

Abstract
Astrocytes play key roles in brain functions through dynamic interactions with neurons. One of their typical features is to express high levels of connexins (Cxs), Cx43 and Cx30, the gap junction (GJ)-forming proteins. Cx30 is involved in basic cognitive processes and shapes synaptic and network activities, as shown by recent studies in transgenic animals. Yet it remains unknown whether astroglial Cx30 expression, localization, and functions are endogenously and dynamically regulated by neuronal activity and could therefore play physiological roles in neurotransmission. We here show that neuronal activity increased hippocampal Cx30 protein levels via a posttranslational mechanism regulating lysosomal degradation. Neuronal activity also increased Cx30 protein levels at membranes and perisynaptic processes, as revealed by superresolution imaging. This translated at the functional level in the activation of Cx30 hemichannels and in Cx30-mediated remodeling of astrocyte morphology independently of GJ biochemical coupling. Altogether, these data show activity-dependent dynamics of Cx30 expression, perisynaptic localization, and functions.
Keywords: astrocytes, connexin, neuroglial interactions, neurons
Introduction
Through dynamic interactions with neurons, astrocytes play critical roles in brain development, activity, and disorders (Clarke and Barres 2013; Perea et al. 2014). Astrocytes sense neuronal inputs through their ion channels, transporters, or membrane receptors, respond by activation transduction pathways, and thereby modulate neighboring neurons. Various mechanisms mediate the astroglial regulation of neuronal activity, such as uptake or release of neuroactive factors, contact-mediated signaling, or plastic physical coverage of neurons (Dallerac et al. 2013; Araque et al. 2014; Bernardinelli et al. 2014). However, molecular description of such modulations is still limited, and both their occurrence and impact during physiological or pathological conditions remain unclear.
A key property of astrocytes is to express high levels of gap junction (GJ) proteins, the connexins (Cxs), which have recently been shown to be involved in synaptic and network activities (Wallraff et al. 2006; Pannasch et al. 2011; Pannasch et al. 2012a; Pannasch et al. 2014). GJ channels are made up of 2 hemichannels (HCs) that align between adjacent cells to form intercellular channels mediating direct cell-to-cell diffusion of ions and small molecules, providing electrical and metabolic coupling (Pannasch and Rouach 2013). The 2 main GJ subunits in mature astrocytes include Cx43, present from embryonic to adult stages, and Cx30, expressed later in development (from P16) (Kunzelmann et al. 1999). Through their role in intercellular communication, Cxs contribute to neurotransmission by both, fueling metabolic active synapses with proper nutrients (Rouach et al. 2008) and by preventing excessive synaptic activity through efficient uptake of synaptically released glutamate and K+ (Pannasch et al. 2011). Recent data have nevertheless unraveled an unexpected complexity of Cx functions, which can be channel (GJ and HC) or nonchannel types, involving for instance protein interactions, cell adhesion, and intracellular signaling (Theis et al. 2005; Elias and Kriegstein 2008). Cx43 is already established as an essential protein in brain development and physiology through channel-dependent and -independent functions (Pannasch and Rouach 2013). In contrast, the specific role of Cx30 in neuronal physiology has largely remained unexplored, although Cx30 was proposed early on to be involved in behavioral and basic cognitive processes (Rampon et al. 2000; Dere et al. 2003).
Recent studies indicate that astroglial networks mediated by GJ channels promote neuronal coordination in the hippocampus (Chever et al. 2016). Remarkably, astroglial Cx30 also tunes hippocampal excitatory synaptic transmission by determining the efficacy of astroglial glutamate clearance through an unprecedented regulation of astroglial morphology, independent of GJ biochemical coupling, which controls the ramification, and insertion of astroglial processes into synaptic clefts (Pannasch et al. 2014). Thus, besides its GJ channel function involved in neuronal network activity (Chever et al. 2016), astroglial Cx30 is also a molecular determinant of astroglial synapse coverage, which controls synaptic efficacy and hippocampal-based memory (Pannasch et al. 2014). These roles of astroglial Cx30 in neurotransmission were revealed by using transgenic mice with Cx30 deletion or mutation to alter channel and nonchannel functions. It is now important to evaluate the physiological relevance of these regulations by determining whether astroglial Cx30 expression and functions are endogenously and dynamically regulated in a native biological context. We therefore examined whether astroglial Cx30 protein levels, localization, and functions are endogenously modulated by neuronal activity. We show here that hippocampal Cx30 protein levels are regulated by neuronal activity via a posttranslational mechanism controlling lysosomal degradation. This translates to an activity-dependent control of Cx30 subcellular recruitment and functions. Hippocampal population bursting indeed promoted Cx30 localization at membrane and perisynaptic processes and induced activation of Cx30 HCs, as well as ramification of astroglial processes independently of GJ-mediated biochemical coupling. Altogether, these data show that Cx30 protein levels, perisynaptic distribution, and functions are activity dependent.
Materials and Methods
Animals
Experiments were carried out according to the guidelines of the European Community Council Directives of 1 January 2013 (2010/63/EU) and of the local animal welfare committee (certificate A751901, Ministère de l’Agriculture et de la Pêche), and all efforts were made to minimize the number of animals used and their suffering. Animals were group housed on a 12 h light/dark cycle. Cx30−/− mice were generated as previously described (Boulay et al. 2013). Cx30T5M/T5M mice (Schutz et al. 2010) were provided by F. Mammano, Venetian Institute of Molecular Medicine, Italy. IP3R2−/− mice (Li et al. 2005) were provided by J. Chen, University of California San Diego, United States of America. Aldhl1:L10a-eGFP mice were obtained from the laboratory of N. Heintz (Rockefeller University, NY, United States of America). All mice were backcrossed to the C57BL6 background. Mice of both sexes and littermates were used at postnatal days 21–30, unless otherwise stated.
Acute Hippocampal Slice Preparation and Pharmacology
Acute transverse hippocampal slices (300–400 μm) were prepared as previously described (Pannasch et al. 2012b). Slices were maintained at room temperature in a submerged storage chamber containing an artificial cerebrospinal fluid (ACSF) including (in mM): 119 NaCl, 2.5 KCl, 2.5 CaCl2, 1.3 MgSO4, 1 NaH2PO4, 26.2 NaHCO3, and 11 glucose, saturated with 95% O2 and 5% CO2 for at least 1 h before further processing. For generation of neuronal bursting activity, slices were stored in Mg2+-free ACSF containing picrotoxin (100 μM). To inhibit action potential firing, tetrodotoxin (TTX, 0.5 μM) was added to ACSF, while for chelating astrocyte intracellular calcium, ACSF was supplemented with BAPTA-AM (25 μM). To dissect the activity-dependent signaling cascade regulating Cx30 expression, the selective group II metabotropic glutamate receptor (mGluR) antagonist LY341495 (20 μM), the competitive purinergic P2Y1 receptor (P2Y1R) antagonist MRS2179 (10 μM) and the selective GABABR antagonist CGP55845 (2 μM) were added to ACSF. Finally, the proteasomal inhibitor MG132 (10 μM) and the lysosomal blocker GPN (200 μM) were used to determine the nature of the protein degradation pathway regulating Cx30 protein levels. Lastly, to selectively block protein synthesis, ACSF was supplemented with cycloheximide (400 μM). All drugs were from Tocris, except for GPN, which was from Sigma.
Antibodies, Immunohistochemistry, and Immunoblotting
All the antibodies used in this study are commercially available and have been validated in previous studies, as reported by the suppliers. The following primary antibodies were used: Cx30 rabbit polyclonal (1:500; 71-2200, Zymed Laboratories), GFAP rabbit polyclonal (1:2000; G3893, Sigma), tubulin rabbit polyclonal (1:10 000, T6199, Sigma), β-actin mouse monoclonal (1:10 000; A5316, Sigma), PSD95 mouse monoclonal (1:500, 610495, BD Biosciences), synapsin I (syn I) mouse monoclonal (1:10 000; 106011, Synaptic Systems). The HRP-conjugated primary anti-GAPDH antibody (1:10000; ab9385, Abcam) has been used as loading control. The following HRP-conjugated secondary antibodies were used: goat anti-rabbit IgG (1:2500; sc-2004, Tebu Santa Cruz), goat anti-mouse IgG (1:2500; sc-2005, Tebu Santa Cruz). The following fluorescent dye-conjugated secondary antibodies were used in appropriate combinations: goat anti-mouse IgG conjugated to Alexa 555 (1:2000; A-21424, Molecular Probes) and goat anti-rabbit IgG conjugated to Alexa 488 or 555 (1:2000; A-11034 and A-21429, Molecular Probes). Immunohistochemistry and quantification were performed as previously described (Pannasch et al. 2011). Briefly, hippocampal acute slices were fixed overnight at room temperature with 4% paraformaldehyde, then washed 3 times with PBS, and preincubated 1 h with PBS-1% gelatin in the presence of 1% Triton X-100. Brain slices were then stained by overnight incubation at 4 °C with primary antibodies and washed 3 times in PBS. Appropriate secondary antibodies were applied for 1–2 h at room temperature. After several washes, hippocampal slices were mounted in Fluoromount or ProLong Gold Antifade reagent and examined with either inverted confocal laser-scanning microscopes (TCS SP5, Leica or custom designed) or custom-designed structured illumination microscopy (SIM) (Young et al. 2016) and stimulated emission depletion (STED) microscopes.
For confocal microscopy (TCS SP5, Leica), stacks of consecutive confocal images taken with a ×63 objective at 600–1000 nm intervals were acquired sequentially with 2 lasers (argon, 488 nm; helium/neon, 543 nm), and Z projections were reconstructed using ImageJ software. LysoTracker red DND99 (1:5000; L7528, Thermo Fisher Scientific) was perfused with oxygen for 10 min on living hippocampal slices, which were then fixed overnight at room temperature with 4% paraformaldehyde and further processed for Cx30 immunohistochemistry as previously described. Finally, the tissue was mounted for confocal imaging and analyzed with ImageJ software: the intensity of Cx30 puncta within “stratum radiatum” LysoTracker-positive structures was measured for each plane of the Z-stack.
