

Abstract
目的
分析新型冠状病毒SARS-CoV-2的进化、变异情况。
方法
从GISAID、NCBI中下载相关病毒全基因组序列,运用生物信息学软件MEGA-X、BEAST、TempEst等软件,构建基因组进化树,推测病毒的时间进化信号,计算病毒出现的tMRCA时间,分析病毒进化的选择压力。
结果
基因组进化树显示SARS-CoV-2与蝙蝠冠状病毒Beta CoV/bat/Yunnan/RaTG13/2013、bat-SL-CoVZC45、bat-SL-CoVZXC21和SARS-CoV等病毒共同构成冠状病毒β属的Sarbecovirus亚属。现在的病毒序列有微弱的时间进化信号,tMRCA平均时间为73 d,95%可信区间(38.9~119.3 d),与BetaCoV/bat/Yunnan/RaTG13/2013病毒不具正性时间进化信号,与bat-SL-CoVZC45和SARS-CoV具有强的正性时间进化关系。病毒在流行期间存在变异,主要是净化选择压力。
结论
病毒SARS-CoV-2可能出现在2019年11月左右,来源于蝙蝠相关冠状病毒。结果将有助于研究病毒SARS-CoV-2的溯源、进化,对疾病进行正确防控具有指导意义。
Keywords: SARS-CoV-2, 冠状病毒, 进化, 变异
Abstract
Objective
To analyze the evolution and variation of SARS-CoV-2 during the epidemic starting at the end of 2019.
Methods
We downloaded the full-length genome sequence of SARS-CoV-2 from the databases of GISAID and NCBI. Using the software for bioinformatics including MEGA-X, BEAST, and TempEst, we constructed the genomic evolution tree, inferred the time evolution signal of the virus, calculated the tMRCA time of the virus and analyzed the selection pressure of the virus during evolution.
Results
The phylogenetic tree showed that SARS-CoV-2 belonged to the Sarbecovirus subgenus of β Coronavirus genus together with bat coronavirus BetaCoV/bat/Yunnan/RaTG13/2013, bat-SL-CoVZC45, bat-SL-CoVZXC21 and SARS-CoV. The genomic sequences of SARS-CoV-2 isolated from the ongoing epidemic showed a weak time evolution signal with an average tMRCA time of 73 days (95% CI: 38.9-119.3 days). No positive time evolution signal was found between SARS-CoV-2 and BetaCoV/bat/Yunnan/RaTG13/2013, but the former virus had a strong positive temporal evolution relationship with bat-SL-CoVZC45 and SARS-CoV. The major cause for mutations of SARS-CoV-2 was the pressure of purification selection during the epidemic.
Conclusion
SARS-CoV-2 may have emerged as early as November, 2019, originating most likely from bat-associated coronavirus. This finding may provide evidence for tracing the sources and evolution of the virus.
Keywords: SARS-CoV-2, coronavirus, evolution, mutation
自2019年12月8日起,在中国武汉爆发了以呼吸道为主要症状的肺炎,经二代测序、病毒分离和鉴定等手段,确定为一种新型的冠状病毒[1-2],世界卫生组织于2020年1月19日将其暂命名为2019新型冠状病毒(2019 Novel Coronavrius, 2019 nCoV)[3],于2020年2月11日正式命名为SARS-CoV-2病毒,引起的疾病称为2019冠状病毒病(COVID-19)[4]。至2020年2月13日,中国新型冠状病毒肺炎确诊59 895例,疑似病例16 067例,死亡1367例;中国以外492例,波及日本、泰国等24个国家[5];随着诊断试剂的足量应用和疫情的进展,确诊病例数量可能更进一步增加,严重威胁到人们的生活和生命健康[6]。因此,在2020年1月30日,世界卫生组织突发事件委员会经过讨论决定:将此次新型冠状病毒感染疫情确认为“国际关注的突发公共卫生事件”[7],以保护其他国家,尤其是为了保护一些医疗系统比较脆弱的国家,以便这些国家对此肺炎疫情进行更好的预防和控制。该病成为2009年H1N1、2014年脊髓灰质炎、2014年埃博拉、2016年寨卡、2019年埃博拉疫情之后[8],第6起国际关注的突发公共卫生事件。由此可见,此次新型冠状病毒疫情的严重性。
在疫情的进行中,中国的科学家对病毒特征、疾病的特征进行了很好的解析和描述[9-10],为疾病的防控打下了坚实的基础。病毒的溯源,推测是由蝙蝠作为病毒库传播而来[11],至于有无中间宿主、中间可能的宿主是什么还待于进一步研究确定。在病毒疫情进行中,有一项重要的研究内容是:病毒进化过程起源时间,疫情进展中病毒的变异情况如何;这个内容对病毒疫情防控也具有重要的意义。本文根据GISAID中提交的序列(截止到2020年1月29日)分析病毒进化的相关情况。
