

Abstract
Hemophilia A and B are inherited X‐linked disorders of hemostasis, associated with an increased bleeding tendency. Patients with severe hemophilia have undetectable clotting factor levels and experience spontaneous bleeds. In patients with nonsevere hemophilia, the clotting factor levels are 2% to 40% of normal and bleeds predominantly occur after provocative events such as trauma and surgery. Despite this milder phenotype, patients with nonsevere hemophilia may suffer from considerable morbidity and have an increased mortality risk. However, many aspects of the course of disease and treatment remain unclear. Information on the factors influencing interindividual differences in bleeding phenotype is lacking, and misdiagnosis may occur due to assay discrepancies in the diagnostic workup. Desmopressin is the preferred treatment modality, but some patients and indications require treatment with clotting factor concentrates. This may elicit inhibitor formation, which is associated with an increased burden of disease and a higher mortality rate. It has been found that patients with nonsevere hemophilia A carry a lifelong risk for this serious complication. In this review, we provide an overview of the current knowledge of the diagnosis and management of nonsevere hemophilia. A report of science presented at the International Society on Thrombosis and Haemostasis 2019 Annual Congress is also provided.
Keywords: diagnosis, hemophilia A, hemophilia B, phenotype, treatment
Essentials.
A State of the Art lecture, “Hemophilia Management: Huge Impact of a Tiny Difference” was presented at the ISTH Congress 2019.
In the diagnostic workup of nonsevere hemophilia, both the one‐stage and chromogenic factor assay should be performed.
Patients with nonsevere hemophilia A have a lifelong risk of inhibitor development to factor VIII concentrate.
Treatment with desmopressin should always be considered in nonsevere hemophilia A.
1. INTRODUCTION
Hemophilia A and B are hereditary X‐linked disorders of hemostasis that are associated with an increased bleeding tendency. They are caused by mutations in the F8 or F9 gene, leading to a deficiency of coagulation factor VIII (FVIII) or IX (FIX), associated with hemophilia A or B, respectively 1. Healthy individuals have a plasma concentration of clotting factor VIII or IX of 50 to 150 IU/dL. In patients with hemophilia, a residual factor level < 1 IU/dL is classified as severe disease, 1 to 5 IU/dL as moderate and > 5 to 40 IU/dL as mild hemophilia.2 The latter 2 are also being referred to as nonsevere hemophilia. The clinical phenotype of hemophilia is associated with the residual factor level and ranges from spontaneous bleeding episodes to minor bleeding after medical procedures or trauma. While patients with severe hemophilia are often confronted with a spontaneous onset of bleeds, especially into joints, most nonsevere hemophilia patients will only suffer from bleeding after a provocative event, such as a trauma or surgical interventions. To prevent or treat such bleeds, the deficient coagulation FVIII or FIX is replaced by intravenous administration of coagulation factor concentrates or by inducing a rise of FVIII by administration of desmopressin in patients with nonsevere hemophilia. Patients with severe hemophilia require multiple prophylactic infusions on a weekly basis to prevent the occurrence of spontaneous and/or life‐threatening bleeds. The majority of patients with nonsevere hemophilia do not require prophylactic infusion of clotting factor concentrates, and they are treated only when bleeding occurs. Nonetheless, patients with nonsevere hemophilia do experience bleeding symptoms and complications from their hemophilia, and health care providers may face several challenges with regard to the management of this patient group. This review presents an overview of the epidemiology, diagnosis, and management of nonsevere hemophilia, including current standards of care and future perspectives for treatment.
1.1. Epidemiology
A recent study that used registry data from 6 high‐income countries estimated a worldwide prevalence of 29.6 persons with hemophilia per 100 000 males for both hemophilia A and B of all severities. These numbers correspond to an expected total number of 1 125 000 patients with hemophilia globally, with 707 000 of them affected by nonsevere hemophilia.3 These numbers are in contrast to the real‐world numbers reported by the World Federation of Hemophilia (WFH). According to their 2017 Global Survey that included data from 117 countries, 64 534 nonsevere hemophilia patients (39 292 mild, 25 242 moderate) have been registered around the world.4 Hence, it is clear that the vast majority of patients with nonsevere hemophilia remain unidentified by hemophilia treatment centers. Large regional differences in the registration are present, as the majority of patients with mild hemophilia have been registered in Europe, followed by North and South America and Asia, while only a small percentage of all patients with mild hemophilia have been registered in African countries5 (unpublished data). In addition, global distribution differs between nonsevere hemophilia A and B. It has been generally accepted that hemophilia B accounts for around 20% of all hemophilia patients, and the WFH Global Survey 2017 indeed demonstrated that 16% of all registered patients with hemophilia were people with hemophilia B.6 This is in line with the results of a study using national registries that estimated a prevalence of 15.1 and 3.5 cases per 100 000 males of nonsevere hemophilia A and B, respectively.3 However, our research group recently identified a gradient in the distribution of nonsevere hemophilia A and B across Europe with a higher proportion of nonsevere hemophilia B (15%‐20%) compared to hemophilia A in the northern European countries, and a lower prevalence (10%) in the southern European countries (unpublished data on file with the authors). These data were collected in the centers that participate in the INSIGHT consortium. The INSIGHT consortium was initiated in 2008 and is a collaboration among 34 hemophilia treatment centers in 10 European countries and Australia, including data from 2711 patients with nonsevere hemophilia A.7, 8
2. CAUSES OF NONSEVERE HEMOPHILIA
Hemophilia A is caused by variants in the gene that encodes coagulation FVIII. There are currently 2015 unique variants of the F8 gene reported in the international database (http://www.factorviii-db.org), corresponding to 5480 individual cases. Hemophilia B is caused by variants in the F9 gene that encodes coagulation FIX. There are currently 1094 unique variants in the F9 gene (http://www.factorix.org), corresponding to 3713 individual cases. In the severe form of the disorder, null mutations (partial‐ or whole‐gene deletions, intron inversions, stop codon, insertions, etc) prevail, whereas in patients with mild hemophilia, missense mutations are most frequently present (89% in hemophilia A and 77% in hemophilia B), as illustrated in Figure 1. Notwithstanding the large genetic heterogeneity, some mutations are shared by several apparently unrelated families as a result of recurrent mutations (RMs) at particular sites prone to spontaneous mutations (eg, C > T transition at CpG dinucleotides) and are defined as “mutation hotspots.” This mechanism appears to be largely responsible for the occurrence of de novo mutations, usually more prevalent in patients with the severe form of the disease. Alternatively, identical mutations may result from a founder mutation that is transmitted through generations and induces a high frequency of specific mutations in different, sometimes isolated patient populations. The offspring affected by the founder mutation are identical by descent (IBD). Without extensive haplotyping it is difficult to ascertain whether a mutation is recurrent or inherited by IBD mechanisms. Haplotyping has revealed that an F8 mutation (p.Val 2035Ala) with an extraordinary high prevalence of mild hemophilia A in a Canadian population could be explained by IBD.9 Similarly, 3 different mutations in the F9 gene (n‐6 G > A or HB Leyden, Gly60 Ser, and Ala271Val) were identified in 24 of 29 Irish kindreds with mild hemophilia B to be the result of IBD, and this explained a prevalence twice as high as reported in other countries.10 When nearly all patients with hemophilia B in Sweden were screened, it became clear that IBD mutations were present in 51% of them, especially in those affected by mild hemophilia.11 A similar pattern was also observed in the Swedish hemophilia A population.12 In a population of Italian patients with moderate hemophilia A of Northern Italy, a founder effect for the c.6046C > T variant (p.Arg2016Trp) in the F8 gene was identified, explaining the genetic origin of hemophilia in 8% of the total hemophilia A population.13 Recently, it has been shown that 6% (61/992) of French patients with mild hemophilia A have a recurrent F8 intronic deletion (c.2113 + 461_2113+461_473del).14 These patients share the same haplotype, suggesting an IBD mechanism, although RM could not be definitely excluded because other, more rare deletions were detected within the same poly(T)‐tail of AluY in F8 intron 13.14 The latter observation shows that discriminating between IBD from RM may sometimes not be so easy when considering gene portions more susceptible to particular mechanisms of gene variations.
