360/535 nm (excitation/emission) so that Mn concentrations could be quantified (Synergy H1 plate reader; Biotek). Mn was quantified based on standard curves previously described (38) and explained in brief below.
Cellular Mn level assay and extraction with Triton/CFMEA
STHdh cells were exposed to varying Mn concentrations at 37 °C or on ice in HBSS for 2 h. Often, other small molecules were co-incubated with the Mn in the HBSS to assess their impact on Mn levels, as in Fig. 1D. After incubation, the cells were washed three times with PBS (without Ca2+ and Mg2+) to remove the extracellular Mn. Extraction buffer (PBS containing 0.1% Triton X-100; Sigma, T8787, and 500 nm Fura-2) was added to lyse the cells. The fluorescence of the Fura-2 in the extraction buffer was then measured at 360/535 nm (excitation/emission) so that Mn could be quantified.
Manganese quantification by Fura-2
A percent maximum of Fura-2 fluorescence 360/535 nm (excitation/emission) was calculated for each condition as the corresponding zero Mn condition was defined as 100%. Background fluorescence was subtracted from all values. The concentration of Mn (in nanomolar) was calculated with the following equation: [Mn] = 1138 × ((1/% maximum) − 1)0.9682, which was generated based on a standard curve of Mn quenching Fura-2 signal (40). The absolute values were calculated by multiplying by the concentration by volume.
Calcium influx quantification
HEK293 cells were plated the day before the experiment at 15,000 cells per well and grown until 100% confluency. Cells were incubated with Fluo-4 AM (2 μm final concentration; ION Biosciences, San Marcos, TX) dissolved first in DMSO and then diluted in Hanks' balanced salt solution plus 20 mm HEPES, pH 7.3, and 0.03% Pluronic F-127 (Sigma) for 1 h at room temperature prior to adding varying concentrations of MESM, ionomycin (Sigma; I0634), calcimycin (Enzo Life Sciences; BML-CA100-0010), and manganese(II) ionophore II (Sigma; 43359), ranging from 3 nm to 30 μm. Using the Panoptic kinetic imaging plate reader (WaveFront Biosciences, Franklin, TN) to measure fluorescence every second for 5 min, 20 μl of ionophore from a donor plate was added to 20 μl/well buffer (HBSS + 20 mm HEPES, pH 7.3; Thermo Fisher Scientific) in the acceptor plate containing cells. Fluorescence was measured at 482 ± 16 nm excitation and 536 ± 20 nm emission. The maximum amplitude was calculated after normalizing for light variations with a static ratio and subtracting background. Percent fluorescence was normalized to the maximum fluoresced wells in each plate (30 μm ionomycin).
MESMER
Cells were exposed to Mn and washed in the same conditions as the CFMEA procedure. The difference was that after washing, efflux buffer was added containing MESM (3 μm unless otherwise indicated) and incubated at 37 °C for 15 min before reading fluorescence at 360/535 nm (excitation/emission).
Continuous Mn treatments with MESMER
STHdh cells were seeded in DMEM on day −1 (10,000 cells/well) and incubated at 33 °C until day 0. On day 0, cells were brought to 37 °C. On day 1, the initial Mn exposure began on the control cells at 1, 10, or 50 μm Mn in DMEM. From this point, every 24 h these cells were washed three times in PBS (without Ca2+ and Mg2+) and then MESMERized with 3 μm MESM in efflux buffer. After 15 min, the efflux buffer was transferred to a separate microplate that would have its fluorescence read at 360/535 nm (excitation/emission). The cells were returned to media containing the same concentration of Mn exposed previously. Every 24 h, for the next 7 days, new cells that were also plated on day −1 but previously unexposed were exposed to Mn in media. The new cells were MESMERized after 24 h, and their efflux buffer fluorescence was read.
Multiple Mn pretreatments
STHdh cells were seeded in DMEM on day 0 (10,000 cells/well) and incubated at 33 °C until day 1. Cells were washed twice with HBSS and exposed to 0, 50, 100, or 200 μm Mn for 2 h at 37 °C. Afterward, the extracellular Mn was washed off with three PBS washes and had extraction buffer added for CFMEA and efflux buffer with 3 μm MESM added for MESMER. After 15 min, the cells that were MESMERized had their fluorescence read and the efflux buffer was then carefully pipetted off and replaced with DMEM before returning to the incubator for 48 h. For the cells lysed open by the CFMEA, a sample was kept of each well to have their DNA content measured. This process was repeated on days 3, 5, and 7, corresponding to pre-exposures 1–3, respectively. Cells that received multiple Mn pretreatments received it every 48 h until lysed by CFMEA.
