

Abstract
The hallmark pathological features of Alzheimer's disease (AD) brains are senile plaques, comprising β-amyloid (Aβ) peptides, and neuronal inclusions formed from tau protein. These plaques form 10–20 years before AD symptom onset, whereas robust tau pathology is more closely associated with symptoms and correlates with cognitive status. This temporal sequence of AD pathology development, coupled with repeated clinical failures of Aβ-directed drugs, suggests that molecules that reduce tau inclusions have therapeutic potential. Few tau-directed drugs are presently in clinical testing, in part because of the difficulty in identifying molecules that reduce tau inclusions. We describe here two cell-based assays of tau inclusion formation that we employed to screen for compounds that inhibit tau pathology: a HEK293 cell-based tau overexpression assay, and a primary rat cortical neuron assay with physiological tau expression. Screening a collection of ∼3500 pharmaceutical compounds with the HEK293 cell tau aggregation assay, we obtained only a low number of hit compounds. Moreover, these compounds generally failed to inhibit tau inclusion formation in the cortical neuron assay. We then screened the Prestwick library of mostly approved drugs in the cortical neuron assay, leading to the identification of a greater number of tau inclusion inhibitors. These included four dopamine D2 receptor antagonists, with D2 receptors having previously been suggested to regulate tau inclusions in a Caenorhabditis elegans model. These results suggest that neurons, the cells most affected by tau pathology in AD, are very suitable for screening for tau inclusion inhibitors.
Keywords: tau protein (tau), Alzheimer disease, aggregation, inhibitor, drug screening, neurodegeneration, drug discovery, amyloid plaque, neurotoxicity
Introduction
Several neurodegenerative diseases, collectively referred to as tauopathies, are characterized by the presence of intracellular inclusions composed of polymeric fibrils of hyperphosphorylated tau protein within the brain (1, 2). These tau aggregates are largely found in neurons, where they are referred to as neurofibrillary tangles (NFTs)2 within the soma and as neuropil threads (NTs) when in dendritic processes. AD is by far the most prevalent tauopathy, afflicting >5 million individuals in the United States (3). Whereas tau inclusions are the primary neuropathological feature in frontotemporal lobar degenerative (FTLD) disorders such as progressive supranuclear palsy, corticobasal syndrome, and Pick's disease (2), individuals with AD also have senile plaque pathology. These extracellular plaques are composed of Aβ peptides, and a prevailing hypothesis in AD research is that Aβ plaques trigger events that ultimately lead to the development of tau pathology and consequent neurodegeneration (i.e. the amyloid cascade hypothesis) (4, 5). The belief that tau inclusions cause neurodegeneration is supported by genetic data showing inherited forms of FTLD result from tau mutations (6, 7) and the strong correlation between the extent of tau pathology and cognitive status in AD (8, 9). The linkage of tau pathology to AD symptomatology is further supported by recent studies showing that AD cognitive status (10) and brain atrophy (11) are correlated with tau PET signal. In fact, a prospective study in which Aβ plaque and tau pathology were both assessed with PET ligands confirmed that cognitive decline was closely associated with tau inclusions, and not plaque changes (12).
Tau is normally a microtubule (MT)-associated protein that appears to affect MT dynamics in axons (13, 14) and may also modulate MT interactions with molecular motors such as kinesin and dynein (15, 16). In humans, tau exists as six alternatively-spliced isoforms, with either 3 or 4 MT-binding repeats and 0, 1, or 2 N-terminal alternatively-spliced exon sequences (2). Tau becomes hyperphosphorylated in all tauopathies, with increased phosphorylation promoting tau disengagement from MTs (17–19) with subsequent misfolding into fibrillar structures that deposit as inclusions. The tau fibrils are hypothesized to mediate a gain–of–function toxicity, and a reduction of tau binding to MTs likely also leads to increased MT dynamicity and altered axonal transport (20, 21) that may contribute to neuronal dysfunction. There is increased interest in developing tau-directed drugs for the treatment of AD and related tauopathies (22, 23), spurred in part by multiple Phase 3 clinical failures of therapeutic candidates designed to lower Aβ levels and/or plaque burden in AD brain. Moreover, there is growing recognition that abundant Aβ pathology forms a decade or more before cognitive symptoms in AD, whereas the development of robust cortical tau pathology is more proximal to symptom onset (24, 25). To date, only a small number of tau-directed drugs have progressed to clinical testing, with the majority being immunotherapeutics (26). Accordingly, there is continued need to identify new small-molecule drug candidates directed toward targets that lead to reduced tau pathology.
A limitation in identifying candidate molecules to reduce tau inclusion formation has been a paucity of robust cell-based assays that model the events leading to tau inclusion formation and clearance. Although we (27, 28) and others (29–31) have previously conducted screens to identify inhibitors of recombinant tau fibril formation, such cell-free assays do not replicate the processes involved in tau fibril formation and degradation within a cellular milieu. There would thus be considerable value in identifying cell-based models of tau inclusion formation that are suitable for compound screening. Although cellular assays of tau inclusion formation have been described and in some cases selectively queried with test compounds (32–36), they have not generally been used for extensive small-molecule screening. An exception is a recent screening of 1649 compounds conducted with the N2A cell line that overexpressed a pro-aggregant repeat domain of tau, leading to the identification of several inhibitors of tau aggregation (37). As described here, we have optimized a unique HEK293 cell assay of tau inclusion formation, as well as a primary rat cortical neuronal assay with AD-like tau pathology, and we show here that both are suitable for compound screening. Notably, the neuronal model shows a time-dependent formation of inclusions composed of endogenously expressed rat tau proteins that elongate from internalized tau “seeds” composed of enriched pathological tau derived from human AD brains (38). This neuronal assay does not rely on overexpression of mutant tau, as has been typically required to induce inclusions in cell lines or iPSC neurons (32, 33, 37, 39), and thus it provides a pathophysiologically-relevant model system in which to survey for inhibitors of tau inclusions. As described below, the National Center for Advancing Translational Sciences (NCATS) Pharmaceutical Collection compound library was screened in the HEK293 cell tau aggregation assay. Confirmed hits from this screen underwent testing in the neuronal assay of tau inclusion formation, yielding a low confirmation rate. This led to a subsequent screening of the Prestwick library of mostly approved drugs in the neuronal tau inclusion assay, which provided a higher number of validated hits with greater potential biological relevance. These findings suggest the importance of assessing compounds in neuronal models that express physiological levels of tau.
Results
Compound screening in a HEK293 cell assay of tau inclusion formation
Our laboratories previously demonstrated (32) that intracellular tau inclusions can be promoted by the cytosolic delivery of pathological seeds composed of pre-formed synthetic fibrils assembled from recombinant tau (tau pre-formed fibrils or PFFs) into HEK293 cells that express the longest form of human recombinant tau containing the P301L mutation found in inherited FTLD (referred to as T40PL) (40). Based on this cellular system of tau inclusion formation, an alternative model was developed in which a HEK293 cell line was created that expresses T40PL with a C-terminal GFP tag (T40PL–GFP) to allow visualization of tau inclusions. T40PL–GFP expression in this clonal line is driven by a tetracycline-regulated promoter such that expression can be induced by addition of doxycycline (Dox) to the culture medium. Aggregates of T40PL–GFP were generated in these HEK293 cells through recombinant tau PFF transduction, and it was found that these intracellular aggregates could be propagated with high efficiency to daughter cells as long as cultures were maintained in the presence of Dox (41), resulting in a clonal line referred to here as T40PL–GFPagg cells. A prior study (41) showed that the tau aggregate burden within T40PL–GFPagg cells could be reduced by Dox withdrawal, with the amount of insoluble tau diminishing over 7 days after cessation of T40PL–GFP expression, such that only small insoluble tau aggregates remain. However, initiation of T40PL–GFP expression through re-introduction of Dox after the 7-day withdrawal period resulted in the rapid reformation of T40PL–GFP inclusions within 2 days, indicating that the remaining small tau aggregates were sufficient to “reseed” new tau aggregate growth upon re-expression of soluble tau (41).
The ability to manipulate the tau aggregate burden within T40PL–GFPagg cells led to the creation of a unique screening assay, as depicted in Fig. 1A, where compounds can be added to Dox-deprived T40PL–GFPagg cells with sparse aggregates prior to re-initiation of T40PL–GFP expression. This allows for an assessment of compound activity on tau inclusion formation or clearance. Preliminary studies revealed that soluble T40PL–GFP could be effectively cleared from tau inclusion-bearing cells with 1% hexadecyltrimethylammonium bromide (HDTA) extraction, followed by 4% paraformaldehyde (PFA) fixation, allowing for quantitative imaging of remaining detergent-insoluble T40PL–GFP (Fig. 1B). This tau aggregation model was optimized for a 1536-well plate format, and the efficiency of the HDTA/PFA extraction and fixation method in removing soluble T40PL–GFP prior to imaging were compared with an alternative method in which soluble T40PL–GFP was released from aggregate-bearing cells with the commercial CellTiter-GloTM (CTG) reagent, with the T40PL–GFP being diluted into the surrounding medium such that it did not interfere with imaging of T40PL–GFP inclusions (Fig. 1B). The addition of the CTG reagent allows for the concurrent assessment of cell toxicity through a determination of a luminescent ATP signal (42), although cell toxicity was ultimately determined by quantifying DAPI-stained cell nuclei that remained after cell extraction. The 1536-well T40PL–GFPagg cellular assay was utilized to screen the NCATS Pharmaceutical Collection of ∼3500 approved drugs and investigational compounds, utilizing the protocol summarized in Fig. 1A and described under “Experimental procedures.” All compounds were screened at five concentrations to generate concentration–response data for each compound, and the insoluble T40PL–GFP-integrated fluorescent signal was normalized to DAPI counts. All compounds were tested in two screening protocols, using the HDTA or CTG extraction procedures to remove soluble T40PL–GFP prior to imaging of the remaining insoluble T40PL–GFP tau aggregates. For the HDTA assay, cells were plated on poly-d-lysine–coated plates to reduce cell loss associated with the wash steps (Fig. S1A). Both extraction methodologies yielded similar plate Z′ values (Fig. S1B) (43), as determined by comparing the normalized large insoluble T40PL–GFP values in wells with Dox reintroduced to those deprived of Dox, as described under “Experimental procedures.” A positive-control compound, BMS-265246, was included at multiple concentrations in each screening plate. This cyclin-dependent kinase inhibitor had been identified during initial assay development to cause a concentration-dependent reduction of soluble T40PL–GFP levels in the T40PL–GFPagg cells, resulting in reduced T40PL–GFP aggregate formation. Although this mechanism of action may be undesirable in a drug candidate, the addition of BMS-265246 to the screening plates provided a quality control standard to confirm proper assay performance. The BMS-265246 inhibition curves from each screening plate are summarized in Fig. S1C, and excellent plate–to–plate reproducibility was observed when using either the HDTA or CTG extraction procedures. Hits from the NCATS screen were defined as compounds that demonstrated a clear concentration-dependent inhibition of T40PL–GFP aggregates using both the HDTA and CTG extraction methods, with tau aggregate inhibitory activity well-separated by at least 1 log unit from cytotoxicity. Generally, compounds that were hits when using the HDTA extraction procedure were also hits when analyzed after CTG extraction, although there were examples where differences in the inhibition curves were observed between the two extraction methods. A total of eight hits were identified from the screening of the ∼3500 compound library that showed concentration-dependent inhibition of tau inclusion formation with minimal toxicity with both extraction procedures (Fig. 2), for a hit rate of ∼0.2%.
