

Abstract
This protocol describes a streamlined method of plasmid DNA extraction by continual thermal lysis, a modification of the basic boiling lysis technique, to simplify the processing of large volumes of Escherichia coli cultures. Fermented bacteria are harvested using a hollow fiber-membrane module and pre-treated with lysozyme prior to passing through a thermal exchange coil set at 70 °C to lyse the cells, and into a juxtaposed cooling coil on ice. The lysed and cooled bacteria are subsequently separated from the lysate by centrifugation and plasmid DNA is precipitated from the supernatant for further purification. The use of peristaltic pumps and two heating coils at constant temperature without the use of centrifugation enable the lysis process to become constant and controllable, providing a flow-through protocol for cell lysis and plasmid DNA extraction. Large volumes of bacterial cultures (20 l) can be processed in 2 h, yielding approximately 100 mg plasmid DNA l−1 culture, making this an attractive protocol for consistent and large-scale preparation of plasmid DNA.
Introduction
Recent progresses in the development of DNA vaccination and gene therapy show great potential for the treatment of disease. As clinical application of these technologies will require large quantities of plasmid DNA at the milligram level, there is an increased demand for large quantities of recombinant plasmid DNA1,2. Thus, developing a large-scale, reproducible and cost-effective protocol for plasmid DNA preparation is now one of the most challenging tasks in DNA vaccine development3,4,5.
Target DNA sequences can be amplified to produce large quantities of purified DNA for use in such applications as gene therapy by ligation into a plasmid vector, which can then be transformed into suitable bacterial cells. These recombinant bacterial strains will replicate the plasmid, producing large quantities of the target DNA6. A typical process of plasmid DNA generation includes the construction and selection of appropriate expression vectors, production of recombinant microorganisms and cell growth, followed by lysis of the cells, and the extraction and purification of plasmid DNA7. When the plasmids are extracted and purified, they usually result in one of two forms: either open-circled or supercoiled. However, when they are used in gene therapy or vaccine immunization, the plasmid with the supercoiled form is preferred. During plasmid DNA extraction, the step of lysing bacterial cells without losing plasmids is probably the most critical for downstream processing8, as the quantity of plasmid DNA recovered is mostly determined by this step after fermentation9,10.
Plasmid DNA extraction from bacterial cells usually begins with a chemical lysis step6, such as alkaline lysis11, which is a widely adopted method in research laboratories. However, during alkaline lysis, precipitates form that contain cell debris, denatured proteins and nucleic acids, which must be removed. Centrifugation on a fixed-angle rotor is the most common operation during this process12,13 and is a rate-limiting step for a large-scale preparation14. An alternative method is boiling lysis, in which the 100 °C thermal treatment disrupts and makes cells release the plasmid DNA15. Although boiling lysis can isolate more plasmids than alkaline lysis, the inconsistent plasmid yield and purity, and the difficulty in operation, make this method undesirable even for laboratory research-scale preparations16. For large-scale production, a high-pressure homogenizer is used to continuously disrupt cells17. However, this often causes problems in collecting fragmented DNA plasmids and more contaminated genomic DNA due to its severe fluid dynamic forces for destruction17. Therefore, the development of a highly efficient continuous lysis and scalable protocol becomes a challenging but important issue.
Based on the conventional boiling lysis protocol18, Lee and Lander have made several modifications to improve the method19,20. Although these modifications significantly improve the efficiency of boiling lysis and the yield of plasmid DNA, they require centrifugation steps at the bacterial-collecting and plasmid-extracting stage, special equipment and reagents. In a recent report21, we described a design for a flow-through protocol from cultured bacteria for the extraction of plasmid DNA. The schematic design of the device and set-up is shown in Figure 1. The use of a 0.2 μm pore size hollow fiber-membrane module to harvest the fermented bacteria will minimize the centrifugation step, which is one of the limiting steps in large-scale preparation. Pumping through the heat-exchanging coils, cells receive uniform thermal treatment regardless of the volume of the sample, which ensures continuity and homogeneity of cell lysis. The diameter of copper coils can vary, depending on the volume of fermentation. This provides versatility for any given volume of samples. This protocol does not aim to increase the yield of plasmid, but to provide consistency between batches, which can otherwise be lacking in boiling lysis methods. A centrifugation step is, however, still required to separate the plasmid-containing supernatant from the lysed bacteria, which will require further optimization. Overall, this protocol provides a scalable, efficient and cost-effective preparation system to meet the needs of large-scale plasmid production for both laboratories and industry. The protocol mainly details the lysis of cells and the extraction of plasmid DNA, and although further purification procedures may be required, they are not explained in detail within this protocol.