Figure 1.
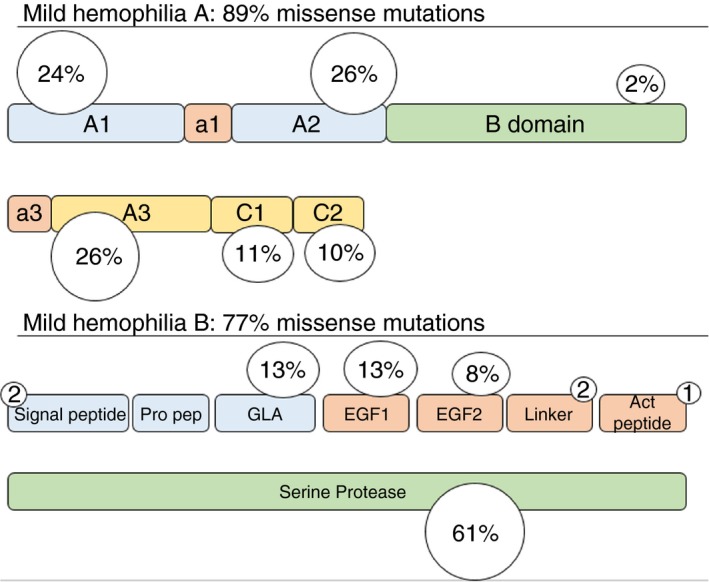
Distribution of mutations in F8 and F9 gene for mild hemophilia A and B, respectively. Figure is based on data from the F8 and F9 international database (http://www.factorviii-db.org and http://www.factorix.org)
3. DIAGNOSIS OF NONSEVERE HEMOPHILIA
The diagnosis and classification of the severity of hemophilia A and B is based on the residual clotting FVIII or FIX activity.15 It is important to acknowledge that mild deficiencies of FVIII and FIX may not prolong the activated partial thromboplastin time (APTT) screening test. Therefore, in patients with suspected mild hemophilia based on a bleeding history or their family history, specific measurement of FVIII or FIX is necessary. FVIII or FIX activity can be measured by using the one‐stage assay, two‐stage assay, or the chromogenic assay. The one‐stage assay is the most commonly used assay, as is evident from a survey performed in 2013 by the ECAT Foundation, which reported that 90% of the participating laboratories primarily used the one‐stage assay.16
The one‐stage assay is an APTT‐based assay in which the FVIII or FIX activity is quantified by comparing the shortening of the APTT in the sample plasma with the reference standard curve. The chromogenic assay is a 2‐step process: In the first step, activated factor X (FXa) is generated by using FVIII or FIX in the sample plasma as a cofactor. In the second step, the produced FXa hydrolyzes the chromogenic substrate, producing a color intensity that is proportional to the amount of FXa in the sample. The product FXa is directly proportional to the FVIII or FIX activity in the citrated plasma sample.17
Several studies have observed discrepancies between the one‐stage and the chromogenic assay in nonsevere hemophilia. Assay discrepancies are reported as the ratio of the one‐stage assay over the chromogenic assay. A significant discrepancy is present when the ratio is < 0.5 or > 2.0 according to a recent Scientific and Standardization Committee (SSC) communication addressing this issue.18 Previous studies in nonsevere hemophilia A have reported an assay discrepancy prevalence of 12% to 40%.19, 20, 21, 22, 23, 24, 25 Studies in hemophilia B are scarce, with 1 study demonstrating a prevalence of 25% assay discrepancy in a nonsevere hemophilia B population.26
In nonsevere hemophilia A, assay discrepancies with higher one‐stage assay results are predominantly seen in patients with mutations in the A1, A2, and A3 domain at the interface between the subunits. This may be explained by the fact that these mutations reduce the stability of the A2 domain in the activated FVIII heterotrimer (FVIIIa), thereby destabilizing FVIIIa. This phenomenon is minimized in the one‐stage assay, since activation of the FVIII protein occurs in the final steps, once calcium is added. However, in the chromogenic assay the inactive FVIII protein is already proteolytically activated in the first step of the incubation. The destabilization of the FVIIIa heterotrimer leads to a lower measurement of FVIII activity in the chromogenic assay compared to the one‐stage assay.17, 24 Higher activity measures with the chromogenic assay are present in patients with mutations that are located in the vicinity of the thrombin cleavage site, reducing the affinity for thrombin. The chromogenic assay will overcome this reduced thrombin affinity, as excess thrombin is added to activate FVIII. In contrast, the one‐stage assay is dependent on the physiological thrombin concentrations, and therefore lower results are found in the one‐stage assay for patients who have mutations that affect thrombin binding.17, 24
In nonsevere hemophilia B, assay discrepancy was predominantly present with higher results with the chromogenic assay.26 The higher chromogenic assay results were found in patients with mutations p.Arg191His and p.Arg191Cys, located at the N‐terminal cleaving site of the linker protein domain. It remains unclear how these mutations affect the FIX activation in vitro. As these patients have a mild bleeding pattern, it has been suggested by the authors that the chromogenic assay is more reflective of the clinical phenotype. However, no firm conclusions can be made due to the retrospective nature of the data and the small number of patients.26
4. BLEEDING PHENOTYPE IN NONSEVERE HEMOPHILIA
Patients with nonsevere hemophilia generally suffer from bleeding after provocative events, such as trauma and/or surgery. In contrast to severe hemophilia, information on the burden of disease in nonsevere hemophilia is limited. In nonsevere hemophilia, few studies have addressed the bleeding phenotype and its association with residual FVIII or FIX levels. In this section, we discuss the currently known data on the type and frequency of bleeding and timing of treatment in nonsevere hemophilia.