Figure 1.
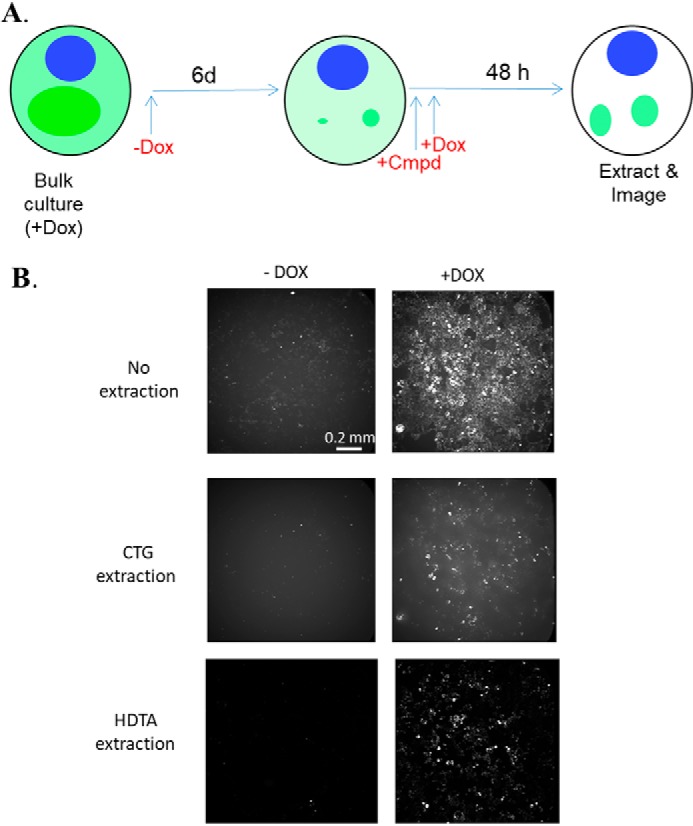
A, schematic summary of the T40PL–GFPagg cell assay used for compound screening. The withdrawal of Dox for a 6-day period results in cessation of soluble tau expression (light green) and appreciable diminution of intracellular tau inclusions (dark green) within the cells, with sufficient insoluble tau remaining to seed inclusion regrowth upon re-initiation of T40PL–GFP expression upon addition of Dox. Compounds were added shortly before the reintroduction of Dox to determine whether they affect the subsequent formation or clearance of tau aggregates, which are visualized after extraction of soluble tau. B, representative whole-well images of T40PL–GFPagg cells at the end of the assay period, either without extraction or after extraction with CTG or HDTA to remove soluble T40PL–GFP to reveal insoluble T40PL–GFP aggregates.
Figure 2.

Concentration–response profiles of hit compounds from the NCATS Pharmaceutical Collection screened in the T40PL–GFPagg cell tau inclusion assay. Data are plotted showing the inhibition of T40PL–GFP aggregates (expressed as % inhibition) after CTG or HDTA extraction. In addition, compound-mediated cytotoxicity is shown as measured by an analysis of DAPI-positive nuclei after each extraction method. All compounds were analyzed as singletons at five concentrations (110 nm to 2.75 μm), with curve fitting as described under “Experimental procedures.”
Optimization of a primary rat cortical neuron assay of tau inclusion formation
The results from the screening of the NCATS Pharmaceutical Collection demonstrated that the HEK293 T40PL–GFPagg cell assay is robust and suitable for assessment of compound libraries. However, a relatively low number of hits were identified. Moreover, whereas transformed cell lines such as HEK293 are convenient to use for screening, they differ considerably from neurons, which are the cell type most affected with tau pathology in disease. In particular, neurons are post-mitotic, with a complex cell physiology that supports the development and maintenance of extended axonal and dendritic processes. Thus, there are likely to be significant differences in the regulation of various cellular pathways, including those involved in proteostasis, in neurons relative to rapidly dividing cells. With the objective of developing a neuronal tau inclusion assay that could be utilized to verify the activity of hits from HEK293 T40PL–GFPagg cell screens, efforts were made to further optimize a primary neuronal assay of tau inclusion formation that was previously described (38).
Prior studies from our laboratories revealed that robust tau inclusions form in primary mouse neuron cultures after incubation with enriched preparations of insoluble pathological tau derived from AD brain tissue (AD-tau) (38). Once internalized by neurons, this AD-tau acted to seed the formation of abundant insoluble tau inclusions, largely within neuritic processes, that were composed of endogenous mouse tau. These insoluble tau aggregates could be readily visualized 14 days after AD-tau addition after extraction of soluble tau and staining with an antibody specific to rodent tau (T49) or by fractionation of neuronal homogenates and measurement of Triton X-100–insoluble tau (38). We adapted this neuron assay to a 384-well plate format, with culturing of mouse or rat neurons for 7 days prior to AD-tau addition, with an additional 14–15 days in culture before assessment of tau inclusions through immunostaining and imaging. Multiple conditions were investigated during optimization of the assay, including the following: 1) determination of the extraction/fixation method that best removes residual soluble tau to provide the greatest differential of insoluble tau staining between AD-tau–treated and -nontreated neurons; 2) comparison of the amount of insoluble tau pathology formed in rat versus mouse primary neurons; 3) assessment of the AD-tau dose required to provide suitable Z′ values for compound screening without reaching saturation of pathology and loss of assay sensitivity; and 4) batch–to–batch consistency of AD-tau in inducing neuronal tau inclusions. In summary, the extraction/fixation method that provided the best insoluble tau signal–to–background readings was 1% HDTA extraction with 4% PFA fixation. Interestingly, rat cortical neurons were found to develop a greater tau inclusion burden than mouse cortical neurons (Fig. 3), and cortical neurons had greater aggregate amounts than hippocampal neurons for each species (data not shown). Based on dose-response comparisons of different AD-tau preparations, little variability was observed among AD-tau batches (Fig. 3). The results of these AD-tau dose-response analyses revealed that addition of 62.5 ng/well of AD-tau to rat cortical neurons in the 384-well format consistently yielded sufficient tau pathology to provide Z′-values of >0.5 when comparing AD-tau–treated versus nontreated neurons, while remaining in the linear range of sensitivity (Fig. 3).
Figure 3.
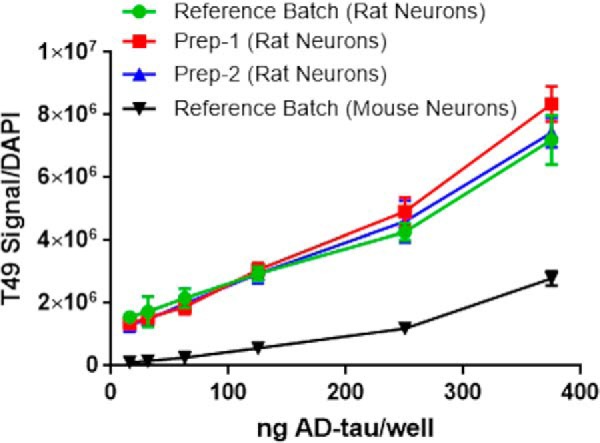
Comparison of the amount of insoluble tau pathology by immunocytochemistry (T49 rodent tau-specific mAb) after addition of varying amounts of a reference batch of AD-tau to rat or mouse cortical neurons, with normalization to DAPI-positive nuclei. Also shown are comparisons of dose-response profiles of two separate additional preparations of AD-tau relative to the reference batch of AD-tau in eliciting tau pathology in rat cortical neurons. A total of 12 wells were tested at each concentration, with error bars representing S.D.
Utilizing the optimized primary rat neuron assay (schematic in Fig. 4A), the available hits from the NCATS Pharmaceutical Collection screen in the HEK293 T40PL–GFPagg tau aggregate assay were evaluated at four concentrations (0.3–10 μm) in triplicate. Because the HDTA extraction and PFA fixation protocol used in the assay negatively affects neuronal morphology, a parallel set of rat cortical neuron cultures were treated with AD-tau and compound but were permeabilized with 0.1% Triton X-100 and fixed with PFA, followed by staining of neuronal nuclei (NeuN antibody) and dendritic processes (MAP2 antibody) to allow assessment of compound neurotoxicity. Somewhat surprisingly, only one of the seven tested hits from the NCATS screen (NCG0024777-04; SNC-80) showed some evidence of a concentration-dependent inhibition of tau inclusions in the neuronal assay that was not also paralleled by a corresponding compound-induced toxicity, as seen by a reduction of DAPI and/or NeuN counts, and/or a reduced MAP2 area (SNC-80 result in Fig. 4B; all other tested compounds summarized in Fig. S2). In contrast, a GSK-3β inhibitor (CHIR-99021) that was tested in the rat cortical assay based on the proposed role of GSK-3β in promoting tau hyperphosphorylation and aggregation (44, 45) routinely caused a concentration-dependent decrease in neuronal tau inclusions that was somewhat separated from toxicity measures (Fig. 4C). In addition, the BMS-265246 compound used as a control in the HEK293 T40PL–GFPagg cell assay also showed evidence of inhibition of tau inclusions in the neuronal assay at nontoxic concentrations (data not shown). Thus, these data reveal that the NCATS compounds that reduced tau aggregates in the HEK293 T40PL–GFPagg cell assay were generally not active in the neuronal assay. This may relate to the considerable differences between post-mitotic primary cortical rat neurons that express endogenous tau and the rapidly dividing HEK293 cells that overexpress mutant human tau.