Figure 1. Schematic diagram of the apparatus for continuous thermal lysis of bacterial cells and isolation of plasmid DNA.
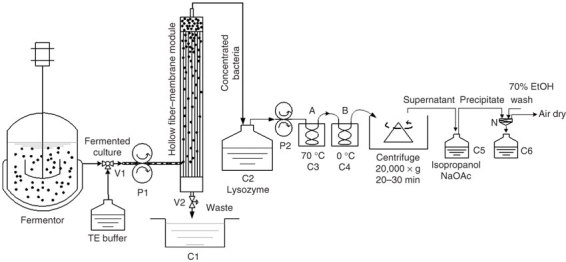
Cells are pumped from the fermentor into the hollow fiber-membrane module by peristaltic pump (P1) via a 3-way inlet valve (V1) and washed once with TE while inside the module. The waste liquid is discharged via an outlet valve (V2) into a waste container (C1). After closing the outlet valve V2, the concentrated cells are pumped from the top outlet of the module into a cell collection container (C2), where the cells are adjusted to the OD 600 nm of 100 by TE and treated with lysozyme before being pumped into the thermal exchange coils by a second peristaltic pump (P2); cells first pass through a thermal exchange coil (A) in a 70 °C water bath (C3) for continual thermal lysis, and are then immediately pumped through a second thermal exchange coil (B) in an ice bath (C4) and are collected in a container, which is also in the ice bath. After thermal treatment, lysed cells are centrifuged and the supernatant is mixed with 0.6 volume of isopropanol and 1/10 volume of NaOAc in container C5. Precipitated plasmid is collected on the 200 mesh nylon filter (N), whereas waste liquid is collected in container C6. Precipitates are washed with 50 ml of 70% EtOH and air-dried.
Materials
Reagents
Yeast extract and tryptone (Oxford Ltd, cat. no. LP0021 and LP0042)
Ampicillin and kanamycin (Amresco, cat. No. 0741 and 0408)
-
Plasmid DNA
Critical
The following plasmids have been extracted using this protocol: pcDNA3, pGEX-4T-1, pVAX1 and pcD-VP1. The latter, encoding a capsid protein VP1 (639 bp) of foot and mouth disease virus (FMDV; described previously22), was extensively used during the optimization of this protocol.
-
Escherichia coli: TOP10 (Invitrogen Inc.) TOP10 provides a high yield of plasmid extraction, in general, from our experience (see Table 1).
Critical
XL1-Blue (Strategen Inc.), DH5a and JM109 can also be used.
KH2PO4 (Beijing Chemical Reagents Company, cat. no. 100017692)
Na2HPO4 (Beijing Chemical Reagents Company, cat. no. K7164901)
(NH4)2SO4 (Beijing Chemical Reagents Company, cat. no. 10002918)
NH4Cl (Beijing Chemical Reagents Company, cat. no. 10001528)
Peptone (Universen Biotech, cat. no. 01-001)
Glycerol (Beijing Chemical Reagents Company, cat. no. 10010618)
MgSO4 (Beijing Chemical Reagents Company, cat. no. 20025128)
1 M Tris-HCl (Sigma, cat. no. T1819)
1 M EDTA, pH 8.0 (Sigma, cat. no. T3913)
-
Lysozyme (Fluca, cat. no. 62790)
Critical
Make up fresh to 1 mg ml−1 in H2O.
