

Abstract
An intermolecular, photocatalytic dicarbofunctionalization (DCF) of olefins enabled by the merger of Giese-type addition with Ni/photoredox dual catalysis has been realized. Capitalizing on the rapid addition of 3° radicals to alkenes and their reluctance toward single electron metalation to Ni complexes, regioselective alkylation and arylation of olefins is possible. This dual catalytic method not only permits elaborate species to be assembled from commodity materials, but also allows quaternary and tertiary centers to be installed in a singular, chemoselective olefin difunctionalization. This multi-component process occurs under exceptionally mild conditions, compatible with a diverse range of functional groups and synthetic handles such as pinacolboronate esters. This technology was directly applied to the synthesis of an intermediate to a preclinical candidate (TK-666) and its derivatives.
Graphical Abstract
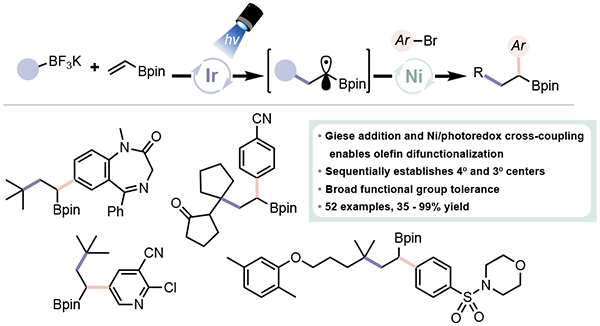
Introduction
Alkenes are versatile commodity feedstocks that can be rapidly converted into functional group-rich species via classical methods (Figure 1).1 They can be readily diversified using an array of vicinal difunctionalization reactions to establish 1,2-substituted alkanes (in some cases with excellent stereochemical fidelity). An attractive subset of vicinal functionalization processes that is of increasing focus are vicinal alkene dicarbofunctionalization (DCF) reactions. DCF allows olefins to be appended to two different carbon fragments, thus enabling a dramatic increase in molecular complexity. Classically, DCF has been accomplished with organometallic nucleophiles and electrophilic olefins or via cycloaddition onto activated alkenes (e.g., Diels-Alder reactions).2 Recently, methods for olefin diarylation or arylation/alkylation via palladium- or nickel catalysis have been developed.3 Although milestones in DCF, these protocols typically rely on stoichiometric reductants to sustain catalytic cycles and, as such, are less than ideal. Further, many still rely on organometallics that display sensitivities to a variety of functional groups. In addition, Csp3-hybridized nucleophiles are rare in transition metal-mediated DCF because of their tendency to undergo facile β-hydride elimination, protonation, and/or dimerization after metalation.4 Additionally, recent advances in radical chemistry have facilitated other DCF approaches (Figure 1).5
Figure 1.
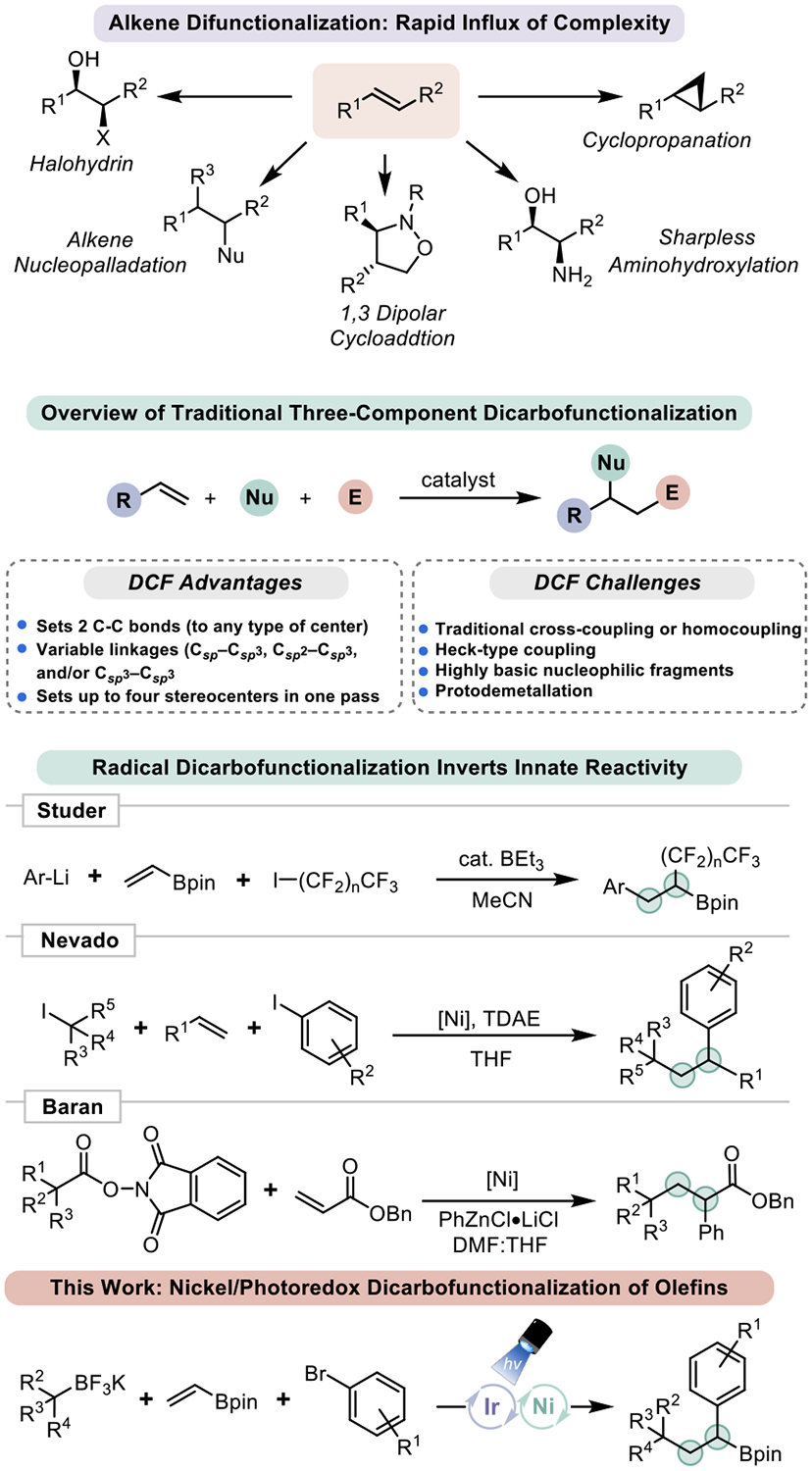
Utility of olefin difunctionalization reactions and comparison of the prior art to Ni/photoredox DCF.
Alkyl radicals can be viewed as surrogates of Csp3 -hybridized nucleophiles that avoid off-target polar reactivity while retaining the ability to form C–C bonds with ole-fins.6 Further, alkyl radicals can be engaged by Ni in place of more traditional Pd catalysts for DCF, mitigating the propensity of β-hydride elimination.7 With these aspects in mind and the reliability of generating alkyl radicals using visible light-activated catalysts,8 the merger of Ni/photoredox dual catalysis9 and Giese-type addition for DCF was considered.10
The crux of this approach is a series of well-orchestrated, radical-mediated bond-forming processes. Specifically, the following series of events (Figure 2) was envisioned: (1) visible light-mediated photoexcitation of the appropriate photocatalyst I to its excited state II; (2) reductive quenching via SET oxidation of radical precursor IV, (3) homolytic fragmentation of IV followed by Giese-type addition11 of the resulting radical V onto an olefin VI to generate VII; (4) Single-electron metalation of VII onto Ni0 species VIII to generate alkylnickel(I) adduct IX;12 (5) Oxidative addition of aryl halides X onto IX to give NiIII complex XI; (6) Reductive elimination to give DCF product XII and NiI species XIII, the latter of which undergoes SET reduction by III to regenerate VIII. Importantly, the process is contingent on radical V adding selectively to the olefin rather than engaging in direct cross-coupling via single electron metalation to Ni0 species VIII. Speculatively, ligation of the olefin VI to Ni0 could enable radical capture and metalation without formally proceeding through VII, thus permitting greater diversity of radical structure.
Figure 2.