For analysis of the Cx30 radial intensity profile, confocal microscopy Z-stacks were processed using the ImageJ software. Briefly, the intensity radial profile plugin ImageJ was used to measure the sum of the pixel values around a circle centered at the cell soma and plotted it as a function of the actual radial distance from this center.
The distribution of pixel intensity values (Cx30 or ethidium bromide [EtBr] signals) for all the quantified images illustrated is represented in Supplementary Figure S3.
For analysis of astrocytic ramification, we used an adaptation of Sholl’s concentric circles technique. Briefly, 5 circles at 5 μm intervals were drawn around each astrocyte; the circle enclosing the soma had a diameter of 7 μm. The number of intersections of astrocytic processes with each circle was quantified.
Immunoblotting and quantification were performed as previously described (Pannasch et al. 2014). Briefly, hippocampal acute slices were collected in a small volume of cold SDS 2% containing a cocktail of protease inhibitors and phosphatase inhibitors (β-glycerophosphate [10 mM] and orthovanadate [1 mM]), to which Laemmli 5× buffer was added. Samples were sonicated, boiled 5 min, and loaded on 10% or 4–12% polyacrylamide gels. Proteins were separated by electrophoresis and transferred onto nitrocellulose membranes. Membranes were saturated with 5% fat-free dried milk in triphosphate buffer solution and incubated overnight at 4 °C with primary antibodies. They were then washed and exposed to peroxidase-conjugated secondary antibodies. Specific signals were revealed with the chemiluminescence detection kit (ECL, GE Healthcare). Semiquantitative densitometric analysis was performed after scanning the bands with the ImageJ software.
Superresolution Imaging
STED-Confocal Imaging
Superresolved and confocal images were acquired on a home built, time-gated STED microscope described previously (Mahou et al. 2015). In brief, the system is based around a commercial point-scanning microscope (RESOLFT, Abberior Instruments) comprising a microscope frame (IX83, Olympus), galvanometer mirrors (Quad scanner, Abberior Instruments), and a detection unit consisting of 2 avalanche photodiodes (SPCM-AQRH, Excelitas Technologies). Images were acquired with a ×100/1.4 NA oil immersion objective lens (UPLSAPO 100X, Olympus). Confocal images of GFP were taken using the in-built laser and detector of the commercial system. Fluorescence excitation was from a 488 nm laser source (Cobolt 06-MLD-488 nm, Cobolot). GFP fluorescence was separated using a dichroic mirror (zt 594 RDC, Chroma) and an emission filter (Brightline HC 550/88, Semrock). STED images were acquired using 638 nm fluorescence excitation and 765 nm depletion. The depletion light was obtained from a titanium-sapphire oscillator (Mai Tai HP, Spectraphysics) operating at 765 nm. Excitation pulses were selected from a supercontinuum generated by pumping a photonic crystal fiber (SCG800, NKT photonics) with the same titanium-sapphire. The depletion beam was shaped using a spatial light modulator (X10468-02, Hamamatsu), which also allowed correction for aberrations. Image acquisition was controlled using the Imspector software (Andreas Schönle, Abberior Instruments GmbH). The pixel dwell time was 10 μs for STED and 7 μs for GFP. A total of 5 line accumulations were acquired. Excitation powers were 20–30 μW (GFP) and 20–30 μW (STED). The depletion laser power was 100–150 mW. The pixel size used throughout was 50 nm (xy) and 1 μm in z. In the STED channel, a time gate of 1.5 ns between excitation and detection is used.
SIM
Images of the sample were collected using 3-color SIM for optical sectioning (Ströhl and Kaminski 2015, 2016). A ×60/1.2 NA water immersion lens (UPLSAPO 60XW, Olympus) focused the structured illumination pattern onto the sample, and the same lens was also used to capture the fluorescence emission light before imaging onto an sCMOS camera (C11440, Hamamatsu) (Young et al. 2016). The wavelengths used for excitation were 488 nm (iBEAM-SMART-488, Toptica), 561 nm (OBIS 561, Coherent), and 640 nm (MLD 640, Cobolt). Images were acquired using custom SIM software described previously (Young et al. 2016). A total of 9 raw images were collected at each plane and were recombined using a custom implementation of a superresolution optical sectioning reconstruction algorithm (O'Holleran and Shaw 2014). For each cell, 12 planes spaced at 0.5 μm were captured, providing a complete 3D reconstruction of the cell. For each cell, the plane with strongest labeling of Cx30 was selected for colocalization analysis between Cx30 and the synaptic markers (VGLUT1 or PSD95). A colocalization analysis was performed using a batch protocol in Icy (de Chaumont et al. 2012). The key steps in the protocol were 1) noise removal, 2) spot detection, and 3) object-based colocalization using Ripley’s K-function analysis.
Brain Slices Biotinylation
Biotinylation of cell surface proteins was performed as previously described (Gabriel et al. 2014). Briefly, after a 3 h preincubation period in either regular ACSF or 0Mg2+-picrotoxin (0Mg-P) containing ACSF, hippocampal acute slices were washed 3 times in ice cold ACSF and then incubated in 0.75 mL of 1 mg/mL sulfo-NHS-SS-biotin on ice for 45 min. Biotinylated brain slices were then washed 3 times in ice cold ACSF and incubated 10 min in ice cold ACSF. Hippocampal slices were next washed 3 times in ice cold quench buffer (ACSF supplemented with 100 mM glycine) and incubated with 0.75 mL quenching buffer on ice 2 times for 25 min. Finally, slices were washed 3 times in ice cold ACSF and pelleted by centrifuging at 200 g/min. The pellet was then resuspended in 400 μL of ice cold RIPA/PI and incubated at 4 °C for 30 min, before centrifuging at 18 000 × g at 4 °C for 15 min. The protein concentration of the supernatant was next determined by BCA protein assay, and the bead/total protein ratio was set empirically at ≈1/4, such that 70 μL of beads were incubated with 275 μg of tissue lysate. Finally, biotinylated proteins were then bound to streptavidin beads by rotating the mix overnight at 4 °C and pelleted at 18 000 × g at RT for 2 min. After washing the beads 3 times with 0.75 mL RIPA, biotinylated proteins were eluted by adding 25 μL of 2× SDS-PAGE reducing sample buffer, vortexed, and rotated at RT for 30 min. Last, biotinylated and total lysate samples were analyzed in parallel by immunoblot as described above.
Isolation of Synaptosomal Crude Membrane Fraction
For the isolation of synaptosomal crude membrane fractions, 8–10 hippocampal acute slices for each condition were first lysed in 500 μL of ice cold homogenization buffer (0.32 M sucrose, 10 mM HEPES, 2 mM EDTA, containing a freshly prepared cocktail of protease inhibitors) using a mechanical Potter-Elvehjem homogenizer. Then, the homogenates were centrifuged at 1000 × g for 15 min at 4 °C. The supernatants were further centrifuged at 200 000 × g for 30 min at 4 °C, while the pellet nuclear fractions were discarded. Subsequently, the new pellets were washed and resuspended in ice cold homogenization buffer, while the supernatant crude cytosols were withdrawn. Finally, crude membrane pellets were centrifuged once more at 200 000 × g for 30 min at 4 °C and resuspended in 100 μL of HEPES lysis buffer (50 mM HEPES, 2 mM EDTA, pH 7.4, containing a freshly prepared cocktail of protease inhibitors). Samples were briefly sonicated, and the protein concentration of synaptosomal crude membrane fraction was next determined by BCA protein assay and further processed for immunoblot as described above.
HC Activity
Basal and evoked HC activity was analyzed as previously described (Chever et al. 2014). Briefly, HC opening was investigated at a depth of 20–40 μm from the surface of acute slices and at a recovery time of at least 2 h after the slicing procedure because in such conditions astrocytes exhibited no sign of reactivity, as previously described (Chever et al. 2014). Living slices were incubated for 1 h in regular ACSF or ACSF containing 0Mg-P (100 μm) within small customized submerged chambers. To investigate the contribution of Cx HCs to dye uptake, independent experiments were performed where slices were preincubated 15 min before and during the application of EtBr (314 Da, 4 μM, 10 min), an HC-permeable fluorescent tracer, with carbenoxolone (CBX, 200 μM). Slices were then rinsed for 15 min in ACSF, fixed for 2 h in 4% paraformaldehyde, immunostained for S100β, and mounted in Fluoromount. Labeled cells were examined in a confocal laser-scanning microscope (TCS SP5, Leica). Stacks of consecutive confocal images with high-bit depth color (16 bit) were taken at 1 μm intervals. A dye uptake analysis was performed in stratum radiatum CA1 astrocytes that were positive for S100β. Fluorescence intensity was digitized into arbitrary units in 65 536 shades of gray with ImageJ software. Dye uptake was evaluated as the difference between the fluorescence measured in astrocytes (20–40 cells per slice), and the background fluorescence measured in the same field where no labeled cells were detected.
Gap Junctional Coupling
Slices were transferred to a submerged recording chamber mounted on a microscope equipped for infrared differential interference microscopy and were perfused with ACSF at a rate of 1.5 mL/min. Somatic whole-cell recordings were obtained from visually and electrophysiologically identified stratum radiatum astrocytes using 4–6 MΩ glass pipettes filled with 105 mM K-gluconate, 30 mM KCl, 10 mM HEPES, 10 mM phosphocreatine, 4 mM ATP-Mg, 0.3 mM GTPTris, and 0.3 mM EGTA (pH 7.4, 280 mOsm). The recorded stratum radiatum astrocytes exhibited typical morphological and electrophysiological properties and were identified by their small soma (5–10 μm), low membrane resistance and resting membrane potentials (≈ − 80 mV), passive membrane properties (linear I–V relationship), and lack of action potentials. For intercellular coupling experiments, the internal solution contained 2-[N-(7-nitrobenz-2-oxa-1,3-diazol-4-yl)amino]-2-deoxyglucose (2-NBDG, 2 mg/ml), and the recorded astrocytes were loaded during 20 min in current-clamp mode. Intercellular diffusion of fluorescent glucose was captured online with a digital camera after 20 min of astrocyte dialysis, and the number of coupled cells was analyzed offline with ImageJ software. Astrocyte recordings were discarded when the resting membrane potential varied by more than 10% or when series resistance varied by more than 20% during the experiment. Recordings were acquired with Axopatch-1D amplifiers, digitized at 10 kHz, filtered at 2 kHz, stored, and analyzed on computer using Pclamp9 and Clampfit9 software.
