

Submission ID#551389
Two Novel Mutations of Major Histocompatibility Class-II Associated Molecules
Lauren Rigg, MD1, Neha Sanan, DO2, Devi Jhaveri, DO3, Haig Tcheurekdjian, MD3
1Internal Medicine Resident, University Hospitals Cleveland Medical Center / Case Western Reserve University
2Adult and Pediatric Allergy / Immunology Fellow, University Hospitals Cleveland Medical Center
3Allergy / Immunology, Allergy Immunology Associates
Introduction/Background: Major Histocompatibility Class II (MHC-II) molecules are transmembrane proteins that are essential to the development of the normal adaptive immune response. The genes that encode the MHCII include the Regulatory Factor X-Associated Ankyrin Containing Protein (RFXANK), Regulatory Factor X-Associated Protein (RFXAP), Regulatory Factor X, 5 (RFX5), and MHC-II transactivator (CIITA) proteins. Homozygous mutations in these genes lead to MHC-II Deficiency Syndrome and have been associated with early onset and severe respiratory and gastrointestinal infections, failure to thrive, and premature death. Herein we report two cases with significant clinical manifestations of immunodeficiency in patients with heterozygous mutations of the RFXANK proteins.
Objectives: To describe two cases of novel RFXANK gene variants and their respective phenotypes.
Methods: The patients were evaluated in the office for possible immune deficiency. A retrospective chart review was conducted examining medical history, diagnosis and response to treatments.
Results/Case Description:
Case 1: A 55-year-old female presented for recurrent mucocutaneous candida infections. Prior treatments included therapeutic and prophylactic fluconazole. Immunodeficiency workup showed a mannose binding lectin deficiency, low lymphocyte response to candida and tetanus antigen testing, and no response to candida skin testing. Genetic testing demonstrated a heterozygous variant in the RFXANK gene (c.612A>G/p.Arg167Cys).
Case 2: An 18-year-old Caucasian female presented for lymphadenopathy, immune thrombocytopenic purpura and recurrent infections since early childhood. Prior treatments included antibiotics, subcutaneous and intravenous immunoglobulin (IVIG) therapy, and Rituximab. Immunodeficiency workup showed decreased immunoglobulin levels, B cells, and T cells. Genetic testing demonstrated a heterozygous variant in the RFXANK gene (c.726C>G/p.Ile242Met).
Conclusions: Homozygous mutations of MHC-II associated molecules lead to a primary immunodeficiency known as MHC-II deficiency. Increasing genetic data is becoming available to physicians and patients including heterozygous mutations. While difficult to categorize, heterozygous mutations of MHC-II related proteins may still present with clinically significant immunodeficiency. As this data is further studied, it may assist in diagnosis and subsequent therapy.
(2) Submission ID#551762
The Effects of Adiantum Capillus Hydro Alcoholic Extract on Some Immunological Parameters in Mice
Mehrdad Modaresi1, Masoomeh Pashaei2
1Faculty Member, Isfahan (Khorasgan) Branch, Islamic Azad University, Isfahan, Iran
2Laboratory employee, Department of Biology, Payam e Noor University, Isfahan Center, Isfahan, Iran
The Adiantum capillus a known medicinal herb in traditional medicine which is widely used in traditional medicine to deal with infection by having chemical compounds that affect the immune system. The current study was carried out to investigate the effects of adiantum hydroalcoholic extract on plasma proteins and electrophoretic pattern of blood in small laboratory mice. Mature female mice (Balb/C) were divided into 5 groups including control, placebo, and 50, 100, and 200mg/kg of extract. The extract was injected intraperitoneal every other day for 20 days. At the end of the experiment, blood samples were taken and used to measure blood proteins and their electrophoretic pattern. Obtained data were analyzed using the SPSS program (p<0.05). According to the results, 100 and 200 mg/kg doses increased the amount of albumin, alpha-1 globulin, beta globulin, and A/G ratio. Therefore, it can be said that the extract has a positive effect on the blood system and plasma proteins and can increase the immune system without the presence of antigenic factors.
(3) Submission ID#554014
Unexpected Diagnosis in a Family with Autoimmune Multilineage Cytopenia and Hypogammaglobulinemia
Yael Gernez, MD, PhD1, Jose Chavez, PhD2, James Bussel, MD3, Charlotte Cunningham-Rundles, MD, PhD4
1Clinical Assistant Professor, Stanford School of Medicine
2Post Doctoral, Division of Clinical Immunology, Icahn School of Medicine, Mount Sinai NY, NY
3Professor in Pediatrics, Department of Hematology and Oncology, Weill Cornell Medicine, NY, USA
4Professor in Medicine, Division of Clinical Immunology, Icahn School of Medicine, Mount Sinai, NY, NY, USA
A 34 y.o. female was referred to our clinic with a history of multilineage cytopenias/Evans syndrome, a history of idiopathic thrombocytopenic purpura, hemolytic anemia, chronic neutropenia, lymphopenia, and hypogammaglobulinemia treated with IVIG.
Our patient was healthy until she was 8 years old; at that time, she developed joint pain, rash, and bruising. She was found to have Evans syndrome with idiopathic thrombocytopenic purpura (ITP), neutropenia, and lymphopenia. She was initially diagnosed with lupus and was given steroids. Her bone marrow biopsy did not conclude myelokathesis. When she was 15 years old, she remained thrombopenic and was started on high dose of immunoglobulin replacement therapy. In 2012 (29 years old), she developed polyarthritis in her upper and lower extremities. In 2013 (30 years old), she had a severe nosebleed, for which she was admitted and treated with Amicar twice; her platelets were found to be 2,000 K/UL. She received rituximab weekly for 4 weeks resulting in an increase of platelet count to 90-100K/UL. She recently (March 2017) had a splenectomy to remove her large spleen, and since then, her platelets have rebounded to 400-500K/UL. In 2015, she was placed on long-term immunoglobulin replacement therapy after being hospitalized for bilateral pneumonia for 5 nights requiring IV antibiotics for treatment. In 2017, she developed and was treated for another pneumonia.
Her family history is characterized by multiple members with autoimmune multilineage cytopenia as well as autoimmune diseases such as multiple sclerosis (mother), thyroiditis and enteropathy.
On physical examination, she did not present with any warts and the remainder of her physical examination being unremarkable, except for her scar from her splenectomy and a cervical lymphadenopathy.
Immunologic evaluations showed IgG 601 mg/dL, IgA <5 mg/dL, and IgM 208 mg/dL. CBC with differential and lymphocyte screen were as follows (cell/mm3): WBC 12.3 x103, Hemoglobin 10.2 g/dl, Platelets 503 x 103; 3 % neutrophils (ANC: 300), 82% lymphocytes, 10% monocytes, 0% eosinophils; absolute total T-cell number was 8884 (750-2500 cells/MCL), CD4+ T-cells 6554 (480-1700cells/MCL), CD8+ T-cells 2185 (180-1000cells/MCL), natural killer cells 206 (135-525 cells/MCL), and absolute number of B cells was 996 (75-375 cells/MCL).
She came to our clinic with her sister, who also had multilineage cytopenia and hypogammaglobulinemia, treated with monthly IVIG; and her nephew whom had neutropenia. Based on this family presentation all three underwent Whole Exome Sequencing (WES). The patient, the patients sister and the patients nephew were all found to have a variant on CXCR4 (frameshift mutation on Chromosome 2, p.Val324fs; RefNt: TCA; AltNt: T). As an important note, the patient had a bone marrow biopsy, which did not conclude myelokathesis.
In summary, our patient with trilineage cytopenia and hypogammaglobulinemia, without any warts or myelokathexis, had WHIM syndrome (Warts, Hypogammaglobulinemia, Immunodeficiency, and Myelokathexis), which was discovered by studying her WES. With the identification of her specific diagnosis, this allowed us to discuss the potential future indication of Plerifaxor (antagonist of the alpha chemokine receptor CXCR4). And equally important, we discussed family planning and future pregnancies given that the mutation is autosomal-dominant.
(4) Submission ID#555017
Risk of Bacterial Infections Among Patients with Secondary Complement Deficiency
Taha Al-Shaikhly, MBChB1, Kathleen Mohan, ARNP2, Matthew Basiaga, DO, MSCE3, Eric Allenspach, MD, PhD4
1Allergy & Immunology Fellow, Division of Allergy & infectious Diseases, University of Washington
2Nurse Practitioner, Department of Immunology, Seattle Children's Hospital
3Assistant Professor, Department of Rheumatology, Seattle Children's Hospital
4Assistant Professor, Department of Immunology, Seattle Children's Hospital
Introduction: Complement component-3 (C3) is shared by the classical, lectin and alternative complement activation pathways. C3, a major opsonin, facilitates phagocytosis of encapsulated microorganisms. Inherited C3 deficiency is rare and is associated with increased risk of bacterial infections. Subjects with connective tissue diseases (CTD) and C3 nephritic factors can have low and occasionally undetectable C3 levels, yet they are at an underappreciated infectious risk. We hypothesize that excessive C3 consumption in secondary complement deficiency disorders (SCD) is associated with higher risk of bacterial infections similar to primary complement deficiency disorders (PCD).
Objectives: To compare the rate of bacterial infections between PCD and SCD patients and evaluate the association between C3 level and bacterial infection risk.
Methods: We performed a retrospective cohort study. Subjects with an undetectable complement activity (CH50) or any of the complement components measured at Seattle Childrens hospital from 2002-2018 were included in our study. We recorded the number of infections, observation periods, diagnosis (PCD, SCD and its underlying etiology), lowest complement component levels, and the immunosuppressive agents used. The date of birth, and date of lowest C3 level were considered as start points to calculate the observation periods for PCD and SCD subjects respectively. Infections requiring hospitalization or parenteral antibiotics were categorized as serious bacterial infections (SBIs). Descriptive analyses were performed to determine medians and ranges for continuous variables. Differences in rates of bacterial infection were assessed using the chi-square and kruskal-wallis tests when appropriate. Among subjects with CTDs, we treated every C3 measurement as a single observation (n=1,197) and studied the association between C3 concentration and the 30-day odds of having a SBI. Multivariable logistic regression was performed to determine infection risk based on C3 level while controlling for contributing factors.
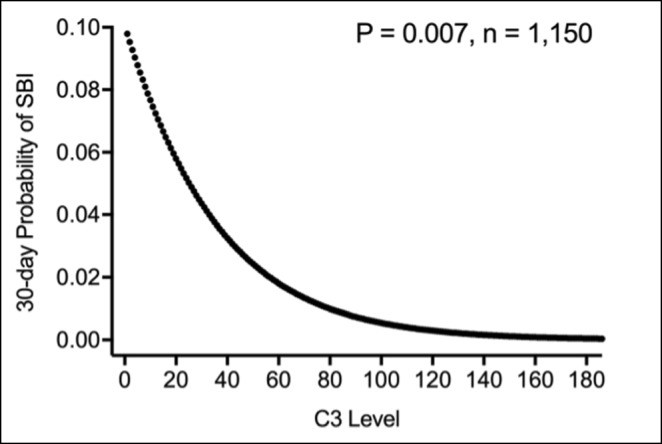
Results: We identified 14 subjects with PCD, and 52 subjects with SCD. SCD consisted of three subgroups (CTD-related (n=44), nephritic factor-related (n=2), and infection-related (n=6)). Collectively, CTD subjects had a lower median rate of SBI compared to PCD subjects (P = 0.004). Subjects with CTD and C3 level <40 have higher rate of bacterial infection (of any severity) (P = 0.002) and of SBI (P = 0.004) when compared to CTD subjects with C3 >=40 at the beginning of observation period (Figure 1). While controlling for immunosuppression level and lupus nephritis diagnosis, C3 levels were predictive of SBI (P = 0.007, Figure 2). CTD subjects with a C3 level < 40 had a significantly higher risk for SBI compared to those > 40 (OR 3.63, 95% CI [1.03-12.7], P = 0.04). Subjects with infection-related hypocomplementemia had an undetectable CH50 lasted for at least 9 days.
Conclusion: Among CTD patients, low C3 levels are predictive of more SBIs in the future. C3 level <40 is associated with higher 30-day odds of having a SBI. Therefore, CTD patients with very low C3 levels should be monitored for early signs of infections and should have a lower threshold for antibiotics initiation. SBI itself can result in an undetectable CH50 and re-testing is warranted before confirming a PCD diagnosis.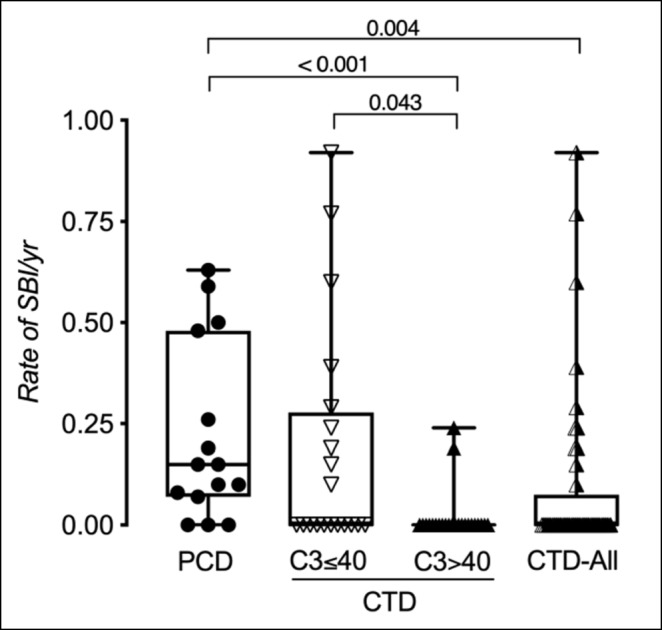
(5) Submission ID#556068
Heterozygous TACI Mutation (TNFRSF13B: A181E) Causing Significant Infections in a Patient with Normal Immunoglobulins
Shan Shan Wu, DO1, Jenny Lee, MD2, Michelle Sergi3, David P. McGarry, DO4, Robert Hostoffer, DO, LhD, FACOP, FACOI, FAAP, FCCP5
1Allergy and Immunology Fellow, University Hospitals Cleveland Medical Center, Cleveland, Ohio
2Internal Medicine/Pediatrics Resident, University Hospitals Cleveland Medical Center/Rainbow Babies and Childrens Hospital, Cleveland, Ohio
3Medical Student, Ohio University Heritage College of Osteopathic Medicine, Warrensville Heights, Ohio
4Allergy and Immunology Fellow, University Hospitals Cleveland Medical Center, Cleveland, Ohio
5Allergy and Immunology Program Director, University Hospitals Cleveland Medical Center, Cleveland, Ohio; Allergy and Immunology Associates Inc. Mayfield Heights, Ohio
Introduction: Common variable immunodeficiency (CVID) is a primary immune deficiency associated with loss of B-cell functions. Genetics of CVID are multifactorial, although both monogenic and polygenic forms have been described in the literature (1). Mutations (heterozygote and homozygote) in TNFRSF13B, the gene that encodes the transmembrane receptor, or TACI, are associated with 8-10% of CVID patients (2). TACI mutation with reduced TACI expression on marginal zone and CD27+ memory B-cells can impair B-cell differentiation, proliferation, and isotype switch (2), contributing to the pathogenicity of CVID. Asymptomatic individuals with normal immunoglobulin levels who have TACI mutation are also reported in the current literature (2). We aim to describe the significance of the heterozygous TNFRSF13B variant in a patient with recurrent sinopulmonary and skin infections without apparent B-cell dysfunction.
Objective: To present a patient with the heterozygous TNFRSF13B variant with the clinical manifestations of those with CVID despite normal immunologic findings inconsistent with CVID.
Method: Immunologic studies for the patient included serum immunoglobulins (IgG, IgA, IgM) and IgG subclasses, B-cell phenotyping, lymphocyte subset markers, mannose-binding lectin, mitogen and antigen stimulation, bacteriophage study, Streptococcus pneumoniae titers to 23 serotypes, and genetic sequence analysis with deletion/duplication testing of 207 genes. Genetic testing was also performed on the patients mother, father, and two sisters.
Results: A 27-year-old male presented with a history of multiple infections since four months old, including recurrent episodes of acute otitis media, bronchitis, sinusitis and pneumonia, viral meningitis, mastoiditis, and cellulitis with abscesses of the axilla, thigh, and perianal region. The patient was found to have the TNFRSF13B, Exon 4.c.542C>A (p.Ala181Glu) heterozygous TACI variant associated with CVID. B-cell phenotyping showed an increase in naive B-cells (CD19+CD27-IgD+) and a decrease in both non-switched, memory B-cells (CD19+CD27+IGD+) and switched, memory B-cells (CD19+CD27+IgD-) with proper levels of transitional B-cells (CD19+CD24+CD38+) and plasmablasts (CD19+CD24-CD38+). This phenotype indicates a dysregulation in B-cell differentiation and proliferation into memory B-cells and impairment in isotype class-switching commonly found in individuals with CVID harboring TACI mutations. Yet the immunoglobulin levels and vaccine response were appropriate, excluding a diagnosis of CVID.
The patients mother, who was asymptomatic, had the same TACI variant. Her immunoglobulins, lymphocyte subset markers, and B-cell phenotype were normal. Sister A has a history of multiple sinopulmonary infections with genetic results pending. The patients father and sister B did not have any immune issues and had no genetic mutations.
Conclusion: CVID is a heterogeneous disease that may be associated with genetic defects. TACI mutations found in a small percentage of individuals with CVID, result in B-cell dysfunction and hypogammaglobinemia. We describe a patient with a TNFRSF13B exon Ala181Glu heterozygous mutation with recurrent infections and normal immunoglobulin levels and vaccine response.
References:
1 Bonilla FA, Barlan I, Chapel H, et al. International Consensus Document (ICON): Common Variable Immunodeficiency Disorders. J Allergy Clin Immunol Pract. 2016;4(1):38-59.
2 Martinez-Gallo M, Radigana L, Belén Almejúne M, et. al. TACI Mutations and Impaired B-cell Function in Subjects with CVID and Healthy Heterozygotes. Allergy Clin Immunol. 2013;131(2):468476.
(6) Submission ID#561686
A Novel Mutation in Zap 70 Leading to an Infant with T+B+NK+ Severe Combined Immunodeficiency
Kelsey Kaman, MD1, Alicia Johnston, MD2, Monique Abrams, MD1
1Pediatric Resident, Baystate Medical Center
2Faculty Advisor, Baystate Medical Center
Introduction: ZAP70 codes for a 619-amino acid enzyme, ZAP70, a member of the Syk-protein tyrosine kinase family that plays an important role in T cell development and activation. ZAP70 is phosphorylated at tyrosine kinase residues upon T cell receptor (TCR) stimulation resulting in TCR-mediated signal transduction with Src family kinases. ZAP70 deficiency results in a rare T+B+NK+ Severe Combined Immunodeficiency (SCID). We report a novel compound heterozygous mutation in ZAP70 leading to presumed absent ZAP70 function in an infant with a normal TREC newborn screen and SCID.
Case Description: The patient is a term, fully immunized female, born to non-consanguineous parents who was hospitalized for RSV bronchiolitis at 2 mo. At 4 mo she developed an erythematous, papular rash on her face and extremities, nonresponsive to topical antifungal therapy. At 6 mo she was re-hospitalized with RSV bronchiolitis and subsequently treated with multiple courses of antibiotics for presumed bacterial pneumonia followed by albuterol and oral steroids for possible reactive airways disease. During this course of treatment, her rash resolved. At 8 mo she presented with failure to thrive (wt <0.1% for age), multifocal pneumonia and respiratory failure requiring intubation. Bronchial alveolar lavage confirmed Pneumocystis jiroveci pneumonia prompting an immune evaluation. Total immunoglobulins were normal for age, however antibody titers to tetanus, diphtheria and Streptococcus pneumoniae were absent. Lymphocyte enumeration revealed elevated CD4 T cells and markedly diminished CD8 T cells, normal B and NK cells. T cell proliferation to mitogens (PHA, PWM) and antigens (Candida, tetanus) was absent, however T cells proliferated normally to stimulation with PMA and ionomycin. TREC number was normal by newborn screening, but was 2 std deviations below the mean and would have resulted in a positive screen upon repeat. Invitae 18 gene SCID panel revealed two variants of unknown significance, c.109C>G (p.Arg37Gly) leading to substitution of Arg with Gly and c.1529_1532dupGCAT (p.Ile511Metfs*65) resulting in a premature translational stop signal expected to disrupt the last 109 amino acids of ZAP70 protein. Parental sequencing revealed these variants to be on opposite chromosomes. The patient was successfully treated for PJP pneumonia and has since successfully engrafted a 9/10 matched unrelated donor stem cell transplant.
Discussion: We report a novel compound heterozygous mutation in ZAP70 which we presume led to T+ B+ NK+ SCID. Our patients clinical presentation of failure to thrive, recurrent lower respiratory tract infections, dermatologic findings and PJP pneumonia are consistent with previously reported cases of ZAP70 SCID. Her paucity of CD8 T cells, abundance of CD4 T cells and absent proliferation to mitogens are also consistent with previously described cases of ZAP70. Normal proliferation of T cells when bypassing the TCR by stimulating cells with ionomycin and PMA confirms a defect in the TCR. We believe this is the second documented case of missed SCID by newborn screen in MA since the implementation of TREC screening in 2008.
(7) Submission ID#564579
A Case of Memory B-cell Dysfunction in a Child with Recurrent Otitis Media
Arjola Cosper, DO MS1, Lisa Barisciano, MD2
1Pediatric Resident (PGY III), Goryeb Children's Hospital
2Attending Physician, Pediatric and Adult Asthma, Allergy and Immunology, LLC
Introduction: Acute Otitis Media (AOM) is one of the most common reasons for antibiotic use in early childhood. We explored the challenges when AOM fails traditional therapies and immunologic evaluation does not identify a commonly described immunodeficiency.
Case Description: An eighteen-month-old male presented with 12 episodes of AOM and recurrent purulent otorrhea requiring intravenous antibiotics. Laboratory evaluation revealed a normal CBC, normal immunoglobulins (IgG 588, IgA 76, IgM 63, IgE 12) and IgG subclasses. Lymphocyte subset panel was normal. Initial responses to DTaP and Prevnar boosters were normal, however, there was rapid decline to tetanus and pneumococcal antibody titers. A sub optimal response to Haemophilus influenza Type B vaccine was noted. Although vaccinated twice for MMR, he never mounted mumps specific IgG. Mitogen response to PHA was normal with decreased responses to ConA and pokeweed and no detectable tetanus nor candida responses. Further investigation revealed decreased non-class and class switched memory B-cells. The patient was recently vaccinated to PCV23 and at the present time has protective titers.
Discussion: It has been previously suggested that decreased memory B cells may contribute to decreased antibody responses to select vaccine antigens resulting in recurrent AOM in children. Our case supports the need to investigate beyond typical immunologic screening for immunodeficiencies.
(8) Submission ID#566756
Coexistence of Lymphoproliferative Syndrome, Neurofibromatosis, Systemic Lupus Erythematosus and Hyper IgM Syndrome in a Patient with MSH6 Mutation
Sukru Cekic1, Yasin Karali1, Sara Sebnem Kilic2
1Fellow of Allergy and Clinical Immunology, Uludag University Faculty of Medicine
2Professor of Allergy and Clinical Immunology, Uludag University Faculty of Medicine
Introduction: DNA mismatch repair (MMR) system corrects replication errors in newly synthesized DNA, and prevent recombination between DNA sequences when they were not identical (1). MSH6 is a part of MMR genes, (2-4).
Case: A ten-year-old girl presented with fever, brown spots on her skin, hair loss, recurrent pulmonary infections, arthritis on the left hand and right ankle. She has also been followed up with NF (Figure 1). There was a first-degree cousin marriage between her parents. Physical examination revealed findings of pneumonia and NF. Anti-nuclear antibody, anti-nDNA, anti-dsDNA, anti-histone, Anti Ro52 and anti-nucleosome antibodies were positive. In her immunologic assessment showed low IgG and IgA levels associated with high IgM level (Table 1). The coexistence of NF, hyper IgM syndrome, SLE, were considered in the patient. Intravenous Ig (400 mg/kg, every 3 weeks) treatment was started due to hypogammaglobinemia. The frame shift mutation in exon 2 of the MSH6 gene was detected in the Boztug's laboratory.
In the follow up period, she admitted at 11 years old with back pain. A mass in the left paravertebral area, related to the spinal canal and neural foramina, was detected At the L4-L5 levels in spinal MRI. The lymphadenopathy around the liver and hilum and the left parietal bone lesions were developed within two months despite surgical excision of primary mass (Figure 2). As a result of PET examination; SUVmax was found to be around 6.5 in the mass lesion in the paravertebral region and SUVmax values did not exceed 2.5 in other lymphadenopathy and masses. Atypical cellular infiltration suggesting neoplastic events, which were including small-medium size atypical pleomorphic mononuclear cells and T cells. Since all these formations did not indicate definite cancer, chemotherapy was not started. Interestingly, although chemotherapy was not given, progression stopped, and partial spontaneous regression was observed.
Discussion: The effect of MSH6 mutations on patients may significantly vary with the inheritance pattern (2). Leukemias or lymphomas are not common in heterozygote MMR gene defects (5,6). However, homozygote mutations in MMR genes show a different pattern. Wimmer and Etzler proposed the new term Constitutional mismatch repair-deficiency syndrome (CMMR-D) for patients who have a homozygous mutation in MMR (3). CMMR-D characterized by development of childhood cancers, mainly hematological malignancies and/or brain tumors, as well as early-onset colorectal cancers, and neurofibromatosis type 1 (3). Bi-allelic germline mutations in any of the MMR genes in which MSH6 is involved increases hematological malignancies by 15% (7,8). MSH6 mutation has been associated with many cancers since its identification. Leukemia, lymphoma, colorectal cancer, endometrial cancer, brain tumors are some of these cancer types (2-4,9).
MSH6 deficiency is an important disease that can affect different systems at the same time. There is a high risk of malignancy in the cases and therefore they must be closely monitored. This case has also shown that atypical lymphoproliferation may occur in MSH6 homozygous mutant cases.
| Table 1. The immunologic assessment of patient | |
| IgG:213 mg/dl (normal rage: 842-1943) | CD3: 83.1% (2717/mm3) |
| IgA: 66,4 mg/dl (normal range: 62-390) | CD4:%38,6 (1262/mm3) |
| IgM: 334 mg/dl (normal range: 54-392) | CD8: %41.5 (1357/mm3) |
| CD19: %14.6 (477/mm3) | |
| HLA-DR+CD19:%14.5 (474/mm3) | |
| CD3-CD16+CD56+:%1.7 (55/mm3) | |
Image 1. Cafe-au-lait spots on the extremities
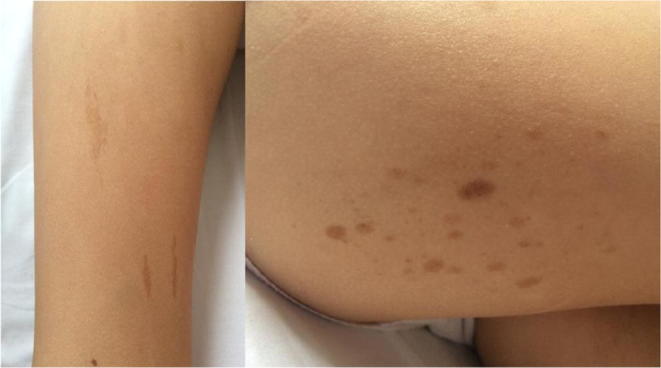
Image 2. Paravertebral mass and parietal bone lesions in the magnetic resonance imaging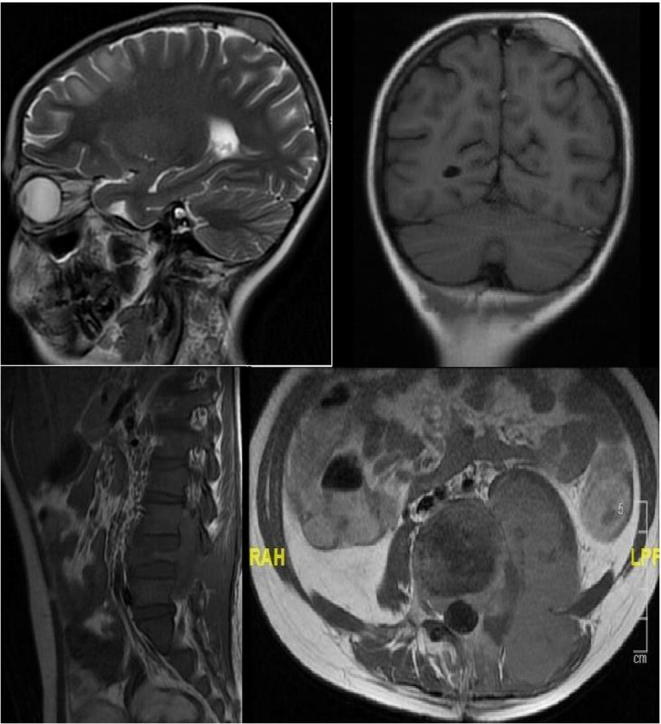
References:
1. Marinus MG. DNA Mismatch Repair. EcoSal Plus. 2012;5(1).
2. Hegde MR, Chong B, Blazo ME, Chin LH, Ward PA, Chintagumpala MM, Kim JY, Plon SE, Richards CS. A homozygous mutation in MSH6 causes Turcot syndrome. Clin Cancer Res. 2005;11(13):4689-93.
3. Wimmer K, Etzler J. Constitutional mismatch repair-deficiency syndrome: have we so far seen only the tip of an iceberg? Hum Genet. 2008;124(2):105-22.
4. Ripperger T, Beger C, Rahner N, Sykora KW, Bockmeyer CL, Lehmann U, Kreipe HH, Schlegelberger B. Constitutional mismatch repair deficiency and childhood leukemia/lymphoma--report on a novel biallelic MSH6 mutation. Haematologica. 2010;95(5):841-4.
5. Lynch HT, de la Chapelle A. Hereditary colorectal cancer. N Engl J Med. 2003;348(10):919-32.
6. Lynch HT, Lynch JF, Lynch PM, Attard T. Hereditary colorectal cancer
syndromes: molecular genetics, genetic counseling, diagnosis and management. Fam Cancer. 2008;7(1):27-39.
7. Bakry D, Aronson M, Durno C, Rimawi H, Farah R, Alharbi QK, Alharbi M, Shamvil A, Ben-Shachar S, Mistry M, Constantini S, Dvir R, Qaddoumi I, Gallinger S, Lerner-Ellis J, Pollett A, Stephens D, Kelies S, Chao E, Malkin D, Bouffet E, Hawkins C, Tabori U. Genetic and clinical determinants of constitutional mismatch repair deficiency syndrome: report from the constitutional mismatch repair deficiency consortium. Eur J Cancer. 2014 Mar;50(5):987-96.
8. Wimmer K, Kratz CP, Vasen HF, Caron O, Colas C, Entz-Werle N, Gerdes AM,
Goldberg Y, Ilencikova D, Muleris M, Duval A, Lavoine N, Ruiz-Ponte C, Slavc I, Burkhardt B, Brugieres L; EU-Consortium Care for CMMRD (C4CMMRD). Diagnostic criteria for constitutional mismatch repair deficiency syndrome: suggestions of the European consortium 'care for CMMRD' (C4CMMRD). J Med Genet. 2014;51(6):355-65.
9. Bougeard G, Charbonnier F, Moerman A, Martin C, Ruchoux MM, Drouot N, Frébourg T. Early onset brain tumor and lymphoma in MSH2-deficient children. Am J Hum Genet. 2003;72(1):213-6.
(9) Submission ID#567651
American Society of Pediatric Hematology and Oncology (ASPHO) Clinical Immunology Special Interest Group (SIG): Expanding Clinical Immunology Education, Research and Care
David K. Buchbinder, MD, MSHS1, Sharat Chandra, MD, MRCPCH2, Blachy J. Davila Saldana, MD3, Rachael F. Grace, MD4, Kim E. Nichols, MD5, Süureyya Savasan, MD6, Nicola A. Wright, MD7, Roshini S. Abraham, PhD8, Shamuganathan Chandarkasan, MD9
1Assistant Clinical Professor, Department of Hematology, Children's Hospital of Orange County, Orange, CA, Department of Pediatrics, University of California at Irvine, Orange, CA
2Assistant Professor, UC Department of Pediatrics, Division of Bone Marrow Transplantation and Immune Deficiency, Cincinnati Childrens
3Blood and Marrow Transplant Specialist, Division of Blood and Marrow Transplantation, Childrens National Medical Center, Department of Pediatrics, The George Washington University, Washington, DC
4Director, Hematology Clinic, Assistant Professor of Pediatrics, Harvard Medical School, Pediatric, Hematology/Oncology, Dana-Farber/Boston Children's Cancer and Blood Disorders
5Director, Cancer Predisposition Division, Division of Cancer Predisposition, St. Jude Children's Research Hospital, Memphis, TN
6Director, Pediatric Blood and Marrow Transplantation Program, Professor of Pediatrics, Bone Marrow Transplant Program, Carman and Ann Adams Department of Pediatrics, Wayne State University School of Medicine, Children's Hospital of Michigan, Detroit, MI
7Associate Professor, Department of Pediatrics, Alberta Children's Hospital, Calgary, Alberta, Canada
8 Department of Pathology and Laboratory Medicine, Nationwide Childrens Hospital, Columbus, OH.
9 Assistant Professor, Division of Bone Marrow Transplant, Aflac Cancer and Blood Disorders Center, Children's Healthcare of Atlanta, Emory University School of Medicine, Atlanta, GA
Background: Advances in inborn errors of human immunity have supported the discovery of new syndromes that are marked by striking features of autoimmunity and immune dysregulation often associated with cytopenias, lymphoproliferation, and a predisposition to reticuloendothelial malignancies leading to evaluation with hematologists/oncologists. Moreover, hematologists/oncologists have also seen an increasing use of effector cell-based therapies, checkpoint inhibitors, immunomodulatory and targeted therapies resulting in autoimmunity and hyperinflammatory complications. A working knowledge of clinical immunology could help practicing hematologists/oncologists in the identification and management of these conditions.
Objectives: To support the advancement of ASPHO members and the field by facilitating education regarding the best practices in diagnosis and management of immunological disorders. To create a platform for the development of collaborative clinical research in patients with hematological/oncological manifestations of immunological disorders or those requiring hematopoietic stem cell transplantation for a underlying immunological disorder.
Design/Methods The ASPHO Clinical Immunology SIG was initiated based on collaboration with the Clinical Immunology Society (CIS). ASPHO members who are pediatric hematology/oncology clinicians, clinical researchers, and trainees are eligible to participate. We have established a steering committee with representatives from across the United States and Canada with diverse clinical and research expertise. Through regular teleconferences and annual in-person meetings, we have developed a platform to provide our members with a network of immunology resources to ensure a strong foundation of knowledge and tools to conduct clinical care and research pertaining to the diagnosis, evaluation, and treatment of patients with immunological disorders.
Results: Prior to the inaugural meeting, we conducted a needs assessment of the ASPHO Clinical Immunology SIG Membership, which defined areas of priority pertaining to clinical immunology including education and research. At the inaugural meeting of the 2018 ASPHO Clinical Immunology Special Interest Group in May 2018, 43 ASPHO Members participated. We currently support over 50 members within our online community. Several educational initiatives have been successfully launched. We have submitted an invited review to Pediatric Blood and Cancer which provides a case-based review of primary immune regulatory disorders. We hosted the first Immunology for Hematology Oncology Practice (I-HOP) Cased-based Webinar Series. This series features case-based discussions of patients with primary immunodeficiency disorders presented by fellow trainees and mentored by senior clinicians. We will also be hosting an ASPHO Webinar focusing on the Laboratory Evaluation of Primary Immunodeficiencies and Immune Dysregulation Syndromes. We have also begun the process of laying the groundwork for clinical research initiatives.
Conclusion: The ASPHO Clinical Immunology SIG seeks to serve as a collaborative resource for pediatric hematology/oncology clinicians and researchers. Through the development of educational and research initiatives, we envision improving the care of patients with immunological disorders that are often managed by pediatric hematologists/oncologists. Moreover, we hope to broaden our understanding and application of clinical immunology within pediatric hematology/oncology. We hope that this successful initiative will serve as a blueprint for the development of future collaborations with other specialty societies and patient groups.
(10) Submission ID#569242
Sepsis as a Sign of Immunodeficiency
Katsiaryna Serhiyenka1, Oxana Romanova, PhD2
1Assistant of professor, Belarussian State Medical University
2Professor, Belarussian State Medical University
Submission Text
Background: T-cell immunity disorders among primary immunodeficiencies (PID) are 9% in the registry of the European Society of Immunodeficiency (ESID) and 10.5% in the United States . T-cell disorders are characterized by the absence or presence of T-lymphocytes. Because T cells are important for the normal functioning of B cells, most PID with a T-cell disorder lead to combined T- and B-cell disorders. Disturbances of the T-cell link of immunity are clinically manifested in early childhood. The most serious form of PID with violation of the T-cell link of immunity is a severe combined immunodeficiency (SCID), the first symptoms of which are already observed in infants and are characterized by the development of life-threatening infections.
Results: Girl N. at the age of 3 months entered the Childrens Infectious Hospital with complaints of cough, high febrile temperature for 5 days, refusal to eat. From the anamnesis of life the girl from the 1st pregnancy, 1 birth, was born full term in 40 weeks gestation, birth weight 4640g. For 3 months of life, a bad increase in body weight was noted and at the time of admission, the weight in 3 months was 5400g. According to the parents, the child had atopic dermatitis. From the anamnesis of the disease on 08.01, the temperature rose to 38.2°C, there was a cough and a mucous discharge from the nose. Then the child refused to eat, the body temperature rose to 39.2°C. January 14 patient was hospitalized.
According to the immunogram, a sharp decrease in CD3 + 26% (58-85%) was detected, activated T-lymphocytes (CD3 + HLA-DR +) were 19.9% (3-15%), T helper / inducers (CD4 + CD8 - 26.6% (30-56%) and T suppressors / cytotoxic (CD8 + CD4-) 0.5% (18-45%), a high ratio of Tx / Tc (CD4 + CD8 +) was detected 53.2% (0.6-2.3), cytotoxic non-T cells (CD3-CD8 +) -1,2, an increase in the number of B-lymphocytes (CD19 +) - 58.9% (7-20%), natural killers (CD16 + CD56 +) - 6.6% (5-25%), natural T-killers (CD3 + CD16 + CD56 +) - 0.3 (0-5%), leukocyte gates (CD45 + CD14-) - 99% (95-100 %). The absolute content of T-lymphocytes was 0.15 x 109/l, B - lymphocytes - 0.35 x 109/l. The number of thymic migrants (CD45 + CD45RA + CD31 +) was not detected (0%).
01/17/2017 CT scan of the chest was diagnosed CT signs of a polysergic two-sided inflammatory process in the lungs.
Blood for sterility - Staphylococcus epidermidis was isolated, CMV DNA was detected in an amount of 7.6 ×106copies/ml.
Despite the therapy, the patient died.
Posthumous diagnosis: Primary immunodeficiency (SCID, T0 B + Nk +). Complications: Sepsis. Septic shock. SPON: ARDS, renal failure, DIS, thrombocytopenia, anemia 3. Two-sided lower-lobe pneumonia. Generalized CMV infection.
Conclusion: The peculiarity of the described clinical case was that the patient's first symptoms of SCID developed in the first months of life and were manifested by a bad weight gain, atopic dermatitis and the development of a life-threatening generalized cytomegalovirus infection and sepsis.
(11) Submission ID#569933
Two Siblings with Autoimmune Polyendocrinopathy-candidiasis-ectodermal Dystrophy-like Phenotype Demonstrating Classic and Atypical Symptoms
Edith Schussler, MD1, Elise Ferre, PA-C, MPH2, Monica Schmitt, CRNP3, Michail Lionakis, MD, Sc.D4
1Assistant Professor of Pediatrics, Division of Pulmonary, Allergy & Immunology, Weill Cornell Medicine
2Physician Assistant, Fungal Pathogenesis Section, Laboratory of Clinical Immunology & Microbiology (LCIM) , National Institute of Allergy & Infectious Diseases (NIAID), NIH
3Nurse Practitioner, Fungal Pathogenesis Section
National Institute of Allergy and Infectious Diseases (NIAID) National Institutes of Health (NIH)
4Chief, Fungal Pathogenesis Section, National Institute of Allergy and Infectious Diseases (NIAID) National Institutes of Health (NIH)
Submission Text
Autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy (APECED) is a rare autosomal recessive disease caused by AIRE gene mutations. Clinical diagnosis is established by the presence of at least two components of the classic triad of chronic mucocutaneous candidiasis, hypoparathyroidism, and Addisons disease. In Europe, the classic presentation is widely recognized and nonendocrine autoimmune manifestations are rarely reported. A recent study of 35 American APECED patients demonstrated a more heterologous presentation, with many non-endocrine manifestations including urticarial eruption, hepatitis, gastritis, intestinal dysfunction, pneumonitis and Sjogrens-like syndrome, all uncommon in European reports. Within the American cohort, 80% of patients developed a mean of three non-triad manifestations before reaching the classic triad. Finding of AIRE mutations and high-titer antiIFN- autoantibodies is seen in both European and American cohorts.
We present the case of two siblings, who demonstrate an APECED-like phenotype with both classical and atypical features. They share the same heterozygous c132+1_132+3delinsCT AIRE mutation.
The older, an eight-year-old boy, with history of prematurity, bronchopulmonary dysplasia and onychomadesis in infancy, came to medical attention at 16 months of age due to failure to thrive (FTT), in addition to fevers and urticarial rash lasting months after his MMR vaccine. The fevers resolved with Anakinra, which was discontinued two years later due to pneumonia. From age 2-4 he developed an ALPs negative lymphadenopathy which self-resolved. Lung issues include chronic cough, initially treated as asthma but with poor bronchodilator response, and frequent lung infections, including 1-2 pneumonias per year. At age five evaluation for FTT revealed growth hormone deficiency. Two years later he was diagnosed with primary Addisons disease. Chronic abdominal discomfort, bloating, cyclical constipation/diarrhea, recurrent rashes, dystrophic nails, and SICCA symptoms are also present.
His sister, age five, shows FTT, but no growth hormone deficiency. At age one, she too developed a fever and rash syndrome lasting 3 months. Severe GERD and constipation started in infancy and are ongoing. At age three she developed a transaminitis, initially diagnosed as EBV, but later thought to be autoimmune hepatitis. She has frequent viral respiratory infections, and pneumonia at age two. She has had a chronic cough, with poor bronchodilator response, for most of her life. Evaluation of seizure at age three showed normal brain activity. Brain MRI revealed partial agenesis of the corpus callosum and microgyria. Her brother has similar MRI findings. Both children have had developmental motor delay and poor tone. Brain dysgenesis and neurodevelopmental delay has not previously been described in APECED.
Although there were both typical and atypical symptoms, the history in combination with genetic findings led to further investigation of an APECED-like syndrome. Autoantibody testing confirmed high-titer antiIFN- autoantibody typical of APECED in both children and high-titer BPIFB1 autoantibodies found almost exclusively in APECED pneumonitis in the brother. Whole exome sequencing and copy number variation analyses are underway to further evaluate the patients condition.
This case demonstrates the importance of clinical presentation in the evaluation of genetic results and in the guidance of therapeutic management.
(12) Submission ID#570047
Different Clinical Manifestations in a Large Cohort of Predominantly Antibody Deficiency Patients with Monogenic Defects
Reza Yazdani, PhD1, Hassan Abolhassani, MD, PhD2, Asghar Aghamohammadi, MD, PhD3
1Fellow, Research Center for Immunodeficiencies, Childrens Medical Center, Tehran University of Medical Sciences, Tehran, Iran
2Postdoctoral, Division of Clinical Immunology, Department of Laboratory Medicine, Karolinska Institute at Karolinska University Hospital Huddinge, Stockholm, Sweden
3 Faculty member, Research Center for Immunodeficiencies, Pediatrics Center of Excellence, Children's Medical Center, Tehran University of Medical Science, Tehran, Iran
BACKGROUND: Predominantly antibody deficiencies (PADs) are the most common primary immunodeficiencies, characterized by hypogammaglobulinemia and inability to generate effective antibody responses.
OBJECTIVE: We intended to report most common monogenic PADs and to investigate how PAD patients who were primarily diagnosed as agammaglobulinemia, hyper IgM syndrome (HIgM) and common variable immunodeficiency (CVID) have different clinical and immunological findings.
METHODS: Stepwise next generation sequencing and Sanger sequencing were performed for confirmation of the mutations in the patients clinically diagnosed as agammaglobulinemia, HIgM and CVID.
RESULTS: Among 550 registered patients, the predominant genetic defects associated with agammaglobulinemia (48 BTK and 6 heavy chain deficiencies), HIgM (21 CD40L and 7 AID deficiencies) and CVID (17 LRBA deficiency and 12 atypical ICF syndromes) were identified. Clinical disease severity was significantly higher in patients with heavy chain and CD40L compared to patients with BTK (P = 0.003) and AICDA (P = 0.009) mutations. Paralysis following live polio vaccination was considerably higher in patients with heavy chain deficiency compared with BTK deficiency (P <0.001). We found a genotype-phenotype correlation among patients with BTK mutations regarding clinical manifestation of meningitis and chronic diarrhea. Surprisingly, we noticed that first presentations in the majority of ICF patients were respiratory complications (P = 0.008), while first presentations in LRBA patients were non-respiratory complications (P = 0.008).
CONCLUSION: This study highlights similarities and differences in clinical and genetic spectrum of the most common PAD-associated gene defects. This comprehensive comparison will facilitate clinical decision making, and improve prognosis and targeted treatment.
(13) Submission ID#574302
Visualizing the Effect of Lymphatic Pump Techniques on Immune System in Normal Subjects “ Randomized Control Trial”
Ahmed Abdelfattah, PhD1, Neveen Abdelraouf, PhD2, Samy Nasef, PhD2, Rania Ali, PhD2
1Lecturer, Faculty of Physical Therapy - Cairo University
2Professor of Physical Therapy - Cairo University Faculty of Physical Therapy
Aim: this study was designed to investigate and compare the efficacy of selected osteopathic lymphatic techniques on the absolute CD4+ count in healthy subjects. Materials and Methods: Forty-five subjects (33 males and 12 female), age varies from 20 to 50 years old. They were allocated to three groups each one has 15 subjects: first one received sternal pump and sternal recoil techniques for 12 sessions, three sessions per week. Second one received thoracic lymphatic pump and splenic pump techniques for 12 sessions, three sessions per week. Third one (control group) didn't receive OMT. Absolute count of CD4 was used to evaluate participants before and after application of the osteopathic techniques. Results: analysis showed significant increase in CD4 count after treatment in the second group also there was no significance in the first and third groups. P-value was 0.05. Conclusion and discussion: thoracic lymphatic and splenic pump manipulative techniques are effective methods of enhancing the immune system in healthy subjects (TLPT & SPT).
Key words: Osteopathy, Lymphatic techniques, Immune system
(14) Submission ID#576532
Infants with Idiopathic Transient and Persistent T Cell Lymphopenia Identified by Newborn Screening a Single Centers Experience from September 2010 December 2017
Artemio M. Jongco, III, MD, PhD, MPH1, Omer Elshaigi2, Foysal Daian2, BS, Emily Bae2, Amanda Innamorato2, Brianne Navetta-Modrov, MD3, Robert Sporter, MD4, David Rosenthal, DO, PhD5, Vincent Bonagura, MD6
1Assistant Professor of Medicine and Pediatrics, Division of Allergy & Immunology, Donald and Barbara Zucker School of Medicine at Hofstra/Northwell; Center for Health Innovations and Outcomes Research, Feinstein Institute for Medical Research, Manhasset, NY
2Research Intern, Division of Allergy & Immunology, Donald and Barbara Zucker School of Medicine at Hofstra/Northwell
3Fellow, Division of Allergy & Immunology, Donald and Barbara Zucker School of Medicine at Hofstra/Northwell
4Allergist, ENT and Allergy Associates
5Assistant Professor of Medicine and Pediatrics, Donald and Barbara Zucker School of Medicine at Hofstra/Northwell
6Professor of Medicine and Pediatrics, Division of Allergy & Immunology, Donald and Barbara Zucker School of Medicine at Hofstra/Northwell
Rationale: Infants with low T cell receptor excision circles (TREC) born in Queens, Nassau, and Suffolk counties are referred to our center for further evaluation. This study elucidates the demographic and laboratory characteristics of referred infants with transient or persistent idiopathic T cell lymphopenia (TCL) without clearly identified genetic or acquired etiology.