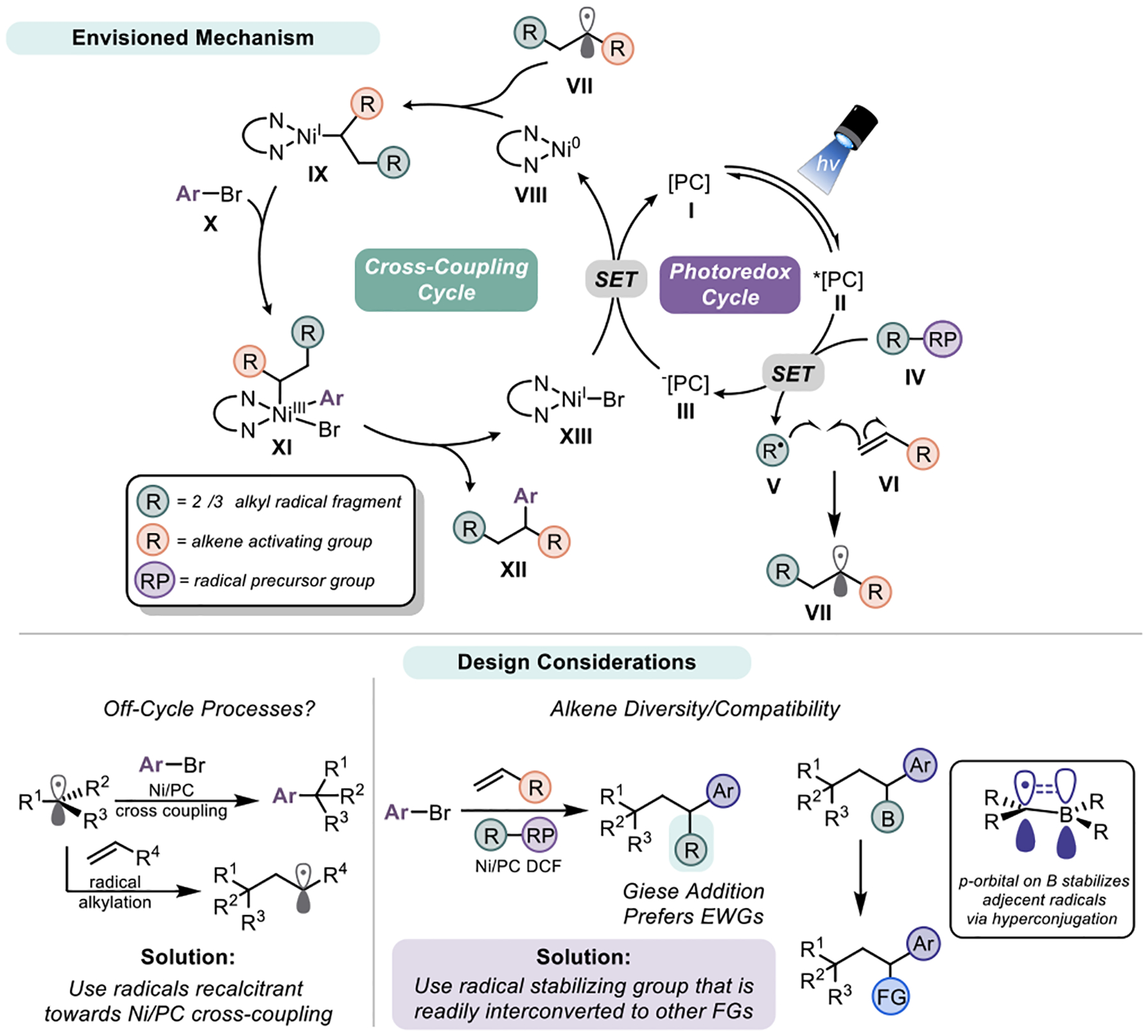
Envisioned mechanism for Ni/photoredox DCF of olefins and design consideration.
Results and Discussion
Initial exploration into the proposed DCF process focused on using 3 as the olefin of choice and a class of radical precursors that would furnish 3° radicals. The former was selected because it is among a privileged class of olefins that are competent radical acceptors13,14 and retain a synthetic handle capable of incredibly diverse downstream functionalization.15 Indeed, because the boronate can be used in myriad carbon-carbon bond-forming reactions or post-transformational functionalizations, this olefin can serve as a masked enol, vinyl fluoride, enamine, etc., thus complementing previous approaches outlined in Figure 1 and demonstrating the valuable modular nature of this protocol.15c Tertiary radicals were used initially because of their reluctance toward single electron metalation onto Ni, which would alleviate the aforementioned challenges concerning competitive (interrupted) cross coupling. Alkyltrifluoroborates16 emerged as the most effective class of radical precursors for such transformations. If not commercial, these boronate species can be accessed from a number of different feedstocks.
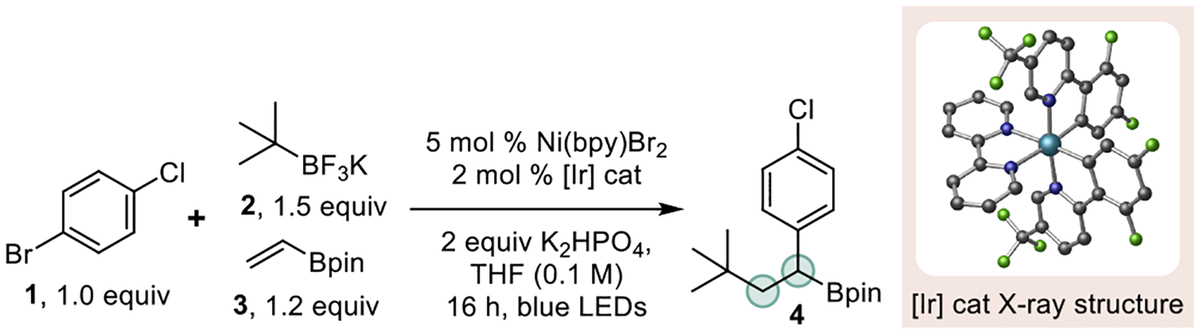
These include carboxylic acids, alkenes, alkyl halides, and even activated C-H bonds.15a-b,16 The reactions to convert these feedstocks to organotrifluoroborates can be carried out in high yield under simple reaction conditions.16b Given the diverse set of materials from which these precursors can be generated (especially for accessing tertiary radicals), we elected to use them to evaluate the proposed DCF process. Model studies utilizing trifluoroborate 2 (+1.26 V vs SCE)17 in tandem with boronate 3 and aryl bromide 1 revealed that Ni/photoredox DCF was viable (Table 1). Examination of solvent and photocatalyst identified THF and [Ir(dFCF3ppy)2bpy] PF6 (E1/2 M*/M− = +1.32 V)18 as a suitable pairing for effecting DCF (see Supporting Information). More rigorous refinement of conditions focused on assessing an array of Ni/ligand combinations, base identity, and stoichiometry (see Supporting Information). The results of these studies led to the conditions provided in Table 1. Interestingly, more intense irradiation (30 W Kessil lamp vs 4 W LED strips, entry 5) resulted in the formation of multiple side products and erosion of product yield. A “zero-precautions” experiment (non-rigorously dried solvent, reaction performed under an air atmosphere) showed a rather significant decrease in yield compared to standard conditions (compare entries 1 and 9), thus emphasizing the need for conducting the reaction free of moisture and air. Control studies confirmed that this DCF process was indeed dual catalytic in nature (Table 1, entries 10–12) and that all the components of the reaction were necessary to ensure successful olefin difunctionalization.
Table 1.
Selected Optimization for Ni/Photoredox DCFa
| ||
|---|---|---|
| entry | deviation from standard conditions | NMR yield of 4 |
| 1 | none | 70% (69%)c |
| 2 | no base | 55% |
| 3 | 0.05 M concentration | 67% |
| 4 | 0.25 M concentration | 39% |
| 5 | Kessil lamp in place of blue LEDs | 60% |
| 6 | 5 mol % [Ir] cat, 3 mol % [Ni] | 59% |
| 7 | 5 mol % [Ir] cat, 10 mol % [Ni] | 66% |
| 8 | CI-4CzIPN in place of [Ir] | (40%)c |
| 9 | “zero precautions” | 44%d |
| 10 | No [Ni] catalyst | 0% |
| 11 | No [Ir] photocatalyst | 0% |
| 12 | No light | 0% |
Optimization performed using 4-chloro-1-bromobenzene (0.1 mmol) for 16 h at 27 °C; [Ir] = [Ir{dF(CF3)2ppy}2(bpy)]PF6.
Yields in parentheses are isolated yield of 4 after purification.
Reaction performed using 2,4,5,6-tetrakis(3,6-dichloro-9H-carbazol-9-yl)isophthalonitrile (Cl-4CzIPN) as the photocatalyst.
Reaction performed under air with non-rigorously dried solvent.
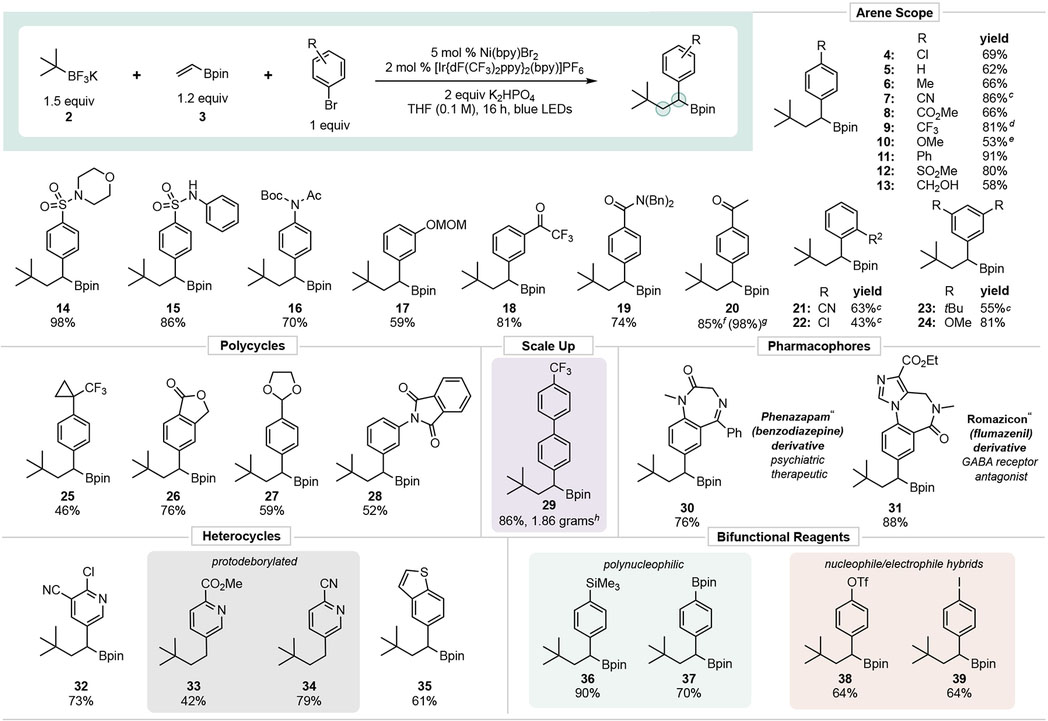
With suitable conditions established, the scope of the DCF process was evaluated. The initial focus was on examining aryl halide diversity (Table 2). A wide array of aryl bromides were competent in the DCF process. Sensitive functional groups, including acidic (13, 15), electrophilic (18, 26) and homolytically labile C-H bonds (17, 27), were all tolerated in the Ni/photoredox DCF process. In general, aryl bromides substituted with electron-withdrawing groups faired best, although electron-neutral and electron-donating groups also gave moderate to good yields. The efficacy of the reaction was not significantly impacted by sterically demanding ortho-substituted aryl bromides (21, 22), albeit these did require extended reaction times. Pharmaceutically relevant moieties with functional group-rich architectures or isosteric moieties could be engaged (25, 30, 31). Hetero-cyclic bromides were also compatible with the DCF process. Some of these substrates yielded a product without the boronate functional group (33, 34), exposing one potential limitation of the method. Reaction monitoring revealed that protodeborylation occurred under the reaction conditions, likely because of the presence of a Lewis basic nitrogen in these systems combined with the electron-withdrawing nature of a-pyridyl groups.19 Finally, a series of bifunctional reagents could be prepared (36–39, and 4), providing linchpins for rapid assembly of complex structures. Compound 39 is of particular interest as it arises from selective mono-functionalization of a diiodide.
