

Abstract
Cellular senescence is terminal cell cycle arrest that represents a prominent response to numerous anticancer therapies. The oncogene inhibitor of the apoptosis-stimulating protein of p53 (iASPP) plays essential roles in regulating cellular drug response by inhibiting apoptosis. However, whether or not it regulates chemotherapy-induced senescence (TIS) in cancer cells remains unclear. Here, using two commonly used cancer cell lines, HCT 116 and MCF-7, along with the xenograft mouse model, we found that iASPP inhibits senescence and also influences the senescence-associated secretory phenotype (SASP), which confers anticancer drug resistance independently of apoptosis. Mechanistically, iASPP is transcriptionally elevated by the p65 subunit of NF-κB in senescent cells and then translocates to the nucleus, where it binds p53 and NF-κBp65. This binding inhibits their transcriptional activities toward p21 and the key SASP factors interleukin (IL)-6/IL-8, respectively, and subsequently prevents senescence. Of note, we observed that iASPP knockdown sensitizes apoptosis-resistant cancers to doxorubicin treatment by promoting senescence both in vitro and in vivo. We conclude that iASPP integrates the NF-κBp65- and p53-signaling pathways and thereby regulates cell fate in response to TIS, leading to chemotherapy resistance. These findings suggest that iASPP inhibition might be a strategy that could help restore senescence in cancer cells and improve outcomes of chemotherapy-based therapies.
Keywords: p53, NF-κB (NF-κB), senescence, interleukin 6 (IL-6), cancer therapy, iASPP, inhibitor of senescence-associated secretion phenotype
Introduction
Senescence is a terminal growth arrest that plays essential roles in limiting the expansion of damaged cells under both physiological and pathological conditions (1). As such, it can serve as a suppressor of carcinogenesis or as a promotor of tissue remodeling in embryogenesis or after wounding. The senescence program can be triggered or accelerated by diverse stresses such as telomere erosion, oncogene activation, and chemoradiotherapy-induced DNA damage (2). The latter is termed therapy-induced senescence (TIS),3 which represents a much less well-understood but practically important type of senescence (3).
Cytotoxic drugs are widely used as anticancer therapies, and resistance is a major obstacle to successful treatment outcomes (4). Apoptosis has long been recognized as an important determinant of drug action; however, recent evidence has also highlighted the clinical significance of senescence. For example, the occurrence of senescence in pre-malignant lesions has been shown to limit tumor progression (5, 6). In addition, although clinically advanced tumors frequently gain the capability to bypass senescence-associated barriers during oncogenic progress, senescence can be triggered by chemotherapies in such tumors both in vitro and in vivo (7, 8). Furthermore, cancer cell senescence has been reported to predict favorable clinical outcomes of anticancer therapies (7, 9). Thus, TIS represents a promising mechanism against cancer, and a number of pro-senescence drugs that aim to selectively enhance senescence in tumor cells have entered clinical trials (10).
Nonetheless, senescence is generally accompanied by a striking increase in protein secretion (termed the senescence-associated secretory phenotype (SASP)), which can elicit contradictory and opposing effects regarding tumor development (11). In some instances, the SASP stimulates tumorigenesis (12), angiogenesis (13), and metastasis (14), and in the others, it simulates the immune response and promotes tumor clearance (15, 16). The SASP can also reinforce senescence in an autocrine fashion or induce senescence in neighboring cells in a paracrine fashion (17–19). Because of its complexity, the promise of pro-senescence therapy can only be realized with a deeper understanding of the precise molecular mechanisms that regulate senescence and the SASP.
Numerous recent studies have provided important insights into the pathways that lead to cellular senescence and the SASP (20). p53 plays key roles in regulating senescence. Genetic disruption of p53-dependent senescence results in poor response to the chemotherapy (9). The SASP is mainly controlled by an independent branch of a regulatory network that involves the activation of NF-κBp65 and classical regulators of inflammation that are associated with NF-κBp65 activity, such as CCAAT/enhancer-binding protein β, interleukin-1α (IL-1α), and p38 mitogen-activated protein kinase (21–23). However, how the NF-κBp65 inflammatory response is regulated during senescence remains largely unknown. In addition, p53 and NF-κBp65 represent primary regulators in response to cellular stresses. The activities of these two signaling systems are often intertwined and produce coordinated or antagonistic effects in a context-dependent manner (24). It is reasonable to propose that the proteins intervening p53 and NF-κBp65 pathways may play fundamental roles in the outcomes of p53 and NF-κBp65 signaling during cellular senescence. We speculate that a newly-identified oncogene, inhibitor of apoptosis-stimulating protein of p53 (iASPP), may be one such candidate as it has the ability to inhibit both p53 and NF-κBp65 (25, 26).
Until now, the major focus of iASPP research in cancer has been its ability to inhibit apoptosis (26–28). Mechanistic studies have revealed that iASPP physiologically binds with p53 and selectively regulates p53's transcriptional activity toward pro-apoptotic genes, such as PIG3 and Bax. For unknown reasons, iASPP has been reported to have no obvious impacts on p53's transcriptional activity toward cell cycle arrest targets, such as p21 (26). In addition, studies have also demonstrated that iASPP exists as dimer in the cytoplasm, which blocks its nuclear translocation signal and also covers the p53-binding sites (29, 30). Intriguingly, we and others have shown that iASPP is predominately located in the cytoplasm and not the nucleus in most types of cancer (28, 30). For this reason, the activity of iASPP in regulating p53 is normally inactive in cancer. Indeed, we have previously shown that caspase-mediated iASPP cleavage during apoptosis can remove the key motif that mediates iASPP's dimerization, leading to iASPP's nuclear translocation and robust inhibitory effects toward the p53-induced gene 3 protein (PIG3) luciferase reporter activity in a p53-dependent manner (31). Similarly, cyclinB1/CDK1-mediated phosphorylation of iASPP at its N terminus disrupts its dimerization in melanoma cells, which also results in iASPP nuclear translocation and p53 inhibition (30). This has inspired us to ask whether iASPP can be activated and subsequently regulate the apoptosis-independent activity of p53 during TIS.
It is also noteworthy that iASPP was initially identified as an NF-κBp65–binding partner in a yeast two-hybrid assay, which was shown to elicit an inhibitory effect on the transcriptional activity of NF-κBp65 via a κB reporter assay (25). Consistent with this, iASPP deficiency in patients and mice results in the elevated expression of inflammation-related genes, some of which are NF-κBp65 targets (32, 33). However, in the epidermis of mice on a 129/C57BL6 background, iASPP deficiency failed to cause a detectable increase in NF-κBp65 activity (34). The biological significance of iASPP–NF-κBp65 interaction is highly cell context–dependent. In addition, NF-κBp65 and iASPP both have significant oncogenic potential in human cancers (27, 35). It remains unknown how iASPP and NF-κBp65 signaling are integrated in the context of cancer.
To this end, we sought to explore the functional interaction between iASPP and p53/NF-κBp65 in the context of TIS. We made an interesting observation that iASPP, a well-studied apoptotic inhibitor, is also a critical regulator of senescence. We found that iASPP is induced by NF-κBp65 during TIS and, in turn, inhibits the senescence program. Mechanistically, enhanced iASPP is translocated into the nucleus, where it associates with nuclear p53 and NF-κBp65, inhibiting cell cycle progression and the SASP (IL-6/IL-8), respectively. Such functions of iASPP contribute to chemoresistance both in vitro and in vivo. These data provide a paradigm about how an oncogene can integrate p53 and NF-κBp65 signaling, influence senescence, and induce chemoresistance, and also suggest that iASPP is a promising target for sensitization of the drug response by modulating senescence and the SASP, in addition to its well-established anti-apoptotic activity.
Results
iASPP is increased during DNA damage–induced senescence and attenuates the senescence program in turn
To understand the role of iASPP in regulating senescence, two commonly used cancer cell lines, HCT 116 and MCF-7, were pulse-exposed to a sublethal dose of the chemotherapeutic drug doxorubicin (Dox, 1 μg/ml) for 2 h, after which the media were replaced, and cells were grown for an additional number of days (Fig. 1a). As shown, the cell number was barely changed after the treatment and cell senescence—as indicated by a larger and flatter morphology, increased SA–β-gal staining (Fig. 1, b and c, and Fig. S1a), reduced LMNB1 expression levels, and elevated levels of p53 and p21 (Fig. 1d and Fig. S1b)—occurred in a time-dependent manner in both cell lines. Consistently, Dox-treated cells exhibited G1-phase arrest compared with the untreated controls (Fig. S1c). Conversely, the numbers of mitotic cells were largely decreased, as revealed by bromodeoxyuridine (BrdU) incorporation assays (Fig. S1d). Furthermore, DNA damage and ROS, as shown by H2Aγ levels (Fig. S1e) and DCFH-DA intensity (Fig. S1f), respectively, were both increased in Dox-treated cells, which was in line with previous reports that accumulated DNA damage and ROS levels contribute to increased TIS (36). These data collectively support an established tenet that chemotherapeutic drugs can induce cell senescence in cancer cells.