1 M NaCl (Beijing Chemical Reagents Company, cat. no. 10019392)
Triton-X-100 (Sigma, cat. no. T8787)
1 M Tris-Acetate (Sigma, cat. no. 93339)
NaOAc (3 M, pH 5.2) (Beijing Chemical Reagents Company, cat. no. 10018728)
Isopropanol (Beijing Chemical Factory, cat. no. 32064)
70% Ethanol (Beijing Chemical Factory, cat. no. 32061)
50% PEG 8000 (in 1.6 M NaCl solution) (Amresco, cat. no. 0159)
10 mg ml−1 Ethidium Bromide (EtBr) (Sigma, cat. no. E7637)
Agar (Sigma, cat. no. 05039)
Agarose (Biowest, cat. no. A6585)
DNA Markers (TaKaRa, cat. no. D501A)
Restriction enzymes (TaKaRa, cat. no. D1040A and D1093A)
Table 1.
Plasmid yields prepared from four different Escherichia coli strains.
| E. coli strain | DH5 | JM109 | TOP10 | XL1-blue |
|---|---|---|---|---|
| OD260/280 | 1.848 | 1.858 | 1.828 | 1.842 |
| pcDNA3d-N (mg) | 1.687 | 1.573 | 1.702 | 1.694 |
Notes: (1) In order to compare the yield from different bacterial strains, these results were obtained following manual plasmid extraction by a standard boiling lysis method18 from 400 ml culture produced overnight in a flask at 37 °C with 200 rpm shaking. The value of OD 600 nm was around 2. (2) Plasmid pcDNA3d-N was obtained by cloning of the N protein of the SARS-CoV coding region into pcDNA3d23.
Equipment
UV/VIS spectrophotometer (Unicon Inc.)
7 l fermentor vessel (Bioflo 110; New Brunswick Scientific) with a proportional integral derivative (PID) cascade controller
0.2 μm pore-size hollow-fiber membrane module (Tianjin Motian Membrane Eng. & Tech. Co. Ltd.), or similar product from other manufacturer
2-way peristaltic pumps (Huxi Instrument Factory)
200 mesh nylon filter (Wangyi, cat. no. D501A)
Thermal exchange coils: made of a copper tube (10 mm inner diameter) and bended into a circular shape with three turns at 80 mm diameter and used for 20 l of the starting fermented culture
The containers C3 and C4 are 25 cm in diameter and 20 cm in depth
Tubing to connect the components of the apparatus: silica tubing with an inner diameter of 10 mm is used for the connection
Reagent setup
Antibiotics Each antibiotic should be individually made into solution in TE, filtered with a 0.2 μm filter and stored at –20 °C.
-
Semi-synthesized medium 5 g l−1 Tryptone, 5 g l−1 yeast extract, 4 g l−1 KH2PO4, 4 g l−1 K2HPO4, 7 g l−1 Na2HPO4, 1.2 g l−1 (NH4)2SO4, 0.2 g l−1 NH4Cl and 100 mg l−1 of antibiotics.
Critical
As both K2HPO4 and Na2HPO4 are easily moisturized in the air, they should be weighed as quickly as possible and dissolved in water first. Chemicals should be dissolved into solution one after other and the tryptone and yeast extract should be the last to be dissolved. After the medium is autoclaved at 121 °C for 20 min and cooled down to room temperature, the filtered antibiotic should be injected via a sterilized inlet valve into the medium.
Feeding medium 71 g l−1 yeast extract, 71 g l−1 peptone, 170 g l−1 glycerol, 5.7 g l−1 MgSO4. The yeast extract, peptone and glycerol should be made in one bottle in water and the MgSO4 made in another bottle in water; they are then autoclaved separately at 121 °C for 20 min and combined together with the same volume after they are cooled to room temperature.
TE buffer 10 mM Tris-HCl, 1 mM EDTA, pH 8.0.
-
10× Lysis buffer 100 μg ml−1 of lysozyme, 1 M of NaCl and 20% of Triton-X-100.
Critical
Lysis buffer must be freshly made.
TAE buffer 40 mM Tris-Acetate, 1 mM EDTA.