Table 2.
|
Values indicate yield of the isolated product. pin = 2,3-dimethylbutane-2,3-diol.
Conditions unless otherwise noted: aryl halide (1 equiv, 0.5 mmol), 3 (1.2 equiv, 0.6 mmol), 2 (1.5 equiv, 0.75 mmol), [Ir(dFCF3ppy)2bpy]PF6 (2 mol %, 0.010 mmol), Ni(bpy)Br2 (5 mol %, 0.025 mmol), K2HPO4 (2 equiv, 1.0 mmol), THF (0.1 M), 16 h, irradiating with blue LEDs (6 W).
Reaction time extended to 48 h.
Ni(dtbbpy)Br2 used as the Ni catalyst.
Ni(phen)Br2 used as the Ni catalyst.
Isolated yield as BF3K salt.
1H NMR yield of boronate ester
Reaction performed on 5 mmol scale of aryl halide.
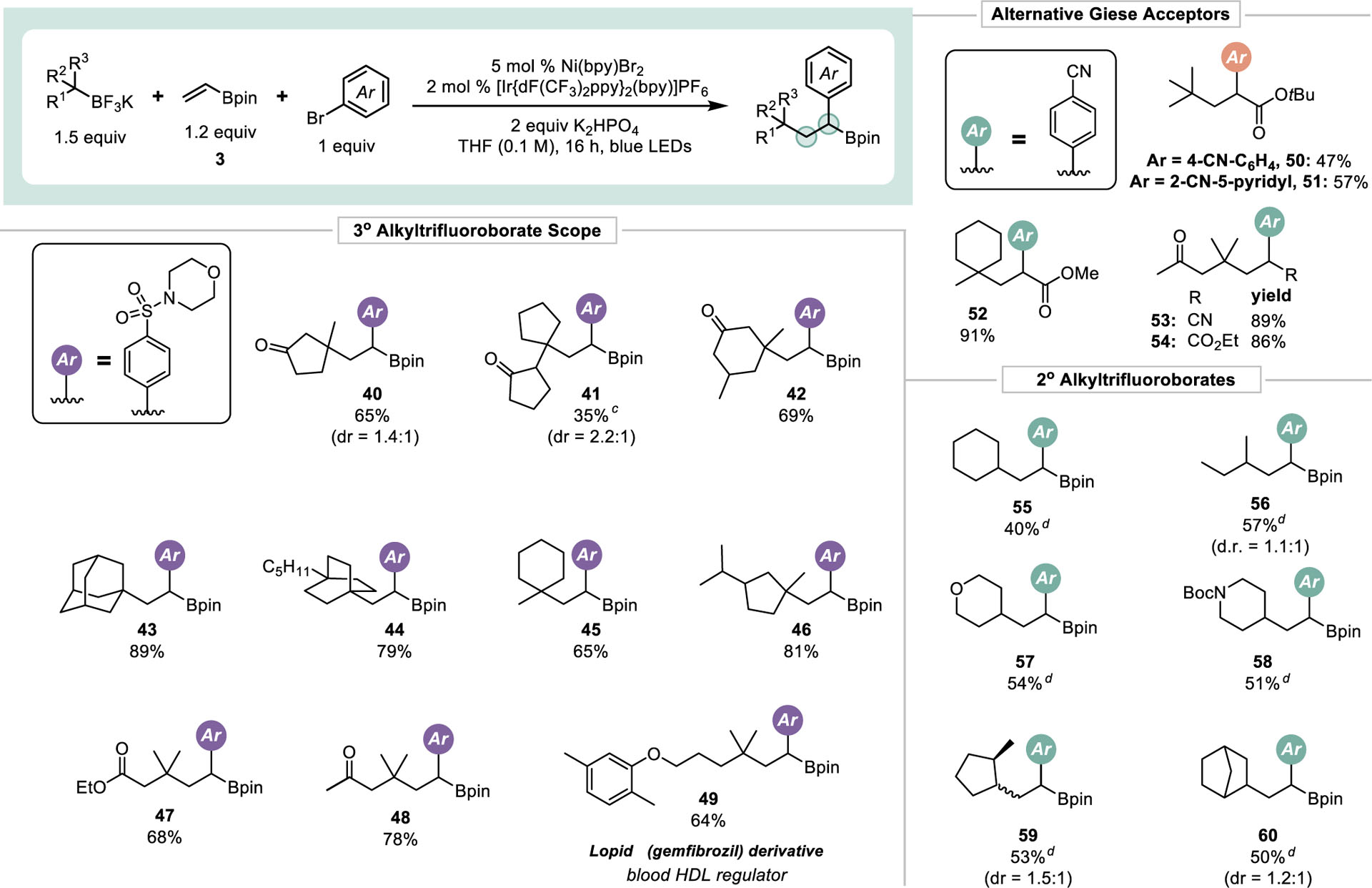
Next, the amenability toward other olefins and radicals was assessed (Table 3). Owing to recently developed protocols, accessing tertiary boronate esters is straightforward, and numerous feedstocks exist for their construction.20 Initial efforts focused on organotrifluoroborates derived from β-borylated carbonyls.20 These radical precursors provided good to moderate yield of the desired products and are especially valuable given that they retain a carbonyl functional handle. Other tertiary alkyltrifluoroborates, prepared via a radical decarboxylation/borylation sequence,21 were competent in the DCF process. Excellent yields were obtained when using polycyclic BF3Ks (43 and 44). Such moieties are of high value given their potential to function as bioisosteres and because of their unique architecture and “freedom to operate” from patent infringement.22
Table 3.
|
Values indicate the yield of the isolated product. pin = 2,3-dimethylbutane-2,3-diol.
Conditions unless otherwise noted: aryl halide (1 equiv, 0.5 mmol), potassium alkyltrifluoroborate (1.5 equiv, 0.75 mmol), Giese acceptor (1.2 equiv, 0.60 mmol), [Ir(dFCF3ppy)2bpy]PF6 (2 mol %, 0.010 mmol), Ni(bpy)Br2 (5 mol %, 0.025 mmol), K2HPO4 (2 equiv, 1.0 mmol), THF (0.1 M), 16 h, irradiating with blue LEDs (6 W).
Reaction time extended to 48 h. dNi(phen)Br2 (5 mol %, 0.025 mmol) and 3 (3.0 equiv, 1.5 mmol) were used.
In addition, a derivative of gemfibrozil, 49, was prepared. Although structurally distinct from the marketed API, this example demonstrates the potential of this reaction to prepare a target core and rapidly elaborate it, which could be of use in a medicinal chemistry setting.
In addition to 3, other electronically deficient alkenes were also successfully incorporated into this reaction protocol. Olefins bearing ester and nitrile functional groups gave high yields similar to that when using 3. The range of functional groups that were successfully incorporated into the olefin acceptor demonstrates the potential applicability of this method to the synthesis of complex molecular architectures.
Despite their known propensity toward Ni/photoredox cross-coupling,23 attempts to utilize 2° alkyltrifluoroborates were made. After a slight modification of the original conditions, these radicals were found to be quite competent in the DCF process (Table 3). A variety of 2° radicals gave the DCF products in synthetically useful yields (55-60), with the major competing pathway being direct cross-coupling to afford materials analogous to that of 4” (Figure 3).
Figure 3.
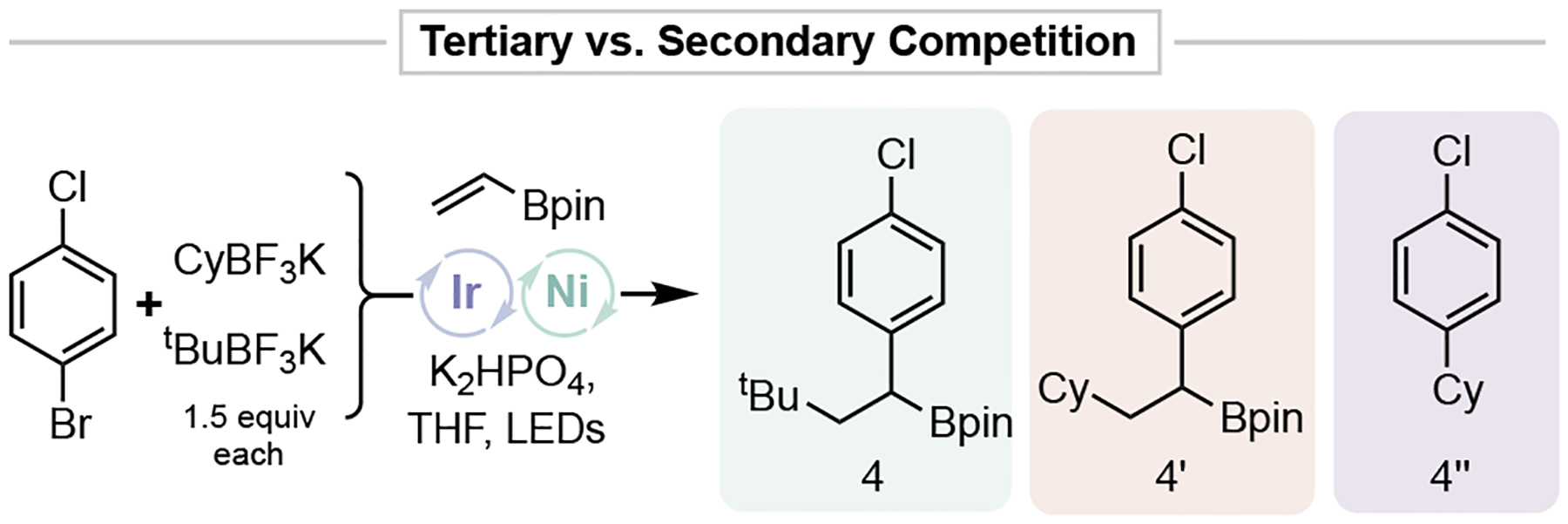
Secondary versus tertiary radical competition.