Figure 1.

iASPP is increased during DNA damage–induced senescence and attenuates the senescence program. a, experimental setup used to trigger TIS in cancer cells. b and c, SA–β-gal staining was performed after triggering senescence in MCF-7 and HCT 116 cells as indicated in a. Representative images are shown in b, and the quantification of SA–β-gal staining-positive cells is shown as a bar graph in c. d and e, expression of iASPP, LMNB1, p21, p53, and H2Aγ as determined by Western blotting in MCF-7 and/or HCT 116 cells with the indicated treatments. Representative blots are shown. β-Actin or α-tubulin was used as an internal control (Ctl). f and g, percentages of senescence and proliferation rates determined by SA–β-gal staining (f) and BrdU assay (g), respectively, 3 days after triggering senescence in HCT 116 cells. Representative images are shown (top). Quantitative data are presented as a bar graph (bottom). Bar = 10 μm in b and f, and bar = 20 μm in g. KD efficiency of iASPP was confirmed by Western blotting. Values are mean ± S.E. from three independent experiments; *, p < 0.05; **, p < 0.01, means compared with DMSO (c, f, and g); #, p < 0.05, and ##, p < 0.01, means compared with Dox-treated control (f and g).
To explore the role of iASPP in senescence, the expression levels of iASPP were examined after triggering senescence. Interestingly, iASPP levels were elevated as early as 24 h after short-term Dox treatment and kept rising with prolonged duration in parallel with the occurrence of senescence (Fig. 1d). In addition to its increase in Dox-induced senescent cancer cells, iASPP induction was also observed in another DNA damage–inducing agent etoposide-induced senescent cancer cells (Fig. S1, g and h). However, although the anti-microtubule drug paclitaxel also induced senescence in HCT 116 cells, no iASPP induction was detected in paclitaxel-induced senescence (Fig. S2a). These data suggest that iASPP may be particularly involved into DNA damage–induced senescence regulation.
To test the role of iASPP in senescence, iASPP expression was inhibited by two independent small interfering RNAs (siRNAs) that specifically target iASPP (Fig. 1e). Remarkably, iASPP inhibition reinforced senescence, as indicated by the increased levels of p21 and H2Aγ and loss of LMNB1 3 and 7 days after Dox exposure (Fig. 1e and Fig. S2b). Re-expression of iASPP in iASPP KD cells restored cell senescence (Fig. S2c). However, neither p21 nor LMNB1 changed with iASPP KD under unstressed conditions (Fig. 1e and Fig. S2, b and c). The numbers of SA–β-gal-positive cells were also elevated (Fig. 1f). Conversely, BrdU incorporation rates were decreased (Fig. 1g). In contrast, siRNA-mediated iASPP KD had no obvious impacts on paclitaxel-induced senescence (Fig. S2d). These data indicate that endogenous iASPP is induced during DNA damage–induced senescence and consequently restrains the senescence program.
iASPP enters the nucleus and inhibits p53-dependent p21 transcription in senescent cells
The fact that iASPP knockdown (KD) facilitated senescence prompted us to investigate the underlying mechanisms behind iASPP's regulation of senescence. It is well-established that the growth arrest of senescent cells is largely dependent on the activation of p53 (37). Therefore, we first asked whether iASPP inhibits senescence by regulating the transcriptional activity of p53. As shown, p53 KD reduced senescence, and importantly, the effects of iASPP KD on SA–β-gal activity and LMNB1 levels were largely diminished by p53 KD (Fig. 2, a and b). As expected, p53 KD reduced p21 levels and, more importantly, also abolished the effect of iASPP on p21 mRNA and protein expression (Fig. 2, b and c). Similar results were obtained via a p21 luciferase reporter assay (Fig. 2d). In agreement with the above data, although iASPP was increased in senescent p53 null H1299 cells, si-mediated iASPP KD had no obvious impacts on the expression levels of p21 and LMNB1 (Fig. 2e). Therefore, iASPP inhibits senescence mainly by inhibiting p53's transcriptional activity toward p21.
Figure 2.

iASPP enters the nucleus and inhibits p53-dependent p21 transcription in senescent cells. a, SA–β-gal staining was performed 3 days after triggering senescence with or without iASPP or p53 KD in HCT 116 cells. Representative images are presented (left) and quantification is shown in the bar graph (right). b, impacts of the two independent siRNA-mediated iASPP KD on senescence were determined by Western blot analysis of p53, p21, and LMNB1 in HCT 116 cells. Representative blots are shown. GAPDH was used as an internal control. c and d, mRNA levels of p21 (c) and the luciferase activity of the p21 promoter luciferase reporter (d) in HCT 116 cells were determined after the indicated treatments. e, expression of iASPP, LMNB1, p21, p53, and H2Aγ as determined by Western blotting in p53 null H1299 cells with the indicated treatments. Representative blots are shown. β-Actin was used as an internal control. f and g, nuclear translocation of iASPP in senescent MCF-7 and HCT 116 cells was determined by nuclear fractionation (f) and immunofluorescence (g). Bar = 20 μm. h, interaction of p53 with iASPP was determined by immunoprecipitation assay of HCT 116 cells. Values are mean ± S.E. from three independent experiments; *, p < 0.05, and **, p < 0.01 (a, c, and d). N.S., not significant.
We next asked how iASPP regulates p53's transcriptional activity toward p21. iASPP had no obvious impacts on p53 protein levels (Figs. 1e and 2b). It has been demonstrated previously that iASPP can bind p53 (26); however, this capability of iASPP depends on its precise nuclear localization (30). To this end, the localization of iASPP was examined in the control and senescent cells. Consistent with our previous report and those of others (28), iASPP was predominately localized in the cytoplasm under basal conditions in the cancer cells examined, as revealed by both cell fractionation and staining (Fig. 2, f and g). Interestingly, dramatic nuclear translocation of iASPP was observed in senescent HCT 116 and MCF-7 cells (Fig. 2, f and g). Immunoprecipitation (IP) results revealed no obvious interaction between iASPP and p53 under unstressed conditions, although such interaction was evident after senescence was triggered (Fig. 2h). iASPP(92–828) is an iASPP mutant that is mainly localized in the nucleus (31). Consistent with the above data, a more dramatic inhibitory effect of iASPP(92–828) than the full-length iASPP on p21 expression was observed after re-expression of exogenous iASPP in iASPP KD cells (Fig. S3a).
NF-κBp65 and Nrf2, two known downstream effectors of iASPP, have also been implicated in regulating cell senescence, at least under certain circumstances. However, neither NF-κBp65 KD nor Nrf2 KD produced a significant effect on senescence (Fig. S3, b and c). In addition, iASPP KD similarly promoted SA–β-gal activity (Fig. S3b) and the expression levels of p21 and LMNB1 (Fig. S3c) regardless of NF-κBp65 or Nrf2 KD. These data collectively indicate that senescence-induced iASPP inhibits senescence mainly by binding with p53 and inhibiting p53's transcriptional activity toward p21 in the nucleus.
iASPP inhibits the expression of IL-6 and IL-8 by binding with and inhibiting NF-κBp65
Given that senescence is generally accompanied by the SASP, we next assessed the effect of iASPP on the SASP. Although SASP's composition can vary, the pro-inflammatory cytokines IL-6 and IL-8 are consistently expressed by senescent cells (17, 39). In line with previous reports, IL-6 and IL-8 were elevated in senescent cells, as shown in a real-time quantitative (q)RT-PCR assay (Fig. 3a). Interestingly, siRNA-mediated iASPP KD augmented the expression of IL-6 and IL-8 (Fig. 3a). As expected, the conditioned medium of senescent cells contained relatively higher levels of IL-6 and IL-8 proteins, as revealed by an ELISA (Fig. 3, b and c). Importantly, the impacts of iASPP KD, and ectopic iASPP expression on IL-6 and IL-8 proteins secreted in the conditioned medium, were also validated (Fig. 3, b and c).
Figure 3.

iASPP inhibits the expression of SASP factors IL-6 and IL-8. a, expression of SASP factors, IL-6 and IL-8, was determined by quantitative (q)RT-PCR in senescent HCT 116 cells, with and without iASPP KD (middle and bottom). The KD efficiency of iASPP was confirmed by Western blotting. GAPDH was used as a loading control (Ctl) (top). b and c, protein levels of IL-6 and IL-8, in the conditioned media from control and senescent cells, were evaluated by ELISA after iASPP KD (b) or overexpression (c). d, κB luciferase reporter activity was determined by luciferase reporter assay in HCT 116 cells after iASPP KD. Values are mean ± S.E. from three independent experiments; *, p < 0.05, **, p < 0.01 and ***, p < 0.001, compared with DMSO; #, p < 0.05, and ##, p < 0.01, compared with Dox-induced senescence control.