LB medium 10 g l−1 tryptone, 5 g l−1 yeast extract, 5 g l−1 NaCl in water. Add the desired antibiotic at the following concentrations: Ampicillin, add 1 ml of 100 mg ml−1 ampicillin per liter of media (final = 100 μg ml−1); Kanamycin, add 1 ml of 50 mg ml−1 kanamycin per liter of media (final = 50 μg ml−1).
LB agar plates Include 15 g agar with the ingredients for 1 l of LB medium. Pour approximately 30 ml LB-agar at 50 °C per plate and store plates at 4 °C. The concentration of antibiotic is the same as for LB medium.
Procedure
Fermentation
Transform a plasmid of interest into competent E. coli TOP10 using an appropriate method (e.g., heat shock or electroporation15).
Propagate bacteria in a suitable antibiotic-selective LB culture15 at 37 °C for 1 h.
-
Plate bacteria onto LB agar plates containing 50 μg ml−1 of kanamycin and incubate at 37 °C overnight.
Critical Step
Do not plate too many cells onto the selective plate. It may be necessary to plate 10 μl and 100 μl of transformed bacteria onto different plates to obtain appropriate colony separation.
-
Pick a single recombinant bacterial colony carrying the target plasmid and seed onto 10 ml of antibiotic-selective LB medium.
Critical Step
Select a single well-separated E. coli colony and use it for seed culturing.
-
Incubate at 37 °C overnight at 200 rpm in an orbital shaker for 10–12 h.
Critical Step
The bacteria should not be used to seed cultures beyond their log phase (if their OD value is more than 0.6), otherwise a low yield of plasmid can occur.
Inoculate 5 ml of the seed bacteria onto 50 ml of LB medium containing an appropriate amount of antibiotics, and incubate at 37 °C for an additional 12 h in the orbital shaker.
Inoculate 50 ml culture into 4 l of semi-synthesized media with antibiotics in an automatically controlled fermentor.
-
Ferment using the following conditions: set the temperature at 37 °C and the dissolved oxygen (DO) at 30%. Maintain the pH at 7.0 by the addition of ammonium hydroxide solution (30%) and 5 M hydrochloric acid (HCl). Set the inflow sparge air to 7 l min−1 and add 400 ml of feeding media at the rate of 20 ml h−1. It takes 10–12 h to complete the fermentation, but it can be varied and is dependent on the bacteria used.
Critical Step
During fermentation, a constant volume of DO should be maintained at 30%; when DO is more than 30%, the bacteria will grow quicker, but the plasmid copies will be lost as the plasmid could not replicate quickly enough to keep up with the rapid cell division. We found the optimal DO value to be 30% in different bacterial strains and different plasmid DNAs.
To monitor cell growth, collect a sample of 2 ml of the culture every 2 h during fermentation to determine the density at OD 600 nm, and stop the fermentation once the density reaches a plateau after two consecutive readings.
Harvesting the bacteria (see Fig. 1)
-
10
After the OD 600 nm value reaches a plateau, pump bacteria into the 0.2 μm pore-size hollow-fiber membrane module using pump P1, by opening inlet valve V1.
Critical Step
The hollow-fiber membrane module should be pre-wetted with one bed volume of TE buffer. The flow rate of fermented culture must be deliberately controlled to allow the liquid to pass into the module and through the fiber membrane, discharging into the waste container (C1), while keeping the retained cells on the outside of the membrane and so inside of the column. The permeate flow control can be achieved by reducing or increasing the feed pump (P1) velocity to reduce or to increase the inlet transmembrane pressure. Outlet valve V2 must be open to allow waste discharge into C1.
-
11
When all the fermented culture has been pumped into the module and discharged into the waste container, leaving the concentrated bacteria within the module, switch inlet valve V1 to allow one bed volume of TE buffer to be pumped into the module by pump P1 to rinse cells as the V2 remains open to discharge the rinsed TE buffer into C1.
-
12
Close outlet valve V2 and increase the feed pump (P1) velocity by 1.5–2 times to increase the inlet pressure of TE, in order to discharge cells from the module through the top outlet into C2 container.