Methods: A retrospective analysis was performed from September 2010 (when TREC screening started) through the end of December 2017. Descriptive statistics were calculated for demographic and laboratory characteristics. T-test or Mann-Whitney tests were used to compare laboratory variables. Pearson or Spearman tests were used to determine correlation between initial TREC levels and T cell counts. By definition, the CD3+, CD4+, and CD8+ populations of transient TCL patients normalize by age 1 year.
Results: Eighteen infants with transient and 17 with persistent TCL were identified. Males comprised 61.1% of the transient and 47.1% of the persistent TCL cohorts. Whites comprised 11.1% of the transient and 35.3% of the persistent TCL cohorts. The mean initial TREC levels did not differ between the transient and persistent cohorts (67.7 vs. 78.5 TRECs/L of blood, P = 0.56). Mean initial absolute counts of CD3+ (2149 vs. 1300 cells/L, P <0.0001), CD4+ (1462 vs. 922 cells/L, P <0.0001), and median initial absolute counts of CD8+ (524 vs. 309 cells/L, P = 0.0075), were higher for transient vs persistent cohorts. Initial TREC level did not correlate with initial CD3+, CD4+, or CD8+ absolute counts. The median age of resolution for the transient cohort was 121.5 days (range 23-244). The absolute CD3+, CD4+, or CD8+ counts rarely exceeded the reported median values for age, and remained closer or below the 5th percentile for age up to 1000 days of life. The majority of both transient and persistent TCL patients demonstrated unremarkable lymphocyte proliferation to mitogens.
Conclusion: Our centers transient TCL cohort appears to be predominantly male and non-white, whereas the persistent TCL cohort is more evenly distributed by sex but still predominantly non-white. The transient cohort had lower initial TREC levels, but higher initial T cell counts. Both cohorts appear to have relatively intact in vitro function.
(15) Submission ID#577904
Primary Immune Deficiency Disease in Patients over Age 60: An Analysis from a Proprietary Immunology Patient Registry
Roger H. Kobayashi, MD1, Daniel Suez, MD2, Ralph Shapiro, MD3, Donald L. McNeil, MD4, Mark R. Stein, MD5, Frank J. Rodino, MHS, PA6, Herbert Lewis, PhD7
1Clinical Professor UCLA School of Medicine National Consultant, Immune Deficiency Foundation Executive Committee: CIIC, Consortium of Independent Immunology Clinics
2President/Director of Daniel Suez, MD Allergy, Asthma & Immunology Clinic, PA, Past President CIIC Consortium of Independent Immunology Clinics
3President/Director of Midwest Immunology Clinic; Past President: CIIC Consortium of Independent Immunology Clinics
4President/Director of Optimed Research LTD Consortium of Independent Immunology Clinics
5Physician, Allergy & immunology Allergy Section, Good Samaritan Medical Center, West Palm Beach, FL, USA
6Founder and President Churchill Outcomes Research, LLC Clinical Assistant Professor Stony Brook University School of Health Technology and Management
Consortium of Independent Immunology Clinics
7Associate Professor - College of Business - Stony Brook University
Consortium of Independent Immunology Clinics
Introduction: Primary immune deficiency disease (PIDD) is typically considered a pediatric illness, although advances in treatment and diagnosis are changing this paradigm. Currently, data on PIDD in older patients are very limited.
Objectives: To characterize the prevalence of PIDD among older individuals using a patient database maintained by the Consortium of Independent Immunology Clinics (CIIC), comprised of 17 specialty immunology outpatient practices in the US.
Methods: Patients with PIDD were identified in the CIIC database using ICD-10 codes D80, D.80.3, D80.4, D80.5, D80.6, D81.1, D81.2, D82.0, D82.3, and D83.0. A total of 235 records from 11 geographically-diverse clinics were identified and characterized by age, gender, and PIDD diagnosis.
Results: Of the 235 PIDD patients in the CIIC registry, 73 (31%) were between 60-87 years of age (see Figure). Within this age group, most patients were female (n=56, 77%). The most common diagnoses among patients >60 years of age included Common Variable Immunodeficiency with Predominant Abnormalities of B-Cell Numbers and Function (D83.0; n=41, 56%) and Antibody Deficiency with Near Normal Immunoglobulins (D80.6; n=14, 19%). In comparison, the registry included 36 (15%) patients aged 0-19 years; this age group was predominantly male (n=23; 64%). The most common ICD-10 codes within the younger cohort were relatively evenly distributed between Hereditary Hypogammaglobulinemia (D80.0), Antibody Deficiency with Near Normal Immunoglobulins (D80.6), and Common Variable Immunodeficiency with Predominant Abnormalities of B-Cell Numbers and Function (D83.0).
Conclusions: Our data suggest that PIDD in patients over age 60 may be more prevalent than previously reported. Additional research is needed to corroborate these findings, further characterize the nature of PIDD in this population, and determine whether there are unique diagnostic and treatment considerations within this demographic.
(16) Submission ID#579038
A Case of C6 Complement Deficiency with a Novel Mutation
Hassan A. Ahmad, MD1, Christopher D. Codispoti, MD, PhD2
1Allergy/Immunology Fellow, Rush University Medical Center
2Assistant Professor, Rush University Medical Center
Introduction/Background: Increased susceptibility to invasive infections with Neisseria has been well documented in patients with deficiency of terminal complement proteins. The molecular attack complex is constructed with complement components C5 to C9. A deficiency in complement C6 has been described previously in both African American and South African populations. Complement C6 deficiency is inherited in a co-dominant pattern, with multiple known mutations. We present a case of a 19-year-old, previously healthy male, who presented with invasive N. Meningitides infection. He was found to have a novel mutation noted on genetic sequencing of the complement C6 gene.
Objective: We present the case of a 19-year-old, previously healthy male, who presented with invasive N. Meningitides infection. On genetic sequencing, he was found to have three mutations of the complement C6 gene. Two of which have been described previously, and a third novel mutation.
Methods: A 19-year-old male with no known history presented to us with a 3-hour history of emesis. He was found to be febrile, and quickly decompensated, developing septic shock. Blood cultures were drawn, and within 12 hours grew N. Meningitides. He was treated with broad spectrum antibiotics upon arrival, and subsequently narrowed to Ceftriaxone. His hospital course was complicated by disseminated intravascular coagulation, as well as acute tubular necrosis, leading to end-stage renal disease for which he is listed for kidney transplant.
Results: On immunodeficiency evaluation, he was noted to have an undetectable CH50 (<13, reference range 31-60). Complement levels returned with C6 of 10.8 (reference range 28-69) and C1r of 41.5% (reference range 61-102%). Complement C6 function screen returned at 0% (reference range 40.7-169%). All other complement levels were within normal limits. Genetic sequencing showed the patient to be compound heterozygous for two of known four variants which have been reported to recur in African patients with complement C6 deficiency. This included c.821del and c.1879del, which are predicted to result in frameshift and premature protein termination. He was also found to be heterozygous for sequence c1202G>A, which results in amino acid substitution p.Arg401Lys. This variant is rare, with one large database reporting it in 6 of 276000 alleles, and not in a homozygous state. It has not been reported in a case of C6 complement deficiency previously.
Conclusions: We present the case of a previously healthy 19-year-old male with invasive meningococcal disease. He is compound heterozygous for two mutations that have been associated with total complement C6 deficiency; however, he was found to have subtotal C6 deficiency. Furthermore, he has a third novel mutation of the complement C6 gene. Further investigation is warranted on the significance of this finding and impact on relevance to possible kidney transplant.
(17) Submission ID#579501
Assay Characteristics of an Automated, Liposome-based Assay for the Measurement of CH50 Complement Activity and Comparison with a Haemolytic Method
Clare E. Tange, PhD1, Kattika Bootdee2, Kritraporn Deesin2, Leigh Williams, PhD3, Asada Leelahavanichkul4, Stephen Harding, PhD5
1Medical Science Liaison, The Binding Site
2Faculty of Medicine, Chulalongkorn University
3Medical Science Liaison, The Binding Site
4Asst Prof, Faculty of Medicine, Chulalongkorn University
5Research and Development Director,The Binding Site
Background: Measuring the function of the classical pathway of complement activation is useful in several disease states, including complement deficiency, autoimmune conditions such as systemic lupus erythematosus and certain forms of nephritis. The original method for assessing classical pathway activity was the haemolytic CH50 method, but this assay can be time consuming and has reagent stability issues due to the use of sheep red blood cells. There can also be high lab-to-lab variability due to differences in the protocols used. Here we report the assay characteristics of an automated, commercial, liposome-based assay to measure CH50 activity. We also compare the results obtained using the traditional haemolytic method with the automated, liposome-based method used on the SPAPLUS turbidimetric analyser.
Methods: A linearity study was performed based on CLSI guideline EP06-A. The linear range of the SPAPLUS CH50 liposome assay was established by analysis of a series of sample dilutions and evaluation of results against pre-defined goals for recovery and %CV. Precision was assessed based on CLSI guideline EP05-A2 over 21 days. 4 samples with different CH50 activities (23.7-65.1 U/mL) were run in duplicate, with two runs per day using 3 reagent lots and 3 different analysers. Interference analysis was performed by spiking haemoglobin, bilirubin, chyle, ascorbic acid or saline (as a control) into samples before measuring the CH50 activity.
For the assay comparison study, sera from 125 routine patient samples were used. Samples were collected from Chulalongkorn Hospital, Faculty of Medicine, Chulalongkorn University, Thailand. CH50 classical pathway activity was assessed using a haemolytic method and also using the liposome based CH50 assay for use on the SPAPLUS turbidimetric analyser (The Binding Site Ltd., Birmingham, UK). C3 protein concentrations were also available for 116 of these samples.
Results: The liposome CH50 assay gives a linear response over the range 11.8-95.5 U/mL, covering the measuring range of the assay (12.0-95.0 U/mL) at the standard analyser dilution (neat). The within run, between run and between day %CVs were all 5.4%. The total %CV was 6.8% in all 4 samples. Minimal interference was observed with the four common interferents tested.
A significant correlation was observed between the two CH50 methods (p<0.0001, r=0.66, y=1.1x±0.1), with 90.4% agreement between the methods in determining whether patients were above or below the lower limit of the assay normal range. The 12 individuals in disagreement had normal CH50 results using the haemolytic method, and low CH50 values in the liposome assay. Of these, C3 values were available for 10/12, and 5 had C3 concentrations below the lower limit of the assay normal range.
Conclusion: The liposome CH50 assay for use on the SPAPLUS analyser has passed assay development guidelines based on those set out by the CLSI for linearity, precision and interference, and there is a strong correlation between this automated assay and the haemolytic CH50 method used here. Five additional patients with low C3 concentrations were defined as having a low CH50 using the SPAPLUS liposome method compared to the haemolytic method.
(18) Submission ID#580179
Frequency of Specific Antibody Deficiency (SAD) and Respiratory Allergy in Patients with Recurrent Sinusitis
Charles Song, MD1, Dennys Estavez, Mr.2, Diana Cherinokova, MD3, Rie Sakai-Bizmark, MD4, Richard Stiehm, MD5
1Chief of Pediatric Allergy and Immunology, Ronald Reagan UCLA Medical Center, UCLA Mattel Children's Hospital
2Research statistician, Harbor-UCLA
3Resident, Harbor-UCLA
4Assistant Professor, Harbor-UCLA
5Professor, Division of Allergy and Immunology, UCLA
Submission Text
Frequency of Specific Antibody Deficiency (SAD) and Respiratory Allergy in Patients with Recurrent Sinusitis
Song CH1, Estavez D1, Chernikova D1, Sakai-Bizmark R1, Stiehm R2
1Harbor UCLA Medical Center, Torrance, CA
2UCLA Childrens Hospital , Los Angeles, CA
Rational: Respiratory allergy and subtle immunodeficiency may lead to recurrent sinusitis. We sought to determine the frequency and relationship of allergic sensitization, allergic respiratory diseases, and specific antibody deficiency (SAD) among patients with recurrent sinusitis and respiratory infections (URI).
Methods: The electronic medical records of 313 ambulatory patients from 6 to 70 years (median age 28 year) with recurrent respiratory infection (sinusitis >1x/y and URI>5x/yr) were screened for SAD, allergic sensitization (to mites, cockroach, cat, dog, and pollens), rhinitis (allergic and non-allergic), and asthma. Patients were divided into sinusitis and non-sinusitis classes; the pneumococcal antibody (PA) responses were categorized into three groups A, B, or C: A. Normal PA levels (defined as 70% of tested serotypes being above or equal to1.3 ug/mL for subjects, 6 years and older), B. Initially low with normal post-vaccination PA levels, and C. SAD with low PA levels even after Pneumovax).
Results: Among the 213 sinusitis patients, 187(88%) had decreased initial protective PA serotypes (groups B & C). Of these, 45 subjects ( C, 21 % of total) had SAD. The prevalence of SAD among the sinusitis patients was significantly higher compared to non-sinusitis group (21% vs.12%, p<0.01). The SAD prevalence increased with age;10 % for 6-19 years olds, 24% for 20 -39 year olds, 21% for 40-59 year olds, and 26 % for 60- 70 year olds.( P <0.05 between 6-19 year olds vs. 20-39 year olds). The initial numbers of protective PA serotypes were highest among Group A and lowest in C (p<0.01). The allergy sensitization was equally high for the both groups (62% vs. 62%). Asthma was common for both groups (43% vs. 42%), and rhinitis was more prevalent among non-sinusitis group (74% vs. 84%, p=0.05).
Discussion: SAD is a common and under-recognized cause of recurrent sinusitis. The prevalence rate increased with increasing age reflecting an aging immune response. Patients with SAD were more likely to present with initial very low number of protective PA serotypes compared to Group B representing a state that had experienced a greater difficulty mounting responses to polysaccharide antigens. The rates of allergy sensitization among both recurrent sinusitis and URI group were equal (62%) and significantly higher than the one reported by NHANES1 (45% among individuals 6 years and older), indicating an association between recurrent sinusitis /URI and allergy. Patients with recurrent sinusitis should be evaluated for both SAD and allergy.
Reference:
1. Arbes Jr SJ,Gergen PJ, Elliott L, Zeldin DC. Pfevalence of positive skin test response to 10 common allergens in the US population;results from the third National Health and Nutrition Examination Survey, J Allergy Clin Immunol 2009;124(3):522-7
(19) Submission ID#583743
Newborn Screening for SCID in Puerto Rico: A Three-year Experience
Giannina Coppola-Fasick, MD1, Yanira M. Arce, MD1, Sonia Ramírez, MS, MT (ASCP)2, Ledith Resto, MS3, Sulay Rivera-Sanchez, MS, PhD4, Sylvette Nazario-Jimenez, MD5, Cristina Ramos-Romey, MD6
1Allergy Immunology Fellow, University of Puerto Rico
2Supervisor Molecular Genetics section of Puerto Rico Newborn Screening Program, University of Puerto Rico
3NBS Follow Up Supervisor of Puerto Rico Newborn Screening Program, University of Puerto Rico
4Associate Director of Puerto Rico Newborn Screening Program, University of Puerto Rico
5Director of Allergy Immunology Program, University of Puerto Rico
6Assistant Director of Allergy Immunology Program, University of Puerto Rico
Background: Severe Combined Immune deficiency (SCID) is the most severe form of inborn immunodeficiencies, which are characterized in most cases by complete absence of T-cell-mediated immunity and by impaired B-cell-function. SCID is a pediatric emergency and is uniformly fatal without hematopoietic cell transplantation. Therefore, early diagnosis is important for prompt treatment. SCID can be detected using T cell receptor excision circle (TREC) assay. Newborn screening for SCID started in 2008 in Wisconsin and was added to the national recommended uniform panel for newborn screened disorders in 2010. There are currently 48 states performing mandatory screening for SCID in the United States. In August 2015 Puerto Rico (PR) added newborn screening for SCID to the mandatory newborn screening panel using dried blood spot specimen. A pilot program was done in 2011, in which one patient was diagnosed with SCID and successfully transplanted. The estimated incidence in PR is of 1:60,000 consistent with USA.
Purpose/Objective: Report and describe the data of the first 3 years of newborn screening for SCID in PR.
Method: We performed a retrospective record review of 15 positive newborn screening cases for SCID from patients born in PR during August 2015-October 2018. Patients received follow up at the Primary Immunodeficiency Clinic at the University of PR and NBS program.
Results: A total of 81,600 infants were screened, identifying 15 cases with low TREC levels. Of these cases, five infants died (four of which were preterm babies), four were lost to follow up, and three had normal follow up TREC levels after surgery for gastroschisis and omphalocele. The other 3 infants were referred to our clinic for diagnostic and follow up evaluation which lead to the identification of one Di George syndrome, one Vici syndrome, and other non SCID lymphopenia who is still undergoing evaluation at our Immunology Clinic.
Conclusion: We identified infants with abnormal TRECs that subsequently lead to diagnosis of non SCID lymphopenia, which may have not been recognized in the past and has enabled us to optimize management and outcomes of these infants. NBS has allowed for the early detection of infants with SCID (as found in our pilot study) and other lymphopenia disorders, which has permitted early diagnosis and management prior to developing symptoms or life-threatening complications.
(20) Submission ID#584818
Loss of Human ICOSL Results in Combined Immunodeficiency
Lucie Roussel, PhD1, Marija Landekic, MSc2, Christina Gavino, MSc3, Alexis Blanchet-Cohen, PhD4, Ming-Chao Zhong, PhD5, Melanie Langelier, MSc6, Denis Faubert, PhD7, André Veillette, MD8, Don Vinh, MD9
1Research Associate, Research Institute - McGill University Health Centre
2Graduate student (PhD trainee), Research Institute - McGill University Health Centre
3Research Assistant, Research Institute - McGill University Health Centre
4Bioinformatics, Institute Recherche Clinique de Montreal
5Research Associate, Institute Recherche Clinique de Montreal
6Research Nurse - Clinical Program Manager, Research Institute - McGill University Health Centre
7Proteomics Director, Institute Recherche Clinique de Montreal
8Director, Molecular Oncology Research Unit, Institute Recherche Clinique de Montreal
9Associate Professor, Clinician-Scientist, McGill University Health Centre
Background: Primary Immunodeficiencies are inborn errors of immunity that represent naturally occurring experimental models to decipher human immunobiology. We present a patient with combined immunodeficiency, who suffered from recurrent respiratory tract and viral infections associated with hypogammaglobulinemia and panlymphopenia. He also had progressive moderate neutropenia, without evidence of bone marrow failure or associated severe prototypical infections.
Methods: Identification of the causal gene was performed by whole exome sequencing, bioinformatics analyses, and Sanger sequencing. The impact of the variant on gene product was assessed by cDNA sequencing and protein detection (flow cytometry, Western blot, confocal microscopy) on various cell lines. The effect of the variant on protein function was assessed by co-culture experiments (Jurkat with lymphoblastoid cells derived from healthy controls or patient) and by transendothelial migration of cells across endothelial cell lines reconstituted with ICOSLG (wild-type vs. variant).
Results: We identified a homozygous mutation in the Inducible T-Cell Costimulator Ligand gene (ICOSLG; c.657C>G; p.N219K). Whereas wild-type ICOSL is expressed at the cell surface, the ICOSL p.N219K mutant abolishes cell surface expression, due to retention of protein in the endoplasmic reticulum/Golgi apparatus. The mutant ICOSL was associated with diminished T cell costimulatory activity and with decreased transendothelial lymphocyte migration. Additionally, endothelial expression of mutant ICOSL compromised neutrophil transmigration, by reducing the proper localization of E-Selectin and ICAM-1 at the cell surface.
Conclusions: Our work identifies human ICOSLG deficiency as a novel cause of a combined immunodeficiency syndrome. Moreover, findings from this natural experiment sheds light on the broad immunologic functions of ICOSLG in human immunobiology.
(21) Submission ID#584884
Safety of Administration of Rotavirus Vaccine in Infants Born to Mothers Receiving Biologic Therapy During Pregnancy: A Retrospective Case Series
Christina Smith, MD1, Niraj C. Patel, MD, MS2, Richard Sigmon, MD3
1Resident Physician, Department of Pediatrics, Levine Children's Hospital, Atrium Health
2Physician, Department of Pediatrics, Division of Infectious Disease and Immunology, Levine Children's Hospital, Atrium Health
3Physician, Department of Medicine, Division of Gastroenterology, Atrium Health
Background/Aims: Rotavirus vaccine is a live viral vaccine that is part of the routine U.S. childhood immunization schedule. Live viral vaccines administered to infants of mothers who received biologic medications during pregnancy can potentially cause vaccine-associated disease. Infant death from disseminated mycobacterial infection after vaccination with bacille Calmette-Guerin (BCG) in infants whose mothers received infliximab during pregnancy has been reported. It is currently recommended that infants born to women who received biologic therapy during pregnancy not receive live viral vaccines, however there is a paucity of information regarding adverse events from live viral vaccines. We report two infants, born to mothers receiving infliximab during pregnancy, who tolerated the complete series of rotavirus vaccine.
Methods: Two infants who received rotavirus vaccine and whose mothers received infliximab (monoclonal antibody against tumor necrosis factor alpha which blocks the inflammatory response) during pregnancy were identified and their charts were reviewed. Each mothers chart was assessed for timing of the biologic doses during pregnancy and concurrent immunosuppressant therapy.
Results: The mother of the first infant had Crohn's Disease and received infliximab every 6 weeks throughout her pregnancy (final infusion at approximately 35 weeks estimated gestational age [EGA]). She did not take additional immunosuppressive drugs throughout her pregnancy. The infant was born at 39 weeks EGA. The infant received rotavirus vaccine at 2, 4, and 6 months of age. The infant did not have coexisting medical conditions or recorded hospitalizations during the first year of life. There were no side effects from rotavirus vaccine documented during well child examinations. The childs growth was normal during the first year of life.
The mother of the second infant also had Crohn's disease and received infliximab infusions every six weeks during pregnancy until 27 weeks EGA. Additionally, she took mesalamine (anti-inflammatory) daily. The infant was born at 33 weeks EGA. The baby had a brief and uncomplicated neonatal intensive care unit stay. She did not have medical conditions diagnosed at the time of birth, or in the first year of life. The child received rotavirus vaccination at 2, 4, and 6 months of chronological age, and the infant did not experience documented adverse reactions. The child presented to the emergency department twice in the first year of life: once for thrush at 10 months of age and once for viral gastroenteritis at 11 months of age. The childs growth curve was unremarkable.
Conclusions: We report two infants, whose mothers received infliximab during pregnancy, who safely tolerated the 3-dose series of rotavirus vaccination. Neither infant in this case series suffered from minor or severe adverse events as a direct consequence of receiving rotavirus vaccine. This suggests that administration of rotavirus vaccine may be safe in infants whose mothers received biologic therapy.
(22) Submission ID#585141
Combined Immune Deficiency in Association with a Single RAG1 Missense Variant in a 28-year-old Female
Charles Song, MD1, Diana Cherinokova, MD2, Joseph A. Church, MD3, Henry Lin, MD4, Christin Deal, MD5, Manish Butte, MD, PhD6
1Chief of Pediatric Allergy and Immunology, Ronald Reagan UCLA Medical Center, UCLA Mattel Children's Hospital
2Resident, Harbor-UCLA
3Professor, Pediatrics, Children's Hospital Los Angeles and Keck School of Medicine of U.S.C.
4Chief, Pediatric Genetics, Harbor-UCLA
5Allergy/Immunology Fellow, UCLA
6Division of Allergy/Immunology Chair, Division of Immunology, Allergy, and Rheumatology, Dept. of Pediatrics and Jeffrey Modell Diagnos-tic and Research Center, University of California, Los Angeles
Introduction: Combined immunodeficiencies (CIDs) can arise from partial loss of function variants in recognized SCID genes, which can lead to relative lymphopenia with poorly functioning and oligoclonal T cells. CIDs have been most commonly associated with variants of the RAG genes, but other genes are also implicated. Clinical symptoms may be less severe, and the onset generally is delayed, compared to typical SCID presentations.
Case Report: A 28-year-old female presented with a history of recurrent and progressively worsening infections involving multiple microorganisms and organs, starting in infancy and requiring frequent hospitalizations. Bacterial or viral infections included rhinosinusitis, otitis media, herpetic stomatitis, dental abscesses, pneumonias, pulmonary mycobacterial abscesses, CMV hepatitis, urinary tract infections, dermal abscesses, and groin hidradenitis. Fungal and yeast infections included cryptococcal meningitis, oral thrush, dermatophytosis of the face, osteomyelitis of a finger, and onychomycosis. Laboratory tests in 2018 showed: mildly low T cell counts (791/uL) with a reversed ratio of CD4/CD8 T cells (0.22); almost absent B cells (2/uL); and low NK cell counts (19/uL). CD4+ T cells were mostly of the memory phenotype (87%). T cell development showed low counts of Th17 cells. T-cell stimulation tests demonstrated poor proliferation responses (<30%) to Concanavalin A, tetanus toxoid, and Candida albicans, with near-normal responses to pokeweed (>13%) and PHA (>84%). She had low Ig levels (IgA 72, IgM 23, IgE <2), except for IgG (872mg /mL; due to replacement since early childhood).
Limited genetic evaluation at age 9 showed a heterozygous variant in the RAG1 gene (g.36595918T>C, c.1064T>C, p.Met355Thr; NM_000448.2).
Discussion: Loss of function variants in RAG1 or RAG2 genes are known to cause a T- B- NK+ type SCID. More than 100 missense variants have been reported for RAG1, with disease-associated variants predominantly in zinc binding regions. The RAG1 missense variant in our patient also lies within the zinc binding region (amino acids 354-383). The variant is rare (mean allele frequency 0.0001521 in gnoMAD) and has been identified in at least one other individual with SCID (T-, B cell-, NK+). Although classified as a variant of unknown significance, occurrence in at least two individuals with deficiencies of T and B cells-- within a functionally important RAG1 domain -- supports an interpretation that the variant may be pathogenic. Most patients with CID with RAG variants are either homozygous for a poorly functional allele or have one nonunfucitonal and a second, poorly functional allele. We detected only a single potentially pathogenic allele. Our patient has decreased NK cells in addition to T and B cell defects. Further genetic studies including whole exome sequencing, are planned to identify further variants in RAG1 or other relevant genes.
(23) Submission ID#586431
Capturing Quality of Life in Patients with Common Variable Immunodeficiency (CVID) Using the Patient-Reported Outcomes Measurement Information System (PROMIS-29) Survey
Shouling Zhang, MD1, Myriam Kline, PhD2, Ramsay Fuleihan, MD3, USIDNET Consortium4, Kathleen E. Sullivan, MD, PhD5, Artemio M. Jongco, III, MD, PhD, MPH6
1Pediatrics Resident, Department of Pediatrics, Donald and Barbara Zucker School of Medicine at Hofstra/Northwell, New Hyde Park, NY
2Associate Research Statistician, Biostatistics Unit, Feinstein Institute for Medical Research, Manhasset, NY
3Professor of Pediatrics, Division of Allergy and Immunology, Northwestern University Feinberg School of Medicine, Chicago, NY
4United States Immunodeficiency Network, National Institute of Allergy and Infectious Diseases (NIAID), Towson, MD. The U.S. Immunodeficiency Network (USIDNET), a program of the Immune Deficiency Foundation (IDF), is supported by a cooperative agreement, U24AI86837, from the National Institute of Allergy and Infectious Diseases (NIAID).
5 Professor, The Children's Hospital of Philadelphia
6Assistant Professor of Medicine and Pediatrics, Division of Allergy & Immunology, Donald and Barbara Zucker School of Medicine at Hofstra/Northwell; Center for Health Innovations and Outcomes Research, Feinstein Institute for Medical Research, Manhasset, NY
Introduction/Background: Common variable immune deficiency (CVID) is the most common antibody deficiency affecting both children and adults. Lifelong immunoglobulin replacement therapy (IGRT) is the mainstay of treatment. Information is limited about health-related quality of life (HRQOL) in patients with CVID receiving IGRT. The Patient Reported Outcomes Measurement Information System (PROMIS) is a validated self-report measure of physical, mental, and social health which can be used to assess HRQOL in patients with primary immunodeficiency diseases (PIDD).
Objectives: The primary objective of this study was to compare patients with and without CVID on HRQOL domains using PROMIS-29 survey data from the United States Immunodeficiency Network (USIDNET) registry. USIDNET maintains a national registry of validated data from PIDD patients through the Immune Deficiency Foundation (IDF). The primary endpoint variables were scores in 7 HRQOL domains: 1) Depression, 2) Anxiety, 3) Physical Function, 4) Pain Interference, 5) Fatigue, 6) Sleep Disturbance, and 7) Social Participation. A secondary objective was to describe and compare patients with or without CVID with respect to IGRT.
Methods: IDF prompts all its electronic personal health record users to answer PROMIS-29 biannually. Only those who have consented to participate in the USIDNET registry are included. Data from Fall 2015 to Spring 2018 were analyzed. Groups were compared using descriptive statistics and the Wilcoxon Mann-Whitney test. A mixed linear model approach compared groups with respect to the endpoint variables while adjusting for time, sex, age, and/or BMI. Simple models were tested followed by the addition of covariates. Interactions that were not significant were removed from models. All analyses use SAS, version 9.4.
Results: Among the 222 PIDD patients in the registry, 173 patients (78%) were diagnosed with CVID. Humoral and cellular immunodeficiencies comprised the remaining non-CVID diagnoses (22%). Patients ranged from 18 to 83 years of age (mean age of 54). The study population was largely female (80%), Caucasian (96%), and with a mean BMI of 29. Twenty-five (11.5%) participants had a family history of PIDD. Results of the PROMIS-29 survey revealed that there was an effect of group (i.e., CVID/non-CVID) on the Fatigue subscale. The CVID group scored 3.05 points higher, on average, than the non-CVID group, after controlling for age and time (p=0.037). No other group differences were found among the remaining subscales. BMI was a significant predictor across all subscales (p<0.01), except for Anxiety (p=0.17). With regards to IGRT, the median IgG dose was less for CVID patients compared to non-CVID patients (17.8g vs. 25.0g, p=0.05), and the median number of days on IGRT was less for the CVID group compared to the non-CVID group (14 vs. 28, p=0.025).
Conclusions: These data suggest that fatigue may be a key factor influencing the quality of life among PIDD patients with CVID. Future prospective longitudinal studies using PROMIS-29 will be needed to confirm this finding. Additional studies elucidating the role of BMI on HRQOL and IGRT dosing are recommended.
(24) Submission ID#586449
Describing Transient T Cell Lymphopenia in the United States Immunodeficiency Network (USIDNET) Following Infants with Low Lymphocytes (FILL) Program and a Single Referral Center from 2010-2017
Shouling Zhang, MD1, Omer Elshaigi2, Foysal Daian, BS2, Emily Bae2, Amanda Innamorato2, Brianne Navetta-Modrov, MD3, Robert Sporter, MD4, David Rosenthal, DO, PhD5, Vincent Bonagura, MD6, Elizabeth A. Secord, MD7, Charlotte Cunningham-Rundles, MD, PhD8, John Routes, MD9, USIDNET Consortium10, Artemio M. Jongco, III, MD, PhD, MPH11
1Pediatrics Resident, Department of Pediatrics, Donald and Barbara Zucker School of Medicine at Hofstra/Northwell, New Hyde Park, NY
2Research Intern, Division of Allergy & Immunology, Donald and Barbara Zucker School of Medicine at Hofstra/Northwell
3Fellow, Division of Allergy & Immunology, Donald and Barbara Zucker School of Medicine at Hofstra/Northwell
4Allergist, ENT and Allergy Associates, NY, NY
5Assistant Professor of Medicine and Pediatrics, Donald and Barbara Zucker School of Medicine at Hofstra/Northwell
6Professor of Medicine and Pediatrics, Division of Allergy & Immunology, Donald and Barbara Zucker School of Medicine at Hofstra/Northwell
7Professor of Pediatrics at Wayne State University, Children's Hospital of Michigan, Division of Allergy, Asthma and Immunology, Children's Hospital of Michigan, Detroit, MI
8Professor in Medicine, Division of Clinical Immunology, Icahn School of Medicine, Mount Sinai, NY, NY, USA
9Chief, Professor, Division of Allergy and Immunology, Children's Hospital of Wisconsin-Milwaukee, Milwaukee, WI
10United States Immunodeficiency Network, National Institute of Allergy and Infectious Diseases (NIAID), Towson, MD.
11Assistant Professor of Medicine and Pediatrics, Division of Allergy & Immunology, Donald and Barbara Zucker School of Medicine at Hofstra/Northwell; Center for Health Innovations and Outcomes Research, Feinstein Institute for Medical Research, Manhasset, NY
Rationale: Infants with low T cell receptor excision circles (TREC) born in Queens, Nassau, and Suffolk counties in New York were referred to Northwell Health for further evaluation after abnormal newborn screens. The demographic and immune parameters of infants with transient T cell lymphopenia (tTCL) without clearly identified genetic or acquired etiology are described. TCL is considered transient if the lymphopenia resolves by 12 months of age. Similar data from the Following Infants with Low Lymphocytes (FILL) program of the United States Immunodeficiency Network (USIDNET) are presented.
Methods: A retrospective analysis of two separate patient cohorts with tTCL are described. Cohorts include patients referred to a single center, Northwell Health, in NY from September 2010 to December 2017 and at USIDNET using data tracked by FILL from June 2011 to July 2018.
Results: Out of 1,234 referrals at Northwell, 18 infants with tTCL were identified. Infants were predominantly male (61.1%) and non-Caucasian (89.9%). Out of 71 FILL participants, 9 infants with tTCL were identified. Infants were predominantly male (55.6%) and non-Caucasian (55.6%). Initial laboratory parameters for the Northwell versus FILL cohorts are summarized: a) median TREC levels: 54.5 vs. 47.0 TREC/L of blood; b) median absolute CD3+ count: 2135 vs. 1166 cells/L; c) median CD4+ count: 1460 vs. 777.0 cells/L; d) median absolute CD8+ count: 524.5 vs. 440.0 cells/L. Initial naïve CD4+ T cell information was available for 0 Northwell and 5 FILL infants (median 52%). Mitogen proliferation studies were performed in 10 (55.6%) Northwell and 6 (66.7%) FILL infants with 90% of these Northwell and 50% of these FILL infants demonstrating normal proliferation. Genetic testing, such as targeted genetic panels or chromosomal microarrays (CMA), was performed in 5 Northwell and 0 FILL infants. No genetic or chromosomal aberrations were identified. Whole exome sequencing (WES) was not performed in either cohort. 11 of 18 (61.1%) Northwell and 7 of 9 (77.8%) FILL infants did not receive the initial rotavirus vaccine. No FILL infants were vaccinated but no adverse effects were reported in 5 of 18 (27.8%) Northwell infants who received the first rotavirus dose. Of these, 3 of 5 (60.0%) had normal mitogen proliferation while 1 (20.0%) had decreased proliferation to phytohemagglutinin.
Conclusions: Identifying biomarkers for tTCL and developing evidence-based guidelines for the diagnosis and management of tTCL are important knowledge gaps. This descriptive study is limited by small sample size and the constraints of registry-based research. Although there appear to be differences between these cohorts, our findings suggest that tTCL may disproportionately affect different segments of the population. tTCL infants with normal mitogen proliferation may be able to tolerate rotavirus vaccination. Thus, routinely checking proliferation studies in all tTCL infants may help risk stratify these patients and minimize vaccine-related adverse events. Currently, there is insufficient evidence to recommend more extensive genetic testing such as genetic panels, CMA, or WES. Systematically collecting information about patient characteristics and outcomes, as well as encouraging increased participation in registries such as FILL, may help address these shortcomings.
(25) Submission ID#586903
An Assay to Measure the Complement Binding Activities of Anti-dsDNA Antibodies in SLE
Clare E. Tange, PhD1, David Taylor2, Marcos López-Hoyos, MD, PhD3, Victor Martínez-Taboada, MD, PhD4, Jaime Calvo-Alén, MD, PhD5, Leigh Williams, PhD1, Stephen Harding, PhDs6
1Medical Science Liaison, The Binding Site
2The Binding Site
3Head of Service, Immunology Service., Hospital Universitario Marqués de Valdecilla
4Staff, Rheumatology Service., Hospital Universitario Marqués de Valdecilla
5Head of Service, Rheumatology Service., Hospital Universitario Araba
6Research and Development Director, The Binding Site
Background: Systemic lupus erythematosus (SLE) is a chronic, inflammatory disease that affects multiple organs. The measurement of anti-dsDNA antibodies (Abs) is a gold standard serological test used in the diagnosis and monitoring of SLE, with higher serum levels associated with worse prognosis. However, not all anti-dsDNA Abs are pathogenic, and some patients have consistently high levels with low disease activity. One mechanism suggested for the pathogenicity of these antibodies is complement activation. Here we describe an assay to measure the C1q binding activities of anti-dsDNA Abs in SLE patients.
Materials & Methods: The concentration of anti-dsDNA Abs was determined using the QuantaLite dsDNA ELISA kit (INOVA) as per the manufacturers instructions. In order to determine the C1q binding capacity of bound Abs, samples were added to the pre-coated plate and incubated. Bound anti-dsDNA Ab/C1q complexes were then detected using a biotinylated anti-C1Q antibody (570 ng/mL) and streptavidin peroxidase (1 mg/mL). Normal reference ranges were developed in serum samples from healthy controls, and upper limits of these normal ranges were used as cut-offs. The dsDNA Abs and C1q binding capacity of bound Abs was then assessed in 49 SLE patients, and compared to other markers and the SLE Disease Activity Index (SLEDAI) score. Results are displayed as absorbance at 450nm (AU).
Results and Conclusions: The 95th percentile ranges for anti-dsDNA Abs (0.068-0.137 AU) and C1Q binding activities (0.207-0.313 AU) were developed from the measurements generated in 17 healthy serum samples. SLE patients with an increased anti dsDNA Ab concentration (>0.137 AU) were then separated into those with low (<0.313 AU) and high (>0.313 AU) C1q binding activities. Patients whose dsDNA Abs had high C1q binding activity were found to have significantly higher SLEDAI scores (mean 6.70 vs 3.19). Serum C1q concentration, serum dsDNA Abs (measured by another method) and serum C3 and C4 concentrations were not significantly different between the two groups. This assay suggests that dsDNA Abs from SLE patients differ in their ability to bind complement, and that high complement binding activity of these antibodies may be linked to a more active form of disease.
(26) Submission ID#587614
Natural History of X-linked Lymphoproliferative Disease, Lessons Learned from a Long-term Survivor
Tiphanie Vogel, MD, PhD1, Mihail Firan, MD2, Joud Hajjar, MD, MS1
1Assistant Professor, Baylor College of Medicine, Texas Childrens Hospital Center for Human Immunobiology and Division of Immunology, Allergy and Rheumatology
2Assistant Professor, Baylor College of Medicine, Departments of Pediatrics, Pathology and Immunology
X-linked lymphoproliferative (XLP) is a primary immunodeficiency, caused by signaling lymphocyte activation molecule (SLAM)-associated protein (SAP) deficiency. Patients with XLP have severe immune dysregulation, usually triggered by EBV infection, leading to fulminant infectious mononucleosis, dysgammaglobulinemia and lymphoproliferation. Without hematopoietic stem cell transplant (HSCT) fatality is reportedly 100% by age 40. We report the natural history of XLP in a patient, and describe the lessons learned.
Our patient was healthy and developed normally until 6-years of age, when he developed progressive respiratory symptoms. Lung biopsy revealed mature lymphoplasmacytic infiltrate in the alveolar septa, consistent with lymphoid interstitial pneumonia (LIP). He received corticosteroids and cyclophosphamide with significant improvement. At age 12, he developed severe infectious mononucleosis (fever, hepatosplenomegaly, lymphadenopathy, lymphocytosis). He had a protracted clinical course, but eventually recovered and seroconverted to a typical convalescent pattern. He subsequently developed hypogammaglobulinemia, and was started on intravenous immunoglobulin (IVIG). During the same year, his 10-year-old brother developed LIP, and subsequently hemophagocytic lymphohistocytosis (HLH) and died within 4 months from overwhelming candidiasis. Unfortunately, his youngest brother (age 7) then developed LIP and died 2 months later from a massive gastrointestinal bleed.
Both siblings were treated with corticosteroids and cyclophosphamide; they did not have detectable EBV infection. At age 13 years, our patient experienced recurrent strokes and was found to have biopsy-proven CNS vasculitis. He was treated with interferon- and recovered with residual left sided weakness, but was lost to follow-up.
He continued on IVIG, with no other immunomodulatory agents for several decades. He had progressive lung disease and recurrent seizures controlled with anti-epileptics. At age 43, he developed sudden vision change, headache and right-sided weakness, followed by a seizure. MRI of the brain revealed small bilateral areas of acute infarction suggestive of a central embolic event, however, no primary thrombus was identified. He did not receive any immunosuppression but was anti-coagulated. Eventually he was discharged home with resolution of weakness to his baseline.
The patient was referred to our clinic after discharge and we re-evaluated him after 31 years. Immune profiles at the time showed therapeutic IgG troughs, low/undetectable IgM/A/E, normal T/B/NK-cell counts, normal spontaneous, but decreased antibody-dependent NK cytotoxicity, 0% SAP protein expression (on CD3+CD8+, CD3-CD56+ and CD3+CD56+ cells), and deletion on the X chromosome encompassing the SH2D1A gene which encodes SAP. His mother was a carrier of the same deletion. His functional status excluded the option of HSCT. A year later, he had rapid deterioration with recurrent lung infections, liver failure, and thrombocytopenia. Bone marrow biopsy revealed Hodgkins Lymphoma. He declined chemotherapy and died few days after diagnosis.
Our case represents a rare patient with XLP surviving to the fifth decade without HSCT, particularly having experienced mononucleosis and non-EBV related CNS vasculitis. Our patient survived decades longer than his brothers (who most likely shared the same genetic defect) without evidence of somatic reversion (0% SAP expression in CD3+CD8+) to explain his milder clinical phenotype. This case may help in understanding the natural history of XLP, and confirms that prognosis remains poor without HSCT.
(27) Submission ID#587907
Abatacept for CTLA-4 Haploinsufficiency Presenting with Severe Bone Marrow Aplasia and Septic Shock - A Case Report
Emilie Proulx, BSc, MD, FRCPC1, Antoine Morin-Coulombe, MD, FRCPC2, Jean-Philippe Drolet, MD, FRCPC3, Vincent Castonguay, MD, FRCPC4
1Fellow-in-training, Clinical immunology, CHU de Québec, Université Laval
2Fellow-in-training, Medical Oncology and Hematology, CHU de Québec, Université Laval
3Allergy and Clinical Immunology, CHU de Québec, Université Laval
4Haematology and Oncology, CHU de Québec
CTLA-4 is a major negative regulator of immune responses, and CTLA-4 haploinsufficiency has been identified as a monogenic cause of primary immunodeficiency in patients presenting with a common variable immunodeficiency (CVID) phenotype with autoimmunity. Here we present the case of PB, a 40-year-old man who had been followed by the immunology service of our center for 17 years. A diagnosis of CVID had first been made when the patient presented with atypical transverse myelitis, low immunoglobulin levels, and lymphopenia. Over the years, his clinical picture was dominated by various forms of autoimmunity, namely inflammatory demyelinating disorder of the central nervous system, autoimmune haemolytic anemia, immune thrombocytopenia, cryptogenic organizing pneumonia, rheumatoid-like polyarthritis, chronic liver transaminitis with biopsy-proven moderate fibrosis, and lymphocytic colitis with malabsorption. Immunoglobulin replacement therapy was started at diagnosis, and autoimmunity was sequentially treated with methotrexate, interferon beta 1-a, cyclophosphamide, mycophenolate mofetil, rituximab, and finally a combination of low-dose prednisone and sirolimus, with stabilization of his neurological condition, the most debilitating complication of his immune dysregulation syndrome. Bone marrow transplant had been offered, but declined by the patient due to perceived good quality of life compared to transplant-associated risks.
The patient was later referred to our hematology ward in July of 2018 for septic shock complicating febrile neutropenia, which was part of a two-month, gradual-onset pancytopenia. The diagnosis of immune-mediated aplastic anemia soon became apparent, as demonstrated by a bone marrow biopsy performed in a peripheral center two days prior to admission. The underlying pneumonia and thereafter biopsy-induced Staphylococcus aureus iliac osteomyelitis and soft-tissue abscess were treated with broad-spectrum antibiotics as well as multiple surgical interventions. The patient was started on eltrombopag, high-dose corticosteroids and cyclosporin A, the latter promptly switched to tacrolimus due to liver enzymes disturbances, all of which resulted in no significant hematologic response despite over seven weeks of treatment (with concurrent treatment of complicating infection, upper gastrointestinal bleeding, and intensive-care-unite myopathy). During that time, genetic confirmation of CTLA-4 haploinsufficiency was received, and the patient was thereafter started on abatacept on day 48 of current hospitalization. Administration of equine anti-thymocyte was initially foregone because of perceived infectious risk in the setting of poor iliac wound healing and superimposed adenovirus viremia; however, given the lack of response, it was given on days 52 through 54 of hospitalization. Haematologic response began on day 67 of hospitalization with a steady rise in all-lineage myelopoiesis up to a complete neutrophil response, platelet near-complete response as well as resolution of transfusion needs by day 101. While waiting for a well-matched bone marrow donor, isolated platelet decrease was observed and attributed to multiple factors, including low-grade thrombotic microangiopathy, inflammatory consumption and drug-related thrombocytopenia, but the patient remained well. To our knowledge, our patients presentation is one of the most severe manifestation of CTLA-4 haploinsufficiency to have responded to targeted therapy with abatacept, as a bridge to hematopoietic stem cell transplantation, with resolution of both immune and infectious complications, showing that genetic diagnosis is helpful in optimizing the management of presumed CVID patients.
(28) Submission ID#588520
Extreme Phenotypes, Identical Mutations: Two Patients with Same Nonsense XLF/Cernunnos Homozygous Mutation
Luis I. Gonzalez-Granado, MD1, Nerea Dominguez-Pinilla, MD2, Melina Soledad Perrig, MD3, Carmen Rodriguez-Vigil, PhD4, Nerea Salmón-Rodriguez, MD5, Cristina Martinez Faci, MD4, Javier Blas-Espada, MD6, Marta López-Nevado, MD6, Raquel Ruiz-Garcia, PhD3, Rebeca Chaparro, MD3, Luis Allende, PhD6, Maria José Recio Hoyas, PhD3
1Consultant. Head of the Primary Immunodeficiencies Unit. Pediatrics, Hospital 12 de Octubre
2Consultant, Hospital 12 de Octubre Health Research Institute (i+12), Madrid, Spain. University Hospital Virgen de la Salud. Pediatric Hematology and Oncology Unit. Toledo. Spain.
3Hospital 12 de Octubre Health Research Institute (i+12), Madrid, Spain, Dept. of Immunology, School of Medicine, Complutense University
4Consultant, University Hospital Miguel Servet. Pediatric Hematology and Oncology Unit. Zaragoza. Spain
5Consultant, Hospital 12 de Octubre Health Research Institute (i+12), Madrid, Spain. University Hospital 12 octubre. Madrid. Spain
6Hospital 12 de Octubre Health Research Institute (i+12), Madrid, Spain, Dept. of Immunology, University Hospital 12 octubre. Madrid. Spain
BACKGROUND: XLF/Cernnunos deficiency is a rare primary immunodeficiency classified within the DNA repair defects. These patients present severe growth retardation, microcephaly, lymphopenia and increased cellular sensitivity to ionizing radiation. Here, we describe two unrelated cases with the same nonsense mutation in the NHEJ1 gene showing significant differences in clinical presentation and immunological profile but a similar DNA repair defect.
METHODS: Missense NHEJ1 mutation was identified by targeted next-generation sequencing with an in-house designed panel of 192 genes.
For foci experiments, primary skin fibroblasts were irradiated with ionizing irradiation (137Cs) or treated with 20mM Etoposide for 1 hour. After irradiation, the cells were seeded at a density of 1x104 cells/mL in T75 flasks in triplicate. To evaluate cell sensitivity to gamma-IR (1 and 3 Gy),adherent cells were trypsinized and counted 11 days later. PBMCs from patient and healthy controls were irradiated with 10Gy, fixed and stained for CD3, CD19 and phospho-histone H2AX. Mean fluorescence intensities (MFI) of gamma-H2AX were evaluated on gated CD3+ lymphocytes.