Cell viability and membrane integrity
Cells were washed twice with HBSS and exposed to varying concentrations (1–30 μm) of MESM and other ionophores or DMSO vehicle in HBSS. After incubating for 2 h at 37 °C, the cells were placed back in DMEM (100 μl per well) with 20 μl of CTB reagent added (Promega; G8081) for 3 h at 37 °C. The CTB assay relies on viable cells to reduce the reagent resazurin to a fluorescent product. After incubation, the fluorescence of each well was recorded at 560ex/590em. Percent fluorescence was normalized to the vehicle as 100%.
In separate experiments, STHdh cells were exposed to ionophore (3 μm) or DMSO (0.03%) for 2 h in HBSS and incubated at 37 °C before adding CytoTox Green reagent (Promega; G8742) at a 1:2000 dilution. The CellTox Green cytotoxicity assay is based on the high fluorescence of a reagent when bound to DNA. The reagent will have higher access to DNA in cells with impaired membrane integrity and will fluoresce more. A positive control of 0.1% detergent was used. After 15 min the fluorescence was recorded at 495ex/525em.
Parallel artificial membrane assay (PAMPA)
The 96 trans-well assay plates (Corning Gentest; 353015) were kept packaged and frozen at −20 °C until 1 h prior to the assay. After warming to room temperature, the membranes were soaked in PBS for another 90 min. Mn was added on the donor side of the sandwich membrane at 2 μm in PBS, with 500 nm Fura-2 in PBS on the opposite side of the sandwich (acceptor side). MESM, calcimycin, VU0026921, or DMSO were added with the Mn on the donor side at 3 μm final so that there were equal volumes of buffer (300 μl) on each side. The trans-wells were added together and incubated for 30 min before 100 μl of the acceptor side was removed and placed in a new 96-well plate and read to assess Fura-2 quenching at 360ex/535em.
ICP-MS
Samples were prepared from STHdh cells plated at 100,000 cells/ml and grown until confluency in 6-well plates. Cells were exposed to 50 μm MnCl2 in DMEM for 2 h before washing three times with PBS. Cells were co-incubated with 3 μm MESM or vehicle for 15 min. The cells were scraped, collected, and spun at 200 × g for 5 min, and then the supernatant was aspirated off the pellet. Cell pellets collected for metal analysis by ICP-MS were resuspended in 100 μl of RIPA buffer containing protease inhibitor (Sigma) and phosphatase inhibitor cocktails 2 and 3 (Sigma). Cell lysates were centrifuged at 4 °C for 10 min at 20,000 × g. 20 μl of the lysates was used for quantifying protein concentration using the BCA assay (Pierce Technologies). The remaining 80 μl of the lysate was diluted (1:10) with 720 μl of concentrated nitric acid (70%) and incubated at 95 °C for 40 min. All the samples were diluted with 2% nitric acid concentration with ultrapure water. To avoid potentially high results, all the samples were further diluted 10-fold. Overall, the dilution factor for these samples is 3150. Measurements were made on an Element 2 ICP-MS instrument (Thermo Fisher Scientific) at the Purdue University Analytical Mass Spectrometry Facility. The samples were analyzed for Mn alone followed by the other metals, including magnesium, calcium, iron, copper, cobalt, zinc, and molybdenum. Fresh samples were prepared for both analyses. Standards of 0.1 ppb was prepared for Mn by diluting 1000 μg/ml Mn in 2% nitric acid (Exaxol Corp., Clearwater, FL). Standards of 0.1, 1, and 10 PPB were prepared for all others by diluting a 68-element standard in 2% nitric acid (Exaxol Corp.). All the samples were calculated as micrograms of metal per mg of protein based on the protein concentration quantified.
DNA quantification
Cells were lysed with 0.1% Triton, and 5 μl from each well was added to 95 μl of Tris-EDTA buffer (Corning; 46-009-CM) with 1:800 PicoGreen reagent (final, Quant-iT PicoGreen dsDNA assay kit; Invitrogen, P7589). Fluorescence of each sample well was measured at 490ex/525em. DNA content for each well was quantified based on an equation of linear regression generated by the fluorescence of a DNA standard curve containing equal amounts of PicoGreen reagent and 0.1% Triton.