In addition to improving the scope of the described process, the competency of these 2° radicals indicates that Giese-type addition is competitive with metalation to Ni or pre-complexation by the olefin with Ni0, thus affording selective sequential olefin difunctionalization.24 A competition experiment between secondary (cyclohexyl) and tertiary (tert-butyl) radicals revealed that DCF is 16 times faster than traditional Ni/photoredox cross-coupling for 3° radicals and 2.5 times faster for secondary radicals, emphasizing the particular efficacy of 3° radicals in Ni/photoredox DCF processes (Figure 3). Attempts to leverage other more stabilized radicals (e.g., benzyl, α-oxy, α-amino, etc.) were met with either a lack of reactivity or exclusive Csp2-Csp3 cross-coupling, indicating poor reactivity in the Giese addition step.24b,c
Attempts to utilize the alkyltrifluoroborate derived from verbenone led to an unexpected radical cascade, giving bis-borylated product 61 (Figure 4). Although radical fragmentations of this type are known,25 this is to the best of our knowledge, the first time that this has been used in a DCF process. The formation of this product provides insight into the relative rate of radical additions to olefins in comparison to nickel single electron metalation. The rate of Giese radical addition to 3 must fall below the rate of radical ring opening in this system (1.1×107 s–1).26 Further, the fact that no direct cross-coupling products were observed is suggestive that the rate of 3° radical single electron metalation to the catalytic nickel complex is quite slow compared to both reaction with 3 and to the cyclization of the borylated 2° alkyl radical intermediate formed as well.
Figure 4.
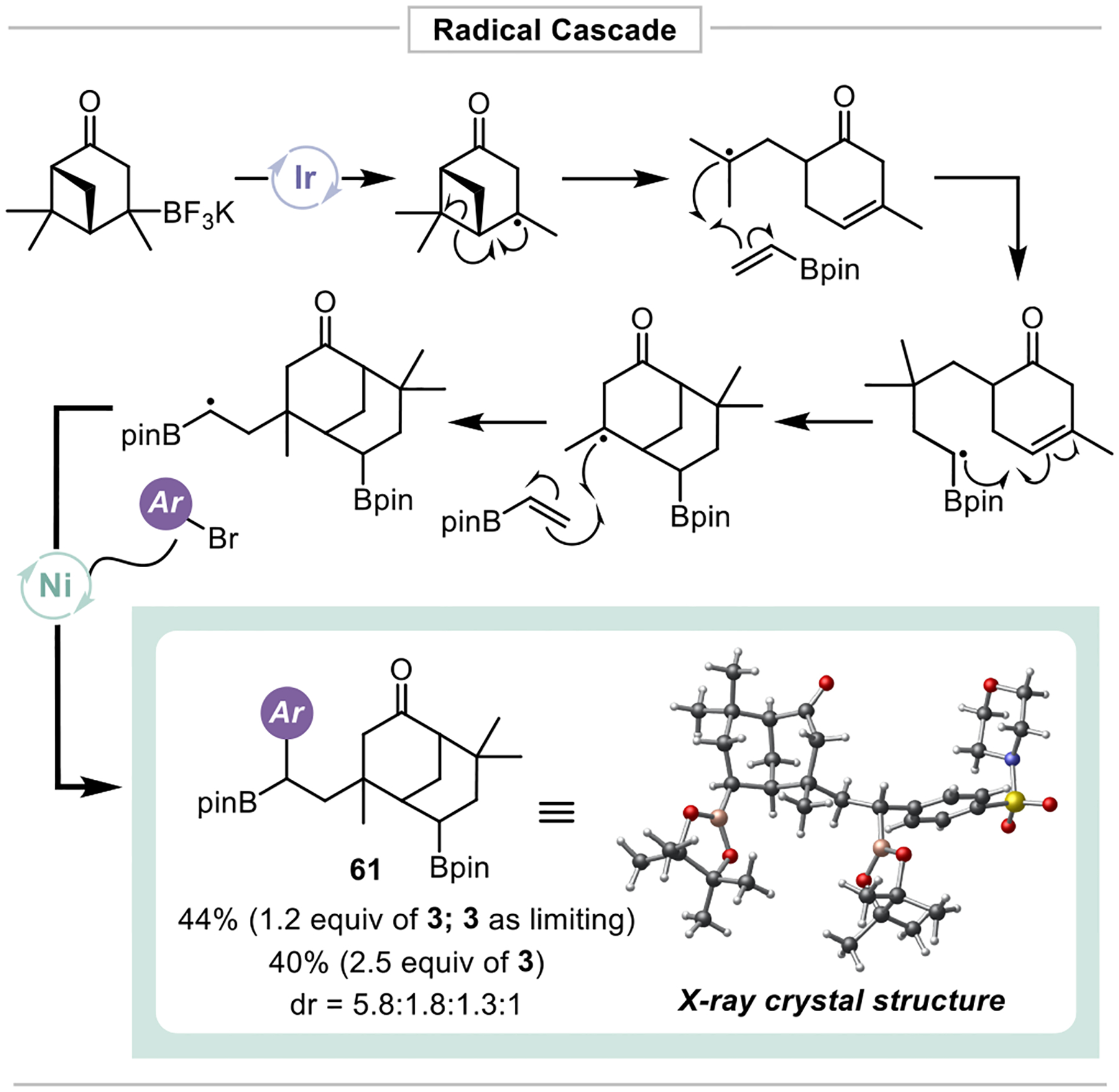
Radical ring opening leads to bis-borylated DCF product.
Although already possessing a broad scope in terms of radical source, aryl halide structure, and olefin type, we sought to gauge the limitations of this method. An HPLC-based robustness assay27 confirmed that Ni/photoredox DCF can be carried out in the presence of a variety of functional groups not explicitly examined in the scope, including protic groups (alcohols and phenols), alkyl halides, aldehydes, and certain Lewis basic heterocycles (see the Supporting Information). Some displayed excellent tolerance, whereas others still allowed DCF with diminished yield. Though robustness assays do have limitations, these experiments provide a general guide for possible applications of the process developed.
To gain a better understanding of the DCF process, additional experiments were conducted (Figure 5). A Ni-mediated Heck reaction28 of the vinyl boronate and aryl bromide followed by radical addition was considered. As anticipated, under similar reaction conditions, no detectable amount of 3 reacted with 1 (see the Supporting Information). Next, an alkyltrifluoroborate bearing a pendent alkene was subjected to Ni/photoredox DCF. After Giese addition, 5-exo-trig cyclization (rate = 2 × 105 s−1)29 of the intermediate radical resulted in formation of 62. Although not definitive, this suggests that the olefin is not bound to Ni during Giese addition and supports the initial mechanistic proposal. This also suggests that the rate of single electron metalation of the resulting α-boryl radical is somewhat slow or reversible. Further experiments are underway to understand the sequence of events in this novel DCF process.
Figure 5.
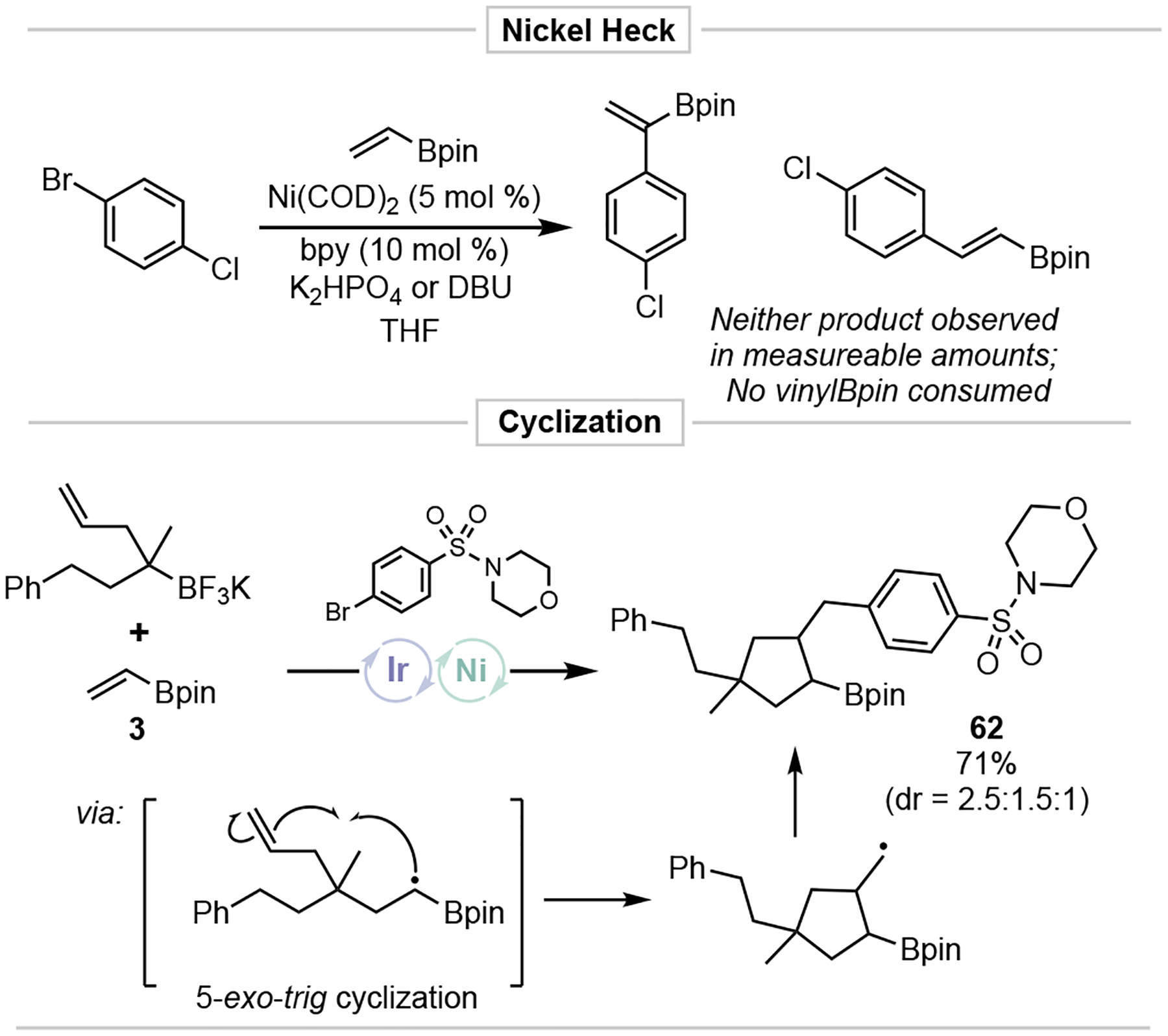
Mechanistic studies.