RESULTS:We report two patients harboring the same homozygous mutation in Cernunnos/XLF/NHEJ1 gene. Strikingly, their clinical phenotype ranges from severe combined immunodeficiency to isolated thrombocytopenia followed until escolar age (Table 1). They harbour the same c.169C>T mutation in NHEJ1 gene but different immunologic features (Table 2). P2 presented with mild T lymphopenia, hypersensitivity and NHEJ repair defect, typical for patients with XLF/NHEJ1 defects. On the other hand, P1 presented a more severe phenotype (T-B-), however hypersensitivity and NHEJ repair defect was similar to P2.Of note, P2 has survived into the first decade of live. Both patients are alive and well after HSCT.
DISCUSSION: Usually the repair defect in these disorders is assessed by immunofluorescence assays of irradiation-induced gamma-H2AX foci using skin fibroblasts. A high throughput, sensitive and reliable assay to quantify gamma-H2AX foci in PBMCs isolated from blood samples would be a valuable tool to diagnose these patients and perform HSCT early. Flow cytometry (FC) can be applied as a rapid diagnostic tool for DNA repair disorders. Patients with the same homozygous mutation (p.R178X) in NHEJ1 gene have been previously reported. Two patients died at 1.5 and 4 years while another of the patients is already 8 years old and is alive (without HSCT). However,none of these patients presented severe T lymphopenia as it has been observed in our first patient.
CONCLUSIONS: The assignment of a timely and accurate diagnosis is of paramount importance in the management of patients with defects in DNA repair. In the era of NBS an abnormal TREC assay should be followed by NGS approach as Cernunnos deficiency may present early in life as SCID,as other RS-SCID defects. Since genetic diagnosis takes time,functional radiosensitivity assays in peripheral blood may lead to the correct diagnosis and avoid exposure to alkylating agents during the conditioning regimen prior to genetic diagnosis. It would also be helpful in cancer patients to individualize and to guide the dosing of ionizing radiation (IR) and/or genotoxic agents to avoid accumulation of cells with genomic instability that could accelerate cancer development.
Table 1. Clinical features of the patients with Cernunnos deficiency
| P1 | P2 | ||
| Origin | Caucasic | Caucasic | |
| Consanguinity | No | No | |
| Age | Onset | 1m | 9m |
| Current | 2y | 7y | |
| Clinical features | Microcephaly | + | + |
| Growth retardation | + | + | |
| Facial dysmorphism | - | + | |
| Additional clinical features | Neurological manifestations | - | - |
| Bone malformation | - | - | |
| Autoimmunity | - | + | |
| Cytopenias | - | + (thrombocytopenia) | |
| Infections | Respiratory tract infections | - | + |
| Bacterial and opportunistic infection | - | - | |
| Urinary tract | - | - | |
| Outcome | Status | Alive and well (HSCT) | Alive and well (HSCT) |
TABLE 2. Immunologic features of the patients
| Parameter |
RefValues (children) |
P1 | P2 |
| Lymphocyte (n°/μL) | 2500-6000 | 809 | 879 |
| T Cells | |||
| CD3+ n°/μL (%) | 1400-4300(52-88) | 60 (7) | 661 (75) |
| TCRαβ (%) | 85-99 | 5 | 54 |
| TCRγδ (%) | 2-15 | 1 | 16 |
| CD3+HLA-DR+ (%) | 0-10 | 22 | 7 |
| DNT (%) | 0-2.5 | 0.2 | 0.7 |
| CD4+ n°/μL (%) | 1000-2500(33-55) | 53 (7) | 304 (35) |
| CD4+CD45RA+CCR7+(Naïve) (%) | 32-82 | 4.1 | 45.4 |
| CD4+CD45RA+CCR7- (CM) (%) | 15-30 | 41.5 | 28.9 |
| CD4+CD45RA-CCR7- (EM) (%) | 8-30 | 53.9 | 23.8 |
| CD4+CD45RA-CCR7+ (E) (%) | 0.4-4 | 0.4 | 1.89 |
| CD4+CD45RA+CD31+ (%) | 44-60 | 2 | |
| CD8+ n°/μL (%) | 400-1400(17-34) | 6 (1) | 264 (30) |
| CD8+CD45RA+CCR7+(Naïve) (%) | 30-80 | 15.3 | 72.0 |
| CD8+CD45RA+CCR7- (CM) (%) | 3-28 | 16.2 | 4.5 |
| CD8+CD45RA-CCR7- (EM) (%) | 17-40 | 59.5 | 16.7 |
| CD4+CD45RA-CCR7+ (TEMRA) (%) | 2-15 | 9 | 6.8 |
| TRECS (copies/punch) | > 10 | < 10 | 50 |
| NK Cells | |||
| CD56+CD3- n°/μL (%) | 100-650(2-20) | 671 (83) | 191 (21.7) |
| B Cells | |||
| CD19+ n°/μL (%) | 400-1500(9-28) | 49 (6) | 22 (2.5) |
| CD19+CD27+ (%) | 7-19 | 32 | |
| CD19+IgD-CD27+ (%Naïve) | 75-89 | 63 | |
| CD19+IgD+CD27+ (%MZ) | 2.6-7.1 | 14.9 | |
| CD19+IgD-CD27+ (%SW) | 4.5-20 | 17.10 | |
| CD19+CD38hiIgM+ (%Transitional) | 3-10 | 13 | |
| Plasmablasts | 0.5-5 | 4.6 | |
| KRECS (copies/punch) | >10 | <10 | 100 |
| Serum Immunoglobulins (mg/dl) | |||
| IgG (mg/dL) | 600-1230 | 446 | 779 |
| IgA (mg/dL) | 30-200 | 18 | <6.67 |
| IgM (mg/dL) | 50-200 | 40 | 109 |
| Specific antibodies | |||
| IgG vs Pneumococcus (mg/dL) | >5.4 | 2.9 | |
| IgG2 vs Pneumococcus (mg/dL) | >2.4 | 0.36 | |
| IgG vs Tetanus toxoid (IU/mL) | >0.1 | 9.10 |
(29) Submission ID#589566
6 Month Old Female with Congenital Onset Indolent Systemic Mastocytosis Successfully Treated with Midostaurin
Christin Deal, MD1, Manish Butte, MD, PhD2, Maria Garcia-Lloret, MD3
1Allergy/Immunology Fellow, UCLA
2Division of Allergy/Immunology Chair, Division of Immunology, Allergy, and Rheumatology, Dept. of Pediatrics and Jeffrey Modell Diagnos-tic and Research Center, University of California, Los Angeles
3Allergy/Immunology Program Director, UCLA
A 6-month-old female presented with symptoms of systemic mastocytosis including history of episodes of cyanosis and irritability, ALTE, chronic diarrhea (6-9 loose stools per day), hematochezia, diffuse skin lesions and bullae and a left axillary mass. Skin biopsy of left axilla was performed by her dermatologist showing presence of mast cells and she was subsequently started on cromolyn, cetirizine, ranitidine, prednisolone, hydroxyzine, diphenhydramine and montelukast without any improvement in symptoms. Axillary mass was initially believed to be abscess formation secondary to the skin biopsy but was found to have complex cystic structure on MRI and ultrasound. The lesion was initially improved by drainage but continued to recur over several weeks despite multiple antibiotic courses and surgical drainages. Cytology of drained fluid showed lymphocytosis.
Bone marrow biopsy showed 5% mast cells and D816V variant of KIT gene. There was no associated hepatosplenomegaly. Germline sequencing of whole blood showed no mutation of KIT indicating congenital onset of acquired indolent systemic mastocytosis. Skin findings consistent with diffuse cutaneous mastocytosis with bullous eruptions and hemorrhage.
She was subsequently hospitalized 2 weeks later for an unresponsive episode and had worsening of her skin lesions. During this hospitalization she started on Midostaurin at 30mg/m2 for systemic mastocytosis. Midostaurin is a novel protein kinase inhibitor that has shown efficacy treatment of patients with D816V c-kit positive advanced systemic mastocytosis (van Anrooij et al., 2018). Dose was increased to 45mg/m2 BID 3 weeks after initiation. She received premedication with ondansetron prior to each dose of Midostaurin. Tryptase levels responded well to Midostaruin and were 109 ug/L on the day of initiation of Midostaurin and were 54.1ug/L at her last check which was approximately 4 weeks after starting Midostaurin (Figure 1). Her skin lesions also significantly improved after starting the medication (Figure 2).
Her hospitalizations were complicated by fluid overload and hypertension. Both fluid overload and hypertension resolved prior to discharge. She remains on 2mg prednisone daily, cetirizine, ranitidine, cromolyn and Benadryl and hydroxyzine PRN. To our knowledge, this is the youngest patient successfully treated with Midostaurin and she is doing very well on therapy with no apparent side effects. She has had resolution of many of her systemic mastocytosis symptoms including skin lesions, axillary mass and improvement in her diarrhea and growth as well as objective improvements in her tryptase levels.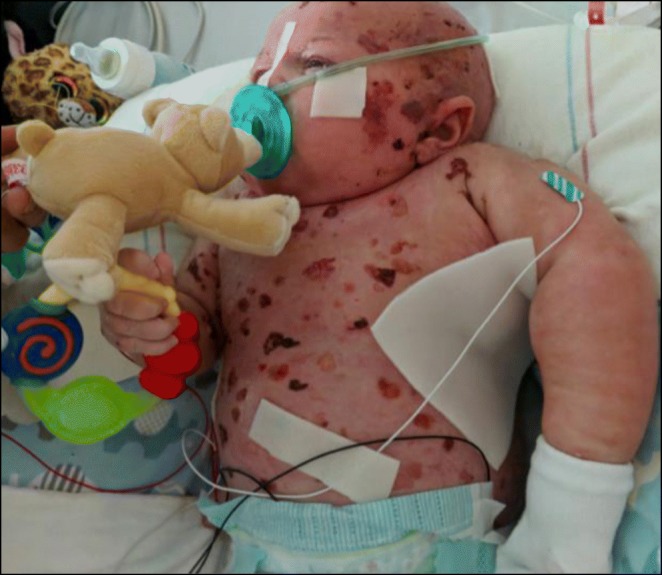
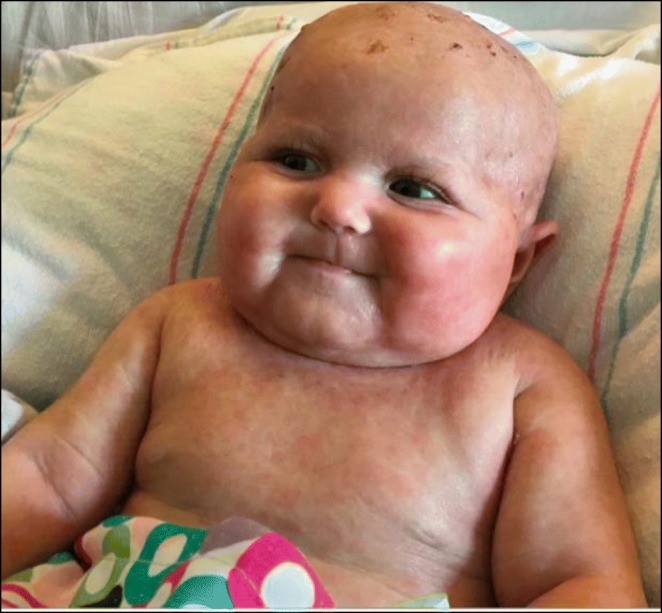
(30) Submission ID#590027
Two-year-old Male with Recurrent Cervical Lymphadenopathy Presenting with Rash
Deborah Bloch, MD1, Meera Patrawala, MD2, Jennifer Shih, MD3, Whitney Sherry, MD4, Shelley Caltharp, MD5, Adina Alazraki, MD6, Jonathan Loewen, MD7, Christina Rostad, MD8,
1Pediatric Infectious Diseases Fellow (PGY-6), Emory University School of Medicine
2Allergy and Immunology Fellow, Emory University School of Medicine
3Assistant Professor of Pediatrics and Internal Medicine, Department of Pediatrics, Division of Allergy and Immunology, Emory University School of Medicine
4Assistant Professor of Pediatric Hospital Medicine, Department of Pediatrics, Emory University School of Medicine
5Adjunct Assistant Professor, Department of Pathology and Laboratory Medicine, Emory University School of Medicine
6Associate Professor, Departments of Pediatrics and Radiology, Division of Pediatric Radiology, Emory University School of Medicine
7Assistant Professor, Departments of Pediatrics and Radiology, Division of Pediatric Radiology, Emory University School of Medicine
8Assistant Professor, Department of Pediatrics, Division of Pediatric Infectious Diseases, Emory University School of Medicine
Case report: A two-year-old male presented to the hospital with a painful, non-pruritic facial and groin rash. The rash started one week prior to presentation. He had no associated fevers. His history was remarkable for failure to thrive (FTT) and chronic bilateral leg pain with antalgic gait. Over the preceding months, he had been diagnosed with hand-foot-mouth disease and varicella. He had also had recurrent cervical lymphadenopathy (LAD) for greater than one year requiring incision and drainage. Gram stain and Gomori Methenamine-Silver Nitrate Stain (GMS) were negative and pathology showed only acute and chronic inflammation with areas of necrosis. His family history was negative for autoimmune disease or immunodeficiency. Infectious exposure history was significant for an incarcerated father with unknown tuberculosis status and history of living in a shelter.
On physical examination, the patient was well appearing with multiple erythematous papules, with superficial erosions and scabbing on the face (Figure 1), lower abdomen, genital area, buttocks and proximal lower extremities. He had large, firm, non-tender submandibular lymph nodes. He also had small palpable axillary and inguinal lymph nodes bilaterally.
His laboratory workup revealed normal white blood cell and platelet counts, but microcytic anemia, an erythrocyte sedimentation rate of 140 mm/hr, and C-reactive protein of 7.5 mg/dL. Full body magnetic resonance imaging (MRI) revealed bilateral cervical, supraclavicular, right hilar and inguinal lymphadenopathy and a patchy right upper lobe consolidation with at least one small area of cavitation (Figure 2) and an adjacent smaller area of ring enhancement. It also revealed three small nonspecific hypodense foci within the right lobe of the liver and borderline splenomegaly. Given these findings, there was concern for granulomatous diseases. The patient underwent a liver biopsy (Figure 3) which showed non-specific evidence of necrotizing granulomatous disease. Microbiological cultures and stains for bacteria, acid-fast bacilli and fungi were negative. His infectious work-up was negative for HSV, tuberculosis, HIV, syphilis, histoplasmosis, and toxoplasmosis. Superficial bacterial cultures from the face and groin grew mixed gram positive and negative organisms, including methicillin-susceptible Staphylococcus aureus (MSSA).
His immunologic workup revealed borderline elevated IgA and IgG with normal IgM, normal T,B, NK-cell counts and pneumococcal and tetanus titers. A dihydrorhodamine (DHR) flow cytometric test was positive, consistent with a diagnosis of chronic granulomatous disease (CGD). Genetic testing confirmed X-linked disease.
He was treated with acyclovir and ceftriaxone with resolution of his rash.
Conclusion: We present a case of a two-year-old male with newly diagnosed X-linked CGD. Though he had been seen by multiple healthcare providers for recurrent lymphadenopathy over the preceding year, he had no other history of recurrent viral or bacterial infections or significant family history that might implicate a primary immunodeficiency. At time of presentation, he had diffuse rash which could have caused his palpable lymphadenopathy on exam. A high index of suspicion for CGD in the setting of recurrent LAD and FTT prompted sending the DHR, which led to the diagnosis.
Figure 1. Facial rash. Patient had similar rash in lower abdomen, genital area, buttocks and proximal posterior lower extremities. Bilateral submandibular fullness can also be appreciated.
Figure 2. Axial post gadolinium images showing abscess within a right submandibular lymph node (arrow) and consolidation in the right upper lobe with central cavitation (*)
Figure 3. Liver biopsy revealing palisading histiocytes surrounding central necrosis, consistent with necrotizing granulomas (400x).
(31) Submission ID#590402
Quality of Life in Adult Patients with Chronic Granulomatous Disease
Samantha Kreuzburg, BA, RN1, Jennifer Treat, PA-C, MSHS2, Dawn Shaw, MBA, MN, RN3, Christa Zerbe, MD, MS4
1Research Nurse Specialist, National Institutes of Health/National Institute of Allergy and Infectious Diseases
2Physician Assistant, Medical Science & Computing
3Protocol Nurse Coordinator II, Leidos Biomedical Research, INC
4Director, Clinical Patient Services, National Institutes of Health/National Institute of Allergy and Infectious Diseases
Chronic Granulomatous Disease (CGD) is an inherited primary immunodeficiency (PID) which results in both inflammatory response dysregulation and an increase in susceptibility to certain bacterial and fungal infections. Without curative treatment such as a bone marrow transplant, it remains a chronic disease with daily medication management, intermittent treatment and life-long surveillance. In general, chronic disease involves physical, psychological and social effects which can affect the patients quality of life. Although some research has been done on how PID affects quality of life, there is little research in the United States about how CGD affects patients quality of life.
To examine the effect of CGD on patients quality of life, as a part of a voluntary research protocol examining the natural history of immune deficiencies, we administered the WHO QOL-BREF instrument to adult CGD patients enrolled on a NIH IRB approved protocol and seen in the Infectious Disease Clinic at the National Institutes of Health (NIH) over a five-month period. The WHO QOL-BREF is comprised of 26 items, which measure the following broad domains: physical health, psychological health, social relationships and environment. Each item is rated on 5-point Likert scale. It has been validated cross culturally and has been widely field tested. The survey was interview administered to 35 patients (23 males, 12 females) with genetically confirmed CGD. The age range was 18 - 60 years old (mean age 37.6 years) with a distribution of 57 % x-linked CGD and 43% autosomal recessive CGD.
Results have been obtained and will be presented.
(32) Submission ID#590834
Health Disparities in CVID: a Report of 1,546 Patients from the USIDNET Registry
Pragya Shrestha, MD1, Anthony Donato, MD, MHPE2, Avni Joshi, MD, MSc3
1Resident Physician, Reading Hospital- Tower Health System
2Associate Program Director, Reading Hospital- Tower Health System
3Assistant Professor, Mayo Clinic, Rochester, MN
Rationale: Common Variable immunodeficiency (CVID) is the most common primary immunodeficiency with an estimated prevalence of 1:25,000. We aimed to analyze the clinical presentations and their associated comorbidities amongst CVID patients in USA.
Methods: Data on 1,546 CVID patients reported in the United States Immunodeficiency Network (USIDNET) from 1992 to 2018 were analyzed based on clinical, immunological and genetic factors. Univariate analysis with Spearman rank coefficients was done to analyze correlations between disease outcomes. Observed survival was estimated using the Kaplan-Meier method.
Results: Among the 1546 patients, 908 (58.7%) were female and 638 (41.3%) were male. Median age at diagnosis was 29 years [mean (SD), 30.1 (20.2); range, 0-82; IQR, 12-47] with median age of onset of 14 years (mean (SD), 20.3 (19.2); range, 0-81; IQR, 3-33). Females showed a longer delay in diagnosis (9.5 vs. 6.6 years, P=0.006). Higher body mass index (BMI) linearly correlated with the age of diagnosis (r= 0.46). In survival analysis, a 5-year delay in age at diagnosis increased the risk of death by 7.4% (HR: 1.07, 95% CI: 0.98-1.18, p=0.14).
Conclusions: Our study suggests a longer delay in diagnosis in female subjects and a strong association with diagnosis of CVID in patients with higher BMI. Females may have a longer period without symptoms leading to a diagnostic delay. Gender- based and disparities-based inquiry into these trends may need additional study.
| Demographics | Number of patients | % USIDNET CVID cohort | 95%Confidence Interval |
| Gender: | |||
| Male | 638 | 41.3% | 0.39-0.43 |
| Female | 908 | 58.7% | 0.56-0.60 |
| Age group: | |||
| <10 years | 16 | 1% | Median age 50 years |
| 10-20 years | 188 | 12% | Mean 47.6 years |
| 20-35 years | 300 | 19% | CI 46.6-48.74 for mean |
| 35-55 years | 415 | 27% | |
| >=55 years | 627 | 41% | |
| Race: | |||
| Caucasian | 1283 | 96% | 0.95-0.97 |
| Others (African-American, Asian, Hispanic) | 34 | 3% | 0.01-0.03 |
| Unknown | 1% | 0.05-0.01 | |
| Living | 1415 | 91.5% | 0.95-0.97 |
| Deceased | 55 | 3.5% | 0.03-0.02 |
| Lost follow up | 76 | 5% | |
| Family history of PIDD | |||
| Yes | 176 | 11.38% | 0.20-0.26 |
| No | 575 | 37.2% | 0.73-0.79 |
| Unknown | 795 | 51.4% |
Table 1: Demographics of CVID patients in USIDNET registry including gender, age groups, race, living status, family history of Primary Immunodeficiency
| Female (N=908) | Male (N=638) | p- value | |
| BMI | 0.00122 | ||
| N | 567 | 364 | |
| Mean (SD) | 26.2 (7.4) | 24.5 (6.5) | |
| Median | 24.9 | 23.7 | |
| Q1, Q3 | 21.3, 29.8 | 19.3, 28.7 | |
| Range | (11.6-60.2) | (13.7-52.8) | |
| Age of onset | <0.00012 | ||
| N | 545 | 436 | |
| Mean (SD) | 23.3 (19.7) | 16.6 (17.8) | |
| Median | 19.0 | 10.0 | |
| Q1, Q3 | 5.0, 37.0 | 2.0, 27.5 | |
| Range | (0.0-81.0) | (0.0-79.0) | |
| Years from onset to diagnosis | 0.00662 | ||
| N | 520 | 405 | |
| Mean (SD) | 9.5 (12.9) | 6.6 (9.8) | |
| Median | 4.0 | 3.0 | |
| Q1, Q3 | 1.0, 13.0 | 1.0, 8.0 | |
| Range | (0.0-64.0) | (0.0-72.0) |
Table 2. Characteristics of Interest- BMI, Age of Onset and Years from onset to diagnosis based on gender among CVID patients in USIDNET registry
(33) Submission ID#591345
Indications of Depressive Disorders in Adults with Primary Immunodeficiency
Christopher Scalchunes, MPA1, Tiffany S. Henderson, PhD2
1Vice President of Research, Immune Deficiency Foundation
2Survey Research Analyst, Immune Deficiency Foundation
The physical well-being of those with primary immunodeficiency (PI) and the physical maladies of those with PI are well-documented. Since the 1950s, advances in identification and treatment of PI has for many led to lives where the physical infections of these groups of diseases are manageable. However, not as well understood are the emotional and mental health aspects of living with PI. As part of a larger survey project The IDF 2017 National Patient Survey, this study aims to quantify any potential mental health issues or challenges faced by adults with PI. Our hypothesis- those with PI, suffer from statistically higher rates of depression when compared to the U.S. general population.
The 2017 IDF National Patient Survey was a nationally distributed, un-incentivized, mail-based survey of 4,500 persons in the IDF patient database identified as being either adults with PI or the parent/caretaker of a child with PI. The questionnaire comprised approximately 44 main questions about PI as well as the validated SF-12v2, Brief Fatigue Inventory and the Patient Health Questionnaire-2 (PHQ-2) instruments. Additional questions asked about current use of prescription medications for anxiety, depression, stress and pain. For the purpose of this study, only adult respondents with PI are included as the basis for analysis.
The two-item Patient Health Questionnaire (PHQ-2) meets the criteria for general screening of depression suggested by the U.S. Preventive Services Task Force. Scored on a scale of 0-6, a score of three or higher is suggested as the cut-point for depressive screening. According to a 2014 AHRQ study that utilized MEPS data, 2,139 of the 23,770 (9%) respondents scored three or greater. In our survey 211 of the 925 (23%) adults scored three or greater (2 <.05.)
Overall, those in our survey scored lower on the SF-12v2 MCS scale when compared to the U.S. population (44.3 v.50.0, p<.05). Further, adults with PI who scored three or higher on the PHQ-2 had an average MCS of 31.8.
Those who met the PHQ threshold in our survey were also more likely to report moderate to severe limitations in normal activities as a result of emotional problems than those that fell below the threshold (74% versus 13%, p <.05).
Not surprisingly, those that met the PHQ threshold reported much higher use of prescription medications for anxiety, depression, stress (69% versus 33% below threshold, p <.05) as well as a higher reported use of prescription pain medications (33% versus 17% below threshold, p <.05). Though moderate to severe fatigue was reported by 68% of those below threshold, 99% of those with PHQ scores at threshold reported experiencing moderate to severe fatigue (p <.05).
Health care providers should consider including the PHQ-2 in the overall health assessments of their patients with PI. Those scoring three or higher should be referred to the appropriate professional for further evaluation.
(34) Submission ID#592136
DLCO Is a Reliable Noninvasive Approach for Pulmonary Monitoring in Patients with HIES
Alyssa Kerber, MD1, Avni Joshi, MD2
1Resident physician, Pediatric and Adolescent Medicine, Mayo Clinic
2Assistant Professor, Allergy and Immunology, Mayo Clinic
A 23-year-old female from Kuwait presented to our multi-disciplinary Primary Immunodeficiency Clinic, for a history of several years of eczema, multiple skin abscesses, recurrent sinusitis, and history of pneumonia complicated by pneumatocele. Based on patient history and outside records, she had been admitted multiple times for cutaneous abscesses on her abdomen, thigh, and gluteal regions, requiring multiple episodes of antibiotics and incision and drainages.
The patients symptoms first started early on in her first year of life, with refractory eczema and recurrent cutaneous infections. During her second year of life, she had recurrent episodes of respiratory infections and was diagnosed with pneumonia complicated by pneumatocele formation, which required surgical intervention. Her medical history is also notable for fungal infections of the nails and multiple episodes of otitis media. There is no family history of recurrent infections or known immunodeficiency.
In 2012 at the age of 17, she was diagnosed with clinical hyper IgE syndrome with eosinophilia and IgE level above 5000 KU/L.
At the time of evaluation at our institution, examination did not reveal evidence of active infection. Her examination was significant for multiple scars to her upper and lower extremities and gluteal region from previous incision and drainages. She had a left thumb nail onychomycosis. She was also noted to have a broad nasal bridge and retained primary teeth.
A CT scan of the chest was obtained, which showed fibrosis and volume loss with underlying bronchiectasis in the right lung. Pulmonary function testing revealed a mild decline in DLCO at 15.99 ml/(min*mmHg).
The patients calculated NIH-HIES score was >60. Due to clinical diagnosis of Hyper-IgE, genetic testing was pursued, which revealed a likely pathogenic variant, p.Trp623Leu (W623L) (TGG>TTG): c.1868 G>T in exon 20 of the STAT3 gene. The W623L missense variant in the STAT3 gene has been previously observed in one individual in association with Hyper-IgE syndrome (Al-Mousa et al., 2016). This variant is not observed in large population cohorts (Lek et al., 2016). The W623L is a semi-conservative amino acid substitution, which may impact secondary protein structure. In-silico analyses supported a deleterious effect, located within the SH2 domain, which is a critical functional domain (Chandesris et al., 2012; Koskela et al., 2012). It was thus determined that this variant is likely pathogenic.
The patients prophylactic treatment was optimized with TMP-SMX (800mg-160mg) twice daily for prevention of infections. She was also started on Hibiclens (chlorhexidine) baths once per week. She was referred to Pulmonology for optimization of pulmonary health in the setting of bronchiectasis and mild decline in DLCO. She was advised to follow-up on a yearly basis to the Primary Immunodeficiency Clinic to assess for recurrent infections and for changes in pulmonary health. Finally, targeted testing and clinical evaluation of both of the patients parents was recommended to determine if W623L was inherited or arose de novo. The pathogenic role of the W623L missense change would be further supported if it had occurred de novo or if it segregates with the disease in the family.
Uploaded File(s)
Uploads
Pulmonary Function Testing Results.pdf
(35) Submission ID#592269
The Effect of Hydroxychloroquine on CTLA4 Expression in Siblings with LRBA (Lipopolysaccharide-responsive and Beige-like Anchor Protein) Deficiency
Nurcicek Padem, MD1, Melanie Makhija, MD2, John Routes, MD3, Jeffrey Woodliff, PhD4, Amer Khojah, MD2
1Fellow, Northwestern University
2Attending, Northwestern University
3Chief, Professor, Division of Allergy and Immunology, Children's Hospital of Wisconsin-Milwaukee, Milwaukee, WI
4Director, Flow Cytometry Core Facility, Medical College of Wisconsin
Introduction: Lipopolysaccharide-responsive and beige-like anchor protein (LRBA) deficiency is a rare autosomal recessive disease of the immune systems characterized by hypogammaglobulinemia and decreased CTLA4 expression on T regulatory cell (T regs) due to defective intracellular trafficking of CTLA4. Previous in vitro study has shown a significant increase of CTLA4 expression on LRBA deficient T cells after overnight culture with chloroquine, an older anti-malarial agent. This effect is likely due to increasing lysosomal pH. However, there is no evidence of such effect in human subjects after administration of weight appropriate doses anti-malarial agents. We are presenting a set of siblings with LRBA deficiency who had CTLA4 expression measured before and four weeks after starting hydroxychloroquine.
Case reports: Case 1 is a 14-year-old East-Indian boy with autoimmune thyroiditis, Type 1 diabetes mellitus (DM), short stature, autoimmune cytopenias, and lymphadenopathy. He was referred to immunology clinic at 9 years of age for suspicion of Autoimmune Lymphoproliferative Disorder. Primary Immunodeficiency Genetic Panel was sent which revealed a homozygous mutation in LRBA gene (c.6480_6481del). This novel variant resulted in a frameshift and created a premature stop codon 18 amino acids downstream from this location which may lead to absent or abnormal protein. Lung CT scan showed interstitial lung disease. Lung biopsy showed interstitial nodular and diffuse lymphoid proliferation. This diagnosis led to the testing of his sister (case 2) given her history of autoimmune illnesses and the family history of consanguinity. Case 2 is a now 13-year-old girl with type 1 DM, autoimmune thyroiditis, lymphadenopathy, psoriatic arthritis, and seizures. Her lung imaging showed pulmonary nodules without interstitial lung disease. Both cases received hydroxychloroquine while waiting for insurance approval of abatacept. CTLA4 expression on Tregs was measured prior to and four weeks after starting hydroxychloroquine treatment. At baseline, 8.6% of Case 1s CD4 cells were Treg (FOXP3+ve, CD25hi) and 51.4% of them expressed CTLA-4 (in contrast to 94.1% Tregs in the healthy control) with mean fluorescence intensity (MFI) of 335. This ratio and MFI did not change after 4 weeks of hydroxychloroquine treatment (6 mg/kg/day). Soluble Interleukin-2 receptor levels were measured: Case 1 had a baseline level of 8510 pg/mL, which decreased to 2228 pg/mL after 4 weeks of hydroxychloroquine treatment. For Case 2: 8.4% of her CD4+ T cells were found to be FOXP3+CD25hi and 36.1% of these Tregs expressed CTLA-4. This ratio increased by 7% after one month of hydroxychloroquine. Increase in MFI was also noted from 298 to 386. Case 2 had a drop in soluble Interleukin-2 receptor level from 1265 pg/mL to 950pg/mL after treatment.
Conclusion: In contrast to the previous in vitro assays, we did not find a significant increase in CTLA4 expression on T regulatory cells in vivo after 4 weeks of 6mg/kg/day hydroxychloroquine. Interestingly, soluble IL-2 receptor levels improved dramatically with hydroxychloroquine.
(36) Submission ID#592574
Human NF-kappaB2 Defect Results in Defective Intrinsic B-cell Differentiation, Function and Class Switching
Shancy Jacob, PhD1, Julie Feusier, MSc2, Krystin Krauel, PhD3, Li Guo, MD/PhD3, Jesse Rowley, PhD4, Robert Campbell, PhD5, Jacob Anderson, BS6, Michael D. Keller, MD7, Lynn Jorde, PhD8, Guy Zimmerman, MD/PhD9, Andrew Weyrich, PhD9, Karin Chen, MD10
1Postdoctoral Research Fellow, Division of Allergy & Immunology, Department of Pediatrics, University of Utah
2Graduate Student, Department of Human Genetics, University of Utah
3Postdoctoral Research Fellow, Department of Internal Medicine, University of Utah
4Assistant Professor, Division of Pulmonary Medicine, Department of Internal Medicine, University of Utah
5Assistant Professor, Division of General Medicine, Department of Internal Medicine, University of Utah
6MD Candidate, Pennsylvania State University College of Medicine
7Assistant Professor, Center for Cancer and Immunology Research, Children's National Health System, Division of Allergy & Immunology, Children's National Health System, Washington, DC
8Professor, Department of Human Genetics, University of Utah
9Professor, Department of Internal Medicine, University of Utah
10Assistant Professor, Department of Pediatrics, Division of Allergy and Immunology, University of Utah
Introduction/Background: Autosomal dominant heterozygous mutations in NFKB2 (encoding for the protein NF-kB2) have been identified in the etiology of a form of primary immunodeficiency disorder that presents with hypogammaglobulinemia, defects in B-cell maturation, endocrinopathy, and autoimmune manifestations. In humans, the effects of altered NF-kB2 and mechanisms of immune system impairment have not been fully delineated.
Objectives: To understand the mechanism of the antibody deficiency in patients with hypomorphic mutations in NFKB2 (c.2564delA; p.Lys855Serfs*7) by evaluating B-lymphocyte proliferation, differentiation, function, and gene expression.
Methods: Immunophenotyping of primary B-cells from subjects with mutant NFKB2 was completed by flow cytometry. Proliferation of B-cells was assessed by CFSE stimulation of primary CD19+ B-cells from healthy and NFKB2 mutant subjects. Differentiation of healthy and affected naïve B-cells (CD27-CD38-) into plasmablasts (CD27+CD38+) following stimulation was assessed by flow cytometry. The supernatant from these cells were assayed for IgA, IgG and IgM production by ELISA. To study the defect in class-switch recombination, naïve B-cells and EBV-transformed B-cells from affected and healthy individuals were stimulated and expression of the AICDA gene was quantified by qPCR. In parallel experiments, EBV B-cells from wildtype and NFNB2 mutant individuals were stimulated and AID (Activation-induced cytidine deaminase) protein levels were determined by western blot.
Results: Patients with hypomorphic mutations in NFKB2 (c.2564delA) had low memory B-cell (CD19+ CD27+ IgD- IgM+) and class-switched memory B-cell (CD19+ CD27+ IgD- IgM-) numbers. In vitro, primary B-cells from these patients demonstrated a 50% reduction in proliferation and cell division in response to CD40L and IL-10 (p =0.01). Compared to healthy naïve B-cells, mutant naïve B-cells had a significant reduction in plasmablast differentiation (p = 0.002) and secreted significantly lower levels of immunoglobulins in response to CD40L and IL-21 stimulation. Mutant naïve B-cells and mutant EBV B-cells failed to increase AICDA expression and AID protein levels in response to CD40L and IL-21 stimulation.
Conclusions: Our studies demonstrate that a hypomorphic NFKB2 mutation in humans affects intrinsic B-cell proliferation and differentiation. The mutation impairs transcription of the AICDA gene that encodes AID, a key protein involved in B-cell class-switch recombination. The NFKB2 gene defect also impairs immunoglobulin production, as seen in common variable immunodeficiency-like cases. These studies provide unique translational insights into physiological activities of NF-kB2 in downstream immunologic outputs in humans, expanding those suggested by experimental observations in mice.
(37) Submission ID#592712
Provider Perceptions of Primary Immunodeficiency Disease Patients Quality of Life, Neurocognition, Physical Well-Being and Psychosocial Health
Thomas F. Michniacki, MD1, Lauren E. Merz, BA2, Roshini S. Abraham, PhD3, Julie Sturza, MPH4, Kelly Walkovich, MD5
1Pediatric Hematology/Oncology Fellow, University of Michigan
2Medical Student, University of Michigan Medical School
3Department of Pathology and Laboratory Medicine, Nationwide Childrens Hospital, Columbus, OH.
4Statistician, University of Michigan Department of Statistics
5Associate Professor, Pediatric Hematology/Oncology, University of Michigan Medical School
Background: Few studies have evaluated the quality of life (QOL) and patient reported outcomes of primary immunodeficiency disease (PIDD) patients, and no studies have assessed medical provider perceptions of their PIDD patients QOL, neurocognition, physical well-being and psychosocial health. Understanding provider beliefs regarding patient reported outcomes is essential to improving clinical management of PIDDs. Here we report our PIDD medical provider survey results.
Methods: Providers were contacted via email with the assistance of the Clinical Immunology Society. Participants completed adult and/or pediatric-based Likert scale survey questions via a secure online survey service. In addition to demographic information, survey questions assessed provider perceptions of patients overall QOL and their impression of the impact of disease or its associated treatment on mental health, physical well-being, neurocognition, social relationships and school/work performance. Clinicians were expected to make their assessments based on their PIDD patient cohort as a whole rather than on specific diagnoses or patients. Given the small sample size, a p-value < 0.1 was considered statistically significant; repeated measures ANOVA and paired t-test analyses were used.
Results: Study participants (n=58) were primarily from the United States (64%), born between 1965-1979 (44%), and trained in allergy/immunology (77%). 85% of survey takers practiced within an academic center, 52% were female and 95% cared for children with 42% of providers concurrently caring for adults. There was a statistically significant difference (p=0.07) in the perceived overall QOL of pediatric versus adult PIDD patients with 41% of providers feeling as though their pediatric patients had a good QOL while only 25% believed their adult patients had a good QOL. Clinicians believed adult PIDD individuals had more difficulties related to associated co-morbidities rather than their actual PIDD compared to pediatric PIDD patients (p=0.046). Providers felt that the neurocognition and school performance of children were more often negatively affected by a PIDD than the neurocognition and work performance of immunodeficient adults (p=0.1). Clinicians believe children with PIDD more frequently had difficulties related to their concentration than memory (p<0.01). 96% of those who care for PIDD adults believe their patients work performance or daily mental functioning is at times negatively impacted. Anxiety symptoms and social relationships were viewed as being more negatively impacted by a PIDD diagnosis or treatment than anger or depressive symptoms in both children and adults (p<0.01). 38% of pediatric clinicians feel their PIDD patients experience anxiety symptoms often or almost always. Of physical health parameters, energy, rather than mobility or pain, was deemed to be more deleteriously influenced by an immunodeficiency in adult and pediatric patients (p<0.01).
Conclusions: Our results show that medical providers perceive the overall QOL of pediatric PIDD patients to be superior to that of adults with PIDD, but most clinicians feel a diagnosis or associated treatment regimen for PIDD can negatively impact the physical well-being, psychosocial health, school/work performance and neurocognition of both children and adults.
(38) Submission ID#592868
Homozygosity for a Novel CARD11 Mutation Causes Severe Combined Immunodeficiency (SCID), Inflammatory Gastrointestinal Disease, and Complete Abrogation of MALT1 Activity
Henry Y. Lu, BSc1, Sneha Suresh, MD, FRCPC2, Lyle McGonigle, MD, FRCPC3, Joanne Luider, BSc, ART, MLT4, Stuart E. Turvey, MBBS, DPhil, FRCPC5
1PhD Candidate, BC Children's Hospital and University of British Columbia
2Assistant Professor, University of Alberta and Alberta Health Services
3Pediatrician, Alberta Health Services
4Laboratory Scientist, Calgary Laboratory Services
5Professor, BC Children's Hospital and University of British Columbia
Introduction/Background: The caspase recruitment domain family member 11 (CARD11)B cell CLL/lymphoma 10 (BCL10)MALT1 paracaspase (MALT1) [CBM] complex is a critical signalling adaptor that regulates lymphocyte activation, proliferation, survival, and metabolism. Primary immunodeficiencies affecting each component (termed CBM-opathies) result in broad clinical manifestations ranging from severe combined immunodeficiency (SCID) to lymphoproliferation. We present the laboratory and clinical findings of two Canadian First Nations patients found to be homozygous for the same novel CARD11 mutation (c.2509C>T; p.R837*).
Results: We have identified an 8-month-old boy who presented with a severe case of entero/rhinovirus bronchiolitis with interstitial lung disease and a 17-year-old boy with a history of severe pulmonary infections (including PJP), chronic sinusitis, candidiasis, invasive bacteremia, and severe ileo-colitis and oral ulceration requiring total colectomy. Both patients possessed absent Tregs, absent memory B cells, and hypogammaglobulinemia. However, only the 8-month-old had poor T cell proliferation to PHA, ConA, and CD3. Both patients were found to be homozygous for the same novel variant of CARD11 (c.2509C>T; p.R837*). The mutation rendered CARD11 protein expression unstable and it was undetectable by immunoblot. To confirm CARD11 deficiency, we stimulated patient B cells with phorbol 12-myristate 13 acetate (PMA) and ionomycin across a time-course and immunoblotted for various signalling proteins in both the NF-B (IKK/, IB, p65) and MAPK (MEK1/2, MKK4, JNK1/2, ERK1/2) pathways as well as various cleavage substrates of the MALT1 paracaspase (RelB, CYLD, BCL10, HOIL1). NF-B and JNK activation were completely absent and MALT paracapase activity was lost, but surprisingly, MKK4 (which acts upstream of JNK) was intact. Furthermore, co-immunoprecipitation experiments revealed that CARD11 was required for optimal MALT1 association with BCL10 in response to stimulation.
Conclusions: These two cases highlight the crucial role of CARD11 in regulating lymphocyte development, function, and humoral responses. In addition, we have identified the oldest known living individual with CARD11 deficiency and he presented uniquely with inflammatory gastrointestinal disease in addition to SCID, further adding to the spectrum of phenotypes associated with CARD11-related primary immunodeficiencies.
(39) Submission ID#593164
NIH Participation to USIDNET Registry (Poster Submission)
Elizabeth K. Garabedian, MSLS, RN1
1Research Nurse, Principal Investigator, National Genome Research Institute, National Institutes of Health
Abstract: The USIDNET Registry began in 1992 with an NIAID contract with the Immune Deficiency Foundation, which continues today. It aims to provide a resource for clinical and lab research through enrollment of known immunodeficiency patients into a national registry, the USIDNET. NIH is a major national and international referral center for clinical trials on inborn errors of immunity, or primary immunodeficiency diseases. It is a mechanism for depositing NIH data into USIDNET. A Registry of patient information may help us understand how many people have each disease. The information may improve how we diagnose and treat these conditions. The patient Registry is designed to obtain longitudinal data on a large number of patients with primary immunodeficiency diseases who come to NIH to participate in research. The data is collected from the NIH electronic medical record system, CRIS and is deposited into a secure registry with restricted and monitored access. All medical information is anonymized for patient privacy.
(40) Submission ID#594012
Heterozygous OAS1 Mutations Cause Spontaneous RNA Cleavage and Apoptosis with Resulting Multisystem Inflammation and Immunodeficiency
Heimall, J1, Magg, T2, Sullivan, KE1, Albert, M2, Griese M2, Conway D3, Gray, PE4, Calderon, B5, Conn, G5, Klein, C2, Hauck, F2
1Allergy Immunology, The Children’s Hospital of Philadelphia, Philadelphia, PA
2Department of Pediatrics, Dr. von Hauner Children’s Hospital, University Hospital, LMU Munich, Germany
3St. Christopher’s Hospital for Children, Philadelphia, PA
4Dept. of Immunology and Infectious diseases, Sydney Children’s Hospital, Sydney, Australia
5Department of Biochemistry, Emory University, Atlanta, GA
OAS1 is an intracellular sensor for dsRNA that generates the second messenger 2'-5'-oligoadenylate to activate RNase-L as a means of antiviral defense. We describe four patients with a complex early-onset autoinflammatory and immunodeficiency disease caused by heterozygous de novo OAS1 mutations.
Patients presented early in life with lung inflammation including pulmonary alveolar proteinosis and interstitial lung disease. They had febrile flares with dermatitis specifically with macular, pustule and bullous features often progressing to ulceration. Infants had episodes of bloody diarrhea in 3 patients (assoc. with villous blunting and cryptitis in two patients and oesophagitis in one patient). Immunoglobulin IgM, IgG, and IgA levels were low while T cell, B cell, and NK cell numbers were generally in the normal range. Exome sequencing identified de novo heterozygous OAS1 missense mutations in all patients.
One patient had a heterozygous de novo OAS1 mutation p.Ala76Val, with mutant OAS1 protein being expressed in ex vivo generated T cell blasts. In sorted primary patient monocytes and B cells, OAS1 p.Ala76Val was associated with spontaneous RNA degradation and apoptosis as determined by RNA chip technology and flow cytometry, respectively, while T cells were not affected. Monocytes displayed disturbed terminal differentiation and functioning as indicated by reduced GM-CSF-R expression and signaling. B-cells display reduced class-switch-recombination. Proliferation of allogeneic T-cells was reduced in response to sorted OAS1 mutated monocytes and B-cells. Activation of interferon response genes in PBMCs was detected.
Two further unrelated patients had a heterozygous de novo OAS1 mutation p.Cys109Tyr, which appeared to compromise protein stability in transformed patient fibroblasts and when transfected. Cells transfected with this mutant protein had reduced 2-5 oligoadenylate synthesis compared to wild type transfected cells. Immortalized fibroblast lines demonstrated higher levels of inflammatory cytokines and spontaneous cleavage of RNAs. A 4th patient with the clinical phenotype had a heterozygous de novo OAS1 variant p.Val121Gly, but has yet to have formal validation of the variant.
Three patients underwent hematopoietic stem cell transplants in an effort to control their diarrhea and skin inflammation. One patient died with ongoing chronic graft versus host disease, while the two others (p.Ala76Val, Cys109Tyr) are alive and reasonably well with a follow-up of 0.5-7 years. The untransplanted patient died as a result of respiratory failure.
In summary, patients with de novo heterozygous OAS1 mutations have chronic ongoing inflammation of multiple organs. This is at least in part due to spontaneous RNA cleavage, apoptosis and production of inflammatory cytokines and type I interferons. This defines a new category of autoinflammatory disorder.
(41) Submission ID#594077
Thinking Outside of Infection: Hemophagocytic Lymphohistiocytosis in a 5-week-old Male with Chronic Granulomatous Disease and Burkholderia Cepacia Sepsis
Jacqueline Squire, MD1, Wil Chamizo, MD2, David M. Berman, DO3, Deepak Chellapandian, MD4, Beatriz Teppa, MD5, Laura Vose, DO6, Jennifer Leiding, MD7
1Allergy and Immunology Fellow, USF - John Hopkin's All Children's Hospital
2Medical director for Pathology and Laboratory medicine, Johns Hopkins All Childrens Hospital.
3Pediatric Infectious Disease, John Hopkins All Children's Hospital
4Bone Marrow Transplant, Johns Hopkins All Childrens Cancer & Blood Disorders Institute
5Pediatric Intensive Care (PICU), John Hopkins All Children's Hospital
6Pediatric Intensive Care (PICU). Co-chair of the Human Values and Ethics committee, John Hopkins All Childrens
7Associate Professor, University of South Florida
Introduction: Increased susceptibility to infections is the most common complication of chronic granulomatous disease (CGD). Hemophagocytic lymphohistiocytosis (HLH) is a severe disorder resulting from hyperinflammation and hypercytokinemia that can lead to multi-organ system dysfunction (1) characterized by certain criteria: fever, splenomegaly, cytopenias, hypofibrinogenemia or hypertriglyceridemia, hyperferritinemia, increased soluble CD25/IL-2Ra, evidence of hemophagocytosis, or decreased/absent NK cell cytotoxicity (2). Secondary HLH occurs infrequently but often is preceded by smoldering infection in CGD (3,4,5). We present a case of HLH in a 38-day old male, the youngest reported case with CGD.
Case: A 38-day old male with previously diagnosed X-linked CGD, due to known family history, presented with fevers. Initial evaluation was unrevealing including chest x-ray, urinalysis, and blood and CSF cultures. He was admitted and treated empirically with cefepime. CT demonstrated multiple multifocal nodules of the lungs and spleen. After lung nodule biopsy was performed, antimicrobial therapy was broadened to IV meropenem, voriconazole, and micafungin. Despite this, he continued to have fever and developed new onset tachycardia, respiratory distress, and lactic acidosis. Further decompensation with vasoactive refractory shock was treated with vasopressors and stress dose hydrocortisone. Additional laboratory evaluation revealed rising liver enzymes (AST 1670u/L, ALT 307u/L), cytopenias (hemoglobin 7 g/dL, ANC 90/uL, platelets 96,000/uL), and coagulopathy (fibrinogen 93-135mg/dL). Splenomegaly was present on abdominal ultrasound. A diagnosis of evolving HLH was considered and dexamethasone was administered. Within 24 hours of clinical decompensation, the patient died of multiorgan failure. Subsequent blood cultures returned with gram-negative rods (and ultimately Burkholderia cepacia). Autopsy confirmed hemophagocytosis within the bone marrow. No mutations were found in genes associated with primary HLH.