Figure 4.
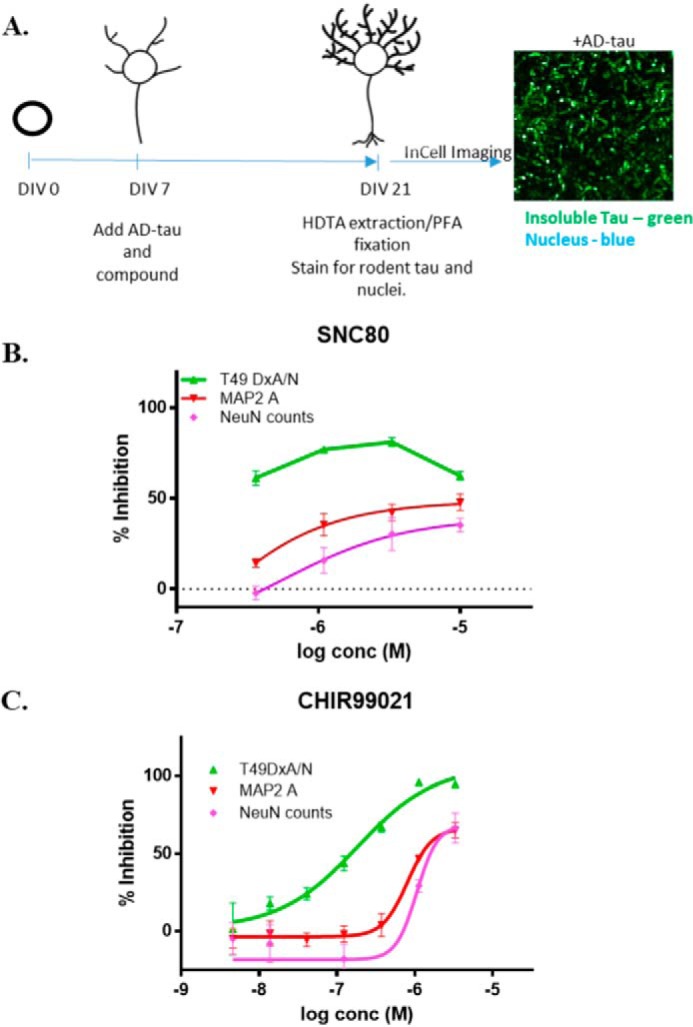
A, schematic representation of the protocol for 384-well compound screening using the optimized rat cortical neuron assay of tau inclusion formation, including a representative image of the tau pathology and DAPI-positive nuclei remaining after HDTA extraction and PFA fixation. B, concentration–response analysis of SNC-80 in the rat cortical tau inclusions assay, with insoluble tau (T49 DxA/DAPI nuclei), MAP2 area, and NeuN counts shown as percent inhibition relative to vehicle-treated neurons. Triplicate wells were tested at each SNC-80 concentration, with error bars representing S.E. Sigmoidal dose-response curves were fit using GraphPad Prism, except for the T49 DxA/N measure in which data points were simply connected by lines due to an incomplete sigmoidal curve in the tested concentration range. C, example concentration–response profile of CHIR-99021 in the rat cortical tau inclusion assay, with insoluble tau (T49 DxA/DAPI nuclei), MAP2 area, and NeuN counts shown. Triplicate wells were tested at each CHIR-99021 concentration, with error bars representing S.E. Sigmoidal dose-response curves were fit using GraphPad Prism.
Compound screening in the primary rat cortical neuron assay of tau inclusion formation
Given the relatively low hit rate from the screening of the NCATS Pharmaceutical Collection in the HEK293 T40PL–GFPagg cell assay, and the poor confirmation of identified hits in the neuronal assay of tau inclusion formation, a compound screen was conducted in the neuronal assay to determine whether a higher hit rate might be observed. Compounds from the Prestwick library of 1280 mostly approved drugs were tested at 10 μm as singletons in the rat cortical neuron assay as depicted in Fig. 4A. The assay performance was good, with Z′-values ≥0.5 typically observed across the plates. Given the low confirmation rate of the hits from the HEK293 T40PL–GFPagg cell assay screen, one potential concern was that compound degradation or metabolism might occur during the 14-day incubation period, perhaps explaining the low confirmation rate of the NCATS hits, although CHIR-99021 and BMS-265246 showed reproducible activity in the assay. Gratifyingly, compound degradation did not appear to be a significant problem, as 117 compounds were identified that caused a >3 S.D. reduction in tau inclusions with <2 S.D. reductions in DAPI counts (Fig. 5). These hits were subsequently retested at 10 μm in triplicate in the same assay, and a total of 43 were confirmed to be active with >3 S.D. reductions of tau pathology, and another 18 showed >2 S.D. tau inclusion reductions, all with a <2 S.D. loss of DAPI counts. It should be noted that a plate hit definition based on a >3 S.D. inhibition can result in varying percent inhibition hit values due to differing plate CV values, such that compounds close to the hit threshold may fail to replicate as >3 S.D. hits on retesting (e.g. a CV value of 10% would result in a hit threshold of 30% inhibition, whereas a CV of 13% would result in a hit threshold of 39% inhibition). Thus, nontoxic compounds with >2 S.D. inhibition of tau inclusions in triplicate testing were retained due to the greater statistical power gained with triplicates compared with singleton analysis.
Figure 5.
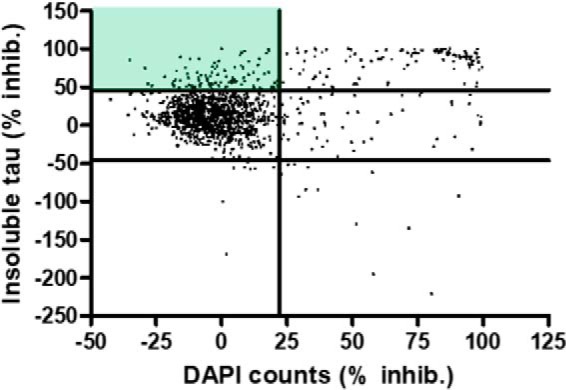
Scatter plot of the results of the Prestwick library screen in the rat cortical tau inclusion assay. Compounds were screened at 10 μm, and each data point represents a single compound, with the percent inhibition of tau aggregates plotted on the y axis and the percent reduction in DAPI-positive nuclei plotted on the x axis. The green shading depicts the quadrant with compounds causing ≥3 S.D. inhibition of tau aggregates and ≤2 S.D. decrease in DAPI counts.
The confirmed hits from the cortical neuron tau inclusion screen were also assessed for untoward effects on neuronal viability and/or morphology. Neuron cultures were treated with AD-tau and 10 μm compound in triplicate as above, but underwent the milder Triton X-100 and PFA permeabilization/fixation protocol to allow visualization of NeuN-positive nuclei and MAP2-positive dendritic processes. This additional analysis revealed that a number of the hits affected dendritic outgrowth and/or reduced neuron counts by >2 S.D., thereby resulting in likely false-positive reductions in tau inclusions. After removal of these compounds, a total of 32 confirmed active and nontoxic compounds were identified from the Prestwick library (2.5% confirmed hit rate; Table S1). An examination of the reported targets of these compounds based on literature review revealed several neurotransmitter receptors and neuronal enzymes, as well as likely nonspecific hits such as topoisomerase inhibitors and antibiotics. A total of 23 of these neuronal screening hits were also within the NCATS Pharmaceutical Collection, and none showed evidence of a concentration-dependent inhibition of tau inclusions in the HEK293 T40PL–GFPagg cell assay screen (Fig. S3). Moreover, two (gefitinib and naftopidil) of the eight hits identified from the NCATS Pharmaceutical Collection screen were within the Prestwick library, and neither of these compounds were found to be active at the 10 μm concentration used in the neuronal tau inclusion screen. Thus, these results further confirm the differences in compound activity observed between the neuronal and HEK293 tau inclusion assays. After prioritizing confirmed hits by removing compounds directed to undesirable targets or those with lower overall tau aggregate inhibition on triplicate testing, 18 compounds that caused >3 S.D. inhibition of tau inclusions upon triplicate testing (highlighted in Table S1) underwent concentration–response analyses, with 13 of these showing well-defined inhibition curves with calculable IC50 values (Table 1).
Table 1.
Summary of confirmed hits with calculable IC50 values from the Prestwick library screen in the rat cortical tau inclusion assay
Data are provided showing the inhibitory activity of each compound on tau inclusions and DAPI nuclei from the original screen (10 μm compound) and tau inclusion, DAPI, MAP2 and NeuN inhibitory activity in the triplicate confirmation analyses (10 μm compound). In addition, the calculated tau aggregate IC50 values and maximal inhibition from concentration–response testing are listed for each compound. Finally, the inhibitory activities of the compounds as measured in the orthogonal tau multimer (mTau8) ELISA are shown, with each compound tested at the approximate IC90 concentration (except Alfuzosin, tested at IC50).

Each of these 13 compounds was subsequently evaluated in an orthogonal assay to confirm that they reduced insoluble neuronal tau. This consisted of testing cellular lysates from AD-tau- and compound-treated rat cortical neurons, as in Fig. 4A except in 96-well plates, in a tau multimer ELISA. This ELISA utilizes a rodent tau-specific mAb as both a capture and detection antibody, such that monomeric tau is not detected due to epitope shielding after monomer binding to the capture antibody. Conversely, rodent tau multimers, including fibrils, are detected in the ELISA. Importantly, the specificity for rodent tau precludes the detection of residual AD-tau that might remain in the culture lysates. Each of the 13 tested compounds showed appreciable inhibition of multimeric tau when tested in triplicate at the ∼EC90 concentration determined from the concentration–response curves for the compounds (Table 1; alfuzosin tested at EC50). In general, there appeared to be a greater compound-mediated inhibition of multimeric tau as measured with the ELISA than had been observed when quantifying tau aggregates via immunostaining and imaging. This could be the result of compound-mediated reduction of diffusible tau multimers that do not persist after extraction prior to immunostaining. Nonetheless, the biochemical assay confirms that the compounds listed in Table 1 indeed reduce multimeric and insoluble tau in the rat cortical neuron assay.
