

Abstract
Myosin-IC (Myo1c) has been proposed to function in delivery of glucose transporter type 4 (GLUT4)–containing vesicles to the plasma membrane in response to insulin stimulation. Current evidence suggests that, upon insulin stimulation, Myo1c is phosphorylated at Ser701, leading to binding of the signaling protein 14-3-3β. Biochemical and functional details of the Myo1c–14-3-3β interaction have yet to be described. Using recombinantly expressed proteins and mass spectrometry–based analyses to monitor Myo1c phosphorylation, along with pulldown, fluorescence binding, and additional biochemical assays, we show here that 14-3-3β is a dimer and, consistent with previous work, that it binds to Myo1c in the presence of calcium. This interaction was associated with dissociation of calmodulin (CaM) from the IQ motif in Myo1c. Surprisingly, we found that 14-3-3β binds to Myo1c independent of Ser701 phosphorylation in vitro. Additionally, in contrast to previous reports, we did not observe Myo1c Ser701 phosphorylation by Ca2+/CaM-dependent protein kinase II (CaMKII), although CaMKII phosphorylated four other Myo1c sites. The presence of 14-3-3β had little effect on the actin-activated ATPase or motile activities of Myo1c. Given these results, it is unlikely that 14-3-3β acts as a cargo adaptor for Myo1c-powered transport; rather, we propose that 14-3-3β binds Myo1c in the presence of calcium and stabilizes the calmodulin-dissociated, nonmotile myosin.
Keywords: myosin, cell motility, 14-3-3 protein, cytoskeleton, calmodulin (CaM), 14-3-3β, cell signaling, molecular motor, myo1c, myosin-IC
Introduction
Myosin-I isoforms are single-headed members of the myosin superfamily that link plasma and intracellular membranes to the actin cytoskeleton (1–3). Cell biological experiments have revealed a range of cellular processes in which myosin-I isoforms participate, including endosomal trafficking, endocytosis, cell–cell and cell–substrate adhesion, and membrane tension (2). Myosin-I isoforms are localized to subcellular regions by motor domain binding to tropomyosin-free actin filaments and by direct binding to the headgroups of negatively charged lipids and phosphoinositides through the pleckstrin homology domain–containing tail (2, 4–6). Numerous proteins that bind myosin-I isoforms have also been identified (reviewed in Ref. 2), but how these proteins participate in regulation, targeting, and/or anchoring of motors is unclear.
A widely expressed myosin-I isoform, myosin-IC (Myo1c), participates in insulin-stimulated translocation of GLUT4-containing vesicles to the plasma membrane in adipocytes (7, 8) and muscle cells (9). Myo1c associates directly with mobile and tethered GLUT4 vesicles, and it is required for insulin-induced vesicle tethering (9). It has been reported that Myo1c-involved GLUT4 transport is affected by myosin's interaction with an adaptor protein, 14-3-3β (10). Additionally, it was suggested that 14-3-3β binding is activated by Ca2+/CaM-dependent2 protein kinase II (CaMKII)–mediated phosphorylation of Myo1c Ser701. 14-3-3 proteins are a family of dimeric adaptors that predominantly bind phosphorylated proteins and mediate interactions with many different proteins (11). Thus, the interaction between Myo1c and 14-3-3β is an intriguing possible mechanical link between contractile and signaling proteins.
The proposed 14-3-3β binding site at Ser701 of Myo1c (10) directly precedes the first calmodulin-binding IQ motif near the fulcrum on the myosin lever arm (Fig. 1A) (12). This is a mechanically important region of the motor that links ATPase activity to the force-generating working stroke and would be an unexpected location for an adaptor that links cellular cargos to transport. Thus, we investigated the biochemical and functional details of the interaction of Myo1c with 14-3-3β. We found that 14-3-3β binding to Myo1c is regulated by Ca2+ and that this association correlates with CaM dissociation from the IQ motif, as suggested previously (10, 12). We also found that Myo1c bound to 14-3-3β does not support actin gliding in motility assays and that phosphorylation did not appear to affect 14-3-3β's ability to bind to the target sequence. These results support a calcium-dependent tethering role for the interaction of 14-3-3β with Myo1c rather than as an adaptor for a mechanically active motor.
Figure 1.

Effects of calcium on binding of 14-3-3β to Myo1c. A, top panel, cartoon representation of the crystal structure (PDB code 4BYF) of the Myo1c motor domain with 1-IQ (light gray) with bound CaM (green). The proposed phosphorylation site, Ser710, is highlighted in red. Bottom panel, schematic of the primary structure of full-length Myo1c and Myo1c-3IQ constructs. B, Coomassie-stained gel showing results of streptavidin bead–mediated pulldown of biotinylated 0.8 μm Myo1c-3IQ–WT and 2.0 μm CaM in the presence or absence of 2.5 μm 14-3-3β, 40 μm free calcium, and 1 mm MgATP. Lane M shows molecular weight markers.
Results
14-3-3β binding to Myo1c is enhanced by calcium and calmodulin dissociation
We investigated the interaction of 14-3-3β with a Myo1c construct that includes the motor domain, a regulatory domain with bound CaM, and a C-terminal Avi tag for site-specific biotinylation (Myo1c-3IQ, Fig. 1A). By analytical ultracentrifugation (see “Experimental procedures”), we determined that 14-3-3β is a dimer under experimental conditions, so dimer concentrations are reported below. Biotinylated Myo1c-3IQ (0.8 μm) was mixed with 2.5 μm 14-3-3β and pulled down by centrifugation of streptavidin-coated beads. 14-3-3β binds to Myo1c-3IQ, and this binding was enhanced in the presence of free calcium. Inclusion of 1 mm MgATP had no detectable effect on the binding (Fig. 1B).