Pause point
The concentrated cells can be stored at −20 °C until use. Normally, fermented bacteria can be concentrated by 5–8-fold after the hollow fiber module.
Thermal lysis of E. coli (see Fig. 1)
-
13
The freshly collected bacteria or the frozen bacteria paste after thawing are re-suspended with TE buffer to OD 600 nm = 100 monitored by a UV/VIS spectrophotometer.
Critical Step
Cells harvested from the fermentation should be diluted to the OD 600 nm of 100 to allow uniform subsequent lysis; a higher density of bacteria may result in an incomplete lysis.
-
14
Treat with 1/10 volume of 10× freshly made lysis buffer at 37 °C for 20 min with gentle stirring.
Critical Step
When the bacteria are treated with the lysis buffer, the stirring should be very gentle and stirring is recommended at 10–20 rpm; otherwise, the cells will be overlysed during the heating process.
-
15
Switch on P2 to pump the bacteria from C2 through a copper-coil (or made of glass; A) that is immersed in a 70 °C water bath (C3) and immediately through another copper-coil (B) that is immersed in an ice bath (C4) to terminate the lysis, before being collected in a container that is set in the ice-cold water bath (see Fig. 2).
Critical Step
Adjust the flow rate using a stop watch so that the bacteria pass through the heated coil within 20 s. We have found this to be an optimal time period for the thermal lysis of cells; however, other times may be tested if an optimal yield can not be obtained. As indicated in Figure 2, the temperatures of 70 °C and 80 °C achieved a higher yield of plasmids with a minimum contamination with genomic DNA. However, 70 °C is probably the most appropriate setting in terms of the balance between energy costs and the quality of plasmids produced. An immediate connection from the heating coil to the coil submerged in ice should be used to avoid excess lysis.
Figure 2. Effect of different lysis temperatures.
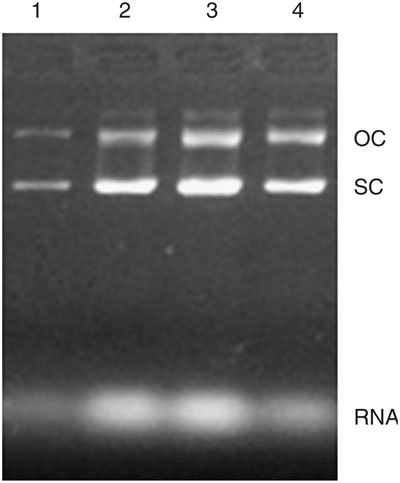
After being pre-treated with lysozyme at 37 °C, the bacteria were heated at various temperatures to lyse for 20 s to determine the yield and quality. One microliter of the extracted plasmid (without purification using PEG 8000) was subjected to electrophoresis on a 0.7% agarose gel. Lane 1, at 60 °C; lane 2, at 70 °C; lane 3, at 80 °C; lane 4, at 90 °C. OC, plasmid in open circle form; SC, plasmid in supercoil form; the genomic DNA contamination is shown as a faint band above the OC; RNA indicates contamination with RNA.
Plasmid isolation (see Fig. 1)
-
16
Lysed bacteria are centrifuged at 12,000g for 30 min.
-
17
To precipitate the plasmid DNA from the supernatant in a floc form, transfer the supernatant into a clean flask (C5) containing 0.1 volume of NaOAc (3 M, pH 5.2) and 0.6 volume of isopropyl alcohol, and incubate at room temperature for 20 min.
Critical Step
To obtain perfect floc precipitates, do not vigorously shake the mixture of plasmid after adding isopropyl alcohol. If a high yield of plasmid is obtained from the precipitation, the incubation time can be reduced, as appropriate, and white floc can be immediately collected by passing through the 200 mesh nylon filter.
-
18
Collect the floc precipitates by passing through a bucket with 200 mesh nylon filter on the bottom.
-
19
Rinse twice with 70% ethanol (EtOH) while the precipitates are still on the filter, and air-dry.
Pause point
The dry precipitates can be stored at 4 °C for weeks.