1. 材料和方法
1.1. 病毒株序列
从<a href="https://www.gisaid.org/" target="_blank">https://www.gisaid.org/</a>上下载关于SARS-CoV-2序列,共计45条全长基因组序列。其中39条序列用于病毒最近共同祖先时间的计算(tMRCA)。其它冠状病毒序列从NCBI序列数据库中下载。
1.2. 进化树构建、序列相似性分析软件
MEGA-X下载于<a href="https://www.megasoftware.net/" target="_blank">https://www.megasoftware.net/</a>;tMRCA相关分析软件组合BEAUti、BEAST、TreeAnnotator下载于<a href="http://www.beast2.org/" target="_blank">http://www.beast2.org/</a>, 从<a href="http://tree.bio.ed.ac.uk/software/" target="_blank">http://tree.bio.ed.ac.uk/software/</a>下载进化树数据显示软件Tracer v1.6、进化树显示软件FigTree、进化分子钟检测软件TempEst。
1.3. 进化树构建方法
将序列导入MEGA-X中,应用软件找到最佳核苷酸替代模型参数,并应用此参数构建进化树和tMRCA的计算,进化树的可靠性检验采用自展法(Bootstrap= 1000次重复);病毒采样时间导入BEAUti中,以2020年1月23日作为0时间,以天为单位,以病毒株BetaCoV/ Wuhan/IPBCAMS-WH-02/2019|EPI_ISL_403931作为局外群(outgroup,树根),进行推算tMRCA。
2. 结果
2.1. 基因组序列基本信息
病毒序列用二代测序或结合三代测序测定,测序长度为29 688~29 899 bp之间[12],全部涵盖了病毒的编码区。病毒编码的结构蛋白S、E、M和N蛋白,非结构蛋白ORF1a、ORF1ab、ORF3等在文献和数据库中有详细的解析[13-14]。
39株SARS-CoV-2全长基因组核苷酸序列经比对后,与SARS冠状病毒(SARS-CoV)、中东呼吸综合征(MERS CoV)的序列相似性分别平均为78.7%和48.7%,与来自于蝙蝠冠状病毒株bat-SL-CoVZC45 (简写CoVZC45)和bat-SL-CoVZXC21 (简写CoVZXC21)较为接近,相似性为87.5%和87.3%,与BetaCoV/bat/ Yunnan/RaTG13/2013 (简写RaTG13)最为接近,相似性为95.9%。其他冠状病毒之间差异较大,在48.0%~ 87.4%之间,也决定了它们在进化上分属于不同的病毒属(表 1)。
1.
39株SARS-CoV-2病毒与其他冠状病毒核苷酸的相似性比较
Comparison of nucleotide similarities between 39 SARS-CoV-2 isolates and other coronaviruses (%)
| Sequence | SARS-CoV-2 | RaTG13 | CoVZC45 | CoVZXC21 | SARS | MERS |
| SARS-CoV-2 | 99.5-100 | 95.7-96.1 | 87.4-87.6 | 87.3-87.4 | 78.6%-78.8 | 48.6-48.8 |
| RaTG13 | - | - | 87.5 | 87.4 | 78.6 | 48.8 |
| CoVZC45 | - | - | - | 97.2 | 80.0 | 48.3 |
| CoVZXC21 | - | - | - | - | 80.1 | 48.2 |
| SARS-CoV | - | - | - | - | - | 48.0 |
各冠状病毒编码蛋白质ORF1ab、S、E、M和N的氨基酸相似性性如表 2~6所示。选用序列MN908947作为参考株进行比较,结果与基因组的核苷酸相似性一致。ORF1ab、S、M和N蛋白与RaTG13、CoVZC45、CoVZXC21相似性最高,其次是SARS-CoV,与MERS CoV相似最低,结合后面进化树分析的结果,在同一亚属内有着高的相似性,不同亚属之间的相似性较低。值得注意的是E蛋白高度保守,SARS-CoV-2与RaTG13、CoVZC45、CoVZXC21完全一致,与SARS-CoV仅有4个氨基酸的差别。
2.
冠状病毒ORF1ab的氨基酸相似性
Amino acid similarities of ORF1ab among different coronaviruses (%)
| Sequence | RaTG13 | CoVZC45 | CoVZXC21 | SARS | MERS |
| SARS-CoV-2 | 98.5 | 95.6 | 95.2 | 86.1 | 45.4 |
| RaTG13 | - | 95.4 | 94.9 | 85.9 | 45.4 |
| CoVZC45 | - | - | 98.0 | 87.0 | 45.5 |
| CoVZXC21 | - | - | - | 86.7 | 45.2 |
| SARS | - | - | - | - | 45.3 |
6.
冠状病毒N蛋白的氨基酸相似性
Amino acid similarities of the N protein among different coronaviruses (%)
| Sequence | RaTG13 | CoVZC45 | CoVZXC21 | SARS | MERS |
| SARS-CoV-2 | 99.0 | 94.2 | 94.2 | 90.2 | 45.9 |
| RaTG13 | - | 94.2 | 94.2 | 90.2 | 45.9 |
| CoVZC45 | - | - | 99.2 | 91.9 | 45.5 |
| CoVZXC21 | - | - | - | 91.7 | 45.5 |
| SARS | - | - | - | - | 45.6 |
3.