MESM molecule synthesis
Mass spectra were obtained on an Agilent series 1200 single quad ChemStation autosampler system using electrospray ionization (ESI) in positive mode. The HPLC was run with Accucore C18 column (2.6 μm, 2.1- × 30-mm column) at 40 °C, an acetonitrile/water with 0.1% trifluoroacetic acid, gradient 40–90% acetonitrile for 1.5 min, and a flow rate of 1.5 ml/min (Fig. S7).
Synthesis of (2-hydroxyphenyl) (piperidin-1-yl)methanethione (1)
2-Hydroxybenzaldehyde (1 ml, 0.0094 mol), sulfur (315 mg, 0.0097 mol, 1.05 eq), and piperidine (926 μl, 0.0094 mol, 1 eq) were added to a flamed-dried microwave vial (20 ml) equipped with magnetic stir bar. The vial was irradiated at 100 °C for 2 h, cooled down to room temperature, and methanol (1 ml) was added. The mixture was heated at 80 °C to dissolve the solid and cooled down to room temperature. The resulting solid was filtered and washed with methanol. The amount obtained was 0.89 g (43%). 1H NMR (DMSO) δ 9.69 (s, 1H), 7.15–7.11 (m, 1H), 7.07 (dd, 1H, J1 = 8 Hz, J 2 = 4 Hz), 6.82–6.78 (m, 2H), 4.46–4.42 (m, 1H), 4.10–4.06 (m, 1H), 1.64 (m, 6H); MS (ESI): mass calculated for C12H15NOS, 211.3; m/z found, 212.3 [M + H]+.
Synthesis of 2-hydroxybenzothiohydrazide (2)
(2-Hydroxyphenyl)(piperidin-1-yl)methanethione (0.89 g, 0.004 mol) and hydrazine hydrate (50% in water, 5 ml) were added to a round bottom flask and heated under argon at 50 °C for 2 h. The reaction mixture was cooled down; water was added (5 ml), and the solution was carefully acidified to pH 6 by addition of acetic acid. The mixture was filtered, and the filtrate was extracted with ethyl acetate (3× 20 ml). The combined extracts were washed with water (30 ml) and concentrated in vacuo to half-volume. Dry ethanol was added, and solvent was removed to half-volume. This step was repeated twice more. The crude hydrazide was used as an ethanol solution (5 ml) for the next step. MS (ESI): mass calculated for C7H8N2OS, 168.2; m/z found, 168.2 [M + H]+, Rf = 0.34.
Synthesis of 5′-(2-hydroxyphenyl)-1-methyl-3′H-spiro[indoline-3,2′-[1,3,4]thiadiazol]-2-one (VU0028386, MESM)
2-Hydroxybenzo-thiohydrazide (ethanol solution, 5 ml) and 1-methylindoline-2,3-dione (220 mg, 1 eq) were placed in a flame-dried microwave vial (20 ml). Dry ethanol (12 ml) and glacial acetic acid (3 drops) were sequentially added. The vial was capped and irradiated at 100 °C for 20 min. The resulting solid was filtered off and washed with ethyl acetate to afford 0.36 g (30%) of VU0028386 (MESM). 1H NMR (DMSO) δ 10.27 (s, 1H), 8.76 (s, 1H), 7.73 (s, 1H), 7.58 (d, 1H, J = 8 Hz), 7.42 (m, 2H), 7.29 (tr, 1H, J = 8 Hz), 7.14 (tr, 1H, J = 8 Hz), 7.06 (d, 1H, J = 8 Hz), 6.96–6.92 (m, 2H), MS (ESI): mass calculated for C16H13N3O2S, 311.3; m/z found, 312.2 [M + H]+, Rf = 1.03.
MESM molecule availability
The molecule is commercially available from ChemBridge (SC-5679472) in small quantities so that interested parties can avoid the cost of synthesis.
Statistical analysis
All data were analyzed statistically by GraphPad Prism 8. The results of the ICP-MS were analyzed using a two-tailed Student's unpaired t test. Statistical significance was determined by paired t tests, ordinary one-way ANOVA, two-way ANOVA with Sidak's multiple comparisons, or Tukey's multiple comparisons where noted. Impact on efflux rates in Fig. 1 and Fig. S1 was determined by an extra sum of squares F-test to compare if one curve could adequately fit both data sets in a one-phase exponential decay model of nonlinear regression.