Previous work showed that calcium binding to CaM weakens CaM affinity for Myo1c (13–15), and given that calcium appears to enhance 14-3-3β binding (Fig. 1B), we investigated the relationship between calmodulin and 14-3-3β binding. We mixed biotinylated Myo1c-3IQ with 0–10 μm 14-3-3β and pulled down the proteins with streptavidin beads in the absence and presence of 100 μm free calcium. In the absence of calcium, 14-3-3β bound weakly to Myo1c-3IQ (Fig. 2A). Addition of calcium resulted in increased binding with increased 14-3-3β concentration (0–10 μm) until binding approached saturation at an apparent ratio of one 14-3-3β dimer per Myo1c-3IQ. The increased 14-3-3β binding correlated with decreased calmodulin binding, with one 14-3-3β dimer displacing one calmodulin (Fig. 2A). Thus, 14-3-3β and CaM appear to compete for Myo1c binding.
Figure 2.

Association of 14-3-3β with Myo1c-3IQ. A, top panels, SYPRO-stained gels showing results of streptavidin bead–mediated pulldown of biotinylated Myo1c-3IQ with various concentrations of 14-3-3β in the absence (1 mm EGTA) and presence of 100 μm free calcium. Bottom panels, the stoichiometry of 14-3-3β and CaM to pulled down Myo1c-3IQ as determined from quantification of gel bands as described under “Experimental procedures.” Plotted values are mean ± S.D. from three to four independent pulldown assays. B, Coomassie-stained gel showing pulldown of biotinylated Myo1c-3IQ with 1.25 μm 14-3-3β under EGTA and Ca2+ conditions. Myo1c-3IQ was treated with or without 0.5 mm TFP.
To further explore the possibility that 14-3-3β competes with calmodulin for Myo1c binding, we treated Myo1c-3IQ with 0.5 mm trifluoperazine (TFP), a calmodulin agonist known to dissociate light chains from the IQ motifs of myosins (16). Upon pulldown of 0.5 μm Myo1c-3IQ in the presence of 1.25 μm 14-3-3β, TFP-mediated dissociation of calmodulin allows 14-3-3β to bind Myo1c robustly in the absence of calcium (Fig. 2B). In the presence of TFP, the molar ratios of 14-3-3β dimer and CaM to heavy chain in the absence of calcium were 1.7 and 0.9, respectively, as determined by densitometry. In the presence of calcium, the ratios of 14-3-3β dimer and CaM to Myo1c were 1.9 and 0.6, respectively. Thus, 14-3-3β competes with calmodulin for Myo1c binding.
Ser701 phosphorylation does not affect 14-3-3β binding to Myo1c
Phosphorylation of Ser701 of Myo1c by CaMKII has been reported to be important for myosin's association with 14-3-3β (10); thus, we examined the effect of CaMKII on Myo1c-3IQ under phosphorylation conditions (see “Experimental procedures”). MS analysis of sf9-purified Myo1c showed partial phosphorylation of Ser373 and Ser572. To dephosphorylate the protein, we preincubated the myosin with λ protein phosphatase and subsequently purified the protein. Dephosphorylated Myo1c-3IQ was incubated with activated CaMKII, and MS analysis showed that Ser73, Ser104, Ser332, and Ser501 of Myo1c were phosphorylated; however, no Ser701 phosphorylation was detected (Fig. S1 and Table S1), in contrast to previous investigations (10). Given the weak affinity of CaM for IQ1 in the presence of calcium (13), it is unlikely that CaM would sterically hinder CAMKII from interacting with Ser701 under the calcium-containing reaction conditions.
It is possible that an unknown kinase participates in the phosphorylation of Ser701 of Myo1c that was not captured in our in vitro assay. Therefore, we expressed unphosphorylatable (S701A) and phosphomimic (S701E) mutants of Myo1c-3IQ and examined 14-3-3β binding using two different pulldown assays. Soluble 14-3-3β was incubated with Myo1c-3IQ–S701A or –S701E attached to streptavidin-coated beads (Fig. 3A), and soluble Myo1c-3IQ–S701A or –S701E was incubated with GST–14-3-3β attached to GSH beads (Fig. 3B). Although mutation of serine to glutamic acid does not always mimic phosphorylation (17), we found in both assays that both mutants bound 14-3-3β, with enhanced binding in the presence of calcium. We also treated both mutants with 0.5 mm TFP and again found 14-3-3β binding (Fig. 3C), supporting the finding that phosphorylation is not required for 14-3-3β binding.
Figure 3.

Effects of phosphorylation of Ser701 on binding of 14-3-3β to Myo1c. A, Coomassie-stained gel of pulldown of 0.8 μm biotinylated Myo1c-3IQ S701A and S701E in the presence of 2.5 μm 14-3-3β in the absence (EGTA) and presence of 40 μm free calcium. ctrl, control. B, GSH–agarose bead–mediated pulldown of GST-14-3-3β in the presence of 1 μm Myo1c-3IQ–S701A or –S701E. # of washes indicates number of times the agarose beads were washed after pulldown. Top panel, pulled down Myo1c-3IQ was detected by Western blotting using anti-FLAG antibody. Bottom panel, GST–14-3-3β was detected by Coomassie staining. C, streptavidin bead–mediated pulldown of 0.5 μm Myo1c-3IQ–S701A and –S701E treated with TFP in the presence of 1.25 μm 14-3-3β.
Given the calcium sensitivity of 14-3-3β binding and its ability to compete with calmodulin, we assessed its ability to bind a peptide with the sequence of the first Myo1c IQ motif (IQ1). Peptides were synthesized without phosphorylation (IQ1-WT) and with Ser701 phosphorylation (IQ1-pSer701; Fig. 4). We determined the stoichiometry of 14-3-3β binding to IQ peptide by titrating both 1 μm and 5 μm IQ1-WT with 0–10 μm 14-3-3β and monitoring changes in intrinsic tryptophan fluorescence. The peptide–14-3-3β complex has a higher fluorescence level than the sum of the individual proteins (Fig. 4A). In both cases, we found that two peptides bind one 14-3-3β dimer, which suggests a tight affinity with dissociation constants well below experimental protein concentrations. We compared the stoichiometry of IQ1-WT and pSer701 interacting with 14-3-3β. Titrations showed that both IQ1-WT and IQ1-pSer701 bound 14-3-3β with a similar stoichiometry of two peptides per 14-3-3β dimer (Fig. 4B). This result differs from 14-3-3β binding experiments with Myo1c-3IQ, which suggests that one Myo1c-3IQ binds one 14-3-3β dimer, which may be the result of steric constraints.