Critical Step
Dipping the bucket into 70% ethanol several times can enable effective rinsing. This will remove contamination as the dipping action will disassociate substances from the plasmid floc. Rinsing with 70% ethanol is important to eliminate impure substances, such as RNAs; sometimes, an additional rinse with chloroform before the 70% ethanol washes will remove some of the protein or lipid contamination.
Plasmid purification
-
20
The precipitates collected are re-suspended with 1–5 ml of TE buffer.
-
21Purification of plasmid DNA can be performed using either option A or B:
-
APurification by PEG precipitation
-
iMix with the same volume of 13% PEG 8000 (in 1.6 M NaCl solution).
-
iiCentrifuge at 15,000g at room temperature for 10 min.
-
iiiDissolve purified plasmid DNA in fresh 1 ml TE buffer and the plasmid can be stored at −20 °C for long-term storage.
-
i
-
BUse a Qiagen Maxiprep column, according to the manufacturer's instructions
-
iThe purification method can be adapted to be used with any available commercial purification column, such as the one made by Qiagen.
-
i
-
A
Agarose gel electrophoresis to measure plasmid quality
-
22
Digest the plasmid with the restriction enzymes EcoRI and XbaI for 2–3 h, and the reaction should be stopped by incubation at 60 °C for 10 min.
-
23
Load the digestion reaction onto 0.7% agarose gel containing 0.05 μg ml−1 of ethidium bromide, covering with TBE buffer to perform electrophoresis for 1 h at 60 V.
Critical Step
The appropriate concentration of agarose gel should be based on the size of the tested plasmid.
-
24
View under 300 nm UV light and record using a digital camera.
-
25
Plasmid quality can also be analyzed by measuring its ratio of OD 260/280 nm using UV/VIS spectrophotometry. The ratio of the OD values at 260 nm versus those at 280 nm should be 1.8–2.0, which is within the range of acceptable purity for plasmids.
Quantitative measures of plasmids using agarose gel electrophoresis
-
26
The plasmid is serially diluted 1:1 with TE buffer and subjected to 0.7% agarose gel electrophoresis (as in Step 23), along with a standard DNA marker with a known concentration. All bands are visualized under UV light after staining with ethidium bromide and visualized on a UV transilluminator. Photographic records obtained from a digital camera (Olympus) are analyzed using Quantity One software (Bio-Rad) by comparing the intensity of the known concentration of marker DNA as a reference with the plasmid to determine the concentration of the tested plasmid.
Troubleshooting
Troubleshooting advice can be found in Table 2.
Table 2.
Troubleshooting table.
| Problem | Possible cause | Solution |
|---|---|---|
| Bacteria overlysed or underlysed | Different bacteria strains may require lyzozyme pretreatment for different periods of time | Adjust the reaction time during the lyzozyme pretreatment |
| Old lyzozyme used | Lyzozyme should be made fresh each time | |
| Wrong pH for lyzozyme | Reaction of lyzozyme is optimal at pH 8.0 | |
| Lyzozyme may vary between different batches | Test new batch before use | |
| The coil temperature is too low | Pre-equilibrate the coil temperature for at least 10 min before the process is started | |
| The water bath is too small | A larger volume of water bath should be used | |
| The ice bath is too small | A larger volume of ice bath should be used | |
| A long connection exists between the heating coil and the cooling coil | A short connecting tube (less than 5 cm in length) should be used between the heating coil and the cooling coil | |
| Low yield of plasmid | Low volume of supernatant obtained from Step 16 | Add TE buffer at half the volume of lysed bacteria and mix well before centrifugation |
| Bacteria strain | Different bacterial strains need to be tested for the best plasmid yield before using the protocol. See Table 1 | |
| Fermentation conditions | The conditions of fermentation need to be taken into account, including the medium selection, dissolved oxygen control, and the volume and rate of batch feeding |
Timing
Collection of bacteria in the hollow fiber membrane module: 20 min
Lysozyme treatment: 20 min
Thermal treatment: 5–10 min
Centrifugation: 30 min
Plasmid precipitation: 20 min
Purification: 20 min
Anticipated results
The plasmid yields from three independent trials are shown in Table 3. In general, this process can yield plasmid at around 100 mg l−1 culture after purification, which presents an attractive protocol for laboratory use and a large-scale process. After purification by PEG 8000 precipitation, most of the plasmids are shown in supercoiled form (Fig. 3), and the ratio of OD 260/280 nm is around 1.8–1.9 (Table 3).