Statistical Analysis
All data are expressed as mean ± standard error of the mean (SEM). Statistical significance for within-group comparisons was determined by 1-way or 2-way analysis of variances (ANOVAs) (followed by Bonferroni’s posttest), whereas unpaired or paired t-tests were used for between-group comparisons. A statistical analysis was performed in GraphPad InStat and Prism software.
Results
Neuronal Activity Increases Cx30 Expression Through a Calcium-Dependent Pathway
To examine the role of neuronal activity on Cx30 protein levels, we investigated the impact of spontaneous action potential firing in acute hippocampal slices using tetrodotoxin (TTX, 0.5 μM, 1 h; Fig. 1A). When action potentials were inhibited, total hippocampal Cx30 protein levels decreased by more than 50%, as assessed by western blotting (n = 7; Fig. 1B,C) and immunohistochemistry (n = 9; Fig. 1D,E). We then investigated the role of network bursting activity generated spontaneously in disinhibited hippocampal slices (0Mg/picrotoxin, 1–3 h) (Fig. 1A). In this case, we found that Cx30 expression levels increased by ~ 40%, as assessed by immunoblotting (+52.6 ± 7%, n = 17, P < 0.001, Mann–Whitney test) and immunohistochemistry (+36.2 ± 10%, n = 9, P = 0.0172, unpaired t-test; 3 h treatment; Fig. 1B–E). This increase in Cx30 protein levels was time dependent and took up to 3 h to occur (Supplementary Fig. S1).
Figure 1.
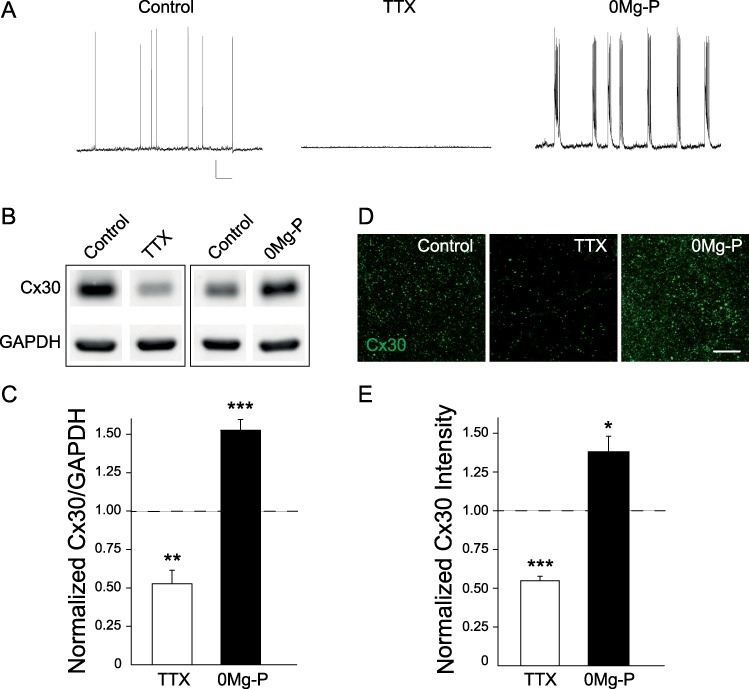
Activity-dependent regulation of Cx30 protein levels. (A) Spontaneous activity of hippocampal CA1 pyramidal cells recorded in current clamp in control, TTX (0.5 μM, 1–3 h) and 0Mg-P (100 μM, 1–3 h) conditions. Scale bar, 20 mV; 6.7 s. (B) Immunoblot detection of Cx30 in hippocampal acute slices in regular ACSF (control), ACSF containing TTX (0.5 μM, 1 h), or 0Mg-P (100 μM, 3 h). GAPDH was used as a loading control. (C) Quantitative analysis of Cx30 protein levels showing that Cx30 expression was reduced in TTX (n = 7) and increased in 0Mg-P (n = 17). Relative expression levels of Cx30 normalized to GAPDH levels; control ratio is set to 1. (D) Immunofluorescent staining for Cx30 in the hippocampal CA1 region from acute slices in regular ACSF (control), ACSF containing TTX (0.5 μM, 1 h) or 0Mg-P (100 μM, 3 h). Scale bar, 25 μm. (E) Quantification of Cx30 staining intensity; control intensity level is set to 1. Cx30 immunostaining intensity was decreased in TTX (n = 9) and increased in 0Mg-P (n = 9) compared to control ACSF (n = 9; ANOVA and Dunnett’s post hoc test). Asterisks indicate statistical significance (*P < 0.05, **P < 0.01, ***P < 0.001).
These data show that Cx30 protein levels are activity dependent and can be bidirectionally regulated by changes in neuronal firing.
Astrocytes integrate neuronal activity in part through intracellular calcium signaling (Bazargani and Attwell 2016). To gain insight into the molecular pathway underlying the activity-dependent regulation of Cx30 expression, we thus investigated the contribution of calcium signaling. Calcium chelation with BAPTA-AM (25 μM, 1 h) strongly decreased hippocampal Cx30 total protein levels (−47 ± 6%, n = 5, P = 0.0019, unpaired t-test Welch corrected; Fig. 2A,B). In addition, genetic deletion of inositol triphosphate type 2 receptors (IP3R2−/− mice), which are enriched in astrocytes and thought to contribute partly to their calcium fluctuations (Srinivasan et al. 2015), inhibited the bidirectional control of Cx30 levels by neuronal activity. It indeed abolished or strongly reduced the effects of bursting activity (n = 5) and action potential silencing (n = 5), respectively, on Cx30 expression (Fig. 2A,B). Calcium increases in astrocytes are largely mediated by G protein-coupled receptors, such as mGluRs, P2Y1Rs, or gamma-aminobutyric acid receptors (GABABRs). Blockade of mGluRs (LY341495, 20 μM, 3 h), but not of P2Y1Rs (MRS2179, 10 μM, 3 h) or GABABRs (CGP55845, 2 μM, 3 h), prevented the increase in Cx30 protein levels induced by bursting activity (n = 7; Fig. 2C,D). These data indicate that mGluR-dependent calcium signaling contributes to the regulation of hippocampal Cx30 protein levels by neuronal activity.
Figure 2.
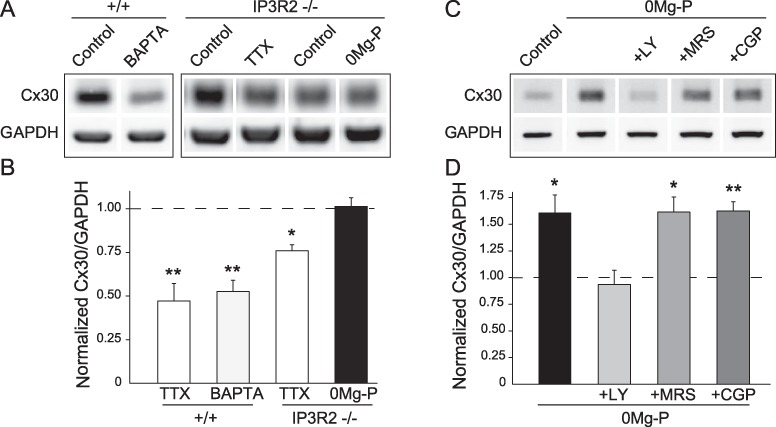
Activity-dependent regulation of Cx30 expression relies on mGluRs and calcium signaling. (A) Immunoblot detection of Cx30 levels in hippocampal acute slices from wild-type (+/+) or IP3R2−/− mice, treated with either BAPTA (25 μM, 1 h), TTX (0.5 μM, 1 h), or 0Mg-P (100 μM, 3 h). GAPDH was used as a loading control. (B) Quantitative analysis of Cx30 expression. Relative expression levels of Cx30 normalized to GAPDH levels showing that Cx30 expression is reduced in the absence of intracellular free calcium (BAPTA, n = 5), and its activity-dependent regulations by TTX (n = 5) and 0Mg-P (n = 5) are reduced or inhibited, respectively, in hippocampal acute slices from IP3R2−/− mice; control ratio is set to 1. (C) Immunoblot detection of Cx30 expression in hippocampal acute slices incubated in 0Mg-P for 3 h with or without antagonists of mGluRs (LY341495, 20 μM), P2Y1Rs (MRS2179, 10 μM), or GABABRs (CGP55845, 2 μM). (D) Quantitative analysis of Cx30 expression. Relative expression levels of Cx30 normalized to GAPDH levels showing that Cx30 expression in 0Mg-P (n = 7) was decreased to control level by inhibition of mGluRs (LY, n = 4) but was unaltered by blockade of P2Y1Rs (MRS, n = 4) or GABABRs (CGP, n = 4; ANOVA and Dunnett’s post hoc test); control ratio is set to 1 (n = 7). Asterisks indicate statistical significance (*P < 0.05, **P < 0.01).
Activity-Dependent Control of Cx30 Expression Is Mediated by the Lysosomal Degradation Pathway
Cx proteins undergo a rapid turnover (Falk et al. 2014). Their expression levels are regulated by synthesis and degradation rates, which occur via the lysosomal and proteasomal pathways. We therefore investigated whether the rapid decrease in hippocampal Cx30 protein levels induced by action potential inhibition resulted from degradation. Immunoblot analysis revealed that inhibition of the lysosomal pathway (GPN, 200 μM, 1 h), but not proteasomal pathway (MG132, 10 μM, 1 h), prevented Cx30 downregulation induced by action potentials blockade (TTX, n = 5; Fig. 3A,B). Accordingly, action potential inhibition strongly increased the number of Cx30 puncta in lysosomal compartments, as revealed by colocalization of Cx30 and the lysosomal dye LysoTracker (+68.4 ± 20.9%, n = 9, P = 0.0005, Mann–Whitney test; Fig. 3C,D). We then examined whether the increase in Cx30 protein levels induced by bursting activity resulted from protein synthesis and/or impaired degradation via the lysosomal pathway. Inhibition of protein synthesis (cycloheximide, 400 μM, 3 h) failed to prevent the upregulation in Cx30 expression, as shown by western blot analysis (n = 3; Fig. 3E,F). However, inhibition of lysosomal degradation mimicked and occluded the effect of bursting activity on Cx30 expression, as it increased by itself Cx30 levels (GPN, n = 5), and prevented bursting activity to further upregulate Cx30 protein levels (0Mg-P + GPN, n = 8; Fig. 3G,H).