It is well-established that NF-κB is a key transcription factor in regulating the SASP, including IL-6 and IL-8 (10). iASPP was initially identified as an NF-κBp65 inhibitor (25). Therefore, we asked whether iASPP regulates the SASP by inhibiting NF-κBp65. As shown, κB reporter activity was increased after senescence, which is consistent with previous reports and our data shown in Fig. 5b that NF-κBp65 activity (p-NF-κBp65) is increased during senescence. Remarkably, iASPP KD elevated Dox-induced κB reporter activity (Fig. 3d). In contrast, ectopic iASPP expression inhibited Dox-promoted κB reporter activity (Fig. 4a). Furthermore, the effects of iASPP on the expression of IL-6 and IL-8 were completely abolished in NF-κBp65 KD cells or in cells treated with the NF-κBp65 chemical inhibitor Bay11-7028 (Fig. 4b, Bay). Under the same conditions, the nuclear iASPP(92–828) mutant inhibited κB reporter activity in a more dramatic manner than full-length iASPP (Fig. 4a), suggesting that the inhibitory effect of iASPP on NF-κBp65 mainly relies on its nuclear portions. In support of this notion, iASPP was present in NF-κBp65 immunoprecipitates in senescent cells, when iASPP nuclear translocation occurred, but not in control cells (Fig. 4c).
Figure 5.

Activation of NF-κBp65 induces iASPP expression during senescence. a, quantification and association of iASPP mRNA and protein after senescence is triggered in MCF-7 and HCT 116 cells. b, expression levels of p53, IκBα, NF-κBp65, phospho-NF-κBp65 (p-NF-κBp65), Nrf2, and Keap1 were determined by Western blotting after senescence was triggered in MCF-7 and HCT 116 cells. GAPDH was used as a loading control. c, iASPP promoter region (−1153 to −10 nucleotides) upstream of the transcription start site contains consensus p53 and NF-κBp65–binding motifs, as predicted by JASPAR software (RRID:SCR_003030 information, 2016 server was used) (shown in the rectangle). The activity of an iASPP promoter luciferase reporter was determined by luciferase assay after NF-κBp65 KD. d, iASPP protein and mRNA levels were determined by Western blotting and qRT-PCR, respectively, after NF-κBp65 KD. e, interaction between NF-κBp65 and the iASPP promoter was analyzed by ChIP. Values are mean ± S.E. from three independent experiments; *, p < 0.05, and **, p < 0.01, compared with DMSO (a and c–e); #, p < 0.05, and ##, p < 0.01, compared with senescent control (c and d). N.S., not significant.
Figure 4.

iASPP inhibits the expression of IL-6 and IL-8 by binding with and inhibiting NF-κBp65. a, κB luciferase reporter activity was determined by luciferase reporter assay in HCT 116 cells after ectopic expression of full-length (FL) or truncated iASPP. b, mRNA levels of IL-6 and IL-8 were determined by qRT-PCR after genetic NF-κBp65 KD or treatment with NF-κBp65 inhibitor (Bay117082, BAY), in control and senescent HCT 116 cells. c, interaction of NF-κBp65 with iASPP in HCT 116 cells was determined by immunoprecipitation. d and e, cells transfected with iASPP or vector were treated with Dox (1 μg/ml) for 2 h followed by 3 days of recovery in fresh medium, which was collected and served as conditioned medium, as indicated in the figure. p21 and LMNB1 protein levels in HCT 116 cells were determined by Western blotting after treatment with conditioned medium from senescent HCT 116 cells, with or without iASPP overexpression, and recombined IL-6 and IL-8 were further added as indicated in the figure. d, quantification of p21 protein levels is shown in the bar graph (e). Values are mean ± S.E. from three independent experiments; *, p < 0.05, and **, p < 0.01, compared with DMSO (a and b), or Dox-induced senescence control (e); #, p < 0.05, and ##, p < 0.01, compared with Dox-induced senescence control (Ctl) (a and b). N.S., not significant.
Nrf2 has been shown to regulate the immune response by regulating the expression of some cytokines, such as IL-6, in an antioxidant-response element (ARE)-binding motif-independent manner (40). As shown, Nrf2 KD indeed reduced IL-6 and IL-8 levels, which was consistent with previous reports (41); however, iASPP elicited similar effects on IL-6 and IL-8 expression regardless of Nrf2 status (Fig. S4a). In addition, p53 KD did not affect iASPP KD-induced IL-6 and IL-8 up-regulation (Fig. S4b), suggesting that iASPP regulates IL-6 and IL-8 mainly by inhibiting NF-κBp65.
IL-6 and IL-8 have been shown to play essential roles in reinforcing senescence via an autocrine mechanism. In line with previous reports (17), p21 was increased, and LMNB1 was decreased by the conditioned medium derived from senescent cells, which contained relatively higher levels of IL-6 and IL-8 proteins (Fig. 4, d and e). Consistent with this, supplementation of recombinant IL-6 and IL-8 in the conditioned medium further elevated p21 expression and reduced LMNB1 expression (Fig. 4, d and e). Furthermore, IL-6 or IL-8 treatment also, at least in part, restored inhibited p21 and elevated LMNB1 expression mediated by iASPP overexpression (Fig. 4, d and e).
These data collectively demonstrate that iASPP inhibits NF-κBp65's transcriptional activity toward pro-inflammatory targets, such as IL-6 and IL-8, which subsequently contributes to the elevated expression of p21 via an autocrine mechanism.
Activation of NF-κBp65 induces iASPP expression during senescence
Because elevated iASPP plays important roles in regulating both senescence and the SASP, we investigated the mechanisms underlying iASPP up-regulation during senescence. As shown in Fig. 5a, both iASPP mRNA and protein levels were elevated and exhibited a positive association during senescence, suggesting that iASPP is mainly regulated at the transcriptional level. In support of this notion, both proteasome inhibitor MG132 and autophagy inhibitor chloroquine failed to affect senescence-regulated iASPP expression (Fig. S5a). To provide further evidence, a reporter plasmid with the iASPP promoter sequence inserted upstream of a luciferase reporter was constructed. Our results showed that iASPP luciferase activity was increased by ∼2-fold in senescent HCT 116 cells (Fig. S5b).
During senescence, multiple transcription factors—such as p53, NF-κBp65, and Nrf2—were activated (Fig. 5b). However, the activation of Nrf2 is unlikely to have contributed to the promotion of iASPP transcription. On the one hand, no classic ARE has been identified in the iASPP promoter. On the other hand, Nrf2 overexpression had no obvious impact on iASPP mRNA or protein expression (Fig. S5c). In contrast, the iASPP promoter contains consensus binding motifs for both p53 and NF-κBp65 (Fig. 5c, upper panel). KD of p53 or NF-κBp65 in senescent cells reduced iASPP luciferase reporter activity to different extents (Fig. S5b). Notably, NF-κBp65 KD had a greater effect on Dox-induced iASPP reporter activity and mRNA expression levels, whereas p53 KD only mildly reduced iASPP luciferase activity, and no statistical significance was obtained under the experimental conditions used (Fig. S5b). In addition, although p53 KD slightly reduced iASPP mRNA, there was no obvious impact on its protein expression (Fig. S5b). Based on these data, we reasoned that senescence-induced iASPP expression mainly relies on the activation of NF-κBp65.
Indeed, ectopic expression of increased amounts of NF-κBp65 promoted iASPP mRNA, iASPP luciferase activity, and iASPP protein levels in a dose-dependent manner (Fig. S5c). KD of NF-κBp65 with two independent siRNAs abolished iASPP expression in both control and senescent cells (Fig. 5d), suggesting that endogenous NF-κBp65 plays important roles in promoting iASPP expression.
To map the binding sites of NF-κBp65 in the iASPP promoter, two mutant iASPP luciferase reporters (M1 and M2) were constructed. The M1 luciferase reporter failed to respond to NF-κBp65 KD (Fig. 5c). Therefore, the results of our luciferase reporter assays were consistent with bioinformatic predictions. The NF-κBp65–binding sites were mapped between nucleotides −860 and −10 upstream of the transcription start site (Fig. 5c). Furthermore, the binding of NF-κBp65 with the iASPP promoter was confirmed via a ChIP assay. The results showed that NF-κBp65 bound with the iASPP promoter and that this interaction was increased in senescent cells (Fig. 5e). These data collectively demonstrate that the elevated expression of iASPP during senescence is mainly due to the activation of transcription factor NF-κBp65 and that iASPP forms a negative regulation loop in regulating NF-κBp65 activity.