Among the 13 compounds with defined IC50 values were a number of reported dopamine D2 receptor antagonists, all of which had well-defined concentration–response curves (Fig. 6). The inhibitory activity observed in the neuronal tau inclusion assay appears to be generally consistent with the reported binding affinities of these compounds. For example, both alizapride and metoclopramide have reported D2 receptor affinities of ∼300 nm (46), which is roughly the IC50 values obtained with these compounds in the neuronal tau assay. We assessed whether these D2 receptor antagonists act to reduce overall tau protein expression by measuring the amount of soluble tau by ELISA in compound-treated rat cortical neuron cultures, and these compounds did not generally affect soluble tau, although bromopride and azaperone caused modest decreases with the latter reaching statistical significance (Fig. S4). To further investigate possible mechanisms by which the D2 receptor antagonists prevent neuronal tau inclusions, we evaluated their ability to directly inhibit tau fibrillization. None of the identified D2 receptor antagonists reduced the in vitro fibrillization of recombinant tau, whereas the positive control compounds oleocanthal (47) and CNDR-51348 (48) both caused concentration-dependent inhibition of tau fibril formation (Fig. S5).
Figure 6.
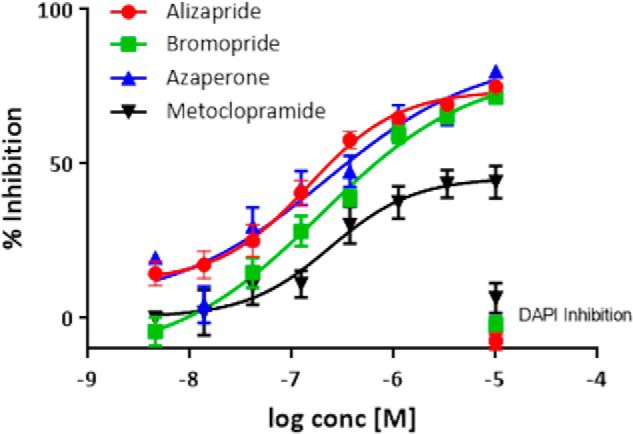
Concentration-response analyses of dopamine D2 receptor antagonists identified as confirmed hits in the rat cortical neuron tau inclusion assay. The D2 receptor antagonists listed in Table 1 caused a concentration-dependent inhibition of tau inclusions without evidence of appreciable toxicity, as shown by the lack of effect on DAPI-positive nuclei at the highest-tested concentration. Moreover, the compounds showed little effects on MAP2 area or NeuN counts when tested at 10 μm concentration (Table 1). Six wells were tested at each compound concentration, with error bars representing S.E. Sigmoidal dose-response curves were fit using GraphPad Prism.
We also examined whether the identified D2 receptor antagonists might inhibit neuronal tau inclusion formation through a reduction of tau phosphorylation. It is known that tau becomes hyperphosphorylated in AD and related tauopathies, and increased phosphorylation can elevate cytosolic tau concentrations through promotion of tau disengagement from microtubules (18, 19, 49). Moreover, phosphorylation may also increase the propensity of tau to fibrillize (50–52). Tau phosphorylation was evaluated in the standard tau neuronal assay (Fig. 4A) after treatment with vehicle or two of the active D2 receptor antagonists (metoclopramide and azaperone) that have differing core chemical scaffolds. At the end of the culture incubation period, the neurons were homogenized in RIPA buffer, and RIPA-soluble and -insoluble fractions were obtained. Tau phosphorylation in the RIPA-soluble tau was examined because it is increased phosphorylation of soluble tau that would likely enhance tau inclusion formation. Moreover, analysis of phosphorylation changes in insoluble tau is confounded by contaminating background tau phosphorylation from the added AD-tau and the fact that neuronal tau inclusions are greatly reduced by treatment with the D2 receptor antagonists (Fig. 6). Although there are >40 described tau phosphorylation sites (53, 54), we chose to specifically assess phosphorylation at Ser-199/Ser-202/Thr-205 (AT8 antibody) and Ser-396/Ser-404 (PHF1 antibody), as these sites are phosphorylated in AD brain tau inclusions, with the extent of phosphorylation at these sites correlating with disease stage (55). Little to no AT8 immunoreactivity was detected in the RIPA-soluble fraction of AD-tau–treated neurons, suggesting that phosphorylation at this site is unlikely to contribute to the observed tau aggregation in the neuron cultures. However, PHF1-positive tau was observed in the RIPA-soluble fraction of neurons treated with or without AD-tau (Fig. S6A). Neurons that also received metoclopramide or azaperone showed no change in PHF1-positive tau relative to those treated with vehicle only (Fig. S6, A and B). Thus, it does not appear that the D2 receptor antagonists decrease tau inclusion formation in the neuronal assay through a reduction of phosphorylation at the Ser-396/Ser-404 or Ser-199/Ser-202/Thr-205 sites. We cannot, however, exclude the possibility that the active D2 receptor antagonists alter phosphorylation at one or more of the other tau phosphorylation sites.
Interestingly, there is prior evidence of D2 receptor antagonists reducing the formation of tau inclusions in a Caenorhabditis elegans model with tau pathology, where azaperone reduced levels of insoluble tau and inhibited neurodegeneration (56). Moreover, genetic knockout of the C. elegans D2-like receptors had a similar tau aggregate-reducing effect (56). Although the detailed mechanism linking D2 receptor function and tau pathology in C. elegans is still being investigated and may be indirect (57), the ability of the neuronal tau inclusion screen to identify these compounds suggests the following: 1) this mechanism may exist in mammalian as well as C. elegans neurons, and 2) the neuronal assay of tau inclusion formation appears to be capable of identifying biologically relevant hits. In this regard, the screening of larger compound libraries in this assay may lead to the identification of additional small molecules and associated molecular targets that regulate the formation or clearance of tau aggregates.
Discussion
The amyloid cascade hypothesis of AD (4, 5) was formulated over 20 years ago based on genetic evidence pointing to a key role of Aβ peptide-containing senile plaques in AD onset. As a result, the vast majority of AD drug candidates that have progressed to Phase 3 testing have been designed to reduce Aβ levels and/or plaque burden, and to date all have failed to show sufficient efficacy to garner regulatory approval. Insights into the possible reasons for these clinical failures have been provided by advances in our understanding of brain changes that occur as AD progresses from prodromal to symptomatic stages. In particular, PET and magnetic resonance imaging (MRI) methodologies that allow for assessment of AD brain function and pathology, as well as cerebrospinal fluid biomarkers that provide potential information about brain changes, reveal that plaque pathology precedes symptom onset by a decade or more (24). Thus, the administration of Aβ-directed drug candidates to patients with even early clinical signs of disease (i.e. mild cognitive impairment) may be ineffective because plaque pathology has largely plateaued and the cascade of events triggered by Aβ accumulation have progressed to a stage where lowering of the plaque burden could be without meaningful benefit. In contrast, immunohistochemical (8, 9) and PET imaging studies (10, 12) provide strong evidence that the development of robust tau pathology in AD brain occurs well after Aβ accumulation, with tau burden showing a strong correlation with patient cognitive status. These findings suggest that tau-directed therapeutics may have greater utility in early symptomatic and late prodromal AD patients than those targeting Aβ, and there has been increased interest in the pharmaceutical sector in identifying mechanisms and molecules that will reduce tau pathology.
A number of strategies have been proposed to reduce the development or consequences of tau pathology (58), but to date most have not resulted in drug candidates entering clinical testing. There are presently multiple immunotherapy trials ongoing in which tau antibodies are being assessed for their ability to slow AD progression (26). In addition, there have been pharmaceutical programs directed to the inhibition of kinases thought to play a role in the hyperphosphorylation of tau (59–61), but this approach has not resulted in clinical success. Finally, the compound LMTX® that was proposed to act as a tau fibrillization inhibitor (62) failed to achieve the co-primary end points in a Phase 3 study in AD patients. There would clearly be value in taking a relatively unbiased approach to identifying new small molecules and associated targets that modulate tau inclusions. In this regard, the development of robust cellular assays that mimic the processes of tau inclusion formation and turnover would be important. Although such assays have been previously developed using cell lines in which mutated and/or truncated tau proteins were overexpressed (32–36), only very recently has such an assay been utilized for extensive compound screening (37). In addition, a model of tau aggregation has been described in human iPS cell-derived neurons that overexpress tau (39), although compound screening was not reported. We report here on two assays of fibrillar tau aggregate formation: one in HEK293 cells that overexpress GFP-tagged T40 tau harboring the P301L mutation found in familial FTLD (41), and the other in primary rat cortical neurons in which fibrillary tau inclusions develop from endogenously expressed rat tau after seeding of the cultures with AD-tau (38). The HEK293 cell assay utilizes cells that maintain intracellular tau aggregates (T40PL–GFPagg cells), and these aggregates can be reduced in number and size by inhibiting tau expression from a tetracycline-inducible promoter through Dox withdrawal. The tau aggregate reformation can be triggered upon re-initiation of tau expression in the T40PL–GFPagg cells, and the addition of compounds prior to tau re-expression allows for the identification of molecules that affect tau inclusion formation or clearance.
The screening of the NCATS Pharmaceutical Collection in the T40PL–GFPagg cell assay resulted in a low hit rate, which could reflect a difficulty in finding molecules that inhibit tau fibrillization or tau aggregate clearance in a cellular milieu. Alternatively, it could be that the T40PL–GFPagg cells, which were selected based on their ability to be propagated while harboring tau aggregates, have an altered cellular physiology that renders them relatively insensitive to compounds that might alter the generation or clearance of tau inclusions. Disappointingly, only one hit from the NCATS Pharmaceutical Collection screen (SNC-80; opioid receptor agonist) showed some evidence of activity in the primary rat cortical assay of tau aggregate formation. As neurons are the primary cell type affected with tau inclusions in AD and related tauopathies, and because the rat cortical neuron assay does not rely on tau overexpression, this model likely represents a more suitable system to assess molecules that might affect tau aggregate formation or clearance. In particular, neurons are highly-specialized post-mitotic cells that are considerably different from rapidly-dividing cell lines such as HEK293 cells, and thus it is not surprising that cellular pathways, including those regulating cellular proteostasis, might differ from those in highly-mitotic cells. Moreover, neurons may show a differential compound toxicity profile than rapidly dividing cells.