Table 3.
Plasmid yields using this protocol.
| Batcha | OD 260/280 | Plasmid (mg l−1)b |
|---|---|---|
| 1 | 1.91 | 110 |
| 2 | 1.84 | 120 |
| 3 | 1.81 | 95 |
aThe host E. coli strain used was TOP10 and 17 l of culture was processed after the OD 600 nm reached a plateau around 30.
bThe yield of plasmid pcD-VP1 was determined after PEG 8000 purification.
Figure 3. Purification by PEG 8000.
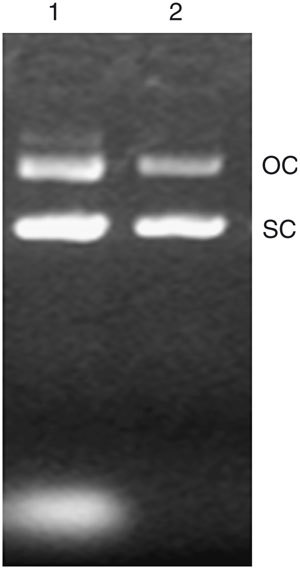
After the cell lysed and the plasmid was extracted, the plasmid was purified by 13% PEG 8000 (in 1.6 M NaCl) precipitation. One microliter of such purified plasmid was subjected to electrophoresis on a 0.7% agarose gel to determine its purity and yield. Lane 1, no purification; lane 2, purified by PEG 8000. OC, plasmid in open circle form; SC, plasmid in supercoil form; the genomic DNA contamination is shown as a band above the OC.
Acknowledgements
This work was supported, in part, by the China High Technology '863' Project (2004AA213102 and 2003AA241110), the China Key Technologies R&D program and a special research fund to B.W. provided by China Agricultural University. We would also like to thank Dr Jane Q.L. Yu for her assistance with the work, and Dr Terry Ng for his critical reading and valuable suggestions for the manuscript.
Competing interests
The authors declare no competing financial interests.
References
- 1.Prather K, Sagar S, Murphy J, Chartrain M. Industrial scale production of plasmid DNA for vaccine and gene therapy: plasmid design, production, and purification. Enzy. Microbial Technol. 2003;33:865–883. doi: 10.1016/S0141-0229(03)00205-9. [DOI] [Google Scholar]
- 2.Luo D, Saltzman WM. Synthetic DNA delivery systems. Nature Biotechnol. 2000;18:33–37. doi: 10.1038/71889. [DOI] [PubMed] [Google Scholar]
- 3.Schleef M. Biotechnology. 1999. [Google Scholar]
- 4.Eastman EM, Durland RH. Manufacturing and quality control of plasmid-based gene expression systems. Adv. Drug Deliv. Rev. 1998;30:33–48. doi: 10.1016/S0169-409X(97)00105-1. [DOI] [PubMed] [Google Scholar]
- 5.Levy MS, O'Kennedy RD, Ayazi-Shamlou P, Dunnill P. Biochemical engineering approaches to the challenges of producing pure plasmid DNA. Trends Biotechnol. 2000;18:296–305. doi: 10.1016/S0167-7799(00)01446-3. [DOI] [PubMed] [Google Scholar]
- 6.Birnboim HC, Doly J. A rapid alkaline extraction procedure for screening recombinant plasmid DNA. Nucl. Acids Res. 1979;7:1513–1523. doi: 10.1093/nar/7.6.1513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ferreia G, Monteiro G, Prazeres D, Cabral J. Downstream processing of plasmid DNA for gene therapy and DNA vaccine application. Trends Biotechnol. 2000;18:380–388. doi: 10.1016/S0167-7799(00)01475-X. [DOI] [PubMed] [Google Scholar]
- 8.Prazeres DM, Ferreira GN, Monteiro GA, Cooney CL, Cabral JM. Large-scale production of pharmaceutical-grade plasmid DNA for gene therapy: problems and bottlenecks. Trends Biotechnol. 1999;17:169–174. doi: 10.1016/S0167-7799(98)01291-8. [DOI] [PubMed] [Google Scholar]