Nonapoptotic functions of iASPP contribute to treatment outcomes in vitro and in vivo
To understand whether iASPP-regulated cell senescence plays roles in influencing drug responses, we generated apoptosis-resistant HCT 116 cells by stably expressing anti-apoptosis Bcl-2, as reported previously (9). Indeed, HCT 116/Bcl-2 cells were more resistant to Dox-induced cell death compared with HCT 116/control cells, as expected (Fig. 6a). iASPP KD sensitized HCT 116 cells to Dox in both apoptotic-resistant and -sensitized HCT 116 cells (Fig. 6a), suggesting that iASPP plays dual roles, i.e. it has both apoptosis-dependent and -independent-activity, in regulating the sensitivity of HCT 116 to Dox. To understand the impacts of iASPP-inhibited senescence on Dox-mediated inhibitory effect on HCT 116, cells were exposed to a low dose (1 μg/ml) of Dox for 2 h and then recovered in fresh medium for 3 days. Cell death rates were first determined by trypan blue staining assay. As shown, under such conditions no obvious cell death was detected, regardless of Dox treatment and iASPP KD (Fig. S6a). Remarkably, iASPP was induced during senescence in HCT 116/Bcl-2 cells (Fig. 6b). Mitotic cells were reduced by iASPP KD in HCT 116/Bcl-2 (Fig. 6b). In addition, iASPP KD produced a similar effect to the expression levels of p21 and LMNB1, as well as the core SASP components IL-6 and IL-8 (Fig. 6, b and c). Therefore, iASPP similarly influences cell senescence in HCT 116 cells after Bcl-2 overexpression.
Figure 6.

Nonapoptotic functions of iASPP contribute to treatment outcomes in vitro and in vivo. a, cell viability of HCT 116/vector and HCT 116/Bcl-2 cells, with or without iASPP KD, was determined by MTT assay with the indicated Dox treatment (top). Overexpression and KD efficiency were confirmed by Western blotting (bottom). b and c, expression of iASPP, p53, p21, and LMNB1 protein levels (b) and IL-6 and IL-8 mRNA levels (c) were determined after triggering senescence in HCT 116/Bcl2 cells, with or without iASPP KD. d–f, tumor volumes on the indicated dates (d) and images and tumor weights of HCT 116/Bcl-2 xenografts of sh-Ctl. and sh-iASPP, with and without Dox treatment (e) are shown. The body weights of nude mice were determined as indicated (f). Values are means ± S.D., n = 6 for each pair. g, expression of iASPP, NF-κBp65, pNF-κBp65, p53, p21, LMNB1, Bcl-2, and cleaved caspase-3 in the indicated xenografts was determined by Western blotting. β-Actin was used as a loading control. h, representative images of ki67 immunohistochemistry staining in the indicated xenografts. i, mRNA levels of iASPP, p21, IL-6, and IL-8 were determined by qRT-PCR in the indicated xenografts. Values are mean ± S.E. from three independent experiments; *, p < 0.05, and **, p < 0.01 (a–e and i). #, p < 0.05 and ##, p < 0.01, compared with DMSO (c and i).
Next, HCT 116/Bcl-2–sh-Ctl and HCT 116/Bcl-2–sh-iASPP cells were paired, and the same number of cells was inoculated into one nude mouse at symmetrical subcutaneous regions (n = 12). Tumor sizes were monitored twice weekly. We randomized mice with detectable tumors (∼100 mm3) into Dox or PBS treatment cohorts (n = 6, each group). iASPP KD reduced tumor growth, as indicated by both tumor volumes and weights in HCT 116/Bcl-2 xenografts. Dox treatment also inhibited tumor growth, with a greater effect observed in iASPP KD cells than in controls (Fig. 6, d and e). Body weight was slightly lower in the Dox treatment group than in the PBS control group (Fig. 6f). p53, p21, and LMNB1 were induced, and the activation of NF-κBp65, as indicated by p-NF-κBp65, was also elevated in Dox-treated xenografts, whereas no obvious caspase-3 activation was detected in the same xenografts (Fig. 6g). Proliferation marker Ki67 was remarkably reduced in Dox-treated xenografts (Fig. 6h). In agreement with the data shown in vitro, the mRNA levels of iASPP, p21, IL-6, and IL-8 were increased by Dox in vivo. Importantly, the effects of iASPP KD were similar to those of Dox alone and also reinforced the effect of Dox (Fig. 6i). These data are analogous to the data presented in vitro and indicate that iASPP KD sensitizes apoptotic-resistant HCT 116/Bcl-2 xenografts in response to Dox by inducing senescence.
Discussion
Although apoptosis is a desirable post-damage response to chemotherapy, it is normally induced at high doses and thus is accompanied by the risk of severe toxicity (42). Instead, modest chemotherapy (cytostatic treatment) inhibits tumors by inducing senescence, offering an opportunity to achieve effective treatment with reduced toxicity (43). Here, we identified novel mechanisms of iASPP that regulate senescence, suggesting that the oncogene can act by limiting the senescence program and skewing the SASP, thus promoting drug resistance in cancers.
iASPP has been recognized as an important oncogene that is overexpressed in multiple types of cancers (27, 44). The promotion of iASPP's expression has been linked to drug resistance (28, 45, 46), further supporting the potential of iASPP as a promising therapeutic target in cancer treatment. Previous research, presented by both us and others, has pinpointed an important function of iASPP in inhibiting drug-induced apoptosis by either p53-dependent or Nrf2-dependent mechanisms (26, 28). Here, we report that iASPP confers drug resistance independently of apoptosis and by inhibiting senescence. This result is unexpected, because iASPP has been reported to selectively regulate p53's transcription activity toward pro-apoptotic genes, while having no obvious impact on cell cycle arrest genes (26). However, our data clearly show that in response to short-term low-dose Dox, iASPP inhibits p21, a well-known cell cycle arrest target of p53, in a p53-dependent manner (Fig. 7). Of note, despite evident p53 activation, cells mainly underwent senescence, but not apoptosis, under the experiment settings used in this study. It is reasonable to propose that p53 predominately binds with its cell cycle arrest targets, such as p21, but not pro-apoptotic target genes under such conditions. Therefore, although iASPP may preferentially inhibit the p53-dependent transcriptional activity of apoptosis targets rather than cell cycle arrest genes, its impact on cell cycle progression is evident when the activation of p53-dependent cell cycle arrest signaling is dominant. Our data suggest that the selectivity of iASPP on p53's transcriptional activity may be determined by which signaling pathway (i.e. pro-apoptosis or cell cycle arrest) is activated. In addition, we cannot exclude the possibility that additional regulators may be activated under the conditions of senescence and then participate in controlling iASPP activity toward p53-dependent cell cycle arrest. As such, a more comprehensive picture of iASPP's functions in regulating p53, and influencing the drug responsiveness and tumor growth of cancer cells, has been provided in this study (47). Interestingly, despite being a commonly recognized apoptosis inhibitor, iASPP knockout MEFs have been reported to enter senescence earlier than WT MEFs (34), and siRNA-mediated iASPP KD has been also shown to induce cell cycle arrest at G1 phase in U251 human glioblastoma cells (48), both via unknown mechanisms. Whether such activity of iASPP is fulfilled by it inhibiting p53 needs further exploration.
Figure 7.
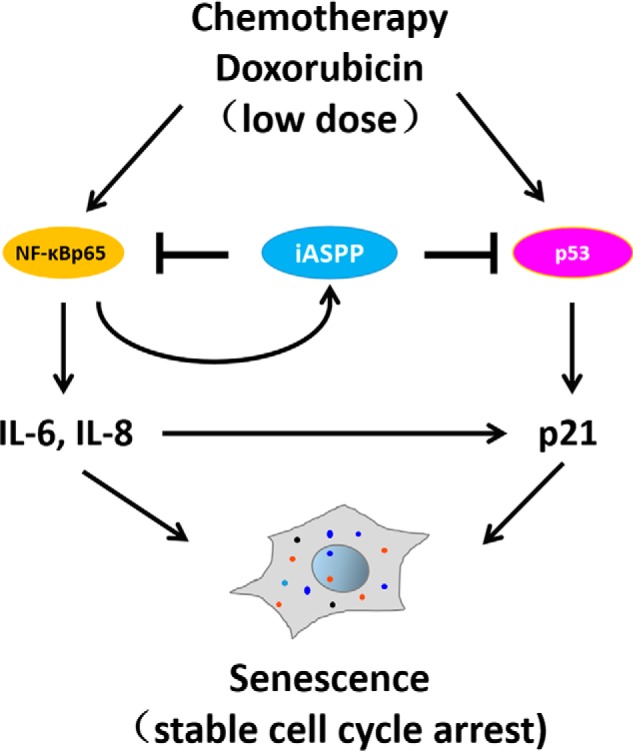
Working model for the role of iASPP in regulating senescence. After the triggering of senescence by a sublethal dose of Dox, iASPP is transcriptionally activated by NF-κBp65. The enhanced iASPP enters into nucleus where it binds with either p53 or NF-κBp65, leading to attenuated expression of p21 and IL-6/IL-8, respectively, both of which contribute to the progress of iASPP-inhibited senescence.