Figure 3.
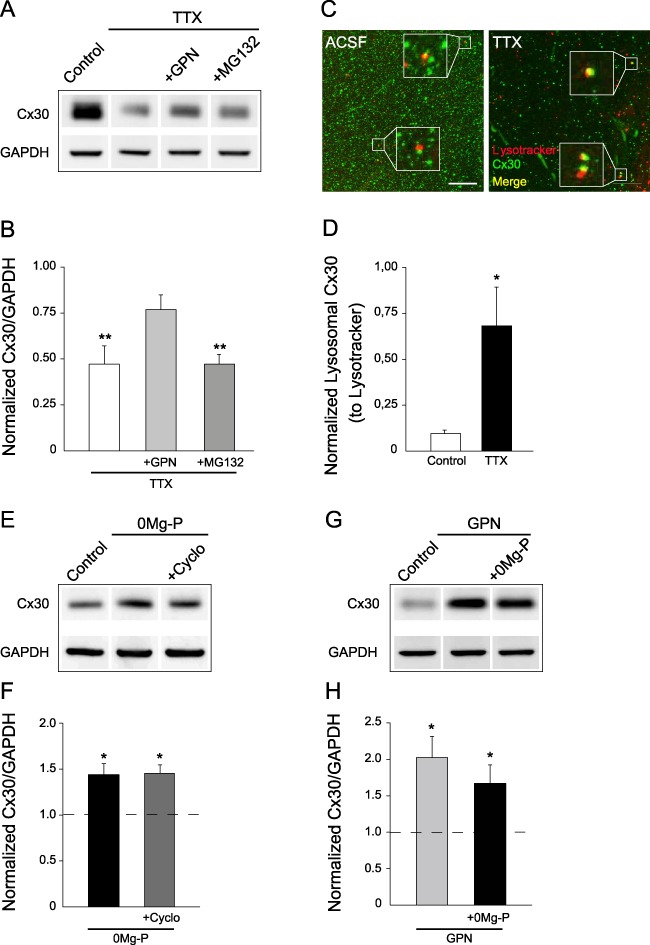
Neuronal activity controls Cx30 protein levels through lysosomal degradation. (A) Immunoblot detection of Cx30 expression in hippocampal acute slices incubated in TTX (0.5 μM, 1 h) with or without inhibitors of the lysosomal (GPN, 200 μM) or proteasomal (MG132, 10 μM) degradation pathways. GAPDH was used as a loading control. (B) Quantitative analysis of Cx30 expression showing that Cx30 levels were restored to control level by lysosomal inhibition (GPN, n = 5) but not by proteasomal inhibition (MG132, n = 5), thereby blocking the effects of action potential inhibition by TTX. Relative expression levels of Cx30 normalized to GAPDH levels; control ratio is set to 1. (C) Immunofluorescent staining of Cx30 (green) associated with LysoTracker labeling (red) in hippocampal CA1 region from acute slices incubated in ACSF or TTX (0.5 μM) for 1 h. Merge images show a net enrichment of Cx30 staining within lysosomes (yellow puncta) in TTX condition. Zooms are shown in white boxes for selected regions of interest highlighting LysoTracker-stained lysosomes contacting Cx30 aggregates. Scale bar, 25 μm. (D) Quantitative analysis of Cx30 intensity within lysosomes. Relative intensity levels of Cx30 staining within LysoTracker particles normalized to LysoTracker staining intensity in control (n = 9) and TTX (n = 9) conditions. (E) Immunoblot detection of Cx30 expression in hippocampal acute slices incubated in 0Mg-P (100 μM, 3 h) with or without cycloheximide, a protein synthesis inhibitor (Cyclo, 400 μM). GAPDH was used as a loading control. (F) Quantitative analysis of Cx30 expression. Relative expression levels of Cx30 normalized to GAPDH levels showing that the activity-dependent upregulation of Cx30 expression (0Mg-P, n = 3) was not blocked by inhibition of protein synthesis (Cyclo, n = 3); control ratio is set to 1. (G) Immunoblot detection of Cx30 expression in hippocampal acute slices incubated with a lysosomal inhibitor (GPN, 200 μM, 3 h) with or without 0Mg-P (100 μM). GAPDH was used as a loading control. (H) Quantitative analysis of Cx30 expression. Relative expression levels of Cx30 normalized to GAPDH levels, showing that blocking lysosomal degradation in control conditions (GPN, n = 5) increased by itself Cx30 expression, and to a similar extent as 0Mg-P. In addition, bursting activity did not further increase Cx30 levels (GPN + 0Mg-P, n = 8); control ratio is set to 1 (n = 3). Asterisks indicate statistical significance (*P < 0.05, **P < 0.01).
Altogether, these data suggest that neuronal activity bidirectionally controls Cx30 proteins levels through regulation of lysosomal degradation.
Activity-Dependent Subcellular Recruitment of Cx30
We investigated whether neuronal activity also altered the subcellular localization of Cx30. For this, we first examined the effect of bursting activity on Cx30 protein levels at membranes using a biotinylation assay in acute hippocampal slices. Strikingly, network activity induced a strong increase (~ + 300%, n = 3; Fig. 4A,B) in Cx30 cell surface expression compared to the enhancement of Cx30 total levels (~ + 35%, n = 3; Fig. 1B,C). We then determined the effect of bursting activity on Cx30 distribution throughout the domain of single astrocytes using confocal imaging of eGFP-labeled astrocytes in acute hippocampal slices of GFAP-eGFP mice (Nolte et al. 2001; Hirrlinger et al. 2004). Network activity induced an overall recruitment of Cx30 toward distal processes of astrocytes, as revealed by quantification of Cx30 (intensity and puncta number) radial distance from soma center (n = 9; Fig. 4C,E).
Figure 4.
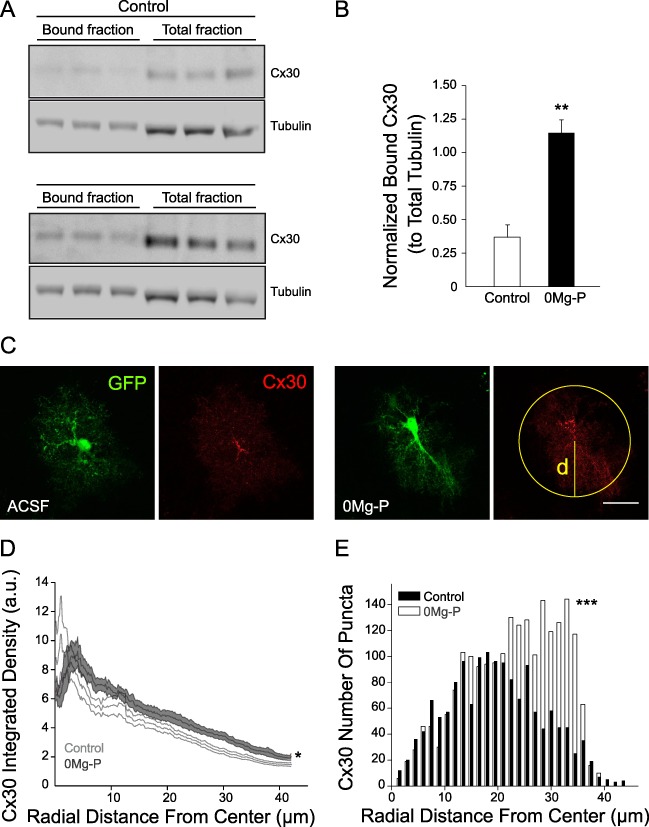
Cx30 subcellular localization is regulated by neuronal activity. (A) Activity-dependent Cx30 plasma membrane trafficking measured by hippocampal acute slices biotinylation. Immunoblot detection of total and surface Cx30 proteins in slices incubated in ACSF or 0Mg-P (100 μM, 3 h). Tubulin was used as a loading control. (B) Quantitative analysis of normalized surface Cx30 protein levels (n = 3). (C) Immunofluorescent staining for Cx30 (red) within eGFP-expressing hippocampal CA1 astrocytes (green) from acute slices incubated in ACSF or 0Mg-P (100 μM, 3 h). Cx30 intensity profile was determined at any given radial position as the sum of the pixel values around a circle centered at the cell soma, as shown in yellow. (D) Quantification of Cx30 radial intensity profile (n = 9 cells). Cx30 intensity was increased in distal astrocytes processes (~ 20–40 μm from the center of the cell soma) in 0Mg-P condition. Scale bar, 25 μm. Asterisks indicate statistical significance (*P < 0.05, **P < 0.01).
Because distal processes of astrocytes are sites of privileged interactions with synapses, we then determined whether neuronal activity altered Cx30 distribution in perisynaptic astroglial processes (PAPs). PAPs are the relevant nanodomains for morphological and functional neuroglial interactions, where synaptic information is integrated, processed, and modulated (Ghezali et al. 2016).