The frequency of bleeding in hemophilia is generally expressed as the annual bleeding rates (ABRs). A previous cohort study in Italy, performed by Tagliaferri et al,27 found a mean ABR of 0.56 (SD ± 0.67) for patients with mild hemophilia A. These results are in line with our own findings of a median ABR of 0.8 (interquartile range, 0.3‐2.5) in patients with mild hemophilia A from the INSIGHT study8 (unpublished data) and other previously published studies of patients with nonsevere hemophilia A, reporting an ABR of 0.5‐0.6.28, 29 In the Italian cohort, most patients experienced mucocutaneous bleeds (80%), followed by muscle bleeds (34%) and joint bleeds (31%). When we focus on the annual joint bleeding rate (AJBR), a rate of 0.08 (SD ± 0.26) is seen in the cohort study of Tagliaferri et al.27 In contrast, a recent study performed in the United States by Soucie et al, demonstrated a higher AJBR of 0.97 in patients with mild hemophilia A. However, for this latter study the data on joint bleeds were collected through patient‐reported forms, and this may have potentially led to an overestimation of the AJBR due to misclassification of bleeds by patients.30
The first detailed study to investigate the association between baseline FVIII levels and the bleeding phenotype was a single‐center study from the Netherlands including 377 patients.31 This study found that the age at first FVIII treatment increased with a higher FVIII level, as the median age at first treatment with FVIII concentrate was 2.9 and 5.5 years in the moderate and mild hemophilia A groups, respectively. A similar trend was seen for the age at the first joint bleed.31 At the age of 20 years, 54% of all patients with mild hemophilia had never experienced a joint bleed. As the study presented data from a single‐center cohort, we investigated the clinical bleeding phenotype in a patient sample that is more broadly representative across Europe. The preliminary results from data collected within the INSIGHT consortium demonstrate that the median age at first treatment with FVIII concentrates increased from 2.5 years in patients with baseline FVIII levels between 2 and 5 IU/dL to a median age of 4.4 years in patients with a baseline FVIII level between 5 and 40 IU/dL.32
Even though nonsevere hemophilia is characterized by a milder bleeding phenotype, patients can still suffer from life‐threatening and fatal bleeds. Intracranial hemorrhage is one of the major causes of fatal bleeding. When compared to the general population a 3.5‐fold higher mortality rate is found for intracranial hemorrhage in our INSIGHT cohort.33 Intracranial hemorrhage remains a serious issue and portrays unmet needs in the management of nonsevere hemophilia.
5. LIFE EXPECTANCY AND QUALITY OF LIFE
5.1. Life expectancy
Over the past decades, the life expectancy and quality of life of patients with hemophilia have improved tremendously, as clotting factor products have become widely available in the Western world. In these areas of the world, the life expectancy is now approaching that of the general male population.34, 35, 36
During the 1980s it became apparent that FVIII and FIX plasma products transmitted viral infections, such as HIV and hepatitis C virus (HCV) infecting the hemophilia population. Although patients with severe hemophilia were mostly affected, this caused an increase in mortality and a reduction in the life expectancy of all patients with hemophilia. A national study performed in the Netherlands between 1992 and 2001, demonstrated that the life expectancy in the nonsevere hemophilia population was 67 and 73 years for moderate and mild hemophilia, respectively; this is 9 and 3 years lower than the life expectancy of 76 years in the general Dutch male population.35 Patients with moderate and mild hemophilia who were not affected by HIV or HCV have comparable life expectancies of 75 years.35
In nonsevere hemophilia, the all‐cause death rate is 19% higher (hazard ratio, 1.19; 95% confidence interval, 1.09‐1.29; P < 0.001) in comparison with the general population. The main causes of death are bleeding and hepatitis‐ and HIV‐related diseases.37, 38 As intracranial hemorrhage is a major cause of fatal bleeding in nonsevere hemophilia, this remains an important concern for hemophilia caregivers.39 The INSIGHT study demonstrated that intracranial hemorrhage was the cause of death in 12% of the 148 patients with nonsevere hemophilia A who died during an observation period of 30 years. The majority (n = 13/17; 77%) of the fatal intracranial hemorrhages occurred spontaneously.
Due to the increased life expectancy, the patient population of nonsevere hemophilia is aging. Therefore, besides hemophilia‐related comorbidities, patients may get other age‐related disorders including cardiovascular disease, diabetes, hypertension, and malignancies. It has been suggested in several reports that hemophilia protects from cardiovascular disease. Indeed, in a report by Darby et al,37 a reduction of 37% in mortality from ischemic heart disease is seen in nonsevere hemophilia when compared to the general population.
5.2. Health‐related quality of life of patients with nonsevere hemophilia
Health outcomes of patients with hemophilia have improved as a consequence of improved hemophilia management, patient education, and awareness. Despite these improved health outcomes, it has been reported that the health‐related quality of life is lower in nonsevere hemophilia when compared to the general population.40, 41, 42 In a Canadian study performed in patients with mild hemophilia A, lower scores are seen in the physical health status, general health, and role emotional domain in the Short Form 36 Health Survey Questionnaire when compared to the general male population. Poor physical health status was associated with joint damage.40 Other patient‐relevant health outcomes including activities (sports, household activities) and participation in work, school, and social interactions were similar to the general population.42 Besides, health‐related quality of life may be affected by treatment complications such as blood‐transmitted viral infections, HCV‐related liver disease, inhibitor development, and the quality of hemophilia care.43
6. TREATMENT
Bleeding in patients with nonsevere hemophilia A and B can be treated or prevented by administration of the deficient clotting factor, FVIII or FIX concentrate, respectively. Due to a baseline plasma concentration of endogenous FVIII or FIX, the target levels can be achieved with lower doses of the concentrate when compared to patients with severe hemophilia. For patients with mild hemophilia A, there is a preferred alternative treatment: desmopressin (DDAVP). DDAVP is a synthetic vasopressin analogue that elicits a 2‐ to 5‐fold rise in FVIII plasma concentrations by inducing the release of von Willebrand factor (VWF) from the storage organelles in the endothelial cells.44 There is a large heterogeneity in response to DDAVP, with some patients not responding at all and some demonstrating an increase in FVIII level of more than 5‐fold baseline level. The half‐life of FVIII following DDAVP administration, generally 6 to 8 hours, rises with age and is dependent on the basal and peak VWF antigen levels.45 Since endothelial storage of VWF may exhaust after 2 to 3 consecutive doses of DDAVP, this is associated with tachyphylaxis (ie, a reduced response on repeated administration). Therefore, DDAVP is not suitable for clinical management of surgical procedures that require prolonged daily administration of FVIII.46
Since DDAVP elicits a rise in the patient's own FVIII, there is no risk of inhibitor development, in contrast to treatment with FVIII concentrates.47 Therefore, the SSC guideline on the management of mild hemophilia A states that DDAVP is the treatment of choice for mild hemophilia A, unless the patient has been shown to be nonresponsive or DDAVP is contraindicated.18 To test responsiveness, all patients with mild hemophilia A should have a trial administration of DDAVP. Procoagulant activity of FVIII (FVIII:C) should be measured 1 hour and, if possible, 4 hours after administration. The FVIII:C should be measured with both the one‐stage and chromogenic assay during this trial to address any assay discrepancies that may be present. Monitoring of the response following DDAVP administration should be with the assay with the lowest FVIII:C baseline level. Because DDAVP also has an antidiuretic effect, hyponatremia and fluid overload may occur rarely, especially in small children with repeated dosing. Fluid intake should be limited during the 12 hours following DDAVP administration to avoid complications. Since thrombotic complications have been reported following the use of DDAVP, there is a relative contraindication to use it in patients with uncontrolled hypertension or recent cardiovascular events such as myocardial infarction or stroke.48, 49 Patients who have an inadequate response to DDAVP or in whom DDAVP is contraindicated should, of course, be treated with an FVIII concentrate.