The screening of the Prestwick library of mostly approved drugs in the rat cortical tau inclusion assay resulted in a significantly greater number of confirmed hits (32 confirmed nontoxic compounds; 2.5% confirmed hit rate) than observed in the T40PL–GFPagg cell tau aggregation screen. An analysis of a subset of confirmed nontoxic hits directed to neuronal targets revealed that most of the compounds showed well-defined concentration-dependent inhibition of neuronal tau inclusions. Of particular note was the finding that multiple dopamine D2 receptor antagonists caused concentration-dependent inhibition of tau aggregates. This receptor has been previously implicated in the regulation of tau aggregates in a C. elegans model of tauopathy (56), where one of the compounds identified here (azaperone) and certain other antipsychotic drugs were shown to suppress tau aggregation and associated neurotoxicity. In addition, genetic knockout of the two C. elegans D2 receptor orthologues reduced tau aggregates and neuron loss (56). More recently, ablation of DOPA decarboxylase in the C. elegans tauopathy model was also shown to improve the behavioral deficits associated with tau pathology (57), suggesting that the effects of D2 receptor antagonists on tau aggregates may result from downstream effects that alter catechol metabolism. Interestingly, another study (63) revealed that haloperidol, an antipsychotic drug that acts as a D2 receptor antagonist, decreased tau phosphorylation in a transgenic mouse model of tauopathy, with this effect perhaps resulting from an inactivation of AMP-activated protein kinase.
Future studies will be necessary to gain a better understanding of the mechanism of D2 receptor antagonist regulation of tau inclusion formation and/or clearance in rat and other mammalian neurons. Our studies reveal that these compounds do not generally decrease tau protein levels and do not directly block tau fibrillization. Moreover, the D2 receptor antagonists do not appear to inhibit tau inclusion formation through reduction of tau phosphorylation at the Ser-199/Ser-202/Thr-205 or Ser-396/Ser-404 sites. However, we cannot exclude the possibility that other tau phosphorylation sites are affected by these compounds, and a comprehensive analysis of all phosphorylation sites would require significant additional experimentation that is beyond the scope of this study.
The identification of targets and or pathways that act downstream of the D2 receptor will be important, as it is unlikely that D2 receptor antagonists themselves will be viable therapeutic candidates for AD. This class of drugs can cause fairly severe side effects, and there is evidence that treatment of AD patients with antipsychotic drugs can increase cognitive decline (64) and perhaps increase mortality (65). Nonetheless, the identification of multiple D2 receptor antagonists, as well as other potentially interesting confirmed hits, in the rat cortical neuron tau inclusion assay described herein reveals the utility of this assay in compound screening. Accordingly, the screening of larger compound collections could lead to the identification of novel small molecules and associated molecular targets that could serve as the basis for drug discovery programs directed to the reduction of tau pathology in AD and related tauopathies.
Experimental procedures
HEK293 T40PL–GFPagg cell 1536-well compound screening
HEK293 T40PL–GFPagg cells were maintained in DMEM (Thermo Fisher Scientific, catalog no. 11995) + 10% tetracycline-screened FBS (HyClone), 100 units/ml penicillin, 100 μg/ml streptomycin, 1% Glutamax, 5 μg/ml blasticidin, and 200 μg/ml Zeocin + 100 ng/ml doxycycline. For use in compound screening, HEK293 T40PL–GFPagg cells were cultured in DMEM + 10% tetracycline-screened FBS + 1% penicillin–streptomycin + 1% GlutaMAX for 6 days (Dox removal stage). Cells were plated into a 1536-well black clear-bottom cyclic olefin imaging plate (Aurora Microplates) at 400 cells per well in a 6.5-μl volume using a Multidrop Combi dispenser (Thermo Fisher Scientific). Plates were either standard tissue culture–treated (when using CellTiterGlo extraction protocol) or coated with poly-d-lysine (hexadecyltrimethylammonium (HDTA) extraction protocol). After overnight incubation, compounds were added at five concentrations (110 nm to 27.5 μm) in singlets using a pin transfer tool (Wako), and 1 μl of 750 ng/ml doxycycline, prepared in DMEM described above, was added to each well (final concentration 100 ng/ml). Control wells without Dox treatment (1 μl DMEM addition) were included on each plate. Cells were returned to a humidified incubator for 48 h. For the CellTiterGlo extraction protocol, 2 μl of CellTiterGlo luminescent cell viability reagent (Promega) containing 10 μg/ml Hoechst 33342 was added to each well, and plates were incubated at room temperature for 5 min. Data were collected using an IN Cell2200 automated wide-field imager at ×10 magnification to acquire the entire well from a single field of view image. The system utilized laser-based autofocus and standard filters to capture tau-GFP (excitation 475/28 and emission 512/23) and Hoechst (excitation 390/18 and emission 432/48). Cell viability was assessed by reading luminescence on a ViewLux Microplate Imager (PerkinElmer Life Sciences) equipped with clear filters. For the HDTA extraction protocol, angled aspirators and dispensers were used for all medium exchanges. First, cells were washed five times by sequentially aspirating medium (leaving 4 μl in well) and adding 4 μl of PBS to each well. On the final wash, 4 μl of PBS remained in the well, and an additional 4 μl of PBS + 2% HDTA was added. Cells were incubated at room temperature for 10 min, and medium was aspirated to a volume of 4 μl per well, followed by addition of 4 μl of 8% paraformaldehyde (prepared in PBS). Plates were incubated at room temperature for 20 min, and five aspiration/wash steps (4 μl) were performed using PBS + 0.05% Tween 20. Then, 4 μl of PBS + 0.05% Tween 20 + 10 μg/ml Hoechst 33342 was added to each well, and plates were incubated at room temperature for 20 min and imaged on an InCell2200 as outlined above. Fluorescence intensity was quantified using InCell Developer Toolbox (GE Healthcare), with intensity and area of large aggregates (4–10 μm diameter size criterion) calculated. Nuclei were identified by using standard top hat segmentation with a size criteria of ∼75 μm in diameter. For assessing compound activity, the feature “sum fluorescence intensity × area of large aggregates” was normalized to cell number (nuclei counts, Hoechst 33342 staining). The screening data were analyzed using software developed internally at NCATS. Percent activity of compounds was normalized to Dox + vehicle (0% activity) and No-Dox (−100% activity) controls on a per-plate basis. The same controls were used for the calculation of the Z′ factor index for each assay. Compound activity was fitted to the Hill equation using in-house software ((NCGC CurveFit, RRID:SCR_018079), and concentration–response curves were generated. Active compounds had defined dose-dependent inhibition of tau aggregation and activity in both HDTA and CTG assays. Visual inspection of curves was performed to ensure compound activity.
Human detergent-insoluble tau (AD-tau) extracts
Detergent-insoluble tau (AD-tau) was prepared essentially according to Guo et al. (38). An intermediate fraction (termed the 1% Sarkosyl pellet in Ref. 38) was determined to be suitable for primary neuron transduction as long as the AD-tau purity was ≥10% of total protein as measured by Tau5 ELISA (below) and BCA (Pierce) protein assays. Sporadic AD cases used to prepare AD-tau were pre-screened to ensure high neuritic tau burden in the frontal cortex.
Rat cortical neuron tau inclusion assay
Primary rat cortical neurons were obtained from a core facility at the University of Pennsylvania and prepared according to previously published methods (66). The cell suspension was plated on poly-d-lysine–coated 384-well plates at 5000 viable cells per well in plating medium (complete Neurobasal medium (Gibco) with 5% FBS), which was replaced at 1 day after plating with complete Neurobasal medium supplemented with 1% penicillin–streptomycin (Gibco), 1% GlutaMAX (Gibco), 2% B-27 supplement (Gibco). The plated neurons were treated with compound and were transduced with AD-tau after 7 days in culture, replacing 50% of the medium with 1:1 preconditioned neuronal medium/fresh complete Neurobasal medium, followed by a 14–15-day incubation period in which cultures were not disturbed. Cells were then extracted and fixed for assessment of insoluble tau formation or fixed and permeabilized for morphology and toxicity assessment. Wells were washed five times with 0.1 ml of PBS using a BioTek ELx405 plate washer, with each aspiration leaving 25 μl of residual PBS in the well. Plates that were extracted for immunocytochemical staining of insoluble tau were washed as above, followed by addition of 25 μl of 2% HDTA in PBS, with incubation for 10 min. This was followed by removal of 50% of the solution (25 μl) and replacement with 25 μl of 8% PFA in PBS, with incubation for 20 min. Plates were then washed and blocked with addition of 25 μl of 6% FBS and 6% BSA for 1 h. All reagents were dispensed with an Evolution P3 dispenser (PerkinElmer Life Sciences). Wells used for assessment of neuronal morphology by fixation–permeabilization received 25 μl of 8% PFA in PBS, followed by removal of 50% of the solution (25 μl) and addition of 25 μl of 0.25% Triton X-100. After blocking, wells were incubated overnight with primary antibody, T49 (mouse anti-rodent tau monoclonal (67)) for extracted cells, and 17028 (rabbit anti-MAP2, (68)) for dendrite outgrowth and anti-NeuN (MAB377; Millipore) for neuron counts for fixed–permeabilized cells. Fluorescently-labeled secondary antibodies (Invitrogen goat anti-mouse 488 and goat anti-rabbit 594) were used for visualization, and DAPI (0.4 μg/ml) was added with secondary antibodies to stain cell nuclei. Plates were imaged on an INCell2200 (GE Healthcare) with a ×10 objective, using nine 1024 × 1024 pixel images per well for 56% coverage of the center of the well to avoid edge fixation artifacts. Data were collected in two wavelengths from HDTA-extracted plates, with nuclear signal (INCell2200 DAPI channel) and mouse tau (INCell2200 FITC channel), or three wavelengths for fixed–permeabilized plates, with nuclear signal (INCell2200 DAPI channel), NeuN (INCell2200 FITC channel), and MAP2 (INCell2200 Texas Red channel). Images were quantified using InCell Developer Toolbox (GE Healthcare). DAPI counts were determined by object size segmentation using kernel size 15 and sensitivity of 1. Then tau staining and MAP2 staining were gated by object intensity. NeuN counts were determined using object intensity followed by clump breaking using a DAPI reference. Neuronal tau inclusion burden was expressed as T49 (area × optical density)/DAPI count. Compound activity was plotted as percent inhibition relative to vehicle-treated AD-tau–transduced control wells, with subtraction of background signal derived from nonAD-tau–treated wells. Compound-mediated toxicity was monitored by three measures: DAPI counts, MAP2 area/DAPI count, and NeuN counts, all expressed as a percent of vehicle-treated AD-tau–transduced control.