We first examined whether Cx30 is present in PAPs. Since PAPs copurify with synapses during synaptosome isolation (Chicurel et al. 1993; Supplementary Fig. S2), we purified hippocampal synaptosomes. The latter were characterized by an enrichment in synaptic markers such as PSD95 and synapsin I (syn I) but not actin (Fig. 5A). Hippocampal synaptosomes were closely associated with PAPs, as shown by the enrichment of the astroglial GLT1 glutamate transporters (total protein levels normalized to actin: synaptosomes: 9.7 ± 0.9, n = 6; whole hippocampi: 5.3 ± 0.9, n = 4; P = 0.0107, unpaired t-test) and the very low levels of the GFAP protein, known to be poorly expressed in PAPs (Lavialle et al. 2011; Carney et al. 2014; Fig. 5A). Remarkably, we found that Cx30 was also enriched in synaptosomal fractions (total protein levels normalized to actin: synaptosomes: 3.3 ± 0.6, n = 6; whole hippocampi: 1.3 ± 0.2, n = 4; P = 0.0262, unpaired t-test), suggesting that PAP membranes do contain high levels of Cx30. To directly visualize Cx30 in PAPs, we then used superresolution imaging of GFP-labeled astrocytes (GFAP-EGFP mice) in hippocampal slices immunostained with Cx30. Consistent with the detection of Cx30 in synaptosomal fractions, STED microscopy in hippocampal sections revealed that Cx30 was abundantly expressed throughout the arborization of fine astroglial processes (Fig. 5B). In addition, SIM, which enables rapid 3D multicolor fluorescence imaging in hippocampal slices, revealed that a significant subset of Cx30 puncta displayed close proximity to synapses (% of Cx30 within 350 nm of synaptic markers: VGLUT1: 11.1 ± 1.2%, n = 8 cells; PSD95: 12.5 ± 1.2%, n = 9 cells), with a similar distance to both presynaptic (VGLUT1) or postsynaptic (PSD95) elements (VGLUT1: 223.6 ± 2.5 nm, n = 1107 puncta; PSD95: 224.3 ± 2.4 nm, n = 1089 puncta, P > 0.05, unpaired t-test; Figs 5C,D and 6B,C). Taken together, these data reveal the presence of Cx30 in PAPs.
Figure 5.
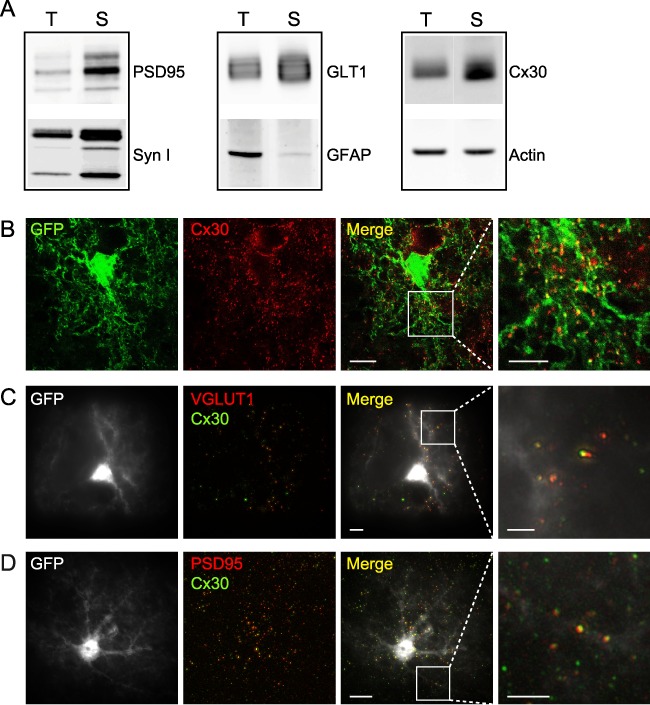
Cx30 is localized in distal perisynaptic astrocyte processes. (A) Western blotting detection of pre- (syn I) and post- (PSD95) synaptic proteins showed an enrichment of plasma membrane synaptic proteins in crude synaptosomal membrane fraction (S) compared to total hippocampal fraction (T), while actin did not change. Astroglial GLT1 glutamate transporters are also enriched in synaptosomal fractions, while GFAP protein levels are very low. Astroglial Cx30 is strongly expressed within crude synaptosomal membrane fraction (n = 6). (B) Confocal/STED images of GFAP-eGFP (green) mouse brain slices immunostained for Cx30 (red). The stack merged image reveals the localization of Cx30 STED-resolved puncta (ATTO 647, 1:250) within distal GFP-labeled astrocyte processes. Scale bar, 10 μM. (C) SIM images of GFAP-eGFP (white) mouse brain slices immunostained for VGLUT1 (red) and Cx30 (green). The single-plane merge image indicates occasional colocalization of Cx30 with the presynaptic protein VGLUT1. Scale bar, 10 μM. (D) SIM imaging of GFAP-eGFP (white) mouse brain slices immunostained for PSD95 (red) and Cx30 (green). The single-plane merge image reveals occasional colocalization of Cx30 with the postsynaptic protein PSD95. Scale bar, 10 μM. White squares in (B–D) indicate the small regions of interest that are magnified. Scale bar, 5 μM.
Figure 6.
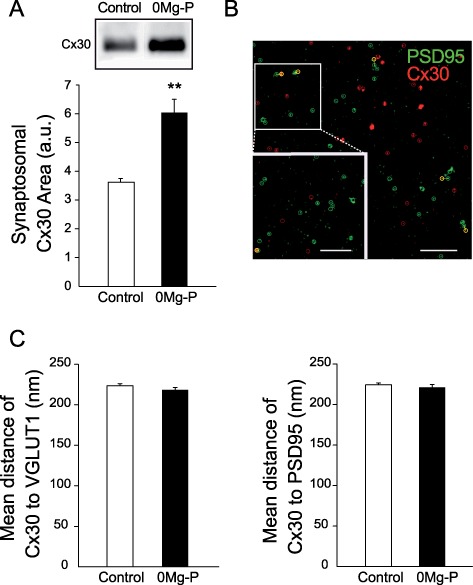
Neuronal activity drives Cx30 to PAPs. (A) Activity-dependent relocalization of astroglial Cx30 proteins assessed by western blotting of crude synaptosomal membrane fractions. Cx30 protein level in crude synaptosomal fraction was increased in 0Mg-P (100 μM, 3 h, n = 3). Quantitative analysis of Cx30 expression in crude synaptosomal membrane fraction. (B) Images showing colocalization analysis of Cx30 and PSD95 puncta using SIM. Scale bar, 2 μM. The white square indicates the magnified small region of interest. Scale bar, 1 μM. (C) Neuronal activity does not alter the distance of Cx30 puncta within PAPs to synapses. Quantitative analysis of the mean distance of Cx30 puncta to pre- (VGLUT1, control: n = 1107, 0Mg-P: n = 862) and postsynaptic (PSD95, control: n = 1089, 0Mg-P: n = 599) markers. Asterisks indicate statistical significance (**P < 0.01).
Remarkably, bursting activity altered the distribution of Cx30, whose level increased in synaptosomal fractions (+65.7 ± 11.5%, n = 3; Fig. 6A), indicating the recruitment in PAPs. However, bursting activity did not alter the distance of Cx30 puncta within PAPs to presynaptic or postsynaptic elements, as revealed by SIM (VGLUT1: 218.6 ± 2.9 nm, n = 862 puncta; PSD95: 221.3 ± 3.5 nm, n = 599 puncta; Fig. 6C).
Altogether, these data show that neuronal activity promotes Cx30 localization at astrocyte membranes and perisynaptic processes.
Neuronal Activity Differentially Regulates Cx30 Functions
Changes in protein expression and localization reportedly lead to functional alterations. We therefore investigated whether the increase of Cx30 protein levels induced by neuronal activity resulted at the functional level in changes in Cx30 channel and nonchannel functions. GJ communication of various neuroactive factors is a typical feature of astrocytes (Pannasch and Rouach 2013). We previously reported that intercellular diffusion of the fluorescent glucose derivative 2-[N-(7-nitrobenz-2-oxa-1,3-diazol-4-yl)amino]-2-deoxyglucose (2-NBDG) into neighboring astrocytes is partially mediated by GJ channels containing the Cx30 subunit and is activity dependent (Rouach et al. 2008). We thus assessed here the effect of bursting activity on Cx30-mediated astroglial GJ communication using a glucose coupling assay. As previously described (Rouach et al. 2008), we found that bursting activity increased the intercellular diffusion of 2-NBDG, dialyzed by patch-clamp recording of stratum radiatum astrocytes, into the GJ astroglial network (+35.8 ± 9.6%, n = 15, P = 0.0164, unpaired t-test; Fig. 7A,B). However, deletion of Cx30 (Cx30−/− mice) did not alter the activity-dependent increase in 2-NBDG trafficking into the GJ-mediated astroglial network (+37.9 ± 9.8%, n = 6, P > 0.05, unpaired t-test; Fig. 7A,B). These data therefore suggest that changes in Cx30 expression and distribution do not contribute to the activity-dependent increase in astroglial GJ-mediated glucose trafficking, which may rather be mediated by Cx43, the other main astrocytic GJ channel subunit.
Figure 7.
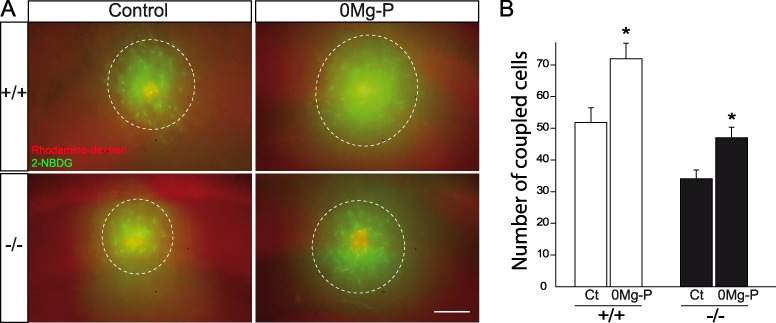
Cx30 GJ channels do not contribute to the activity-dependent increase in glucose trafficking within astroglial networks. (A) Sample images of functional coupling between CA1 astrocytes visualized by diffusion of 2-NBDG (green) via patch-clamp whole-cell recording for 20 min, together with dextran tetramethylrhodamine (red) to identify the recorded astrocyte. The upregulation of 2-NBDG interastrocytic trafficking induced by 0Mg-P (100 μM, 3 h) is independent of Cx30 GJ channels as it is unchanged in Cx30−/− mice. Scale bar, 100 μm. (B) Graph summarizing the extent of 2-NBDG astrocytic coupling during spontaneous and neuronal bursting conditions (+/+: Control (Ct): n = 21, 0Mg-P: n = 15; −/−: Ct: n = 11, 0Mg-P: n = 6). Asterisks indicate statistical significance (*P < 0.05).