Discussion: Patients with CGD are susceptible to infectious complications and auto-inflammation most commonly involving the lungs, GI, and GU systems (6,7). Patients with CGD can be at increased risk of hyperinflammatory syndromes secondary to infections and chronic inflammation. As shown in the included case, HLH can present in infancy and can be deadly. Early consideration and directed treatment of HLH is imperative, even in the setting of sepsis.
References:
1. Janka GE, Lehmberg K. Hemophagocytic syndromes An update. Blood Rev. 2014 Jul;28(4):135-42
2. Henter J, et al. HLH-2004: Diagnostic and Therapeutic Guidelines for Hemophagocytic Lymphohistiocytosis. Pediatr Blood Cancer. 2007 Feb;48(2):124-31.
3. Bode et al. The syndrome of hemophagocytic lymphohistiocytosis in primary immunodeficiencies: implications for differential diagnosis and pathogenesis. Haematologica. 2015 Jul;100(7):978-88.
4. Parekh C, Hofstra T, Church JA, Coates TD. Hemophagocytic lymphohistiocytosis in children with chronic granulomatous disease. Pediatr Blood Cancer. 2011 Mar;56(3):460-2
5. Valentin G, Thomas TA, Nguyen T, Lai YC. Chronic granulomatous disease presenting as hemophagocytic lymphohistiocytosis: a case report. Pediatrics. 2014 Dec;134(6):e1727-30
6. Henrickson SE, et al. Noninfectious Manifestations and Complications of Chronic Granulomatous Disease. J Pediatric Infect Dis Soc. 2018 May 9;7(suppl_1):S18-S24.
7. Magnani A, et al. Inflammatory manifestions in a single-center cohort of patients with chronic granulomatous disease. J Allergy Clin Immunol. 2014 Sep;134(3):655-662.e8
(42) Submission ID#594080
Benign Reactive Gamma Delta T Cells Proliferation in Spleen The Mirage Effect
Snegha Ananth, MD1, David J Haile, MD2, Andrea Yunes, MD3
1Resident, UTHSCSA
2Attending, Audie L Murphy VA Hospital, San Antonio
3Pathologist, Audie L Murphy VA Hospital, San Antonio
Malignant proliferation of Gamma-Delta T cells include hepato-splenic T-cell lymphoma (HSTL), primary cutaneous T-cell lymphoma and T-cell large granular lymphocytic leukemia (T-LGL). The former two have often been associated with splenomegaly and cytopenias. However, reactive proliferation of Gamma-Delta T cells in spleen mimicking malignancy has only been reported once and has a significant risk of misdiagnosis.
A 30-year-old female presented with two years of unintentional weight loss, persistent leukopenia and thrombocytopenia, with leucocytes around 1-2 X 10^9/L and platelets around 100 X 10^9/L. She also had associated macrocytic anemia (Hemoglobin=10-11g/dl) with laboratory evidence of DAT (Direct Anti-globin Test) negative hemolysis. Physical examination and computed tomography (CT) imaging showed splenomegaly. There was no hepatomegaly or lymphadenopathy. Serum liver function test, auto-immune studies, hemolysis and hereditary diseases workup, viral and bacterial serologies were all normal or negative, except for mild hyperbilirubinemia and LDH elevation. Bone marrow examination performed four months prior to the splenectomy revealed mildly hypocellular marrow (50%) with trilineage hematopoiesis. Flow cytometric analysis and cytogenetics of the bone marrow aspirate and peripheral blood were normal except for small population of large granular lymphocyte and mild low absolute B cell counts in peripheral blood. A laparoscopic splenectomy was performed for diagnostic and therapeutic purposes due to patients worsening LUQ pain. There was no other treatment given prior to surgery. 24 hours post- splenectomy her leucocytes increased to 13.1 and platelets to 247. Her three-month post-splenectomy WBC count and platelet count was 8.9 and 391, respectively. Hemoglobin also improved to 14.9. Pathology showed red pulp expansion by small lymphocytes (Fig. 1) and subsequent IHC (Immunohistochemistry) was positive for CD3 (Fig. 2), CD2, CD7, TIA-1 and negative for CD8, CD5 and CD56. CD4 was difficult to interpret. EBER was negative. Flow cytometry (Fig. 3) showed increased gamma-delta T-cell population (20%) with positive CD3, CD2 and CD 7 and negative CD 5, CD4 and CD 8. Molecular studies by PCR didnt reveal any T-cell receptor gamma or beta gene rearrangement. Cytogenetics was negative for isochromosome 7q or any other abnormalities. She was symptom free at 6 months from her splenectomy.
The morphology and immuno-phenotype of these Gamma-Delta T cells show significant overlap with the malignant cells seen in HSTL and T- LGL, such as loss or downregulation of CD5, CD4 and CD8. Awareness of this reactive condition is necessary to prevent making a wrong diagnosis of a malignant disease with a potentially benign, spontaneously resolving disease. Additional studies of similar cases is needed in order to establish more definitive criterion to separate benign from malignant processes and delineate the role of Gamma-Delta T cells.
Uploaded File(s)
Uploads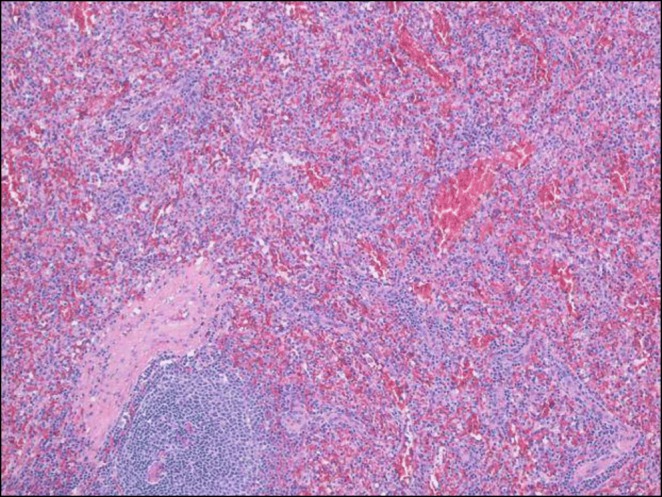
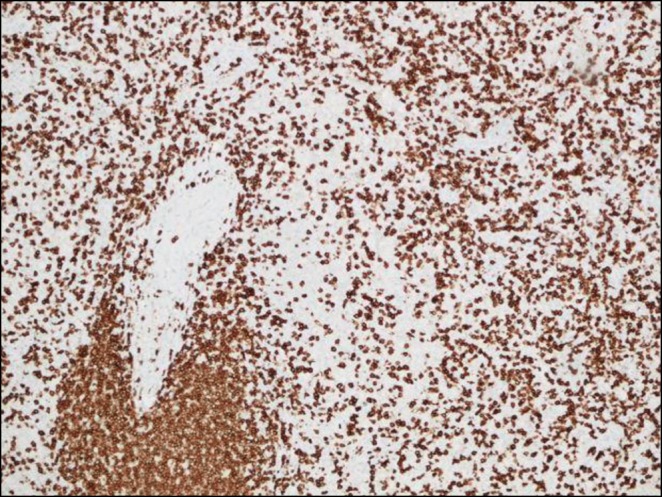
Fig 3. Flow cytomtery.pptx
(43) Submission ID#594181
Sexual Dimorphism and AIRE-AIRE Interactors-miRNA Coexpression Networks in the Infant Human Thymus
Silvia Y. Bando, PhD1, Fernanda B. Bertonha, PhD1, Lucila H. B. Oliveira, PhD2, Carlos Alberto Moreira-Filho, PhD3, Magda Carneiro-Sampaio, MD, PhD4
1Scientific Researcher, Department of Pediatrics, Faculdade de Medicina da Universidade de Sao Paulo, Sao Paulo, Brazil.
2Postdoctoral Fellow, Department of Pediatrics, Faculdade de Medicina da Universidade de Sao Paulo, Sao Paulo, Brazil.
3Associate Professor, Department of Pediatrics, Faculdade de Medicina da Universidade de Sao Paulo, Sao Paulo, Brazil.
4Full Professor, Department of Pediatrics, Faculdade de Medicina da Universidade de Sao Paulo, Sao Paulo, Brazil
Background: Sex steroids in the human thymic environment influence AIRE expression as well as interactions with its partners, i.e. genes coding for AIRE interactors. Here we investigated the effects of sex steroids on these interactions during minipuberty the surge of sex hormones that occur along the first six months of life - and up to 18 months of life. We employed a network-based approach for investigating AIRE-interactors gene-gene relationships and how abundantly co-expressed thymic miRNAs covariate with those genes. AIRE-interactors networks allowed the measuring of gender-related differences in gene-gene expression correlation disclosing relevant differences between minipuberty groups.
Methods: Total RNA was extracted from thymic surgical explants obtained from male (M) and female (F) infants - aged 0-6 months (groups MM and MF, for minipuberty) and 7-18 months (group NM and NF, for non-puberty) and used in DNA microarray assays. Gene coexpression network (GCN) analyses were performed for AIRE and its interactors and for miRNA-gene coexpression analysis. The set of genes coding for the AIRE-targeted proteins was previously identified in TECs by Abramson et al. (Cell 140:123-35, 2010). AIRE-interactors networks were obtained for all groups (link strength cut-off for gene-gene > |0.80| and for miRNA-gene < -0.80). AIRE expression in mTECs was quantified by immunohistochemistry. These methodologies are described in Moreira-Filho et al. (Sci Rep 8:13169, 2018).
Results: The MM x MF networks comparison showed that 16 abundantly expressed miRNAs are interacting with the different AIRE interactor genes in both networks. It is interesting to note that network topology were more similar between NM and NF groups, although AIRE interacts with only one distinct miRNA in each network (miR-150-5p in the NM group or miR-7977 in the NF group). Conversely, in the non-puberty networks the sets of miRNAs and their interacting genes are distinct for each network. Immunohistochemistry analysis revealed a higher percentage of mTEC AIRE positive cells in the minipuberty groups: i.e. there is a significant difference between MM x NM (p = 0.0006) and between MF x NF (p = 0.0060).
Conclusions Minipuberty and genomic mechanisms shape thymic sexual dimorphism along the first 6 months of life. This process does not involve changes in AIRE expression between genders, but differences in the interactions of AIRE with its partners that persist throughout the non-puberty period, probably regulated by miRNAs and also by genetic and epigenetic factors.
FAPESP 2014/50489-9
(44) Submission ID#595214
Rescue Therapy with Granulocyte Transfusions (GT) as a Bridge to Hematopoietic Stem Cell Transplant (HSCT) in a Chronic Granulomatous Disease (CGD) Patient with Severe Disseminated Aspergillosis
Sneha Suresh, MD1, Wendy Vaudry, MD2, Marta Rojas-Vasquez, MD3, Sunil Desai, MD4, Luis Murguia-Favela, MD5, Rashid Alobaidi, MD3, Victor Lewis, MD6, Jean Jacques De Bruycker, MD, FRCPC7
1Assistant Professor, University of Alberta
2Professor, University of Alberta
3MD, University of Alberta
4Clinical Professor, University of Alberta
5Clinical Assistant Professor, University of Calgary
6Associate Professor, University of Calgary
7Clinical Assistant Professor, CHU Sainte-Justine, University of Montreal
Introduction: Neutrophils are presumed to defend against Aspergillus species by releasing reactive oxygen species (ROS) and neutrophil extracellular traps (NETs) to degrade fungal hyphae. Triazole antifungals synergistically enhance neutrophil mediated hyphal degradation. Patients with CGD are particularly susceptible to Aspergillus species likely due to their inability to create ROS and NETs, and in severe cases may not be amenable to antifungal therapy alone.
Objective: We present a case of severe disseminated aspergillosis in a patient with CGD in whom GT served as an important adjunct to antifungal therapy and bridge to transplant.
Results: A 6-year-old boy with known CGD, lost to follow up and non-adherent to prophylaxis, presented acutely with right-sided hemiparesis. Neuroimaging revealed an embolic left middle cerebral artery infarction and cardiac magnetic resonance imaging showed extensive vegetations involving both right and left ventricles and atria, with an ejection fraction of 28%. The patient was admitted to intensive care, started on liposomal amphotericin B, meropenem and vancomycin, and underwent debulking of the intracardiac masses on post admission day (PAD) 1. Operative findings showed severe constrictive pericarditis with multiple abscesses and intracardiac vegetations. Thorough debridement of the vegetations was undertaken, however some deep seated abscesses in the myocardium were not amenable. Operative cultures were positive for Aspergillus fumigatus. Clinical status remained precarious, with ongoing requirement for inotropic and ventilator support. Antimicrobial therapy was refined to voriconazole, with amphotericin B remaining on board until therapeutic levels of voriconazole were achieved.
As effective neutrophils are integral in the immune response against Aspergillus, the decision was made to start granulocyte transfusions to aid in clinical stabilization prior to HSCT. Interferon gamma infusions were not administered because of the risks of adverse effects and potentially increasing transplant rejection.
GTs were started on PAD 6, at a dose of approximately 1X10^10 granulocytes, three times a week. The patient tolerated the infusions well, with no allergic or inflammatory response. Neutrophil oxidative burst measured one hour post infusion showed 23.9% mean fluorescent intensity, compared to a baseline of 0% (Figure 1). Clinical improvement was seen, with inotrope cessation on PAD 12 and extubation to BiPaP on PAD 41.
Human leukocyte Antigen (HLA) allosensitizaton was tested on PAD 12, 6 days after the first GT, with no evidence of HLA antibodies. A total of 28 GTs were given over 3 months, prior to proceeding to a 10/10 HLA matched related donor transplant (PAD 69), with two transfusions given before neutrophil engraftment (ANC 500) on Day +14. The patient is now stable 13 months post transplant, with no evidence of graft rejection. He remains on chronic suppressive antifungal therapy, to continue until full lymphoid reconstitution.
Conclusion: GT may be a useful adjunct to antifungal therapy in patients with impaired neutrophil function with severe invasive aspergillosis, and potentially provide a life sustaining bridge to HSCT.
Uploaded File(s)
Uploads
(45) Submission ID#595387
Investigational Cultured Thymus Tissue Transplantation (RVT-802) Following Failed Stem Cell Transplantation in Athymic Patients
Tyler R. Yates, MD1, Jennifer Puck, MD2, Morna J. Dorsey, MD, MMSc3, Ziad Khatib, MD4, Karin Chen, MD5, Vivian Hernandez-Trujillo, MD6, William Blouin, CPNP7, Ralph Quinones, MD8, Erwin W. Gelfand, MD9, M. Louise. Markert, MD, PhD10
1Immunology Fellow, Department of Pediatrics, Division of Allergy, Immunology, and Pulmonology, Duke University Medical Center, Durham, NC
2Pediatric Immunologist, Department of Pediatrics, Division of Allergy, Immunology, and Bone Marrow Transplant, University of California San Francisco, San Francisco, CA
3Pediatric Immunologist and Allergist, Department of Pediatrics, Division of Allergy, Immunology, and Bone Marrow Transplant, University of California San Francisco, San Francisco, CA
4Pediatric Hematologist and Oncologist, Department of Oncology, Nicklaus Childrens Hospital/Miami Childrens Health System, Miami, FL
5Assistant Professor, Department of Pediatrics, Division of Allergy and Immunology, University of Utah
6Pediatric Immunologist and Allergist, Division of Pediatric Allergy and Immunology, Department of Pediatrics, Nicklaus Childrens Hospital, Miami, FL
7Certified Nurse Practioner, Division of Pediatric Allergy and Immunology, Department of Pediatrics, Nicklaus Childrens Hospital, Miami, FL
8Pediatric Hematologist and Oncologist, Department of Pediatrics, Section of Hematology, Oncology, and BMT, University of Colorado School of Medicine and Childrens Hospital of Colorado, Aurora, CO
9Pediatric Immunologist and Allergist, National Jewish Health, Immunodeficiency Diagnosis and Treatment Program, Department of Pediatrics, Denver, CO
10Professor of Pediatrics and Immunology, Department of Pediatrics, Division of Allergy, Immunology, and Pulmonology, Duke University Medical Center, Durham, NC
Introduction: Transplantation of cultured thymus tissue (RVT-802) is an investigational therapy that has led to generation of naive T cells in 61/86 (71%) of subjects with complete DiGeorge anomaly (cDGA). Two children with cDGA and one with a FOXN1 mutation were treated with RVT-802 after prior failed hematopoietic transplants.
Methods: Subjects were enrolled in IRB protocol 00051692 for RVT-802. RVT-802 was implanted into the quadriceps with immunosuppression.
Results: Subject 1 was normal at 22q11.2 but had hypocalcemia, an ASD, PDA, and abnormal ears. The subject received a cord blood transplant mismatched at HLA-B and HLA-C alleles at age 3 months. Subsequently mild graft-versus-host disease (GVHD) developed and was treated with antithymocyte globulin, steroids and cyclosporine. Donor T cells developed in low numbers. Twelve years later, the subject developed Epstein Barr virus lymphoma and suffered two relapses. While in remission, subject 1 received unmatched RVT-802. Two weeks after RVT-802 implantation, the subject developed an adenovirus infection resulting in skin and gut GVHD, presumably from activation of the cord blood T cells. Subject 1 was treated with corticosteroids, cyclosporine, cidofovir and infliximab. Four years post RVT-802, subject 1 is healthy with 609 genetically recipient T cells/mm3 and 40% naïve CD4 T cells.
Subject 2 was normal at 22q11.2 but had an ASD, PDA, hypoparathyroidism, and no T cells at birth. His genetic defect is unknown. Subject 2 was treated with a RIC myeloablative, allogenic, unrelated, 10/10 cord blood transplant, and a subsequent myeloablative, unrelated 9/10 cord blood transplant. Hematopoietic chimerism was established without T cell development. RVT-802 expressed the one allele in the recipient that was not expressed by the second cord donor. The post-thymic transplant course included immune thrombocytopenia requiring rituximab and splenectomy and generalized adenopathy for 3 years but no GVHD. He failed weaning of immunoglobulin replacement. Three years post RVT-802, he has 930 CD3, 750 CD4, and 105 CD8 T cells/mm3. He is active in school.
Subject 3 had absent TRECs on newborn screening with 7 CD3+ T cells/mm.3 A single mutation in FOXN1 was identified; she has sparse scalp hair. Subject 3 received a 9/10 matched unrelated umbilical cord transplant. The post-transplant course was complicated by significant morbidity, and no naïve T cell development. RVT-802 expressed the one allele in the recipient that was not in the cord blood donor. The subject did not develop GVHD, is healthy and at 9 months has 98 naïve CD4+ T cells. She had resolution of longstanding norovirus and sapovirus gastroenteritis.
Conclusion: RVT-802 can improve T cell immunity after poor or failed correction with allogeneic hematopoietic transplants. In subject 1, GVHD post RVT-802 was related to an acute viral infection; cord T cells attacked HLA mismatches in the recipient. Subjects 2 and 3 were given RVT-802 matched to recipient alleles that were not expressed in the hematopoietic donor. We hypothesize that thymocytes developing in RVT-802, if strongly reactive to the recipient-mismatched allele, are deleted by the bone-marrow-donor dendritic cells (that acquire recipient MHC from the recipient-allele-matched thymic epithelial cells) thereby preventing GVHD.
(46) Submission ID#595739
A Novel Mutation in the Cytotoxic T-lymphocyte Antigen4 (CTLA-4) Gene with Cytopenias, Interstitial Lung Disease, Hypogammaglobulinemia and Recurrent Bacterial Endocarditis
Victoria Dimitriades, MD1, Samantha Swain, MD2
1Associate Clinical Professor of Pediatrics, Division of Pediatric Allergy, Immunology & Rheumatology, University of California Davis Health
2Assistant Professor of Medicine, Division of Allergy and Clinical Immunology, University of California Los Angeles
Rationale: CTLA4 haploinsufficiency is an autosomal dominant immune dysregulation syndrome characterized by variable phenotypes. Here we present a young woman diagnosed with Evans Syndrome and lymphoproliferation as a child, found to have a novel CTLA4 variant as a young adult, and who developed hypogammaglobulinemia and a bacterial endocarditis while stabilized on CTLA-4 replacement therapy.
Methods: Sequencing of 207 genes, including CTLA4, in Primary Immunodeficiency Panel.
Results: Our patient was diagnosed with Evans Syndrome at age 2 with manifestations of anemia and thrombocytopenia recalcitrant to treatment over many years with steroids, cyclosporine, and vincristine. Bone marrow biopsy reportedly showed normal trilineage maturation and her symptoms responded for a short time to splenectomy at age 14. Symptoms recurred at age 16 when she was also found to have pulmonary reticular opacities, prominent lymph nodes, and elevated B cells. Repeat bone marrow and lymph node biopsies at that time were unrevealing. Minor responses to treatment with IVIg, rituximab, mycophenolate mofetil and GCSF were noted. At age 17, she developed varicella-related encephalitis shortly after vaccination. With a strong suspicion of an immune dysregulation syndrome, immune evaluation revealed normal immunoglobulins with good vaccine responses, elevated B cell numbers, normal T cell numbers, and normal mitogen proliferation. CTLA4 sequencing revealed a mutation in exon 2 [c.420C>A, p.Tyr140*] causing a premature translational stop signal, which was consistent with previously reported cases of CTLA4 haploinsufficiency. She was started on rapamycin initially for her cytopenias but was then transitioned successfully to abatacept with almost complete resolution of her anemia, neutropenia, and pulmonary opacities. After 6 months of stable control, she developed a precipitous drop in her platelets and was eventually diagnosed with Streptococcus viridans endocarditis of her native mitral valve. This responded to antimicrobial therapy, but eventually needed surgical intervention due to ongoing insufficiency. Around this time, she was also found to be newly hypogammaglobulinemic, necessitating ongoing IgG supplementation therapy. During successful replacement of her mitral valve with a biosynthetic prosthesis, it was noted that her aortic valve also had evidence of previous disease, implicating a prior endocarditis as part of her clinical syndrome as well.
Conclusions: In this patient, the presentation of recalcitrant cytopenias, lymphadenopathy, elevated B cells, vaccine-induced viral infections and lung findings precipitated concern for immune dysregulation syndromes and allowed for identification of a novel deleterious CTLA4 mutation. In addition to previously reported clinical findings, our patient presents with the first reported case of repeated endocarditis in the setting of CTLA4 insufficiency disease. Given the finding in this patient of prior (unrecognized) disease, regularly screening patients with CTLA4 insufficiency for evidence of cardiac affectation may be prudent.
(47) Submission ID#596393
Alpha Fetoprotein Levels in Ataxia Telangiectasia as Related to Age, Disease Characteristics and Outcomes
Ariela Agress, MD1, Howard M. Lederman, MD, PhD2, Rong Guo, MS3, Jennifer Wright, RN4
1Pediatric Resident, Westchester Medical Center
2Professor of Pediatrics, Medicine and Pathology, Division of Pediatric Allergy and Immunology at Johns Hopkins University School of Medicine
3Statistician, Division of General Internal Medicine and Health Services Research, UCLA
4Clinical Research Nurse, Johns Hopkins University
Background: The relationship between elevated serum alpha fetoprotein (AFP) concentration and age, mortality, genotype and neurologic outcome in Ataxia Telangiectasia (A-T) patients has remained inconclusive over the past decades, leaving AFP as a useful marker for disease diagnosis without further clinical significance.
Objective: To examine the relationship between AFP levels and age, mortality, genotype and neurologic outcome using a data set larger than any prior study.
Methods: We retrospectively collected data on 280 A-T patients at Johns Hopkins Medical Center (0- 34 years of age) with both classical (predicted protein null) and variant A-T. This included 459 serum AFP measurements (179 serial levels in 50 A-T patients, max observations 9 per patient). Mixed model compound symmetry covariance was used for statistical analysis to examine the effect of age at visit on AFP levels. Subgroup analysis by mutation type, mortality, feeding/swallowing scores as a surrogate for neurologic function, x-ray induced in vitro chromosomal breakage and serum transaminase levels were similarly analyzed.
Results: Significant association between age and AFP level was found such that for every 1 year increase in age, AFP level increases 20 ng/mL (p<0.0001). Subgroup analysis by mutation type found that the 12 patients with missense mutations showed a negative linear relationship between log AFP levels and age (r= -0.10, p=0.03). We found greater AFP levels in patients who subsequently died, after controlling for age (least square mean AFP level in log scale 0.67 greater in deceased patients versus living patients, p=0.002). We found a significant decline in feeding score by 0.18 units (score range 0-5) per 100 ng/mL AFP increase (p=0.05) after adjusting for age. There was no significant relationship between AFP levels and serum transaminase levels.
Conclusion: AFP increases with age in A-T patients, though this may not apply to patients with missense mutations. There is a statistically significant increase in mortality and worsened swallowing scores with increasing AFP levels, but this remains to be proven clinically significant.
(48) Submission ID#596397
Pill Endoscopy as a Diagnostic Tool for an Abdominal Exacerbation in a Pediatric Patient with Hereditary Angioedema: A Case Report
Yatyng Chang, MD1, Melissa Cardenas-Morales, MD2, Luis Caicedo Oquendo, MD3, Jose Calderon, MD4, William Blouin, APRN5, Vivian Hernandez-Trujillo, MD4
1Pediatric Resident, Nicklaus Children's Hospital
2Allergy Immunology Fellow, Nicklaus Children's Hospital
3Gastroenterology Attending, Nicklaus Children's Hospital
4Allergy Immunology Attending, Nicklaus Children's Hospital
5Nurse Practitioner, Nicklaus Children's Hospital
Introduction: Hereditary Angioedema (HAE) is a chronic illness characterized by recurrent attacks of angioedema and results in frequent Emergency Department (ED) visits per year. Here we present a pediatric HAE patient who had recurrent abdominal attacks in which constipation, secondary to the ADHD medication dexmethylphenidate (Focalin), appears to be a trigger. Of importance, this is the first pediatric patient with HAE to be described as having safely undergone a capsule endoscopy for direct visualization of the gastrointestinal tract. This was done to decrease the risks associated with the more invasive procedure of traditional endoscopy and colonoscopy.
Case Presentation: The patient was an 8-year-old male with Hereditary Angioedema who presented with 1 day history of diffuse abdominal pain and nausea. In the ED, patient was in no acute distress. Abdominal ultrasound showed severe circumferential thickening of the wall of multiple bowel loops and a large amount of simple ascites. X-ray revealed stool in the colon. He was admitted for pain control and hydration.
In the next year, he visited the ED five more times for exacerbations of angioedema of his hand, penis, and bowel. Each time, he presented he had underlying abdominal pain and constipation. He was seen by Gastroenterology and had a workup that was negative for helicobacter pylori, parasites, and other gastrointestinal infections. To further evaluate his abdominal pain, capsule endoscopy was performed and well tolerated. During an admission in January 2016 he received a full inpatient bowel cleanout, after which, his angioedema finally improved. Of note, he was diagnosed with ADHD and started on dexmethylphenidate (Focalin) just prior to this period of recurrent angioedema attacks, and he did not have attacks during the summer months when he was off the medication.
Discussion: Abdominal pain is a common complaint in pediatric hospitals, and further workup consists of endoscopy and colonoscopy. This may be easily accomplished in the general population, however, in patients with HAE, these procedures carry greater risk and may be avoided, leading to delayed diagnosis and treatment (2,4). A newer and less commonly used alternative for direct visualization of the gastrointestinal tract is capsule endoscopy. Some benefits are that it does not require sedation, is less invasive, and is less likely to be irritating to the mucosa (3). Additionally, since psychological stress may be a trigger for angioedema attacks, the decreased stress associated with a noninvasive procedure such as capsule endoscopy, makes it safer to use (1). Limitations of capsule endoscopy include dependence on battery life and its inability to biopsy or administer therapy if needed (3).
Hereditary Angioedema treatment consists primarily of avoiding triggers and managing acute episodes. In this first case of HAE in a pediatric patient where capsule endoscopy was used, the procedure was well tolerated without any complications. Recognizing constipation as a trigger and capsule endoscopy as a safe method of direct visualization of the gastrointestinal tract will help others to control and decrease the severity of their HAE attacks as well.
References
(1) Aygoren-Pursun E, Saguer IM, Kreuz W, et al. Risk of angioedema following invasive or surgical procedures in HAE type I and II – the natural history. Allergy European Journal of Allergy and Clinical Immunology 2013; 68:1034-1039. (https://onlinelibrary.wiley.com/doi/epdf/10.1111/all.12186)
(2) Nzeako U, Longhurst H. Many faces of angioedema: focus on the diagnosis and management of abdominal manifestations of hereditary angioedema. European Journal of Gastroenterology and Hepatology 2012; 24(4):353-359.
(3) Robertson KD, Bhimji SS. Capsule Endoscopy. StatPearls Publishing LLC 2018; Last updated February 27, 2018. https://www.ncbi.nlm.nih.gov/books/NBK482306/.
(4) Soni P, Kumar V, Alliu S, et al. Hereditary Angioedema (HAE): a cause for recurrent abdominal pain. BMJ Case Reports 2016; Published November 14 2016. https://www.ncbi.nlm.nih.gov/pubmed/27873761
Capsule Endoscopy Images
(49) Submission ID#596915
Hepatic Complications of CVID
Kristine Vanijcharoenkarn, MD1, Merin Kuruvilla, MD2, Frances Lee, MD3, Ekemini Ogbu, MD4, Srihari Veeraraghavan, MD3, Jennifer Shih, MD5
1Allergy and Immunology Fellow, Emory University School of Medicine
2Assistant Professor of Medicine, Emory University School of Medicine
3Associate Professor of Medicine, Emory University School of Medicine
4Pediatric Rheumatology Fellow, Emory University School of Medicine
5Assistant Professor of Pediatrics and Internal Medicine, Department of Pediatrics, Division of Allergy and Immunology, Emory University School of Medicine
A 45 year old male with past medical history of common variable immune deficiency (CVID) and related autoimmune complications, including granulomatous-lymphocytic interstitial lung disease (GLILD), hepatosplenomegaly, leukopenia, and thrombocytopenia tolerated monthly subcutaneous immunoglobulin replacement as outpatient for several years with infrequent infectious complications. Four months ago, he was found to have elevated liver enzymes on routine chemistry. A liver biopsy two months later showed pathology consistent with nodular regenerative hyperplasia (NRH) without overt cirrhosis. A hepatic venous pressure gradient (HVPG) of 21 mmHg was found, consistent with portal hypertension. His hepatitis viral markers were negative, he did not drink, and portal venogram was negative for thrombosis. In early October, the patient was admitted to the hospital with anasarca and tense ascites. He underwent a diagnostic and therapeutic large volume paracentesis and was also found to have spontaneous bacterial peritonitis (SBP) and bacteremia with Group B streptococcus.
The patients course was complicated by polymicrobial peritonitis, VRE bacteremia, fungemia, variceal hemorrhage, hepatic encephalopathy, and hepatorenal syndrome. His hepatic complications from portal hypertension were out of proportion to his liver parenchymal disease. Transjugular intrahepatic portosystemic shunt (TIPS) was considered to alleviate portal hypertension but was not feasible due to his degree of encephalopathy. Immunosuppressants such as high dose steroids were given while in the hospital with plans to start rituximab to treat patients GLILD after he had recovered from the acute infections. Unfortunately, after two months in the hospital, the patient succumbed to sepsis and progressive liver failure.
This case emphasizes the importance of systematic screening and continued vigilance for hepatic complications in patients of CVID as studies have shown that NRH of the liver is present in more than 80% of CVID patients who undergo a liver biopsy (PMID: 23219764). A cross-sectional study of patients with primary hypogammaglobulinemia and hepatic dysfunction found that histological findings of NRH were present in 84% of CVID patients and was associated with portal hypertension in 75% of cases (PMID: 17998147). Another study estimated the minimal prevalence of NRH in CVID patients as 12% (PMID: 18647320), stating that this was likely a gross underestimate as NRH may also be present in patients with normal liver function tests that are not routinely biopsied. Therefore, liver enzyme levels may not anticipate the severity of liver involvement. There is currently no treatment for CVID-related liver disease. Other causes of non-cirrhotic portal hypertension, including hepatic veno-occlusive disease and Budd-Chiari Syndrome should be ruled out or treated in CVID patients presenting with hepatic disease. In the case of hepatic NRH in CVID patients, early detection could lead to earlier interventions (such as TIPS prior to hepatic encephalopathy), to mitigate complications.
(50) Submission ID#597549
Epigenetic Immune Cell Quantification for Diagnosis and Monitoring of Patients with Primary Immune Deficiencies and Immune Regulatory Disorders
Janika Schulze, MSc1, Jeannette Werner, PhD2, Konstantin Schildknecht, MSc3, Uma Lakshmanan4, Andreas Grützkau, PhD5, Julia Chu6, Yael Gernez, MD7, Carsten Speckmann, MD8, Katja G. Weinacht, MD9, Alice Bertaina, MD10, Udo Baron, PhD11, Stephan Borte, MD, PhD12, Sven Olek, PhD13, Rosa Bacchetta, MD14
1Research Scientist, Epimune GmbH
2VP Research & Development, Epimune GmbH
3Statistician, Epiontis GmbH, part of Precision for Medicine
4Staff, Stanford University
5Head Immunomonitoring, Deutsches Rheumaforschungszentrum (DRFZ)
6Instructor, Pediatrics - Stem Cell Transplantation, Stanford University
7Clinical Assistant Professor, Pediatrics - Immunology And Allergy, Stanford University
8Assistant Medical Director - Pediatric Immunology, University Hospital Freiburg
9Assistant Professor Of Pediatrics (Stem Cell Transplantation And Regenerative Medicine), Stanford University
10Associate Professor Of Pediatrics (Stem Cell Transplantation), Lucile Salter Packard Children's Hospital
11 Principal Scientist, Epiontis GmbH - part of Precision for Medicine
12Clinical Research Director, ImmunoDeficiencyCenter Leipzig, Municipal Hospital St. Georg Leipzig
13Managing Director, Epiontis GmbH -a Precision for Medicine Company
14Associate Professor, Department of Pediatrics, Stem Cell Transplantation, at the Lucile Salter Packard Childrens Hospital, Stanford school of medicine, Stanford, CA, USA
We describe the application of epigenetic quantification of T regulatory (Treg) cells in addition to CD3+, CD4+, CD8+ T cells, B cells, NK cells, monocytes and neutrophils from as little as 50 μl of fresh, frozen or dried blood. The method yields identical results to flow cytometry from fresh blood samples of a healthy donor cohort, with the advantage of being more sensitive and precise with limited amount of blood and minimal sample preparation (Sci Transl Med 2018). We have used this method 1) to immunophenotype patients with early onset immune regulatory disorders (PIRD) and primary immune deficiency (PID), and 2) to evaluate cell subsets reconstitution early after hematopoietic stem cell transplantation (HSCT).
Patients with Immune Dysregulation, Polyendocrinopathy, Enteropathy, X-Linked (IPEX) and IPEX-like PIRD were evaluated by analyzing the Treg-Specific Demethylated Region (TSDR) of the FOXP3 locus in the total of CD3+ T-cells. Despite the dysfunctional FOXP3 mutated protein, IPEX patients exhibited elevated Treg/CD3+ cell ratios which seemed to correlate with disease severity. In contrast, most of the patients with IPEX-like symptoms without FOXP3 mutations exhibited decreased Treg/CD3+ cell ratios - in line with the possible central pathogenic role of Treg function and number in PIRD.
Using epigenetic quantification of CD3+/B- and NK cells, 23 out of 24 confirmed SCID and XLA cases were correctly identified within a cohort of 250 newborn dried blood spot (DBS) samples (96% sensitivity, 100% specificity). The method identified one delayed onset SCID as well as a XLA case that were missed by combined TREC/KREC testing. Epigenetic immune cell quantification missed one SCID case with maternal engraftment that was identified by combined TREC/KREC testing. Abnormally elevated Treg/CD3+ ratio was also detected in a DBS from a newborn who was subsequently confirmed to be affected with IPEX Syndrome.
When applied to serial blood samples during engraftment and reconstitution post-HSCT, the epigenetic method allowed identification of the different blood cell subsets, including Treg cells, at earlier time points than flow cytometry according to current clinical practice. This opens the way to a better understanding of the correlation between early immune reconstitution events and Graft vs. Host Disease or viral reactivation, earlier than with the current methods, in different types of HSCT.
These studies underscore the suitability of epigenetic immune cell quantification for accurately measuring multiple immune cell types from limited blood sample sources. We propose this method as uniquely suitable for novel molecular diagnostic applications in settings with limited fresh blood sample or limited cell number, at the point of care as well as for newborn screening.
(51) Submission ID#597700
Patient with Hypohidrotic Ectodermal Dysplasia and Recurrent Infections Mimicking NEMO-Deficiency Syndrome
Roman Deniskin, MSc, MSc, MD, PhD1, Tara Rosenberg, MD2, Nicholas Rider, DO3
1Resident, Baylor College of Medicine/Texas Children's Hospital
2Surgical Director, Vascular Anomalies Center, Baylor College of Medicine/Texas Children's Hospital
3Clinic Chief of Allergy and Immunology, Baylor College of Medicine/Texas Children's Hospital
We evaluated a 5-year-old male with hyperpyrexia, hypertrichosis, conical hypodontia, and a history of illnesses concerning for NEMO-deficiency syndrome. Starting at six months of age, he suffered recurrent episodes of acute otitis media (non-typeable Hib and Actinobacter Iwolffli), pneumonia, and RSV bronchiolitis. Whole exome sequencing demonstrated a de novo heterozygous c.1259G>A (p.R420Q) mutation in the EDA-receptor (EDAR) gene not present in the parental DNA. His physical exam findings and mutation were consistent with hypohidrotic ectodermal dysplasia (HED), a rare genetic condition characterized by abnormal development of skin, teeth, hair, and sweat glands. HED is caused by defects in the ectodysplasin-A (EDA)-NFkB signaling pathway but is not typically associated with immune deficiency. Consistent with this, immunophenotyping showed normal sub-populations of T-, B-, and NK-cells. Immunoglobulin and complement levels were quantitatively appropriate. He had normal mitogen-induced lymphocyte proliferation and normal antibody response to pneumococcal vaccination. NK-cell studies demonstrated robust cytotoxicity. However, nasal mucosa biopsy showed diffuse squamous metaplasia and the absence of ciliated epithelial cells. We hypothesize that recurrent infections in our patient arose from impaired mucociliary clearance due to a ciliary defect. This case raises the possible association between EDAR variants and ciliary dysfunction. It also underscores the importance of evaluating the immune status of HED patients with recurrent infections which could mimic NEMO-deficiency and have broad implications about clinical management.
(52) Submission ID#597704
Show Me the Phenotype: The Ordering Clinicians Role in Genetic Variant Interpretation for Primary Immunodeficiency Diseases
Jennifer Holle, MS, CGC1, Rebecca Truty, PhD2, Shiloh Martin, MD, PhD3, Hui Yu, PhD4, Michael Anderson, PhD5, Britt Johnson, PhD, FACMG6
1Genetic Counselor, Invitae
2Scientist, Invitae
3Scientist, Invitae
4Scientist, Invitae
5Scientist, Invitae
6Lab Director, Invitae
The rapid pace of new gene discovery and phenotype expansion for Primary Immunodeficiency Diseases (PIDDs) creates challenges for genetic testing and variant interpretation. Whereas well-described clinical case reports in published literature have traditionally served as the source of phenotypic data used for variant interpretation, for PIDDs the causal variants are often private to the patients family and thus the sole source of phenotypic information for a novel genetic variant is frequently the history provided by the clinician on the test requisition form. Taking into account such heterogeneous information during variant interpretation requires establishing objective criteria for its inclusion as part of the variant interpretation process. To this end, we adapted our laboratorys pre-existing, evidence-based variant classification framework, called Sherloc, by developing point-based criteria for the inclusion of clinical information such as a patients phenotype, familial segregation patterns, and whether the variant is inherited or de novo in the patient. As part of this process, we defined clinical criteria for 154 PIDD genes. Here, we illustrate the application of this method and the importance of integrating clinical information into variant interpretation.
Between April 2017 and October 2018, our commercial diagnostic laboratory performed 4057 immunological genetic tests, and information about the patients clinical history was provided in 2849 (70%) of these orders. Restricting our analysis to just the 154 genes for which case report information is currently used in variant interpretation, these tests revealed 3868 variants, 370 (10%) of which were classified as pathogenic or likely pathogenic (P/LP). Information from case report descriptions, segregation patterns, and de novo status were applied for 32%,15% and 4% of P/LP variants, respectively. In 37 (10%) cases, the clinical information provided by the clinician on the test requisition form was used as evidence in the classification of the patients variant as P/LP. Ten variants were initially classified as being of uncertain significance and reclassified following receipt of further clinical information or testing of additional relatives. In addition, 35 suspicious variants of uncertain significance were identified in which one or two additional patient case reports would allow for reclassification from uncertain significance to P/LP. These data illustrate the importance of providing good quality clinical information to the genetic testing laboratory both at the time of sample submission and following the receipt of genetic test results.
(53) Submission ID#597776
A 30-year Prospective Study Reveals Risk Factors for Malignancies and Early Death in Cartilage-hair Hypoplasia
Svetlana Vakkilainen, MD1, Mervi Taskinen, MD, PhD2, Paula Klemetti, MD, PhD3, Outi Mäkitie, MD, PhD4
1Fellow in Pediatric Infectious Diseases, Children's Hospital, Pediatric Research Center, University of Helsinki and HUS Helsinki University Hospital, and Folkhälsan Institute of Genetics, Helsinki, Finland
2Senior Consultant, Children's Hospital, Pediatric Research Center, University of Helsinki and HUS Helsinki University Hospital
3Consultant, Children's Hospital, Pediatric Research Center, University of Helsinki and HUS Helsinki University Hospital
4Professor, Children's Hospital, HUSLAB, University of Helsinki and HUS Helsinki University Hospital, and Folkhälsan Institute of Genetics, Helsinki, Finland, and Karolinska Institutet and Karolinska University Hospital, Stockholm, Sweden
Background: Cartilage-hair hypoplasia (CHH) is a skeletal dysplasia with combined immunodeficiency, variable clinical course and increased risk of malignancy, mostly non-Hodgkin lymphoma and basal cell carcinoma. There is a paucity of long-term follow-up data, as well as knowledge on prognostic factors in CHH.
Objective: We conducted a prospective cohort study in Finnish patients with CHH to describe clinical course and analyze risk factors for adverse outcomes.
Methods: We recruited 80 Finnish patients with CHH in 1985-1991 and performed clinical follow-up in 2011-2015. We obtained health information from Finnish National Medical Databases (covering time period of 1969-2016), the Finnish Cancer Registry (1953-2016) and the Cause-of-Death Registry of the Statistics Finland (1971-2016) and analyzed all patients’ health records. Standardized mortality ratios (SMRs) were calculated based on the population data. Primary outcomes included immunodeficiency-related death (from infections, respiratory diseases or malignancies), the development of lymphoma and the development of skin cancer.
Results: The study cohort included 35 males and 45 females. Median age at recruitment was 14.6 yrs (range 2 weeks – 49.6 yrs) and median duration of follow-up for the surviving patients was 29.2 yrs (range 25.6 – 31.0 yrs). Half of the patients (46/80, 57%) had no symptoms of immunodeficiency, while 15 (19%) and 19 (24%) patients manifested symptoms of humoral or combined immunodeficiency respectively, including six cases of late-onset immunodeficiency. In a significant proportion of patients (17/79, 22%), clinical features of immunodeficiency progressed over time. Of the 15 patients with non-skin cancer, eight had no preceding symptoms of immunodeficiency. Altogether 20 patients had deceased (SMR=7.0, 95% confidence interval (CI)=4.3-11) including deaths due to pneumonia (n=4), malignancy (n=7, SMR=10, 95%CI=4.1-21) and lung disease (n=4, SMR=46, 95%CI=9.5-130). Malignancy was diagnosed in 21/80 (31%) patients, mostly lymphoma (n=9) and skin cancer (n=15). Severe short stature at birth (compared to normal, SMR/SMR ratio=5.4, 95%CI=1.5-20), symptoms of combined immunodeficiency (compared to asymptomatic, SMR=19 (95%CI=8.0-36) vs SMR=4.8 (95%CI=2.3-8.9), Hirschsprung disease (odds ratio (OR) 7.2, 95%CI=1.04-55), pneumonia in the first year of life or recurrently in adulthood (OR=7.6/19, 95%CI=1.3-43/2.6-140), and autoimmunity (OR=39, 95%CI=3.5-430) in adulthood associated with early mortality. In addition, recurrent pneumonia in childhood was associated with the development of lymphoma, while warts and actinic keratosis were associated with the development of skin cancer. Birth length standard deviation score correlated significantly with the age at the diagnosis of first malignancy (p=0.0029), lymphoma (p=0.011) and skin cancer (p=0.014), demonstrating that patients with shorter birth length developed malignancies at an earlier age.
Conclusions: Patients with CHH have high mortality due to infections and malignancies, but also from lung disease. Some subjects present with late-onset immunodeficiency or malignancy without preceding symptoms of immune defect, warranting careful follow-up and screening for cancer even in asymptomatic patients. We provide clinicians with the risk factors for adverse outcomes to assist in management decisions.
(54) Submission ID#599129
A Novel Genetic Etiology for FAS-associated Protein with Death Domain Deficiency
Lisa A. Kohn, MD, PhD1, Caroline Y. Kuo, MD2
1Fellow, Pediatrics in the Division of Allergy, Immunology, and Rheumatology at UCLA
2Assistant Clinical Professor of Pediatrics, University of California, Los Angeles
Autoimmune Lymphoproliferative Syndromes (ALPS and related disorders) are characterized by insufficient apoptosis due to defects in the FAS apoptosis pathway. FADD deficiency (OMIM 602457) is an autosomal recessive disorder resulting from a mutation in FAS-associated protein with death domain (FADD), the adaptor protein involved in Fas signaling to Caspases 8 and 10. We present a case of FADD deficiency identified by whole exome sequencing with a novel genetic mutation
We describe two brothers with recurrent febrile episodes accompanied by seizures and respiratory compromise. The older sibling initially presented with status epilepticus following the Measles Mumps Rubella vaccination later experiencing similar episodes until his demise at 18 months of age.
The younger sibling, who is unvaccinated, presented at 14 months with fever, rash, vomiting, and diarrhea. He developed status epilepticus with respiratory depression that required intubation. He also had enlarged cervical lymph nodes that regressed with antibiotics and steroids. He recovered from that episode but subsequently had a series of similar illnesses with fevers, altered mental status and seizures. With the exception of elevated HHV6 IgG, extensive infectious workup up in all instances was negative.
Previously described FADD deficiency patients demonstrate an ALPS like phenotype with increased circulating double negative T cells, lymphocyte apoptosis defects, elevated Fas ligand and IL10, encephalopathy, functional asplenism but no splenomegaly or lymphadenopathy. Our patients clinical and laboratory findings were similar. He had normal IgG and IgA, decreased IgM, and lack of isohemagglutinins. Absolute CD3+ count is elevated, with elevated percent of CD3+ TCR+ CD4- CD8-. Normal mitogen and antigen T lymphocyte stimulation, but with defect in pokeweed induced B cell proliferation. Fas ligand and IL10 level are increased (See Table 1). No hepatosplenomegaly, but Howell Jolly bodies were detected in peripheral blood indicating functional hyposplenism.
Whole-exome sequencing revealed two different genetic alterations in the FADD gene: a maternally inherited nonsense mutation predicted to severely truncate the protein and a paternally inherited missense mutation in codon 105. Although this paternal mutation has not been described as pathogenic, a different variant in same nucleotide of FADD has been associated with FADD deficiency (Reference1).
There are very few cases in the literature of FADD deficiency patients and the overall prognosis is poor compared to classical ALPS patients, as these patients are at significant risk of deadly sepsis from encapsulated organisms or death from neurologic complications. Of the FADD deficiency patients described in the literature, several died prior to 5 years old. While pneumococcal prophylaxis may reduce the risk of sepsis, hematopoietic stem cell transplant has been reported for patients with FADD deficiency (Reference2), and is being considered for our patient.