7. INHIBITOR DEVELOPMENT IN HEMOPHILIA A AND B
7.1. FVIII and FXI inhibitors: incidence and clinical relevance
The development of neutralizing antibodies (inhibitors) against FVIII or FIX is the most severe complication of hemophilia treatment because they neutralize the procoagulant activity of the coagulation factor, thereby rendering replacement therapy ineffective.50, 51 In clinical practice, both FVIII and FIX inhibitors are measured by a Bethesda or Nijmegen‐modified Bethesda assay, that quantifies the neutralizing capacity of the antibodies.52
Inhibitor development occurs far more frequently in hemophilia A compared to hemophilia B. The cumulative incidence of inhibitor development is 30% in severely affected patients and about 13% in patients with nonsevere hemophilia A (Figure 2).7, 53, 54 In hemophilia B, inhibitor development is a rare event, occurring in 1.5% to 3% of all patients.55
Figure 2.
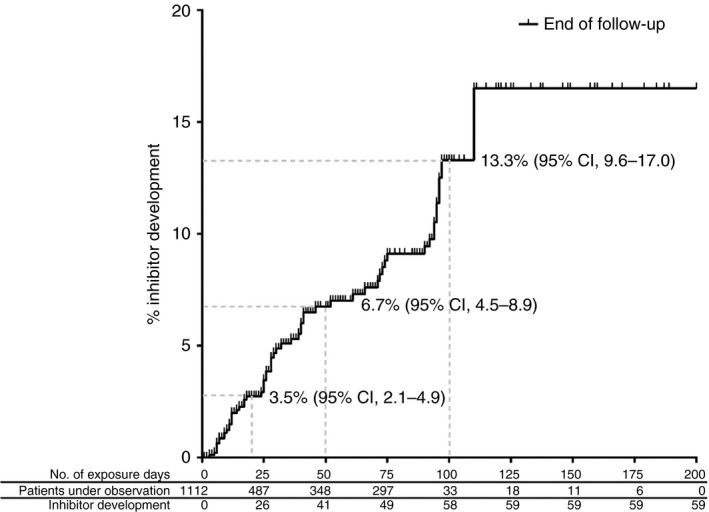
Cumulative inhibitor incidence in 1112 nonsevere hemophilia A patients, according to cumulative exposure days to factor VIII concentrates. This research was originally published in Blood Online. Eckhardt CL, van Velzen AS, Peters M, et al. Factor VIII gene (F8) mutation and risk of inhibitor development in nonsevere hemophilia A. Blood. 2013;122(11):1954‐62
7.2. Risk factors for FVIII and FIX inhibitor development
The risk of inhibitor development varies between individuals and depends on the interaction of multiple genetic and nongenetic risk factors.
Research on FVIII inhibitor development has mostly focused on patients with severe hemophilia A; therefore, our knowledge on risk factors and treatment strategies for patients with nonsevere hemophilia A is still limited. The INSIGHT study established that FVIII inhibitor development in nonsevere hemophilia A is associated with a far more severe clinical outcome than previously acknowledged, illustrated by a 10‐fold increase in bleeding rate and a 5‐fold increase in mortality rate.38, 56 In hemophilia B, morbidity is also related to the occurrence of allergic and/or anaphylactic reactions and nephrotic syndrome.57
The causative FVIII gene (F8) mutation is an important genetic risk factor for inhibitor development in nonsevere hemophilia A. The INSIGHT study identified 19 out of a total of 214 missense mutations that were associated with inhibitor development (Figure 3).7 These missense mutations may provoke antibody development because they encode an alternative peptide sequence within the endogenous FVIII protein compared to the wild‐type sequence. When the patient is treated with FVIII concentrate, he is exposed to the wild‐type sequence that may be recognized as nonself by the immune system and elicit an antibody response. Indeed, it has been demonstrated that antibodies can distinguish the therapeutic wild‐type FVIII from the patient's endogenous FVIII.58, 59
Figure 3.