Assessment of neuronal multimeric tau
Neuronal cultures that were utilized to determine the extent of compound-mediated reduction of multimeric tau as determined by ELISA were plated on 96-well plates at 17,500 cells/well in 100 μl of plating medium (as above), with medium replacement after 1 day to complete neurobasal medium. AD-tau transduction was scaled based on the fibril concentration from the 384-well plate to 2.5 μg/well (1.25 ng/μl). Timing of AD-tau and compound addition was as described for the 384-well plate assay. Following 14–15 days of incubation after AD-tau addition, the plated cells were extracted with 50 μl of 0.1% RIPA (50 mm Tris, pH 8.0, 150 mm NaCl, 5 mm EDTA, 0.5% sodium deoxycholate, 1% Nonidet P-40, and 0.1% SDS) after washing the wells with PBS on the BioTek ELx405 plate washer, fully evacuating the wells with a hand-held aspiration wand and then addition of 50 μl/well of RIPA, pH 8.0, with extensive trituration. Maxisorp plates (Thermo Fisher Scientific) were coated with 2.5 μg/ml mTau8 IgG fraction (mouse anti-rodent tau, kindly provided by Janssen Pharmaceutica) and blocked with 1% Blockace in PBS until needed. Cell RIPA extracts were diluted 5-fold in 0.2% BSA in PBS to achieve a linear range of detection and added to the ELISA plates, with overnight incubation. Plates were washed in PBS and then incubated in biotinylated mTau8 followed by incubation with streptavidin–horseradish peroxidase conjugate. Visualization was done using TMB A+B reagents and stopping with 10% phosphoric acid. Plates were read at A450, and data were normalized to BCA protein content and plotted as percent inhibition relative to fibril-transduced vehicle controls.
Soluble tau ELISA
Neuronal cultures were grown on 96-well plates and treated with AD-tau and compounds as described for the tau multimer ELISA. These underwent the ELISA conditions as described above, except that plates were coated with 2.5 μg/ml Tau5 IgG fraction (mouse anti-pan tau, Thermo Fisher Scientific), and the detection antibodies were a mixture of biotinylated BT2 and HT7 (Thermo Fisher Scientific, 31.2 and 62.5 ng/ml respectively).
Tau fibrillization assay
The testing of the compound effect on heparin-induced tau fibrillization was performed essentially as described previously (27). Briefly, 0.1 μl of compounds in 100% DMSO were dispensed with a pin tool into wells of 384-well plates containing 15 μl of 33.3 μm K18P301L tau in 100 mm sodium acetate buffer, pH 7.0. After incubating at room temperature for 30 min, 10 μl of 100 μm heparin was added to induce fibrillization (final concentrations of 20 μm tau and 40 μm heparin with 250-fold dilution of compound). The assay plate was incubated overnight at room temperature, followed by the addition of thioflavin T (12.5 μm final concentration). Thioflavin T signal was read 30 min later at an excitation of 450 nm and an emission of 510 nm, with a 495-nm cutoff, on a Spectramax M5 spectrophotometer. Data were represented as percent inhibition relative to vehicle control.
Phospho-tau analysis
Rat cortical neurons were cultured as in the 384-well plate tau inclusion assay, but scaled for 12-well plates (Costar) resulting in 100,000 cells per well (26,000 cells per cm2) in 1 ml of plating medium. Cultures were grown for 7 days and then treated with metoclopramide or azaperone at concentrations selected from concentration–response data to result in maximal inhibition (1 μm final concentration for both compounds) or 50% inhibition (50 nm for metoclopramide and 300 nm for azaperone). Plates were incubated for 30 min with compound and then transduced with control medium or 1.25 μg of sonicated AD-tau fibrils. After an additional 15 days in culture, cells were washed with ice-cold PBS and then scraped in 100 μl of RIPA buffer per well and sonicated to ensure disruption of cellular contents. Sonicated samples were centrifuged at 100,000 × g for 30 min. Pellets were resuspended in 100 μl of RIPA as supernatants and sonicated with 20 pulses to fully disperse the pelleted material. Compound inhibition of tau inclusion formation was confirmed for the neurons receiving 1 μm D2 receptor antagonist and AD-tau by analysis of aliquots from the pellet fraction in the mTau8 rodent tau-specific multimer ELISA.
Aliquots from the RIPA homogenate supernatant fraction were run on 10% polyacrylamide gels (Bio-Rad Protean III) and then blotted onto 0.22-μm pore size nitrocellulose (Bio-Rad). Blots were blocked using LiCOR blocking buffer for 1 h at room temperature and then incubated in primary antibody overnight. Mouse tau was identified using an in-house rabbit polyclonal anti-rodent tau antibody (R2295M) that had been pre-adsorbed to remove antibody recognizing human tau (38). Phospho-tau was identified with AT8 (Thermo Fisher Scientific; MN1020) mAb or PHF1 mAb (prepared in-house). Phospho-tau signal was quantified on a LiCOR imager and normalized to the mouse tau signal, utilizing Image Studio software with local background signal correction. All treatments were run in triplicate wells, with each of the triplicate samples run on separate gels such that each experimental condition was represented once on each gel.
Author contributions
A. C., M. J. H., J. A., S. A. T., and K. R. B. data curation; A. C., M. J. H., S. A. T., A. Z., A. S., and K. R. B. formal analysis; A. C. supervision; A. C., M. J. H., J. A., S. A. T., A. Z., and A. S. investigation; A. C., M. J. H., J. A., A. Z., A. S., A. B., and C. D. methodology; M. J. H., D. M., and K. R. B. project administration; M. J. H., A. B., C. D., and D. M. writing-review and editing; A. Z., A. S., A. B., D. M., and K. R. B. conceptualization; C. D. validation; D. M., J. Q. T., V. M. L., and K. R. B. resources; D. M., J. Q. T., V. M. L., and K. R. B. funding acquisition; K. R. B. writing-original draft.
Supplementary Material
Acknowledgments
We thank Carleen Klumpp-Thomas for assistance with HEK293 T40PL–GFPagg cell assay automation and the NCATS compound management group for sourcing, quality control, and plating compounds for the HEK293 T40PL–GFPagg cell screen. In addition, we acknowledge the assistance of the University of Pennsylvania core facility that supplied embryonic neurons.
This work was supported by Janssen Pharmaceutica NV and in part by the Intramural Research Program, National Center for Advancing Translational Sciences, National Institutes of Health. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Figs. S1–S6 and Table S1.
- NFT
- neurofibrillary tangle
- AD
- Alzheimer's disease
- Aβ
- β-amyloid
- NT
- neuropil thread
- FTLD
- frontotemporal lobar degeneration
- MT
- microtubule
- NCATS
- National Center for Advancing Translational Sciences
- HDTA
- hexadecyltrimethylammonium bromide
- PFA
- paraformaldehyde
- DAPI
- 4′,6-diamidino-2-phenylindole
- PET
- positron emission tomography
- DMEM
- Dulbecco's modified Eagle's medium
- FBS
- fetal bovine serum
- Dox
- doxycycline
- PFF
- pre-formed fibril
- CTG
- CellTiter-GloTM
- CV
- column volume.