References:
1. Bolze A et al. Whole-exome-sequencing-based discovery of Human FADD Deficiency. Am J Hum Genet. 2010
2. Savic S, et al. A new case of Fas-associated death domain protein deficiency and update on treatment outcomes. JACI. 2015.
| 6 years old | Reference Range | |
| Total Lymphocytes (cells/μl) | 8740 | 1500-7000 |
| CD3+ (T cells) absolute | 5947 | 1253-2216 |
| CD16+CD56+ (NK cells) absolute | Not done | |
| CD19+ (B cells) absolute | 2186 | 214-624 |
|
Phenotype of CD3+ (% T cells) |
||
| TCR αβ+ T cells | 64% | 59-81% |
|
TCRαβ+ CD4-CD8- |
4.7% | 0.3-1.7% |
| TCR γδ+ T cells | 3.6% | 1.2-12.7% |
|
TCR γδ+ CD4-CD8- |
2.5% | 0.7-7.3% |
| HLADR+ activated T cells | 14% | 3-25% |
| Phenotype of B cells | ||
| Total B cells CD19+ | 25% | 8-22% |
| CD5+ | 29% | 6-33% |
| CD27+ B cells | 6% | 11-51% |
| Serum immunoglobulins (g/L) | ||
| IgG | 1,068 | 397-1,652 |
| IgA | 204 | 50-240 |
| IgM | 27 (low) | 40-140 |
| Isohemmaglutins (Blood type A+) | ||
| A1 | <1:2 | <1:2 |
| A2 | <1:2 | <1:2 |
| B | <1:2 | <1:2 |
| Other studies | ||
| IL10 level | 9 pg/ml | <=6 pg/ml |
| Soluble FasL | 758 | 70-308 |
| Vitamin B12 level | 1415 | 254-1060 |
(55) Submission ID#599209
Infusion Parameters and Key Characteristics of Pediatric and Adolescent Patients with Primary Immunodeficiency Initiated on Ig20Gly in a Patient Program
Lisa Meckley, PhD1, Yanyu Wu, PhD2, Spiros Tzivelekis, MSc3, Andre Gladiator, PhD4
1Director, GHEORE, Shire
2Lead, Health Economics and Outcomes Analytics, Shire
3ORE Lead, ORE Immunology & Opthalmolgy, Shire
4Global Medical Lead Immunology - Global Medical Affairs, Shire
Rationale: HCUVP is a patient product-introduction program that provides Cuvitru® (immune globulin subcutaneous [human], 20% solution [Ig20Gly]) free of charge for the first 4 infusions to eligible patients with primary immunodeficiency disease (PID). Using patient data from this ongoing program, our analysis described the clinical characteristics and infusion parameters of pediatric and adolescent patients who were initiated on Ig20Gly through HCUVP.
Methods: HCUVP eligibility criteria were: patients aged 2 years old, with a primary ICD-10-CM code verifying diagnosis of PID, and no current or prior use of Ig20Gly at program initiation. Data from patients who received the first Ig20Gly infusion between January 1, 2017, and September 1, 2017 were included. Data from patients receiving infusions after October 31, 2017 were censored. Descriptive statistics were calculated for patients demographic and clinical characteristics and prescribed and actual infusion characteristics by age group (<18 years and 18 years).
Results: In total, 817 patients who completed all 4 infusions were included in the analysis, of whom 97 were aged <18 years. Among those who previously received immunoglobulin (IG) therapy, a greater percentage of patients aged <18 years were treated with intravenous IG therapy (n=46; 73%) compared with adult patients (n=222; 62%) before initiating Ig20Gly. Nine patients aged <18 years were treatment naïve. The mean infusion volume per site was lower among patients aged <18 years (25 years: 17.9 mL; 611 years: 26.4 mL; and 1217 years: 34.6 mL) than among patients aged 18 years (1864 years: 38.5 mL and 65 years: 38.9 mL). However, the mean infusion rate per site was similar between patients aged <18 years (25 years: 45.9 mL/h; 611 years: 47.3 mL/h; and 1217 years: 40.7 mL/h) and patients aged 18 years (1864 years: 43.3 mL/h and 65 years: 44.0 mL/h). In addition, by the final infusion, fewer patients aged <18 years were infused weekly (n=18 [19%] patients) compared with patients aged 18 years (n=232 [32%] patients). Conversely, a greater percentage of patients aged <18 years were infused biweekly (n=35 [36%] patients) compared with patients 18 years (n=168 [23%] patients).
Conclusion: The results provide insights into the clinical and infusion characteristics of pediatric and adolescent patients who have received Ig20Gly and clinical use of Ig20Gly outside of a controlled clinical trial setting.
Funding: This research was sponsored by Shire.
(56) Submission ID#599435
XMEN: MAGT1 Mutation Associated Immunodeficency. Case Report of an Atypical Presentation
Carlos A. Verdugo, Medical Doctor1, Alejandra V. King, MD2
1Immunology Resident, Universidad de Chile
2Staff, Immunology Unit Luis Calvo Mackenna Children's Hospital, Clínica Alemana de Santiago
XMEN disease (X-linked Immunodeficency with Magnesium defect, Epstein-Barr virus infection and Neoplasia) is a primary immune deficiency caused by mutations in MAGT1 and characterized by chronic infection with Epstein-Barr virus (EBV), EBV-driven lymphoma, CD4 T-cell lymphopenia, and dysgammaglobulinemia. MAGT1 gene codifies to MagT1 protein, a Mg2+- selective transporter, expressed in the human immune system, specifically in the spleen and the thymus. Functional studies have established the key role of MAGT1 in T cells and natural killer (NK) cell activation. Upon CD4+ T-cell receptor stimulation, MAGT1 mediates a transient Mg2+ influx that is necessary for phospholipase C gamma 1 (PLCy1) activation, which drives Ca2+ rise and downstream signaling. This Mg2+ influx also regulates cytotoxic functions of NK and CD8 T cells through NKGD2, reason why these patients have impaired cytolytic responses against EBV. Eleven male XMEN patients have been described. We present the case of a 1-year old Hispanic infant with a pathogenic variant in MAGT1 gene that clinically manifested with early Pneumocystis jirovecii and cytomegalovirus (CMV) interstitial pneumonia, and EBV chronic infection with good response to intravenous immunoglobulins supplementation without hematopoietic stem cell transplantation or gene therapy. Laboratory study highlights low levels of NKG2D ligands. The objective of this case report is to broaden the spectrum of clinical presentation of XMEN disease, that manifests initially as a Combined Immune Deficiency (CID) and evolved with a favorable course of the disease with intravenous immunoglobulins supplementation therapy and chemoprophylaxis with trimethoprim-sulfamethoxazole.
(57) Submission ID#599449
Smoldering Hemophagocytic Lymphohistiocytosis Secondary to Compound Heterozygous Variants in SLCA7 Treated with Anakinra
Nicholas L. Hartog, MD1, Beth Kurt, MD2, Stacie Adams, MD3, Johanna Zea-Hernandez, MD4, Surender Rajasekaran, MD5
1Allergy and Immunology, Helen DeVos Children's Hospital and Michigan State University
2Hematology and Oncology, Helen DeVos Children's Hospital and Michigan State University
3Biochemical Genetics, Helen DeVos Children's Hospital and Michigan State University
4Pediatric Pulmonary, Helen DeVos Children's Hospital and Michigan State University
5Pediatric Intensive Care Unit, Helen DeVos Children's Hospital and Michigan State University
Introduction: Lysinuric protein intolerance (LPI) is a recessively inherited disorder of the cationic amino acids transporter subunit y+LAT1 caused by variants in the SLC7A7 gene. The disease is characterized by protein-rich food intolerance has a heterogeneous presentation. The clinical findings are a result of depletion of lysine, ornithine, and arginine. Symptoms can include hyperammonemia, failure to thrive, protein aversion, neurologic disease, and lung disease. There is also evidence that inflammatory manifestations are mediated through upregulation of NFB, IL1, and TNF that occur independent of intracellular arginine levels and can lead to life-threatening episodes of hemophagocytic lymphohistiocytosis (HLH).
Case Presentation: A 17-year-old male presented with history of anxiety, depression, eating disorder, delayed puberty and complex partial seizures. Due to poor nutrition and failure to thrive, a gastrostomy tube was placed. Following commencement of enteral feeds, he presented with altered mental status, bilateral mydriasis, hyperreflexia, and agitation which lead to a PICU admission. Ammonia peaked as high as 181 μmol/L and episodes ceased with cessation of enteral feedings. Prior to enteral feeds, he had been self-restricting protein in his diet. Biochemical testing was consistent with LPI and Illumina next-generation sequencing revealed compound heterozygous variants in SLC7A7 (p.S396Lfs*122 and p.E465Dfs*54). Hyperammonemia resolved quickly with cessation of protein intake and high rate dextrose infusion without the need for ammonia scavenging agents. He was subsequently started on protein-restricted enteral feeds.
At diagnosis he did not have any respiratory symptoms, CT scan of chest showed patchy areas of groundglass opacification that was suggestive of early pulmonary alveolar proteinosis (PAP). Bronchoalveolar lavage demonstrated foamy, cloudy pink fluid and elevated bronchioalveolar macrophages on cell differential.
His clinical course and SLC7A7 genotype led to suspicion for smoldering HLH. The findings of elevated ferritin, hypertriglyceridemia, decreased fibrinogen, splenomegaly, elevated IL-2 receptor, decreased NK cell function, along with hemophagocytosis on bone marrow biopsy confirmed the diagnosis. Because of his PAP and HLH, in addition to dietary modifications, a trial of IL-1 beta inhibition (anakinra) at 3 mg/kg/day was initiated.
Follow up CT scan of chest 2 months after initiation of anakinra showed complete resolution of pulmonary groundglass opacifications and PAP. Bone marrow evaluation showed continued hemophagocytosis in spite of the normalization in ferritin, soluble IL-2 receptor, NK function, and triglycerides levels. Overall, he is significantly improved on daily anakinra and no longer meets criteria for HLH or PAP.
Discussion: Recent data has shown in y+LAT1 models that THP-1 macrophages and A549 airway epithelial cells upregulate IL1 and TNF regardless of intracellular arginine content. This suggests that inflammatory manifestations may continue independent of dietary modifications. We present a 17 year old patient with newly diagnosed LPI who was treated dietary modification and anti-IL1 therapy resulting in resolution of HLH and PAP. More research is needed to see if long-term IL1 blockade that can consistently control both the immunologic and pulmonary manifestations of LPI and positively impact morbidity and mortality.
(58) Submission ID#599526
Bartonella Endocarditis in a Child with Probable ALPS
Keerti Dantuluri, MD1, James A. Connelly, MD2, Donna Hummell, MD3, Leigh Howard, MD, MPH4, Yasmin Khan, MD5, Daniel Dulek, MD6
1Pediatric Infectious Diseases Clinical Fellow, Vanderbilt University Medical Center
2Assistant Professor Hematology/Oncology/Bone Marrow Transplant, Vanderbilt University Medical Center
3Professor Allergy and Immunology, Vanderbilt University Medical Center
4Assistant Professor Pediatric Infectious Diseases, Vanderbilt University Medical Center
5Assistant Professor Pediatric Allergy/Immunology, Vanderbilt Children's Hospital
6Assistant Professor Pediatric Infectious Diseases, Vanderbilt University Medical Center
Learning Objective: Recognize that symptoms of Bartonella endocarditis and associated complications can share features of certain immunocompromising conditions.
Case Description: An 8-year-old Caucasian boy with history of repaired pulmonary atresia and aortic root dilation was diagnosed with pancytopenia and splenomegaly during a brief hospitalization for atypical pneumonia. Pancytopenia persisted, splenomegaly worsened, and five months after presentation, he developed hypertension and renal insufficiency. He was diagnosed with hypocomplementemic, diffuse sclerosing and crescentic glomerulonephritis and was started on mycophenolate mofetil with improvement in kidney function and stabilization of cytopenias. As part of a comprehensive immune work-up, ALPS (autoimmune lymphoproliferative syndrome) panel was sent and demonstrated elevated double-negative T (DNT) cells with 3 out of 4 positive immunologic criteria for ALPS. Neither targeted sequencing for ALPS and ALPS-like disorders nor whole exome sequencing revealed pathogenic mutations.
By age 10, the patient remained on mycophenolate, but developed failure to thrive, with weight dropping from 37th percentile to less than 3rd percentile. He was hospitalized again for low-grade fever, increased work of breathing, left shoulder pain and fatigue and was found to have right lower lobe pneumonia. Pancytopenia worsened, and he was started on cefepime and azithromycin without improvement in symptoms. Echocardiogram revealed vegetations in his pulmonary conduit and bilateral branch pulmonary arteries, but multiple blood cultures were negative. Upon further history, the patient reported contact with kittens. Bartonella henselae titers and polymerase chain reaction (PCR) from blood were sent and were both positive. He completed a 2-week course of gentamicin, 1-month course of ceftriaxone, and was transitioned to doxycycline and rifabutin. After initiating antimicrobial therapy, his weight and energy significantly improved, his blood Bartonella PCR became negative, and his splenomegaly resolved. Approximately one year later, the patient underwent pulmonary artery conduit replacement and Bartonella PCR testing of the tissue specimen was positive. He has had sustained weight increase, resolution of hypocomplementemia and splenomegaly, decrease in DNT cell frequency from >2% to 0.9%, and improvement though not resolution of cytopenias. He currently remains on doxycycline and rifabutin and continues treatment with mycophenolate.
Discussion: ALPS is characterized by defective lymphocyte apoptosis and clinical features such as lymphadenopathy, splenomegaly, hepatomegaly, cytopenias, and glomerulonephritis. The hallmark laboratory finding is expansion of DNTs. Our patient met criteria for a probable ALPS diagnosis based on the presence of both required criteria (chronic splenomegaly and elevated DNT cells) and secondary additional criteria (typical immunologic findings noted on ALPS panel). Pediatric cases of Bartonella henselae endocarditis have been associated with splenomegaly, cytopenias, and glomerulonephritis which mimic many features of monogenic immune dysregulatory disorders. The diagnosis of Bartonella endocarditis in our patient therefore raises the question of whether his immunosuppression predisposed him to infection or if his entire clinical presentation can be explained by Bartonella endocarditis. Physicians taking care of patients with immune dysregulatory disorders should consider Bartonella endocarditis in the differential diagnosis of onset or exacerbations of immune dysregulation.
(59) Submission ID#599570
Body Temperature in Patients with Primary Immunodeficiency
Shouling Zhang, MD1, Tiffany S. Henderson, PhD2, Christopher Scalchunes, MPA3, Kathleen E. Sullivan, MD, PhD4, Artemio M. Jongco, III, MD, PhD, MPH5
1Pediatrics Resident, Department of Pediatrics, Donald and Barbara Zucker School of Medicine at Hofstra/Northwell, New Hyde Park, NY
2Survey Research Analyst, Immune Deficiency Foundation
3Vice President of Research, Immune Deficiency Foundation
4Professor, The Children's Hospital of Philadelphia
5Assistant Professor of Medicine and Pediatrics, Division of Allergy & Immunology, Donald and Barbara Zucker School of Medicine at Hofstra/Northwell; Center for Health Innovations and Outcomes Research, Feinstein Institute for Medical Research, Manhasset, NY
Rationale: While fever is considered a sign of infection, many individuals with primary immunodeficiency (PI) anecdotally report a lower than normal average body temperature. On Immune Deficiency Foundation (IDF) Friends and IDF PI CONNECT Research Forum online, PI patients report a diminished fever response even when other signs of infection are present. There is limited knowledge about the average body temperature in persons with PI. However, the implications of missing an infection in those with PI is well established.
Methods: Study investigators partnered with patient investigators to design a prospective cohort study to determine whether body temperature differed between persons living with and without PI. Three hundred fifty adults with PI were recruited from IDF and one adult household member without PI was also recruited. McKesson digital oral thermometers (Model 01-413BGM) were provided and used to record temperatures in all participants three times a day for five consecutive days. Descriptive statistics were calculated. Median body temperatures were compared between the two cohorts at each time point using Mann-Whitney test.
Results: Data from 254 households were used for analysis (72.6% participation rate). The PI population was largely female (85.8%) with a median age of 49 years and largely Caucasian population (97.6%). The non-PI population was largely male (66.9%) with a median age of 53 years and largely Caucasian population (92.9%). PI diagnoses included CVID (74.8%), hypogammaglobulinemia (12.6%), IgG subclass deficiency (4.7%), selective IgA deficiency (3.1%), specific antibody deficiency (3.1%), agammaglobulinemia (0.4%), chronic granulomatous disease (0.4%), combined immunodeficiency (0.4%), and complement deficiency (0.4%). A total of 123 individuals with PI (48.4%) reported a lower than normal non-sick body temperature, while 108 individuals with PI (42.5%) reported a normal (between 97°F - 99°F) non-sick body temperature. A total of 172 individuals with PI (67.7%) reported absence of fever with infection, while 50 individuals (19.7%) reported a normal fever response with infection. The median body temperature was significantly higher for the PI patients in the morning, but not evening or bedtime, reading in 4 of the 5 days (Monday: PI = 97.5°F vs. non-PI = 97.2°F, p = 0.0291; Tuesday: PI = 97.4°F vs. non-PI = 97.2°F, p = 0.0020; Wednesday: PI = 97.5°F vs. non-PI = 97.2°F, p = 0.0009; Thursday: PI = 97.4°F vs. non-PI = 97.2°F, p = 0.0575; Friday: PI = 97.4°F vs. non-PI = 97.2°F, p= 0.0008).
Conclusions: Despite the limitations of this non-clinical study, individuals with PI are knowledgeable about their conditions and can offer unique insights and direction to researchers. This study demonstrates that collaboration with patient advocacy groups may facilitate patient-centered and patient-driven research with high participation among the target population.
(60) Submission ID#599598
A 2-year-old Male with Compound Heterozygous Familial Mediterranean Fever (FMF)
Shouling Zhang, MD1, Zoya Treyster, MD2, Vincent Bonagura, MD3
1Pediatrics Resident, Department of Pediatrics, Donald and Barbara Zucker School of Medicine at Hofstra/Northwell
2 Fellow, Division of Allergy & Immunology, Donald and Barbara Zucker School of Medicine at Hofstra/Northwell
3Professor of Medicine and Pediatrics, Division of Allergy & Immunology, Donald and Barbara Zucker School of Medicine at Hofstra/Northwell
Introduction: Familial Mediterranean Fever (FMF) is a hereditary condition characterized by recurrent episodes of painful inflammation caused by mutations in the pyrin (MEFV) gene. Alterations in the MEFV gene affect pyrin production leading to recurrent fevers and painful inflammation in the peritoneum, synovium, and pleura. Amyloidosis may also develop as a complication. Arabic, Turkish, Armenian, and Sephardic Jewish populations are most commonly affected. Homozygosity for MEFV mutations are associated with a more severe course. There is a paucity of information regarding pediatric FMF in the literature.
Case: We present a case of a 2-year-old male with minor speech delay diagnosed with compound heterozygous FMF. Patient was initially referred due to recurrent fevers and infections. At 4 months of age, he was hospitalized with septic shock requiring intubation secondary to adenovirus. At 5 months of age, the patient began to have recurrent fevers every 3 to 4 weeks, leading to multiple blood draws and courses of antibiotics prior to referral. At 11, 12, and 22 months of age, he developed three separate episodes of febrile seizures. A total of 10-15 lifetime episodes of acute otitis media occurred prior to bilateral myringotomy tube placement. Four episodes of Streptococcus pyogenes pharyngitis confirmed by throat culture preceded tonsillectomy. No oral ulcers, joint pain, or abdominal pain were reported. No other infections such as pneumonia, sinusitis, UTI, non-viral gastroenteritis, fungal infections, or skin infections were reported. Both parents are Ashkenazi Jewish and a maternal history of early miscarriage was noted. Family history was negative for immunodeficiency, malignancy, and autoimmunity.
The patients vital signs and physical exam were unremarkable. Serology indicated leukocytosis of 18.53 K/L with elevated monocytes of 1390 cells/L, elevated eosinophils of 1200 cells/L, and slightly elevated CD8 T cell count of 2653 cells/L. Neutrophil, CD4 T cell, B cell, NK cell enumeration, and immunoglobulin panel were normal for age. Tetanus, diphtheria, rubella, Streptococcus pneumoniae, and Haemophilus influenzae B titers were protective. Genetic analysis identified that the patient was compound heterozygous for the E148Q and V726 mutations in the MEFV gene.
Family was instructed to keep a fever diary. Colchicine 0.6mg once a day was given initially, then increased to 1.2mg once a day for inadequate response. Loose stools were observed while patient was maintaining a lactose free diet so he was switched to colchicine 0.6mg BID with resolution of loose stools. Apart from two occasions when his colchicine dose was missed, the patient remained afebrile at his follow up visits.
Conclusion: We present a pediatric case of compound heterozygous FMF (E148Q and V726 MEFV mutations) in an otherwise healthy 2-year-old male of Ashkenazi Jewish background, initially symptomatic at 5 months of age. Individuals who are compound heterozygous for the E148Q and a second MEVF mutation are generally symptomatic, although severity cannot be predicted. Additional pediatric research on symptomatic heterozygous and compound heterozygous FMF is recommended.
(61) Submission ID#599700
CD27 Deficiency Causes Human NK Cell Deficiency with Specific Loss of the CD56(bright) Subset: A Single Case Report
Natalia Chaimowitz, MD, PhD1, Sarah Nicholas, MD2, Leonora Noroski, MD, MPH3, Lisa R. Forbes, MD4, Sara Nandiwada, Ph.D., D(ABMLI)5, Nicholas Rider, DO6
1Fellow, Texas Childrens Hospital/Baylor College of Medicine
2Assistant Professor, Baylor College of Medicine/Texas Children's Hospital
3Associate Professor of Pediatrics, Baylor College of Medicine/Texas Children's Hospital
4Assistant Professor, Department of Pediatrics, Baylor College of Medicine, Houston, TX, USA
5Assistant Professor of Pediatrics, Baylor College of Medicine/ Texas Children's Hospital
6Associate Professor of Pediatrics, Texas Childrens Hospital/Baylor College of Medicine
Natural killer (NK) cells are innate lymphocytes that play a key role in defense against virally-infected cells and in tumor surveillance. NK cells can be divided in two subsets. The majority of NK cells in peripheral blood expressed intermediate levels of CD56 and are referred to as CD56(dim). These NK cells are responsible for NK cell cytotoxicity. A minor population of NK cells express very high expression of CD56 and are referred to as CD56(bright). These NK cells are responsible for cytokine production and are precursors to CD56(dim) NK cells. A few immunodeficiencies have been described in which there are abnormal NK cell subsets, such as autosomal dominant GATA2 deficiency where CD56(bright) NK cells are absent and IRF8 where there is a paucity of CD56(dim) NK cells and relative expansion of CD56(bright) NK cells.
Here we present a patient with an absence in CD56(bright) NK cells secondary to CD27 deficiency. Our patient is a 6-year-old African American female born to non-consanguineous parents. The patients past medical history is significant for chronic lung disease secondary to prematurity, recurrent acute otitis media, failure to thrive and congenital hypothyroidism. Family history is significant for an older sister that presented at age 3 with EBV-associated Hodgkin lymphoma whose treatment was complicated by chronic activated EBV infection and who ultimately underwent hematopoietic stem cell transplantation (HSCT). Our patient presented with pancytopenia, fever, lymphadenopathy and splenomegaly. She was found to have EBV viremia with greater than 550,000 copies in whole blood by PCR. She was treated with two doses of rituximab followed by etoposide and dexamethasone as a bridge to HSCT. Whole exome sequencing demonstrated a homozygous mutation in CD27. CD27 is a member of the tumor necrosis factor receptor family and influences the function of T cells, B cells and NK cells. In NK cells, CD27 is primarily expressed in CD56(bright) NK cells. CD27 deficiency is an autosomal recessive disorder associated with persistent symptomatic EBV viremia, including EBV-driven hemophagocytosis and lymphoma, hypogammaglobulonemia and specific antibody deficiency. Our patients immune evaluation prior to initiation of chemotherapy and immunosuppression was notable for very elevated IgG, IgA and IgM. Despite hypergammaglobulonemia patient had only 3 out of 11 protective titers against streptococcus pneumoniae. The patient had pan-lymphopenia with appropriate percentages of lymphocyte subsets. Assessment of her B cell subsets showed a slight increase in the percentage of transitional B cells/plasmablast and a nearly complete absence of CD27-expressing B cells. Her NK cell phenotyping demonstrated a complete loss of CD56(bright) NK cells with reduced NK cell cytotoxicity, comparable to what has been previously reported in patients with GATA2 deficiency. Previous reports of patients with CD27 deficiency denote normal NK cell numbers with normal to moderately reduced NK cell cytotoxicity, however, CD27 deficiency causing a specific loss of the CD56(bright) NK cell subset has not been previously reported. CD27 deficiency should be consider in patients with EBV driven disease and abnormal NK cell studies.
(62) Submission ID#599897
Allogeneic Hematopoietic Stem Cell Transplant Outcomes for Patients with Dominant-Negative IKFZ1/IKAROS Mutations
Erinn S. Kellner, MD1, Christa Krupski, MD2, Hye Sun Kuehn, PhD3, Sergio D. Rosenzweig, MD/PhD4, Nao Yoshida, MD/PhD5, Seiji Kojima, MD/PhD6, David Boutbol, MD7, Sylvain Latour, PhD8, Vincent Barlogis, MD/PhD9, Claire Galambrun, MD10, Asbjørg Stray-Pedersen, MD/PhD11, Hans Christian Erichsen, MD/PhD12, Rebecca A. Marsh, MD13
1Clinical Immunodeficiency Fellow, Division of Bone Marrow Transplantation and Immune Deficiency, Cincinnati Childrens Hospital Medical Center
2Instructor, Division of Bone Marrow Transplantation and Immune Deficiency, Cincinnati Childrens Hospital Medical Center
3unknown, Immunology Service, Department of Laboratory Medicine, Clinical Center
4Chief, Immunology Service, Department of Laboratory Medicine, NIH Clinical Center, Bethesda, MD, USA
5unknown, Department of Hematology and Oncology, Childrens Medical Center, Japanese Red Cross Nagoya First Hospital
6Professor, Department of Pediatrics, Nagoya University Graduate School of Medicine, Nagoya, Japan
7unknown, Laboratory of Lymphocyte Activation and Susceptibility to EBV Infection, Inserm UMR 1163 and Clinical Immunology Department, Hôpital Saint Louis, Assistance Publique Hôpitaux de Paris (APHP) Université Paris Diderot
8unknown, Laboratory of Lymphocyte Activation and Susceptibility to EBV Infection, Inserm UMR 1163 and University Paris Descartes Sorbonne Paris Cité, Imagine Institut
9unknown, Department of Pediatric Immunology, La Timone Hospital
10unknown, Department of Pediatric Hematology and Oncology, Timone Enfants Hospital and Aix-Marseille University
11unknown, Norwegian National Unit for Newborn Screening, Oslo University Hospital
12unknown, Section of Specialized Pediatric Medicine, Oslo University Hospital
13Associate Professor, Division of Bone Marrow Transplantation and Immune Deficiency, Cincinnati Childrens Hospital Medical Center
Introduction/Background: The transcription factor IKAROS is encoded by the IKZF1 gene and plays a crucial role in lymphopoiesis. Somatic, and more recently also germline mutations of IKZF1 are associated with a hematologic malignancies, most notably B-cell precursor acute lymphoblastic leukemia. Germline mutation in IKZF1 was first reported as a monogenic cause of human disease characterized by marrow failure and immune deficiency in a single neonate in 2012. Subsequently, mutations leading to haploinsufficiency were discovered to underlie a proportion of patients with CVID and low B cell numbers, and dominant-negative mutations have been observed to cause more severe combined immune deficiency phenotypes. At this time, there is very little known regarding allogeneic hematopoietic cell transplantation (HCT) outcomes for patients with severe dominant-negative IKZF1 mutations. Concerningly, IKAROS deficiency has been observed to have a negative impact on graft versus host disease in mouse models.
Objective: To describe allogeneic stem cell transplant outcomes in patients with the dominant-negative IKAROS mutation.
Methods: We collected transplant data from 4 patients who underwent allogeneic HCT at transplant centers around the world.
Results: Patients underwent allogeneic HCT using a variety of conditioning regimens. Patients received bone marrow (N=3) or cord blood (N=1) grafts from an HLA-matched sibling donor (N=1) or single allele HLA-mismatched unrelated donor (N=3). Neutrophil engraftment occurred between Day +12 and +51 post-transplant. Platelet engraftment occurred between Day +8 and +167 except in one patient who did not have return of normal platelet counts due to underlying liver dysfunction. All patients were documented to have greater than 99% whole blood donor chimerism at a median of 28 days (range 12-51 days) following transplant and maintained >95% donor chimerism until last follow-up. Only one patient developed grade II acute GVHD. No patients developed chronic GVHD. One patient died approximately 1 year post transplant related to cryptosporidium cholangitis which existed prior to HCT. At the most recent follow up of the 3 surviving patients (range: 0.99-7.2y), IVIG had been discontinued, antimicrobial prophylaxis had been stopped, and patients had received routine vaccinations. They all had excellent performance status.
Conclusions: Allogeneic HCT may be a safe option to consider for patients with dominant-negative IKAROS mutation as there does not appear to be an increased risk of death or GVHD. Moreover, 3-out-of-4 of the transplanted patients are alive and well and show no features of the disease. However, because of the limited number of patients evaluated and the retrospective nature of this analysis, our data do not allow firm conclusions to be made, and further studies will be needed to evaluate outcomes in larger cohorts.
(63) Submission ID#600169
Plastic Bronchitis and Secondary T-cell Lymphopenia
Saara Kaviany, DO1, Yasmin Khan, MD2, Dan Dulek, MD3, Michael O'Connor, MD4, Stacy Dorris, MD4, John Newman, MD5, Rizwan Hamid, MD6, John Phillips, MD6, James A. Connelly, MD7
1Clinical Fellow, Vanderbilt Children's Hospital
2Assistant Professor Pediatric Allergy/Immunology, Vanderbilt Children's Hospital
3Assistant Professor Pediatric Infectious Disease, Vanderbilt Children's Hospital
4Assistant Professor Pediatric Pulmonology, Vanderbilt Children's Hospital
5Professor Allergy/Immunology, Vanderbilt University Medical Center
6Professor Genetics, Vanderbilt University Medical Center
7Assistant Professor Hematology/Oncology/Bone Marrow Transplant, Vanderbilt University Medical Center
Introduction: When evaluating patients with T- cell lymphopenia, we often are concerned about defects in lymphocyte production and function, especially in the setting of frequent infections. Here we outline a case demonstrating T-cell lymphopenia due to increased loss, which should be considered in the differential diagnosis.
Case Report: We report a 13-year-old male who initially presented with recurrent, right-sided pneumonias requiring frequent hospital admissions including severe episodes necessitating intensive care unit admission. His work up for the pneumonias included a bronchoscopy revealing normal anatomy with minimal inflammation, and a chest CT with mild peribronchial wall thickening.
As his pulmonary disease progressed, he developed a persistent, productive cough with expectorated mucous plugs that were plastic-like in appearance. While his pulmonary symptoms responded to steroids, his mucous plug production persisted. Sputum cultures were intermittently positive, isolating Cryptococcus neoformans and Aspergillus niger. He underwent VATS and wedge biopsy, concerning for recurrent aspiration. An immunologic evaluation initially demonstrated normal T- and B-cell counts, but serial evaluation of his lymphocyte population demonstrated low CD4+ cells (ranging 151-367 cells/cumm), and low normal CD8 cells (ranging 101-177 cells/cumm) with normal B- and NK-cell numbers.
Further T-cell evaluation revealed normal ratios of naive and memory populations (CD4CD45RA+ 61%, CD4CD45RO+ 39%, CD8CD45RA+ 74%, CD8CD45RO 33%), normal TREC (7768 copies per 10^6 CD3 cells) and normal thymic emigrants (CD4CD31CD45RA+: 158, normal 150-1500), indicative of sufficient thymopoiesis. Mitogen and antigen stimulation assays demonstrated normal responses to phytohemagglutin, concanavalin A, and pokeweed mitogen, with a low lymphocyte response to Candida. He had normal quantitative immunologlobulins, normal diphtheria, tetanus and streptococcus pneumonia titers. His dihydrorhodamine flow cytometry and FISH for chromosome 21q11.2 deletion were negative.
Given normal function and thymic output, his immunologic profile was concerning for T-cell loss. Our patient was registered with the Undiagnosed Disease Network, and had a second review of his lung biopsy, concerning for plastic bronchitis. Subsequent lymphatic imaging demonstrated abnormal lymphatics within the bilateral clavicular space, right greater than left, with questionable partial thoracic duct, explaining his unilateral symptoms. He was diagnosed with plastic bronchitis secondary to abnormal lymphatic drainage, with lymphatic fluid filling his airways and secondary T-cell loss.
Discussion: Plastic bronchitis is a rare and potentially fatal disorder, seen commonly after the Fontan procedure for congenital heart disease. This process has resulted in T-cell loss into the airway and subsequent T-cell lymphopenia.
In patients with Fontan-related protein losing enteropathy, multiple immune abnormalities have been described including reduced immunoglobulins, lymphopenia, and selective CD4 lymphocyte deficiency. Similar findings have been reported in patients with lymphatic malformations. Although the impact of T-cell loss on adaptive immunity is not entirely known, there is no indication of increased risk for atypical infections.
Given his normal mitogen assay, our patient did not start prophylactic antibiotics. He continues to have symptomatic episodes with lymphopenia, but has had no opportunistic infections, and remains stable with an aggressive pulmonary regimen. We conclude by reiterating the importance of considering T-cell loss in patients presenting with lymphopenia, particularly with evidence of normal thymopoeisis and T-cell function.
(64) Submission ID#600267
Granulomatous Disease and Lymphoma in a Cohort of 1395 Patients with CVID in the USIDNET Registry
Joao Pedro Matias Lopes, MD1, Nicole Ramsey, MD, Phd1, Ramsay Fuleihan, MD2, Kathleen E. Sullivan, MD, PhD3, Avni Joshi, MD4, Daniel Suez, MD5, Patricia Lugar, MD, MS6, John Routes, MD7, Charlotte Cunningham-Rundles, MD, PhD8
1Allergy and Immunology Fellow, Icahn School of Medicine at Mount Sinai
2Professor of Pediatrics, Division of Allergy and Immunology, Northwestern University Feinberg School of Medicine, Chicago, NY
3Professor, The Children's Hospital of Philadelphia
4Assistant Professor, Allergy and Immunology, Mayo Clinic
5President, Allergy, Asthma & Immunology Clinic, PA
6Assistant Professor, Allergy and Immunology, Duke Health
7Chief, Professor, Division of Allergy and Immunology, Children's Hospital of Wisconsin-Milwaukee, Milwaukee, WI
8Professor in Medicine, Division of Clinical Immunology, Icahn School of Medicine, Mount Sinai, NY, NY, USA
Introduction: Granulomatous disease (GD) has been described with a variable incidence (8.0-22.0%) in patients with common variable immunodeficiency (CVID). An increase in malignancies has been reported in CVID patient cohorts, particularly for lymphoma, reported in 1.6-8.2% of the CVID patients depending on the cohorts. Prior analysis of a cohort of 436 CVID patients included 59 patients with GD (GD+). In these, there was a suggestion of more cases of lymphoma (12.5%) when compared to cases without (GD-) (5.0%) although the difference was not statistically significant (p=.07).
Objectives: Compare the frequency of lymphoma in GD+ and GD- patients in the CVID patient cohort from the USIDNET Registry.
Methods: We submitted a query to the USIDNET registry requesting de-identified data for patients with the diagnosis of CVID, through August 2018. Statistical analysis was performed on SPSS, with comparisons done with Pearson chi-square or Fisher's exact test, depending on the sample sizes, using an alpha level of .05.
Results: A cohort of 1395 CVID patients from the USIDNET registry was analyzed. Ninety-one patients (6.5%) were GD+. Overall, 152 patients (10.9%) had a malignancy diagnosis, 47 of these (3.4%) with lymphoma. Lymphoma was present in 6/91 GD+ patients (6.6%) versus 41/1304 GD- patients (3.1%) (p=.12). Overall malignancy was present in 15/91 GD+ (16.5%) versus 137/1304 (10.5%) (p=.08).
Discussion: In the cohort of 1395 CVID patients from the USIDNET registry, we found a frequency of lymphoma of 3.4%, which is in the range of previously described cohorts. The frequency of lymphoma was 6.6% in patients with GD, higher than the 3.1% frequency for GD- patients, but these differences were not statistically significant. Our identified frequency of lymphoma in GD+ patients was lower than the one previously identified in the 436 CVID patient cohort, but with similar proportional differences between GD+ and GD- patients. Despite no statistical significance, the frequency of lymphoma, as shown here and elsewhere, was higher in CVID patients GD+ than GD- in both studies, with no full understanding of this increased risk of lymphoma. Expanding this analysis to larger groups of CVID patients may help to confirm, or deny a more robust association, which may have a meaningful impact in the outcomes of this particular population.
(65) Submission ID#600312
Four Patients with Refractory Pericarditis Treated with Concurrent Hyaluronidase-facilitated Subcutaneous Immunoglobulin and Anti-interleukin 1 Therapy
Melissa D. Gans, MD1, Rachel Eisenberg, MD2, Arye Rubinstein, MD, Ph.D.2
1Fellow in Allergy & Immunology, Montefiore Medical Center
2Attending in Allergy & Immunology, Montefiore Medical Center
Introduction: Patients with refractory pericarditis have been treated with intravenous immunoglobulin (IVIG) or interleukin 1 receptor antagonist (Anakinra) with limited and transient benefit. Separate or combined therapy with subcutaneous immunoglobulin (SCIG) and interleukin (IL) 1 inhibitor (Rilonacept) for refractory pericarditis in a cohort of patients has not been previously described.
Case Descriptions: 4 patients were referred for recurrent pericarditis refractory to traditional therapies at ages ranging from 16 to 54 years. They all had multiple serious sequelae of their pericarditis and abnormal immune parameters including hypogammaglobulinemia, poor responses to vaccines, poor mitogen induced lymphocyte proliferation, and/or B cell lymphopenia. The patients had varied past medical histories and associated conditions. Patients were started on IG, with some initiated on IVIG, though all were transitioned to hyaluronidase-facilitated SCIG (HYQVIA). Patients were then started on either Anakinra or Rilonacept with 3 patients continuing on Rilonacept and 1 remaining on Anakinra. All patients had complete or near complete resolution of their pericarditis on dual therapy for greater than 1 year. The markedly elevated IL1 prior to therapy seen in all of the patients normalized post-therapy. Some patients had elevated IL6 prior to therapy that also improved post-therapy. 1 patient who has also been diagnosed with Familial Mediterranean Fever (FMF) has stopped both therapies for greater than 1 year with no further episodes of her pericarditis.
Discussion: 4 patients with recurrent refractory pericarditis and signs of immunodeficiency and autoinflammatory disease on laboratory testing responded to dual therapy with HYQVIA and Rilonacept or Anakinra resulting in resolution of pericarditis. Inflammasome and immune abnormalities may be implicated or associated with recurrent pericarditis and may respond to targeted therapies.
| Patient #1 | Patient #2 | Patient #3 | Patient #4 | Reference Range | |
| Age at first pericarditis episode (years) | 16 | 54 | 57 | 21 | N/A |
| Pericarditis pre-therapy | 6 acute episodes over 6 years | Constrictive pericarditis for 6 years | 2 episodes over 1 year | Constrictive pericarditis for 12 years, signs of pericarditis on imaging starting at 5 years old | N/A |
| Pericarditis complications | Pulmonary edema, ventricular tachycardia, myocarditis, depressed ejection fracture | Empyema, congestive heart failure | Cardiac arrest, pacemaker placed for complete heart block | Pericardiocentesis | N/A |
| Therapies failed | NSAIDs, steroids, azathioprine | NSAIDs, steroids, colchicine | NSAIDs, steroids, methotrexate Adalimumab, Mycophenolic acid, rituximab | NSAIDs, steroids, colchicine | N/A |
| Age at starting therapy (years) |
22 (IG,) 22 (Rilonacept) |
60 (IG) 63 (Anakinra) |
54 (IG) 64 (Rilonacept) |
33 (IG) 33 (Anakinra) 34 (Rilonacept) |
N/A |
| Pericarditis post-therapy | No subsequent episodes | Resolved on imaging | No subsequent episodes | 1 episode on Anakinra, none on Rilonacept; stopped IG and Rilonacept for past 18 months with no episodes | N/A |
| Time on therapy (years) |
2 (IG) 2 (Rilonacept) |
4 (IG) 1 (Anakinra) |
11 (IG) 1 (Rilonacept) |
2 (IG) 1 (Rilonacept) |
N/A |
| Associated conditions | None | Protein loosing enteropathy | Rheumatoid arthritis, ITP, thyroid disease, CIDP | Familial Mediterranean Fever | N/A |
| Immunodeficiency diagnosis | Selective antibody deficiency with B cell lymphopenia | Hypogammaglobulinemia with poor T cell function | CVID with poor T cell function | Selective antibody deficiency | N/A |
|
CD3+ cells (cells/μL, %) |
1009, 89% | 226, 53% | 1588, 93% | 1897, 80% | 1087-2198, 65-80% |
|
CD4+ cells (cells/μL, %) |
657, 56% | 66, 15% | 1105, 65% | 946, 39% | 677-1401, 38-55% |
|
CD8+ cells (cells/μL, %) |
342, 29% | 139, 32% | 472, 28% | 878, 37% | 212-1007, 15-38% |
|
CD19+ cells (cells/μL, %) |
29, 3% | 148, 37% | 31, 2% | 280, 12% |
180-492, 8-23% |
|
CD16+56+ cells (cells/μL, %) |
73, 7% | 34, 8% | 60, 3% | 181, 8% |
97-421, 5-17% |
| IgG (mg/dL) | 995 | 423 | 492 | 1000 | 844-1912 |
| IgA (mg/dL) | 193 | 124 | 225 | 137 | 68-423 |
| IgM (mg/dL) | 83.2 | 25.5 | 15.3 | 72 | 50-196 |
| Streptococcal pneumonia titers post-vaccination | poor | adequate | poor | poor | adequate |
| Mitogen induced lymphocyte proliferation | adequate | poor | poor | adequate | adequate |
| IL1β (pg/mL) |
39.8 (pre) <3.9 (post) |
>250 (pre) <3.9 (post) |
150.4 (pre) <3.9 (post) |
69.6 (pre) <3.9 (post) |
<3.9 |
| IL6 (pg/mL) | 0.92 (pre) |
373.76 (pre) 4.8 (post) |
3547 (pre) | 85 (off tx) | 0.31-5 |
| Tumor necrosis factor-α (pg/mL) | <1 (pre) | 2 (pre) |
1.8 (pre) 2.84 (post) |
1.9 (pre) <5 (off tx) |
1.2-15.3 |
| Interferon-Υ (pg/mL) | <5 (pre) | N/A | N/A | <5 (off tx) | <=5 |
| Genetic testing | negative | negative | not performed | MEFV V726A heterozygous | negative |
Table 1. Clinical characteristics and immune evaluation for 4 patients.
(66) Submission ID#600335
Characterization of Gut Inflammation and Autoimmunity in Mice Carrying Rag1 Hypomorphic Mutations
Riccardo Castagnoli, MD1, Marita Bosticardo, PhD2, Rosita Rigoni, PhD3, Elena Fontana, PhD4, Ottavia M Delmonte, MD, PhD5, Lisa M Ott de Bruin, MD6, John P Manis, MD7, Cristina Corsino8, Yu Han, PhD9, Emilia Falcone, MD10, Anna Villa, MD11, Luigi D. Notarangelo, MD, PhD12
11 Laboratory of Clinical Immunology and Microbiology, Division of Intramural Research, National Institute of Allergy and Infectious Diseases, National Institutes of Health, Bethesda, MD. 2 Department of Pediatrics, University of Pavia, Pavia, Italy
2Staff Scientist, Laboratory of Clinical Immunology and Microbiology, IDGS, DIR, NIAID, NIH, Bethesda, MD, USA
33 San Raffaele Telethon Institute for Gene Therapy (SR-Tiget), Division of Regenerative Medicine, Stem Cells and Gene Therapy, San Raffaele Scientific Institute, Milan, Italy
44 Humanitas Clinical and Research Institute, Rozzano, Italy. 8 Milan Unit, Istituto di Ricerca Genetica e Biomedica, Consiglio Nazionale delle Ricerche, Milan, Italy
5Staff Clinician, 1 Laboratory of Clinical Immunology and Microbiology, Division of Intramural Research, National Institute of Allergy and Infectious Diseases, National Institutes of Health, Bethesda, MD
65 Department of Pediatric Immunology, Wilhelmina Children's Hospital, Utrecht University Medical Center, Utrecht, The Netherlands
76 Department of Laboratory Medicine, Boston Children's Hospital, Harvard Medical School, Boston, MA
81 Laboratory of Clinical Immunology and Microbiology, Division of Intramural Research, National Institute of Allergy and Infectious Diseases, National Institutes of Health, Bethesda, MD
9Research Associate, Immunopathogenesis Section, Laboratory of Clinical Infectious Diseases, National Institute of Allergy and Infectious Diseases, National Institutes of Health, Bethesda, MD, USA
107 Immunopathogenesis Section, Laboratory of Clinical Infectious Diseases, National Institute of Allergy and Infectious Diseases, National Institutes of Health, Bethesda, MD, USA
11Clinician, Head of Unit, San Raffaele Telethon Institute for Gene Therapy (SR-Tiget), Division of Regenerative Medicine, Stem Cells and Gene Therapy, San Raffaele Scientific Institute, Milan, Italy. 8 Milan Unit, Istituto di Ricerca Genetica e Biomedica, CNR, Milan, Italy
12Chief, Laboratory of Clinical Immunology and Microbiology, IDGS, DIR, NIAID, NIH, Bethesda, MD, USA
Hypomorphic Recombination Activating Gene 1 (RAG1) mutations result in residual T- and B-cell development in both humans and mice and have been found in patients presenting with delayed-onset combined immune deficiency with granulomas and/or autoimmunity (CID-G/AI). Recent studies have shed light on how hypomorphic RAG1 mutations alter the primary repertoire of T and B cells, but less is known about their effect on immune dysregulation in targeted organs. In order to investigate the role of these mutations in determining intestinal disease, we set out to evaluate gut immunity and microbiota interplay in Rag1 mutant hypomorphic mice.
We evaluated two mouse models carrying homozygous Rag1 mutations (R972Q and R972W), corresponding to human mutations (R975Q and R975W, respectively) described in patients with CID-G/AI. Both mutations fall in the coding flanksensitive region of the RAG1 C-terminal domain. On the basis of aminoacid properties and in vitro studies, the R972Q mutation has demonstrated a moderate effect on Rag1 protein stability while the R972W mutation resulted highly disruptive.
Analysis of intestinal pathology in Rag1 mutant mice (NIAID animal protocol LCIM 6E) revealed different degrees of spontaneous colitis, with the most severe inflammatory infiltrate observed in mice carrying the most disruptive mutation, R972W. Colonic inflammation was characterized by crypt elongation, epithelial hyperplasia, and an abundant inflammatory infiltrate extending to the colonic lamina propria, with occasional crypt abscesses. A significant increase in activated CD44hiCD62LCD4+ T cells expressing the gut homing receptor 47 was observed in mesenteric lymph nodes (MLNs) of both mutant strains, and was especially prominent in R972W mutant mice. Additionally, the proportion of MLN CD4+ T regulatory (Treg) cells was increased in both mouse models. Finally, MLN of mutant mice contained a high number of myeloid cells (CD11b+) along with a decreased number of B220+ B cells, and these abnormalities were also more prominent in R972W than in R972Q mice.
In summary, we have shown that Rag1 mutant hypomorphic mice present with different degrees of inflammatory bowel disease, with the mouse model carrying the most disruptive mutation presenting with the most severe phenotype. We are currently performing studies to evaluate the impact of Rag1 mutations on microbiome composition and diversity in these mouse models of CID-G/AI.