In various cell types, Cx30 can also form HCs, which mediate direct exchange with the extracellular space (Majumder et al. 2010; Svenningsen et al. 2013). Although functional Cx30 HCs have not yet been described in astrocytes, we investigated whether bursting activity may induce their activation. An EtBr uptake assay in stratum radiatum astrocytes from wild-type acute hippocampal slices showed that the majority of S100β-positive astrocytes exhibited a basal EtBr labeling in control conditions (Fig. 8A). Such labeling is currently thought to be partly mediated (~30%) by active uptake through Cx43 HCs in basal conditions (Chever et al. 2014). We found that bursting activity increased activation of Cx HCs by ~ 55% (+ 56.8 ± 25.2%, n = 9, P = 0.0412, unpaired t-test; Fig. 8B,C), as suggested by inhibition of the EtBr uptake increase by action potentials blockade with TTX (0.5 μM, 1 h; n = 12) or by HCs blockade with CBX (200 μM, 15 min; n = 12; Fig. 8B,C). Remarkably, knocking out Cx30 in mice also fully inhibited the activity-dependent increase in HCs activity (n = 16; Fig. 8B,C), suggesting that bursting activated Cx30 HCs in astrocytes.
Figure 8.
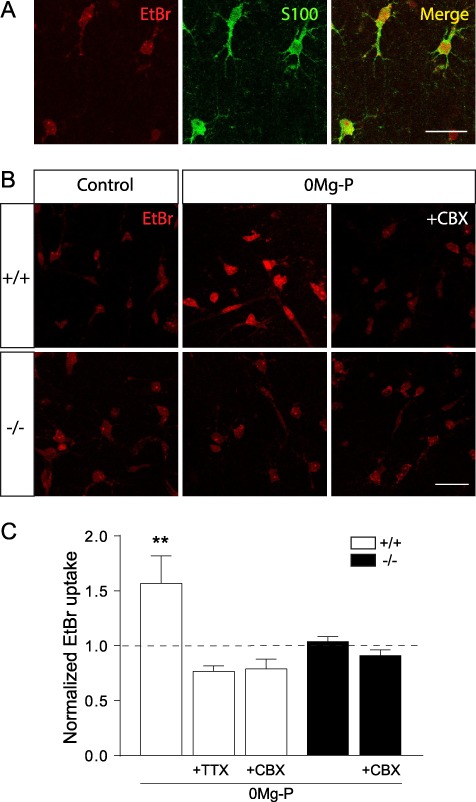
Bursting induces activation of astroglial Cx30 HCs. (A) Representative basal EtBr (red) in CA1 stratum radiatum astrocytes labeled with S100β (green) in hippocampal slices. Scale bar, 25 μm. (B) Representative pictures showing that EtBr uptake through Cx HCs is increased in 0Mg-P (100 μM, 3 h) compared to control conditions, as this effect is abolished by CBX (200 μM, 15 min) in wild-type mice (+/+). Cx30 is mediating the activity-dependent increase in astroglial EtBr uptake, as such increase is inhibited in Cx30−/− mice (−/−). (C) Quantitative analysis of astroglial EtBr uptake normalized to basal condition in acute hippocampal slices treated with 0Mg-P from wild-type (+/+: Ct, n = 12; 0Mg-P, n = 9; 0Mg-P + TTX, n = 12; 0Mg-P + CBX, n = 12) and Cx30−/− mice (−/−: Ct, n = 16; 0Mg-P, n = 16; 0Mg-P + CBX, n = 17). Asterisks indicate statistical significance (**P < 0.01).
Cx30 also exhibits functions independent of GJ biochemical coupling, which regulate the morphology of astrocytes by controlling the ramification of their distal processes (Pannasch et al. 2014). We therefore examined whether bursting activity also altered ramification of astrocytes. As previously described (Pannasch et al. 2014), we observed an enhancement in the ramification of astroglial distal processes from Cx30−/− mice in control conditions (+/+: 13.1 ± 1.1, n = 14 vs. −/−: 19.3 ± 2, n = 17 for processes located 22 μm from the soma, P = 0.0109, unpaired t-test Welch corrected; +/+: 8.9 ± 0.8, n = 14 vs. −/−: 15.6 ± 1.7, n = 17 for processes located at 27 μm from the soma, P = 0.0015, unpaired t-test Welch corrected). We found that activity enhanced ramification of distal astroglial processes, as a Sholl analysis revealed a selective increase in the number of processes located at large distances from the soma (+23.2 ± 5.8%, P = 0.0294 and + 41.9 ± 7.9%, P = 0.0026, n = 14 for ACSF and n = 21 for 0Mg-P, Mann–Whitney test, for processes located at 22 μm and 27 μm from the soma, respectively; Fig. 9A,B). Cx30 was involved in this effect, as its deletion inhibited the activity-dependent ramification of astroglial processes (n = 17; Fig. 9A,B). To determine whether the activity-dependent control of astroglial ramification mediated by Cx30 relied on its GJ-mediated biochemical coupling, we investigated astrocyte ramification in Cx30 T5M mice, in which the replacement of a threonine by a methionine at position 5 of Cx30 leads to defective channel pores inhibiting intercellular biochemical coupling with no alteration in membrane targeting (Grifa et al. 1999; Schutz et al. 2010). We found that, despite the loss of Cx30 channel-mediated biochemical coupling, Cx30T5M mice still displayed the activity-dependent ramification of astrocyte distal processes (n = 16; Fig. 9B).
Figure 9.
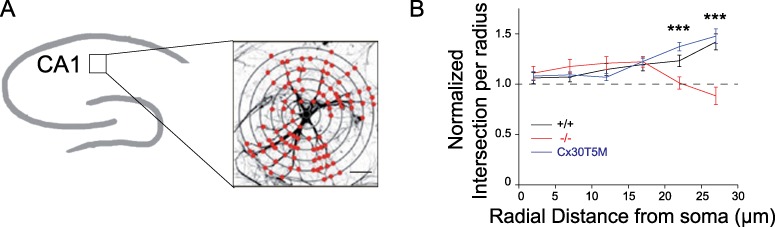
Cx30 controls the activity-dependent remodeling of distal astroglial processes independently of GJ-mediated biochemical coupling. (A) Schematic representation of Sholl analysis for intersection quantification in a GFAP-labeled astrocyte from the hippocampal CA1 area. Scale bar, 10 μm. (B) Quantification of the branching pattern of CA1 hippocampal astrocytes normalized to control condition in acute slices treated by 0Mg-P (100 μM, 3 h) from wild-type (n = 21), Cx30T5M (n = 14), and Cx30−/− (n = 17) mice. Neuronal bursting increased GFAP branching within almost all concentric radii between 18 and 28 μm in wild-type astrocytes, while this activity-dependent remodeling of astroglial distal processes was inhibited Cx30−/− mice but not in Cx30T5M mice. Asterisks indicate statistical significance (***P < 0.001).
Altogether, these data indicate that neuronal activity promotes Cx30 functions independent of GJ biochemical coupling.
Discussion
Our study reveals that Cx30 expression, distribution, and functional properties are activity dependent. Indeed, we found that neuronal activity tightly controls Cx30 protein levels via calcium signaling and inhibition of lysosomal degradation. Moreover, network bursting orchestrates Cx30 localization by promoting expression at astroglial membranes and perisynaptic processes. This resulted in specific regulation of Cx30 functions, as bursting activity induced activation of Cx30 HCs, and promoted structural remodeling of astroglial processes independently of Cx30 GJ-mediated biochemical coupling.
Activity-Dependent Regulation of Cx30 Protein Levels
We show here that spontaneous spiking activity, as well as network bursting, upregulates Cx30 protein levels. This novel activity-dependent regulation of Cx30 is mediated by a posttranslational mechanism, which differs from the transcriptional controls of Cx30 previously reported (Rampon et al. 2000; Princen et al. 2001; Condorelli et al. 2002; Bernard et al. 2011; Ernst et al. 2011; Liu et al. 2013). Several mechanisms thus control Cx30 abundance at the mRNA and protein levels, suggesting that tight and dynamic regulations of Cx30 play important roles in normal and pathological conditions. Interestingly, we found that the increase in Cx30 expression is mediated by an mGluR signaling pathway. This suggests that Cx30 expression may be specifically regulated by excitatory transmission through activation of astroglial mGluRs coupled to intracellular calcium signaling and downstream pathways. This hypothesis is consistent with our finding that Cx30 control of neurotransmission also appears to be selective of excitatory synapses (Pannasch et al. 2014). Astroglial Cx30 indeed alters excitatory synaptic transmission in hippocampal CA1 pyramidal cells by controlling the insertion of astroglial processes into synaptic clefts but has no effect on inhibitory synaptic transmission (Pannasch et al. 2014). Taken together, our findings suggest mutual regulations between glutamatergic activity and astroglial Cx30. Noteworthy, Cx30 transcription has also been reported to be calcium dependent in the inner ear, through a P2Y-mediated IP3 pathway (Ortolano et al. 2008). However, in our study, P2YRs were not involved in the activity-dependent regulation of Cx30 levels.
We also show here that neuronal bursting regulates Cx30 protein levels by inhibiting its degradation via lysosomal pathways and not by promoting its synthesis. This is in line with the long lifespan (>12 h) of Cx30 (Kelly et al. 2015) and previous data showing that Cxs are indeed degraded by lysosomal pathways (Falk et al. 2014). Since Cxs have also been reported to be degraded by the proteasomal and autophagosomal pathways, our data suggest that neuronal activity specifically alters the lysosomal degradation pathway. This likely occurs through the activity-dependent astroglial calcium signaling mediated by IP3 receptors, as lysosomal function critically depends on lysosome uptake of calcium released by IP3 receptors from ER (Lopez Sanjurjo et al. 2014; Garrity et al. 2016).