Author contributions
K. J. H., C. D. W., and A. B. B. conceptualization; K. J. H., R. C. B., and F. M. Y. data curation; K. J. H., R. C. B., and F. M. Y. formal analysis; K. J. H., M. A., and A. B. B. funding acquisition; K. J. H. and A. B. B. investigation; K. J. H., P. J., R. N., R. C. B., F. M. Y., P. C., G. A. S., C. D. W., and A. B. B. methodology; K. J. H. writing-original draft; K. J. H., P. J., R. N., K. K., M. A., G. A. S., C. D. W., and A. B. B. writing-review and editing; K. K., G. A. S., C. D. W., and A. B. B. resources; A. B. B. supervision; A. B. B. project administration.
Supplementary Material
Acknowledgments
The Panoptic plate reader was purchased from funds obtain through NIH Grant 1S10OD025281-01. The Panoptic is housed and managed in the Vanderbilt Institute of Chemical Biology's High-throughput Screen Facility.
This work has been supported in part by National Institutes of Health NIEHS Grants RO1 ES010563, RO1 ES016931, T32 ES007028, and T15 LM007450 (to A. B. B., K. J. H., and M. A.). C. D. W. is an owner of WaveFront Biosciences and ION Biosciences, maker of the Panoptic plate reader and Fluo-4 AM, respectively. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Figs. S1–S9.
- Mn
- Manganese
- HD
- Huntington's disease
- MESMER
- manganese-extracting small molecule estimation route
- MESM
- manganese-extracting small molecule
- ICP
- inductively-coupled plasma
- CFMEA
- cellular Fura-2 Mn extraction assay
- DMEM
- Dulbecco's modified Eagle's medium
- FBS
- fetal bovine serum
- HBSS
- Hanks' balanced salt solution
- ANOVA
- analysis of variance
- CTB
- Cell-Titer Blue
- PAMPA
- parallel artificial membrane permeability assay
- AAS
- atomic absorption spectroscopy
- hiPSC
- human-induced pluripotent stem cell
- NPC
- neuroprogenitor
- ESI
- electrospray ionization.
References
- 1. Horning K. J., Caito S. W., Tipps K. G., Bowman A. B., and Aschner M. (2015) Manganese is essential for neuronal health. Annu. Rev. Nutr. 35, 71–108 10.1146/annurev-nutr-071714-034419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bowman A. B., and Aschner M. (2014) Considerations on manganese (Mn) treatments for in vitro studies. Neurotoxicology 41, 141–142 10.1016/j.neuro.2014.01.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Madejczyk M. S., and Ballatori N. (2012) The iron transporter ferroportin can also function as a manganese exporter. Biochim. Biophys. Acta 1818, 651–657 10.1016/j.bbamem.2011.12.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Li X., Xie J., Lu L., Zhang L., Zhang L., Zou Y., Wang Q., Luo X., and Li S. (2013) Kinetics of manganese transport and gene expressions of manganese transport carriers in Caco-2 cell monolayers. BioMetals 26, 941–953 10.1007/s10534-013-9670-y [DOI] [PubMed] [Google Scholar]
- 5. Yin Z., Jiang H., Lee E.-S., Ni M., Erikson K. M., Milatovic D., Bowman A. B., and Aschner M. (2010) Ferroportin is a manganese-responsive protein that decreases manganese cytotoxicity and accumulation. J. Neurochem. 112, 1190–1198 10.1111/j.1471-4159.2009.06534.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Jin L., Frazer D. M., Lu Y., Wilkins S. J., Ayton S., Bush A., and Anderson G. J. (2019) Mice overexpressing hepcidin suggest ferroportin does not play a major role in Mn homeostasis. Metallomics 11, 959–967 10.1039/C8MT00370J [DOI] [PubMed] [Google Scholar]