Figure 4.
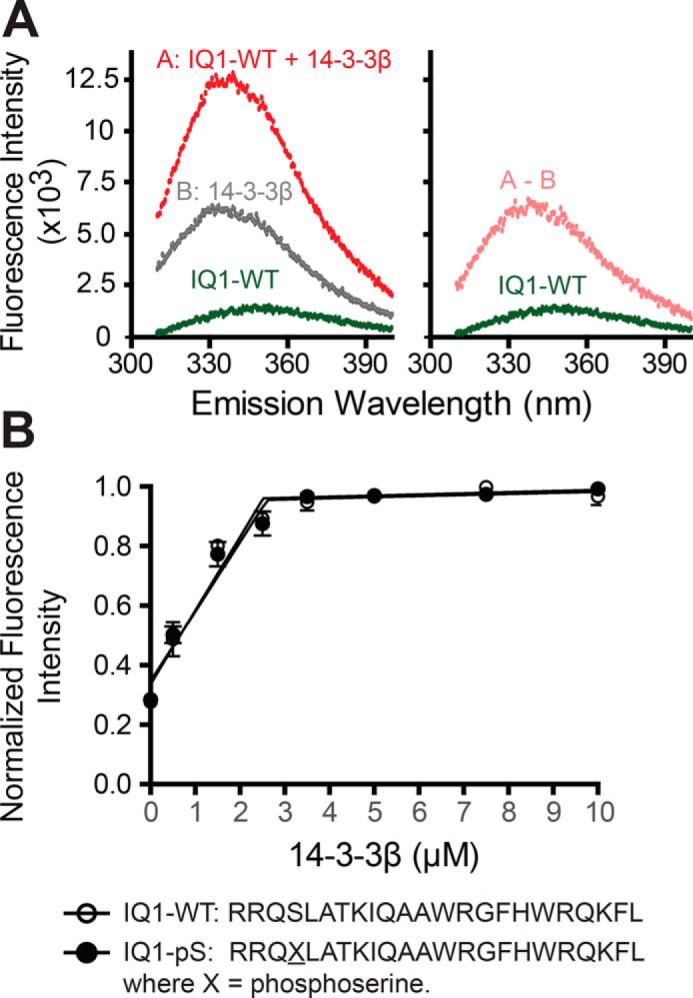
Determination of the binding stoichiometry of IQ peptides and 14-3-3β. A, steady-state fluorescence emission spectra of 14-3-3β with IQ1 peptide (λexcitation = 295 nm). Left panel, fluorescence emission spectra of 3.5 μm 14-3-3β dimer with 5 μm IQ1 peptide. Right panel, corrected fluorescence spectra of IQ1 peptide from subtracting the contribution of the spectra of samples containing only 3.5 μm 14-3-3β (A-B). B, 5 μm IQ peptides (IQ1-WT and IQ1-pSer701) were titrated with 0–10 μm 14-3-3β. Values (350 nm) are mean ± S.D. from three independent assays. Linear regression of the nonsaturated and saturated parts of the data points reveals saturation of IQ peptides at ∼2.5 μm 14-3-3β dimer.
14-3-3β does not affect Myo1c motor function
To determine whether 14-3-3β affects Myo1c motor function, we measured the ATPase activity of Myo1c in the presence of 50 μm actin and calcium. Although micromolar concentrations of calmodulin slightly activate the actin-activated ATPase (<2-fold) (13), we found no effect of 0–7.5 μm 14-3-3β on this activity (Fig. 5A).
Figure 5.

14-3-3β does not affect Myo1c motor function. A, ATPase activity of Myo1c-3IQ–WT in the presence of 14-3-3β. Steady-state ATPase activity (37 °C) was measured in KMg25 containing 50 μm free Ca2+ using the NADH-coupled assay. The values are mean ± S.D. from three independent assays. B, determination of in vitro actin gliding velocity of Myo1c-3IQ with 0–15 μm of 14-3-3β in the presence of EGTA. The inhibited motility was not rescued for Myo1c-3IQ–WT with 10 μm of 14-3-3β in the presence of 20 μm free Ca2+.
We determined whether modification of Ser701 affects the speeds of actin gliding. Because calcium inhibits actin gliding (13), Myo1c-3IQ–WT, –S701A, and –S701E were assayed under EGTA conditions. The three different myosin constructs powered actin gliding at the same speed, and 0–15 μm 14-3-3β did not affect this gliding rate (Fig. 5B). Although micromolar concentrations of calmodulin are able to restore actin gliding in the presence of calcium (13), 10 μm 14-3-3β was not able to rescue the inhibited motility of Myo1c (Fig. 5B).
Discussion
Our results show that in vitro binding of dimeric 14-3-3β to Myo1c is independent of phosphorylation and that it is enhanced by calcium-mediated dissociation of calmodulin from the motor's lever arm. We also found that 14-3-3β does not affect the ATPase activity of Myo1c in the presence of calcium and that 14-3-3β bound to myosin in the presence of calcium does not support actin filament gliding.
14-3-3β binding to nonphosphorylated IQ motifs
Pulldown assays show that 14-3-3β does not compete effectively with CaM for binding to Myo1c-3IQ in the absence of calcium but displaces a single CaM in the presence of calcium. Previous work with this Myo1c-3IQ construct showed that the IQ motif nearest the motor domain (IQ1) has the weakest affinity among the three for CaM in the presence of calcium (13). Taken together with the result that 14-3-3β has a very tight affinity for the IQ1 peptide (Fig. 4B), we propose that one 14-3-3β dimer is binding to IQ1 in the presence of calcium. However, we note that, when two CaMs are dissociated from Myo1c-3IQ by the agonist TFP, two 14-3-3β dimers are able to interact with the myosin.