The ability of iASPP to inhibit p53 relies on its precise nuclear localization (30). However, iASPP is predominately located in the cytoplasm in human cancers (28), suggesting that its oncogenic activity cannot be attributed to its inhibitory effect on p53 under steady-state conditions. Indeed, the KD of endogenous iASPP leads to enhanced apoptosis in unstressed cells, which is due to Nrf2 inhibition but not p53 activation (28). It is well-known that p53 remains intact in ∼50% of cancers. Under basal conditions, WT p53 is maintained at low levels due to rapid protein degradation (49). Nonetheless, although it can be stabilized and translocated to the nucleus by DNA-damaging drugs, nuclear p53's transcriptional activity can be largely impaired in cancer cells (50). Here, we made an interesting observation that nuclear translocation of endogenous iASPP occurs during senescence and is accompanied by the activation of p53. These observations provide a potential explanation for why iASPP is simultaneously translocated from the cytoplasm into the nucleus after treatment, where it exerts its inhibitory activity on p53 activation leading to the promotion of drug resistance, by inhibiting apoptosis or interfering with senescence in a cell context-dependent manner.
NF-κBp65 was the first protein to be identified to bind with iASPP (25). However, the biological significance of the iASPP/NF-κBp65 interaction, particularly in cancer cells, remains largely unknown. The activation of NF-κBp65 is generally associated with the promotion of tumor growth and progression (35). iASPP has the ability to inhibit NF-κBp65 (25), which likely contradicts the well-established oncogenic function of iASPP. We envision that iASPP may form a negative feedback loop with NF-κBp65 during senescence. It is a direct transcription target of NF-κBp65 and inhibits NF-κBp65-dependent IL-6/IL-8 expression during senescence. There is evidence that inhibition of NF-κBp65 results in a more evident drug resistance phenotype due to a reduction of the SASP (51), which is consistent with the results reported here. In addition, conditioned medium supplemented with IL-6 or IL-8 can rescue iASPP overexpression–inhibited p21 expression. Therefore, iASPP may elicit its oncogenic function by inhibiting NF-κBp65–induced IL-6 and IL-8. In keeping with this idea, both cytokines have been reported to reinforce senescence, as indicated by increased p21 (18). However, we found that NF-κBp65 KD had no obvious effect on the expression of p21 in our current experimental settings. This discrepancy may be due to multiple SASP factors being subjected to regulation by NF-κBp65 (52), which may elicit diverse effects regarding cell cycle progression and thus compromise the effects of IL-6 and IL-8. It will be interesting to further explore whether iASPP also regulates additional SASP factors besides IL6 and IL8 and, if so, how they contribute to the biological effects of iASPP–NF-κBp65.
It has been reported that activated p53 and NF-κBp65 can induce iASPP expression (53–55), suggesting that iASPP is a potential target for both transcription factors. In keeping with this, consensus binding motifs for both transcription factors have been identified in iASPP's promoter region. However, the induction of iASPP during senescence is mainly dependent on the activation of NF-κBp65 and not p53. We cannot exclude the possibility that NF-κBp65 and p53 have different binding affinities for the iASPP promoter, and it is also possible that additional cofactors are involved in modulating the selectivity of NF-κBp65 and p53 in regulating iASPP transcription during senescence. Future work is need to test these hypotheses.
More recent studies conducted by us have yielded insights into novel mechanisms of iASPP oncogene action in the cytoplasm (28). As such, iASPP physiologically binds with Keap1, a master inhibitor of Nrf2, and then prevents Keap1 from binding Nrf2, leading to the promotion of Nrf2 stabilization and activation. Nrf2 is a central regulator of antioxidative signaling. The newly-identified iASPP–Nrf2 axis is linked to enhanced antioxidative capability and confers the apoptosis resistance elicited by chemotherapeutic drugs in human cancers. Nonetheless, Nrf2 has been reported to regulate the inflammatory and immune responses (38, 40). However, our current data suggest that the inhibitory effects of iASPP on growth arrest and IL-6/IL-8 expression are not likely linked to its ability to activate Nrf2. The involvement of iASPP–Nrf2 in the SASP in general and their contribution to drug treatment outcomes require investigation in the future.
Collectively, the p53 and NF-κBp65–signaling pathways contribute to the integrated cell senescence and/or SASP core components of IL-6/IL-8 expression. How these complex signal systems are regulated and integrated is not completely understood. Our study has identified a novel biological function for a previously identified antiapoptotic gene, iASPP, in senescence and the IL-6/8 expression, which not only provides important insights into how p53 and NF-κBp65 signaling systems coordinate in the context of senescence, but also adds to our understanding of drug resistance. These findings suggest that iASPP inhibition could be a powerful strategy to restore senescence, and it may represent a new target to shape SASP- and chemotherapy-based therapeutic opportunities.
Experimental procedures
Cell lines and treatments
The human breast cancer cell lines MCF-7(ATCC) and primary mouse embryonic fibroblasts were maintained in DMEM (Gibco). The colorectal cancer cell line HCT 116 (ATCC) was maintained in RPMI 1640 medium (Gibco) supplemented with 10% (v/v) fetal bovine serum (Biological Industries) and 2 mm l-glutamine. All cells were grown at 37 °C in the humidified incubator (Thermo Fisher Scientific) with 5% CO2 and have not been passaged longer than 3 months before the experiments. Cell lines were routinely tested to exclude mycoplasma contamination and were characterized by the Genetic Testing Biotechnology Corp. (Suzhou, China) using short tandem repeat markers. siRNA oligonucleotides or plasmids were introduced into cells by Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions. Cells were subjected to the indicated analysis 72 h after transfection with or without additional treatments with doxorubicin (Selleck Chemicals) or etoposide (MedChemExpress, MCE). For drug treatments, HCT 116 and MCF-7 were pulse-exposed to doxorubicin (1 μg/ml) or etoposide (10 μm) for 2 h, after which the media were replaced, and cells were grown for an additional number of days. Unless otherwise specified, senescent cells obtained at 3 days after drug treatment were used for mechanistic studies. The detailed information of plasmids, chemicals, and siRNA sequences are list in Table S1.
In vivo xenograft mouse study
HCT 116 cells were infected with PLKO.1-iASPP (short hairpin RNA, shRNAiASPP) lentivirus to knock down iASPP expression (CHT 116/sh-iASPP), with a blank lentivirus as a control (HCT 1116/sh-Ctl). These two stable cell lines were both infected with Bcl-2 lentivirus (Bcl-2) and its blank lentivirus control (vector) to avoid the influence of apoptosis induced by doxorubicin. The female nude mice between 4 and 5 weeks old were purchased from Beijing HFK Bioscience Co., Ltd. (Table S1), 1 × 107 paired cells were inoculated subcutaneously into either side of the flank of the same female nude mouse. The tumor volumes were measured every week and calculated as length × width2 × 0.5 (cm3). When the tumor volume reached about 200 mm3, the mice were randomly divided into four groups and subjected to treatment of DMSO and Dox alone (10 mg/kg, intraperitoneally) once to set up a senescence model. The tumor size and the body weights of the mice were measured weekly. After injection for 2 weeks, the mice were anesthetized and culled. The tumor was carefully removed, photographed, and weighed. All animal procedures were performed according to protocols approved by the Rules for Animal Experiments published by the Chinese Government (Beijing, China) and approved by the Research Ethics Committee of Harbin Institute of Technology, China.
SA–β-Gal staining
Senescence was confirmed by SA–β-Gal assay. Briefly, HCT 116 cells were treated as indicated or transfected with two independent siRNAs that specifically target iASPP. After treatment of doxorubicin, cells were washed with cold PBS three times and then fixed with 0.2% glutaraldehyde (prepared in PBS) for 5 min. The cells were then washed again two times with PBS. After the last wash, staining solution was added (1 mg/ml 5-bromo-4-chloro-3-inolyl β-d-galactoside (X-gal) in staining solution (dimethyl formamide (20 mg/ml stock), 40 mm citric acid/sodium phosphate, pH 6.0, 5 mm potassium ferrocyanide, 5 mm potassium ferricyanide, 150 mm NaCl, 2 mm MgCl2), and the cells were incubated in a 37 °C incubator without CO2 for 18 h. After incubation, the cells were washed two times with PBS and photographed. The percentage of stained cells was counted. The number of blue structures was counted in 20 fields, and the blue cells percentage was made into bar graph.
Immunohistochemistry
The tissue microarray was subjected to antigen retrieval by boiling in 0.01 mol/liter citrate buffer for 5 min. Slides were then incubated with anti-Ki-67 antibody at room temperature for 1 h. The antibody dilution ratio was 1 to 100. After incubation, the tissue were washed three times with PBS. The second antibody was then incubated and washed twice with PBS. Detection was carried out by the REAL EnVision detection system (Dako) with diaminobenzidine peroxidase serving as chromogen. After that, the slides were briefly counterstained with hematoxylin before mounting. The images were captured under the same conditions, and the intensity was evaluated with a 3-tier grading system (negative or weak, moderate, and strong staining intensity).