(67) Submission ID#600337
Immunomodulatory Effects of Immunoglobulin Replacement Therapy on T-cells in Patients with Hypogammaglobulinemia
Tri M. Dinh, BSc1, Jun Oh, MSc2, Bill Cameron, MD, FRCPC, FACP3, Seung-Hwan Lee, PhD4, Juthaporn Cowan, MD, PhD, FRCPC5
1Honour's Research Student Associate, University of Ottawa
2PhD Candidate, University of Ottawa
3Medical Director for Clinical Research, Ottawa Health Research Institute
4Associate Professor, University of Ottawa
5Associate Scientist and Assistant Professor, University of Ottawa, Ottawa Health Research Institute
Background: Hypogammaglobulinemia or low serum immunoglobulin G (IgG) levels either inherited (primary) or acquired (secondary) is associated with increased infection rates. Primary (1°) hypogammaglobluinemia can be caused by many primary immune deficiencies (PID) including combined variable immune deficiency (CVID), while secondary (2°) hypogammaglobluinemia can be caused by many acquired conditions such as lymphomas, leukemias, or chemotherapies and other immunosuppressive drugs. Immunoglobulin replacement therapy (IRT) has been the mainstay of treatment in patients with hypogammaglobulinemia by reducing infection through replenishing the quantitative IgG. There are other applications of Ig therapy such as in autoimmune diseases, where the mechanism of action is thought to be Ig mediated immunomodulation. Innate immune cells have shown to be involved in such mechanism, but whether IRT modulates adaptive immune cells in patients with hypogammaglobulinemia is not well known.
Hypothesis: IRT has an immunomodulatory effect on T-cell function and proliferation in patients with hypogammaglobulinemia.
Methods: Blood from thirty patients with 1°(n=12) or 2° (n=18) hypogammaglobulinemia recruited from the Immunodeficiency Clinic at the Ottawa Hospital was drawn for peripheral blood mononuclear cell (PBMC) isolation, before starting IRT and minimum 8 weeks after starting IRT. Data regarding IgG level, number and type of infections after receiving IRT was collected. PBMCs were analyzed using flow cytometry for quantitation of T-cell subset. Cultured and anti-CD3/CD28 stimulated PBMC were also analyzed for extracellular and intracellular cytokine production, measured by ELISA and flow cytometry, respectively. Combined Cytomegalovirus, Epstein-Barr Virus and Influenza virus (CEF) peptides were used to study specific T-cell responses. Anti-CD3/CD28 stimulated PBMC were used for CellTrace T-cell proliferation assays. Data was grouped based on nature of hypogammaglobulinemia i.e. 1° or 2°. Results were compared between before and after IRT using Wilcoxon matched-pairs signed rank test.
Results: IRT was not found to significantly alter proportion of Treg, CD4+, or CD8+ T-cell populations or activation state as measured by CD45RA/R0 expression. However, IRT was found to significantly increase expression of intracellular IFN-y in CD4+ and CD8+ T-cells post-CD3/CD28 stimulation in 2° (p = 0.007), but not in 1° hypogammaglobulinemia patients. There was no change in extracellular IL-10 and IL-17 cytokine production in both groups. In contrast, CD8+ T-cells in 1° hypogammaglobulinemia patients showed significantly higher expression of intracellular IFN-y and TNF-a post-CEF viral peptide stimulation (p = 0.027). CD3+ and CD8+ T-cell proliferation after CD3/CD28 stimulation was found to be decreased after IRT for both groups (p = 0.025 & p = 0.049).
Conclusions: Our results suggest that IRT can alter CD4+ and CD8+ T-cell function with differential effect in patients with 1° or 2° hypogammaglobulinemia in addition to replenishing serum IgG level. More experiments assessing cytotoxicity of T-cells will be conducted to further study T-cell subset function as well as B-cell function. These laboratory results will be analyzed for association with clinical outcomes.
Uploaded File(s)
Uploads

(68) Submission ID#600349
Severe Congenital Neutropenia Caused by ELANE Gene Mutation in a Malaysian Girl
Siti Mardhiana Binti. Mohamad, MD, PhD1, Intan Juliana Abdul Hamid, MD, MMed, PhD2, Asrul Abdul Wahab, MD, MPath3, Adli Ali, MD,MMed4, Choo Chong Ming, MBBS, MMed5, Florence Bakon, MD, MMed6, Amir Hamzah Abdul Latiff, MBBS, MMed, MRCP, FACAAI, FAAAA7, Lokman Mohd Noh, MBBS, DCH,MRCP,FRCPE,Cert. Fellowship Immunology8
1Clinical Scientist, Primary Immunodeficiency Diseases Group, Regenerative Medicine Cluster, Institut Perubatan & Pergigian Termaju, Universiti Sains Malaysia
2Paediatric Immunologist, Primary Immunodeficiency Diseases Group, Regenerative Medicine Cluster, Institut Perubatan & Pergigian Termaju, Universiti Sains Malaysia
3Immunopathologist, Universiti Kebangsaan Malaysia, Kuala Lumpur, Malaysia
4Paediatrician, Universiti Kebangsaan Malaysia, Kuala Lumpur, Malaysia
5Paediatric Infectious Diseases, Hospital Sultan Abdul Halim, 08000 Sungai Petani, Kedah
6Paediatrician, Kuching Specialist Hospital, Kuching, Sarawak
7Clinical Immunologist and Allergy, Allergy and Immunology Centre, Pantai Hospital, Kuala Lumpur, Malaysia
8Paediatric Immunologist Consultant, Hospital Kuala Lumpur, Jalan Pahang, 50586 Kuala Lumpur, Wilayah Persekutuan
Background: Severe congenital neutropenia (SCN) is a rare immunodeficiency disorder characterised by the extremely low absolute neutrophils count (ANC) less than 0.5x109/L. The clinical feature of SCN is recurrent bacterial infections and the patients the risk of leukemia development. The incidence of SCN is estimated to be 1 in 200 000 individuals. Mutations in more than 20 genes have been described causing SCN and it is either recessive, dominant or X-linked inheritance.
Case presentation: We described an 11 years old Malaysian girl who presented with recurrent abscesses over the whole part of the body, recurrent oral candidiasis, growth failure and recurrent pneumonias since 4 months old. She also had history of a few episodes of acute tonsillitis, chronic suppurative otitis media and herpes zoster infections. Throughout her age, she had persistent neutropenia less than 0.5x109/L but in few occasions, her ANC elevated up to more than 1.0x109/L . She was treated as autoimmune neutropenia, respectively due to few positive results of autoimmunity workout such as antinuclear antibodies (ANA) and double stranded DNA (dsDNA) but eventually later to be negative. Later at the age 9 years old, whole exome sequencing was performed and confirmed by Sanger sequencing, found a heterozygous variant in ELANE gene(c.640G>T; p.Gly214Ter), an autosomal dominant which was described to cause SCN. Both parents do not carry this mutation, hence, it is a de novo mutation. Currently, she had few on and off recurrent infections. Despite that, she is relatively well and on prophylaxis antibiotic.
Conclusion: To our knowledge, we report for the first time a Malaysian girl with SCN, with confirmed mutational analysis of the ELANE gene. The delayed diagnosis might be due to the insufficient awareness of the phenotypic presentation of this rare disease. Moreover, the genetic analysis is not available in Malaysia and need to be done outside of the country. This case demonstrates the importance of the genetic analysis which may help in improving the diagnosis and management of the patient.
(69) Submission ID#600360
Autologous Ex Vivo Lentiviral Gene Therapy for the Treatment of Severe Combined Immune Deficiency Due to Adenosine Deaminase Deficiency
Donald B. Kohn, MD, MS, BS1, Kit L. Shaw, PhD2, Elizabeth K. Garabedian, MSLS, RN3, Denise A. Carbonaro-Sarracino, PhD4, Theodore B. Moore, MD5, Satiro De Oliveira, MD6, Gay M. Crooks, MBBS7, John Tse, PharmD8, Sally Shupien, BA9, Dayna Terrazas, RN10, Alejandra Davila, BS11, Amalia Icreverzi, PhD12, Allen Yu, BS11, Provaboti Barman, PhD13, Maritess Coronel, MS14, Beatriz Campo Fernandez, PhD, MSc, BSc15, Ruixue Zhang, Master16, Roger Hollis, PhD17, Chilenwa Uzowuru, MSc, BSc18, Hilory Ricketts19, Jinhua Xu-Bayford, Degree (graduate diploma)20, Valentina Trevisan, MD21, Serena Arduini, PhD22, Frances Lynn, MSc23, Mahesh Kudari, MBBS, MA24, Andrea Spezzi, MD, MBBS25, Lilith Reeves, MS, MT(ASCP)26, Kenneth Cornetta, MD27, Robert Sokolic, MD, FACP28, Roberta Parrott, BS29, Rebecca Buckley, MD30, Claire Booth, MBBS, PhD, MSc31, Fabio Candotti, MD, PhD32, Harry L. Malech, MD33, Adrian J. Thrasher, MBBS, PhD34, H Bobby Gaspar, MD, PhD35
1Professor of Microbiology, Immunology and Molecular Genetics (MIMG) and Pediatrics, University of California, Los Angeles
2Study Manager for Gene Therapy Clinical Trials, University of California, Los Angeles
3Research Nurse, Principal Investigator, National Genome Research Institute, National Institutes of Health
4Senior Scientist, University of California, Los Angeles; Orchard Therapeutics, Boston MA
5Professor of Pediatrics, Chief of Pediatric Hematology/Oncology and Director of the Pediatric Blood and Marrow Transplant Program at UCLA, University of California, Los Angeles
6Assistant Professor, Pediatrician and Cancer Immunotherapy Researcher, University of California, Los Angeles
7Professor, Pathology & Laboratory Medicine; Paediatric oncologist, Division of Stem Cell Transplantation and Regenerative Medicine, Department of Pediatrics, Stanford School of Medicine, Stanford, CA
8Clinical Pharmacist, University of California, Los Angeles
9Clinical Trials staff, University of California, Los Angeles
10Clinical Research Nurse Coordinator, University of California, Los Angeles
11SRAII, University of California, Los Angeles
12 Head of Clinical Manufacturing and Gene Therapy, University of California, Los Angeles
13Lead Manufacturing and QA at UCLA GMP (Human Gene and Cell Therapy Facility), University of California, Los Angeles
14Regulatory and data manager, University of California, Los Angeles
15Associate Project Scientist, University of California, Los Angeles
16SRA, University of California, Los Angeles
17Project Scientist VII, University of California, Los Angeles
18Clinical Trial coordinator, University College London; Great Ormond Street Hospital NHS Trust
19Data Manager, University College London; Great Ormond Street Hospital NHS Trust
20Gene Therapy and Immunology CNS Team lead, University College London; Great Ormond Street Hospital NHS Trust
21Clinical Research Fellow, University College London; Great Ormond Street Hospital NHS Trust
22Clinical Development Scientist, Orchard Therapeutics, London, UK
23Biostatistician, Orchard Therapeutics, London, UK
24Senior Director, Clinical Development, Orchard Therapeutics, London, UK
25Chief Medical Officer, Orchard Therapeutics, London, UK
26Assistant Professor; Translational Core Director, Cincinnati Childrens Hospital Medical Center
27Professor of Clinical Medical & Molecular Genetics, Indiana University School of Medicine
28Hematologist/Oncologist at Lifespan Cancer Institute, National Genome Research Institute, National Institutes of Health; Comprehensive Cancer Center at Rhode Island Hospital
29Research Associate, Duke University
30Sidbury Professor of Pediatrics, in the School of Medicine; Professor of Immunology, Duke University
31Consultant Paediatric Immunologist and senior clinical lecturer at Great Ormond Street Hospital for Children NHS Foundation Trust, University College London; Great Ormond Street Hospital NHS Trust
32Associate Professor of Medicine and Head Physician, Division of Immunology and Allergy; Director, Vaccine and Immunotherapy Center, University Hospital of Lausanne, Switzerland, National Genome Research Institute, National Institutes of Health; Current Address: Division of Immunology and Allergy, University Hospital of Lausanne, Lausanne, Switzerland
33Chief, Genetic Immunotherapy Section, Laboratory of Clinical Immunology and Microbiology, IDGS, DIR, NIAID, NIH, Bethesda, MD, USA
34Professor of Paediatric Immunology and Wellcome Trust Principal Research Fellow, University College London; Great Ormond Street Hospital NHS Trust
35Professor of Paediatrics and Immunology, University College London; Great Ormond Street Hospital NHS Trust; Orchard Therapeutics, London, UK
Background: ADA-SCID is a rare genetic disorder which causes severe combined immunodeficiency. Historically, ADA-SCID has been treated using enzyme replacement therapy (ERT) followed by allogeneic hematopoietic stem cell (HSC) transplant (HSCT) from a matched related donor (MRD) or, if none is identified, a non-MRD (matched/mismatched unrelated or mismatched related donor). We developed a self-inactivating lentiviral vector (LV), in which a codon optimized human ADA cDNA is driven by the short form of the elongation factor-1alpha (EFS) promoter (EFS-ADA LV). The drug product (OTL-101), composed of autologous HSCs transduced ex vivo with the EFS-ADA LV, was evaluated in a prospective, historically-controlled Phase I/II clinical trial in ADA-SCID pediatric subjects. We report safety and efficacy at 24 months in 20 ADA-SCID subjects treated with lentiviral gene therapy (GT) compared to a historical cohort of 26 ADA-SCID patients treated with HSCT.
Methods: Twenty subjects (9 male, 11 female; 4 mo 4.3 yrs) were treated with GT. Autologous CD34+ HSCs were isolated from bone marrow and pre-stimulated with cytokines before transduction with EFS-ADA LV. Busulfan was administered at a single dose (4 mg/kg) prior to infusion of OTL-101. The control group included 26 patients (0.2 mo 9.8 yrs) treated with allogeneic HSCT (MRDs n=12, non-MRDs n=14) at Great Ormond Street Hospital, UK (n=16) or Duke University Childrens Hospital, USA (n=10) between 20002016.
Results: At 24 months, overall survival (OS) and event-free survival (EvFS), defined as survival in the absence of ERT reinstitution or rescue allogeneic HSCT) were statistically significantly higher in the GT group compared with the HSCT group (Table). Successful engraftment of genetically modified HSC was observed in all GT subjects at 6 months, which persisted over 24 months, based on vector gene marking in granulocytes (median 0.085 copies/cell [range 0.04-2.50] at 24 months) and peripheral blood mononuclear cells (median 0.843 copies/cell [range 0.13-1.86] at 24 months), and was associated with increased red blood cell ADA enzyme activity and metabolic detoxification from deoxyadenosine nucleotides.
Over 24 months, none of the GT subjects required PEG-ADA ERT reinstitution and 90% were able to stop receiving immunoglobin replacement therapy (IgRT), whereas 38% HSCT patients required rescue HSCT or reinstitution of PEG-ADA ERT, and 52% were able to stop receiving IgRT (Table). Nine subjects in the GT group experienced a serious adverse event (SAE), most frequently infections and gastrointestinal events; only one was considered treatment-related. In the GT group, there were no events of autoimmunity during the study. Due to the autologous nature of the product, there was no incidence of graft vs host disease (GvHD) in the GT group; whereas 5 patients in the HSCT group experienced acute GvHD and 3 experienced chronic GvHD events, one of whom died.
Conclusions: Treatment with lentiviral GT for ADA-SCID is well tolerated and has a favorable benefit-risk profile at 24 months based on sustained gene correction and restoration of immune function, as well as improved OS and EvFS compared with HSCT (MRD or non-MRD) at 24 months.
Grant Support:
Supported by a research grant from the NIAID, NIH (U01 AI100801), the National Gene Vector Biorepository (5P40HL116242), the California Institute for Regenerative Medicine (CL1-00505-1.2, FA1-00613-1), Medical Research Council (MR/K015427/1), and the National Institute for Health Research Biomedical Research Centre at Great Ormond Street Hospital for Children NHS Foundation Trust and University College London.
(70) Submission ID#600370
Autologous ex vivo lentiviral gene therapy for the treatment of Severe Combined Immune Deficiency due to Adenosine Deaminase Deficiency (ADA-SCID) Improves B Cell Function
Donald B. Kohn1, Kit L. Shaw1, Elizabeth Garabedian2, Denise A. Carbonaro-Sarracino1,3, Theodore B. Moore1, Satiro De Oliveira1, Gay M. Crooks1, John Tse4, Sally Shupien1, Dayna Terrazas1, Alejandra Davila1, Amalia Icreverzi1, Allen Yu1, Provaboti Barman1, Maritess Coronel1, Beatriz Campo Fernandez1, Ruixue Zhang1, Roger Hollis1, Chilenwa Uzowuru5, Hilory Ricketts5, Jinhua Xu Bayford5, Valentina Trevisan5, Serena Arduini3, Frances Lynn3, Mahesh Kudari3, Andrea Spezzi3, Lilith Reeves6, Kenneth Cornetta7, Robert Sokolic2,*, Roberta Parrott8, Rebecca Buckley8, Claire Booth5, Fabio Candotti2,**, Harry Malech9, Adrian J. Thrasher5, and H. Bobby Gaspar3,5
1University of California, Los Angeles
2National Genome Research Institute, National Institutes of Health
3Orchard Therapeutics Limited
4Department of Pharmaceutical Services, Ronald Reagan Medical Center, UCLA
5University College London/Great Ormond Street Hospital
6Cincinnati Children’s Hospital Medical Center
7Indiana University School of Medicine
8Duke University
9National Institute of Allergy and Infectious Disease, National Institutes of Health
*Current Address: Comprehensive Cancer Center at Rhode Island Hospital
**Current Address: Division of Immunology and Allergy, University Hospital of Lausanne, Lausanne, Switzerland
Grant Support:
Supported by a research grant from the NIAID, NIH (U01 AI100801), the National Gene Vector Biorepository (5P40HL116242), the California Institute for Regenerative Medicine (CL1-00505-1.2, FA1-00613-1), Medical Research Council (MR/K015427/1), and the National Institute for Health Research Biomedical Research Centre at Great Ormond Street Hospital for Children NHS Foundation Trust and University College London.
Background: ADA-SCID is a rare genetic disorder that causes severe combined immunodeficiency, with minimal or absent B cell function. Prior to, and often after, treatment with allogeneic hematopoietic stem cell (HSC) transplant (HSCT) or autologous ex vivo HSC gene therapy (GT), patients are managed with enzyme replacement therapy (ERT) and immunoglobulin (Ig) replacement therapy (IgRT). We evaluated a GT treatment with autologous HSCs transduced ex vivo with a self-inactivating lentiviral vector (LV), in which a codon optimized human ADA cDNA is driven by an internal short form of the elongation factor-1alpha (EFS) promoter (“EFS-ADA LV”). At 24 months follow-up, 20 pediatric ADA-SCID subjects treated with GT were compared to a historical cohort of 26 ADA-SCID patients treated with HSCT. Here, we report on B cell reconstitution in these cohorts.
Methods: Twenty subjects (9 male, 11 female) aged 4 mo – 4.3 yrs received GT. Autologous CD34+ HSCs were isolated from bone marrow and pre-stimulated with cytokines before transduction with EFS-ADA LV. Genetically modified cells were administered after conditioning with single dose busulfan (4 mg/kg). The control group included 26 patients aged 0.2 mo to 9.8 yrs treated with HSCT at Great Ormond Street Hospital (UK) (n=16) or Duke University Children’s Hospital (US) (n=10) between 2000 - 2016. The HSCT patients received an allogeneic transplant from matched related donors (MRDs) (n=12) or non-MRDs (n=14). Subjects continued to receive IgRT post-GT until a clinical decision was made to stop, factoring in B cell reconstitution, general medical condition and seasonal infections.
Results: By Month 12, in the GT group, 45% had stopped treatment with IgRT compared to 38% in the HSCT group overall. By Months 18 and 24, higher proportions of GT-treated subjects had stopped IgRT (70% and 90%, respectively) compared with MRD HSCT patients (55% and 70%, respectively) and non-MRD HSCT patients (42% at both timepoints) (Table).
In the GT group, vector gene marking was detectable in peripheral blood mononuclear cells within 3 months and persisted at 24 months post-infusion (median 0.843 copies/cell [range 0.13-1.86]), suggesting successful gene modification. As evidence of B cell reconstitution, IgA and IgM levels in peripheral blood sera more than doubled by 18 months, from 18.5 mg/dL (range 8 to 95) to 48.0 mg/dL (range 20 to 110) and 32.5 mg/dL (range 16 to 107) to 69.0 mg/dL (range 20 to 180), respectively. Additionally, antibody response following tetanus vaccination, was evaluated in 3 subjects. All 3 subjects mounted a protective response to the vaccine (median antibody response 3.2 IU/mL [range 0.1 to 3.5]), based on a normal threshold of 0.01 IU/mL (Hammarlund Clin Infect Dis 2016) and a laboratory reference range (0.10 to 2.9 IU/mL).
Conclusions: GT with autologous HSCs transduced ex vivo with EFS-ADA LV resulted in B cell reconstitution, as evidenced by doubled IgA and IgM production at 18 months, cessation of IgRT in 90% of patients by 24 months, and protective specific antibody responses to tetanus vaccine in patients that were evaluated.
(71) Submission ID#600374
Lentiviral Vector Gene Therapy for X-linked Chronic Granulomatous Disease Corrects Neutrophil Function
Harry L. Malech, MD1, Claire Booth, MBBS, PhD, MSc2, Elizabeth M. Kang, MD3, Sung-Yun Pai, MD4, Kit L. Shaw, PhD5, Giorgia Santilli, PhD6, Myriam Armant, PhD7, Karen F. Buckland, PhD, BSc8, Uimook Choi, PhD, BSc9, Suk See De Ravin, MD, PhD10, Morna J. Dorsey, MD, MMSc11, Caroline Y. Kuo, MD12, Diego Leon-Rico, PhD, MS, BSc13, Christine Rivat, PhD8, Katie Snell, Dip (children's nursing), BSc14, Jinhua Xu-Bayford, Degree (graduate diploma)15, Emma C. Morris, MB BChir, MA, PhD, MRCP, FRCPath16, Dayna Terrazas, RN17, Leo D. Wang, MD, PhD18, Geraldine Honnet, MD19, Peter Newburger, MD, BA20, Frederic D. Bushman, PhD, BA21, Manuel Grez, PhD, BSc22, H Bobby Gaspar, MD, PhD23, David A. Williams, MD24, Anne Galy, PhD25, Donald B. Kohn, MD, MS, BS26, Adrian J. Thrasher, MBBS, PhD27
1Chief, Genetic Immunotherapy Section, Laboratory of Clinical Immunology and Microbiology, IDGS, DIR, NIAID, NIH, Bethesda, MD, USA
2Consultant Paediatric Immunologist and senior clinical lecturer at Great Ormond Street Hospital for Children NHS Foundation Trust, University College London; Great Ormond Street Hospital NHS Trust
3Staff Clinician and Chief of the Hematotherapeutics Unit of Genetic Immunotherapy Section, National Institute of Allergy and Infectious Diseases, National Institutes of Health
4Associate Professor of Pediatrics, Harvard Medical School, Boston Childrens Hospital, Harvard Medical School
5Study Manager for Gene Therapy Clinical Trials, University of California, Los Angeles
6Non-clinical lecturer in gene therapy, University College London; Great Ormond Street Hospital NHS Trust
7Instructor in Pediatrics, Boston Childrens Hospital, Harvard Medical School
8Healthcare Scientist, University College London; Great Ormond Street Hospital NHS Trust
9Staff Scientist, National Institute of Allergy and Infectious Diseases, National Institutes of Health
10Clinician, Laboratory of Clinical Immunology and Microbiology, IDGS, DIR, NIAID, NIH, Bethesda, MD, USA
11Pediatric Immunologist and Allergist, Department of Pediatrics, Division of Allergy, Immunology, and Bone Marrow Transplant, University of California San Francisco, San Francisco, CA
12Assistant Clinical Professor of Pediatrics, University of California, Los Angeles
13Senior Research Associate, University College London; Great Ormond Street Hospital NHS Trust
14Lead gene therapy research nurse, University College London; Great Ormond Street Hospital NHS Trust
15Gene Therapy and Immunology CNS Team lead, University College London; Great Ormond Street Hospital NHS Trust
16Haematologist and Professor of Haematology, University College London; Great Ormond Street Hospital NHS Trust
17Clinical Research Nurse Coordinator, University of California, Los Angeles
18Assistant Professor, Department of Immuno-Oncology, City of Hope National Medical Center
19Director of Development, Genethon, Evry, France
20Professor of Hematology, University of Massachusetts, Worcester
21Professor of Microbiology, University of Pennsylvania
22Research Group Leader, Georg-Speyer Haus, Frankfurt, Germany
23Professor of Paediatrics and Immunology, University College London; Great Ormond Street Hospital NHS Trust; Orchard Therapeutics, London, UK
24President, Dana-Farber/Boston Children's Cancer and Blood Disorders Center, Boston Childrens Hospital, Harvard Medical School
25Head of Inserm Unit 951, Genethon, Evry, France
26Professor of Microbiology, Immunology and Molecular Genetics (MIMG) and Pediatrics, University of California, Los Angeles
27Professor of Paediatric Immunology and Wellcome Trust Principal Research Fellow, University College London; Great Ormond Street Hospital NHS Trust
Background: X-linked Chronic Granulomatous Disease (XCGD) results from mutations in CYBB encoding the gp91phox subunit of phagocyte NADPH-oxidase. Attempts to treat XCGD with gene therapy (GT) using transduced autologous hematopoietic stem cells (HSC) transduced ex vivo with a gammaretroviral vector have met with limited efficacy due to transient engraftment of gene corrected HSCs, gene silencing, and vector insertion-mediated activation of oncogenes leading to myelodysplasia. We developed a novel self-inactivating (SIN) lentiviral vector (G1XCGD LV) with a chimeric cathepsin G/cFes myeloid-specific promoter driving gp91phox expression from a codon optimized cDNA. Following transplant of G1XCGD LV ex vivo transduced autologous HSCs into busulfan-conditioned XCGD patients, there was long-term restoration of oxidase activity in peripheral blood polymorphonuclear neutrophils (PMN) at 12 months in 6 of 9 severely affected XCGD patients without evidence of genotoxicity. Here we present data about the multiple assays used to assess quality and quantity of restoration of PMN oxidase activity.
Methods: Similar trials of GT with G1XCGD LV were initiated in the UK (n=3, plus 1 compassionate use patient) and USA (n=5). All patients had histories of inflammatory disease and severe, persistent infections (some non-responsive to conventional therapy at time of GT). G-CSF plus Plerixafor-mobilized CD34+ HSCs were transduced with ex vivo G1XCGDLV. Subjects received myeloablative conditioning with single-agent busulfan, targeted to net area-under-the-curve of 70,000 ng/mL*hr. Freshly prepared or cryopreserved quality-tested genetically-modified HSC, manufactured on-site, were administered intravenously. PMN oxidase activity post-GT was assessed by p-nitroblue tetrazolium (NBT) reduction, dihydrorhodamine (DHR) flow cytometry assay, and quantitative Ferricytochrome C Assay (FerriC) measurement of superoxide generation.
Results: We report results for 7 patients (aged 2-27 years) with 1-2.5 years of follow-up; two additional patients were treated but died within three months of GT from complications deemed related to pre-existing disease-related co-morbidities (severe pulmonary disease and anti-platelet antibodies). Within 1 month post-GT, oxidase (+) PMN were present in peripheral blood based on NBT testing and DHR flow cytometry. Expression of the corrective transgene was confirmed by flow cytometry using antibody detection of gp91phox. Quantitative biochemical measurements of oxidase activity were also confirmed in some samples using the FerriC assay, demonstrating quantitative levels of superoxide production per corrected cell that were within the normal range. Functional testing of oxidase burst activity using DHR fluorescent assays was applied serially to follow levels of corrected PMN where oxidase activity per corrected cell also were in the normal range. All patients had >15% PMN DHR+ within one month, which remained stable for most patients over the follow-up period (Figure). Follow-up demonstrated sustained stable persistence of 12-46% oxidase burst positive neutrophils in 6 of 7 surviving subjects at 12 months, with restoration to clinically beneficial levels (defined as 10% of PMN being DHR+) in these patients as of December 2018.
Conclusion: These results demonstrate corrected PMN function within 1 month in X-CGD patients treated with autologous GT. PMN oxidase activity was sustained at levels which restore biochemical function and provide clinically beneficial levels of immunity for 12 months in 6/7 patients.
Grant Support:
Supported by research grants from the: California Institute of Regenerative Medicine (CLIN2-08231; FA1-00613-1), the Gene Therapy Resource Program from NHLBI, NIH (CRB-SSS-S-15-004351 1840), the
NIAID Intramural Program, NET4CGD (FP7 EU grant agreement no. 305011), the Wellcome Trust (104807/Z/14/Z), and the National Institute for Health Research Biomedical Research Centre at Great Ormond Street Hospital for Children NHS Foundation Trust and University College London.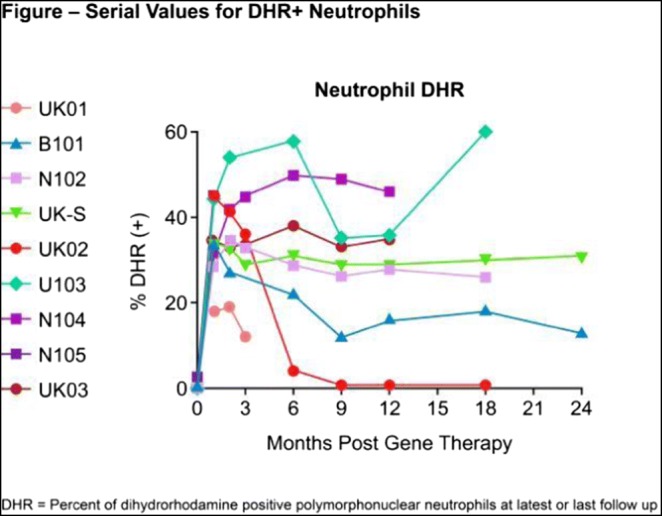
(72) Submission ID#600426
A New High Concentration Immunoglobulin Product for Subcutaneous Administration (IGSC 20%)
William Alonso1, John Lang2, Pete Vandeberg3
1Principe Process Development Scientist II, Grifols Bioscience Research Group
2Principle Research Scientist I, Grifols Bioscience Research Group
3R&D Program Director, Grifols Bioscience Research Group
Introduction/Background: Grifols has developed a new 20% immunoglobulin liquid product for subcutaneous administration (IGSC 20%). The formulation for IGSC 20% was developed based on the knowledge acquired from the formulation of Grifols currently licensed 10% Immune Globulin (Human), Gamunex®-C; however, the protein concentration was increased from 10% to 20% to facilitate efficient subcutaneous administration. Gamunex-C has an extensive record of safety and tolerability when administered intravenously and subcutaneously for greater than 15 years in diverse patient populations. The IGSC 20% manufacturing process employs the same purification steps as Gamunex-C and was demonstrated to be robust and to provide an IgG product with the required potency, purity, and quality. The formulation excipient characteristics and compatibility with the drug product have been well established. Glycine has been an excipient of Intramuscular Immune Globulin (Human) for fifty years and Intravenous Immune Globulin (IGIV) for over twenty years. The IGSC 20% formulation has low buffering capacity, and a low pH was selected to achieve a product with low aggregates, low fragments and viscosity suitable for subcutaneous administration. To improve visual clarity, the IGSC 20% formulation contains a small amount of polysorbate 80 (PS80), which is widely used in biopharmaceutical products. Subcutaneous administration of the IGSC 20% formulation has been well tolerated in clinical studies.
Objectives: The goal was to provide the PID population with a new 20% immunoglobulin liquid product for subcutaneous administration (IGSC 20%).
Methods: IGSC 20% is manufactured using the current manufacturing process for Gamunex-C, followed by an additional concentration step so that the product can be formulated at a higher protein concentration. IGSC 20% and Gamunex-C batches were produced at full industrial scale and then subjected to a series of analytical testing including assessment of purity, composition and neutralizing activity.
Results: The IGSC 20% and Gamunex-C manufacturing processes and formulations have preserved the IgG integrity, molecular characteristics and potency. The manufacturing processes have eliminated lipids, alcohols, and acetate and coagulation factor impurities, including FXIa, which were undetectable by either specific or global methods. The IGSC 20% and Gamunex-C batches were 100% gamma globulin by agarose membrane electrophoresis, and have a subclass distribution similar to normal plasma and acceptable specific antibody content. IGSC 20% was shown to be primarily monomer plus dimer IgG (99±1%) with minimal aggregate or fragment, which confirms that appropriately gentle processing conditions were used during the concentration of 10% IgG solutions to 20% IgG.
Conclusions: IGSC 20% is a highly concentrated IgG solution with characteristics comparable to Gamunex-C, but with twice the IgG concentration in order to facilitate subcutaneous administration with reduced volumes and shorter infusion times. Analytical testing demonstrates suitable potency, purity, and neutralizing activity for a number of specific antigens.
Funding: This study was funded and conducted by Grifols, a manufacturer of 20% immunoglobulin for subcutaneous administration.
Disclosure: All authors are employees of Grifols.
(73) Submission ID#600434
Miller-Dieker Syndrome May Be Another Syndromic Primary Immunodeficiency
Erika Tsutsui, MD1, Deepti Deshpande, MD, MPH2, Yesim Demirdag, MD3
1Resident, The University of Tokyo Hospital
2Fellow, Division of Allergy, Immunology and Rheumatology, Department of Pediatrics, Columbia University Medical Center
3Faculty, Division of Allergy, Immunology and Rheumatology, Department of Pediatrics, Columbia University Medical Center
INTRODUCTION: Miller-Dieker Syndrome (MDS) is a contiguous gene deletion on chromosome 17p13.3, characterized by lissencephaly, distinctive facial features and severe intellectual disability and seizures. Frequent respiratory tract infections and seizures cause recurrent hospitalizations in these children and are typically considered a result of neurological impairment and poor airway clearance. Evaluation of these patients for immunodeficiency is not a common clinical practice. Here we report combined immune deficiency in 2 patients with MDS and recurrent respiratory tract infections.
CASE PRESENTATION
Case 1: A boy with MDS was initially referred at age 2 months for an abnormal newborn screen with low T cell receptor excision circles (TREC) for severe combined immunodeficiency (SCID). Initial evaluation revealed moderate CD3+ and CD4+ T cell lymphopenia (figure 1). Initial immunoglobulins levels were normal. He was placed on anti-seizure medications. He later developed recurrent and severe respiratory tract infections starting in infancy. At 12 months of age, he developed hypogammaglobulinemia (figure 2). In addition, T cell counts progressively decreased and stayed around 600 cells/ul. Immunoglobulin replacement therapy started at 18 months of age. Hospitalizations due to respiratory tract infections significantly decreased.
Case 2: A 3-year-old boy with MDS had recurrent bacterial and viral respiratory infections which required numerous hospitalizations including intensive care unit stays. Newborn screening for SCID was negative. He had been on anti-seizure medications. Immunologic evaluation at 3 years of age revealed low total CD3+ cells and CD8+ T cells (CD3+: 1284cells/uL[normal range 1400-3700cells/uL], CD8+:278cells/uL[normal range 490-1300cells/uL]), hypogammaglobinemia (IgG: 252mg/dL[normal range 453-916mg/dL]), and non-protective IgG levels to tetanus, varicella and pneumococcus serotypes. Immunoglobulin replacement therapy started at 3 years of age which resulted in reduced frequency and severity of respiratory infections, and improved quality of life.
DISCUSSIONS: T cell lymphopenia and hypogammaglobulinemia were seen in both our cases of Miller-Dieker Syndrome. To our knowledge, immune deficiency has never been reported in MDS. One of our cases suggests that low T cell counts may start as early as at birth and may be detected by newborn screening. Hypogammaglobulinemia may be primary or secondary due to antiepileptics. Both children had reduced frequency and severity of respiratory infections and improved quality of life after immunoglobulin replacement highlighting the importance of screening and early management of immunodeficiency.
CONCLUSION: Miller-Dieker Syndrome is likely another syndromic primary immune deficiency disorder. A high index of suspicion with early screening and management of immunodeficiency may be beneficial for children with Miller-Dieker Syndrome.
Uploaded File(s)
Uploads

(74) Submission ID#600442
Safety and Pharmacokinetics of IGSC 20% in Subjects with Primary Immunodeficiency in an Open-label, Multicenter, Phase 3 Study
John Sleasman, MD1, Amy Darter, MD2, William Lumry, MD3, Iftikhar Hussain, MD4, H. James Wedner, MD5, James Harris, III, MD6, Kecia Courtney7, Elsa Mondou, MD8, Jiang Lin, PhD9, Mark R. Stein, MD10
1Professor of Pediatrics, Division of Allergy, Immunology, and Pulmonary Medicine, Duke University School of Medicine
2Physician, Oklahoma Institute of Allergy & Asthma Clinical Research, LLC
3Physician, Allergy and Asthma Specialists
4President and Principal Investigator, Vital Prospects Clinical Research Institute, P.C., and Allergy, Asthma and Immunology Center, P.C.
5Chief, Division of Allergy and Immunology, Washington University Physicians, Washington University School of Medicine in St. Louis
6Physician, Allergy & Immunology, The South Bend Clinic Center for Research
7Director, Clinical Development, Grifols Bioscience Research Group
8Medical Director II, Grifols Bioscience Research Group
9Biostatistician III, Grifols Bioscience Research Group
10Physician, Allergy & immunology, Allergy Section, Good Samaritan Medical Center, West Palm Beach, FL, USA
This prospective, multi-center, open-label study assessed the pharmacokinetic (PK), safety, and tolerability of Immune Globulin Subcutaneous (Human), 20% Caprylate/Chromatography Purified (IGSC 20%) in subjects with primary immunodeficiency (PI). The objectives were to determine a weekly subcutaneous (SC) dose of IGSC 20% that is noninferior to the intravenous (IV) dose of Immune Globulin Injection (Human), 10% Caprylate/Chromatography Purified (IGIV-C 10%) and to determine the steady state trough IgG levels after IGSC 20% and IGIV-C 10% infusions. There were 3 possible phases. If not on a qualifying IgG regimen at enrollment, subjects (n=44) were required to enter the Run-In Phase, receiving IGIV-C 10% to achieve steady-state before entering the IV Phase to determine steady-state area-under-the-curve (AUC) of IV infusions. Subjects with a qualifying IGIV-C 10% regimen (300-800 mg/kg) (n=9) directly entered the IV Phase for steady-state IV PK assessments. Upon completion of the IV PK assessments subjects entered the SC Phase, receiving weekly doses of IGSC 20% for up to24 weeks, with steady-state AUC determined at the 13th dose. IGSC 20% was not associated with any reports of serious local infusion site reactions (ISRs). The majority of local ISRs were mild-to-moderate. IGSC 20% (at a dose conversion factor of 1.37) provided equivalent exposure to IGIV-C 10% as assessed by steady-state AUC0-7 days, with 33% higher mean IgG trough values, lower fluctuations in IgG concentrations and the flexibility of at home administration. IGSC 20% was well tolerated with a safety profile comparable to IGIV-C 10%.
ClinicalTrials.gov Identifier: NCT02604810
Disclosure: Kecia Courtney, Elsa Mondou, and Jiang Lin are employees of Grifols, a manufacturer of IGSC 20%. Grifols is the sponsor of this study.
(75) Submission ID#600556
Deficiency of Adenosine Deaminase 2: An Expanding Spectrum of Disease
Jenna Bergerson, MD/MPH1, Karyl Barron, MD2, Deborah Stone, MD3, Patrycja Hoffmann, MSN, FNP4, Natalia Sampaio Moura, BS5, Oskar Schnappauf, PhD6, Ivona Aksentijevich, MD7, Daniel Kastner, MD, PhD8, Amanda Ombrello, MD3
1Staff Clinician, Laboratory of Clinical Immunology and Microbiology, NIAID, NIH, Bethesda, MD, USA
2Deputy Director, DIR, NIAID
3Staff Clinician, NIH/NHGRI/Inflammatory Disease Section
4Commissioned Corps, NIH/NHGRI/Inflammatory Disease Section
5Post Baccalaureate IRTA, NIH/NHGRI/Inflammatory Disease Section
6Postdoctoral Fellow, NIH/NHGRI/Inflammatory Disease Section
7Staff Scientist, NIH/NHGRI/Inflammatory Disease Section
8Scientific Director, NHGRI, NIH/NHGRI/Division of Intramural Research
Background: In 2014 two reports described the deficiency of adenosine deaminase 2 (DADA2) as early-onset lacunar strokes, intermittent fevers, livedoid rash, and early onset polyarteritis nodosa (PAN). Since these first reports, the clinical spectrum has dramatically expanded to include antibody deficiency, liver disease, vasculopathy, pure red cell aplasia, cytopenias, and lymphoproliferative disease.
Methods: Forty-two patients were enrolled in an IRB approved study at the NIH. Sequencing of ADA2, the gene encoding adenosine deaminase 2 (ADA2), was performed in all patients. Information was obtained by chart review of all clinical, serologic, and radiographic testing.
Results: All 42 patients had germline biallelic loss of function mutations in ADA2, leading to absent or significantly decreased protein expression and function of ADA2. The cohort comprises 20 females (48%) and 22 males (52%). There were 6 sibling pairs and 2 families with 3 affected individuals. Twenty-seven patients had a history of at least one ischemic stroke and 6 experienced a hemorrhagic stroke. The average age at the time of first stroke is 5.6 years (range 4 months - 24 years), and the average number of strokes is 3 (range 1-11). No new strokes have occurred in patients on anti-TNF therapy. Skin manifestations occurred in 86% of patients and include livedo (74%), cutaneous vasculitis resembling PAN (64%), and Raynauds (19%). Hepatomegaly (43%) and splenomegaly (55%) were also notable. Portal hypertension was observed in 6 (14%) patients, with 1 patient requiring a spleno-renal shunt for a massive variceal bleed. Abdominal MRA revealed arteritis and aneurysm in 7/13 patients evaluated; 3 patients developed bowel necrosis. Peripheral vasculopathy was seen in 3 patients, with one requiring amputation of gangrenous digits.
The most common immune abnormality seen in this cohort is hypogammaglobulinemia (62%); 20 patients have low IgG, 20 patients have low IgM, and 14 patients have low IgA. Ten of these patients are on immunoglobulin replacement. Specific antibody responses to vaccines were inadequate in 5/16 patients challenged. Lymphocyte phenotyping revealed decreased class-switched memory B cells in 23/32 patients (72%) tested. However, there was no relationship between absolute number of class switched memory B cells and hypogammaglobulinemia or infection frequency. Hematologic abnormalities include transfusion depended anemia (7%), neutropenia (7%), lymphopenia (5%), and thrombocytopenia (2%). Seven patients developed pancytopenia, 1 presented with pure red cell aplasia, and 1 developed aplastic anemia. Three patients have undergone bone marrow transplant, with two of those patients requiring a second transplant for graft failure.
Conclusions: The spectrum of DADA2 has expanded from strokes, intermittent fever, and cutaneous manifestations to include portal and systemic hypertension, immune deficiency, cytopenias, vascular abnormalities, and bone marrow failure. While initiation of anti-TNF therapy improves inflammatory markers, and no new strokes have occurred while on therapy, cytopenias do not seem to improve. Bone marrow transplantation should be considered in patients with findings of bone marrow failure, although transplant of our patients has been complicated by immune mediated neutropenia. Disease manifestations are heterogenous, making a comprehensive evaluation critical to our understanding of this disease.
(76) Submission ID#600606
Guidance for the Care of Patients Undergoing Cultured Thymus Tissue Transplantation (RVT-802)
Stephanie E. Gupton, MSN, CPNP1, Elizabeth A. McCarthy, RN, MSN2, Mary Louise. Markert, MD, PhD3
1Nurse Practitioner, Department of Pediatrics, Division of Allergy, Immunology and Pulmonary, Duke University Medical Center
2Research Program Leader, Sr., Department of Pediatrics, Division of Allergy, Immunology and Pulmonary, Duke University Medical Center
3Professor of Pediatrics and Immunology, Department of Pediatrics, Division of Allergy, Immunology and Pulmonary, Duke University Medical Center
Cultured thymus tissue transplantation (RVT-802) is an investigational therapy used to treat athymia or other conditions with severely diminished thymic function. Since 1993, 97 transplants of RVT-802 have been performed under the direction of Dr. M. Louise Markert. The overall survival rate after RVT-802 is 71% with most deaths secondary to pre-existing infections, cardiac defects and/or respiratory conditions. With the advent of widespread newborn screening for primary immunodeficiency, the average number of patients referred for RVT-802 implantation is 18 per year. Given the increase in neonatal diagnosis of athymia, clinical care is provided by the referring medical centers prior to RVT-802 implantation and patients return to the referring centers earlier after RVT-802. This creates the need for clear, concise guidelines for the care of these patients.
Primary goals of pre-transplantation clinical care are (1) management of pre-existing medical needs such as feeding difficulties, airway obstruction, congenital cardiac defects and developmental disabilities; (2) management of symptoms related to oligoclonal recipient T cell expansion (autologous GVHD/atypical complete DiGeorge anomaly) and (3) prevention of infections. Most deaths in the pre and early post-transplantation period are secondary to pre-existing infections. Necessary surgical and medical procedures (ie cardiac surgery, hearing aids) should not be delayed.
For the first 6 to 9 months after RVT802, patients have profoundly low naïve T cell numbers and may require immunosuppression to prevent rejection of RVT-802 by oligoclonal recipient T cells. Immunosuppression needs to be closely monitored and titrated for desired effect while minimizing side effects such as renal toxicity, electrolyte abnormalities and hypertension. T cell counts should be performed every 3 months and are used to guide weaning of immunosuppression. Most patients with successful transplants develop greater than 100/mm3 naïve T cells by 12 months post RVT-802. Infection prevention, clinical stability and optimal nutrition are critical for lasting engraftment. Clinical guidelines have been developed to address immunosuppression, management of autologous GVHD symptoms (gut, skin and liver), preservation of renal function, and developmental considerations.
After the development of naïve T cells, patients should continue to be monitored regularly by an immunologist. Patients may develop autoimmune complications such as thyroid disease and transient cytopenias. While risk of complications related to viral infections is greatly decreased after development of naïve T cells, patients with comorbidities (central venous access device dependence, tracheostomy, chronic lung disease) continue to require complex care from multidisciplinary teams. Medical conditions associated with athymia but not alleviated by thymus transplantation, such as hypoparathyroidism or cardiac defects, may require lifelong medical care. Lastly, patients must be evaluated for readiness for killed and live vaccines.
Transplant outcomes are influenced by the clinical condition at the time of RVT-802 implantation and optimization of immunosuppression, nutrition and clinical stability in the first 9 months following RVT-802. Clinical care that maintains a well-nourished, clinically stable, infection free patient yields the best chance for successful T cell development. Guidance documents supporting these goals ensure patients are best prepared to receive RVT-802 and develop long lasting thymic function.
(77) Submission ID#600641
Severe Inflammatory Episodes Associated with COG4-Congenital Disorder of Glycosylation (CDG-IIj) Presenting as Hemophagocytic Lymphohistiocystis (HLH)
Jeffrey Lo, MD1, William J. Brucker, MD, PhD2, John Prensner, MD, PhD3, Anita Pai, MD4, Mary Beth Son, MD5, Christine K. Lee, MD6, Matthew M. Heeney, MD7, Olaf Bodamer, MD, PhD8, Christina S.K. Yee, MD, PhD9
1Pediatric Fellow, Division of Immunology, Boston Childrens Hospital/Harvard Medical School
2Pediatric Fellow, Division of Genetics and Genomics, Boston Childrens Hospital/Harvard Medical School
3Pediatric Fellow, Division of Hematology/Oncology, Dana-Farber/Boston Children's Cancer and Blood Disorders Center/Harvard Medical School
4Pediatric Fellow, Division of Gastroenterology, Hepatology, and Nutrition, Boston Childrens Hospital/Harvard Medical School
5Assistant Professor of Pediatrics, Program Director Pediatric Rheumatology, Division of Immunology Boston Childrens Hospital/Harvard Medical School
6Instructor of Pediatrics, Division of Gastroenterology, Hepatology, and Nutrition, Boston Childrens Hospital/Harvard Medical School
7Assistant Professor of Pediatrics, Associate Chief Hematology, Dana-Farber/Boston Children's Cancer and Blood Disorders Center/Harvard Medical School
8Associate Professor of Pediatrics, Associate Chief of Genetics and Genomics, Division of Genetics and Genomics, Boston Childrens Hospital/Harvard Medical School
9Instructor of Pediatrics, Division of Immunology, Boston Childrens Hospital/Harvard Medical School
Hemophagocytic lymphohistiocytosis (HLH) is a life-threatening disease of immune dysregulation characterized by unchecked inflammatory responses leading to end-organ dysfunction. Primary HLH results from inherited mutations that impair capacity for immune regulation whereas secondary HLH arises from inappropriate response to an immune stimulus such as infection, malignancy or autoimmunity. We report a 9-month-old male who presented with symptoms of HLH as an initial manifestation of congenital disorder of glycosylation (CDG) due to mutations in the gene Component of Oligomeric Golgi Complex 4 (COG4) resulting in COG4-CDG (CDG-IIj).