With a better understanding of the limits of this reaction in the context of scope and reaction rate, selective olefin functionalization was attempted. An allylic aryl bromide readily underwent selective DCF, giving 63. This is notable for two reasons: 1) No reaction of the electron-rich olefin was observed, despite their propensity toward oxidation30 2) No H-atom abstraction of the benzyl-stabilized allylic methylene was observed.9f Using a functionalized acrylate containing a pendent alkene, selective 1,2 difunctionalization of the electron- deficient olefin was observed, yielding 64 (Figure 6). This example demonstrates remarkable selectivity given the lack of steric bias in this system, and relies exclusively on the innate polarity-matching of oddelectron species. In addition, selective functionalization was possible in several other poly-olefin systems (65-68). Those derived from steroids or containing strained olefins readily underwent selective DCF. More complex terpenebased species were similarly tolerated and did not display any off-target Giese products, although the yields were somewhat diminished because of incomplete conversion in the standard reaction time.
Figure 6.
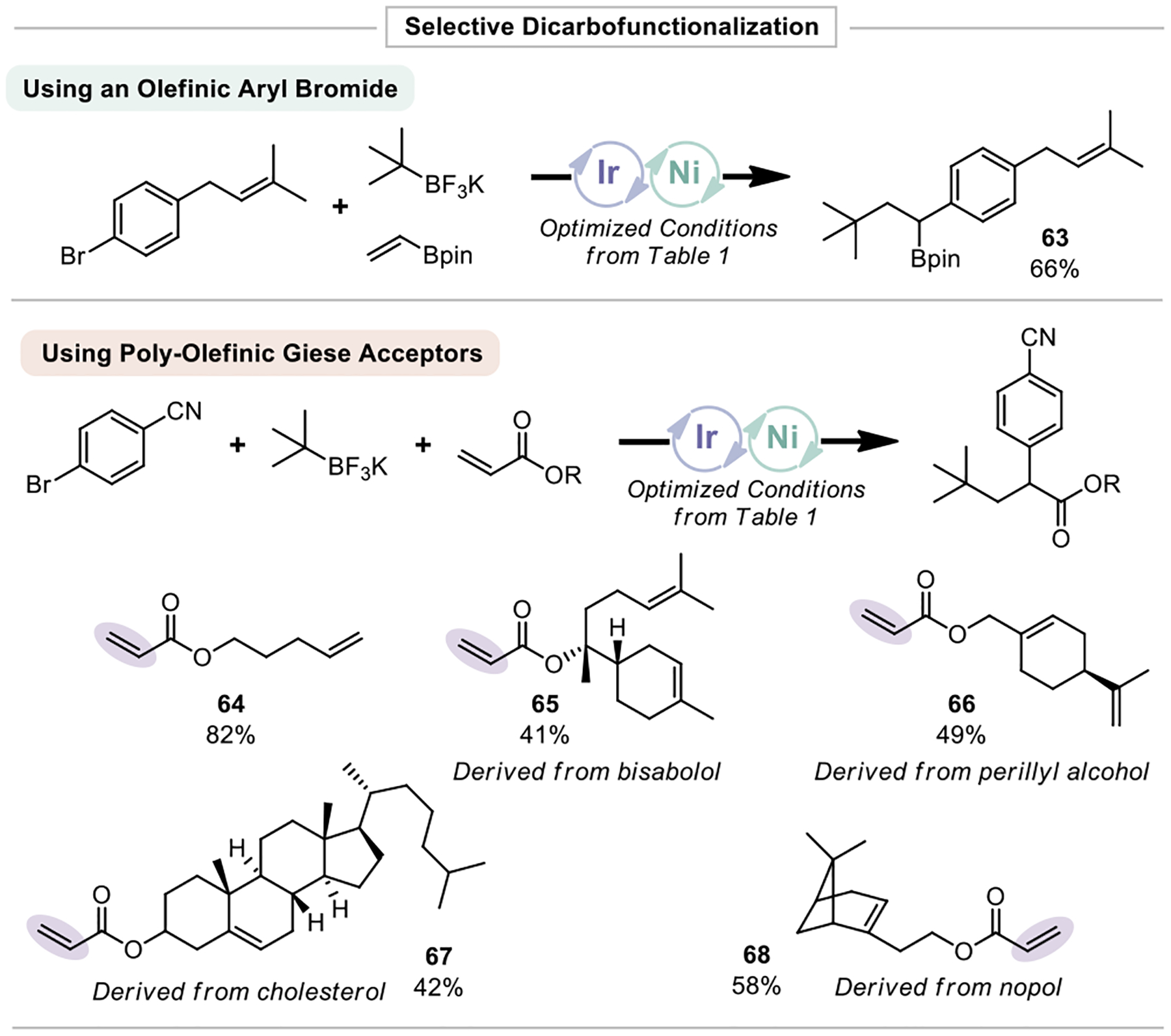
Selective dicarbofunctionalization of olefins using Ni/photoredox dual catalysis
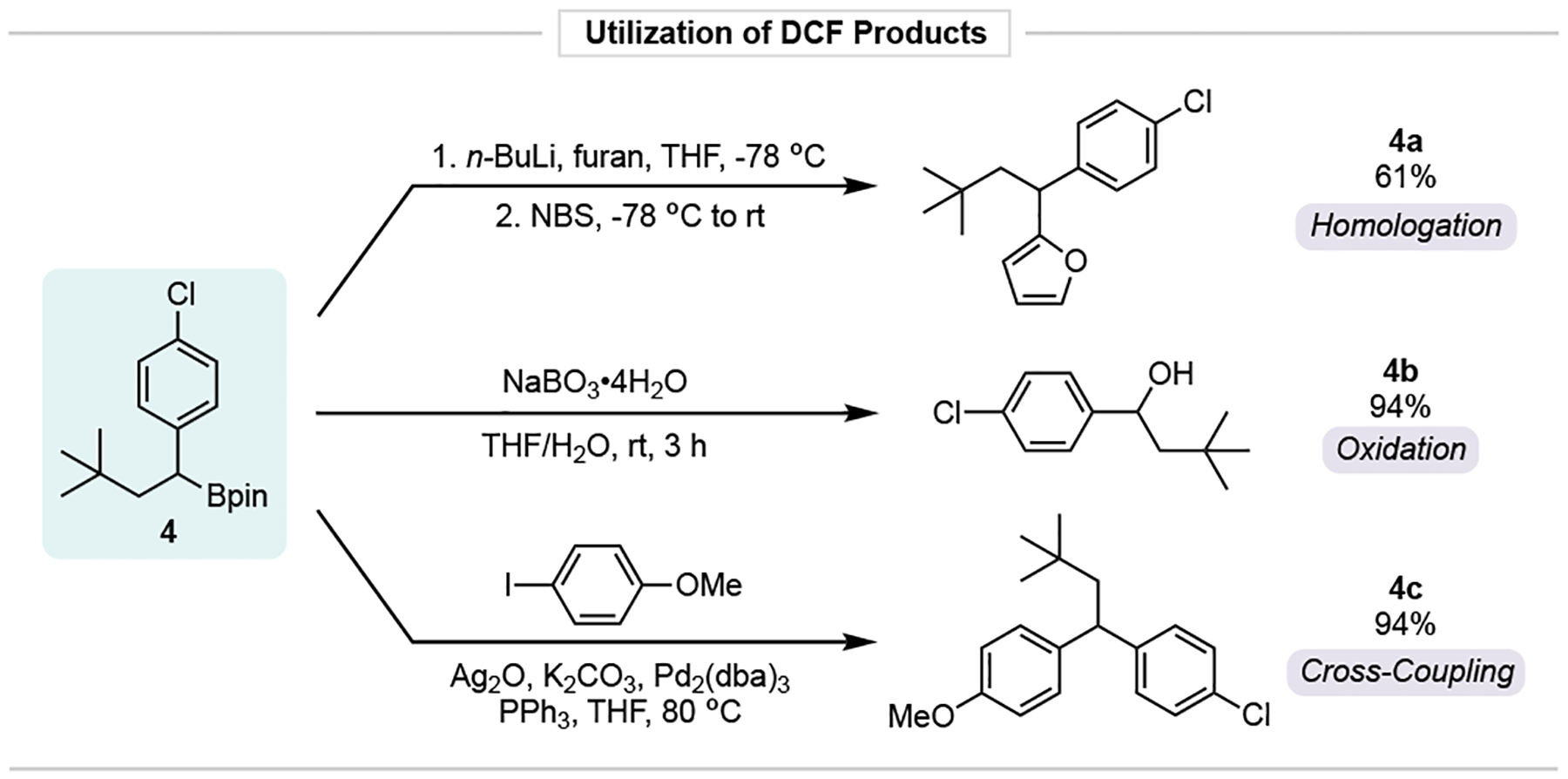
Finally, to demonstrate the inherent synthetic value of the developed DCF process and, more specifically, the boronate functional group installed in DCF products, several derivatization experiments were undertaken (Figure 7), using 4 as a model substrate. Oxidation with perborate yielded the benzylic alcohol 4b in excellent yield. Using a recently developed procedure by Aggarwal,31 a new C-C bond was forged via Matteson-type lithiation/migration sequence to give 4a, whereas traditional Pd-mediated cross coupling gave 4c in excellent yield.32
Figure 7. Utilization of DCF products to enhance molecular complexity and application to the synthesis of a preclinical candidate.