Western blotting
Cells were lysed in urea buffer containing 2 m thiourea, 4% CHAPS, 40 mm Tris base, 40 mm DTT, 2% Pharmalyte and sonicated to shear DNA. Protein expression were detected by ECL and visualized by Image Studio system (ECL, LI-COR, Lincoln, GA). ImageJ software (National Institutes of Health, Bethesda, MD) was used to quantify protein expression. The antibody dilution ratios used were as follows: 1 to 2000 for anti-iASPP, anti-p53, anti-LMNB1, anti-Keap1, anti-NF-κBp65, anti-Myc, anti-Bcl2, anti-GAPDH, anti-α-tubulin, anti-β-actin, and anti-histone 3.1; 1 to 1000 for anti-Nrf2, anti-p-NF-κBp65, anti-H2Aγ, and anti-cleaved caspase3; and 1to 500 for p21 (Table S1).
BrdU staining
BrdU incorporation assay was carried out by following the protocol provided by Cell Signaling Technology. Briefly, BrdU was diluted to a final concentration of 0.03 mg/ml with fresh DMEM and then applied to the cells grown on slices. Cells were incubated with 1.5 m HCl followed by a 5-min fixation in 70% cold ethanol. After a 3% BSA block overnight and immunostained with anti-BrdU antibody was then conducted as shown above. The antibody dilution ratio is 1 to 1000. The next day, the antibodies were washed three times with PBS, stained with fluorescent secondary antibody, incubated at room temperature for 1 h, and washed with PBS three times. Nucleus was visualized by DAPI staining. The representative images were captured by a Zeiss LSM510 confocal microscope (Carl Zeiss, Heidelberg, Germany). The primer sequences are listed in Table S1.
ROS measurement
Cells were incubated with 10 μm DCFH-DA (Sigma) for 30 min at 37 °C. Cells were then washed with PBS and trypsinized. Trypsinized cells were resuspended in ice-cold PBS and kept as a single cell suspension on ice until rapid analysis by FACS. The data are reported as the fold changes in mean fluorescence intensity normalized to the fluorescence intensity of untreated control cells.
Cell cycle analysis
When cells reached 70–80% confluence, they were washed with PBS, detached with 0.25% trypsin, and fixed with 75% ethanol overnight. After treatment with 1 mg/ml RNase A (Sigma) at 37 °C for 30 min, cells were resuspended in 0.5 ml of PBS and stained with propidium iodide in the dark for 30 min. Fluorescence was measured with a FACScan flow cytometry system (BD Biosciences).
Immunoprecipitation
Cells were lysed in NETN buffer (50 mm Tris-HCl, pH 8.0, 150 mm NaCl, 1% Nonidet P-40,1 mm EDTA, and fresh proteinase inhibitor mixture (MedChemExpress, HY-K0010) before use). The resulting lysate were then pre-cleaned by 25 μl of protein G–Sepharose beads (GE Healthcare) at 4 °C. Immunoprecipitation mixtures, including protein lysates, blocked protein G–Sepharose, and the indicated antibodies or IgG control derived from the same species as the indicated antibody, were incubated on a rotating wheel at 4 °C overnight. Sepharose beads with the bound immunoprecipitates were collected and subjected to four washes with the cold NETN buffer and then analyzed by WB assay. The detailed information of the antibodies are listed in Table S1.
Cell fraction
Cytoplasmic lysis buffer (10 mm HEPES, pH 7.9,10 mm KCl, 1.5 mm MgCl2, 0.5 mm β-mercaptoethanol) was applied to cells, followed by moderate vortexing for 15 s and 15–20 min incubation on ice. Additional 5 μl of 10% Nonidet P-40 (Amersco) was then added to the mixture followed by another round of vortexing and incubation. The cytoplasmic fraction was obtained by collecting supernatants after centrifugation at 16,000 × g for 10 min at 4 °C. The resulting pellet was lysed in the nuclear fraction buffer (10 mm HEPES, pH 7.6, 1 mm DTT, 7.5 mm MgCl2, 0.2 mm EDTA, 0.3 m NaCl, 1 m urea, 1% Nonidet P-40). The supernatant was collected as the nuclear fraction by centrifugation at 16,000 × g for 10 min at 4 °C.
Luciferase activity
iASPP promoter regions at −1153 to −10 for WT, −1153 to −860 for MT1, and −859 to −10 for MT2 were cloned into pGL3-basic luciferase reporter plasmid. The same amount of iASPP promoter–luciferase reporter plasmid together with NF-κBp65 plasmids/siRNA target NF-κBp65 or p53 was transfected into the indicated cancer cells. Each of the transfections included the same amount of Renilla, which was used to standardize the control. 48–72 h after transfection, the cells were treated with Dox or DMSO control for 24 h. Finally, the luciferase activities were detected with the luciferase assay system (Promega) according to the manufacturer's instructions. The relative luciferase activities were normalized with the Renilla luciferase activities.
The same amount of p21 promoter–luciferase reporter plasmid together with siRNA specifically targeting iASPP or p53 were introduced into the cells in the presence or absence of Dox. The luciferase activities were detected with the luciferase assay system (Promega) according to the manufacturer's instructions. The relative luciferase activities were normalized with the Renilla luciferase activities.
RNA extraction and quantitative (q)RT-PCR
Total RNA isolated with TRIzol (Invitrogen), by following the manufacturer's protocol, was subjected to reverse transcription with GoScriptTM reverse transcription system (Promega) and analyzed by the qRT-PCR with SYBR Premix Ex Tag II (TaKaRa). Gene expression levels relative to GAPDH control were calculated by 2−ΔΔCt method. The primer sequences are list in Table S1.
Immunofluorescence assay
MCF-7 or HCT 116 cells grown on coverslips treated with DMSO and Dox were fixed with 4% paraformaldehyde. The expression of iASPP was detected by typical immunofluorescence assay. Nucleus was visualized by DAPI staining. The representative images were captured by a Zeiss LSM510 confocal microscope (Carl Zeiss, Heidelberg, Germany).
ChIP
Briefly, asynchronously-growing cells were incubated with formaldehyde to yield protein–DNA cross-link complexes. The cross-linked chromatin was then purified, diluted with lysis buffer at 1:5, and sheared by sonication. After preclearing with protein G–agarose beads, the chromatin was divided equally into two groups for further immunoprecipitation reaction with anti-NF-κBp65 and nonspecific immunoglobulin G (IgG) derived from same species. The immunoprecipitates were pelleted by centrifugation and then incubated at 65 °C to reverse the protein–DNA cross-linking. The same amount of precipitated DNA fragments was subjected to PCR analysis at 58 °C for 33 cycles by 2× GoldStar Best MasterMix (Cwbiotech) and visualized by running 1% agarose gel.
MTT
Colorimetric MTT assay was performed to measure cell viability. Equal number of cells was plated in 96-well plates. After 0, 24, 48, and 72 h of incubation, cells were treated with 5 mg/ml MTT solution for 4 h at 37 °C. Formazan was dissolved with 100 μl of DMSO after removing the supernatant. Absorbance at 490 nm was measured using a Microplate Reader (Tecan Austria GmbH 5082, Grodig, Austria).
ELISA
IL-6 and IL-8 concentrations of cell culture medium were measured by ELISA using the Quantikine human IL-6 or IL-8 ELISA kit according to the manufacturer's instructions (R&D Systems, Minneapolis). The sample absorbance was determined at 450 and 540 nm. Each experiment was repeated three times.
Statistical analysis
Statistical analysis was done by GraphPad software, version 5. Data are presented as the mean ± S.E. of the means or standard deviations. Student's t test was applied to assess the statistical significance. Correlations were calculated according to Spearman or Pearson correlation. p value <0.05 was considered significant.
Author contributions
H. L., W. Z., K. Z., D. Z., and S. Z. investigation; H. L. and W. Z. methodology; Y. H. conceptualization; H. L., and W. Z. formal analysis; Y. H. supervision; Y. H. funding acquisition; Y. H. writing-original draft; Y. H. writing-review and editing.
Supplementary Material
Acknowledgments
We thank all members of the Gene Function Study Group, Harbin Institute of Technology, for their discussion and technological support of this work.
This work was supported by National Natural Science Foundation of China Grants 31871389, 31301131, and 31741084 and Basic Science Foundation of Science and Technology Innovation Commission in Shenzhen Grant JCYJ20170811154452255. The authors declare that they have no conflicts of interest with the contents of this article.
This article contains Figs. S1–S6 and Table S1.
- TIS
- chemotherapy-induced senescence
- Dox
- doxorubicin
- iASPP
- inhibitor of apoptosis-stimulating protein of p53
- SASP
- senescence-associated secretory phenotype
- GAPDH
- glyceraldehyde-3-phosphate dehydrogenase
- qRT-PCR
- quantitative RT-PCR
- MTT
- 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2-H-tetrazolium bromide
- IP
- immunoprecipitation
- DAPI
- 4′,6-diamidino-2-phenylindole
- ROS
- reactive oxygen species
- DCFH-DA
- dichlorodihydrofluorescein diacetate
- ARE
- antioxidant-response element
- DMEM
- for Dulbecco's modified Eagle's medium.