References
- 1. Ballatore C., Lee V. M., and Trojanowski J. Q. (2007) Tau-mediated neurodegeneration in Alzheimer's disease and related disorders. Nat. Rev. Neurosci. 8, 663–672 10.1038/nrn2194 [DOI] [PubMed] [Google Scholar]
- 2. Lee V. M., Goedert M., and Trojanowski J. Q. (2001) Neurodegenerative tauopathies. Annu. Rev. Neurosci. 24, 1121–1159 10.1146/annurev.neuro.24.1.1121 [DOI] [PubMed] [Google Scholar]
- 3. Alzheimer's Association. (2015) 2015 Alzheimer's disease facts and figures. Alzheimers Dement. 11, 332–384 10.1016/j.jalz.2015.02.003 [DOI] [PubMed] [Google Scholar]
- 4. Hardy J., and Selkoe D. J. (2002) The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science 297, 353–356 10.1126/science.1072994 [DOI] [PubMed] [Google Scholar]
- 5. Selkoe D. J., and Schenk D. (2003) Alzheimer's disease: molecular understanding predicts amyloid-based therapeutics. Annu. Rev. Pharmacol. Toxicol. 43, 545–584 10.1146/annurev.pharmtox.43.100901.140248 [DOI] [PubMed] [Google Scholar]
- 6. Hutton M., Lendon C. L., Rizzu P., Baker M., Froelich S., Houlden H., Pickering-Brown S., Chakraverty S., Isaacs A., Grover A., Hackett J., Adamson J., Lincoln S., Dickson D., Davies P., et al. (1998) Association of missense and 5′-splice–site mutations in tau with the inherited dementia FTDP-17. Nature 393, 702–705 10.1038/31508 [DOI] [PubMed] [Google Scholar]
- 7. Hong M., Zhukareva V., Vogelsberg-Ragaglia V., Wszolek Z., Reed L., Miller B. I., Geschwind D. H., Bird T. D., McKeel D., Goate A., Morris J. C., Wilhelmsen K. C., Schellenberg G. D., Trojanowski J. Q., and Lee V. M. (1998) Mutation-specific functional impairments in distinct Tau isoforms of hereditary FTDP-17. Science 282, 1914–1917 10.1126/science.282.5395.1914 [DOI] [PubMed] [Google Scholar]
- 8. Arriagada P. V., Growdon J. H., Hedley-Whyte E. T., and Hyman B. T. (1992) Neurofibrillary tangles but not senile plaques parallel duration and severity of Alzheimers disease. Neurology 42, 631–639 10.1212/WNL.42.3.631 [DOI] [PubMed] [Google Scholar]
- 9. Wilcock G. K., and Esiri M. M. (1982) Plaques, tangles and dementia–a quantitative study. J. Neurol. Sci. 56, 343–356 10.1016/0022-510X(82)90155-1 [DOI] [PubMed] [Google Scholar]
- 10. Cho H., Choi J. Y., Hwang M. S., Lee J. H., Kim Y. J., Lee H. M., Lyoo C. H., Ryu Y. H., and Lee M. S. (2016) Tau PET in Alzheimer disease and mild cognitive impairment. Neurology 87, 375–383 10.1212/WNL.0000000000002892 [DOI] [PubMed] [Google Scholar]
- 11. Wang L., Benzinger T. L., Su Y., Christensen J., Friedrichsen K., Aldea P., McConathy J., Cairns N. J., Fagan A. M., Morris J. C., and Ances B. M. (2016) Evaluation of tau imaging in staging Alzheimer disease and revealing interactions between β-amyloid and tauopathy. JAMA Neurol. 73, 1070–1077 10.1001/jamaneurol.2016.2078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hanseeuw B. J., Betensky R. A., Jacobs H. I. L., Schultz A. P., Sepulcre J., Becker J. A., Cosio D. M. O., Farrell M., Quiroz Y. T., Mormino E. C., Buckley R. F., Papp K. V., Amariglio R. A., Dewachter I., Ivanoiu A., et al. (2019) Association of amyloid and tau with cognition in preclinical Alzheimer disease: a longitudinal study. JAMA Neurol. 76, 915–924 10.1001/jamaneurol.2019.1424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Drechsel D. N., Hyman A. A., Cobb M. H., and Kirschner M. W. (1992) Modulation of the dynamic instability of tubulin assembly by the microtubule-associated protein tau. Mol. Biol. Cell 3, 1141–1154 10.1091/mbc.3.10.1141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Gustke N., Trinczek B., Biernat J., Mandelkow E. M., and Mandelkow E. (1994) Domains of tau-protein and interactions with microtubules. Biochemistry 33, 9511–9522 10.1021/bi00198a017 [DOI] [PubMed] [Google Scholar]
- 15. Dixit R., Ross J. L., Goldman Y. E., and Holzbaur E. L. (2008) Differential regulation of dynein and kinesin motor proteins by tau. Science 319, 1086–1089 10.1126/science.1152993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Vershinin M., Carter B. C., Razafsky D. S., King S. J., and Gross S. P. (2007) Multiple-motor based transport and its regulation by Tau. Proc. Natl. Acad. Sci. U.S.A. 104, 87–92 10.1073/pnas.0607919104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Bramblett G. T., Trojanowski J. Q., and Lee V. M. (1992) Regions with abundant neurofibrillary pathology in human brain exhibit a selective reduction in levels of binding-competent-tau and accumulation of abnormal tau-isoforms (A68 proteins). Lab. Invest. 66, 212–222 [PubMed] [Google Scholar]
- 18. Alonso A. C., Zaidi T., Grundke-Iqbal I., and Iqbal K. (1994) Role of abnormally phosphorylated tau in the breakdown of microtubules in Alzheimer Disease. Proc. Natl. Acad. Sci. U.S.A. 91, 5562–5566 10.1073/pnas.91.12.5562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wagner U., Utton M., Gallo J. M., and Miller C. C. (1996) Cellular phosphorylation of tau by GSK-3β influences tau binding to microtubules and microtubule organisation. J. Cell Sci. 109, 1537–1543 [DOI] [PubMed] [Google Scholar]
- 20. Brunden K. R., Zhang B., Carroll J., Yao Y., Potuzak J. S., Hogan A. M., Iba M., James M. J., Xie S. X., Ballatore C., Smith A. B. 3rd., Lee V. M., and Trojanowski J. Q. (2010) Epothilone D improves microtubule density, axonal integrity, and cognition in a transgenic mouse model of tauopathy. J. Neurosci. 30, 13861–13866 10.1523/JNEUROSCI.3059-10.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Barten D. M., Fanara P., Andorfer C., Hoque N., Wong P. Y., Husted K. H., Cadelina G. W., Decarr L. B., Yang L., Liu V., Fessler C., Protassio J., Riff T., Turner H., Janus C. G., et al. (2012) Hyperdynamic microtubules, cognitive deficits, and pathology are improved in tau transgenic mice with low doses of the microtubule-stabilizing agent BMS-241027. J. Neurosci. 32, 7137–7145 10.1523/JNEUROSCI.0188-12.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Brunden K. R., Trojanowski J. Q., and Lee V. M. (2009) Advances in tau-focused drug discovery for Alzheimer's disease and related tauopathies. Nat. Rev. Drug Discov. 8, 783–793 10.1038/nrd2959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lane R. F., Shineman D. W., Steele J. W., Lee L. B., and Fillit H. M. (2012) Beyond amyloid: the future of therapeutics for Alzheimer's disease. Adv. Pharmacol. 64, 213–271 10.1016/B978-0-12-394816-8.00007-6 [DOI] [PubMed] [Google Scholar]
- 24. Jack C. R. Jr., Knopman D. S., Jagust W. J., Petersen R. C., Weiner M. W., Aisen P. S., Shaw L. M., Vemuri P., Wiste H. J., Weigand S. D., Lesnick T. G., Pankratz V. S., Donohue M. C., and Trojanowski J. Q. (2013) Tracking pathophysiological processes in Alzheimer's disease: an updated hypothetical model of dynamic biomarkers. Lancet Neurol. 12, 207–216 10.1016/S1474-4422(12)70291-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Jack C. R. Jr., Knopman D. S., Jagust W. J., Shaw L. M., Aisen P. S., Weiner M. W., Petersen R. C., and Trojanowski J. Q. (2010) Hypothetical model of dynamic biomarkers of the Alzheimer's pathological cascade. Lancet Neurol. 9, 119–128 10.1016/S1474-4422(09)70299-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hoskin J. L., Sabbagh M. N., Al-Hasan Y., and Decourt B. (2019) Tau immunotherapies for Alzheimer's disease. Expert Opin. Investig. Drugs 28, 545–554 10.1080/13543784.2019.1619694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Crowe A., Ballatore C., Hyde E., Trojanowski J. Q., and Lee V. M. (2007) High throughput screening for small molecule inhibitors of heparin-induced tau fibril formation. Biochem. Biophys. Res. Commun. 358, 1–6 10.1016/j.bbrc.2007.03.056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Crowe A., Huang W., Ballatore C., Johnson R. L., Hogan A. M., Huang R., Wichterman J., McCoy J., Huryn D., Auld D. S., Smith A. B. 3rd., Inglese J., Trojanowski J. Q., Austin C. P., Brunden K. R., and Lee V. M. (2009) The identification of aminothienopyridazine inhibitors of tau assembly by quantitative high-throughput screening. Biochemistry 48, 7732–7745 10.1021/bi9006435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Honson N. S., Johnson R. L., Huang W., Inglese J., Austin C. P., and Kuret J. (2007) Differentiating Alzheimer disease-associated aggregates with small molecules. Neurobiol. Dis. 28, 251–260 10.1016/j.nbd.2007.07.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Pickhardt M., Gazova Z., von Bergen M., Khlistunova I., Wang Y., Hascher A., Mandelkow E. M., Biernat J., and Mandelkow E. (2005) Anthraquinones inhibit tau aggregation and dissolve Alzheimer's paired helical filaments in vitro and in cells. J. Biol. Chem. 280, 3628–3635 10.1074/jbc.M410984200 [DOI] [PubMed] [Google Scholar]
- 31. Taniguchi S., Suzuki N., Masuda M., Hisanaga S., Iwatsubo T., Goedert M., and Hasegawa M. (2005) Inhibition of heparin-induced tau filament formation by phenothiazines, polyphenols, and porphyrins. J. Biol. Chem. 280, 7614–7623 10.1074/jbc.M408714200 [DOI] [PubMed] [Google Scholar]
- 32. Guo J. L., and Lee V. M. (2011) Seeding of normal tau by pathological tau conformers drives pathogenesis of Alzheimer-like tangles. J. Biol. Chem. 286, 15317–15331 10.1074/jbc.M110.209296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Pickhardt M., Lawatscheck C., Borner H. G., and Mandelkow E. (2017) Inhibition of tau protein aggregation by Rhodanine-based compounds solubilized via specific formulation additives to improve bioavailability and cell viability. Curr. Alzheimer Res. 14, 742–752 10.2174/1567205014666170202103136 [DOI] [PubMed] [Google Scholar]
- 34. Lim S., Haque M. M., Kim D., Kim D. J., and Kim Y. K. (2014) Cell-based models to investigate tau aggregation. Comput. Struct. Biotechnol. J 12, 7–13 10.1016/j.csbj.2014.09.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Khlistunova I., Biernat J., Wang Y., Pickhardt M., von Bergen M., Gazova Z., Mandelkow E., and Mandelkow E. M. (2006) Inducible expression of tau repeat domain in cell models of tauopathy–aggregation is toxic to cells but can be reversed by inhibitor drugs. J. Biol. Chem. 281, 1205–1214 10.1074/jbc.M507753200 [DOI] [PubMed] [Google Scholar]