The success of the model perborate oxidation of 4 led us to pursue the preparation of a key intermediate (69) in the synthesis of a gram-positive bacterial thymidylate kinase inhibitor (TK-666).33 This particular TMK inhibitor shows picomolar activity against antibiotic resistant S. pneumoniiae, S. aureus, and Enterococcus. Incorporation of DCF in the early steps of this synthesis provided a dramatic increase in the yield of an analogous benzylic alcohol intermediate. Further, alteration of the alkyl group installed has been shown to have a dramatic effect on potency. As such, the modular nature DCF process described here is highly suitable for rapid exploration of the arene and alkyl group chemical space because of their commercial availability and/or ease of synthetic preparation. To demonstrate this, we utilized an advanced aryl bromide in DCF with various alkyltrifluoroborates to prepare novel TK-666 derivative analogues (70-72). Further oxidation of 70 gave the corresponding benzylic alcohol (70a) that would serve as an intermediate toward a chloroarene variant of TK-666. In addition to the oxidation protocol described here, these DCF-derived alkylboronates open up exploration into the role of the benzylic functional group (given the vast array of functional group transformations available from the boronate ester) in the context of medicinal chemistry.
Conclusion
In summary, a dicarbofunctionalization of olefins enabled by Ni/photoredox dual catalysis has been realized. The DCF process accommodates a broad palette of aryl halides, olefins, and radical architectures. This three-component process enables two C–C bonds to be formed in a single step (Csp3–Csp3 and Csp3–Csp2), sets both quaternary and tertiary centers, all while retaining the mild, functional group tolerant nature characteristic of Ni/photoredox dual catalysis. The inherent mechanistic underpinnings enable selective olefin functionalization in polyolefinic systems based on olefin electronics. In addition, the amenability of vinyl boronates is particularly attractive from the standpoint of late-stage functionalization and enabled the preparation of an intermediate to a preclinical antibiotic. Overall, the developed DCF process allows a rapid buildup of molecular complexity from simple building blocks. The implications of the developed process have sparked additional mechanistic investigations whose findings will be reported in due course.
Supplementary Material
Figure 8.
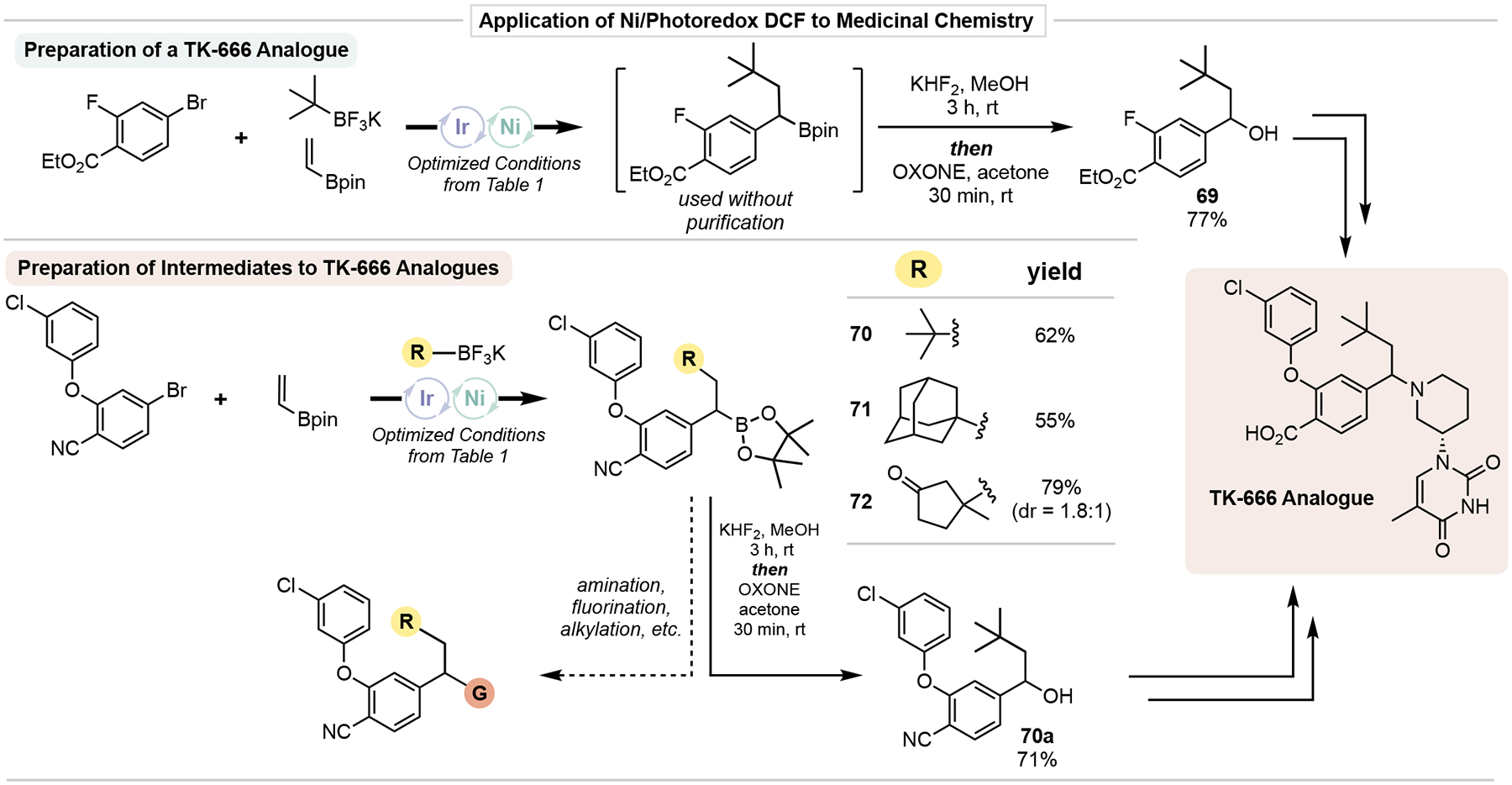
Utilization of Ni/photoredox DCF to prepare medicinally-relevant molecular architectures.
ACKNOWLEDGMENT
The authors are grateful for the financial support provided by NIGMS (R35 GM 131680 to G.A.M.). C.B.K. acknowledges start-up funds from Virginia Commonwealth University (VCU). M.W.C. and J.S.C. are grateful for NSF Graduate Research Fellowships. We thank Mr. Kevin Burns (VCU), Mr. Anthony Le (VCU), and Ms. Saskia Engle (VCU) for technical assistance. We thank Dr. Charles W. Ross, III (University of Pennsylvania) for obtaining HRMS data. We gratefully acknowledge Dr. Mike Gao and Dr. Pat Carroll for acquiring X-ray crystal structures. We acknowledge Frontier Scientific and Johnson-Matthey for fine chemicals donations and Kessil Lighting for lights used in this study.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website.
Experimental details and spectral data (PDF)
The authors declare no competing financial interest.