- 7. Williams B. B., Kwakye G. F., Wegrzynowicz M., Li D., Aschner M., Erikson K. M., and Bowman A. B. (2010) Altered manganese homeostasis and manganese toxicity in a Huntington's disease striatal cell model are not explained by defects in the iron transport system. Toxicol. Sci. 117, 169–179 10.1093/toxsci/kfq174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Leyva-Illades D., Chen P., Zogzas C. E., Hutchens S., Mercado J. M., Swaim C. D., Morrisett R. A., Bowman A. B., Aschner M., and Mukhopadhyay S. (2014) SLC30A10 is a cell surface-localized manganese efflux transporter, and Parkinsonism-causing mutations block its intracellular trafficking and efflux activity. J. Neurosci. 34, 14079–14095 10.1523/JNEUROSCI.2329-14.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Quadri M., Federico A., Zhao T., Breedveld G. J., Battisti C., Delnooz C., Severijnen L. A., Di Toro Mammarella L., Mignarri A., Monti L., Sanna A., Lu P., Punzo F., Cossu G., Willemsen R., et al. (2012) Mutations in SLC30A10 cause Parkinsonism and dystonia with hypermanganesemia, polycythemia, and chronic liver disease. Am. J. Hum. Genet. 90, 467–477 10.1016/j.ajhg.2012.01.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Tuschl K., Clayton P. T., Gospe S. M. Jr., Gulab S., Ibrahim S., Singhi P., Aulakh R., Ribeiro R. T., Barsottini O. G., Zaki M. S., Del Rosario M. L., Dyack S., Price V., Rideout A., Gordon K., et al. (2012) Syndrome of hepatic cirrhosis, dystonia, polycythemia, and hypermanganesemia caused by mutations in SLC30A10, a manganese transporter in man. Am. J. Hum. Genet. 90, 457–466 10.1016/j.ajhg.2012.01.018 ; Correction (2016) Am. J. Hum. Genet. 99, 521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lechpammer M., Clegg M. S., Muzar Z., Huebner P. A., Jin L.-W., and Gospe S. M. Jr. (2014) Pathology of inherited manganese transporter deficiency. Ann. Neurol. 75, 608–612 10.1002/ana.24131 [DOI] [PubMed] [Google Scholar]
- 12. He L., Girijashanker K., Dalton T. P., Reed J., Li H., Soleimani M., and Nebert D. W. (2006) ZIP8, member of the solute-carrier-39 (SLC39) metal-transporter family: characterization of transporter properties. Mol. Pharmacol. 70, 171–180 10.1124/mol.106.024521 [DOI] [PubMed] [Google Scholar]
- 13. Koike A., Sou J., Ohishi A., Nishida K., and Nagasawa K. (2017) Inhibitory effect of divalent metal cations on zinc uptake via mouse Zrt-/Irt-like protein 8 (ZIP8). Life Sci. 173, 80–85 10.1016/j.lfs.2016.12.006 [DOI] [PubMed] [Google Scholar]
- 14. Lin W., Vann D. R., Doulias P. T., Wang T., Landesberg G., Li X., Ricciotti E., Scalia R., He M., Hand N. J., and Rader D. J. (2017) Hepatic metal ion transporter ZIP8 regulates manganese homeostasis and manganese-dependent enzyme activity. J. Clin. Invest. 127, 2407–2417 10.1172/JCI90896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Choi E. K., Nguyen T. T., Gupta N., Iwase S., and Seo Y. A. (2018) Functional analysis of SLC39A8 mutations and their implications for manganese deficiency and mitochondrial disorders. Sci. Rep. 8, 3163 10.1038/s41598-018-21464-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Park J. H., Hogrebe M., Fobker M., Brackmann R., Fiedler B., Reunert J., Rust S., Tsiakas K., Santer R., Grüneberg M., and Marquardt T. (2018) SLC39A8 deficiency: biochemical correction and major clinical improvement by manganese therapy. Genet. Med. 20, 259–268 10.1038/gim.2017.106 [DOI] [PubMed] [Google Scholar]
- 17. Aydemir T. B., and Cousins R. J. (2018) The multiple faces of the metal transporter ZIP14 (SLC39A14). J. Nutr. 148, 174–184 10.1093/jn/nxx041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zhao N., Zhang A. S., Wortham A. M., Jue S., Knutson M. D., and Enns C. A. (2017) The tumor suppressor, P53, decreases the metal transporter, ZIP14. Nutrients 9, E1335 10.3390/nu9121335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Aydemir T. B., Kim M.-H., Kim J., Colon-Perez L. M., Banan G., Mareci T. H., Febo M., and Cousins R. J. (2017) Metal transporter Zip14 (Slc39a14) deletion in mice increases manganese deposition and produces neurotoxic signatures and diminished motor activity. J. Neurosci. 37, 5996–6006 10.1523/JNEUROSCI.0285-17.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Xin Y., Gao H., Wang J., Qiang Y., Imam M. U., Li Y., Wang J., Zhang R., Zhang H., Yu Y., Wang H., Luo H., Shi C., Xu Y., Hojyo S., Fukada T., Min J., and Wang F. (2017) Manganese transporter Slc39a14 deficiency revealed its key role in maintaining manganese homeostasis in mice. Cell Discov. 3, 17025 10.1038/celldisc.2017.25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zogzas C. E., and Mukhopadhyay S. (2017) Inherited disorders of manganese metabolism. Adv. Neurobiol. 18, 35–49 10.1007/978-3-319-60189-2_3 [DOI] [PubMed] [Google Scholar]