冠状病毒S蛋白的氨基酸相似性
Amino acid similarities of the S protein among different coronaviruses (%)
| Sequence | RaTG13 | CoVZC45 | CoVZXC21 | SARS | MERS |
| SARS-CoV-2 | 97.4 | 80.1 | 79.5 | 75.9 | 28.9 |
| RaTG13 | - | 80.1 | 79.5 | 76.5 | 29.1 |
| CoVZC45 | - | - | 98.6 | 75.5 | 29.4 |
| CoVZXC21 | - | - | - | 75.3 | 29.3 |
| SARS | - | - | - | - | 28.9 |
4.
冠状病毒E蛋白的氨基酸相似性
Amino acid similarities of the E protein among different coronaviruses (%)
| Sequence | RaTG13 | CoVZC45 | CoVZXC21 | SARS | MERS |
| SARS-CoV-2 | 100.0 | 100.0 | 100.0 | 94.7 | 35.3 |
| RaTG13 | - | 100.0 | 100.0 | 94.7 | 35.3 |
| CoVZC45 | - | - | 100.0 | 94.7 | 35.3 |
| CoVZXC21 | - | - | - | 94.7 | 35.3 |
| SARS | - | - | - | - | 35.3 |
5.
冠状病毒M蛋白的氨基酸相似性
Amino acid similarities of the M protein among different coronaviruses (%)
| Sequence | RaTG13 | CoVZC45 | CoVZXC21 | SARS | MERS |
| SARS-CoV-2 | 0.981 | 0.986 | 0.986 | 0.891 | 0.387 |
| RaTG13 | - | 0.986 | 0.986 | 0.909 | 0.393 |
| CoVZC45 | - | - | 1 | 0.896 | 0.387 |
| CoVZXC21 | - | - | - | 0.896 | 0.387 |
| SARS | - | - | - | - | 0.411 |
2.2. 进化分析
应用MEGA-X软件寻找最佳核苷酸替代模型模块,以BIC (Bayesian Information Criterion)最小值为最佳核苷酸替代模型参数,显示TN93模型为最优。以TN93替代模型、NJ (Neighbor joint)法构建全基因组进化树,结果如下图 1A所示。根据国际病毒分类委员会的分类,目前冠状病毒的分类是Alpha、Beta、Gamma和Delta[15]。从图 1中,我们可以看到SARS-CoV-2与BetaCoV/bat/Yunnan/RaTG13/2013、bat-SL-CoVZC45和bat-SL-CoVZXC21形成了sarbecovirus属的一个分支,而SARS-CoV等病毒构成了sarbecovirus属的另一个分支。
1.
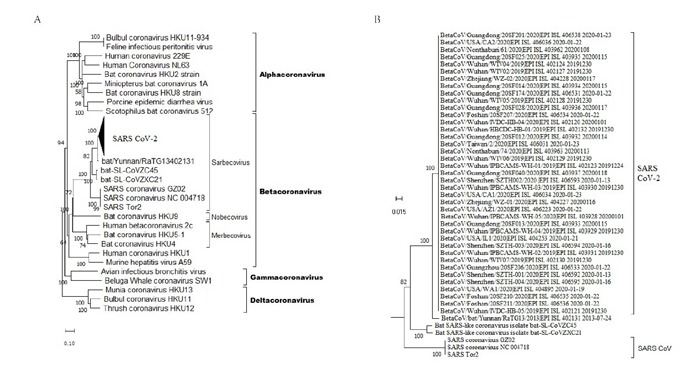
SARS-CoV-2的全基因组进化分析
Phylogenetic analysis of the full-length genome of SRAS-CoV-2. A: Whole genome evolutionary tree of SARS-CoV-2 virus strains(compressed in triangle); B: Unfolded evolutionary tree of 38 isolates of SARS-CoV-2 virus.
2.3. tMRCA分析
应用MEGA-X软件构建进化树,导入进化时间信号分析软件TempEst中,分析是否具有进化上的时间信息。如图 2A所示,显示有微弱正向进化信号,粗估时钟速率是2.24×10-6substitution/site/day,tMRCA为-131 d,R2=0.08。在此基础上,我们应用BEAST软件进行精细评估tMRCA。如图 2B所示,tMRCA平均时间是73.0 d (2019年11月10日),95%可信区间是38.9~119.3 d,后验概率是100%。结果提示病毒的出现日期在2019年9月23日~2019年12月15日之间。这与文献中报道的第1例病例在2019年12月1日出现也是相吻合的。
2.