Dimeric 14-3-3 proteins largely interact with phosphorylated ligands, forming a symmetric bowl-shaped structure consisting of helices that create an amphipathic ligand binding groove in which a positively charged cluster and a hydrophobic “roof” coordinate an interaction with phosphate (11, 18). Mechanisms of phosphorylation-independent binding of ligands to 14-3-3 appear to be more variable, with some proteins showing similar binding interactions with phosphorylated ligands (18–20) whereas others have unique modes of interaction, e.g. the α2 helix of carbohydrate response element–binding protein (ChREBP) (21). Comparison of the amino acid sequence of the α2 helix of ChREBP with Myo1c shows that the sequence of the key interacting amino acids of the α2 helix is similar to IQ1 of Myo1c (Fig. S2). Thus, Myo1c-3IQ may bind to 14-3-3β in a similar manner as ChREBP (Fig. S2). Finally, we note that residue Ser701 does not overlap with the calmodulin binding site on IQ1 (6, 12), and it points away from the conserved positively charged cluster (Arg58-Arg129-Tyr130), again suggesting that this residue does not have a role in 14-3-3β binding.
Cellular role of the 14-3-3β-Myo1c interaction
Although it has been demonstrated that Myo1c and 14-3-3β affect GLUT4 translocation in response to insulin stimulation (10), the molecular and functional details of these interactions are not clear. 14-3-3β binds to Myo1c in the presence of calcium, and it has been proposed that calcium signaling beneath the plasma membrane in both muscle and adipocyte cells is important for insulin-stimulated GLUT4 transport (22, 23). However, Myo1c-powered transport of GLUT4 in response to an increase in calcium concentration is unlikely. Calcium binding to Myo1c-bound CaM results in CaM dissociation, which structurally compromises the motor's lever arm. Although actin-activated ATPase activity increases in the presence of calcium, motility is inhibited (1, 13–15, 24). Micromolar concentrations of CaM can rescue motility in the presence of calcium in vitro; however, the free calmodulin concentration in cells is less than 200 nm in the presence of calcium (25, 26). IQ1 would be calmodulin-free under these conditions (13), and our results show that 14-3-3β does not rescue this calcium-inhibited Myo1c motility (Fig. 5B). Thus, our in vitro results indicate that it is unlikely that 14-3-3β acts as a cargo adaptor for Myo1c-powered transport of membranes.
14-3-3β bound to the first IQ motif may act as an adaptor that links Myo1c to other proteins, allowing the motor to act as a tether. Notably, several proteins other than calmodulin have been identified to bind to the IQ regions of Myo1c (27–30) and myosin-V (31), suggesting that this region not only acts as myosin's lever arm but is also an important signaling hub.
Finally, it has been shown in vitro that dissociation of light chains from IQ motifs within the lever arm results in aggregation of myosin-I isoforms (32–34). Thus, we suggest the possibility that 14-3-3β acts as a stabilizing protein to prevent myosin aggregation during the calcium-regulated state. This role is consistent with the a chaperone-like activity that has been proposed for some 14-3-3 isoforms (35).
Experimental procedures
Expression and purification of Myo1c-3IQ
A Myo1c construct (Myo1c-3IQ, isoform b, NP_032685.1) containing the N-terminal motor domain, three CaM-binding IQ motifs, and C-terminal Avi (GLNDIFEAQKIEWHE) and FLAG tags (DYKDDDDK) was expressed and purified with CaM as described previously (5, 36). Biotinylation of Myo1c-3IQ was performed as described previously (37). The protein was stored in liquid nitrogen after dialyzing in KMg25 buffer (10 mm Mops (pH 7.0), 25 mm KCl, 1 mm MgCl2, 1 mm EGTA, and 1 mm DTT). The plasmids of Myo1c-3IQ mutants, Myo1c-3IQ–S701A and –S701E, were generated via a QuikChange kit (Stratagene), and the proteins were expressed and purified as described for Myo1c-3IQ.
Expression and purification of 14-3-3β
A plasmid (pGEX-2TK) containing human 14-3-3β, originally generated by Yaffe et al. (38), was purchased from Addgene (plasmid 13276) and transformed into BL21 (DE3) pLysS cells. To express GST-tagged 14-3-3β, the cultured cells were induced with 0.2 mm isopropyl 1-thio-β-d-galactopyranoside and cultured at room temperature for ∼10 h. Cell pellets from 1 liter of culture were suspend with 25 ml of 1× Bugbuster protein extraction reagent (Millipore Sigma) containing 1 mm PMSF, 0.01 mg/ml leupeptin and aprotinin, and 0.01 mg/ml lysozyme and mixed and stayed at room temperature for 15 min. The lysate was centrifuged at 43,000 × g for 30 min at 4 °C after sonication six times (15 s on, 15 s off). 1 ml of GSH-Sepharose 4B was mixed with the supernatant and rotated at 4 °C for 1 h. The Sepharose was washed with 10 ml of 1× PBS five times, loaded onto a column, and washed with 5 ml of 1× thrombin buffer (20 mm Tris-HCl (pH 8.4), 150 mm NaCl, and 2.5 mm CaCl2) twice. The Sepharose was resuspended with ∼3 ml of 1× thrombin buffer containing ∼30 units of thrombin (Heametech) and rotated at 4 °C overnight. The cleaved protein was dialyzed in KMg25 buffer overnight, centrifuged, and loaded onto a MonoQ 5/50 GL column (GE Healthcare) that was prewashed with buffer A (10 mm Tris-HCl (pH 7.5), 50 mm KCl, 1 mm EGTA, and 1 mm DTT). A gradient of buffer A (0–0.6 m KCl) was flowed through the MonoQ column with FPLC (Amersham Biosciences) at 1 ml/min. The purified 14-3-3β protein was collected, dialyzed in KMg25 buffer overnight, aliquoted, flash-frozen, and stored in liquid nitrogen.