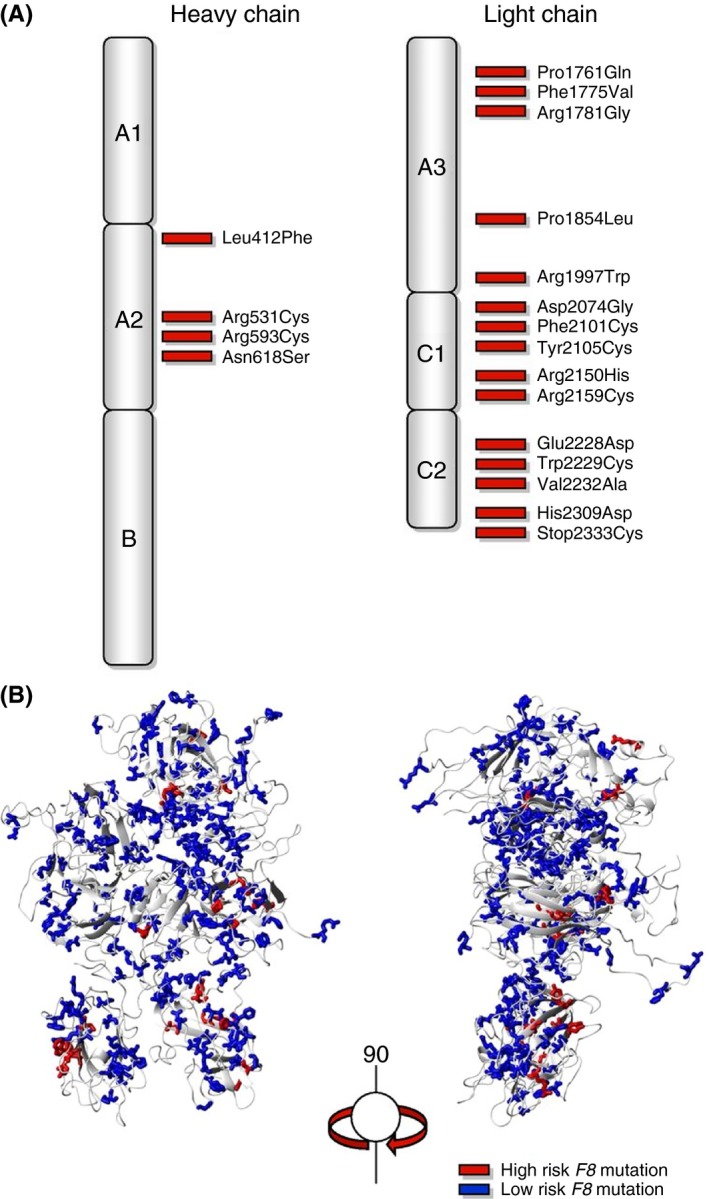
Distribution of F8 missense mutations associated with inhibitor development. (A) Two‐dimensional and (B) 3‐dimensional structure of the factor VIII protein. This research was originally published in Blood Online. Eckhardt CL, van Velzen AS, Peters M, et al Factor VIII gene (F8) mutation and risk of inhibitor development in nonsevere hemophilia A. Blood. 2013;122(11):1954‐62
With regard to clinical risk factors, both surgery and a high dose of FVIII concentrate have been identified as risk factors for inhibitor development in nonsevere hemophilia A.8 The association between treatment‐related risk factors and inhibitor development may be partly explained by the danger theory. This theory states that the FVIII protein itself is not sufficient to activate antigen‐presenting cells, but that substances released by damaged tissue, so‐called “danger signals,” are required to elicit an effective anti‐FVIII immune response.60
Our knowledge of risk factors for inhibitors in hemophilia B is limited to a small number of studies. Thus far, inhibitors have been reported predominantly in patients with severe hemophilia B with null mutations in the FIX (F9) gene (ie, deletion, stop codon) 55, 61 and compared to patients with hemophilia A, mild hemophilia B seems to carry a very low inhibitor risk.62, 63
7.3. The immunological characteristics of FVIII and FXI inhibitors
Currently, our knowledge of the immunological pathways driving inhibitor responses is limited to studies performed in hemophilia A, predominantly in patients with severe hemophilia A. These studies indicate that the response against FVIII depends on the help of CD4 + T cells. In this T‐cell–dependent pathway, presentation of internalized FVIII‐derived peptides that are bound by the major histocompatibility complex class II molecules is required for the activation of FVIII‐specific CD4 + T‐helper cells, which can subsequently stimulate B cells to produce high‐affinity anti‐FVIII antibodies, mostly from IgG1 and IgG4 isotypes.64, 65, 66
To install preventive measures, we need to identify patients who are at risk of developing inhibitors by increasing our understanding of the underlying immunological pathways. In this regard, emerging data in patients with severe hemophilia A indicate that high‐affinity anti‐FVIII antibodies precede the appearance of FVIII inhibitors. Interestingly, these antibodies could already be detected 1.5 years before the first positive Bethesda assay.67 This finding suggests that high‐affinity FVIII‐specific IgG4 antibodies are associated with FVIII inhibitors through a distinct immune regulatory pathway and could serve as a biomarker for the early detection of evolving FVIII inhibitor responses.
8. NONREPLACEMENT THERAPY, WHAT TO EXPECT
Novel treatment agents are on the horizon and the bispecific antibody emicizumab is already available in Europe and the United States. Its advantages in severe hemophilia A are evident as the route of administration is subcutaneous and the half‐life is 28 days, securing steady levels instead of peaks and troughs. However, presently there is very little known about the efficacy of emicizumab in patients with nonsevere hemophilia A. As the bispecific antibody will compete with the endogenous FVIII for FIXa and FX, the additional effect of emicizumab depends on the residual endogenous FVIII activity and the affinity of this endogenous FVIII for the tenase complex.
9. ISTH MELBOURNE REPORT
At the ISTH 2019 meeting in Melbourne, 2 abstracts were presented on nonsevere hemophilia A that we would like to mention here. The research group of Chai‐Adisaksopha presented data on the Patient Reported Outcomes, Burdens and Experiences (PROBE) questionnaire for patients with nonsevere hemophilia. The health status of people living with nonsevere hemophilia was compared to the male control population, with both groups containing 183 participants. Results showed that patients with nonsevere hemophilia experienced acute and chronic pain more often, and pain medication was used more frequently when compared to controls. Patients with nonsevere hemophilia had a significantly higher number of sick days (mean 44.9 vs. 3.7 days; P < 0.001), and the overall health score was worse in nonsevere hemophilia as compared with controls (0.71 vs. 0.89; P < 0.001).68
Zanon et al69 presented data from the Italian EMO.REC registry demonstrating similar risks for the occurrence of intracranial hemorrhage in adults with mild hemophilia compared to adults with severe and moderate hemophilia. In mild hemophilia, hypertension was shown to be a major risk factor for intracranial hemorrhage.
10. FUTURE PERSPECTIVES
Previous research in patients with nonsevere hemophilia shows that unmet needs in the treatment and management are still present and that patients may experience a high burden of disease, especially when an inhibitor develops. Further research is needed to provide a better understanding of the bleeding phenotype in patients with nonsevere hemophilia, especially to identify factors that drive the intraindividual variation in bleeding phenotype. An international multicenter study, the DYNAMO study, is currently being performed within the INSIGHT consortium to address this question (ClinicalTrials.gov Identifier: NCT03623295). Patients with nonsevere hemophilia have a lifelong risk of developing an inhibitor directed toward the coagulation factor used for treatment. Clinical risk factors for inhibitor development have previously been identified in severe hemophilia, but the specific immunological mechanism(s) of inhibitor development in nonsevere hemophilia A remain unclear. Currently, research is being performed by the INSIGHT consortium, the FLOW study, with the aim to elucidate the immunological mechanisms of inhibitor development in nonsevere hemophilia A.
11. CONCLUSION
In conclusion, the field of nonsevere hemophilia still faces many challenges in diagnosis and management, requiring further research to answer the unmet needs in this population. In the diagnostic workup, it is advised to perform both the one‐stage and the chromogenic assay to prevent misdiagnoses. Patients with nonsevere hemophilia have a lifelong risk for inhibitor development, especially patients with genotypes that are known to have a high risk for this complication. Therefore, DDAVP should always be considered where possible to prevent unnecessary exposure to clotting factor concentrates. In the future, nonreplacement treatment options such as emicizumab could provide an alternative. However, this needs to be further evaluated.