- 36. Bandyopadhyay B., Li G., Yin H., and Kuret J. (2007) Tau aggregation and toxicity in a cell culture model of tauopathy. J. Biol. Chem. 282, 16454–16464 10.1074/jbc.M700192200 [DOI] [PubMed] [Google Scholar]
- 37. Pickhardt M., Tassoni M., Denner P., Kurkowsky B., Kitanovic A., Möhl C., Fava E., and Mandelkow E. (2019) Screening of a neuronal cell model of tau pathology for therapeutic compounds. Neurobiol. Aging 76, 24–34 10.1016/j.neurobiolaging.2018.11.026 [DOI] [PubMed] [Google Scholar]
- 38. Guo J. L., Narasimhan S., Changolkar L., He Z., Stieber A., Zhang B., Gathagan R. J., Iba M., McBride J. D., Trojanowski J. Q., and Lee V. M. (2016) Unique pathological tau conformers from Alzheimer's brains transmit tau pathology in nontransgenic mice. J. Exp. Med. 213, 2635–2654 10.1084/jem.20160833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Medda X., Mertens L., Versweyveld S., Diels A., Barnham L., Bretteville A., Buist A., Verheyen A., Royaux I., Ebneth A., and Cabrera-Socorro A. (2016) Development of a scalable, high-throughput-compatible assay to detect tau aggregates using iPSC-derived cortical neurons maintained in a three-dimensional culture format. J. Biomol. Screen 21, 804–815 10.1177/1087057116638029 [DOI] [PubMed] [Google Scholar]
- 40. Goedert M. (2005) Tau gene mutations and their effects. Mov. Disord. 20, Suppl. 12, S45–S52 10.1002/mds.20539 [DOI] [PubMed] [Google Scholar]
- 41. Guo J. L., Buist A., Soares A., Callaerts K., Calafate S., Stevenaert F., Daniels J. P., Zoll B. E., Crowe A., Brunden K. R., Moechars D., and Lee V. M. (2016) The dynamics and turnover of tau aggregates in cultured cells: insights into therapies for tauopathies. J. Biol. Chem. 291, 13175–13193 10.1074/jbc.M115.712083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Titus S. A., Southall N., Marugan J., Austin C. P., and Zheng W. (2012) High-throughput multiplexed quantitation of protein aggregation and cytotoxicity in a Huntington's disease model. Curr. Chem. Genomics 6, 79–86 10.2174/1875397301206010079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Zhang J. H., Chung T. D., and Oldenburg K. R. (1999) A simple statistical parameter for use in evaluation and validation of high throughput screening assays. J. Biomol. Screen. 4, 67–73 10.1177/108705719900400206 [DOI] [PubMed] [Google Scholar]
- 44. Bhat R. V., Budd Haeberlein S. L., and Avila J. (2004) Glycogen synthase kinase 3: a drug target for CNS therapies. J. Neurochem. 89, 1313–1317 10.1111/j.1471-4159.2004.02422.x [DOI] [PubMed] [Google Scholar]
- 45. Mazanetz M. P., and Fischer P. M. (2007) Untangling tau hyperphosphorylation in drug design for neurodegenerative diseases. Nat. Rev. Drug Discov. 6, 464–479 10.1038/nrd2111 [DOI] [PubMed] [Google Scholar]
- 46. Chivers J. K., Gommeren W., Leysen J. E., Jenner P., and Marsden C. D. (1988) Comparison of the in vitro receptor selectivity of substituted benzamide drugs for brain neurotransmitter receptors. J. Pharm. Pharmacol. 40, 415–421 10.1111/j.2042-7158.1988.tb06306.x [DOI] [PubMed] [Google Scholar]
- 47. Li W., Sperry J. B., Crowe A., Trojanowski J. Q., Smith A. B. 3rd., and Lee V. M. (2009) Inhibition of tau fibrillization by oleocanthal via reaction with the amino groups of tau. J. Neurochem. 110, 1339–1351 10.1111/j.1471-4159.2009.06224.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Crowe A., James M. J., Lee V. M., Smith A. B. 3rd, Trojanowski J. Q., Ballatore C., and Brunden K. R. (2013) Aminothienopyridazines and methylene blue affect tau fibrillization via cysteine oxidation. J. Biol. Chem. 288, 11024–11037 10.1074/jbc.M112.436006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Merrick S. E., Trojanowski J. Q., and Lee V. M. (1997) Selective destruction of stable microtubules and axons by inhibitors of protein serine/threonine phosphatases in cultured human neurons (NT2N cells). J. Neurosci. 17, 5726–5737 10.1523/JNEUROSCI.17-15-05726.1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Alonso A. C., Grundke-Iqbal I., and Iqbal K. (1996) Alzheimer's disease hyperphosphorylated tau sequesters normal tau into tangles of filaments and disassembles microtubules. Nat. Med. 2, 783–787 10.1038/nm0796-783 [DOI] [PubMed] [Google Scholar]
- 51. Necula M., and Kuret J. (2004) Pseudophosphorylation and glycation of tau protein enhance but do not trigger fibrillization in vitro. J. Biol. Chem. 279, 49694–49703 10.1074/jbc.M405527200 [DOI] [PubMed] [Google Scholar]
- 52. Despres C., Byrne C., Qi H., Cantrelle F. X., Huvent I., Chambraud B., Baulieu E. E., Jacquot Y., Landrieu I., Lippens G., and Smet-Nocca C. (2017) Identification of the tau phosphorylation pattern that drives its aggregation. Proc. Natl. Acad. Sci. U.S.A. 114, 9080–9085 10.1073/pnas.1708448114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Morishima-Kawashima M., Hasegawa M., Takio K., Suzuki M., Yoshida H., Titani K., and Ihara Y. (1995) Proline-directed and non-proline-directed phosphorylation of Phf-Tau. J. Biol. Chem. 270, 823–829 10.1074/jbc.270.2.823 [DOI] [PubMed] [Google Scholar]
- 54. Hanger D. P., Betts J. C., Loviny T. L., Blackstock W. P., and Anderton B. H. (1998) New phosphorylation sites identified in hyperphosphorylated tau (paired helical filament-tau) from Alzheimer's disease brain using nanoelectrospray mass spectrometry. J. Neurochem. 71, 2465–2476 10.1046/j.1471-4159.1998.71062465.x [DOI] [PubMed] [Google Scholar]
- 55. Koss D. J., Jones G., Cranston A., Gardner H., Kanaan N. M., and Platt B. (2016) Soluble pre-fibrillar tau and β-amyloid species emerge in early human Alzheimer's disease and track disease progression and cognitive decline. Acta Neuropathol. 132, 875–895 10.1007/s00401-016-1632-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. McCormick A. V., Wheeler J. M., Guthrie C. R., Liachko N. F., and Kraemer B. C. (2013) Dopamine D2 receptor antagonism suppresses tau aggregation and neurotoxicity. Biol. Psychiatry 73, 464–471 10.1016/j.biopsych.2012.08.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Kow R. L., Sikkema C., Wheeler J. M., Wilkinson C. W., and Kraemer B. C. (2018) DOPA decarboxylase modulates tau toxicity. Biol. Psychiatry 83, 438–446 10.1016/j.biopsych.2017.06.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Khanna M. R., Kovalevich J., Lee V. M., Trojanowski J. Q., and Brunden K. R. (2016) Therapeutic strategies for the treatment of tauopathies: hopes and challenges. Alzheimers Dement. 12, 1051–1065 10.1016/j.jalz.2016.06.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Bhat R. V., Andersson U., Andersson S., Knerr L., Bauer U., and Sundgren-Andersson A. K. (2018) The conundrum of GSK3 inhibitors: is it the dawn of a new beginning? J. Alzheimers Dis. 64, S547–S554 10.3233/JAD-179934 [DOI] [PubMed] [Google Scholar]
- 60. Martin L., Latypova X., Wilson C. M., Magnaudeix A., Perrin M. L., Yardin C., and Terro F. (2013) Tau protein kinases: involvement in Alzheimer's disease. Ageing Res. Rev. 12, 289–309 10.1016/j.arr.2012.06.003 [DOI] [PubMed] [Google Scholar]
- 61. Tell V., and Hilgeroth A. (2013) Recent developments of protein kinase inhibitors as potential AD therapeutics. Front. Cell. Neurosci. 7, 189 10.3389/fncel.2013.00189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Harrington C. R., Storey J. M., Clunas S., Harrington K. A., Horsley D., Ishaq A., Kemp S. J., Larch C. P., Marshall C., Nicoll S. L., Rickard J. E., Simpson M., Sinclair J. P., Storey L. J., and Wischik C. M. (2015) Cellular models of aggregation-dependent template-directed proteolysis to characterize tau aggregation inhibitors for treatment of Alzheimer disease. J. Biol. Chem. 290, 10862–10875 10.1074/jbc.M114.616029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Koppel J., Jimenez H., Adrien L., Greenwald B. S., Marambaud P., Cinamon E., and Davies P. (2016) Haloperidol inactivates AMPK and reduces tau phosphorylation in a tau mouse model of Alzheimer's disease. Alzheimers Dement. 2, 121–130 10.1016/j.trci.2016.05.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Vigen C. L., Mack W. J., Keefe R. S., Sano M., Sultzer D. L., Stroup T. S., Dagerman K. S., Hsiao J. K., Lebowitz B. D., Lyketsos C. G., Tariot P. N., Zheng L., and Schneider L. S. (2011) Cognitive effects of atypical antipsychotic medications in patients with Alzheimer's disease: outcomes from CATIE-AD. Am. J. Psychiatry 168, 831–839 10.1176/appi.ajp.2011.08121844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Maust D. T., Kim H. M., Seyfried L. S., Chiang C., Kavanagh J., Schneider L. S., and Kales H. C. (2015) Antipsychotics, other psychotropics, and the risk of death in patients with dementia: number needed to harm. JAMA Psychiatry 72, 438–445 10.1001/jamapsychiatry.2014.3018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Dichter M. A. (1978) Rat cortical neurons in cell culture: culture methods, cell morphology, electrophysiology, and synapse formation. Brain Res 149, 279–293 10.1016/0006-8993(78)90476-6 [DOI] [PubMed] [Google Scholar]
- 67. Kosik K. S., Orecchio L. D., Binder L., Trojanowski J. Q., Lee V. M., and Lee G. (1988) Epitopes that span the tau molecule are shared with paired helical filaments. Neuron 1, 817–825 10.1016/0896-6273(88)90129-8 [DOI] [PubMed] [Google Scholar]
- 68. Volpicelli-Daley L. A., Luk K. C., Patel T. P., Tanik S. A., Riddle D. M., Stieber A., Meaney D. F., Trojanowski J. Q., and Lee V. M. (2011) Exogenous α-synuclein fibrils induce Lewy body pathology leading to synaptic dysfunction and neuron death. Neuron 72, 57–71 10.1016/j.neuron.2011.08.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.