References
- 1. He S., and Sharpless N. E. (2017) Senescence in health and disease. Cell 169, 1000–1011 10.1016/j.cell.2017.05.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Campisi J., and d'Adda di Fagagna F. (2007) Cellular senescence: when bad things happen to good cells. Nat. Rev. Mol. Cell Biol., 8, 729–740 10.1038/nrm2233 [DOI] [PubMed] [Google Scholar]
- 3. Pérez-Mancera P. A., Young A. R., and Narita M. (2014) Inside and out: the activities of senescence in cancer. Nat. Rev. Cancer 14, 547–558 10.1038/nrc3773 [DOI] [PubMed] [Google Scholar]
- 4. Hussain S., Singh A., Nazir S. U., Tulsyan S., Khan A., Kumar R., Bashir N., Tanwar P., and Mehrotra R. (2019) Cancer drug resistance: a fleet to conquer. J. Cell. Biochem. 120, 14213–14225 10.1002/jcb.28782 [DOI] [PubMed] [Google Scholar]
- 5. Michaloglou C., Vredeveld L. C., Soengas M. S., Denoyelle C., Kuilman T., van der Horst C. M., Majoor D. M., Shay J. W., Mooi W. J., and Peeper D. S. (2005) BRAFE600-associated senescence-like cell cycle arrest of human naevi. Nature 436, 720–724 10.1038/nature03890 [DOI] [PubMed] [Google Scholar]
- 6. Braig M., Lee S., Loddenkemper C., Rudolph C., Peters A. H., Schlegelberger B., Stein H., Dörken B., Jenuwein T., and Schmitt C. A. (2005) Oncogene-induced senescence as an initial barrier in lymphoma development. Nature 436, 660–665 10.1038/nature03841 [DOI] [PubMed] [Google Scholar]
- 7. te Poele R. H., Okorokov A. L., Jardine L., Cummings J., and Joel S. P. (2002) DNA damage is able to induce senescence in tumor cells in vitro and in vivo. Cancer Res. 62, 1876–1883 [PubMed] [Google Scholar]
- 8. Roberson R. S., Kussick S. J., Vallieres E., Chen S. Y., and Wu D. Y. (2005) Escape from therapy-induced accelerated cellular senescence in p53-null lung cancer cells and in human lung cancers. Cancer Res. 65, 2795–2803 10.1158/0008-5472.CAN-04-1270 [DOI] [PubMed] [Google Scholar]
- 9. Schmitt C. A., Fridman J. S., Yang M., Lee S., Baranov E., Hoffman R. M., and Lowe S. W. (2002) A senescence program controlled by p53 and p16INK4a contributes to the outcome of cancer therapy. Cell 109, 335–346 10.1016/S0092-8674(02)00734-1 [DOI] [PubMed] [Google Scholar]
- 10. Acosta J. C., and Gil J. (2012) Senescence: a new weapon for cancer therapy. Trends Cell Biol. 22, 211–219 10.1016/j.tcb.2011.11.006 [DOI] [PubMed] [Google Scholar]
- 11. Rao S. G., and Jackson J. G. (2016) SASP: tumor suppressor or promoter? Yes! Trends Cancer 2, 676–687 10.1016/j.trecan.2016.10.001 [DOI] [PubMed] [Google Scholar]
- 12. Yoshimoto S., Loo T. M., Atarashi K., Kanda H., Sato S., Oyadomari S., Iwakura Y., Oshima K., Morita H., Hattori M., Hattori M., Honda K., Ishikawa Y., Hara E., and Ohtani N. (2013) Obesity-induced gut microbial metabolite promotes liver cancer through senescence secretome. Nature 499, 97–101 10.1038/nature12347 [DOI] [PubMed] [Google Scholar]
- 13. Gunaratna R. T., Santos A., Luo L., Nagi C., Lambertz I., Spier M., Conti C. J., and Fuchs-Young R. S. (2019) Dynamic role of the codon 72 p53 single-nucleotide polymorphism in mammary tumorigenesis in a humanized mouse model. Oncogene 38, 3535–3550 10.1038/s41388-018-0630-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kim Y. H., Choi Y. W., Lee J., Soh E. Y., Kim J. H., and Park T. J. (2017) Senescent tumor cells lead the collective invasion in thyroid cancer. Nat. Commun. 8, 15208 10.1038/ncomms15208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Freund A., Orjalo A. V., Desprez P. Y., and Campisi J. (2010) Inflammatory networks during cellular senescence: causes and consequences. Trends Mol. Med. 16, 238–246 10.1016/j.molmed.2010.03.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zitvogel L., Kepp O., and Kroemer G. (2011) Immune parameters affecting the efficacy of chemotherapeutic regimens. Nat. Rev. Clin. Oncol. 8, 151–160 10.1038/nrclinonc.2010.223 [DOI] [PubMed] [Google Scholar]
- 17. Acosta J. C., O'Loghlen A., Banito A., Guijarro M. V., Augert A., Raguz S., Fumagalli M., Da Costa M., Brown C., Popov N., Takatsu Y., Melamed J., d'Adda di Fagagna F., Bernard D., Hernando E., and Gil J. (2008) Chemokine signaling via the CXCR2 receptor reinforces senescence. Cell 133, 1006–1018 10.1016/j.cell.2008.03.038 [DOI] [PubMed] [Google Scholar]
- 18. Ortiz-Montero P., Londoño-Vallejo A., and Vernot J. P. (2017) Senescence-associated IL-6 and IL-8 cytokines induce a self- and cross-reinforced senescence/inflammatory milieu strengthening tumorigenic capabilities in the MCF-7 breast cancer cell line. Cell Commun. Signal. 15, 17 10.1186/s12964-017-0172-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Coppé J. P., Patil C. K., Rodier F., Sun Y., Muñoz D. P., Goldstein J., Nelson P. S., Desprez P. Y., and Campisi J. (2008) Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol. 6, 2853–2868 10.1371/journal.pbio.0060301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Tchkonia T., Zhu Y., van Deursen J., Campisi J., and Kirkland J. L. (2013) Cellular senescence and the senescent secretory phenotype: therapeutic opportunities. J. Clin. Invest. 123, 966–972 10.1172/JCI64098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Salminen A., Kauppinen A., and Kaarniranta K. (2012) Emerging role of NF-κB signaling in the induction of senescence-associated secretory phenotype (SASP). Cell. Signal. 24, 835–845 10.1016/j.cellsig.2011.12.006 [DOI] [PubMed] [Google Scholar]
- 22. Rodier F., Coppé J. P., Patil C. K., Hoeijmakers W. A., Muñoz D. P., Raza S. R., Freund A., Campeau E., Davalos A. R., and Campisi J. (2009) Persistent DNA damage signalling triggers senescence-associated inflammatory cytokine secretion. Nat. Cell Biol. 11, 973–979 10.1038/ncb1909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Freund A., Patil C. K., and Campisi J. (2011) p38MAPK is a novel DNA damage response-independent regulator of the senescence-associated secretory phenotype. EMBO J. 30, 1536–1548 10.1038/emboj.2011.69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Schneider G., Henrich A., Greiner G., Wolf V., Lovas A., Wieczorek M., Wagner T., Reichardt S., von Werder A., Schmid R. M., Weih F., Heinzel T., Saur D., and Krämer O. H. (2010) Cross-talk between stimulated NF-κB and the tumor suppressor p53. Oncogene 29, 2795–2806 10.1038/onc.2010.46 [DOI] [PubMed] [Google Scholar]
- 25. Yang J. P., Hori M., Sanda T., and Okamoto T. (1999) Identification of a novel inhibitor of nuclear factor-κB, RelA-associated inhibitor. J. Biol. Chem. 274, 15662–15670 10.1074/jbc.274.22.15662 [DOI] [PubMed] [Google Scholar]
- 26. Bergamaschi D., Samuels Y., O'Neil N. J., Trigiante G., Crook T., Hsieh J. K., O'Connor D. J., Zhong S., Campargue I., Tomlinson M. L., Kuwabara P. E., and Lu X. (2003) iASPP oncoprotein is a key inhibitor of p53 conserved from worm to human. Nat. Genet. 33, 162–167 10.1038/ng1070 [DOI] [PubMed] [Google Scholar]
- 27. Trigiante G., and Lu X. (2006) ASPP [corrected] and cancer. Nat. Rev. Cancer 6, 217–226 10.1038/nrc1818 [DOI] [PubMed] [Google Scholar]
- 28. Ge W., Zhao K., Wang X., Li H., Yu M., He M., Xue X., Zhu Y., Zhang C., Cheng Y., Jiang S., and Hu Y. (2017) iASPP is an antioxidative factor and drives cancer growth and drug resistance by competing with Nrf2 for Keap1 binding. Cancer Cell 32, 561–573.e6 10.1016/j.ccell.2017.09.008 [DOI] [PubMed] [Google Scholar]
- 29. Lu M., Zak J., Chen S., Sanchez-Pulido L., Severson D. T., Endicott J., Ponting C. P., Schofield C. J., and Lu X. (2014) A code for RanGDP binding in ankyrin repeats defines a nuclear import pathway. Cell 157, 1130–1145 10.1016/j.cell.2014.05.006 [DOI] [PubMed] [Google Scholar]