RELATIONSHIP DISCLOSURES
GC received fees to act as a speaker or to participate in advisory board meetings from Ablynx, CSL Behring, Kedrion, Novo Nordisk, Shire/Takeda, Sobi, Roche, Uniqure, and Werfen and has received unrestricted research grants from CSL Behring, Pfizer, and Sobi. The institution of KF has received unrestricted research grants from CSL Behring and Novo Nordisk and her institution received consultancy fees from Grifols, Takeda, and Novo Nordisk. FK, AZ, AA, and SC report nothing to disclose.
AUTHOR CONTRIBUTIONS
FK, AZ, AA, SC, GC, and KF reviewed the literature and cowrote the manuscript. FK edited the final version of this manuscript. FK, AZ, AA, SC, GC, and KF reviewed and approved the final version of the manuscript.
Kloosterman F, Zwagemaker A-F, Abdi A, Gouw S, Castaman G, Fijnvandraat K. Hemophilia management: Huge impact of a tiny difference. Res Pract Thromb Haemost. 2020;4:377–385. 10.1002/rth2.12314
Handling Editor: Fiona Newall
REFERENCES
- 1. Fijnvandraat K, Cnossen MH, Leebeek FW, Peters M. Diagnosis and management of haemophilia. BMJ (Clinical Research Ed). 2012;344:e2707. [DOI] [PubMed] [Google Scholar]
- 2. White GC 2nd, Rosendaal F, Aledort LM, Lusher JM, Rothschild C. Ingerslev J. Definitions in hemophilia. Thromb Haemost. 2001;85:560. [PubMed] [Google Scholar]
- 3. Iorio A, Stonebraker JS, Chambost H, Makris M, Coffin D, Herr C, et al. Establishing the prevalence and prevalence at birth of hemophilia in males: a meta‐analytic approach using national registries. Ann Intern Med. 2019;171(8):540. [DOI] [PubMed] [Google Scholar]
- 4. World Federation of Hemophilia . World Federation of Hemophilia Annual Global Survey 2017, Interactive Graphs. [Google Scholar]
- 5. Personal calculations from data on mild hemophilia from the World Federation of Hemophilia Annual Global Surveys 2010–2017. The most recent registration numbers per country were used. [Accessed 2019 October 20] Available from https://www1.wfh.org/GlobalSurvey/Public_AGS/AGS_Patients_Severity_EN.aspx [Google Scholar]
- 6. World Federation of Hemophilia . World Federation of Hemophilia Annual Global Survey Report 2017. 2019. [Accessed 2019 October 20] Available from http://www1.wfh.org/publications/files/pdf-1714.pdf [Google Scholar]
- 7. Eckhardt CL, van Velzen AS, Peters M, Astermark J, Brons PP, Castaman G, et al. Factor VIII gene (F8) mutation and risk of inhibitor development in nonsevere hemophilia A. Blood. 2013;122:1954–62. [DOI] [PubMed] [Google Scholar]
- 8. van Velzen AS, Eckhardt CL, Peters M, Leebeek FWG, Escuriola‐Ettingshausen C, Hermans C, et al. Intensity of factor VIII treatment and the development of inhibitors in non‐severe hemophilia A patients: results of the INSIGHT case‐control study. J Thromb Haemost. 2017;15:1422–9. [DOI] [PubMed] [Google Scholar]
- 9. Xie YG, Zheng H, Leggo J, Scully MF, Lillicrap D. A founder factor VIII mutation, valine 2016 to alanine, in a population with an extraordinarily high prevalence of mild hemophilia A. Thromb Haemost. 2002;87:178–9. [PubMed] [Google Scholar]
- 10. Jenkins PV, Egan H, Keenan C, O'Shea E, Smith OP, Nolan B, et al. Mutation analysis of haemophilia B in the Irish population: increased prevalence caused by founder effect. Haemophilia. 2008;14:717–22. [DOI] [PubMed] [Google Scholar]
- 11. Hallden C, Martensson A, Nilsson D, Sall T, Lind‐Hallden C, Liden AC, et al. Origin of Swedish hemophilia B mutations. J Thromb Haemost. 2013;11:2001–8. [DOI] [PubMed] [Google Scholar]
- 12. Hallden C, Nilsson D, Sall T, Lind‐Hallden C, Liden AC, Ljung R. Origin of Swedish hemophilia A mutations. J Thromb Haemost. 2012;10:2503–11. [DOI] [PubMed] [Google Scholar]
- 13. Garagiola I, Seregni S, Mortarino M, Mancuso ME, Fasulo MR, Notarangelo LD, et al. A recurrent F8 mutation (c.6046C>T) causing hemophilia A in 8% of northern Italian patients: evidence for a founder effect. Molecular genetics & genomic. Medicine. 2016;4:152–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Jourdy Y, Janin A, Fretigny M, Lienhart A, Negrier C, Bozon D, et al. Recurrent F8 intronic deletion found in mild hemophilia A causes alu exonization. Am J Hum Genet. 2018;102:199–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Blanchette VS, Key NS, Ljung LR, Manco‐Johnson MJ, van den Berg HM, Srivastava A. Definitions in hemophilia: communication from the SSC of the ISTH. J Thromb Haemost. 2014;12:1935–9. [DOI] [PubMed] [Google Scholar]
- 16. Potgieter JJ, Damgaard M, Hillarp A. One‐stage vs. chromogenic assays in haemophilia A. Eur J Haematol. 2015;94(Suppl 77):38–44. [DOI] [PubMed] [Google Scholar]
- 17. Peyvandi F, Oldenburg J, Friedman KD. A critical appraisal of one‐stage and chromogenic assays of factor VIII activity. J Thromb Haemost. 2016;14:248–61. [DOI] [PubMed] [Google Scholar]
- 18. Makris M, Oldenburg J, Mauser‐Bunschoten EP, Peerlinck K, Castaman G, Fijnvandraat K. The definition, diagnosis and management of mild hemophilia A: communication from the SSC of the ISTH. J Thromb Haemost. 2018;16:2530–3. [DOI] [PubMed] [Google Scholar]
- 19. Cid AR, Calabuig M, Cortina V, Casana P, Haya S, Moret A, et al. One‐stage and chromogenic FVIII: C assay discrepancy in mild haemophilia A and the relationship with the mutation and bleeding phenotype. Haemophilia. 2008;14:1049–54. [DOI] [PubMed] [Google Scholar]
- 20. Poulsen AL, Pedersen LH, Hvas AM, Poulsen LH, Thykjaer H, Ingerslev J. Assay discrepancy in mild haemophilia A: entire population study in a National Haemophilia Centre. Haemophilia. 2009;15:285–9. [DOI] [PubMed] [Google Scholar]
- 21. Trossaert M, Boisseau P, Quemener A, Sigaud M, Fouassier M, Ternisien C, et al. Prevalence, biological phenotype and genotype in moderate/mild hemophilia A with discrepancy between one‐stage and chromogenic factor VIII activity. J Thromb Haemost. 2011;9:524–30. [DOI] [PubMed] [Google Scholar]