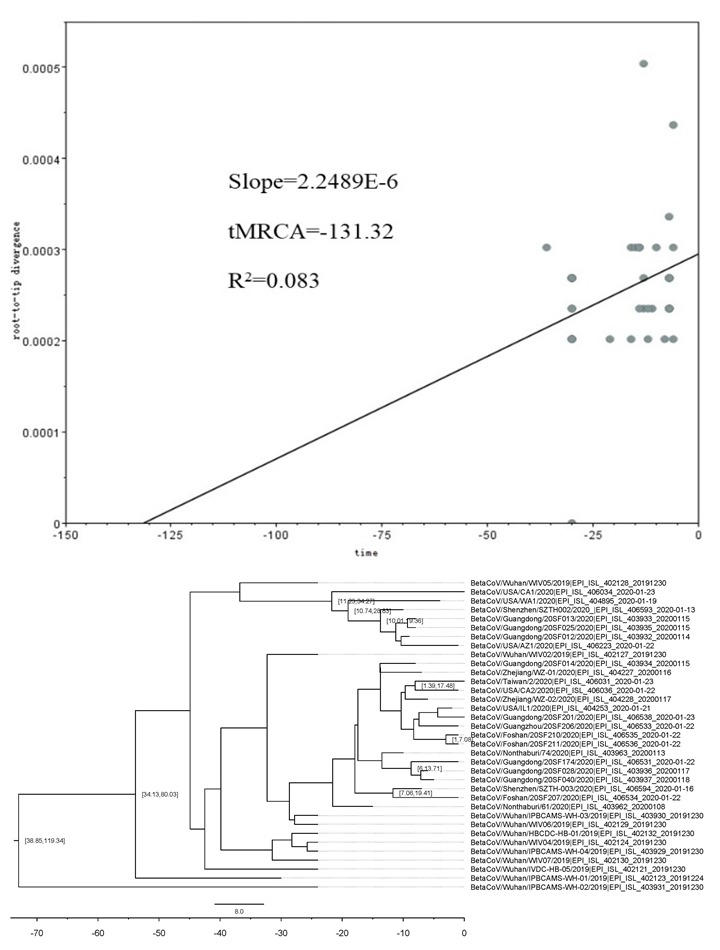
SARS-CoV-2病毒的tMRCA计算
tMRCA of SARS-CoV-2. A: Calculation of evolutionary time signals of SARS-CoV-2 virus. The point indicates the genetic distance of each isolate of the virus from the reference strain (outgroup, tree root) on the time scale; B: Analysis of tMRCA of SARSCoV-2 virus using BEAST.
2.4. SARS-CoV-2与其他冠状病毒的关系
应用进化时间信号分析软件TempEst检测SARSCoV- 2与BetaCoV/bat/Yunnan/RaTG13/2013、bat- SL-CoVZC45、SARS-CoV之间是否有进化上的时间关系。结果显示:SARS-CoV-2与BetaCoV/bat/Yunnan/ RaTG13/2013不存在时间进化关系,斜率为-1.8×10- 5 substitution/site/day,R2=0.998 (图 3A),说明在自然界自然进化中是很难实现的,然而与CoVZC45、SARS-CoV确存在着明显的正性时间进化信号(图 3B、C)。
3.
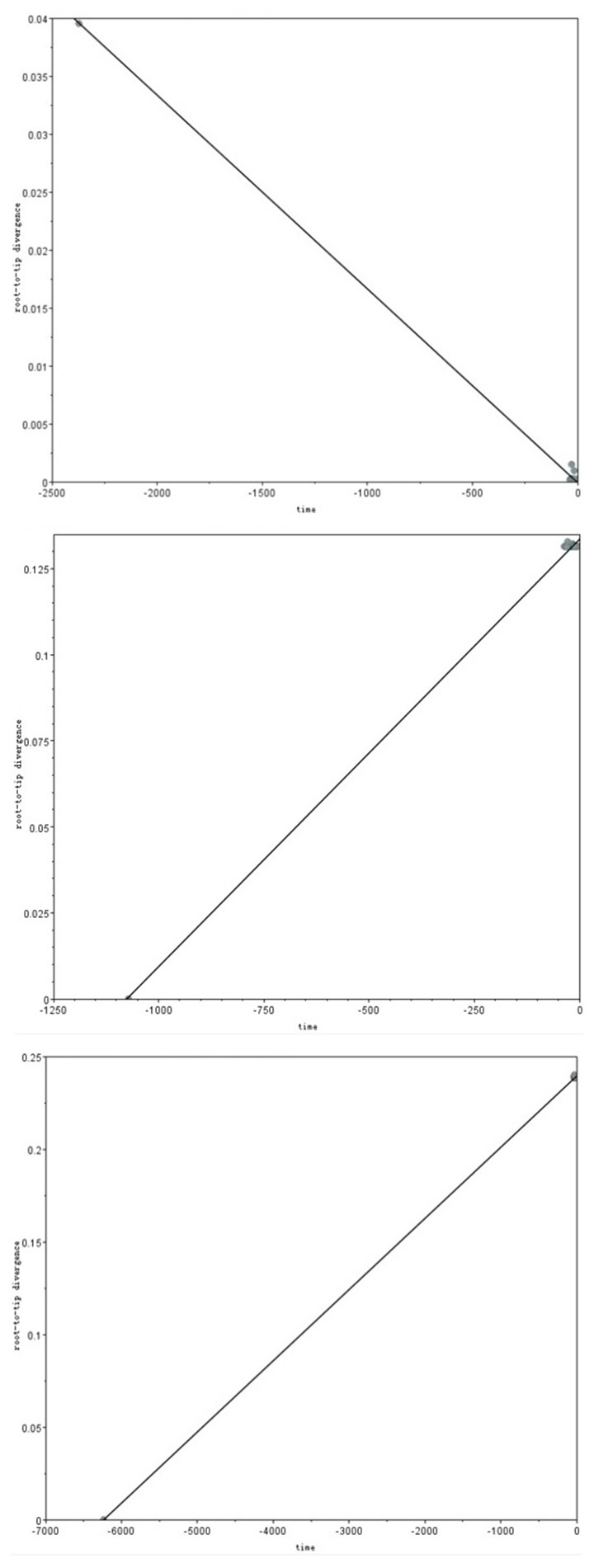
SARS-CoV-2病毒与其他冠状病毒进化时间信号检测
Temporal signal test of molecular phylogenies of SARS-CoV-2 together with other coronaviruses. A, B, and C: Evolution time signal detection results of SARS-CoV-2 virus strain and RaTG13, CoVZC45, and SARS-CoV viruses, respectively.