Analytical ultracentrifugation
Analytical ultracentrifugation experiments were performed with an XL-A analytical ultracentrifuge (Beckman-Coulter) and a TiAn60 rotor with six-channel charcoal-filled epon centerpieces and quartz windows as described previously (39). Briefly, sedimentation equilibrium data of 14-3-3β were collected at 4 °C with detection at 280 nm for three concentrations (0.3, 0.55, and 0.8 mg/ml) at three successive speeds (22, 24, and 27,000 rpm), allowing the samples to reach equilibrium (20 h). 14-3-3β migrated as a single component under all conditions, and the acquired data were analyzed using global fits with strict mass conservation using the program SEDPHAT (40). We determined that 14-3-3β is a very tight dimer (Kd < 0.1 nm) by analytical ultracentrifugation. The 14-3-3β protein concentrations reported in this paper refer to the concentration of 14-3-3β dimer.
Phosphorylation of Myo1c-3IQ and MS analysis
To prepare Myo1c-3IQ for identification of the CaMKII-phosphorylated sites via MS, biotinylated Myo1c-3IQ (200 μl of ∼5 μm) was incubated in dephosphorylation buffer (50 mm HEPES (pH 7.5), 100 mm NaCl, 2 mm DTT, 1.5 mm MnCl2, 0.01% Brij35, and 6 μl of λ protein phosphatase (400 units/μl, New England Biolabs)) for 2.5 h at 30 °C and then mixed with 25 μl of streptavidin-agarose beads and rotated at 4 °C for 1.5 h. The beads were washed with 200 μl of phosphorylation buffer (50 mm Tris-HCl (pH 7.5), 10 mm MgCl2, 2 mm DTT, 0.1 mm EDTA, 0.01% Brij35, 20 mm NaCl, 5 mm Mg2+-ATP, and 1 mm CaCl2) 15 times, resuspended with 200 μl of phosphorylation buffer containing 3 μl of activated CaMKII (500 units/μl, New England Biolabs), and incubated at 30 °C for 2 h. The supernatant was discarded after brief centrifugation, and 200 μl of fresh phosphorylation buffer and 3 μl of activated CaMKII were added and incubated for another 2 h. The phosphorylation buffer was replaced with another 200 μl of fresh buffer containing 3 μl of activated CaMKII and rotated with beads at 4 °C overnight. After washing five times with 200 μl of KMg25 buffer, the beads were resuspended with 100 μl of 1× SDS loading buffer and boiled. Phosphorylated Myo1c-3IQ was subjected to SDS-PAGE and detected by Pro-Q Diamond Phosphoprotein Gel Stain and SYPRO Ruby Protein Gel Stain (Thermo Fisher Scientific), respectively. Nonphosphorylated (∼25 μg) and ∼60 μg of phosphorylated Myo1c-3IQ were subjected to Bio-Rad 4–20% gradient SDS-PAGE, stained with Coomassie Brilliant Blue G-250.
The gel band was destained with 100 mm ammonium bicarbonate:acetonitrile (50:50). The band was reduced in 10 mm DTT and 100 mm ammonium bicarbonate for over 60 min in 52 °C. The band was then alkylated with 100 mm iodoacetamide in 100 mm ammonium bicarbonate at room temperature for 1 h in the dark. The protein in the gel band was digested by incubation with trypsin overnight. The supernatant was removed and kept in fresh tubes. Additional peptides were extracted from the gel by adding 50 μl of 50% acetonitrile and 1% TFA and shaking for 10 min. The supernatants were combined and dried. The dried samples were reconstituted in 0.1% formic acid for MS analysis.
Desalted peptides were analyzed with a Q-Exactive HF mass spectrometer (Thermo Scientific) attached to an UltiMate 3000 Nano UPLC system (Thermo Scientific). Peptides were eluted with a 25-min gradient from 2% to 32% acetonitrile and to 98% acetonitrile over 5 min in 0.1% formic acid. Data-dependent acquisition mode with a dynamic exclusion of 45 s was enabled. One full MS scan was collected with a scan range of 350–1200 m/z, resolution of 70 K, maximum injection time of 50 ms, and automatic gain control of 1e6. Then a series of MS2 scans was acquired for the most abundant ions from the MS1 scan (top 15). Ions were filtered with charge 2–5. An isolation window of 1.4 m/z was used with quadruple isolation mode. Ions were fragmented using higher-energy collisional dissociation with a collision energy of 28%. Orbitrap detection was used with a resolution of 35 K, maximum injection time of 54 ms, and automatic gain control of 5e4.
Database search criteria were as follows: taxonomy, Mus musculus (55,029 sequences; July 15, 2019); carboxyamidomethylated (+57 Da) at cysteine residues for fixed modifications; oxidized at methionine (+16 Da) residues; phosphorylation (+79.9 Da) at serine, threonine, and tyrosine residues for variable modifications; two maximum allowed missed cleavages; 10 ppm MS tolerance; and 0.02-Da MS/MS tolerance. Only peptides resulting from trypsin digestion were considered. The target–decoy approach was used to filter the search results, in which the false discovery rate was less than 1% at the peptide and protein levels.
Pulldown assays
Pulldown assays of biotinylated Myo1c-3IQ constructs in the presence of 14-3-3β were performed as follows. 100–200 μl of 0.25–0.8 μm biotinylated Myo1c-3IQ constructs and 14-3-3β in KMg25 buffer containing 0.5 mg/ml BSA and 2 μm CaM was mixed with 10 μl of streptavidin-agarose beads and rotated at 4 °C for 1 h. Unbound proteins were washed out with 100–200 μl of KMg25 buffer five times. Beads were boiled at 100 °C for 5 min after adding 35 μl of 1× SDS loading buffer and then centrifuged. 15 μl of supernatant was subjected to SDS-PAGE (4% to 20% gradient gel) and visualized by Coomassie Brilliant Blue staining or SYPRO Ruby Protein Gel Stain. To calculate the molar ratio of 14-3-3β or CaM versus Myo1c-3IQ heavy chain, the concentration of Myo1c-3IQ heavy chain and 14-3-3β was determined by monitoring the fluorescence intensity of protein bands on the gel stained with SYPRO Ruby Protein Gel Stain and quantified using ImageJ software. The concentrations of Myo1c-3IQ heavy chain, 14-3-3β, and CaM were determined by comparing the intensities with known concentrations of BSA, 14-3-3β, and CaM on the same gel. The total concentration of CaCl2 to add to KMg25 buffer to achieve the indicated free calcium concentrations was determined using the program MaxChelator (41).