- 22. Taylor C. A., Hutchens S., Liu C., Jursa T., Shawlot W., Aschner M., Smith D. R., and Mukhopadhyay S. (2019) cro SLC30A10 transporter in the digestive system regulates brain manganese under basal conditions while brain SLC30A10 protects against neurotoxicity. J. Biol. Chem. 294, 1860–1876 10.1074/jbc.RA118.005628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Jenkitkasemwong S., Akinyode A., Paulus E., Weiskirchen R., Hojyo S., Fukada T., Giraldo G., Schrier J., Garcia A., Janus C., Giasson B., and Knutson M. D. (2018) SLC39A14 deficiency alters manganese homeostasis and excretion resulting in brain manganese accumulation and motor deficits in mice. Proc. Natl. Acad. Sci. 115, E1769–E1778 10.1073/pnas.1720739115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Vollet K., Haynes E. N., and Dietrich K. N. (2016) Manganese exposure and cognition across the lifespan: contemporary review and argument for biphasic dose–response health effects. Curr. Environ. Heal. Rep. 3, 392–404 10.1007/s40572-016-0108-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Torres-Agustín R., Rodríguez-Agudelo Y., Schilmann A., Solís-Vivanco R., Montes S., Riojas-Rodríguez H., Cortez-Lugo M., and Ríos C. (2013) Effect of environmental manganese exposure on verbal learning and memory in Mexican children. Environ. Res. 121, 39–44 10.1016/j.envres.2012.10.007 [DOI] [PubMed] [Google Scholar]
- 26. Haynes E. N., Sucharew H., Kuhnell P., Alden J., Barnas M., Wright R. O., Parsons P. J., Aldous K. M., Praamsma M. L., Beidler C., and Dietrich K. N. (2015) Manganese exposure and neurocognitive outcomes in rural school-age children: the communities actively researching exposure study (Ohio). Environ. Health Perspect. 123, 1066–1071 10.1289/ehp.1408993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Roth J., Ponzoni S., and Aschner M. (2013) in Metallomics and the Cell (Banci L., ed) pp. 169–201, Springer, Netherlands: 10.1007/978-94-007-5561-1 [DOI] [Google Scholar]
- 28. Herrero Hernandez E., Discalzi G., Valentini C., Carmellino C., Rossi L., Venturi F., Chiò A., Sacchetti A., and Pira E. (2006) Follow-up of patients affected by manganese-induced Parkinsonism after treatment with CaNa 2 EDTA. NeuroToxicology 27, 333–339 10.1016/j.neuro.2005.09.003 [DOI] [PubMed] [Google Scholar]
- 29. Bryan M. R., Uhouse M. A., Nordham K. D., Joshi P., Rose D. I. R., O'Brien M. T., Aschner M., and Bowman A. B. (2018) Phosphatidylinositol 3-kinase (PI3K) modulates manganese homeostasis and manganese-induced cell signaling in a murine striatal cell line. NeuroToxicology 64, 185–194 10.1016/j.neuro.2017.07.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Bichell T. J., Wegrzynowicz M., Tipps K. G., Bradley E. M., Uhouse M. A., Bryan M., Horning K., Fisher N., Dudek K., Halbesma T., Umashanker P., Stubbs A. D., Holt H. K., Kwakye G. F., Tidball A. M., et al. (2017) Reduced bioavailable manganese causes striatal urea cycle pathology in Huntington's disease mouse model. Biochim. Biophys. Acta 1863, 1596–1604 10.1016/j.bbadis.2017.02.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Tidball A. M., Bryan M. R., Uhouse M. A., Kumar K. K., Aboud A. A., Feist J. E., Ess K. C., Diana Neely M. D., Aschner M., and Bowman A. B. (2015) A novel manganese-dependent ATM-p53 signaling pathway is selectively impaired in patient-based neuroprogenitor and murine striatal models of Huntington's disease. Hum. Mol. Genet. 24, 1929–1944 10.1093/hmg/ddu609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Pan Z., Choi S., and Luo Y. (2018) Mn2+ quenching assay for store-operated calcium entry. Methods Mol. Biol. 1843, 55–62 10.1007/978-1-4939-8704-7_4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Forbes J. R., and Gros P. (2003) Iron, manganese, and cobalt transport by Nramp1 ( Slc11a1) and Nramp2 ( Slc11a2) expressed at the plasma membrane. Blood 102, 1884–1892 10.1182/blood-2003-02-0425 [DOI] [PubMed] [Google Scholar]