A 9-month-old male with history of mild motor delay presented with 3 days of fever, vomiting, and diarrhea. Initial evaluation identified highly elevated ferritin and triglycerides, transaminitis, coagulopathy, and hyperammonemia. He subsequently developed generalized seizures. Liver and bone marrow biopsies demonstrated erythrophagocytosis consistent with HLH. Immunologic evaluation was notable for mild hypogammaglobulinemia, neutropenia, thrombocytopenia, and anemia. Serum CD25 levels and NK functional studies were later found to be normal.
The patient was initially treated with ammonia-scavenger therapy and fresh frozen plasma (FFP) for coagulopathy with subsequent intravenous immunoglobulin and dexamethasone several days later. Within 24 hours after starting FFP, the patients ferritin level declined sharply. Hyperammonemia and transaminitis also resolved, and his fever curve improved. Additional immunosuppression was considered, but not initiated due to the patients ongoing clinical improvement.
Over the next 3 months, the patient experienced two further acute episodes of fever, liver dysfunction, coagulopathy, and sepsis physiology. The second episode was successfully treated with FFP, though no clear infectious trigger was identified. The third episode occurred 4 days after routine vaccinations. The patient had prolonged hypotension requiring ionotropic support that resolved after receiving daily FFP, and hypoxia with pleural effusions that resolved after a single treatment with protein C concentrate.
As the patient had met 5/8 clinical diagnostic criteria for HLH, but also had a history of hyperammonemia, he underwent concurrent biochemical and genetic evaluation for both primary HLH and inborn errors of metabolism. Whole exome sequencing identified compound heterozygous mutations in COG4, part of an oligomeric protein complex involved in Golgi apparatus structure and function. COG4 mutations have previously been reported in two patients with autosomal recessive COG4-CDG (CDG-IIj), who were described to have similar clinical symptoms of hypotonia, seizures, coagulopathy, and liver dysfunction, as well as recurrent infections. Subsequent immune phenotyping while the patient was healthy was notable for slightly low numbers of NK cells, but normal CD107a mobilization and perforin/granzyme B expression in vitro.
Our patient represents a novel presentation of CDG due to COG4 defect with associated immune dysfunction manifesting as recurrent episodes of inflammatory crisis with features of HLH. CDG and inborn errors of metabolism should be considered during diagnostic evaluation for patients with HLH symptoms, as CDG patients may develop acute episodes of severe inflammation, in the absence of cellular regulatory defects, for which FFP and protein C concentrate may have therapeutic value.
(78) Submission ID#600705
Natural History of Anti-Interferon-gamma Autoantibody-associated Immunodeficiency Syndrome in Thailand
Gloria H. Hong, BA1, Ploenchan Chetchotisakd, MD2, Siriluck Anunnatsiri, MD2, Piroon Mootsikapun, MD2, Lindsey B. Rosen, BS3, Christa S. Zerbe, MD, MS4, Steven M. Holland, MD5
1NIH Medical Research Scholar, Laboratory of Clinical Immunology and Microbiology, National Institute of Allergy and Infectious Diseases, National Institutes of Health
2Faculty of Medicine, Division of Infectious Diseases and Tropical Medicine, Department of Medicine, Khon Kaen University, Thailand
3NIH-Oxford Scholar, Laboratory of Clinical Immunology and Microbiology, National Institute of Allergy and Infectious Diseases, National Institutes of Health
4Senior Research Physician, Laboratory of Clinical Immunology and Microbiology, NIAID, NIH
5Director, Division of Intramural Research, Laboratory of Clinical Immunology and Microbiology, National Institute of Allergy and Infectious Diseases, National Institutes of Health
Introduction/Background: Autoantibodies to interferon-gamma (IFN-g) are associated with disseminated nontuberculous mycobacterial (NTM) and other opportunistic infections in previously healthy adults, predominantly in or from Southeast Asia. Although the clinical manifestations of this acquired immunodeficiency syndrome have been reported, its natural history is not well understood.
Objectives: To characterize demographic data, recurrence of infections, clinical outcomes, and autoimmunity-related complications in patients with anti-IFN-g autoantibodies.
Methods: Eighty-one HIV uninfected voluntary participants (40 with disseminated NTM infection and 41 with another opportunistic infection with or without NTM infection) at Srinagarind Hospital in eastern Thailand were enrolled in an Institutional Review Board-approved protocol (09-I-N060) beginning in 2010 and followed annually until November 2018. Demographic information and clinical histories were recorded on standard forms at each visit and plasma samples were obtained. Serial plasma samples are being analyzed for anti-IFN-g antibody levels.
Results: Seventy-four out of 81 patients (91%) had anti-IFN-g autoantibodies. The median [interquartile range, IQR] age of patients with anti-IFN-g autoantibodies was 50 [46,56] years. Forty-seven patients (64%) were female. At the time of diagnosis, 36 patients (49%) with anti-IFN-g autoantibodies had disseminated NTM infection, 35 patients (47%) had another opportunistic infection with NTM infection, and 3 patients (4%) had another opportunistic infection without NTM infection. Mycobacterium abscessus was the most commonly isolated organism and lymph nodes (69 patients, 93%) were the most commonly involved site.
During the follow-up period, 25 patients (34%) with anti-IFN-g autoantibodies had at least one recurrence of culture-proven infection. After a median [IQR] follow-up time of 85 [42,96] months, 41 patients (55%) with anti-IFN-g autoantibodies had inactive disease after prolonged antibiotic treatment, 6 patients (8%) had active/progressive disease, and 18 patients (24%) had died. Of the 14 deaths with identifiable causes, 10 (71%) were related to infections. The rate of death per person-year was 0.044. The most common autoimmunity-related complication was Sweets syndrome, seen in 29 patients (39%) with anti-IFN-g autoantibodies. Sixteen of those patients (55%) had recurring Sweets syndrome. Additionally, 14 patients (19%) developed lymphatic obstruction, which continued to recur in 12 patients (86%).
Seven patients (9%) in this study did not have anti-IFN-g autoantibodies. The median [IQR] age of autoantibody-negative patients was 38 [27,54] years and 3 patients (43%) were female. None of the autoantibody-negative patients developed new infections during follow-up. At the end of the follow-up period, none of the patients had active/progressive disease and 2 patients (29%) had died.
Conclusions: Ninety-one percent of HIV uninfected Thai patients with disseminated NTM infection with or without other opportunistic infections had detectable anti-IFN-g autoantibodies. About one third of patients with autoantibodies to IFN-g had recurrent infections during follow-up. After approximately 7 years of follow-up, 55% of patients with anti-IFN-g autoantibodies had inactive disease following multi-drug antibiotic therapy while 8% had active/progressive disease and 24% had died. Patients with anti-IFN-g autoantibodies are at risk for recurrent infections and autoimmunity-related complications. Therefore, long-term follow-up is recommended. Life-long secondary antibiotic prophylaxis may be required to prevent recurrence of infection in the setting of persistent anti-IFN-g autoantibodies.
(79) Submission ID#600727
Artificial Thymic Organoids Represent a Reliable and Quick Tool to Study T Cell Differentiation in Human Bone Marrow Samples from Patients with Severe T Cell Immunodeficiency
Marita Bosticardo, PhD1, Francesca Pala, PhD2, Enrica Calzoni, MD3, Cameron Gardner, BSc4, Kerry Dobbs, BSc5, Suk See De Ravin, MD, PhD6, Nicholas Hartog, MD7, M. Louise. Markert, MD, PhD8, Katja G. Weinacht, MD, PhD9, Harry L. Malech, MD10, Christopher Seet, MD, PhD11, Amelie Montel-Hagen, PhD12, Gay M. Crooks, MBBS13, Luigi D. Notarangelo, MD, PhD14
1Staff Scientist, Laboratory of Clinical Immunology and Microbiology, IDGS, DIR, NIAID, NIH, Bethesda, MD, USA
2Post doctoral Fellow, Laboratory of Clinical Immunology and Microbiology, IDGS, DIR, NIAID, NIH, Bethesda, MD, USA
3Graduate Student, Laboratory of Clinical Immunology and Microbiology, IDGS, DIR, NIAID, NIH, Bethesda, MD, USA
4Graduate Student, Laboratory of Clinical Immunology and Microbiology, IDGS, DIR, NIAID, NIH, Bethesda, MD, USA; Department of Medicine, University of Oxford, Oxford, UK
5Biologist, Laboratory of Clinical Immunology and Microbiology, NIAID, NIH, Bethesda, MD, USA
6Clinician, Laboratory of Clinical Immunology and Microbiology, IDGS, DIR, NIAID, NIH, Bethesda, MD, USA
7Assistant Professor, Michigan State University, College of Human Medicine, Grand Rapids, MI
8Professor of Pediatrics and Immunology, Department of Pediatrics, Division of Allergy, Immunology, and Pulmonology, Duke University Medical Center, Durham, NC
9Assistant Professor, Division of Stem Cell Transplantation and Regenerative Medicine, Department of Pediatrics, Stanford School of Medicine, Stanford, CA
10Chief, Genetic Immunotherapy Section, Laboratory of Clinical Immunology and Microbiology, IDGS, DIR, NIAID, NIH, Bethesda, MD, USA
11Fellow, Department of Pathology and Laboratory Medicine, DGSOM, UCLA, Los Angeles, CA
12Associate Project Scientist, Department of Pathology and Laboratory Medicine, DGSOM, UCLA, Los Angeles, CA
13Professor, Pathology & Laboratory Medicine; Paediatric oncologist, Division of Stem Cell Transplantation and Regenerative Medicine, Department of Pediatrics, Stanford School of Medicine, Stanford, CA
14Chief, Laboratory of Clinical Immunology and Microbiology, IDGS, DIR, NIAID, NIH, Bethesda, MD, USA
The study of early T cell development in patients with severe T cell immunodeficiencies is challenging because of the rarity of these diseases, the difficulty to obtain hematopoietic stem cells (HSCs), and limitations in the assays to assess in vitro differentiation of HSCs to mature T cells. We recently developed a serum-free system that allows faithful analysis of sequential steps of T cell differentiation. In this system, artificial thymic organoids (ATOs) are generated, based on the 3D aggregation and culture of a delta-like canonical Notch ligand 4 (DLL4)-expressing stromal cell line (Ms5-Dll4) with CD34+ cells isolated from bone marrow (BM) samples of normal donors (ND). In this project, we set out to evaluate the possibility of using the ATO system to study T cell differentiation in patients carrying T cell defects, in order to define the exact steps of T cell development affected by different genetic defects. Using the ATO system, we studied in vitro T cell differentiation from CD34+ cells obtained from patients carrying defects that are intrinsic to hematopoietic cells (RAG1, RAG2, AK2, IL2RG) or that affect thymus development (DiGeorge syndrome, DGS). The AK2-deficient patient showed a markedly decreased viability in CD34+ cells and a very early defect in T cell development, already at the pro-T cell stage. This defect was very similar to that observed in a patient carrying a null IL2RG mutation who was reported to show autologous reconstitution after unconditioned haploidentical HSC transplantation. In contrast, CD34+ cells from a patient carrying a missense IL2RG mutation and with a leaky SCID phenotype were capable of differentiating into mature T cells in vitro, although with 100-fold decreased efficiency as compared to normal donors (ND). Interestingly, in the patient carrying the null IL2RG mutation, we noticed very few cells that could reach full maturation, with an absolute number of CD3+TCRab+ cells around 1000-times less than in ND. At variance with pro-T cells (that failed to express the gc protein), these mature T cells did express normal levels of gc, suggesting that they may have derived from residual CD34+ cells from the BM donor. In addition, CD34+ cells from the patients carrying RAG1 and RAG2 hypomorphic mutations were able to differentiate to CD4+CD8+ double positive cells, but not to CD3+TCRab+ cells. Finally, the DGS patient showed a completely normal in vitro T cell differentiation, confirming that T cell deficiency reflected thymic abnormalities. In summary, our data show that the ATO system could be extremely useful in determining whether the lack of T cells in patients with unknown gene defects reflect hematopoietic or thymic intrinsic problems, and may therefore provide critical evidence in deciding whether HSC or thymus transplantation is warranted, even without knowing the actual gene defect.
Supported by the Intramural Research Program, DIR, NIAID, NIH
Protocol 18-I-N128
(80) Submission ID#600761
Ataxia Telangiectasia with Chronic Skin Granulomas Preventable with SCID Newborn Screening?
Sara Seghezzo, MD1, Dana Feigenbaum, MD2, Sonal D. Shah, MD3, Erin F. Mathes, MD4, Morna J. Dorsey, MD, MMSc5, Jennifer Puck, MD6,
1Clinical Fellow, Department of Pediatrics, Division of Allergy, Immunology, and Bone Marrow Transplant, University of California, San Francisco
2Resident, Department of Dermatology, University of California San Francisco, San Francisco, CA
3Assistant Professor, Department of Dermatology, University of California San Francisco, San Francisco, CA
4Associate Professor, Department of Dermatology, University of California San Francisco, San Francisco, CA
5Pediatric Immunologist and Allergist, Department of Pediatrics, Division of Allergy, Immunology, and Bone Marrow Transplant, University of California San Francisco, San Francisco, CA
6Pediatric Immunologist, Department of Pediatrics, Division of Allergy, Immunology, and Bone Marrow Transplant, University of California San Francisco, San Francisco, CA
Introduction: Ataxia-Telangiectasia (AT) is an autosomal recessive disorder caused by mutations in the Ataxia Telangiectasia Mutated (ATM) gene, which aids in detection and repair of DNA damage. AT is characterized by progressive cerebellar ataxia, oculomotor apraxia, choreoathetosis, conjunctival telangiectasias, variable degrees of T-cell lymphopenia (TCL) and immune compromise. Patients are at an increased risk for malignancy, particularly leukemia and lymphoma, and are unusually sensitive to ionizing radiation. With the advent of TREC-based newborn screening (NBS) for SCID, AT patients are being recognized with asymptomatic TCL in early infancy.
Objectives: We present an older child with AT and chronic granulomatous lesions and discuss how this may be avoided in individuals with AT diagnosed following abnormal NBS.
Case Report: A 12 y/o male was born at term following an uncomplicated twin pregnancy and delivery, prior to institution of SCID NBS. He demonstrated mild gross motor and speech delay as an infant and was diagnosed with AT at age 3. He had received all routine immunizations, including live vaccinations. He developed granulomatous skin lesions at age 1, initially small papules on his cheeks and ears, which subsequently formed large disfiguring plaques on sun-exposed areascheeks, arms and hands (Fig 1). Following an extensive workup, his lesions were found to be secondary to a mutated vaccine-strain Rubella (RA27/3) based on 739bp genotyping, previously described in other immunocompromised individuals [Perelygina/Sullivan et al. JACI 2016]. His lesions have been refractory to multiple treatments including nitazoxanide. He is currently on daily oral and topical steroids, TMP/SMX and IVIG. Retrieval of his NBS for TREC determination revealed that he would have screened positive [Mallot/Puck et al. J Clin Immunol 2013]. When first measured at age 3, CD3 T-cells were low, 443/ul, with CD4 227/ul and CD8 140/ul. B and NK cell numbers were normal.
Since April 2017, 4 cases of AT were seen at UCSF in infants with non-SCID TCL on NBS. These 3 males and 1 female were all born at term and discharged from well-infant nurseries. AT was diagnosed at 2-7 months of age. Their initial TRECs ranged from 5-12/ul (normal with PerkinElmer Enlite kit >18), and all had low T-cells on initial flow cytometry (242-1612 CD3/ul, ref range>2500) with decreased CD4 (146-1178/ul) and CD8 (87-403/ul) T-cells; however naïve T-cells were present, ruling out typical SCID and raising concern for non-SCID TCL. Three infants also demonstrated low B-cells (<20-77/ul), while NK cells were normal in all. Two are currently receiving IVIG, one of whom is also on TMP/SMX. All have avoided not only rotavirus but also MMR and varicella live vaccinations.
Conclusions: AT is now often diagnosed in infants with low TRECs on SCID NBS, prior to neurologic manifestations. Benefits of early diagnosis include avoidance of live vaccines, including MMR, which led to the debilitating granulomas in our older patient. Additionally, patients receive prompt immunologic monitoring and treatment, avoidance of unnecessary radiation, specialty referrals and family genetic counseling. While there is no cure for AT, ongoing research may bring neuroprotective treatments in the future.
(81) Submission ID#600763
Comorbidities, Concomitant Medications, Infusion Parameters, and Tolerability in Advanced Age Patients with Primary Immunodeficiency Diseases Treated with Ig20Gly
Mark R. Stein, MD1, Daniel Suez, MD2, Iftikhar Hussain, MD3, Sudhir Gupta, MD4, Amy Darter, MD5, Ping Wang, PhD6, Barbara McCoy, PhD7, Leman Yel, MD8
1Physician, Allergy & immunology, Allergy Section, Good Samaritan Medical Center, West Palm Beach, FL, USA
2President, Allergy, Asthma & Immunology Clinic, PA
3President and Principal Investigator, Vital Prospects Clinical Research Institute, P.C., and Allergy, Asthma and Immunology Center, P.C.
4Professor, University of California at Irvine, Irvine, CA, USA
5Medical Director, Oklahoma Institute of Allergy & Asthma Clinical Research
6Project Lead, Biostatistics & Statistical Programming, Shire
7Clinical Scientist Lead – Immunology, Shire
8Sr Medical Director, Global Development Leader, IG, Clinical Research Immunology, Shire, Cambridge, MA, USA
Introduction: Subcutaneous immune globulin 20%, Ig20Gly, was well tolerated in the phase 2/3 North American study in patients with primary immunodeficiency diseases (PIDD). Here we assess comorbidities, use of concomitant medications, infusion parameters, and tolerability in advanced age patients (60 y) treated with Ig20Gly in the North American study.
Methods: Patients aged 2 years with PIDD received weekly Ig20Gly infusions at volumes 60 mL/site and rates 60 mL/h/site for ~1.3 years in the North American study (NCT01218438). The medical history at baseline, medical conditions that were ongoing (defined as comorbid events), use of concomitant medications, adverse events (AEs), tolerability, and infusion parameters were assessed by age: in advanced age patients (60 y; n=14), adult (16<60 y; n=39), and pediatric/adolescent patients (<16 y; n=21).
Results: The mean number of medical history events at baseline was higher in advanced age patients (28.7 events/patient; 402 events in 14 patients) versus adult (16.8 events/patient; 657 events in 39 patients), and pediatric/adolescent patients (6.5 events/patient; 137 events in 21 patients). Of these, the medical conditions that were ongoing at baseline (comorbid events) were also higher in the advanced age patients (20.9 events/patient; 292 events in 14 patients) versus adult (12.4 events/patient; 482 events in 39 patients), and pediatric/adolescent patients (3.4 events/patient; 71 events in 21 patients). In the advanced age patients, neurological comorbidities (51 events) were the most common, followed by those related to eyes, ears, nose, and throat (49 events), gastrointestinal (43 events), and musculoskeletal comorbidities (43 events). Concomitant medications were given to treat a preexisting condition in all patients in the advanced age group (225 medications in 14 patients). Despite the higher mean number of comorbid conditions, infusion parameters in the advanced age patients were comparable to those in the adult age group. Median maximum infusion rates and infusion volumes/site were comparable in the advanced age patients (60 mL/h/site; 47.5 mL/site) and adults (60 mL/h/site; 44 mL/site); lower infusion rates and volumes/site were reported in the pediatric/adolescent patients (30 mL/h/site; 26.8 mL/site). Infusions were well tolerated in all patients. Percentages of infusions associated with causally related AEs were low in advanced age patients (all [1.3 %], local [0.4%], systemic [0.9%]), adults (all [3.5%], local [1.1%], systemic [2.7%]) and pediatric/adolescent patients (all [2.8%], local [2.7%], systemic [0.4%]). Larger infusion volumes and faster infusion rates were not associated with increases in causally related local AEs in the advanced age group, consistent with the trends seen in the pediatric/adolescent and adult patients.
Conclusions: Despite the higher mean number of comorbidities in advanced age patients with PIDD, Ig20Gly was infused at relatively high rates and volumes and was well tolerated.
(82) Submission ID#600823
Interim Analysis of Infusion Characteristics and Adverse Events During Facilitated Subcutaneous Immunoglobulin Treatment for Primary Immunodeficiency Diseases: Global Post Authorization Safety Study
Arye Rubinstein, MD, Ph.D.1, Tracy Bridges, MD2, H. James Wedner, MD3, Donald McNeil, MD4, Richard L. Wasserman, MD, PhD5, Raffi Tachdjian, MD6, Katharina Fielhauer, MA7, Heinz Leibl, PhD8, Leman Yel, MD9
1Attending in Allergy & Immunology, Montefiore Medical Center
2Allergy / Immunology, Allergy & Asthma Clinics-Ga
3Chief, Division of Allergy and Immunology, Washington University Physicians, Washington University School of Medicine in St. Louis
4President, Founder and Principal Investigator, Optimed Research, LTD
5Allergist/immunologist, Allergy Partners of North Texas Research, Dallas, TX, USA
6Assistant Clinical Professor of Medicine and Pediatrics in the Division of Allergy and Clinical Immunology, Ronald Reagan Medical Center, UCLA School of Medicine
7Clinical Scientist, Shire
8Sr Medical Director, Global Development Leader, IG, Clinical Research Immunology, Shire, Vienna, Austria
9Sr Medical Director, Global Development Leader, IG, Clinical Research Immunology, Shire, Cambridge, MA, USA
Introduction: HyQvia (IGHy; immunoglobulin infusion 10% with recombinant human hyaluronidase [rHuPH20]) is an immunoglobulin (IG) replacement therapy approved for patients with primary immunodeficiency diseases (PIDD) that allows larger infusion volumes, up to 600 mL/site, and has improved IG bioavailability compared with conventional subcutaneous IG products. A post-authorization safety study is being conducted in the United States to acquire long-term safety data on IGHy and to assess prescribed administration regimens in routine clinical practice. Infusion characteristics and treatment-related adverse events from an interim analysis are reported here.
Methods: Patients aged 16 years with PIDD receiving IGHy were included in this ongoing, prospective, non-interventional, open-label, uncontrolled, multicenter study. As a part of routine clinical practice, patients are treated with IGHy according to standard medical care and their treatment regimen is at the discretion of the treating physician. Adverse events (AEs) are collected from enrollment to study completion/discontinuation using a subject diary and assessed at every study visit (every 3 months or standard practice). AEs are assessed based on seriousness, severity, and causal relatedness to IGHy. The presence of anti-rHuPH20 antibody is evaluated on a voluntary basis. Treatment preferences for various attributes of IG therapy were assessed annually using a treatment preference questionnaire.
Results: A total of 175 patients were enrolled at 26 US study sites (data cut-off date: August 21, 2017). Infusions were self-administered at home (56%) or at the clinical site (44%) most commonly using 4-week infusion intervals (56.6%). The mean maximum IG infusion rate was 302.8 mL/h and the mean IG dose was 418 mg/kg bodyweight/4weeks. The mean number of infusion sites used for administration was 1.9 and mean infusion duration was 2.8 hours. Most infusions (97.3%) were administered without a rate reduction, interruption, or discontinuation due to AEs. There were no serious AEs (SAEs) related to IGHy. Sixteen patients experienced a causally related non-serious local AE (9.1%; 0.43 events/patient-year, 0.07 events per infusion) and 25 patients experienced a causally related non-serious systemic AE (14.3%, 0.88 events/patient year, 0.14 events per infusion). Seven of 113 patients who were tested for anti-rHuPH20 antibody had 1 positive binding antibody test to rHuPH20 (titer 1:160; maximum titer 1:10240 at enrollment, 1:5120 during the study); no neutralizing rHuPH20 antibodies were detected. Of the patients who responded to the treatment preference questionnaire at the end of year 1, the majority (38/52 [73.1%]) preferred to receive their IG therapy at home; 21.2% (11/52) preferred the doctors office; 3 patients preferred treatment at the hospital, had no preference, or indicated other. Almost all patients (51/52 [98.1%]) indicated a preference to continue treatment with IGHy.
Conclusion: This interim analysis of 175 patients with PIDD treated with IGHy in routine clinical practice supports previous observations that IGHy is a well-tolerated and preferred therapy with no reports of treatment-related SAEs or neutralizing anti-rHuPH20 antibodies.
(83) Submission ID#600846
Lymphocyte Radiosensitivity in Cartilage Hair Hypoplasia
Jennifer R. Yonkof, MD1, Sharat Chandra, MD, MRCPCH2, Matthew J. Smith, MS3, Roshini S. Abraham, PhD4
1Fellow, Department of Pediatrics, Division of Allergy and Immunology, Nationwide Children's Hospital
2Assistant Professor, UC Department of Pediatrics, Division of Bone Marrow Transplantation and Immune Deficiency, Cincinnati Childrens
3Research Technologist, Division of Hematology, Department of Medicine, Mayo Clinic
4Department of Pathology and Laboratory Medicine, Nationwide Childrens Hospital, Columbus, OH.
Background: Cartilage hair hypoplasia (CHH) is an autosomal recessive chondrodysplasia associated with variable immunodeficiency. Pathogenic defects in RMRP, encoding the untranslated RNA subunit of ribonucleoprotein endoribonuclease complex (RMRP), result in reduced mRNA and rRNA cleavage. RMRP c.70A>G is the most common variant, increased in Finnish and Amish populations. While cellular immunodeficiency is associated with increased morbidity and mortality, there is no established correlation between clinical and immunological phenotype. Lymphocyte radiosensitivity has not been described.
Case: A full-term Amish female infant had low TREC copies on newborn SCID screen. Flow cytometry at 3 months-old demonstrated severe T and B cell lymphopenia (CD3+T-cells 413 cells/mcL, range: 2,300-6,500 cells/mcl; CD19+B-cells 214 cells/mcL, range: 600-3,000 cells/mcL) with normal NK quantitation (CD16/56+ 340 cells/mcl, range: 100-1,300 cells/mcL) and CD4+ memory T-cell expansion (33.2%) relative to the naïve subset (67.0%). T-cell functional mitogen responses were normal. She was diagnosed with CHH with homozygous RMRP c.70A>G mutation. Lymphocyte subset (T, B and NK cells) radiosensitivity was evaluated by flow cytometric analysis of phosphorylated (p) ATM, SMC1 and gamma-H2AX after low-dose (2Gy) irradiation. An increase in gamma-H2AX level was observed in a subset of non-irradiated T cells (17.66% v. 1.36% gamma-H2AX+) and NK cells (23.07% v. 1.04% gamma-H2AX+) in the patient, suggestive of a constitutive defect in DNA repair. The relative distribution of T, B and NK cells expressing pATM, pSMC1 and gamma-H2AX at 1 hour post-irradiation (IR) was not significantly different from the experimental healthy control (EHC) or pediatric reference range (pRR). However, the kinetics of dephosphorylation at 24 hours post-IR was altered with residual gamma-H2AX expression in a subset of the patients T cells (delta 3.84%, mode ratio mean fluorescence intensity (MFI)=2.58; EHC: delta 0.10%, mode ratio MFI=1.39; pRR: delta 2.16%, mode ratio MFI=2.42). A similar finding was observed in a subset of patient B-cells for gamma-H2AX (delta 11.35%, mode ratio MFI=1.48; EHC: delta 0.82%, mode ratio MFI=0.86; pRR: delta 1.95%, mode ratio MFI=1.19). The frequency of the patient's lymphocytes with residual gamma-H2AX persistence at 24h post-IR was prominent, with 8.29% T-cells demonstrating persistence of gamma-H2AX (compared to 0.82% in the EHC, and 2.60% in the pRR), and 18.02% B-cells gamma-H2AX+ (compared to 1.80% in the EHC, and 2.96% in the pRR). There has been lack of follow-up, but verbal report suggests no significant immunological or infectious concerns at 1 year of age.
Discussion: Lymphocyte radiosensitivity is a novel finding in CHH with T and B cell lymphopenia. The ability of RMRP to associate with telomerase reverse transcriptase (TERT) and function as an RNA-dependent RNA polymerase, yielding distinct silencing RNA sequences, may underlie radiosensitivity in RMRP mutants. Systematic characterization of lymphocyte radiosensitivity and immunological phenotype could provide useful information on whether this could serve as a biomarker for the magnitude or complexity of immunodeficiency. Assessment of radiosensitivity has implications in conditioning regimen selection for patients requiring allogeneic hematopoietic cell transplantation. We recommend lymphocyte radiosensitivity assessment in CHH infants identified by NBS SCID and CHH patients with significant immunodeficiency and/or malignancy.
(84) Submission ID#600887
Novel Primary Immunodeficiency with Lymphoproliferative Disease Due to Biallelic Defects in NCKAP1L
William A. Comrie, PhD1, M. Cecilia Poli, MD, PhD2, Douglas B. Kuhns, PhD3, Jason W. Caldwell, DO4, Morgan Similuk, ScM5, Alexandre F. Carisey, PhD6, Lisa R. Forbes, MD2, Emily M. Mace, PhD7, Tram N. Cao, MS8, Zeynep H. Coban-Akdemir, PhD9, Shalini N. Jhangiani, PhD10, Donna M. Muzny, MSc11, Richard A. Gibbs, PhD12, James R. Lupski, MD, PhD12, V. Koneti Rao, MD, FRCPA13, Jordan S. Orange, MD, PhD14, Ivan K. Chinn, MD2, Michael J. Lenardo, MD15
1Postdoctoral Research Fellow, Molecular Development of the Immune System Section, Laboratory of Immune System Biology, NIAID, National Institutes of Health, Bethesda, MD, USA
2Assistant Professor, Department of Pediatrics, Baylor College of Medicine, Houston, TX, USA
3Principal Scientist, Neutrophil Monitoring Lab, National Cancer Institute-Frederick, Frederick National Laboratory for Cancer Research, Frederick, MD, USA
4Assistant Professor, Section of Pulmonary, Critical Care, Allergic and Immunological Diseases, Wake Forest University School of Medicine, Winston-Salem, NC, USA
5Genetic Counselor, Molecular Development of the Immune System Section, Laboratory of Immune System Biology, NIAID, National Institutes of Health, Bethesda, MD, USA
6Postdoctoral Research Fellow, Department of Pediatrics, Baylor College of Medicine, Houston, TX, USA
7Assistant Professor, Department of Pediatrics, Columbia University Irving Medical Center, New York, NY
8Research Coordinator, Department of Pediatrics, Baylor College of Medicine, Houston, TX, USA
9Postdoctoral Research Fellow, Department of Molecular and Human Genetics, Baylor College of Medicine, Houston, TX
10Project Manager, Department of Molecular and Human Genetics, Baylor College of Medicine, Houston, TX
11Assistant Professor, Department of Molecular and Human Genetics, Baylor College of Medicine, Houston, Texas, USA
12Professor, Department of Molecular and Human Genetics, Baylor College of Medicine, Houston, Texas, USA
13Staff Physician, Molecular Development of the Immune System Section, Laboratory of Immune System Biology, NIAID, National Institutes of Health, Bethesda, MD, USA
14Professor and Chair, Department of Pediatrics, Columbia University Irving Medical Center, New York, NY
15Senior Investigator, Molecular Development of the Immune System Section, Laboratory of Immune System Biology, NIAID, National Institutes of Health, Bethesda, MD, USA
BACKGROUND: Three children from 2 non-consanguineous families and different ethnic backgrounds developed lymphoproliferative disease by 2 years of age. They also had recurrent infections, including pneumonia and bronchiectasis, otitis media, and skin pustules. Immune phenotyping revealed low CD4+ T cell percentages, an accumulation of memory-like CD8+ T cells, impaired T cell proliferation, and low total NK cell numbers.
METHODS: The affected individuals, unaffected parents, and other unaffected family members underwent exome sequencing.
RESULTS: All 3 affected cases had rare and bioinformatically damaging biallelic variants, with appropriate familial segregation, in NCKAP1L, which encodes Hem1. Hem1 is an essential component of the WAVE2 Regulatory Complex (WRC). Immunoblotting confirmed destabilization of the WRC in all patients. Immunofluorescence microscopy demonstrated defective F-actin and WAVE2 localization to immune synapses in NK cells. Significant abnormalities were identified in patient lymphocyte and neutrophil migration and morphology, consistent with altered WRC-mediated cytoskeletal dynamics. All patients exhibited impaired inside-out integrin activation. Knockdown of Hem1 produced deficient proliferative responses and mTORC2-mediated AKT activation in control T cells.
CONCLUSIONS: The immunologic and clinical phenotype in the affected individuals recapitulates the phenotype observed in Hem1-deficient mice. Biallelic defects in NCKAP1L therefore result in a novel human primary immunodeficiency disease characterized by lymphoproliferation and susceptibility to infections.
(85) Submission ID#600899
Prevalence of Hypogammaglobulinemia in Newly Diagnosed Lymphoma
Namrata Singh, MD, MSCI, FACP1, Sarah Mott, MS2, Ashley McCarthy, MPH3, Aaron Knaack, BA4, James Cerhan, MD, PhD5, Zuhair Ballas, MD6, Brian Link, MD6
1Clinical Assistant Professor, University of Iowa Hospitals and Clinics
2 Biostatistician, College of Public Health
3Manager, Holden Comprehensive Cancer Center
4Division Coordinator, University of IOwa Hospitals and CLinics
5Professor, Mayo Clinic
6Professor, University of Iowa Hospitals and Clinics
Background: Concurrent existence/significance of immunodeficiency with new onset lymphoproliferative disease remains understudied. Just two studies to date have evaluated the prevalence of hypogammaglobulinemia in chronic lymphocytic leukemia (CLL) and neither studied prevalence and impact of IgE deficiency on outcomes in CLL [1, 2]. Therefore, the objective of this study was to examine the prevalence of hypogammaglobulinemia, examining all isotypes, in newly diagnosed CLL patients and to test the hypothesis that patients with hypogammaglobulinemia have a distinct clinical profile and outcome.
Methods: Using the banked sera of 150 newly diagnosed, treatment-naïve, CLL adult patients from the Lymphoma Molecular Epidemiology Resource (L-MER), Ig (IgG, IgA, IgM and IgE) levels were measured. The L-MER was initiated as an observational cohort study of prospectively enrolled newly diagnosed lymphoma patients evaluated at the Mayo Clinic (Rochester, MN) and the University of Iowa (Iowa City, IA) [3]. IgG/A/M levels were measured using immunoturbidimetric assay whereas the IgE level was determined using electrochemiluminescence immunoassay. The associations between Ig deficiencies and clinical factors were evaluated with Wilcoxon rank sum and chi-squared (Fishers exact, where appropriate) tests. Cox regression models were used to assess the effects of clinical variables on overall survival (OS). Time was calculated from biopsy to death due to any cause; patients still alive were censored at last contact. All tests were two-sided and assessed for significance at the 5% level using SAS v9.4 (SAS Institute, Cary, NC).
Results: The mean age (SD) of the selected CLL cohort was 63.8 (11.0) years with a male predominance (69.3%). 96.2% of the patients were white. With a median follow-up of five years, there were 50 deaths. Hypogammaglobulinemia in newly diagnosed, treatment-naïve CLL was common in our cohort with 88 (58.7%) patients having a measurable isotype deficiency. The most common Ig deficiency was IgM (44.0%, 95% CI 35.9-52.3%), followed by IgG (34.7%, 95% CI 27.1-42.9%), IgE (16.7%, 95% CI 11.1-23.6%) and IgA (12.0%, 95% CI 7.3-18.3%). Multiple deficiencies in the same patient were common (Figure 1). IgA and IgE deficiency were associated with higher Rai stages (grading system for CLL) at presentation (p<0.01 and 0.04 respectively) as well as with higher white blood cell counts at presentation (p=0.02 and 0.01 respectively). A higher proportion of IgA deficient patients needed second treatment during follow-up (61% compared to 36%, p=0.04). When comparing predictors of overall survival, higher Rai stage [3-4 vs 0, Hazard ratio (HR) 2.43, 95% CI 1.08-5.46, p=0.03] and age (HR 1.08, 95% CI 1.05-1.12, p<0.01) correlated with worse overall survival. Individual immunoglobulin deficiencies did not correlate with overall survival.
Conclusions: A significant proportion of treatment-naïve patients with CLL have underlying Ig deficiencies- both in isolation and a combination of different isotypes. A deficiency of IgA or IgE was associated with severe disease at presentation. The underlying relationship between these two immunologic disorders deserves further study.
Figure 1. Distribution of immunoglobulin deficiencies among patients with chronic lymphocytic leukemia (CLL).
(86) Submission ID#600905
Clinical Features and Management of Patients with Rheumatoid Arthritis and a Coexisting Immunodeficiency Disorder
Ruth Fernandez, MD1, Scott Vogelgesang, MD2, Bharat Kumar, MD, MME3, Zuhair Ballas, MD2, Namrata Singh, MD, MSCI, FACP4
1Fellow, NYU Langone Health System
2Professor, University of IOwa Hospitals and Clinics
3Clinical Assistant Professor, UIHC
4Clinical Assistant Professor, University of Iowa Hospitals and Clinics
Background: Patients with primary immunodeficiency (PID) have an increased risk of developing autoimmune diseases, including rheumatoid arthritis (RA). Management of these patients is challenging as immunomodulators can further increase their risk for infections. Additionally, patients with RA that undergo therapy with drug modifying anti-rheumatic drugs (DMARDs) may develop a secondary immunodeficiency. There are few studies reviewing the characteristics of patients with a PID who later develop RA, and no studies have been reported comparing these patients to those who develop an immunodeficiency after starting DMARD therapy for RA.
Methods: 65 patients were identified as having inflammatory arthritis and a concomitant immunodeficiency (ID) at our institution between 1/1/2000-10/03/2017 using ICD-9 and 10 codes. Manual chart review was performed to confirm and identify the timing of diagnosis of these disorders. Patients were excluded if either there was no definitive diagnosis of ID or RA (clinically diagnosed by a practicing allergist/immunologist and meeting ACR 2010 criteria for RA with a score of 6 or higher, respectively), or rituximab was administered prior to diagnosis of ID . Clinical symptoms, treatment, and laboratory data were extracted. Fishers exact test was used to compare the categorical variables between the groups; t-test was used to compare the continuous variables.
Results: 10 patients met the inclusion criteria. 5 patients were diagnosed with an ID and developed RA later in life (group 1), and 5 patients were diagnosed with RA and subsequently developed a clinically significant ID (group 2). The mean ages of diagnosis of ID and RA in group 1 patients were 32.0 years (SD ± 26.9) and 42.6 years (SD ± 19.0), respectively. In group 2, the mean age of diagnosis of RA was 37.8 (SD ± 14.2), compared to 54.8 years (SD ± 12.7) for the diagnosis of ID. Most patients in both groups were female (60% in group 1 and 80% in group 2). All patients in both groups had a humoral ID, including common variable immunodeficiency (CVID) (40% of group 1 patients), specific antibody deficiency (SAD) (20% of group 1 and 60% of group 2 patients), and hypogammaglobulinemia (20% of group 1 and 40% of group 2 patients). All patients in group 2 were seropositive for rheumatoid factor (RF) or anti-cyclic citrullinated peptide (anti-CCP), whereas only 20% of patients in group 1 were positive for RF or anti-CCP (Table 1). Most patients in both groups were treated with immunoglobulin replacement therapy. Treatment of RA in both groups was similar, but combination DMARD therapy was not used in group 1 patients in contrast to group 2 patients.
Conclusions: Our study indicates that even though clinical characteristics and management are similar in patients with coexisting ID and RA, RF and anti-CCP are usually negative in patients who develop RA after ID, possibly due to impaired antibody production in immunodeficient patients.
Table 1. Comparison of the clinical and laboratory features of patients with an immunodeficiency disorder diagnosed prior to development of rheumatoid arthritis (Group 1) to those diagnosed with an immunodeficiency disoder after diagnosis of rheumatoid arthritis (Group 2)
|
Group 1 n (%) |
Group 2 n (%) |
P-value | |
|
Sample Size Demographics |
5 (100%) | 5 (100%) | |
| Race/Ethnicity | |||
| White, not Hispanic | 5 (100.0) | 5 (100.0) | 1.0000 |
| Age at the time of diagnosis of RA (years) | 42.6 ± 19.0 | 37.8 ± 14.2 | 0.6964 |
| Age at the time of diagnosis of ID (years) | 32.0 ± 26.9 | 54.8 ± 12.7 | 0.1638 |
| Gender | 1.0000 | ||
| Male | 2 (40.0) | 1 (20.0) | |
| Female | 3 (60.0) | 4 (80.0) | |
| Deceased | 2 (40.0) | 1 (20.0) | 1.0000 |
| Positive RF | 1 (20.0) | 4 (80.0) | 0.2063 |
| Positive anti-CCP | 1 (33.3) | 2 (66.7)* | 1.0000 |
| Positive RF or anti-CCP | 1 (20.0) | 4 (80.0) | 0.0476 |
RA: Rheumatoid arthritis; ID: Immunodeficiency disorder; RF: Rheumatoid factor; anti-CCP: Cyclic citrullinated peptide antibody
(87) Submission ID#600937
A Case of Complement Factor D Deficiency with Streptococcus Pneumoniae Pneumonia with Associated Lung Abscess and Empyema
Ashleah Courtney, MD, MSPH1, Matthew Bell, MD2, Diana K. Bayer, DO3, Sheva Chervinskiy, DO4
1Pediatric Resident, Arkansas Children's Hospital, University of Arkansas for Medical Sciences
2Assistant Professor of Allergy and Immunology, Arkansas Children's Hospital, University of Arkansas for Medical Sciences
3Clinical Assistant Professor of Allergy/Immunology, Division of Allergy/Immunology and Pulmonary, Department of Pediatrics, University of Iowa Stead Family Childrens Hospital
4Assistant Professor of Allergy and Immunology, Arkansas Children's Hospital, University of Arkansas Medical Sciences
Introduction/Background: Complement deficiencies are relatively rare, comprising less than 1% of primary immunodeficiencies. They are associated with increased risk for infections with encapsulated organisms and autoimmunity. Of all complement deficiencies, the rarest are defects in the alternative complement pathway. Properdin deficiency is the most commonly described alternative pathway deficiency, with Factor B and Factor D deficiency more rarely described. Fewer than 5 patients with factor D deficiency have been reported with all reported cases being children of consanguineous parents who succumbed to meningococcal sepsis.
Objectives: To describe a case of Factor D deficiency associated with recurrent respiratory infections with Streptococcus pneumoniae pneumonia with associated lung abscess and empyema.
Methods: Retrospective chart review was conducted. Laboratory investigations included lymphocyte immunophenotyping by flow cytometry, lymphocyte proliferation to mitogen, quantitative serum immunoglobulins, vaccine titers, complement assays and functional evaluation, and genetic evaluation by next generation sequencing.
Results: A 2 year old Marshallese male was transferred from an outside hospital to our facility for further evaluation of worsening pneumonia and was found to have right-sided pleural effusion and pulmonary abscess in the right lower lobe. The abscess was drained and was found to be positive for Streptococcus pneumoniae via polymerase chain reaction. He improved after chest tube placement and treatment with intravenous antibiotics. His medical history was significant for recurrent acute otitis media and prior hospitalization out-of-state for pneumonia with empyema secondary to Streptococcus pneumoniae, which required chest tube placement and admission to the pediatric intensive care unit at 18 months of age. Immunologic work up revealed age-appropriate lymphocyte subpopulations, lymphocyte proliferative responses to mitogens, quantitative immunoglobulin levels, pneumococcal/tetanus/diphtheria titers, and CH50 complement assay. AH50 complement assay was decreased to 44 units/mL. Complement testing was repeated - with normal CH50 and AH50 of 0 units/mL. Further evaluation revealed normal levels of Factors B, H, I and properdin. Factor D level was 0.12 mcg/mL, and Factor D function was decreased to 2 units/mL, indicating a diagnosis of Factor D deficiency. Sequencing of the CFD gene revealed a previously undescribed homozygous deletion (c.721_723del and p.Lys241del). The parents were not agreeable to personally undergoing genetic evaluation to determine if this was a de novo mutation. The patient was managed with pneumococcal and meningococcal immunizations, prophylactic amoxicillin and intravenous gamma globulin (IVIG) without any further infections. Unfortunately, after two IVIG infusions, he was lost to follow up.
Conclusion: Factor D deficiency is an extremely rare alternative complement pathway deficiency, described in less than 5 patients. All infections described thus far have been secondary to Neisseria meningitidis. This case represents not only a novel mutation in the CFD gene leading to Factor D deficiency, but also the first description of a patient with Factor D deficiency developing invasive infection secondary to Streptococcus pneumoniae.
(88) Submission ID#600938
Hexaviral-Specific T-cells for Treatment and Prevention of Viral Infections Post Hematopoietic Stem Cell Transplant
Michael D. Keller, MD1, Katherine Harris, MD2, Patrick Hanley, PhD3, Blachy J. Davila Saldana, MD4, Allistair Abraham, MD5, Nan Zhang, PhD6, Gelina Sani, BS7, Haili Lang, MS8, Richard Childs, MD9, Richard Jones, MD10, Catherine Bollard, MD11,
1Assistant Professor, Center for Cancer and Immunology Research, Children's National Health System, Division of Allergy & Immunology, Children's National Health System, Washington, DC
2Clinical Fellow, Children's National Health System
3Director, Stem cell Therapy Lab, Children's National Health System
4Blood and Marrow Transplant Specialist, Division of Blood and Marrow Transplantation, Childrens National Medical Center, Department of Pediatrics, The George Washington University, Washington, DC
5Assistant Professor, Children's National Health System
6Lead Cell Therapy Technician, Children's National Health System
7Technician, Children's National Health System
8Staff Scientist, Children's National Health System
9Clinical Director, NHLBI
10Professor, Johns Hopkins
11Director, Center for Cancer and Immunology Research, Children's National Health System
Background: Viral infections are a significant cause of morbidity and mortality in patients with primary immunodeficiency disorders and following hematopoietic stem cell transplantation. Adoptive immunotherapy using virus specific T-cells (VSTs) has been shown to prevent and treat viral infections in immunocompromised hosts. Human Parainfluenza Virus-3 (HPIV3) is a common cause of severe respiratory illness in immunocompromised patients and has no approved antiviral therapies and has not previously been used as a target for T cell therapeutics.
Objective: The primary aim was to determine whether donor derived hexaviral specific T-cells are effective in preventing and treating CMV, EBV, AdV, BK virus, HHV-6, and HPIV3.
Patients and Methods: This was a first in man study where we studied the antiviral effect in 8 patients who received hexa-valent VSTs after stem cell transplant on a Phase I trial. Assessment of virus-specific immunity was measured by IFN-g ELIspot. Viral loads for the primary targeted viruses were measured at specific time points post VST infusion.
Results: Three patients were treated for active CMV and had resolution of viremia. Two patients treated for active BK virus had complete resolution of symptoms and viremia, while one had resolution of hemorrhagic cystitis but fluctuating viral loads in the blood and urine. Two patients were treated prophylactically. One patient did not develop any infections, while the other developed EBV viremia requiring rituximab. Two patients received VSTs under expanded access for emergency treatment 1 patient was treated disseminated adenoviremia and the second patient was treated for HPIV3 pneumonia. These critically ill patients demonstrated partial clinical improvements, but VST persistence was likely hindered by concomitant steroid use which resulted in incomplete antiviral responses. ELISpot showed evidence of antiviral T-cell activity in 3 of 4 evaluable patients by 3 months post-infusion, with in vivo VST expansion detectable in 2 patients.
Conclusions: Preliminary results show that hexaviral specific VSTs are safe and may be effective in preventing and treating multiple viral infections. Further studies are warranted to determine if VSTs are effective against active HPIV3 infections.