Pulldown assays of GST-14-3-3β with Myo1c-3IQ mutants (S701A and S701E) were performed as follows. 100 μl of 0.25 μm GST–14-3-3β and 1 μm Myo1c-3IQ in 1× PBS solution was mixed with 10 μl of GSH-Sepharose (Amersham Biosciences) and rotated at 4 °C for 1 h. The unbound proteins were washed out with 100 μl of 1× PBS. The bound proteins were eluted by 30 μl of elution buffer (5 mm Tris-HCl (pH 8.0), 0.2 m NaCl, 10 mm GSH, and 1 mm DTT). 10 μl of 4× SDS loading buffer was added to the eluted solution and boiled. 15 μl of supernatant was subjected to SDS-PAGE, visualized by Coomassie Brilliant Blue staining, and blotted with anti-FLAG antibody.
Steady-state fluorescence binding assay
Steady-state fluorescence measurements were taken with a Photon Technology International fluorometer at room temperature. Two IQ1 peptides (IQ1-WT and IQ1-pSer701) were synthesized by Peptide 2.0 Inc. (Chantilly, VA) and dissolved in KMg25 buffer. IQ1 (5 μm) peptide and 0–10 μm 14-3-3β were mixed, and binding was determined by monitoring changes in the steady-state fluorescence of the intrinsic tryptophans of IQ peptides and 14-3-3β. Tryptophan residues were excited at 295 nm, and fluorescence emission spectra were collected in the range 310–400 nm, with 2-nm excitation and emission monochromator slits. Because two intrinsic tryptophans (Trp61 and Trp230) exist within 14-3-3β, the final steady-state fluorescence spectra were corrected by subtracting the contribution of the fluorescence spectra of samples containing only 14-3-3β (0–10 μm) from the corresponding total fluorescence spectra.
ATPase activity measurement and motility assay
Steady-state ATPase activity was measured in KMg25 buffer (containing 50 μm free calcium) at 37 °C using the NADH-coupled assay as described previously (42). The final protein concentrations after mixing were 100 nm Myo1c-3IQ, 10 μm CaM, 50 μm phalloidin–actin, and 0–7.5 μm 14-3-3β.
In vitro motility assays were performed in standard motility chambers that were assembled with double sticky tape and silicon vacuum grease (43). Coverslips were coated with 1% collodion (Electron Microscopy Sciences). Streptavidin (1 mg/ml, 20 μl) was flowed into the chamber and incubated for 2 min. The surface was then blocked with 100 μl of 1 mg/ml BSA for 2 min. Biotinylated myo1c-3IQ (600 nm, 50 μl) was added to the chamber and allowed to bind to the immobilized streptavidin for 2 min. Activation solution (100 μl; 10 mm Mops (pH 7.0), 25 mm KCl, 1 mm MgCl2, 1 mm EGTA, 5 mm Mg2+-ATP, 20 mm DTT, and 2 μm CaM), 1 nm Rhodamine phalloidin–F-actin, 0–15 μm 14-3-3β, 1 mg/ml BSA, 5 mg/ml glucose, and 1× GOC mixture (0.2 mg/ml glucose oxidase and 0.04 mg/ml catalase) were flowed into the chamber. The chamber was sealed with silicon grease and mounted on the microscope with a heated objective (37 °C). The chamber was allowed to equilibrate to temperature for 8 min before image acquisition. The velocity of actin filament gliding was measured using the manual tracking plugin in ImageJ.
Author contributions
H.-H. J. and E. M. O. conceptualization; H.-H. J. data curation; H.-H. J. and E. M. O. formal analysis; H.-H. J. validation; H.-H. J. investigation; H.-H. J. and E. M. O. methodology; H.-H. J. writing-original draft; H.-H. J. and E. M. O. project administration; H.-H. J. and E. M. O. writing-review and editing; E. M. O. supervision; E. M. O. funding acquisition.
Supplementary Material
Acknowledgments
We thank Tianming Lin and Dr. Daniel Safer for technical assistance and Dr. Aaron Snoberger for comments on the manuscript. We also thank Dr. Hyoungjoo Lee for assistance with mass spectra data analysis and Dr. Kushol Gupta for assistance with analytical ultracentrifuge data analysis. Mass Spectrometry proteomics resources and services are provided by the Quantitative Proteomics Resource Core at the School of Medicine in the University of Pennsylvania.
This work was supported by NIGMS, National Institutes of Health Grant R37 GM057247 (to E. M. O.) and National Science Foundation CMMI Grant 15-48571 (to E. M. O.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Figs. S1 and S2 and Table S1.
The raw mass spectrometry data can be found in the MassIVE database under ID MassIVE MSV000084445.
- CaM
- calmodulin
- CaMKII
- Ca2+/calmodulin-dependent protein kinase II
- TFP
- trifluoperazine
- ChREBP
- carbohydrate response element–binding protein.