- 34. Lockwich T., Mertz L. M., and Ambudkar I. S. (1993) Involvement of carboxyl groups in the divalent cation permeability of rat parotid gland basolateral plasma membrane. Mol. Cell. Biochem. 126, 143–150 10.1007/BF00925692 [DOI] [PubMed] [Google Scholar]
- 35. Lukács G. L., and Kapus A. (1987) Measurement of the matrix free Ca2+ concentration in heart mitochondria by entrapped fura-2 and quin2. Biochem. J. 248, 609–613 10.1042/bj2480609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Denny M. F., Hare M. F., and Atchison W. D. (1993) Methylmercury alters intrasynaptosomal concentrations of endogenous polyvalent cations. Toxicol. Appl. Pharmacol. 122, 222–232 10.1006/taap.1993.1191 [DOI] [PubMed] [Google Scholar]
- 37. Komagiri Y., Nakamura K., and Kubokawa M. (2011) A nicardipine-sensitive Ca2+ entry contributes to the hypotonicity-induced increase in [Ca2+]i of principal cells in rat cortical collecting duct. Cell Calcium 49, 35–42 10.1016/j.ceca.2010.11.006 [DOI] [PubMed] [Google Scholar]
- 38. Kwakye G. F., Li D., Kabobel O. A., and Bowman A. B. (2011) Cellular fura-2 manganese extraction assay (CFMEA). Curr. Protoc. Toxicol. 2011, Chapter 12, Unit 12.18 10.1002/0471140856.tx1218s48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kumar K. K., Lowe E. W. Jr., Aboud A. A., Neely M. D., Redha R., Bauer J. A., Odak M., Weaver C. D., Meiler J., Aschner M., and Bowman A. B. (2014) Cellular manganese content is developmentally regulated in human dopaminergic neurons. Sci. Rep. 4, 6801 10.1038/srep06801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kumar K. K., Aboud A. A., Patel D. K., Aschner M., and Bowman A. B. (2013) Optimization of fluorescence assay of cellular manganese status for high throughput screening. J. Biochem. Mol. Toxicol. 27, 42–49 10.1002/jbt.21457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kumar K. K., Aboud A. A., and Bowman A. B. (2012) The potential of induced pluripotent stem cells as a translational model for neurotoxicological risk. Neurotoxicology 33, 518–529 10.1016/j.neuro.2012.02.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Reed P. W., and Lardy H. A. (1972) A23187: a divalent ionophore. J. Biol. Chem. 247, 6970–6977 [PubMed] [Google Scholar]
- 43. Pfeiffer D. R., Reed P. W., and Lardy H. A. (1974) Ultraviolet and fluorescent spectral properties of the divalent cation ionophore A23187 and its metal ion complexes. Biochemistry 13, 4007–4014 10.1021/bi00716a029 [DOI] [PubMed] [Google Scholar]
- 44. Pfeiffer D. R., and Deber C. M. (1979) Isosteric metal complexes of ionophore A23187. A basis for cation selectivity. FEBS Lett. 105, 360–364 10.1016/0014-5793(79)80649-3 [DOI] [PubMed] [Google Scholar]
- 45. Kauffman R. F., Taylor R. W., and Pfeiffer D. R. (1980) Cation transport and specificity of ionomycin. J. Biol. Chem. 255, 2735–2739 [PubMed] [Google Scholar]
- 46. Gee K. R., Brown K. A., Chen W. N., Bishop-Stewart J., Gray D., and Johnson I. (2000) Chemical and physiological characterization of fluo-4 Ca2+-indicator dyes. Cell Calcium 27, 97–106 10.1054/ceca.1999.0095 [DOI] [PubMed] [Google Scholar]
- 47. Freedman J. C. (2012) Cell Physiology Source Book, Sperelakis N. ed. 4th Ed., pp. 61–66, Academic Press, Orlando: 10.1016/b978-0-12-387738-3.00004-4 [DOI] [Google Scholar]
- 48. Delnooz C. C., Wevers R. A., Quadri M., Clayton P. T., Mills P. B., Tuschl K., Steenbergen E. J., Bonifati V., and van de Warrenburg B. P. (2013) Phenotypic variability in a dystonia family with mutations in the manganese transporter gene. Mov. Disord. 28, 685–686 10.1002/mds.25390 [DOI] [PubMed] [Google Scholar]