2.5. SARS-CoV-2进化中的变异
我们将38株序列进行比较,核苷酸之间变化差异非常小,以最早测定的序列BetaCoV/Wuhan/ IPBCAMS- WH- 01/2019|EPI_ISL_402123为参考序列,共计有117处变异,如图 4所示,变异位点没有聚集性,相对均匀地分布整个基因组。从中也可以看出,有3个位点的变异(发生在ORF1ab上),稳定地表达在其它36株病毒中,同时有多个位点在病毒株序列中聚集。
4.
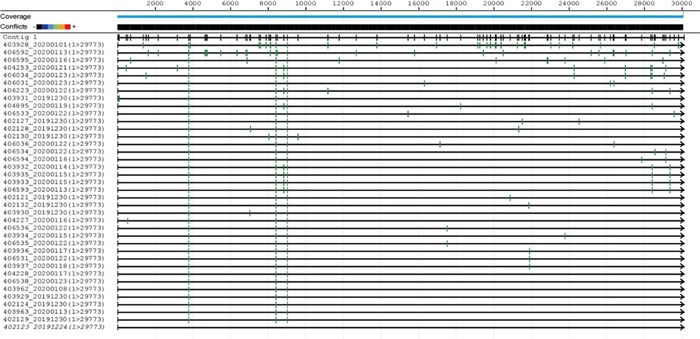
38株病毒序列变异
Variation of 38 SARS-CoV-2 strains. Each green vertical line represents the the location of variation in the genome, using the earliest BetaCoV/Wuhan/IPBCAMS-WH-01/2019|EPI_ISL_402123 20191224 as the reference sequence.
我们将变异序列导入<a href="http://www.datamonkey.org/" target="_blank">http://www.datamonkey.org/</a>中,分析其变异是否存在选择压力。如<xref ref-type="table" rid="Table7">表 7</xref>所示,参与病毒复制相关蛋白有48处变异位点,采用固定效应似然比检验模型(FEL)发现有4处存在净化选择,而采用相对保守的方法单一似然祖先计数(SLAC)法只能检出一个位点。在S蛋白、M蛋白和N蛋白中只能用FEL法检出具有统计学意义的2~3种净化选择,在N蛋白中检出1种正向选择进化。
7.
SARS-CoV-2病毒主要蛋白的选择进化压力分析
Analysis of selection pressure of the main proteins of SARS-CoV-2
| Gene | Synonymous number | Non-synonymous number | Site | FEL | SLAC | |||
| dN/dS | P | dN/dS | P | |||||
| S2839 | 0 | 0.018 | 0 | 0.057 | ||||
| E377 | 0 | 0.071 | - | - | ||||
| ORF1ab | 16 | 32 | S6927 | 0 | 0.061 | - | - | |
| T1171 | 0 | 0.081 | - | - | ||||
| K921 | 0 | 0.028 | - | - | ||||
| S | 5 | 7 | K417 | 0 | 0.08 | - | - | |
| G669 | 0 | 0.092 | - | - | ||||
| A69 | 0 | 0.052 | ||||||
| M | 2 | 1 | G79 | 0 | 0.093 | - | - | |
| I415D | Infinity | 0.066 | - | - | ||||
| N | 4 | 4 | A173 | 0 | 0.096 | - | - | |
| F274 | 0 | 0.096 | - | - | ||||
3. 讨论
自2019年12月初起,从武汉流行的新现的冠状病毒[8],至2020年2月,引发了至目前为止全中国6万人的感染,病死人数1100余人,对人们健康危害已经远超2003年前后的SARS流行[16-17]。该病毒由于与SARSCoV病毒有较高的相似性,与SARS-CoV同属于β冠状病毒属的Sarbecovirus亚属(图 1),以及对人们的健康产生严重影响,国际病毒命名委员会将其命名为SARSCoV-2,与SARS-CoV是姊妹病毒[18-19]。世界卫生组织将该病毒引起的疾病称为COVID-19。这也是目前发现第7种能使人致病的冠状病毒[20]。
该病毒具有比SARS-CoV更强的传播力、更长的潜伏期,这是病毒广泛感染人的基础[21-22]。尽管病毒的基因组在疾病流行期间很快被解析[2],有多篇文献报道了其基因组特征[23-24],但这些特征还难以解释病毒的强传播力[8]。影响病毒播散的另外一些重要因素是病毒的溯源,包括:病毒出现的时间,是否有中间宿主,传染源是什么,以及传染源是否一直存在。科学家根据现存数据库中病毒序列信息,以及当前的溯源研究,推测是来源于菊头蝙蝠[25]。
关于病毒出现的可能时间,我们应用进化分析软件TempEst分析当前基因组数据,结果提示已经出现了分子钟事件信号,这是后面进行tMRCA计算前提。在此基础上,我们应用BEAST软件推算病毒可能出现的时间为38.9~119.3 d (从1月23日往前推),也就是在2019年9月23日~2019年12月15日之间。这也提示目前武汉市高感染原因之一。我们注意到,本文中关于tMRCA的推算因为时间跨度太短(只有30 d),可能结果并不是很准确,但也提供了一定的信息。
关于病毒的来源,目前与SARS-CoV-2相似性比较高从蝙蝠中分离到的病毒是RaTG13 (云南,2013年)、bat- SL- CoVZC45和bat- SL- CoVZXC21(2017年,浙江),其中相似性最高的是RaTG13病毒株。如果SARS-CoV-2是来自于RaTG13病毒株,那么RaTG13病毒就应与SARS-CoV-2具有时间进化信号。我们检测了两种病毒时间进化上的信息,发现呈负相关(进化速率是负的)(图 3A);如果SARS-CoV-2演化于RaTG13病毒,应该是正性进化速率,因此我们推测:SARS-CoV-2来源于RaTG13病毒的可能性不大,尽管它们之间有较高的相似性。我们随后演算了SARS-CoV-2与bat-SLCoVZC45和SARS-CoV之间的进化关系,发现它们具有很明显的正性相关关系(图 3B、C)。提示SARS-CoV-2的产生与bat-SL-CoVZC45等冠状病毒有一定的关系。这一点仍需更多的实验进行支持。
病毒在流行过程中,是否产生对抗外界压力的适应力,也是影响病毒传播能力一个重要因素[26],从临床的数据显示,病毒的传播确是不断增加[27]。我们通过全基因组数据选择进化压力分析,结果显示病毒表现一种进化选择,在高度保守的基因内,发生多频次的同义替代,这种替代远高于非同义替代,这些替代的位置可能对病毒的生命活力具有重要的影响。继续加强这些位点的监测,将有助于解析SARS-CoV-2强传播能力,对后期的防控具有一定的指导作用。
致谢
衷心感谢众多研究者将研究数据提交到GISAID中,为本文奠定了基础!
Biography
周烨真,硕士,E-mail: 1360855328@qq.com
Funding Statement
国家自然科学基金(31670168)