Activity-Dependent Recruitment of Astroglial Cx30
Bursting activity redistributed Cx30 to specific astroglial compartments. Cx30 indeed accumulated at plasma membranes, suggesting its functional activation. Cx30 levels were also enriched in perisynaptic processes, the relevant nanodomains for functional neuroglial interactions. Since one important function of Cx30 is to regulate astroglial synapse coverage (Pannasch et al. 2014), enhanced levels of Cx30 in PAPs may serve to locally decrease astroglial contacts with synapses. Such reduced coverage should promote excitatory synaptic efficacy, as Cx30 ensures efficient transmission by preventing astrocytic processes from entering synaptic clefts and clearing glutamate (Pannasch et al. 2014). This hypothesis is supported by recent studies showing that hippocampal long-term potentiation (LTP) decreases astrocytic coverage of dendrites (Perez-Alvarez et al. 2014) and that consolidation of contextual memory decreases astroglial synapse coverage in the amygdala (Ostroff et al. 2014). Overall, these findings suggest reciprocal regulations between glutamatergic activity and astroglial Cx30, since enhanced excitatory activity may promote Cx30 protein levels, which would in turn enhance excitatory activity. Interestingly, this positive feedback loop may sustain the development of aberrant bursting leading to epileptiform activity, and chronic seizures. This hypothesis is consistent with the upregulation of Cx30 expression in kainate or kindling models of epilepsy (Condorelli et al. 2002; Akbarpour et al. 2012).
Neuronal Activity Differentially Regulates Cx30 Functions
We show here that neuronal activity promoted several Cx30 functions independent of GJ biochemical coupling, namely Cx30-mediated HC activity and remodeling of astroglial morphology. However, neuronal activity did not alter Cx30-mediated GJ metabolic coupling, since Cx30-based GJ channels did not contribute to the activity-dependent increase in glucose trafficking into astroglial networks. This suggests that it is the regulation of Cx43, but not Cx30, that is involved in the activity-dependent plasticity of astroglial metabolic networks.
Our data show for the first time that Cx30 HCs are functional in astrocytes and open as a result of bursting activity. This is consistent with findings reporting on the activity of Cx30 HCs in various cell types (Majumder et al. 2010; Svenningsen et al. 2013) and the opening of Cx HCs by calcium signaling (Decrock et al. 2011). The differential regulation of Cx30 GJ and HC functions by neuronal activity is also consistent with several studies reporting activation of Cx HCs with no alteration or opposite regulation of GJ function (Retamal et al. 2007; Abudara et al. 2015). This further suggests that the activity-dependent regulatory pathway differentially affects Cx30 channel functions. Cx hexamers can form functional HCs, as well as GJ channels, by docking of HCs from adjacent cells. Neuronal activity is thus likely to control the fate of the Cx hexamer pool toward functional HCs rather than GJ channels through posttranslational regulations that may alter their assembly, trafficking, or activity.
Recent data show that Cx43 HCs can be active in both physiological (Stehberg et al. 2012; Chever et al. 2014) and pathological conditions (Giaume et al. 2013). Cx30 HCs are likely activated by sustained neuronal activity as we show here that they are opened in response to network bursting, while previously we showed that Cx30 HCs are not active in basal conditions (Chever et al. 2014; Pannasch et al. 2014). Moreover, Cx30 HCs may mediate specific signaling as the permeability of Cx30 and Cx43 HCs to several physiologically relevant molecules such as glutamate or glucose, differ (Hansen et al. 2014). Since Cx HCs can release or uptake various neuroactive substances, their opening should efficiently modulate the activity of adjacent synapses. This would particularly hold true at perisynaptic processes, as suggested by the activity-dependent recruitment of Cx30 in PAPs. However, there is presently no tool to specifically inhibit Cx30 HCs without altering GJ coupling. Thus, addressing the specific role of Cx30 HCs in neuroglial signaling will require the development of pharmacological or molecular tools targeting selectively Cx30 connexons.
Cx30 interacts with elements of the cytoskeleton (Qu et al. 2009), and its function, independent of GJ biochemical coupling, has been shown to regulate cell morphology, adhesion, and migration (Pannasch et al. 2014). We demonstrate here that bursting activity also induced a Cx30-mediated remodeling of astroglial morphology independently of GJ biochemical intercellular coupling. We found that Cx30 controlled the increased ramification of astrocytes induced by bursting epileptiform activity. This may translate into loss of astroglial domain organization, as reported in several mice models of epilepsy (Oberheim et al. 2008). Such structural alteration most probably perturbs proper synaptic processing within astroglial territories.
Altogether, these data reveal that astroglial Cx30 is endogenously regulated by neuronal activity, especially in perisynaptic nanodomains regulating the moment-to-moment synaptic transmission. Such regulations may thus actively shape synaptic network activity underlying physiological brain rhythms or aberrant bursting during epilepsy.
Supplementary Material
Funding
European Union’s Horizon 2020, European Research Council (COG-683154) (N.R.); Marie Sklodowska-Curie Research and Innovation Programme EU-GliaPhD (722053) (N.R.); Agence Nationale de la Recherche (ANR-15-CE16-0001) (N.R.); City of Paris (Emergence Program) (N.R.); Institut National de la Sante et de la Recherche Medicale (N.R.); Centre National de la Recherche Scientifique (N.R.); College de France (N.R.); Paris 6 University Doctoral School Ecole Doctorale Cerveau, Cognition, Comportement (G.G.); Labex Memolife (G.G. and F.V.); Observatoire B2V des Memoires (F.V.); UK Engineering and Physical Science Research Council; Infinitus China, Ltd; Wellcome Trust (C.F.K.); Medical Research Council (C.F.K.).
Notes
We thank F. Mammano for providing the Cx30T5M mice and J. Chen for providing the IP3R2−/− mice. Conflict of Interest: None declared.
References
- Abudara V, Roux L, Dallerac G, Matias I, Dulong J, Mothet JP, Rouach N, Giaume C. 2015. Activated microglia impairs neuroglial interaction by opening Cx43 hemichannels in hippocampal astrocytes. Glia. 63:795–811. [DOI] [PubMed] [Google Scholar]
- Akbarpour B, Sayyah M, Babapour V, Mahdian R, Beheshti S, Kamyab AR. 2012. Expression of connexin 30 and connexin 32 in hippocampus of rat during epileptogenesis in a kindling model of epilepsy. Neurosci Bull. 28:729–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Araque A, Carmignoto G, Haydon PG, Oliet SH, Robitaille R, Volterra A. 2014. Gliotransmitters travel in time and space. Neuron. 81:728–739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bazargani N, Attwell D. 2016. Astrocyte calcium signaling: the third wave. Nat Neurosci. 19:182–189. [DOI] [PubMed] [Google Scholar]
- Bernard R, Kerman IA, Thompson RC, Jones EG, Bunney WE, Barchas JD, Schatzberg AF, Myers RM, Akil H, Watson SJ. 2011. Altered expression of glutamate signaling, growth factor, and glia genes in the locus coeruleus of patients with major depression. Mol Psychiatry. 16:634–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernardinelli Y, Randall J, Janett E, Nikonenko I, Konig S, Jones EV, Flores CE, Murai KK, Bochet CG, Holtmaat A et al. . 2014. Activity-dependent structural plasticity of perisynaptic astrocytic domains promotes excitatory synapse stability. Curr Biol. 24:1679–1688. [DOI] [PubMed] [Google Scholar]
- Boulay AC, del Castillo FJ, Giraudet F, Hamard G, Giaume C, Petit C, Avan P, Cohen-Salmon M. 2013. Hearing is normal without connexin30. J Neurosci. 33:430–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carney KE, Milanese M, van Nierop P, Li KW, Oliet SH, Smit AB, Bonanno G, Verheijen MH. 2014. Proteomic analysis of gliosomes from mouse brain: identification and investigation of glial membrane proteins. J Proteome Res. 13:5918–5927. [DOI] [PubMed] [Google Scholar]
- Chever O, Dossi E, Pannasch U, Derangeon M, Rouach N. 2016. Astroglial networks promote neuronal coordination. Sci Signal. 9:ra6. [DOI] [PubMed] [Google Scholar]
- Chever O, Lee CY, Rouach N. 2014. Astroglial connexin43 hemichannels tune basal excitatory synaptic transmission. J Neurosci. 34:11228–11232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chicurel ME, Terrian DM, Potter H. 1993. mRNA at the synapse: analysis of a synaptosomal preparation enriched in hippocampal dendritic spines. J Neurosci. 13:4054–4063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke LE, Barres BA. 2013. Emerging roles of astrocytes in neural circuit development. Nat Rev Neurosci. 14:311–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Condorelli DF, Mudo G, Trovato-Salinaro A, Mirone MB, Amato G, Belluardo N. 2002. Connexin-30 mRNA is up-regulated in astrocytes and expressed in apoptotic neuronal cells of rat brain following kainate-induced seizures. Mol Cell Neurosci. 21:94–113. [DOI] [PubMed] [Google Scholar]
- Dallerac G, Chever O, Rouach N. 2013. How do astrocytes shape synaptic transmission? Insights from electrophysiology. Front Cell Neurosci. 7:159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Chaumont F, Dallongeville S, Chenouard N, Herve N, Pop S, Provoost T, Meas-Yedid V, Pankajakshan P, Lecomte T, Le Montagner Y et al. . 2012. Icy: an open bioimage informatics platform for extended reproducible research. Nat Methods. 9:690–696. [DOI] [PubMed] [Google Scholar]
- Decrock E, Vinken M, Bol M, D'Herde K, Rogiers V, Vandenabeele P, Krysko DV, Bultynck G, Leybaert L. 2011. Calcium and connexin-based intercellular communication, a deadly catch? Cell Calcium. 50:310–321. [DOI] [PubMed] [Google Scholar]
- Dere E, De Souza-Silva MA, Frisch C, Teubner B, Sohl G, Willecke K, Huston JP. 2003. Connexin30-deficient mice show increased emotionality and decreased rearing activity in the open-field along with neurochemical changes. Eur J Neurosci. 18:629–638. [DOI] [PubMed] [Google Scholar]