REFERENCES
- 1.Representative review on reaction of olefins that dramatically increase molecular complexity:; (a) Kolb HC; VanNieuwenhze MS; Sharpless KB Catalytic Asym metric Dihydroxylation Chem. Rev 1994, 94, 2483–2547. [Google Scholar]; (b) Hashimoto T; Maruoka K Recent Advances of Catalytic Asymmetric 1,3-Dipolar Cycloadditions Chem. Rev 2015, 115, 5366–5412. [DOI] [PubMed] [Google Scholar]; (c) Wu W; Lina Z; Jiang H Recent advances in the synthesis of cyclopropanes Org. Biomol. Chem 2018, 16, 7315–7329. [DOI] [PubMed] [Google Scholar]; (d) McDonald RI; Liu G; Stahl SS Palladium(II)-Catalyzed Alkene Functionalization via Nucleopalladation: Stereochemical Pathways and Enantioselective Catalytic Applications Chem. Rev 2011, 111, 2981–3019. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Cresswell AJ; Eey ST-C; Denmark SE Catalytic, Stereoselective Dihalogenation of Alkenes: Challenges and Opportunities Angew. Chem. Int. Ed 2015, 54, 15642–15682. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Heravi MM; Lashaki TB; Fattahia B; Zadsirjan V Application of asymmetric Sharpless aminohydroxylation in total synthesis of natural products and some synthetic complex bio-active molecules RSC Adv. 2018, 8, 6634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.(a) Chapdelaine MJ; Hulce M Tandem Vicinal Difunctionalization: β-Addition to α,β-Unsaturated Carbonyl Substrates Followed by α-Functionalization Org. React 1990, 38, 227–294. [Google Scholar]; (b) Ihara M; Fukumoto K Syntheses of Polycyclic Natural Products Employing the Intramolecular Double Michael Reaction Angew. Chem. Int. Ed. Engl 1993, 32, 1010–1022. [Google Scholar]; (c) Bian M; Li LK; Ding HF, Recent Advances on the Application of Electrocyclic Reactions in Complex Natural Product Synthesis. Synthesis 2017, 49, 4383–4413. [Google Scholar]
- 3.(a) Anthony D; Lin Q; Baudet J; Diao TN, Nickel-Catalyzed Asymmetric Reductive Diarylation of Vinylarenes. Angew. Chem. Int. Ed 2019, 58, 3198–3203. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Derosa J; Kleinmans R; Tran VT; Karunananda MK; Wisniewski SR; Eastgate MD; Engle KM Nickel-Catalyzed 1,2-Diarylation of Simple Alkenyl Amides J. Am. Chem. Soc 2018, 140 ,17878–17883. [DOI] [PubMed] [Google Scholar]; (c) Liao LY; Jana R; Urkalan KB; Sigman MS A Palladium-Catalyzed Three-Component Cross-Coupling of Conjugated Dienes or Terminal Alkenes with Vinyl Triflates and Boronic Acids J. Am. Chem. Soc 2011, 133, 5784–5787. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Urkalan KB; Sigman MS Palladium-Catalyzed Oxidative Intermolecular Difunctionalization of Terminal Alkenes with Organostannanes and Molecular Oxygen Angew. Chem. Int. Ed 2009, 48, 3146–3149. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Liao L; Jana R; Urkalan KB; Sigman MS A Palladium-Catalyzed Three-Component Cross-Coupling of Conjugated Dienes or Terminal Alkenes with Vinyl Triflates and Boronic Acids J. Am. Chem. Soc 2011, 133, 5784–5787. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Liu Z; Zeng T; Yang KS; Engle KM β,γ-Vicinal Dicarbofunctionalization of Alkenyl Carbonyl Compounds via Directed Nucleopalladation J. Am. Chem. Soc 2016, 138, 15122–15125. [DOI] [PubMed] [Google Scholar]; (e) Stokes JB; Liao L; de Andrade AM; Wang Q; Sigman MS A Palladium-Catalyzed Three-Component-Coupling Strategy for the Differential Vicinal Diarylation of Terminal 1,3-Dienes Org. Lett 2014, 16, 4666–4669. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Wu X; Lin H-C; Li M-L; Li L-L; Han Z-Y; Gong L-Z Enantioselective 1,2-Difunctionalization of Dienes Enabled by Chiral Palladium Complex-Catalyzed Cascade Arylation/Allylic Alkylation Reaction J. Am. Chem. Soc 2015, 137, 13476–13479. [DOI] [PubMed] [Google Scholar]; (g) Shrestha B; Basnet P; Dhungana RK; KC S; Thapa S; Sears JM; Giri R Ni-Catalyzed Regioselec tive 1,2-Dicarbofunctionalization of Olefins by Intercepting Heck Intermediates as Imine-Stabilized Transient Metal-lacycles J. Am. Chem. Soc 2017, 139, 10653–10656. [DOI] [PubMed] [Google Scholar]; (h) KC S; Basnet P; Thapa S; Shrestha B; Giri R Ni-Catalyzed Regioselective Dicarbofunctionalization of Un-activated Olefins by Tandem Cyclization/Cross-Coupling and Application to the Concise Synthesis of Lignan Natural Products J. Org. Chem 2018, 83, 2920–2936. [DOI] [PubMed] [Google Scholar]
- 4.(a) Rilatt I; Jackson RFW Kinetic Studies on the Stability and Reactivity of β-Amino Alkylzinc Iodides Derived from Amino Acids J. Org. Chem 2008, 73, 8694. [DOI] [PubMed] [Google Scholar]; (b) Lozada J; Liu Z; Perrin DM Base-Promoted Protodeboronation of 2,6-Disubstituted Arylboronic Acids J. Org. Chem 2014, 79, 5365–5368. [DOI] [PubMed] [Google Scholar]; (c) Zou G; Reddy YK; Falck JR Ag(I)-promoted Suzuki–Miyaura cross-couplings of n-alkylboronic acids Tetrahedron Lett. 2001, 42, 7213–7215. [Google Scholar]; (d) Li L; Wang C-Y; Huang R; Biscoe MR Stereoretentive Pd-catalysed Stille cross-coupling reactions of secondary alkyl azastannatranes and aryl halides Nat. Chem 2013, 5, 607–612. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Tamao K; Kiso Y; Sumitani J; Kumada M Alkyl Group Isomerization in the Cross-Coupling Reaction of Secondary Alkyl Grignard Reagents with Organic Halides in the Presence of Nickel-Phosphine Complexes as Catalysts J. Am. Chem. Soc 1972, 94, 9268–9269. [Google Scholar]
- 5.(a) Qin T; Cornella J; Li C; Malins LR; Edwards JT; Kawamura S; Maxwell BD; Eastgate MD; Baran PS A general alkyl-alkyl cross-coupling enabled by redox-active esters and alkylzinc reagents Science 2016, 352, 801–805. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) García-Domínguez A; Li Z; Nevado C Nickel-Catalyzed Reductive Dicarbofunctionalization of Alkenes J. Am. Chem. Soc 2017, 139, 6835–6837. [DOI] [PubMed] [Google Scholar]; (c) Kischkewitz M; Okamoto K; Mück-Lichtenfeld C; Studer A Radical-polar crossover reactions of vinylboron ate complexes Science 2017, 355, 936–938. [DOI] [PubMed] [Google Scholar]; (d) Li Z; García-Domínguez A; Nevado C Nickel-Catalyzed Stereoselective Dicarbofunctionalization of Alkynes Angew. Chem. Int. Ed 2016, 55, 6938–6941. [DOI] [PubMed] [Google Scholar]
- 6.Yan M; Lo JC; Edwards JT; Baran PS Radicals: Reactive Intermediates with Translational Potential J. Am. Chem. Soc 2016, 138, 12692–12714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tasker SZ; Standley EA; Jamison TF Recent Advances in Homogeneous Nickel Catalysis Nature 2014, 509, 299–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.(a) Prier CK; Rankic DA; MacMillan DWC Visible Light Photoredox Catalysis with Transition Metal Complexes: Applications in Organic Synthesis Chem. Rev 2013, 113, 5322–5363. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Romero NA; Nicewicz DA Organic Photoredox Catalysis Chem. Rev 2016, 116, 10075–10166. [DOI] [PubMed] [Google Scholar]; (c) Matsui JK; Lang SB; Heitz DR; Molander GA Photoredox-Mediated Routes to Radicals: The Value of Catalytic Radical Generation in Synthetic Methods Development ACS Catal. 2017, 7, 2563–2575. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Shaw MH; Twilton J; MacMillan DWC Photoredox Catalysis in Organic Chemistry J. Org. Chem 2016, 81, 6898–6926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.For seminal reports, see:; (a) Tellis JC; Primer DN; Molander GA Single-electron transmetalation in organoboron cross-coupling by photoredox/nickel dual catalysis Science 2014, 345, 433–436. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Zuo Z; Ahneman DT; Chu L; Terrett JA; Doyle AG; MacMillan DWC Merging photoredox with nickel catalysis: Coupling of α-carboxyl sp3 -carbons with aryl halides Science 2014, 345, 437–440. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) For reviews see:Tellis JC; Kelly CB; Primer DN; Jouffroy M; Patel NR; Molander GA Single-Electron Transmetalation via Photoredox/Nickel Dual Catalysis: Unlocking a New Paradigm for sp3−sp2 Cross-Coupling Acc. Chem. Res 2016, 49, 1429–1439. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Gui Y-Y; Sun L; Lu Z-P; Yu D-G Photoredox Sheds New Light on Nickel Catalysis: From Carbon−Carbon to Carbon−Heteroatom Bond Formation Org. Chem. Front 2016, 3, 522–526. [Google Scholar]; (e) Skubi KL; Blum TR; Yoon TP Dual Catalysis Strategies in Photochemical Synthesis Chem. Rev 2016, 116, 10035–10074. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Milligan JA; Phelan JP; Badir SO; Molander GA Recent Advances in Alkyl Carbon-Carbon Bond Formation by Nickel/Photoredox Cross-Coupling Angew. Chem. Int. Ed 2019, 58, 6152–6163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.While this manuscript being revised, two reports detailing conceptually similar transformations using Ni/photoredox dual catalysis were reported, see:; (a) Guo L; Tu H-Y; Zhu S; Chu L Selective, Intermolecular Alkylarylation of Alkenes via Photoredox/Nickel Dual Catalysis Org. Lett 2019, 21, 4771–4776. [DOI] [PubMed] [Google Scholar]; (b) García- Domínguez A; Mondal R; Nevado C Dual Photoredox/Nickel-Catalyzed Three-Component Carbofunctionalization of Alkenes 2019, 58, 12286–12290. [DOI] [PubMed] [Google Scholar]