Supported by National Natural Science Foundation of China (31670168)
Contributor Information
周 烨真 (Yezhen ZHOU), Email: 1360855328@qq.com.
赵 卫 (Wei ZHAO), Email: zhaowei@fimmu.com.
张 宝 (Bao ZHANG), Email: zhang20051005@126.com.
References
- 1.Wu F, Zhao S, Yu B, et al. A new coronavirus associated with human respiratory disease in China. https://www.ncbi.nlm.nih.gov/pubmed/32015508. Nature. 2020 doi: 10.1038/s41586-020-2008-3. [Wu F, Zhao S, Yu B, et al. A new coronavirus associated with human respiratory disease in China[J]. Nature, 2020, 10.1038/ s41586-020-2008-3.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhou P, Yang XL, Wang XG, et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. https://www.ncbi.nlm.nih.gov/pubmed/32015507. Nature. 2020 doi: 10.1038/s41586-020-2012-7. [Zhou P, Yang XL, Wang XG, et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin[J]. Nature, 2020, 10.1038/s41586-020-2012-7.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rubin EJ, Baden LR, Morrissey S, et al. Medical Journals and the 2019-nCoV Outbreak. http://cn.bing.com/academic/profile?id=3d2f310d5fd9ca41904de4521d724fcc&encoded=0&v=paper_preview&mkt=zh-cn. N Engl J Med. 2020 doi: 10.1056/NEJMe2001329. [Rubin EJ, Baden LR, Morrissey S, et al. Medical Journals and the 2019-nCoV Outbreak[J]. N Engl J Med, 2020, 10.1056/NEJMe2001329.] [DOI] [PubMed] [Google Scholar]
- 4.Guarner J. Three emerging coronaviruses in two decades. https://academic.oup.com/ajcp/article/153/4/420/5735509. Am J Clin Pathol. 2020;aqaa029 doi: 10.1093/ajcp/aqaa029. [Guarner J. Three emerging coronaviruses in two decades[J]. Am J Clin Pathol, 2020, aqaa029.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Stein RA. The 2019 Coronavirus: Learning Curves, Lessons, and the Weakest Link. https://www.ncbi.nlm.nih.gov/pubmed/32052918. Int J Clin Pract. 2020:e13488. doi: 10.1111/ijcp.13488. [Stein RA. The 2019 Coronavirus: Learning Curves, Lessons, and the Weakest Link[J]. Int J Clin Pract, 2020, e13488.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Velavan TP, Meyer CG. The COVID-19 epidemic. https://onlinelibrary.wiley.com/doi/pdf/10.1111/tmi.13383. Trop Med Int Health. 2020 doi: 10.1111/tmi.13383. [Velavan TP, Meyer CG. The COVID-19 epidemic[J]. Trop Med Int Health, 2020, 10.1111/tmi.13383.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.World Health Organization. Statement on the second meeting of the international health regulations (2005) emergency committee regarding the outbreak of novel coronavirus (2019-nCoV). Retrieved from <a href="https://www.who.int/news-room/detail/30-01-2020-statementon-the-second-meeting-of-the-international-health-regulations-(2005)-emergency-committee-regarding-the-outbreak-of-novel-coronavirus-(2019-ncov)" target=_blank>https://www.who.int/news-room/detail/30-01-2020-statementon-the-second-meeting-of-the-international-health-regulations-(2005)-emergency-committee-regarding-the-outbreak-of-novel-coronavirus-(2019-ncov)</a>.