- 30. Lu M., Breyssens H., Salter V., Zhong S., Hu Y., Baer C., Ratnayaka I., Sullivan A., Brown N. R., Endicott J., Knapp S., Kessler B. M., Middleton M. R., Siebold C., Jones E. Y., et al. (2016) Restoring p53 function in human melanoma cells by inhibiting MDM2 and cyclin B1/CDK1-phosphorylated nuclear iASPP. Cancer Cell 30, 822–823 10.1016/j.ccell.2016.09.019 [DOI] [PubMed] [Google Scholar]
- 31. Hu Y., Ge W., Wang X., Sutendra G., Zhao K., Dedeić Z., Slee E. A., Baer C., and Lu X. (2015) Caspase cleavage of iASPP potentiates its ability to inhibit p53 and NF-κB. Oncotarget 6, 42478–42490 10.18632/oncotarget.6478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Falik-Zaccai T. C., Barsheshet Y., Mandel H., Segev M., Lorber A., Gelberg S., Kalfon L., Ben Haroush S., Shalata A., Gelernter-Yaniv L., Chaim S., Raviv Shay D., Khayat M., Werbner M., Levi I., et al. (2017) Sequence variation in PPP1R13L results in a novel form of cardio-cutaneous syndrome. EMBO Mol. Med. 9, 319–336 10.15252/emmm.201606523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Herron B. J., Rao C., Liu S., Laprade L., Richardson J. A., Olivieri E., Semsarian C., Millar S. E., Stubbs L., and Beier D. R. (2005) A mutation in NFkB interacting protein 1 results in cardiomyopathy and abnormal skin development in wa3 mice. Hum. Mol. Genet. 14, 667–677 10.1093/hmg/ddi063 [DOI] [PubMed] [Google Scholar]
- 34. Notari M., Hu Y., Koch S., Lu M., Ratnayaka I., Zhong S., Baer C., Pagotto A., Goldin R., Salter V., Candi E., Melino G., and Lu X. (2011) Inhibitor of apoptosis-stimulating protein of p53 (iASPP) prevents senescence and is required for epithelial stratification. Proc. Natl. Acad. Sci. U.S.A. 108, 16645–16650 10.1073/pnas.1102292108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Karin M. (2006) Nuclear factor-κB in cancer development and progression. Nature 441, 431–436 10.1038/nature04870 [DOI] [PubMed] [Google Scholar]
- 36. Kuilman T., Michaloglou C., Mooi W. J., and Peeper D. S. (2010) The essence of senescence. Genes Dev. 24, 2463–2479 10.1101/gad.1971610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Hainaut P., and Wiman K. G. (2009) 30 years and a long way into p53 research. Lancet Oncol. 10, 913–919 10.1016/S1470-2045(09)70198-6 [DOI] [PubMed] [Google Scholar]
- 38. Mohan S., and Gupta D. (2018) Crosstalk of toll-like receptors signaling and Nrf2 pathway for regulation of inflammation. Biomed. Pharmacother. 108, 1866–1878 10.1016/j.biopha.2018.10.019 [DOI] [PubMed] [Google Scholar]
- 39. Kuilman T., Michaloglou C., Vredeveld L. C., Douma S., van Doorn R., Desmet C. J., Aarden L. A., Mooi W. J., and Peeper D. S. (2008) Oncogene-induced senescence relayed by an interleukin-dependent inflammatory network. Cell 133, 1019–1031 10.1016/j.cell.2008.03.039 [DOI] [PubMed] [Google Scholar]
- 40. Kobayashi E. H., Suzuki T., Funayama R., Nagashima T., Hayashi M., Sekine H., Tanaka N., Moriguchi T., Motohashi H., Nakayama K., and Yamamoto M. (2016) Nrf2 suppresses macrophage inflammatory response by blocking proinflammatory cytokine transcription. Nat. Commun. 7, 11624 10.1038/ncomms11624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Wruck C. J., Streetz K., Pavic G., Götz M. E., Tohidnezhad M., Brandenburg L. O., Varoga D., Eickelberg O., Herdegen T., Trautwein C., Cha K., Kan Y. W., and Pufe T. (2011) Nrf2 induces interleukin-6 (IL-6) expression via an antioxidant response element within the IL-6 promoter. J. Biol. Chem. 286, 4493–4499 10.1074/jbc.M110.162008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Oronsky B., Ao-Ieong E. S. Y., Yalcin O., Carter C. A., and Cabrales P. (2019) Cardioprotective effect of phase 3 clinical anticancer agent, RRx-001, in doxorubicin-induced acute cardiotoxicity in mice. Mol. Pharm. 16, 2929–2934 10.1021/acs.molpharmaceut.9b00150 [DOI] [PubMed] [Google Scholar]
- 43. Ewald J. A., Desotelle J. A., Wilding G., and Jarrard D. F. (2010) Therapy-induced senescence in cancer. J. Natl. Cancer Inst. 102, 1536–1546 10.1093/jnci/djq364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Li Y., Ahmad A., and Sarkar F. H. (2015) ASPP and iASPP: Implication in cancer development and progression. Cell. Mol. Biol. 61, 2–8 [PubMed] [Google Scholar]
- 45. Xiong Y., Sun F., Dong P., Watari H., Yue J., Yu M. F., Lan C. Y., Wang Y., and Ma Z. B. (2017) iASPP induces EMT and cisplatin resistance in human cervical cancer through miR-20a-FBXL5/BTG3 signaling. J. Exp. Clin. Cancer Res. 36, 48 10.1186/s13046-017-0520-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Jia Y., Peng L., Rao Q., Xing H., Huai L., Yu P., Chen Y., Wang C., Wang M., Mi Y., and Wang J. (2014) Oncogene iASPP enhances self-renewal of hematopoietic stem cells and facilitates their resistance to chemotherapy and irradiation. FASEB J. 28, 2816–2827 10.1096/fj.13-244632 [DOI] [PubMed] [Google Scholar]
- 47. Morris E. V., Cerundolo L., Lu M., Verrill C., Fritzsche F., White M. J., Thalmann G. N., ten Donkelaar C. S., Ratnayaka I., Salter V., Hamdy F. C., Lu X., and Bryant R. J. (2014) Nuclear iASPP may facilitate prostate cancer progression. Cell Death Dis. 5, e1492 10.1038/cddis.2014.442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Li G., Wang R., Gao J., Deng K., Wei J., and Wei Y. (2011) RNA interference-mediated silencing of iASPP induces cell proliferation inhibition and G0/G1 cell cycle arrest in U251 human glioblastoma cells. Mol. Cell. Biochem. 350, 193–200 10.1007/s11010-010-0698-9 [DOI] [PubMed] [Google Scholar]
- 49. Haupt Y., Maya R., Kazaz A., and Oren M. (1997) Mdm2 promotes the rapid degradation of p53. Nature 387, 296–299 10.1038/387296a0 [DOI] [PubMed] [Google Scholar]
- 50. Muller P. A., and Vousden K. H. (2014) Mutant p53 in cancer: new functions and therapeutic opportunities. Cancer Cell 25, 304–317 10.1016/j.ccr.2014.01.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Chien Y., Scuoppo C., Wang X., Fang X., Balgley B., Bolden J. E., Premsrirut P., Luo W., Chicas A., Lee C. S., Kogan S. C., and Lowe S. W. (2011) Control of the senescence-associated secretory phenotype by NF-κB promotes senescence and enhances chemosensitivity. Genes Dev. 25, 2125–2136 10.1101/gad.17276711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Coppé J. P., Desprez P. Y., Krtolica A., and Campisi J. (2010) The senescence-associated secretory phenotype: the dark side of tumor suppression. Annu. Rev. Pathol. 5, 99–118 10.1146/annurev-pathol-121808-102144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Lu B., Guo H., Zhao J., Wang C., Wu G., Pang M., Tong X., Bu F., Liang A., Hou S., Fan X., Dai J., Wang H., and Guo Y. (2010) Increased expression of iASPP, regulated by hepatitis B virus X protein-mediated NF-κB activation, in hepatocellular carcinoma. Gastroenterology 139, 2183–2194.e5 10.1053/j.gastro.2010.06.049 [DOI] [PubMed] [Google Scholar]
- 54. Laska M. J., Vogel U. B., Jensen U. B., and Nexø B. A. (2010) p53 and PPP1R13L (alias iASPP or RAI) form a feedback loop to regulate genotoxic stress responses. Biochim. Biophys. Acta 1800, 1231–1240 10.1016/j.bbagen.2010.09.002 [DOI] [PubMed] [Google Scholar]
- 55. Kenzelmann Broz D., Spano Mello S., Bieging K. T., Jiang D., Dusek R. L., Brady C. A., Sidow A., and Attardi L. D. (2013) Global genomic profiling reveals an extensive p53-regulated autophagy program contributing to key p53 responses. Genes Dev. 27, 1016–1031 10.1101/gad.212282.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.