- 11.(a) Giese B Formation of CC Bonds by Addition of Free Radicals to Alkenes Angew. Chem. Int. Ed. Engl 1983, 22, 753–764. [Google Scholar]; (b) Qin T; Malins LR; Edwards JT; Merchant RR; Novak AJE; Zhong JZ; Mills RB; Yan M; Yuan C; Eastgate MD; Baran PS Nickel- Catalyzed Barton Decarboxylation and Giese Reactions: A Practical Take on Classic Transforms Angew. Chem. Int. Ed 2017, 56, 260–265. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) ElMarrouni A; Ritts CB; Balsells J Silyl-mediated photoredox-catalyzed Giese reaction: addition of non-activated alkyl bromides Chem. Sci 2018, 9, 6639–6646 [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Seath CP; Vogt DB; Xu Z; Boyington AJ; Jui NT Radical Hydroarylation of Functionalized Olefins and Mechanistic Investigation of Photocatalytic Pyridyl Radical Reactions J. Am. Chem. Soc 2018, 140, 15525–15534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gutierrez O; Tellis JC; Primer DN; Molander GA; Kozlowski MC Nickel-Catalyzed Cross-Coupling of Photoredox Generated Radicals: Uncovering a General Manifold for Stereoconvergence in Nickel-Catalyzed Cross-Couplings J. Am. Chem. Soc 2015, 137, 4896–4899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.(a) Guennouni N; Lhermitte F; Cochard S; Carboni B Radical reactions in organoboron chemistry II — Inter- and intramolecular addition of carbon centered radicals to alkenylboranes Tetrahedron 1995, 51, 6999–7018 [Google Scholar]; (b) Lee E; Zong K; Kang HY; Lim J; Kim JY Radical Cyclization Studies of Alkenylboronates Bull. Korean Chem. Soc 2000, 21, 765–766. [Google Scholar]; (c) Lopez-Ruiz H; Zard SZ A flexible access to highly functionalised boronates Chem. Commun 2001, 2618–2619. [Google Scholar]; (d) Heinrich MR; Sharp LA; Zard SZ A convergent approach to γ-carbonyl vinyl boronates Chem. Commun 2005, 3077–3079. [DOI] [PubMed] [Google Scholar]; (e) Noble A; Mega RS; Pflästerer D; Myers EL; Aggarwal VK Visible-Light-Mediated Decarboxylative Radical Additions to Vinyl Boronic Esters: Rapid Access to g-Amino Boronic Esters Angew. Chem. Int. Ed 2018, 57, 2155–2159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Walton JC; McCarroll AJ; Chen Q; Carboni B; Nziengui R The Influence of Boryl Substituents on the Formation and Reactivity of Adjacent and Vicinal Free Radical Centers J. Am. Chem. Soc 2000, 122, 5455–5463. [Google Scholar]
- 15.(a) Fyfe JWB; Watson AJB Recent Developments in Organoboron Chemistry: Old Dogs, New Tricks Chem 2017, 3, 31–55. [Google Scholar]; (b) Darses S; Genet J-P Potassium Organotrifluoroborates: New Perspectives in Organic Synthesis Chem. Rev 2008, 108, 288–325. [DOI] [PubMed] [Google Scholar]; (c) Larouche-Gauthier R; Elford TG; Aggarwal VK Ate Complexes of Secondary Boronic Esters as Chiral Organometallic-Type Nucleophiles for Asymmetric Synthesis J. Am. Chem. Soc 2011, 133, 16794–16797. [DOI] [PubMed] [Google Scholar]
- 16.(a) Molander GA Organotrifluoroborates: Another Branch of the Mighty Oak J. Org. Chem 2015, 80, 7837–7848. [DOI] [PubMed] [Google Scholar]; (b) Molander GA; Jean-Gérard L Cross-Coupling Reactions of Organotrifluoroborate Salts Org. React 2012, 79, 1. [Google Scholar]
- 17.Primer DN; Molander GA Enabling the Cross-Coupling of Tertiary Organoboron Nucleophiles through Radical-Mediated Alkyl Transfer J. Am. Chem. Soc 2017, 139, 9847–9850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kelly CB; Patel NR; Primer DN; Jouffroy M; Tellis JC; Molander GA Preparation of visible-light-activated metal complexes and their use in photoredox/nickel dual catalysis Nat. Protoc 2017, 12, 472–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.(a) Knapp DM; Gillis EP; Burke MD A General Solution for Unstable Boronic Acids: Slow-Release Cross-Coupling from Air-Stable MIDA Boronates J. Am. Chem. Soc 2009, 131, 6961–6963. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Dick GR; Woerly EM; Burke MD A General Solution for the 2-Pyridyl Problem Angew. Chem. Int. Ed 2012, 51, 2667–2672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.(a) Schiffner JA; Muther K; Oestreich M Enantioselective Conjugate Borylation. Angew. Chem. Int. Ed 2010, 49, 1194–1196. [DOI] [PubMed] [Google Scholar]; (b) Kobayashi S; Xu PY; Endo T; Ueno M; Kitanosono T Chiral Copper(II)-Catalyzed Enantioselective Boron Conjugate Additions to α,β-Unsaturated Carbonyl Compounds in Water. Angew. Chem. Int. Ed 2012, 51, 12763–12766. [DOI] [PubMed] [Google Scholar]; (c) Molander GA; McKee SA, Copper-Catalyzed β-Boration of α, β-Unsaturated Carbonyl Compounds with Tetrahydroxydiborane Org. Lett 2011, 13, 4684–4687. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Xu L Decarboxylative Borylation: New Avenues for the Preparation of Organoboron Compounds Eur. J. Org. Chem 2018, 3884–3890. [Google Scholar]; (e) Cheng Y; Mück-Lichtenfeld C; Studer A Metal-Free Radical Borylation of Alkyl and Aryl Iodides Angew. Chem. Int. Ed 2018, 57, 16832–16836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fawcett A; Pradeilles J; Wang Y; Mutsuga T; Myers EL; Aggarwal VK Photoinduced decarboxylative borylation of carboxylic acids Science 2017, 357, 283–286. [DOI] [PubMed] [Google Scholar]
- 22.(a) Dandapani S; Marcaurelle LA Accessing New Chemical Space for ‘Undruggable’ Targets Nature Chem. Biol 2010, 6, 861–863. [DOI] [PubMed] [Google Scholar]; (b) López-Vallejo F; Giulianotti MA; Houghten RA; Medina-Franco JL Expanding the Medicinally Relevant Chemical Space with Compound Libraries Drug Discov. Today 2012, 17, 718–726. [DOI] [PubMed] [Google Scholar]; (b) Meanwell NA Synopsis of Some Recent Tactical Appli cation of Bioisosteres in Drug Design J. Med. Chem 2011, 54, 2529–2591. [DOI] [PubMed] [Google Scholar]; (c) Auberson YP; Brocklehurst C; Furegati M; Fessard TC; Koch G; Decker A; La Vecchia L; Briard E Improving Nonspecific Binding and Solubility: Bicycloalkyl Groups and Cubanes as para- Phenyl Bioisosteres Chem. Med. Chem 2017, 12, 590–598. [DOI] [PubMed] [Google Scholar]
- 23.Primer DN; Karakaya I; Tellis JC; Molander GA Single-Electron Transmetalation: An Enabling Technology for Secondary Alkylboron Cross-Coupling J. Am. Chem. Soc 2015, 137, 2195–2198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.(a) Desnoyer AN; He W; Behyan S; Chiu W; Love JA; Kennepohl P The Importance of Ligand- Induced Backdonation in the Stabilization of Square Planar d10 Nickel π-Complexes Chem. Eur. J 2019, 25, 5259–5268; [DOI] [PubMed] [Google Scholar]; Such issues with selectivity are well-known in the area of reductive cross-coupling, see:; (b) Weix DJ Methods and Mechanisms for Cross-Electrophile Coupling of Csp2 Halides with Alkyl Electrophiles Acc. Chem. Res 2015, 48, 1767. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Shu W; García-Domínguez A; Quirós MT; Mondal R; Cárdenas DJ; Nevado C Ni-Catalyzed Reductive Dicarbofunctionalization of Nonactivated Alkenes: Scope and Mechanistic Insights J. Am. Chem. Soc 2019,141 13812–13821. [DOI] [PubMed] [Google Scholar]
- 25.Fernández-Mateos A; Teijón PH; González RR Radical reactions on pinene-oxide derivatives induced by Ti(III) Tetrahedron 2011, 67, 9529–9534. [Google Scholar]
- 26.Fernandez-Mateos A; Herrero Teijon P; Rabanedo Clemente R; Gonzalez RR A Radical Clock for Reactions of Epoxy Derivatives Induced by Titanocene Chloride Synlett 2008, 3208–3212. [Google Scholar]
- 27.Newcomb M; Choi SY; Horner JH Adjusting the Top End of the Alkyl Radical Kinetic Scale. Laser Flash Photolysis Calibrations of Fast Radical Clocks and Rate Constants for Reactions of Benzeneselenol J. Org. Chem 1999, 64, 1225–1231. [Google Scholar]
- 28.Carreras J; Caballero A; Pérez PJ Alkenyl Boronates: Synthesis and Applications Chem. Asian J 2019, 14, 329–343. [DOI] [PubMed] [Google Scholar]
- 29.Beckwith ALJ; Schiesser CH Regio- and stereo-selectivity of alkenyl radical ring closure: A theoretical study Tetrahedron 1985, 41, 3925–3941 [Google Scholar]
- 30.Nicewicz DA; Hamilton DS Organic Photoredox Caalysis as a General Strategy for Anti-Markovnikov Alkene Hydrofunctionalization Synlett 2014, 25, 1191–1196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bonet A; Odachowski M; Leonori D; Essafi S; Aggarwal VK Enantiospecific sp(2)-sp(3) coupling of secondary and tertiary boronic esters Nat. Chem 2014, 6, 584–589. [DOI] [PubMed] [Google Scholar]
- 32.Imao D; Glasspoole BW; Laberge VS; Crudden CM Cross Coupling Reactions of Chiral Secondary Organoboronic Esters With Retention of Configuration J. Am. Chem. Soc 2009, 131, 5024–5025. [DOI] [PubMed] [Google Scholar]
- 33.Martínez-Botella G; Breen JN; Duffy JES; Dumas J; Geng B; Gowers IK; Green OM; Guler S; Hentemann MF; Hernandez-Juan FA; Joseph-McCarthy D; Kawatkar S; Larsen NA; Lazari O; Loch JT; Macritchie JA; McKenzie AR; Newman JV; Olivier NB; Otterson LG; Owens AP; Read J; Sheppard DW; Keating TA Discovery of Selective and Potent Inhibitors of Gram-Positive Bacterial Thymidylate Kinase (TMK) J. Med. Chem 2012, 55, 10010–10021. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.