References
- 1. Greenberg M. J., and Ostap E. M. (2013) Regulation and control of myosin-I by the motor and light chain-binding domains. Trends Cell Biol. 23, 81–89 10.1016/j.tcb.2012.10.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. McIntosh B. B., and Ostap E. M. (2016) Myosin-I molecular motors at a glance. J. Cell Sci. 129, 2689–2695 10.1242/jcs.186403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. McConnell R. E., and Tyska M. J. (2010) Leveraging the membrane: cytoskeleton interface with myosin-1. Trends Cell Biol. 20, 418–426 10.1016/j.tcb.2010.04.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Hokanson D. E., and Ostap E. M. (2006) Myo1c binds tightly and specifically to phosphatidylinositol 4,5-bisphosphate and inositol 1,4,5-trisphosphate. Proc. Natl. Acad. Sci. U.S.A. 103, 3118–3123 10.1073/pnas.0505685103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hokanson D. E., Laakso J. M., Lin T., Sept D., and Ostap E. M. (2006) Myo1c binds phosphoinositides through a putative pleckstrin homology domain. Mol. Biol. Cell 17, 4856–4865 10.1091/mbc.e06-05-0449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lu Q., Li J., Ye F., and Zhang M. (2015) Structure of myosin-1c tail bound to calmodulin provides insights into calcium-mediated conformational coupling. Nat. Struct. Mol. Biol. 22, 81–88 10.1038/nsmb.2923 [DOI] [PubMed] [Google Scholar]
- 7. Bose A., Guilherme A., Robida S. I., Nicoloro S. M., Zhou Q. L., Jiang Z. Y., Pomerleau D. P., and Czech M. P. (2002) Glucose transporter recycling in response to insulin is facilitated by myosin Myo1c. Nature 420, 821–824 10.1038/nature01246 [DOI] [PubMed] [Google Scholar]
- 8. Bose A., Robida S., Furcinitti P. S., Chawla A., Fogarty K., Corvera S., and Czech M. P. (2004) Unconventional myosin Myo1c promotes membrane fusion in a regulated exocytic pathway. Mol. Cell Biol. 24, 5447–5458 10.1128/MCB.24.12.5447-5458.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Boguslavsky S., Chiu T., Foley K. P., Osorio-Fuentealba C., Antonescu C. N., Bayer K. U., Bilan P. J., and Klip A. (2012) Myo1c binding to submembrane actin mediates insulin-induced tethering of GLUT4 vesicles. Mol. Biol. Cell 23, 4065–4078 10.1091/mbc.e12-04-0263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Yip M. F., Ramm G., Larance M., Hoehn K. L., Wagner M. C., Guilhaus M., and James D. E. (2008) CaMKII-mediated phosphorylation of the myosin motor Myo1c is required for insulin-stimulated GLUT4 translocation in adipocytes. Cell Metab. 8, 384–398 10.1016/j.cmet.2008.09.011 [DOI] [PubMed] [Google Scholar]
- 11. Aitken A. (2006) 14-3-3 proteins: a historic overview. Semin. Cancer Biol. 16, 162–172 10.1016/j.semcancer.2006.03.005 [DOI] [PubMed] [Google Scholar]
- 12. Münnich S., Taft M. H., and Manstein D. J. (2014) Crystal structure of human myosin 1c: the motor in GLUT4 exocytosis: implications for Ca2+ regulation and 14-3-3 binding. J. Mol. Biol. 426, 2070–2081 10.1016/j.jmb.2014.03.004 [DOI] [PubMed] [Google Scholar]
- 13. Manceva S., Lin T., Pham H., Lewis J. H., Goldman Y. E., and Ostap E. M. (2007) Calcium regulation of calmodulin binding to and dissociation from the myo1c regulatory domain. Biochemistry 46, 11718–11726 10.1021/bi700894h [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Zhu T., Beckingham K., and Ikebe M. (1998) High affinity Ca2+ binding sites of calmodulin are critical for the regulation of myosin Iβ motor function. J. Biol. Chem. 273, 20481–20486 10.1074/jbc.273.32.20481 [DOI] [PubMed] [Google Scholar]
- 15. Zhu T., Sata M., and Ikebe M. (1996) Functional expression of mammalian myosin Iβ: analysis of its motor activity. Biochemistry 35, 513–522 10.1021/bi952053c [DOI] [PubMed] [Google Scholar]
- 16. Trybus K. M., Waller G. S., and Chatman T. A. (1994) Coupling of ATPase activity and motility in smooth muscle myosin is mediated by the regulatory light chain. J. Cell Biol. 124, 963–969 10.1083/jcb.124.6.963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Chen Z., and Cole P. A. (2015) Synthetic approaches to protein phosphorylation. Curr. Opin. Chem. Biol. 28, 115–122 10.1016/j.cbpa.2015.07.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Yang X., Lee W. H., Sobott F., Papagrigoriou E., Robinson C. V., Grossmann J. G., Sundström M., Doyle D. A., and Elkins J. M. (2006) Structural basis for protein-protein interactions in the 14-3-3 protein family. Proc. Natl. Acad. Sci. U.S.A. 103, 17237–17242 10.1073/pnas.0605779103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Petosa C., Masters S. C., Bankston L. A., Pohl J., Wang B., Fu H., and Liddington R. C. (1998) 14-3-3ζ binds a phosphorylated Raf peptide and an unphosphorylated peptide via its conserved amphipathic groove. J. Biol. Chem. 273, 16305–16310 10.1074/jbc.273.26.16305 [DOI] [PubMed] [Google Scholar]
- 20. Ottmann C., Yasmin L., Weyand M., Veesenmeyer J. L., Diaz M. H., Palmer R. H., Francis M. S., Hauser A. R., Wittinghofer A., and Hallberg B. (2007) Phosphorylation-independent interaction between 14-3-3 and exoenzyme S: from structure to pathogenesis. EMBO J. 26, 902–913 10.1038/sj.emboj.7601530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ge Q., Huang N., Wynn R. M., Li Y., Du X., Miller B., Zhang H., and Uyeda K. (2012) Structural characterization of a unique interface between carbohydrate response element-binding protein (ChREBP) and 14-3-3β protein. J. Biol. Chem. 287, 41914–41921 10.1074/jbc.M112.418855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lanner J. T., Bruton J. D., Katz A., and Westerblad H. (2008) Ca2+ and insulin-mediated glucose uptake. Curr. Opin. Pharmacol. 8, 339–345 10.1016/j.coph.2008.01.006 [DOI] [PubMed] [Google Scholar]