- 8.Wang FS, Zhang C. What to do next to control the 2019-nCoV epidemic? Lancet. 2020;395(10222):391–3. doi: 10.1016/S0140-6736(20)30300-7. [Wang FS, Zhang C. What to do next to control the 2019-nCoV epidemic[J]? Lancet, 2020, 395(10222): 391-3.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen Y, Liu Q, Guo D. Emerging coronaviruses: Genome structure, replication, and pathogenesis. J Med Virol. 2020 doi: 10.1002/jmv.25681. [Chen Y, Liu Q, Guo D. Emerging coronaviruses: Genome structure, replication, and pathogenesis[J]. J Med Virol, 2020, 10.1002/jmv.25681.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen ZM, Fu JF, Shu Q, et al. Diagnosis and treatment recommendations for pediatric respiratory infection caused by the 2019 novel coronavirus. https://link.springer.com/article/10.1007%2Fs12519-020-00345-5. World J Pediatr. 2020 doi: 10.1007/s12519-020-00345-5. [Chen ZM, Fu JF, Shu Q, et al. Diagnosis and treatment recommendations for pediatric respiratory infection caused by the 2019 novel coronavirus[J]. World J Pediatr, 2020, 10.1007/s12519-020-00345-5.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Benvenuto D, Giovanetti M, Ciccozzi A, et al. The 2019-new coronavirus epidemic: Evidence for virus evolution. http://cn.bing.com/academic/profile?id=189ca22dd3b52cdfa346766bcd17b8da&encoded=0&v=paper_preview&mkt=zh-cn. J Med Virol. 2020 doi: 10.1002/jmv.25688. [Benvenuto D, Giovanetti M, Ciccozzi A, et al. The 2019-new coronavirus epidemic: Evidence for virus evolution[J]. J Med Virol, 2020, 10.1002/jmv.25688.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li X, Wang W, Zhao X, et al. Transmission dynamics and evolutionary history of 2019-nCoV. https://www.ncbi.nlm.nih.gov/pubmed/32027035. J Med Virol. 2020 doi: 10.1002/jmv.25701. [Li X, Wang W, Zhao X, et al. Transmission dynamics and evolutionary history of 2019-nCoV[J]. J Med Virol, 2020, 10.1002/jmv.25701.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wu A, Peng Y, Huang B, et al. Genome composition and divergence of the novel coronavirus (2019-nCoV) originating in China. https://www.ncbi.nlm.nih.gov/pubmed/32035028. Cell Host Microbe. 2020:S1931–3128(20)30072-X. doi: 10.1016/j.chom.2020.02.001. [Wu A, Peng Y, Huang B, et al. Genome composition and divergence of the novel coronavirus (2019-nCoV) originating in China[J]. Cell Host Microbe, 2020, S1931-3128(20)30072-X.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ceraolo C, Giorgi FM. Genomic variance of the 2019-nCoV coronavirus. https://www.biorxiv.org/content/10.1101/2020.02.02.931162v2.full.pdf. J Med Virol. 2020 doi: 10.1002/jmv.25700. [Ceraolo C, Giorgi FM. Genomic variance of the 2019-nCoV coronavirus[J]. J Med Virol, 2020, 10.1002/jmv.25700.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Phan T. Novel coronavirus: From discovery to clinical diagnostics. Infect Genet Evol. 2020;79:104211. doi: 10.1016/j.meegid.2020.104211. [Phan T. Novel coronavirus: From discovery to clinical diagnostics[J]. Infect Genet Evol, 2020, 79: 104211.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Riou J, Althaus CL. Pattern of early human-to-human transmission of Wuhan 2019 novel coronavirus (2019-nCoV), December 2019 to January 2020. https://www.ncbi.nlm.nih.gov/pubmed/32019669. Euro Surveill. 2020;25(4):2000058. doi: 10.2807/1560-7917.ES.2020.25.4.2000058. [Riou J, Althaus CL. Pattern of early human-to-human transmission of Wuhan 2019 novel coronavirus (2019-nCoV), December 2019 to January 2020[J]. Euro Surveill, 2020, 25(4): 2000058.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cowling BJ, Leung GM. Epidemiological research priorities for public health control of the ongoing global novel coronavirus (2019-nCoV) outbreak[J]. Euro Surveill, 2020, <a href="http://dx.doi.org/10.2807/1560-7917" target="_blank">10.2807/1560-7917</a>. ES.2020.25.6.2000110.