- 22. Duncan EM, Rodgers SE, McRae SJ. Diagnostic testing for mild hemophilia a in patients with discrepant one‐stage, two‐stage, and chromogenic factor VIII: C assays. Semin Thromb Hemost. 2013;39:272–82. [DOI] [PubMed] [Google Scholar]
- 23. Bowyer AE, Van Veen JJ, Goodeve AC, Kitchen S, Makris M. Specific and global coagulation assays in the diagnosis of discrepant mild hemophilia A. Haematologica. 2013;98:1980–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Pavlova A, Delev D, Pezeshkpoor B, Muller J, Oldenburg J. Haemophilia A mutations in patients with non‐severe phenotype associated with a discrepancy between one‐stage and chromogenic factor VIII activity assays. Thromb Haemost. 2014;111:851–61. [DOI] [PubMed] [Google Scholar]
- 25. Provaznikova D, Houskova K, Radovska A, Salaj P, Hrachovinova I. Novel mutations associated with a discrepancy between one‐stage and chromogenic FVIII activity assays. Haemophilia. 2015;21:e330–e332. [DOI] [PubMed] [Google Scholar]
- 26. Kihlberg K, Strandberg K, Rosen S, Ljung R, Astermark J. Discrepancies between the one‐stage clotting assay and the chromogenic assay in haemophilia B. Haemophilia. 2017;23:620–7. [DOI] [PubMed] [Google Scholar]
- 27. Tagliaferri A, Di Perna C, Riccardi F, Pattacini C, Rivolta GF, Franchini M. The natural history of mild haemophilia: a 30‐year single centre experience. Haemophilia. 2012;18:166–74. [DOI] [PubMed] [Google Scholar]
- 28. Aznar JA, Lucia F, Abad‐Franch L, Jimenez‐Yuste V, Perez R, Batlle J, et al. Haemophilia in Spain. Haemophilia. 2009;15:665–75. [DOI] [PubMed] [Google Scholar]
- 29. Batty P, Austin SK, Khair K, Millar CM, Palmer B, Rangarajan S, et al. Treatment burden, haemostatic strategies and real world inhibitor screening practice in non‐severe haemophilia A. Br J Haematol. 2017;176:796–804. [DOI] [PubMed] [Google Scholar]
- 30. Soucie JM, Monahan PE, Kulkarni R, Konkle BA, Mazepa MA. The frequency of joint hemorrhages and procedures in nonsevere hemophilia A vs B. Blood Adv. 2018;2:2136–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Den Uijl IE, Mauser Bunschoten EP, Roosendaal G, Schutgens RE, Biesma DH, Grobbee DE, et al. Clinical severity of haemophilia A: does the classification of the 1950s still stand? Haemophilia. 1950s;17:849–53. [DOI] [PubMed] [Google Scholar]
- 32. Kloosterman FK, Abdi A, Eckhardt CL, Male C, Castaman G, Fischer K, et al. Expected timing of initial exposure to factor VIII treatment in non‐replacement therapies in hemophilia A [abstract]. Special Issue: Abstracts of the XXVII Congress of the International Society on Thrombosis and Haemostasis, July 6–10, 2019 ed: Res Pract Thromb Haemost; 2019. p. 280–1. [Google Scholar]
- 33. Loomans JI, Eckhardt CL, Reitter‐Pfoertner SE, Holmstrom M, van Gorkom BL, Leebeek FWG, et al. Mortality caused by intracranial bleeding in non‐severe hemophilia A patients. J Thromb Haemost. 2017;15:1115–22. [DOI] [PubMed] [Google Scholar]
- 34. Larsson SA. Life expectancy of Swedish haemophiliacs, 1831–1980. Br J Haematol. 1985;59:593–602. [DOI] [PubMed] [Google Scholar]
- 35. Plug I, Van Der Bom JG, Peters M, Mauser‐Bunschoten EP, De Goede‐Bolder A, Heijnen L, et al. Mortality and causes of death in patients with hemophilia, 1992–2001: a prospective cohort study. J Thromb Haemost. 2006;4:510–6. [DOI] [PubMed] [Google Scholar]
- 36. Lovdahl S, Henriksson KM, Baghaei F, Holmstrom M, Nilsson JA, Berntorp E, et al. Incidence, mortality rates and causes of deaths in haemophilia patients in Sweden. Haemophilia. 2013;19:362–9. [DOI] [PubMed] [Google Scholar]
- 37. Darby SC, Kan SW, Spooner RJ, Giangrande PL, Hill FG, Hay CR, et al. Mortality rates, life expectancy, and causes of death in people with hemophilia A or B in the United Kingdom who were not infected with HIV. Blood. 2007;110:815–25. [DOI] [PubMed] [Google Scholar]
- 38. Eckhardt CL, Loomans JI, van Velzen AS, Peters M, Mauser‐Bunschoten EP, Schwaab R, et al. Inhibitor development and mortality in non‐severe hemophilia A. J Thromb Haemost. 2015;13:1217–25. [DOI] [PubMed] [Google Scholar]
- 39. Jardim LL, van der Bom JG, Caram‐Deelder C, Gouw SC, Leal Cherchiglia M, Meireles RS. Mortality of patients with haemophilia in Brazil: First report. Haemophilia. 2019;25:e146–e152. [DOI] [PubMed] [Google Scholar]
- 40. Walsh M, Macgregor D, Stuckless S, Barrett B, Kawaja M, Scully MF. Health‐related quality of life in a cohort of adult patients with mild hemophilia A. J Thromb Haemost. 2008;6:755–61. [DOI] [PubMed] [Google Scholar]
- 41. Miners AH, Sabin CA, Tolley KH, Jenkinson C, Kind P, Lee CA. Assessing health‐related quality‐of‐life in individuals with haemophilia. Haemophilia. 1999;5:378–85. [DOI] [PubMed] [Google Scholar]
- 42. Plug I, Peters M, Mauser‐Bunschoten EP, de Goede‐Bolder A, Heijnen L, Smit C, et al. Social participation of patients with hemophilia in the Netherlands. Blood. 2008;111:1811–5. [DOI] [PubMed] [Google Scholar]
- 43. Posthouwer D, Plug I, van der Bom JG, Fischer K, Rosendaal FR, Mauser‐Bunschoten EP. Hepatitis C and health‐related quality of life among patients with hemophilia. Haematologica. 2005;90:846–50. [PubMed] [Google Scholar]
- 44. Mannucci PM. Desmopressin (DDAVP) in the treatment of bleeding disorders: the first 20 years. Blood. 1997;90:2515–21. [PubMed] [Google Scholar]
- 45. Castaman G, Mancuso ME, Giacomelli SH, Tosetto A, Santagostino E, Mannucci PM, et al. Molecular and phenotypic determinants of the response to desmopressin in adult patients with mild hemophilia A. J Thromb Haemost. 2009;7:1824–31. [DOI] [PubMed] [Google Scholar]