- 23. Whitehead J. P., Molero J. C., Clark S., Martin S., Meneilly G., and James D. E. (2001) The role of Ca2+ in insulin-stimulated glucose transport in 3T3-L1 cells. J. Biol. Chem. 276, 27816–27824 10.1074/jbc.M011590200 [DOI] [PubMed] [Google Scholar]
- 24. Lewis J. H., Greenberg M. J., Laakso J. M., Shuman H., and Ostap E. M. (2012) Calcium regulation of myosin-I tension sensing. Biophys. J. 102, 2799–2807 10.1016/j.bpj.2012.05.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Black D. J., Tran Q. K., and Persechini A. (2004) Monitoring the total available calmodulin concentration in intact cells over the physiological range in free Ca2+. Cell Calcium 35, 415–425 10.1016/j.ceca.2003.10.005 [DOI] [PubMed] [Google Scholar]
- 26. Persechini A., and Cronk B. (1999) The relationship between the free concentrations of Ca2+ and Ca2+-calmodulin in intact cells. J. Biol. Chem. 274, 6827–6830 10.1074/jbc.274.11.6827 [DOI] [PubMed] [Google Scholar]
- 27. Cyr J. L., Dumont R. A., and Gillespie P. G. (2002) Myosin-1c interacts with hair-cell receptors through its calmodulin-binding IQ domains. J. Neurosci. 22, 2487–2495 10.1523/JNEUROSCI.22-07-02487.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Tang N., Lin T., Yang J., Foskett J. K., and Ostap E. M. (2007) CIB1 and CaBP1 bind to the myo1c regulatory domain. J. Muscle Res. Cell Motil. 28, 285–291 10.1007/s10974-007-9124-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Phillips K. R., Tong S., Goodyear R., Richardson G. P., and Cyr J. L. (2006) Stereociliary myosin-1c receptors are sensitive to calcium chelation and absent from cadherin 23 mutant mice. J. Neurosci. 26, 10777–10788 10.1523/JNEUROSCI.1847-06.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Chen X. W., Leto D., Chiang S. H., Wang Q., and Saltiel A. R. (2007) Activation of RalA is required for insulin-stimulated Glut4 trafficking to the plasma membrane via the exocyst and the motor protein Myo1c. Dev. Cell 13, 391–404 10.1016/j.devcel.2007.07.007 [DOI] [PubMed] [Google Scholar]
- 31. Watanabe M., Nomura K., Ohyama A., Ishikawa R., Komiya Y., Hosaka K., Yamauchi E., Taniguchi H., Sasakawa N., Kumakura K., Ushiki T., Sato O., Ikebe M., and Igarashi M. (2005) Myosin-Va regulates exocytosis through the submicromolar Ca2+-dependent binding of Syntaxin-1A. Mol. Biol. Cell 16, 4519–4530 10.1091/mbc.e05-03-0252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Howe C. L., and Mooseker M. S. (1983) Characterization of the 110-kdalton actin-calmodulin-, and membrane-binding protein from microvilli of intestinal epithelial cells. J. Cell Biol. 97, 974–985 10.1083/jcb.97.4.974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Swanljung-Collins H., and Collins J. H. (1991) Ca2+ stimulates the Mg2+-ATPase activity of brush border myosin I with three or four calmodulin light chains but inhibits with less than two bound. J. Biol. Chem. 266, 1312–1319 [PubMed] [Google Scholar]
- 34. Gillespie P. G., and Cyr J. L. (2002) Calmodulin binding to recombinant myosin-1c and myosin-1c IQ peptides. BMC Biochem. 3, 31 10.1186/1471-2091-3-31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Sluchanko N. N., and Gusev N. B. (2012) Oligomeric structure of 14-3-3 protein: what do we know about monomers? FEBS Lett. 586, 4249–4256 10.1016/j.febslet.2012.10.048 [DOI] [PubMed] [Google Scholar]
- 36. El Mezgueldi M., Tang N., Rosenfeld S. S., and Ostap E. M. (2002) The kinetic mechanism of Myo1e (human myosin-IC). J. Biol. Chem. 277, 21514–21521 10.1074/jbc.M200713200 [DOI] [PubMed] [Google Scholar]
- 37. Lin T., Tang N., and Ostap E. M. (2005) Biochemical and motile properties of Myo1b splice isoforms. J. Biol. Chem. 280, 41562–41567 10.1074/jbc.M508653200 [DOI] [PubMed] [Google Scholar]
- 38. Yaffe M. B., Rittinger K., Volinia S., Caron P. R., Aitken A., Leffers H., Gamblin S. J., Smerdon S. J., and Cantley L. C. (1997) The structural basis for 14-3-3:phosphopeptide binding specificity. Cell 91, 961–971 10.1016/S0092-8674(00)80487-0 [DOI] [PubMed] [Google Scholar]
- 39. Gupta K., Martin R., Sharp R., Sarachan K. L., Ninan N. S., and Van Duyne G. D. (2015) Oligomeric properties of survival motor neuron.Gemin2 complexes. J. Biol. Chem. 290, 20185–20199 10.1074/jbc.M115.667279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Vistica J., Dam J., Balbo A., Yikilmaz E., Mariuzza R. A., Rouault T. A., and Schuck P. (2004) Sedimentation equilibrium analysis of protein interactions with global implicit mass conservation constraints and systematic noise decomposition. Anal. Biochem. 326, 234–256 10.1016/j.ab.2003.12.014 [DOI] [PubMed] [Google Scholar]
- 41. Patton C., Thompson S., and Epel D. (2004) Some precautions in using chelators to buffer metals in biological solutions. Cell Calcium 35, 427–431 10.1016/j.ceca.2003.10.006 [DOI] [PubMed] [Google Scholar]
- 42. De La Cruz E. M., and Ostap E. M. (2009) Kinetic and equilibrium analysis of the myosin ATPase. Methods Enzymol. 455, 157–192 10.1016/S0076-6879(08)04206-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Greenberg M. J., Shuman H., and Ostap E. M. (2017) Measuring the kinetic and mechanical properties of non-processive myosins using optical tweezers. Methods Mol. Biol. 1486, 483–509 10.1007/978-1-4939-6421-5_19 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.