- Elias LA, Kriegstein AR. 2008. Gap junctions: multifaceted regulators of embryonic cortical development. Trends Neurosci. 31:243–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernst C, Nagy C, Kim S, Yang JP, Deng X, Hellstrom IC, Choi KH, Gershenfeld H, Meaney MJ, Turecki G. 2011. Dysfunction of astrocyte connexins 30 and 43 in dorsal lateral prefrontal cortex of suicide completers. Biol Psychiatry. 70:312–319. [DOI] [PubMed] [Google Scholar]
- Falk MM, Kells RM, Berthoud VM. 2014. Degradation of connexins and gap junctions. FEBS Lett. 588:1221–1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabriel LR, Wu S, Melikian HE. 2014. Brain slice biotinylation: an ex vivo approach to measure region-specific plasma membrane protein trafficking in adult neurons. J Vis Exp 86:51240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrity AG, Wang W, Collier CM, Levey SA, Gao Q, Xu H. 2016. The endoplasmic reticulum, not the pH gradient, drives calcium refilling of lysosomes. Elife. 5:e15887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghezali G, Dallerac G, Rouach N. 2016. Perisynaptic astroglial processes: dynamic processors of neuronal information. Brain Struct Funct. 221:2427–2442. [DOI] [PubMed] [Google Scholar]
- Giaume C, Leybaert L, Naus CC, Saez JC. 2013. Connexin and pannexin hemichannels in brain glial cells: properties, pharmacology, and roles. Front Pharmacol. 4:88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grifa A, Wagner CA, D'Ambrosio L, Melchionda S, Bernardi F, Lopez-Bigas N, Rabionet R, Arbones M, Monica MD, Estivill X et al. . 1999. Mutations in GJB6 cause nonsyndromic autosomal dominant deafness at DFNA3 locus. Nat Genet. 23:16–18. [DOI] [PubMed] [Google Scholar]
- Hansen DB, Braunstein TH, Nielsen MS, MacAulay N. 2014. Distinct permeation profiles of the connexin 30 and 43 hemichannels. FEBS Lett. 588:1446–1457. [DOI] [PubMed] [Google Scholar]
- Hirrlinger J, Hulsmann S, Kirchhoff F. 2004. Astroglial processes show spontaneous motility at active synaptic terminals in situ. Eur J Neurosci. 20:2235–2239. [DOI] [PubMed] [Google Scholar]
- Kelly JJ, Shao Q, Jagger DJ, Laird DW. 2015. Cx30 exhibits unique characteristics including a long half-life when assembled into gap junctions. J Cell Sci. 128:3947–3960. [DOI] [PubMed] [Google Scholar]
- Kunzelmann P, Schroder W, Traub O, Steinhauser C, Dermietzel R, Willecke K. 1999. Late onset and increasing expression of the gap junction protein connexin30 in adult murine brain and long-term cultured astrocytes. Glia. 25:111–119. [DOI] [PubMed] [Google Scholar]
- Lavialle M, Aumann G, Anlauf E, Prols F, Arpin M, Derouiche A. 2011. Structural plasticity of perisynaptic astrocyte processes involves ezrin and metabotropic glutamate receptors. Proc Natl Acad Sci U S A. 108:12915–12919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Zima AV, Sheikh F, Blatter LA, Chen J. 2005. Endothelin-1-induced arrhythmogenic Ca2+ signaling is abolished in atrial myocytes of inositol-1,4,5-trisphosphate(IP3)-receptor type 2-deficient mice. Circ Res. 96:1274–1281. [DOI] [PubMed] [Google Scholar]
- Liu X, Petit JM, Ezan P, Gyger J, Magistretti P, Giaume C. 2013. The psychostimulant modafinil enhances gap junctional communication in cortical astrocytes. Neuropharmacology. 75:533–538. [DOI] [PubMed] [Google Scholar]
- Lopez Sanjurjo CI, Tovey SC, Taylor CW. 2014. Rapid recycling of Ca2+ between IP3-sensitive stores and lysosomes. PLoS One. 9:e111275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahou P, Curry N, Pinotsi D, Kaminski Schierle G, Kaminski CF. 2015. Stimulated emission depletion microscopy to study amyloid fibril formation. Proc. SPIE. 9331, Single Molecule Spectroscopy and Superresolution Imaging VIII. 9331OU.(San Francisco, USA).
- Majumder P, Crispino G, Rodriguez L, Ciubotaru CD, Anselmi F, Piazza V, Bortolozzi M, Mammano F. 2010. ATP-mediated cell–cell signaling in the organ of Corti: the role of connexin channels. Purinergic Signal. 6:167–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nolte C, Matyash M, Pivneva T, Schipke CG, Ohlemeyer C, Hanisch UK, Kirchhoff F, Kettenmann H. 2001. GFAP promoter-controlled EGFP-expressing transgenic mice: a tool to visualize astrocytes and astrogliosis in living brain tissue. Glia. 33:72–86. [PubMed] [Google Scholar]
- O'Holleran K, Shaw M. 2014. Optimized approaches for optical sectioning and resolution enhancement in 2D structured illumination microscopy. Biomed Opt Express. 5:2580–2590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oberheim NA, Tian GF, Han X, Peng W, Takano T, Ransom B, Nedergaard M. 2008. Loss of astrocytic domain organization in the epileptic brain. J Neurosci. 28:3264–3276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortolano S, Di Pasquale G, Crispino G, Anselmi F, Mammano F, Chiorini JA. 2008. Coordinated control of connexin 26 and connexin 30 at the regulatory and functional level in the inner ear. Proc Natl Acad Sci U S A. 105:18776–18781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostroff LE, Manzur MK, Cain CK, Ledoux JE. 2014. Synapses lacking astrocyte appear in the amygdala during consolidation of Pavlovian threat conditioning. J Comp Neurol. 522:2152–2163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pannasch U, Derangeon M, Chever O, Rouach N. 2012a. Astroglial gap junctions shape neuronal network activity. Commun Integr Biol. 5:248–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pannasch U, Freche D, Dallerac G, Ghezali G, Escartin C, Ezan P, Cohen-Salmon M, Benchenane K, Abudara V, Dufour A et al. . 2014. Connexin 30 sets synaptic strength by controlling astroglial synapse invasion. Nat Neurosci. 17:549–558. [DOI] [PubMed] [Google Scholar]
- Pannasch U, Rouach N. 2013. Emerging role for astroglial networks in information processing: from synapse to behavior. Trends Neurosci. 36:405–417. [DOI] [PubMed] [Google Scholar]
- Pannasch U, Sibille J, Rouach N. 2012b. Dual electrophysiological recordings of synaptically-evoked astroglial and neuronal responses in acute hippocampal slices. J Vis Exp. 69:e4418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pannasch U, Vargova L, Reingruber J, Ezan P, Holcman D, Giaume C, Sykova E, Rouach N. 2011. Astroglial networks scale synaptic activity and plasticity. Proc Natl Acad Sci U S A. 108:8467–8472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perea G, Sur M, Araque A. 2014. Neuron-glia networks: integral gear of brain function. Front Cell Neurosci. 8:378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Alvarez A, Navarrete M, Covelo A, Martin ED, Araque A. 2014. Structural and functional plasticity of astrocyte processes and dendritic spine interactions. J Neurosci. 34:12738–12744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Princen F, Robe P, Gros D, Jarry-Guichard T, Gielen J, Merville MP, Bours V. 2001. Rat gap junction connexin-30 inhibits proliferation of glioma cell lines. Carcinogenesis. 22:507–513. [DOI] [PubMed] [Google Scholar]
- Qu C, Gardner P, Schrijver I. 2009. The role of the cytoskeleton in the formation of gap junctions by Connexin 30. Exp Cell Res. 315:1683–1692. [DOI] [PubMed] [Google Scholar]
- Rampon C, Jiang CH, Dong H, Tang YP, Lockhart DJ, Schultz PG, Tsien JZ, Hu Y. 2000. Effects of environmental enrichment on gene expression in the brain. Proc Natl Acad Sci U S A. 97:12880–12884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Retamal MA, Froger N, Palacios-Prado N, Ezan P, Saez PJ, Saez JC, Giaume C. 2007. Cx43 hemichannels and gap junction channels in astrocytes are regulated oppositely by proinflammatory cytokines released from activated microglia. J Neurosci. 27:13781–13792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rouach N, Koulakoff A, Abudara V, Willecke K, Giaume C. 2008. Astroglial metabolic networks sustain hippocampal synaptic transmission. Science. 322:1551–1555. [DOI] [PubMed] [Google Scholar]
- Schutz M, Scimemi P, Majumder P, De Siati RD, Crispino G, Rodriguez L, Bortolozzi M, Santarelli R, Seydel A, Sonntag S et al. . 2010. The human deafness-associated connexin 30 T5M mutation causes mild hearing loss and reduces biochemical coupling among cochlear non-sensory cells in knock-in mice. Hum Mol Genet. 19:4759–4773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srinivasan R, Huang BS, Venugopal S, Johnston AD, Chai H, Zeng H, Golshani P, Khakh BS. 2015. Ca(2+) signaling in astrocytes from Ip3r2(−/−) mice in brain slices and during startle responses in vivo. Nat Neurosci. 18:708–717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stehberg J, Moraga-Amaro R, Salazar C, Becerra A, Echeverria C, Orellana JA, Bultynck G, Ponsaerts R, Leybaert L, Simon F et al. . 2012. Release of gliotransmitters through astroglial connexin 43 hemichannels is necessary for fear memory consolidation in the basolateral amygdala. FASEB J. 26:3649–3657. [DOI] [PubMed] [Google Scholar]
- Ströhl F, Kaminski CF. 2015. A joint Richardson—Lucy deconvolution algorithm for the reconstruction of multifocal structured illumination microscopy data. Methods Appl Fluoresc. 3:014002. [DOI] [PubMed] [Google Scholar]
- Ströhl F, Kaminski CF. 2016. Frontiers in structured illumination microscopy. Optica. 3:667–677. [Google Scholar]
- Svenningsen P, Burford JL, Peti-Peterdi J. 2013. ATP releasing connexin 30 hemichannels mediate flow-induced calcium signaling in the collecting duct. Front Physiol. 4:292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theis M, Sohl G, Eiberger J, Willecke K. 2005. Emerging complexities in identity and function of glial connexins. Trends Neurosci. 28:188–195. [DOI] [PubMed] [Google Scholar]
- Wallraff A, Kohling R, Heinemann U, Theis M, Willecke K, Steinhauser C. 2006. The impact of astrocytic gap junctional coupling on potassium buffering in the hippocampus. J Neurosci. 26:5438–5447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young LJ, Strohl F, Kaminski CF. 2016. A guide to structured illumination TIRF microscopy at high speed with multiple colors. J Vis Exp. 111:53988. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.