- 49. Park J. H., Hogrebe M., Grüneberg M., DuChesne I., von der Heiden A. L., Reunert J., Schlingmann K. P., Boycott K. M., Beaulieu C. L., Mhanni A. A., Innes A. M., Hörtnagel K., Biskup S., Gleixner E. M., Kurlemann G., et al. (2015) SLC39A8 deficiency: a disorder of manganese transport and glycosylation. Am. J. Hum. Genet. 97, 894–903 10.1016/j.ajhg.2015.11.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Oakes S. G., Martin W. J. 2nd., Lisek C. A., and Powis G. (1988) Incomplete hydrolysis of the calcium indicator precursor fura-2 pentaacetoxymethyl ester (fura-2 AM) by cells. Anal. Biochem. 169, 159–166 10.1016/0003-2697(88)90267-9 [DOI] [PubMed] [Google Scholar]
- 51. Moynier F., and Fujii T. (2017) Calcium isotope fractionation between aqueous compounds relevant to low-temperature geochemistry, biology and medicine. Sci. Rep. 7, 44255 10.1038/srep44255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Skulan J., and DePaolo D. J. (1999) Calcium isotope fractionation between soft and mineralized tissues as a monitor of calcium use in vertebrates. Proc. Natl. Acad. Sci. U.S.A. 96, 13709–13713 10.1073/pnas.96.24.13709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Zogzas C. E., and Mukhopadhyay S. (2018) Putative metal binding site in the transmembrane domain of the manganese transporter SLC30A10 is different from that of related zinc transporters. Metallomics 10, 1053–1064 10.1039/C8MT00115D [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Stillwell W. (2016) An Introduction to Biological Membranes, pp. 423–451, Elsevier, London [Google Scholar]
- 55. Das S., Carmona A., Khatua K., Porcaro F., Somogyi A., Ortega R., and Datta A. (2019) Manganese mapping using a fluorescent Mn2+ sensor and nanosynchrotron X-ray fluorescence reveals the role of the Golgi apparatus as a manganese storage site. Inorg. Chem. 58, 13724–13732 10.1021/acs.inorgchem.9b01389 [DOI] [PubMed] [Google Scholar]
- 56. Gavin C. E., Gunter K. K., and Gunter T. E. (1990) Manganese and calcium efflux kinetics in brain mitochondria. Relevance to manganese toxicity. Biochem. J. 266, 329–334 10.1042/bj2660329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Liccione J. J., and Maines M. D. (1988) Selective vulnerability of glutathione metabolism and cellular defense mechanisms in rat striatum to manganese. J. Pharmacol. Exp. Ther. 247, 156–161 [PubMed] [Google Scholar]
- 58. Morello M., Canini A., Mattioli P., Sorge R. P., Alimonti A., Bocca B., Forte G., Martorana A., Bernardi G., and Sancesario G. (2008) Sub-cellular localization of manganese in the basal ganglia of normal and manganese-treated rats An electron spectroscopy imaging and electron energy-loss spectroscopy study. NeuroToxicology 29, 60–72 10.1016/j.neuro.2007.09.001 [DOI] [PubMed] [Google Scholar]
- 59. Kalia K., Jiang W., and Zheng W. (2008) Manganese accumulates primarily in nuclei of cultured brain cells. NeuroToxicology 29, 466–470 10.1016/j.neuro.2008.02.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Bakthavatsalam S., Sarkar A., Rakshit A., Jain S., Kumar A., and Datta A. (2015) Tuning macrocycles to design “turn-on” fluorescence probes for manganese(II) sensing in live cells. Chem. Commun. 51, 2605–2608 10.1039/C4CC09542A [DOI] [PubMed] [Google Scholar]
- 61. Tidball A. M., Neely M. D., Chamberlin R., Aboud A. A., Kumar K. K., Han B., Bryan M. R., Aschner M., Ess K. C., and Bowman A. B. (2016) Genomic instability associated with p53 knockdown in the generation of Huntington's disease human induced pluripotent stem cells. PLoS ONE 11, e0150372 10.1371/journal.pone.0150372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Joshi P., Bodnya C., Ilieva I., Neely M. D., Aschner M., and Bowman A. B. (2019) Huntington's disease associated resistance to Mn neurotoxicity is neurodevelopmental stage and neuronal lineage dependent. NeuroToxicology 75, 148–157 10.1016/j.neuro.2019.09.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.