- 46. Castaman G. Desmopressin for the treatment of haemophilia. Haemophilia. 2008;14(Suppl 1):15–20. [DOI] [PubMed] [Google Scholar]
- 47. Castaman G, Fijnvandraat K. Molecular and clinical predictors of inhibitor risk and its prevention and treatment in mild hemophilia A. Blood. 2014;124:2333–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Bond L, Bevan D. Myocardial infarction in a patient with hemophilia treated with DDAVP. N Engl J Med. 1988;318:121. [PubMed] [Google Scholar]
- 49. Byrnes JJ, Larcada A, Moake JL. Thrombosis following desmopressin for uremic bleeding. Am J Hematol. 1988;28:63–5. [DOI] [PubMed] [Google Scholar]
- 50. Ananyeva NM, Lacroix‐Desmazes S, Hauser CA, Shima M, Ovanesov MV, Khrenov AV, et al. Inhibitors in hemophilia A: mechanisms of inhibition, management and perspectives. Blood Coagul Fibrinolysis. 2004;15:109–24. [DOI] [PubMed] [Google Scholar]
- 51. Santoro C, Quintavalle G, Castaman G, Baldacci E, Ferretti A, Riccardi F, et al. Inhibitors in Hemophilia B. Semin Thromb Hemost. 2018;44:578–89. [DOI] [PubMed] [Google Scholar]
- 52. Verbruggen B, Novakova I, Wessels H, Boezeman J, van den Berg M, Mauser‐Bunschoten E. The Nijmegen modification of the Bethesda assay for factor VIII: C inhibitors: improved specificity and reliability. Thromb Haemost. 1995;73:247–51. [PubMed] [Google Scholar]
- 53. Wight J, Paisley S. The epidemiology of inhibitors in haemophilia A: a systematic review. Haemophilia. 2003;9:418–35. [DOI] [PubMed] [Google Scholar]
- 54. Gouw SC, van den Berg HM, Fischer K, Auerswald G, Carcao M, Chalmers E, et al. Intensity of factor VIII treatment and inhibitor development in children with severe hemophilia A: the RODIN study. Blood. 2013;121:4046–55. [DOI] [PubMed] [Google Scholar]
- 55. DiMichele D. Inhibitor development in haemophilia B: an orphan disease in need of attention. Br J Haematol. 2007;138:305–15. [DOI] [PubMed] [Google Scholar]
- 56. van Velzen AS, Eckhardt CL, Streefkerk N, Peters M, Hart DP, Hamulyak K, et al. The incidence and treatment of bleeding episodes in non‐severe haemophilia A patients with inhibitors. Thromb Haemost. 2016;115:543–50. [DOI] [PubMed] [Google Scholar]
- 57. Ewenstein BM, Takemoto C, Warrier I, Lusher J, Saidi P, Eisele J, et al. Nephrotic syndrome as a complication of immune tolerance in hemophilia B. Blood. 1997;89:1115–6. [PubMed] [Google Scholar]
- 58. James EA, Kwok WW, Ettinger RA, Thompson AR, Pratt KP. T‐cell responses over time in a mild hemophilia A inhibitor subject: epitope identification and transient immunogenicity of the corresponding self‐peptide. J Thromb Haemost. 2007;5:2399–407. [DOI] [PubMed] [Google Scholar]
- 59. Jacquemin M, Vantomme V, Buhot C, Lavend'homme R, Burny W, Demotte N, et al. CD4+ T‐cell clones specific for wild‐type factor VIII: a molecular mechanism responsible for a higher incidence of inhibitor formation in mild/moderate hemophilia A. Blood. 2003;101:1351–8. [DOI] [PubMed] [Google Scholar]
- 60. Matzinger P. The danger model: a renewed sense of self. Science (New York, NY). 2002;296:301–5. [DOI] [PubMed] [Google Scholar]
- 61. Chitlur M, Warrier I, Rajpurkar M, Lusher JM. Inhibitors in factor IX deficiency a report of the ISTH‐SSC international FIX inhibitor registry (1997–2006). Haemophilia. 2009;15:1027–31. [DOI] [PubMed] [Google Scholar]
- 62. Miller CH, Benson J, Ellingsen D, Driggers J, Payne A, Kelly FM, et al. F8 and F9 mutations in US haemophilia patients: correlation with history of inhibitor and race/ethnicity. Haemophilia. 2012;18:375–82. [DOI] [PubMed] [Google Scholar]
- 63. Radic CP, Rossetti LC, Abelleyro MM, Candela M, Perez Bianco R, de Tezanos PM, et al. Assessment of the F9 genotype‐specific FIX inhibitor risks and characterisation of 10 novel severe F9 defects in the first molecular series of Argentinian patients with haemophilia B. Thromb Haemost. 2013;109:24–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Wroblewska A, Reipert BM, Pratt KP, Voorberg J. Dangerous liaisons: how the immune system deals with factor VIII. J Thromb Haemost. 2013;11:47–55. [DOI] [PubMed] [Google Scholar]
- 65. Pashov AD, Calvez T, Gilardin L, Maillere B, Repesse Y, Oldenburg J, et al. In silico calculated affinity of FVIII‐derived peptides for HLA class II alleles predicts inhibitor development in haemophilia A patients with missense mutations in the F8 gene. Haemophilia. 2014;20:176–84. [DOI] [PubMed] [Google Scholar]
- 66. Ettinger RA, James EA, Kwok WW, Thompson AR, Pratt KP. HLA‐DR‐restricted T‐cell responses to factor VIII epitopes in a mild haemophilia A family with missense substitution A2201P. Haemophilia. 2010;16:44–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Hofbauer CJ, Whelan SF, Hirschler M, Allacher P, Horling FM, Lawo JP, et al. Affinity of FVIII‐specific antibodies reveals major differences between neutralizing and nonneutralizing antibodies in humans. Blood. 2015;125:1180–8. [DOI] [PubMed] [Google Scholar]
- 68. Chai‐Adisaksopha C, Skinner M, Page D, Stonebraker JS, Noone D, Curtis R, et al. Health status of people living with non‐severe Hemophilia ‐ Insights from the patient reported outcomes, burdens and experiences (PROBE) study [Abstract]. Special Issue: Abstracts of the XXVII Congress of the International Society on Thrombosis and Haemostasis, July 6–10, 2019 ed: Res Pract Thromb Haemost; 2019. p. 100. [Google Scholar]
- 69. Zanon E, Pasca S, Pollio B, Santoro C, Castaman G, Demartis F, et al. Final data from the Italian Registry on intracranial haemorrhage in haemophilia patients: EMO.REC registry (2009–2018) [Abstract]. Special Issue: Abstracts of the XXVII Congress of the International Society on Thrombosis and Haemostasis, July 6–10, 2019 ed: Res Pract Thromb Haemost; 2019. p. 331. [Google Scholar]