- 9.Levy M. Effect of shear on plasmid DNA solution. Bioprocess. Eng. 1999;20:7–13. doi: 10.1007/s004490050552. [DOI] [Google Scholar]
- 10.Ciccolini LA, Shamlou PA, Titchener-Hooker NJ, Ward JM, Dunnill P. Time course of SDS-alkaline lysis of recombinant bacterial cells for plasmid release. Biotechnol. Bioeng. 1998;60:768–770. doi: 10.1002/(SICI)1097-0290(19981220)60:6<768::AID-BIT13>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- 11.Lahijani R, Hulley G, Soriano G, Horn NA, Marquet M. High-yield production of pBR322-derived plasmids intended for human gene therapy by employing a temperature-controllable point mutation. Hum. Gene Ther. 1996;7:1971–1980. doi: 10.1089/hum.1996.7.16-1971. [DOI] [PubMed] [Google Scholar]
- 12.Prazeres DM, Schluep T, Cooney C. Preparative purification of supercoiled plasmid DNA using anion-exchange chromatography. J. Chromatogr. A. 1998;806:31–45. doi: 10.1016/S0021-9673(97)01254-5. [DOI] [PubMed] [Google Scholar]
- 13.Kelly B, Hatton T. The fermentation/downstream processing interface. Bioseparation. 1991;1:333–349. [Google Scholar]
- 14.Theodossiou I. The processing of plasmid based gene from Escherichia coli: primary recovery by filtration. Bioprocess. Eng. 1997;16:175–183. [Google Scholar]
- 15.Sambrook J, Fritsch E, Maniatis T. Molecular Cloning: A Laboratory Manual. 1989. [Google Scholar]
- 16.Wang B, Merva M, Williams WV, Weiner DB. Large-scale preparation of plasmid DNA by microwave lysis. Biotechniques. 1995;18:554–555. [PubMed] [Google Scholar]
- 17.Clemson M, Kelly WJ. Optimizing alkaline lysis for DNA plasmid recovery. Biotechnol. Appl. Biochem. 2003;37:235–244. doi: 10.1042/BA20030002. [DOI] [PubMed] [Google Scholar]
- 18.Holmes DS, Quigley M. A rapid boiling method for the preparation of bacterial plasmids. Anal. Biochem. 1981;114:193–197. doi: 10.1016/0003-2697(81)90473-5. [DOI] [PubMed] [Google Scholar]
- 19.Lee, A.L. & Sagar, S. A Method for Large-Scale Plasmid Purification (Patent 6,197,553, 2001) (Merck Co. Inc., USA).
- 20.Lander R, Winters M, Meacle F, Buckland B, Lee AL. Fractional precipitation of plasmid DNA from lysate by CTAB. Biotechnol. Bioeng. 2004;79:776–784. doi: 10.1002/bit.10335. [DOI] [PubMed] [Google Scholar]
- 21.Zhu K, et al. A continuous thermal lysis procedure for the large-scale preparation of plasmid DNA. J. Biotechnol. 2005;118:257–264. doi: 10.1016/j.jbiotec.2005.05.003. [DOI] [PubMed] [Google Scholar]
- 22.Jin H, et al. Effect of chemical adjuvants on DNA vaccination. Vaccine. 2004;22:2925–2935. doi: 10.1016/j.vaccine.2003.12.026. [DOI] [PubMed] [Google Scholar]
- 23.Jin H, et al. Induction of Th1 type response by DNA vaccinations with N, M, and E genes against SARS-CoV in mice. Biochem. Biophys. Res. Commun. 2005;328:979–986. doi: 10.1016/j.bbrc.2005.01.048. [DOI] [PMC free article] [PubMed] [Google Scholar]