- 18.Chen L, Liu W, Zhang Q, et al. RNA based mNGS approach identifies a novel human coronavirus from two individual pneumonia cases in 2019 Wuhan outbreak. Emerg Microbes Infect. 2020;9(1):313–9. doi: 10.1080/22221751.2020.1725399. [Chen L, Liu W, Zhang Q, et al. RNA based mNGS approach identifies a novel human coronavirus from two individual pneumonia cases in 2019 Wuhan outbreak[J]. Emerg Microbes Infect, 2020, 9 (1): 313-9.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Malik YS, Sircar S, Bhat S, et al. Emerging novel Coronavirus (2019-nCoV) - Current scenario, evolutionary perspective based on genome analysis and recent developments. https://www.tandfonline.com/doi/full/10.1080/01652176.2020.1727993. Vet Q. 2020:1–12. doi: 10.1080/01652176.2020.1727993. [Malik YS, Sircar S, Bhat S, et al. Emerging novel Coronavirus (2019-nCoV) - Current scenario, evolutionary perspective based on genome analysis and recent developments[J]. Vet Q, 2020, 1-12.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhu N, Zhang D, Wang W, et al. A Novel Coronavirus from Patients with Pneumonia in China, 2019. http://cn.bing.com/academic/profile?id=431bc637c5a6a390019d1cba74099467&encoded=0&v=paper_preview&mkt=zh-cn. N Engl J Med. 2020 doi: 10.1056/NEJMoa2001017. [Zhu N, Zhang D, Wang W, et al. A Novel Coronavirus from Patients with Pneumonia in China, 2019[J]. N Engl J Med, 2020, 10.1056/NEJMoa2001017.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wu JT, Leung K, Leung GM. Nowcasting and forecasting the potential domestic and international spread of the 2019-nCoV outbreak originating in Wuhan, China: a modelling study. https://www.thelancet.com/journals/lancet/article/PIIS0140-6736(20)30260-9/fulltext. Lancet. 2020:S0140–6736(20): 30260-9. doi: 10.1016/S0140-6736(20)30260-9. [Wu JT, Leung K, Leung GM. Nowcasting and forecasting the potential domestic and international spread of the 2019-nCoV outbreak originating in Wuhan, China: a modelling study[J]. Lancet, 2020, S0140-6736(20): 30260-9.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen N, Zhou M, Dong X, et al. Epidemiological and clinical characteristics of 99 cases of 2019 novel coronavirus pneumonia in Wuhan, China: a descriptive study. Lancet. 2020;395(10223):507–13. doi: 10.1016/S0140-6736(20)30211-7. [Chen N, Zhou M, Dong X, et al. Epidemiological and clinical characteristics of 99 cases of 2019 novel coronavirus pneumonia in Wuhan, China: a descriptive study[J]. Lancet, 2020, 395(10223): 507-13.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chan JF, Kok KH, Zhu Z, et al. Genomic characterization of the 2019 novel human-pathogenic coronavirus isolated from a patient with atypical pneumonia after visiting Wuhan. Emerg Microbes Infect. 2020;9(1):221–36. doi: 10.1080/22221751.2020.1719902. [Chan JF, Kok KH, Zhu Z, et al. Genomic characterization of the 2019 novel human-pathogenic coronavirus isolated from a patient with atypical pneumonia after visiting Wuhan[J]. Emerg Microbes Infect, 2020, 9(1): 221-36.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Paraskevis D, Kostaki EG, Magiorkinis G, et al. Full-genome evolutionary analysis of the novel corona virus (2019-nCoV) rejects the hypothesis of emergence as a result of a recent recombination event. Infect Genet Evol. 2020;79:104212. doi: 10.1016/j.meegid.2020.104212. [Paraskevis D, Kostaki EG, Magiorkinis G, et al. Full-genome evolutionary analysis of the novel corona virus (2019-nCoV) rejects the hypothesis of emergence as a result of a recent recombination event[J]. Infect Genet Evol, 2020, 79: 104212.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lu R, Zhao X, Li J, et al. Genomic characterisation and epidemiology of 2019 novel coronavirus: implications for virus origins and receptor binding. https://www.cdc.gov/coronavirus/2019-ncov/downloads/genomic-characterization-of-2019-nCoV-Lancet-1-29-2020.pdf. Lancet. 2020;S0140-6736(20):30251–8. doi: 10.1016/S0140-6736(20)30251-8. [Lu R, Zhao X, Li J, et al. Genomic characterisation and epidemiology of 2019 novel coronavirus: implications for virus origins and receptor binding[J]. Lancet, 2020, S0140-6736(20): 30251-8.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wilder-Smith A, Freedman DO. Isolation, quarantine, social distancing and community containment: pivotal role for old-style public health measures in the novel coronavirus (2019-nCoV) outbreak. J Travel Med. 2020;taaa020 doi: 10.1093/jtm/taaa020. [Wilder-Smith A, Freedman DO. Isolation, quarantine, social distancing and community containment: pivotal role for old-style public health measures in the novel coronavirus (2019-nCoV) outbreak[J]. J Travel Med, 2020, taaa020. doi:10.1093/jtm/taaa020.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liu Y, Gayle AA, Wilder-Smith A, Rocklöv J. The reproductive number of COVID-19 is higher compared to SARS coronavirus. J Travel Med. 2020;taaa021 doi: 10.1093/jtm/taaa021. [Liu Y, Gayle AA, Wilder-Smith A, Rocklöv J. The reproductive number of COVID-19 is higher compared to SARS coronavirus[J]. J Travel Med, 2020, taaa021. doi:10.1093/jtm/taaa021.] [DOI] [PMC free article] [PubMed] [Google Scholar]