Table 1: Demographics and Outcomes for Recipients of Hexaviral Specific T-cells
| Patient # | Infections Pre-VSTs | Prior antiviral therapy | CMV | EBV | Adv | BKV | HHV6 | HPIV3 | Outcome |
| 1 | - | Cidofovir (CMV cleared prior to VSTs) | - | - | - | - | - | - |
Free of viral infections; Alive and well |
| 2 | CMV, BKV | Cidofovir | CR | - | CR | CR | - | - | Transient Adv detection post-infusion; cleared. Alive and well |
| 3 | CMV, BKV | Ganciclovir, Cidofovir | CR | - | - | PR | - | - |
BK hemorrhagic cystitis resolved post-infusion,with fluctuating BK viremia/viruria. Alive and well |
| 4 | CMV, BKV | Cidofovir | CR | - | - | CR | - | - | Alive and well |
| 5 (EIND) | AdV | Cidofovir | - | - | PR | - | - | - | Died, veno-occlusive disease |
| 6 | - | Valacyclovir | - | NR | - | - | - | - | EBV reactivation without PTLD, treated with rituximab. Alive and well. |
| 7 (EIND) | HPIV3 | - | - | - | - | - | - | PR | Transient radiographic improvement by 1 month. |
CR: complete response (resolution of viral infection); PR: partial response; NR: no response
(89) Submission ID#600976
Predictors of Fatigue in Common Variable Immunodeficiency
Joud Hajjar, MD, MS1, Carleigh Kutac, MPH2, Tiffany S. Henderson, PhD3, Christopher Scalchunes, MPA4
1Assistant Professor, Baylor College of Medicine, 1Texas Childrens Hospital Center for Human Immunobiology and Division of Immunology, Allergy and Rheumatology
2Biostatistician, Baylor College of Medicine
3Survey Research Analyst, Immune Deficiency Foundation
4Vice President of Research, Immune Deficiency Foundation
Introduction: We previously reported that fatigue is increased in common variable immunodeficiency (CVID). However, in previous studies, fatigue was not defined using validated tools. Our aim from this study is to identify the prevalence of patient-reported fatigue, using validated questionnaires, and determine the factors predisposing to fatigue in CVID
Methods: Data from CVID who responded to the IDF 2017 patient national survey a were analyzed. Fatigue was measured using the Brief Fatigue Inventory (BFI) questionnaire, which includes seven items to identify fatigue, and measure fatigue severity. A total of 555 patients with CVID and responses to BFI were enrolled. Demographics, co-morbidities, immunoglobulin replacement therapy (IgGRT) route and dose, co-morbidities, infections, depression, quality of life (QOL) (using the SF-12v2) and disability were compared between fatigued and non-fatigued. Logistic regression was used to identify the significant variables.
Results: The overall prevalence of fatigue was 83.24% (437/555), 69.55% of fatigued patients reported having moderate/severe fatigue. Significant predictors of fatigue were: Asthma (p=0.014), Patients Current Health status (p<0.001), history of Bronchitis (p=0.002), Sinus Infections (p<0.001), Pneumonia (p=0.028), Female sex (p<0.001), Employment Status (employed vs disabled; p<0.001), Household Income (p<0.001), BMI (p=0.024), abnormal digestive function (p<0.001), Permanent lung impairment (p<0.002), Recurrent Diarrhea (p<0.001), Inflammatory Bowel Disease (p=0.022), Depression (p<0.001), Noticeable wear-off of immunoglobulins (p<0.001), SF-12 Physical Component Score (PCS) (p<0.001), and SF-12 Mental Component Score (MCS) (p<0.001).
Increased fatigue severity corresponded with significantly lower PCS and MCS scores. Using a univariate linear regression model for PCS and MCS score prediction, and the BFI variable (fatigue= none, mild, moderate, severe) as the independent variable, there was a significant increase in both PCS and MCS predicted scores as the severity of fatigue decreased (p<0.001). Improvement of fatigue scores from severe to moderate/mild predicted improvement in QoL scores, PSC scores by (5.3 and 12.7 points respectively) and MSC scores (4.9 and 9.9, respectively).
Conclusion: Moderate to severe fatigue is significantly increased in CVID patients, increased fatigue severity predicts lower PCS and MCS, and improving fatigue score could lead to improvement in QoL and patient-reported health outcomes in CVID patients.
(90) Submission ID#600981
Rapid Response of CVID Skin Granulomatous Disease to Infliximab
Maria Gabriela Torre, MD1, Eloisa Malbran, MD2, Maria Cecilia Juri, MD3, Alejandro Malbran, Phd4
1Staff, Unidad de Alergia, Asma e Inmunologia Clinica; British Hospital; Army Hospital
2Staff, Unidad de Alergia, Asma e Inmunologia Clinica; Sanatorio Mater Dei
3Staff, Unidad de Alergia, Asma e Inmunologia Clinica; British Hospital; Hospital de Clinicas Jose de San Martin
4Director; Head of Department, Unidad de Asma, Alergia e Inmunologia Clinica; British Hospital
Granulomas are the most significant day-to-day problem for CVID patient management. Currently, there are limited options for their treatment and the optimal therapy is unknown. In case reports and small series, Infliximab has been reported effective while others found it useless.
We here describe a 26yo white male referred for monthly IVIG in august 2016. At age 1, he developed large areas of erythematous polymorphic plaques in his cheeks, arms and legs. A skin biopsy showed tuberculoid granulomas negative for bacteria, BAAR and fungi, with infiltrating CD4+ lymphocytes. A prolonged course of steroids did not improve his skin. He also had multiple pneumonias and bronchiectasis, and oral candidiasis. He received all vaccines, including BCG with no complications. With low immunoglobulins and a poor response to pneumococcal polysaccharides and tetanus toxoid he was diagnosed as CVID and placed on IVIG at 7yo with excellent infectious control since then. At age 8, his skin lesions persisted and deepened to the bone on his left leg. Broad spectrum antibiotics for 3 months were unsuccessful. At 16yo to 18yo, skin grafts were performed on his arms, legs and both cheeks. Two ulcers persisted on his left leg until August 2018 that increased in size, deepened and became erythematous and extremely painful (Fig. 1). In September, two new ulcers appeared on his right cheek and right gluteus, respectively. One week later a third ulcer was found on his left calf. On September 28th, Infliximab 5mg/kg (300mg) was administered. On the second Infliximab dose, October 12th, the pain was completely gone and all ulcers were shrinking, and those ones in the cheek, gluteus and calf almost completely resolved. By the third dose, on November 23rd the ulcers in his right leg were almost closed (Fig. 2). Infliximab 300mg treatment continues every 8 weeks. Lab test remained unchanged from 2016 till 2018, when his wounds got worsened. (Table 1)
Granulomatous disease in CVID is a challenge. Both B and T cell directed therapies are encouraged. We add a new case of an Infliximab responsive patient to others already reported.
Fig 1. Before Infliximab After second dose Infliximab
Fig. 2
Table 1
| 08/2016 | 08/2017 | 12/09/2018 | 20/09/2018 | 28/09/2018 | |
| IgG (600-1600mg/dl) | 868 | 885 | 928 | ||
| IgA (70-400mg/dl) | 41 | 31 | 30 | ||
| IgM (50-300mg/dl) | 9 | 12 | 11 | ||
| IgE (1-100mg/dl) | 2 | 3 | 4 | ||
| CD3 % (67-75) | 96 | 98 | 98 | ||
| CD4 % (36-46) | 47 | 51 | 50 | ||
| CD8 % (31-40) | 43 | 45 | 44 | ||
| CD56/CD16 % (10-19) | 3 | 1 | |||
| CD19 % (11-16) | 0 | 1 | 1 | ||
| CD20 % (11-16) | 0 | 1 | 1 | ||
| Leucocytes (3700-9500/ul) | 4000 | 3900 | 4100 | 3500 | 3600 |
| Beta2 micro globulin (0.8-2.3mg/l) | 4.1 | 4.3 | |||
| Sedimentation ((0-15mm) | 12 | 14 | 28 | 21 | 13 |
| C3 (80-160mg/dl) | 147 | 134 | 177 | 171 | 162 |
| C4 (15-45mg/dl) | 52 | 51 | 50 | 51 | 51 |
(91) Submission ID#601004
A Novel Form of Partial Recessive IFN-gamma R2 Deficiency Caused by a Mutation of the Initiation Codon Presenting with a Severe Phenotype
Ayse Metin, MD, PhD1, Saliha Knk Yüksel, MD2, Belgin Gulhan, MD2, Aslnur Ozkaya Parlakay, MD2, Carmen Oleaga Quintas, MSci3, Jacinta Bustamante, MD, PhD4
1Prof. of Pediatric Immunology, MD, PhD, SBU, Ankara Children's Health and Diseases Hematology Oncology Training and Research Hospital, Ankara, Turkey
2Pediatric Infectious diseases Unit, Ankara Children's Health and Diseases Hematology Oncology Training and Research Hospital, Ankara Turkey
3M. Sci, Laboratory of Human Genetics of Infectious diseases, Necker Branch, INSERM UMR 1163, Imagine Institute, Necker hospital for Sick children, Paris, France
4Senior Investigator, Laboratory of Human Genetics of Infectious diseases, Necker Branch, INSERM UMR 1163, Imagine Institute, Necker hospital for Sick children, Paris, France
Introduction: Mendelian susceptibility of mycobacterial disease (MSMD) is characterized by infections caused by weakly virulent mycobacteria in otherwise healthy individuals. Known genetic etiologies disrupt IFN-gamma (IFN-g) immunity like IFNGR2. Germline bi-allelic mutations of IFNGR2 can underlie partial or complete deficiency of IFN-g receptor2 (IFN-gR2). Patients with partial IFN-gR2 deficiencies express dysfunctional molecule on the cell surface. We describe here a novel mutation within the start of IFNGR2 in two siblings born to a consanguineous parents and their clinical presentation.
Methods: We studied 2 siblings fron one kindred from Turkey by whole- exom sequencing (WES). Candidate mutation of IFNGR2 was analzed experimentally.
Conclusion: We describe a novel mutation in both siblings at the first codon of IFNGR2 that define a new form of partial recessive IFN-gR2 deficiency. The low amounts of full length IFN-gR2 confer a more severe clinical phenotype in these siblings than that of patients with other forms of partial recessive IFN-gR2 deficiency due to surface expressed dysfunctional receptors.
(92) Submission ID#601013
Maternal Diabetes Causing Atypical, Complete DiGeorge Syndrome
Richa Panara, MD1, Kiran Patel, MD2, Lisa Kobrynski, MD, MPH3, Gerald Lee, MD2, Kristine Vanijcharoenkarn, MD4, Meera Patrawala, MD4, Jennifer Shih, MD5
1Resident Physician, Department of Internal Medicine, Emory University School of Medicine
2Assistant Professor of Allergy, Department of Pediatrics, Emory University School of Medicine
3Associate Professor of Allergy, Department of Pediatrics, Emory University of School of Medicine
4Allergy and Immunology Fellow, Emory University School of Medicine
5Assistant Professor of Pediatrics and Internal Medicine, Department of Pediatrics, Division of Allergy and Immunology, Emory University School of Medicine
INTRODUCTION: DiGeorge Syndrome (DGS) is a primary immunodeficiency characterized by thymic hypoplasia, cardiac defects and hypoparathyroidism. Approximately 90% of cases of DGS are due to a chromosomal microdeletion at 22q11.2, however non-genetic etiologies that have been described in the literature include maternal diabetes, fetal alcohol syndrome, and prenatal exposure to isotretinoins.
CASE PRESENTATION: We describe an 8 month-old male born at 38 weeks to a woman with insulin dependent diabetes mellitus. At delivery, examination revealed microcephaly, small upper lips, small nares and pyriform aperture stenosis. Chest imaging revealed no thymic tissue, right-sided rib and spinal anomalies. Renal ultrasound was normal.
Newborn Screen showed critically low levels of TREC. Flow cytometry confirmed low T-cells: CD4 count 1 cell/ uL, CD8 count 7 cell/uL, CD3 count 8 cells/uL/ <1%, CD19 at 1398 cells/uL/79%. Helper and suppressor t-cells <1%. CD4/CD8 ratio 0.6. Lymphocyte proliferation to mitogens was absent to phytohemagglutinin (PHA) and decreased to pokeweed mitogen (PWM). Calcium and parathyroid hormone were low. Quantitative immunoglobulins: IGG 488mg/dL, IgA <8mg/dL, IgM 40mg/dL.
DNA microarray for 22q deletion, TBX1 testing, and CHD7 sequencing was negative. FISH for trisomy 13, 18, 21 was normal. Given the negative genetics, the cause of DGS in this case is likely maternal diabetes.
Patient has had a complicated course with recent admission for hypocalcemia requiring IV calcium infusions complicated by acute liver dysfunction with coagulopathy, MRSA and pseudomonas bacteremia, and tube feeding intolerance with abdominal distention and diarrhea. He was noted to have lymphadenopathy, generalized rash and eosinophilia, concerning for oligoclonal expansion of T-cells. Repeat testing showed increased total T cells from 43 cells/uL to 342 cells/uL with an increase in CD4 cells from 21 cells/uL to 293 cells/uL that were 96% memory. Cytokine panel IL2 receptor 1059 pg/ml suggested T-cell activation. EGD and colonoscopy biopsies showed descending colon and rectum colitis.
DISCUSSION: Maternal diabetes mellitus has been described as a possible etiology of DGS in several case reports. Infants born to insulin-dependent diabetic mothers are eight times more likely to have major malformations than infants of non-diabetic mothers. The cause of these defects is hypothesized to be from alteration of neural crest migration and development. Since 10% of DGS are not due to a chromosomal microdeletion at 22q11.2, it is prudent to consider maternal diabetes mellitus as an etiology for DGS, especially when the microdeletion is not found, but clinical presentation is suggestive of DGS.
(93) Submission ID#601038
Novel Mutation in the WAS Gene Causing a Phenotypic Presentation of Wiskott- Aldrich Syndrome
Elisa Ochfeld, MD1, Melanie Makhija, MD2
1Allergy/ Immunology Fellow, Northwestern University
2Attending, Northwestern University
Introduction/ Background: Wiskott- Aldrich Syndrome (WAS) is a rare X- linked disorder characterized by immunodeficiency, thrombocytopenia and atopic dermatitis. The phenotype is variable and depends on the type and location of the mutation in the WAS gene (located on Xp11.23). The treatment of choice for WAS is hematopoietic stem cell transplant (HSCT).
Objective: To present a clinical case with a novel genetic mutation causing WAS.
Results: We describe a now 5 month-old infant who presented at birth with petechiae and bruising of his legs and abdomen after an elective cesarean section at 38 weeks gestation. He was found to have severe neonatal thrombocytopenia based on complete blood count (CBC). He received a platelet transfusion on day of life (DOL) 1, was discharged home with stable platelets on DOL 5 but had worsening thrombocytopenia on DOL 8 requiring re-admission for further work up (platelet count 40,000). Platelet count reached a nadir of 26,000 on DOL 11. He was initially thought to have neonatal alloimmune thrombocytopenia. Parental testing showed no platelet abnormalities or platelet alloantibodies. He re-presented for acute lower GI bleeding on DOL 19. His platelet morphology at that time showed small platelets, though previously his platelet size was reportedly normal. Genetic testing for WAS revealed a variant of unknown significance (VUS) in the WAS gene (WAS NM_000377.2. Exon 2. C.151GT. P.VAL51Phe. Hemizygous. VUS. Chr X: g: 48542690G>T). This mutation, WAS c.151G > T (p. Val51Phe) is a missense variant in exon 2 which changes an amino acid valine at codon 51 to phenylalanine. This is a region essential for interaction with the WASP interacting protein (Rajmohan, 2009). This variant has not been reported in the literature as pathogenic and has not been reported as a known variant in the general population. In silico computational evidence predict the variant to be damaging (Polyphen2, Mutation Taster, SIFT, HSF). Further immune evaluation was performed. Flow cytometry revealed a low absolute CD3 count, low absolute CD4 count, low absolute CD8 count, CD4/CD8 ratio of 5:1, with normal NK cell count and low absolute CD19 count. His immunoglobulins were abnormal, with low IgM and elevated IgA with normal IgG and slightly elevated IgE. WAS protein expression testing revealed decreased expression of WAS protein on lymphocytes (0.24 ratio, normal range 0.71-1.31). At the time of his initial presentation to immunology at age 3 months, he had not developed atopic dermatitis nor any significant infections. However, at 4 months he was admitted for otitis externa with adjacent facial cellulitis, which responded to IV antibiotics. During that admission he was given IVIG and started on Bactrim prophylaxis. He has since developed significant seborrheic dermatitis skin lesions. Family history was not significant for immunodeficiency. No known WAS in family, no consanguinity. Maternal genetic testing was positive for the same missense mutation in the WAS gene. The patient is currently undergoing stem cell transplantation with a matched sibling donor.
Conclusion: We report a new mutation in the WAS gene that causes a phenotypic presentation of Wiskott-Aldrich Syndrome.
(94) Submission ID#601045
Common Variable Immunodeficiency (CVID) Associated with Variants in Neuroblastoma Amplified Sequence (NBAS) Gene
Joseph A. Church, MD1, Dominic Lenz, MD2, Christian Staufner, MD3, Georg Hoffmann, MD4
1Professor, Pediatrics, Children's Hospital Los Angeles and Keck School of Medicine of U.S.C.
2Resident, Department of General Pediatrics, University Hospital Heidelberg, Heidelberg Germany
3Consultant, Department of General Pediatrics, University Hospital Heidelberg, Heidelberg, Germany
4Professor of Pediatrics, Department of General Pediatrics, University Hospital Heidelberg, Heidelberg, Germany
Over 20 genes have been reported to cause monogenic CVID.
A 4 year old girl presented with recurrent pneumonias and a diagnosis of CVID. The parents sought a second opinion. Born at 33 weeks gestational age, she was "always smaller and sicker than her friends," and in the prior 8 months she had 3 episodes of pneumonia with fever to 104F requiring emergency department treatment. Two of these were associated with RSV and metapneumovirus, respectively.
Laboratory evaluation confirmed low levels of IgG (326 mg/dL) IgA (7) and IgM (6), and poor antibody responses to tetanus, Hib, MMR and VZV antigens. Her response to Pneumovax was marginal (+12 of 23 serotypes tested) and transient (#2 of 23 after 4 months). CBC including smear and chemistry panel were normal. Results of routine lymphocyte subset analysis were normal except for relatively low NK cells (3%). On examination she appeared well. Ht and wt were at the 20th%iles. There was no evidence of active infection, hepatosplenomegaly, lymphadenopathy or skin rash. She had a small VSD noted since birth, and bilateral optic atrophy identified during routine ophthalmology follow-up for prematurity. Ig replacement was initiated and continues as weekly infusions of SCIG, maintaining IgG levels over 1000 mg/dL.
At 4.5 years she was hospitalized for pneumonia, and transiently elevated transaminases (AST 117 U/L, ALT 552) were noted for the first and only time.
Exome sequencing revealed compound heterozygous, likely pathogenic, variants limited to the C-terminal domain of the NBAS gene. The product of this gene is a component of the syntaxin 18 complex that is implicated in Golgi to ER retrograde transport.
Mutations in NBAS are associated with Infantile Liver Failure Syndrome 2 (ILFS2, OMIM#616483) and Short Stature, Optic Atrophy and Pelger-Huet Anomaly in neutrophils (SOPH syndrome). Hyopgammaglobulinemia and low NK cell numbers have been reported in affected patients, and NBAS deficiency is listed in the IUIS 2017 PID Committee report, but under "Defects in intrinsic and innate immunity."
Clinical features reported in NBAS deficiency include familial consanguinity, fever-related recurrent liver crises, short stature (<3%ile), skeletal dysplasia, cervical instability (small C1-C2) and retinal dystrophy. To the present the patient has not exhibited these findings.
Our patient's NBAS variants are being studied for functional validity. A skin biopsy was performed and fibroblasts cultured for analysis. In contrast to our patient's variants that are both in the C-terminal domain, most reported cases have at least 1 mutation affecting domains further toward the N-terminus. To the present our patient's fibroblasts do not show a clear reduction in NBAS protein, and protein interaction partners were not diminished. Additional studies planned include challenging the fibroblasts with elevated temperature to measure any reduction in variant NBAS expression and function.
(95) Submission ID#601049
An Unexpected Diagnosis in a Premature Infant with Persistent Fever, Respiratory Distress and Significant Neutrophilia: Congenital Tuberculosis
Idil Ezhuthachan, MD1, Sara Sussman, MD2, Renata Schillizzi, NP3, Shanika Uduwana, MD1, Lily Glater, MD4, Blanka Kaplan, MD5, Todd Sweberg, MD6, Maria Santiago, MD7, Stefan Hagmann, MD7, Vincent Bonagura, MD8, Gina Coscia, MD5
1Fellow, Northwell Health
2Fellow, Department of Pediatrics, Zucker School of Medicine at Hofstra Northwell School of Medicine
3Nurse Practitioner, Northwell Health
4Pediatric Intensivist, Northwell Health Cohen Children's Medical Center
5Assistant Professor of Medicine and Pediatrics, Northwell Health
6Assistant Professor of Pediatrics, Northwell Health
7Associate Professor of Pediatrics, Northwell Health
8Professor of Medicine and Pediatrics, Division of Allergy & Immunology, Donald and Barbara Zucker School of Medicine at Hofstra/Northwell
Congenital tuberculosis (CTB) is a rare disease most often associated with maternal genitourinary (GU) tuberculosis (TB) or disseminated TB. Due to infertility caused by GU TB, CTB is rarely reported even in endemic countries. Infants can acquire TB hematogenously via the placenta or umbilical vein or by fetal aspiration of infected amniotic fluid. Presenting symptoms include respiratory distress, fever, hepatosplenomegaly, poor feeding, lethargy, and low birth weight.
We report a premature female infant conceived via in vitro fertilization (IVF), who was born to Indian immigrant parents at 29 weeks of gestation due to preterm premature rupture of membranes. Maternal history was significant for pulmonary TB at 9 years of age. She denied abdominal or GU symptoms. Infants NICU course was complicated by opacifications in the right lung and leukocytosis with neutrophil predominance, identified during evaluation of frequent apnea and bradycardia episodes at 1 month of age. Clinical improvement was noted after treatment with vancomycin, amikacin and piperacillin-tazobactam; however, leukocytosis of unknown etiology persisted. At 1.5 months of age she was discharged to inpatient rehabilitation.
At 3 months of age, she was readmitted for fever and respiratory distress. During this admission, an immune evaluation was undertaken due to persistence of symptoms along with unresolved leukocytosis with a peak of 58,000 cells/l with neutrophilia to 42,850 cells/l, and chest CT evidence of progressive multifocal lung disease worse in the right upper lobe despite empiric treatment with broad-spectrum antibiotics. Infectious work-up was negative, including acid-fast bacilli testing from bronchoalveolar lavage. Due to the pronounced and persistent leukocytosis and neutrophilia, a primary immune defect was suspected. Immune evaluation included: normal immunoglobulins (Ig) G, A, and E, elevated IgM, vaccine-specific antibody titers protective to diphtheria and 9 of 13 Streptococcus pneumonia strains, mildly elevated T and B cells, a normal flow cytometry for dihydrorhodamine, myeloperoxidase stain and glucose-6-phosphate dehydrogenase level, as well as a peripheral smear with no giant azurophilic granules. Her primary immunodeficiency genetic panel was unrevealing. She underwent lung biopsy via video-assisted thoracoscopic surgery, which showed non-caseating granulomas and eventual growth of multi-drug-resistant Mycobacterium tuberculosis (MTB). Upon treatment with an appropriately adjusted anti-tuberculosis regimen, she showed rapid clinical and laboratory improvement. Endometrial samples obtained from mother showed GU TB, confirming the diagnosis of CTB.
The slow-growing nature of MTB that resulted in delayed diagnosis, along with the presence of non-caseating granulomas and persistent neutrophilia, prompted an immune work up that was completely normal. This case demonstrates the importance of considering CTB in the differential diagnosis of an infant presenting with severe lung infection, persistent neutrophilia, suboptimal response to broad-spectrum antibiotics and relevant epidemiologic risk factors. Furthermore, in the setting of appropriate parental exposures and infertility prompting the use of IVF, maintaining a high level of suspicion of CTB can aid in earlier diagnosis of affected neonates.
(96) Submission ID#601056
Herpes Simplex Virus Whole Genome Sequencing for Antiviral Resistance in a Child with DOCK8 Deficiency and Chronic Infection
Sean Stout, MD1, Alexander Greninger, MD, PhD, MS, M.Phil.2, Rangaraj Selvarangan, PhD3, Alexandra F. Freeman, MD4, Brandon Newell, MD5, Erin Stahl, MD6, Dwight Yin, MD, MPH7
1Pediatric Resident, Department of Pediatrics, Children's Mercy Hospital
2Department of Laboratory Medicine, University of Washington School of Medicine
3Department of Pathology and Laboratory Medicine, Children's Mercy Hospital; School of Medicine, University of Missouri-Kansas City
4Director, Primary Immune Deficiency Clinic, Laboratory of Clinical Immunology and Microbiology, NIAID, NIH, Bethesda, MD, USA
5Division of Dermatology, Department of Pediatrics, Children's Mercy Hospital; School of Medicine, University of Missouri-Kansas City
6Section of Ophthalmology, Department of Surgery, Children's Mercy Hospital; School of Medicine, University of Missouri-Kansas City
7Division of Infectious Diseases, Department of Pediatrics, Children's Mercy Hospital; School of Medicine, University of Missouri-Kansas City
Background: Patients with dedicator of cytokinesis 8 (DOCK8) deficiency are prone to severe, recurrent or chronic mucocutaneous herpes simplex virus (HSV) infections that may develop antiviral resistance. We present the case of a child with DOCK8 deficiency and chronic, resistant HSV-1 mucocutaneous infections to illustrate the potential clinical utility of an investigational viral whole genome sequencing approach to detecting active and latent HSV resistance mutations longitudinally.
Methods: We abstracted clinical and laboratory data in a 14 year old boy with DOCK8 deficiency with repeated viral culture growth of HSV-1. HSV-1 DNA from seven positive viral culture specimens collected during 2015-2018 were sequenced on an Illumina® MiSeq to >150X depth. Consensus genomes were called using an established HSV genome pipeline, and reads were mapped to the HSV-1 strain reference genome (NC_001806). Sequence variants were checked against an online database of UL23 (thymidine kinase) and UL30 (DNA polymerase) variants associated with antiviral resistance.
Results: The patient had low CD4+ T cells (initial 248 cells/mm3), eosinophilia, elevated IgE (initial 9660 kU/L), severe eczematous dermatitis, chronic obstructive and interstitial lung disease, growth delay, and presented with recurrent infections including Staphylococcus aureus, Candida albicans, JC virus, and HSV-1. He was receiving prophylaxis with subcutaneous immunoglobulin G, trimethoprim-sulfamethoxazole, and acyclovir. Family declined hematopoietic stem cell transplantation. Mucocutaneous HSV-1 infections extensively involved his face, trunk, and extremities with more severe infections involving his cornea, lips, perineum, scalp, and periorbital regions. Scalp and periorbital lesions were proliferative and edematous papules and plaques consistent with HSV vegetans (Figures 1-2) and confirmed by scalp biopsy. Although early HSV-1 infections responded to oral or IV acyclovir, clinical response decreased over time, requiring advancement of therapies to high-dose acyclovir IV, foscarnet IV, cidofovir IV, topical cidofovir cream, and/or interferon-alpha with variable clinical response.
Phenotypic testing detected acyclovir resistance in HSV isolated from four samples while the patient was on acyclovir and no resistance in a sample while not on acyclovir. Phenotypic foscarnet resistance was detected in one sample without prior patient exposure to foscarnet. Viral whole genome sequencing detected the UL23 variant R176Q (associated with acyclovir resistance) on all specimens, whether on acyclovir or not, and the UL30 variant T821M (associated with acyclovir and cidofovir resistance) only when on cidofovir. When phenotypic testing and genome sequencing were discordant, clinical response appeared to be more consistent with genome sequencing results.
Conclusions: This patient with DOCK8 deficiency illustrates the potential severity of chronic, resistant mucocutaneous HSV-1 infection. Viral genome sequencing for antiviral resistance mutations may provide additional information about the presence of clinically significant variants, which may result from detecting smaller or latent HSV-1 sub-populations.
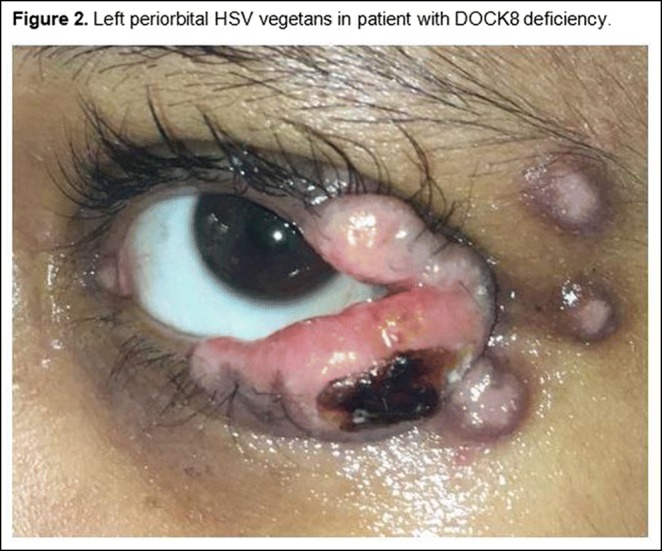
(97) Submission ID#601066
Variable Phenotypes in Three Patients with Two Novel STAT3 Gain of Function Mutations
Musa G. Bolkent1, Melissa Elder, MD, PhD2, Tiphanie Vogel, MD, PhD3, Akaluck Thatayatikom, MD, RhMSUS4
1Fellow, University of Florida
2Division Chief, University of Florida
3Assistant Professor, Baylor College of Medicine, 1Texas Childrens Hospital Center for Human Immunobiology and Division of Immunology, Allergy and Rheumatology
4Associate Professor, University of Florida
Introduction: Gain-of-function (GOF) mutations in signal transducer and activator of transcription 3 (STAT3) cause autosomal dominant, early-onset autoimmune disease. STAT3 GOF syndrome is characterized by lymphadenopathy (LAD), hepatosplenomegaly (HSM), autoimmune cytopenias, hypogammaglobulinemia and other solid organ autoimmunities, including interstitial lung disease, enteropathy, endocrinopathy, arthritis, and scleroderma-like skin changes. We report three children with two different novel missense STAT3 GOF mutations.
Objectives: To describe novel GOF mutations of STAT3 that expand our current understanding of the clinical manifestations of the disease.
Methods: Molecular investigation was performed in suspected patients using whole exome sequencing (WES). The STAT3 mutations caused GOF were tested using a luciferase assay.
Results: Results of WES identified three children with novel STAT3 mutations at the same amino acid (Table 1). Standardized luciferase assay confirmed the mutations caused GOF in the STAT3 protein. Case#1 was a 15-year-old Caucasian male who initially presented with recurrent otitis media, persistent HSM, LAD, and hypogammaglobinemia (IgG <170 mg/dL) at 2 years of age. He was diagnosed with common variable immunodeficiency (CVID) and chronic arthritis when he was 6 and 9 years of age, respectively. Subsequently, he developed hepatitis and recurrent pneumonia with Mycobacterium avium complex (MAC). His arthritis partially responded to anti-tumor necrosis factor (TNF) agents and tofacitinib, but did not respond to anti-interleukin-6 treatment. A combination of anti-TNF inhibitor, tofacitinib, and low dose prednisone was required to control his arthritis. Case#2 was an 18-year-old Caucasian male who initially developed thrombocytopenia, hypogammaglobulinemia (IgG <110 mg/dL), recurrent otitis media, pneumonia, Crohn's disease, celiac disease, LAD and failure to thrive at 2 years of age with more recent development of HSM. He required only immunoglobulin replacement therapy. Case#3 is a 9-year-old Caucasian male, the half-brother of case#2, who initially presented with recurrent pleural effusion and bilateral pulmonary infiltrates, HSM, LAD, abdominal distension and ascites at 7 years of age. A transbronchial lung biopsy revealed chronic eosinophilic pneumonitis. Liver biopsy showed increased eosinophils in the sinusoids with diffuse enlargement of hepatocytes, but without hepatitis. Colon biopsy revealed minimal colonic eosinophilia. His pulmonary infiltrates and pleural effusion responded to prednisone, and he has not required additional treatment for past 1.5 years.
Conclusions: The clinical manifestations of the same genetic variant may be variable and unpredictable even in the same family. STAT3 GOF syndrome should be considered in children with multisystem autoimmune diseases, LAD, HSM and low switched memory B cells regardless of presence of hypogammaglobulinemia or history of recurrent infections.
Table.1 Patient Characteristics
| Pt | Age at onset (years) | STAT3 GOF mutation | Class switched memory B cells | Infections | GI | MSK* | Lympho-proliferation | Other |
| #1 | 2 | c.1165 A>T (p.T389S) | <1% | Sinusitis, otitis media, pneumonia | Hepatitis | Chronic arthritis | LAD, HSM | Uveitis |
| #2 | 2 | c.1165 A>G (p.T389A) | 1% | Pneumonia | Crohn’s disease | None | LAD, HSM |
*ITP, Portal HTN |
| #3 | 7 | c.1165 A>G (p.T389A) | <1% | none | Abdominal distensionwith colonic eosinophilia | None | LAD, HSM | Chronic eosinophilic pneumonitis |
(98) Submission ID#601069
Immunophenotype and Metabolic Characteristics of EBV-Specific T Cells Generated Using Different Manufacture Approaches
Danielle E. Arnold, MD1, David M. Barrett, MD, PhD2
1Fellow In Training, Children's Hospital of Philadelphia
2Attending Physician, Children's Hospital of Philadelphia
Background: Patients with primary immune deficiencies characterized by severe T lymphopenia and/or poor T cell function and patients post-hematopoietic cell transplantation are at high risk of severe viral infections. Antiviral medications are expensive, not always effective and associated with significant toxicity and/or long-term side effects. As such, there has been increasing interest in the use of donor-derived or third-party virus-specific T cells (VSTs), and several studies have demonstrated efficacy of VSTs generated using various manufacture strategies. However, in depth immunologic and metabolic characterization of VSTs has not been reported, limiting correlative investigations into efficacy.
Methods: EBV-VSTs were generated from apheresis T cells collected from healthy donors using three methods: (1) stimulation and expansion with HLA-matched EBV-lymphoblastoid cell lines (LCLs) purchased from Astarte Biologics or Sigma-Aldrich over a period of 4 weeks, (2) stimulation with EBV PepTivator from Miltenyi followed by expansion over 9-12 days with different cytokines, and (3) stimulation with EBV PepTivator followed by isolation of activated cells using the IFN-gamma capture system from Miltenyi. Immunophenotyping by flow cytometry was performed using the Miltneyi MACSQuant Analyzer. The NanoString nCounter system was used to measure gene expression for metabolic pathway analysis, and the Agilent Seahorse XF cell mito stress test system was used to measure mitochondrial respiration.
Results: EBV-VSTs generated using LCLs or PepTivator plus IL-15 both resulted in a high percentage of CD8 T cells skewed to the effector memory and terminal effector memory phenotype with high expression of the exhaustion markers PD-1, TIM-3, and LAG-3. Conversely, EBV-VSTs generated using PepTivator plus IL-4 and IL-7 and the IFN-gamma capture system resulted in a mixed CD4 and CD8 T cell population with a high number of central memory T cells and lower percentage of cells positive for PD-1, TIM-3, and LAG-3. Stimulation with PepTivator followed by expansion with IL-2 resulted in an intermediate immunophenotype. NanoString results demonstrated upregulation of the glycolytic pathway in EBV-VSTs stimulated with PepTivator followed by expansion with IL-2 or IL-15 compared to EBV-VSTs generated using the other manufacture approaches. The Seahorse mito stress test demonstrated that the PepTivator plus IL-2 EBV-VSTs had a significantly lower spare respiratory capacity than other EBV-VSTs and a low extracellular acidification rate despite upregulation of the glycolytic pathway. The Peptivator plus IL-4 and IL-7 EBV-VSTs had the highest basal oxygen consumption rate, ATP-linked respiration, and extracellular acidification rate.
Conclusions: Manufacture of EBV-VSTs using the various approaches currently employed clinically results in T cell pools with different immunophenotypes and different metabolic profiles. EBV-VSTs stimulated with PepTivator followed by expansion in IL-4 and IL-7 and EBV-VSTs isolated using the IFN-gamma capture system have immunophenotypes and metabolic phenotypes suggestive of potential greater in vivo persistence, whereas EBV-VSTs expanded in IL-2 and IL-15 have characteristics correlated with increased effector function. However, these VSTs are more likely to be short-lived and to have impaired metabolic fitness. These phenotypes will enable better correlation with clinical results and suggest combinatorial approaches depending on clinical indication.
(99) Submission ID#601079
Efficacy, Tolerability and Safety of Cutaquig®, a New Subcutaneous Human Immunglobulin 16.5% in Adult Patients with Primary Immunodeficiencies
Latysheva E.1, Rodina Y.2, Sizyakina L.3, Totolian A.4, Tuzankina I.5
1National Research Center Institute of Immunology FMBA, Russia
2Dmitry Rogachev National Research Center of Pediatric Hematology, Oncology and Immunology, Moscow
3State Medical University, Rostov
4Pasteur Institute, Saint-Petersburg
5Institute of Immunology and Physiology of the Ural Branch of the Russian Academy of Sciences, Yekaterinburg
Introduction: Majority of patients with primary immunodeficiencies (PID) require life-long replacement therapy with immunoglobulins (Ig) to prevent severe infections and irreversible complications. In addition to safety and efficacy, tolerability and convenience of administration of Ig products are essential factors for patients. A new 16.5% Ig preparation octanorm (Octapharma, Lachen; tradename cutaquig® in North America) has been developed for subcutaneous administration (SCIG) derived from the established manufacturing process of Octapharmas intravenous Ig (IVIG) brand octagam®.
Objectives: Primary outcome was assessment of the efficacy of octanorm in preventing serious bacterial infections. Main secondary endpoints included (among others) evaluation of tolerability and safety of octanorm, the number and rate of other infections, number of days missed at work, and use of antibiotics.
Methods: A prospective, open-label, non-controlled, single-arm phase 3 study involving 25 adult patients with PID was conducted at 5 Russian centers. Patients treated with at least 4 infusions of IVIG prior to enrollment and with IgG trough levels 5.0 g/L underwent an 8-week wash-in/wash-out period followed by a 24-week efficacy period. During the study, patients received weekly administrations of octanorm at the same monthly dose as during previous IVIG treatment (monthly IVIG dose divided by 4 for weekly dose). In total, each patient received 32 SCIG infusions.
Results: Twenty-four patients completed the study. One patient terminated early (after infusion 7, during wash-in/wash-out phase; personal reasons). Mean age was 35.24 years (range 18-64 years). Fifteen patients (60%) were female and 10 patients (40%) male.
No serious bacterial infections were recorded. During the efficacy period a total of 26 non-serious infections was observed in 14 patients. Seventeen infections in 11 patients were of mild and 9 infections in 5 patients of moderate intensity. The infection rate per person-year was 2.37.
In total 25 patients received 775 infusions of study drug. The average dose of cutaquig® was 0.11 g/kg/week. During the entire study, 59 systemic adverse events were reported (including 34 infections). Three of these systemic adverse events were rated as related to study drug, all were non-serious. There was no serious or significant adverse event nor was there an adverse event leading to withdrawal. Infusion site reactions were reported for 15% of infusions.
Serum IgG trough levels were nearly constant during the efficacy period. Median IgG trough levels were 8.15 g/L at Screening, 9.52 g/L at the end of wash-in/wash-out period and 10.71 g/L at the Termination Visit. One patient had a trough level 5g/L at 2 visits during the efficacy period and the dosing was subsequently adjusted for this patient.
During the primary treatment period 10 patients (41.7%) used antibiotics in 19 treatment episodes (total of 229 treatment days; range 5-76 days) and 3 patients had 4 absences from work or school due to infections (total of 14 days of absence).
Conclusion: This study demonstrated that the new subcutaneous human normal immunoglobulin 16.5% is well tolerated, safe and effective in adult patients with PID.
(100) Submission ID#601082
Infective Endocarditis, Osteomyelitis of Skull and Invasive Aspergillosis in a Child with Chronic Granulomatous Disease
Gummadi Anjani, MBBS;MD Pediatrics1, Amit Rawat, MD (Pathology) PDCC (Laboratory Immunology) PDCC (Nephropathology) MAMS2, Ankur Jindal, MD;DM (pediatric clinical immunology and rheumatology)3, Pandiarajan Vignesh, MD;DM (pediatric clinical immunology and rheumatology)3, Ankita Singh, MD4, Surjit Singh, MD; DCH (Lon.); FRCP (Lon.); FRCPCH (Lon.); FAMS5
1Fellow in pediatric clinical immunology and rheumatology, Postgraduate institute of medical education and research
2Professor of Pediatric Allergy and Immunology, Paediatric Allergy Immunology Unit, Department of Paediatrics, Advanced Paediatric Centre, Postgraduate Institute of Medical Education & Research
3Assisstant professor, department of pediatrics, Postgraduate Institute of Medical Education & Research
4fellow in pediatric clinical immunology and rheumatology, Postgraduate Institute of Medical Education & Research
5Head, Department of Pediatrics and Chief, Allergy Immunology Unit, Advanced Pediatrics Centre,Principal Investigator, Indian Council of Medical Research (ICMR) Centre for Advanced Research in Primary Immunodeficiency DiseasesVice-President, Indian Rh, Postgraduate Institute of Medical Education & Research
Background: Children with chronic granulomatous disease (CGD) are at high risk for fungal infections (especially with Aspergillus species) and these infections usually have contiguous site involvement. Most patients have pulmonary presentation. Infective endocarditis and fungal osteomyelitis of skull are distinctly unusual. We report one such case.
Case: A 6-year-old boy, born out of a non-consanguineous marriage, presented with soft tissue swellings of skull for 2 months. His past history was significant with an episode of pneumonia at 1 year and recurrent soft tissue swellings all over the body since 1½ years of age. On examination he was wasted, had signs of micronutrient deficiency, rickets, pallor, cervical lymphadenopathy and two abscesses, 12x4 cm on right temporo-parietal region and 4x3 cm over left frontal region. He was also found to have hyperdynamic precordium with an ejection systolic murmur. Investigations revealed hemoglobin 85g/L; platelet count 7.34x109/L; total leukocyte count 13x109/L(N60/L23/M13/E1); elevated C-reactive protein( 244 mg/L) and a raised erythrocyte sedimentation rate(104 mm 1sthr). Chest x ray revealed cardiomegaly (cardiothoracic ratio 67%) and 2D echocardiography showed vegetation of 6x3 mm over the anterior mitral leaflet suggestive of infective endocarditis. Blood and urine cultures were sterile. Culture from pus over the temporo-parietal abscess showed growth of Aspergillus fumigatus. Human immunodeficiency virus serology was non-reactive. Immunoglobulin profile revealed elevated IgG 21.20g/L (5.40-16.10g/L) and IgA 5.66 g/L(0.5-2.4g/L); IgM was 1.63 g/L(0.50-1.8g/L). In view of strong suspicion of CGD, nitroblue tetrazolium dye reduction test (NBT) was carried out- it revealed no reduction and Dihydrorhodamine (DHR) assay showed a low stimulation index (4.34). Flow cytometry for gp 47 phox and gp 67 phox was normal and DHR of mother did not reveal X linked carrier state. Contrast enhanced computerized tomography (CECT) of head showed osteomyelitis of the calvarial bones. Contrast enhanced magnetic resonance imaging (CEMRI) brain showed heterogeneously enchancing soft tissue lesion in the scalp at right fronto-parietal region and left frontal region with underlying bony destruction suggestive of osteomyelitis. He was given intravenous antimicrobials (ceftriaxone, gentamycin, cloxacillin, voriconazole). After 6 weeks of therapy, he showed resolution of findings on MRI brain and a repeat 2D echocardiography showed significant decrease in size of mitral leaflet vegetation.
Conclusion: This case highlights a rare presentation of CGD with infective endocarditis and skull osteomyelitis due to Aspergillus fumigatus. To the best of our knowledge, this has not been reported previously.
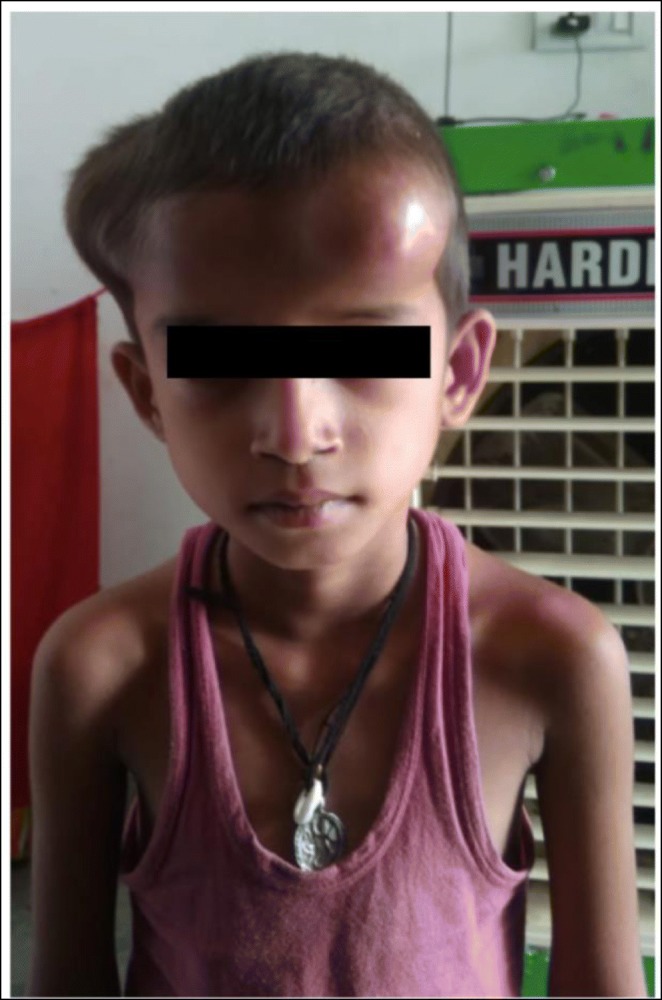
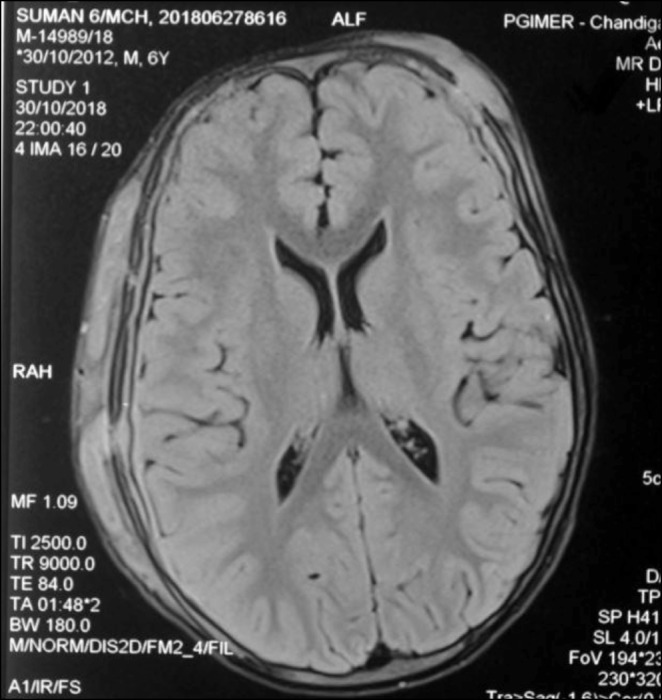
(101) Submission ID#601094
Enigmas of IL-12R1 Deficiency: Contemporary of Two Disease, Mendelian Susceptibility to Mycobacterial Disease and Crohn Disease
Roya Sherkat, MD 1, Razieh Khoshnevisan, MD2, Nioosha Nekoei, PhD3, Christoph Klein, MD, PhD4, Daniel Kotlarz, MD , PhD5, Mahdieh Behnam, MSc6, Soodabeh Rostami, PhD7, Majid Yaran, PhD8, Hamid Tavakoli, MD9, Abbas Rezaei, PhD10
1Associate Prof. ,Head of Acquired Immunodeficiency Research Center, Acquired Immunodeficiency Research Center, Isfahan University of Medical Sciences , Isfahan , Iran
2Phd, Acquired Immunodeficiency Research Center, Isfahan University of Medical Sciences , Isfahan , Iran
3Research Assistant, Acquired Immunodeficiency Research Center, Isfahan University of Medical Sciences , Isfahan , Iran
4Director, Dr. von Hauner Children's Hospital, University Hospital, LMU, Munich Germany
5Senior Scientist, Department of Pediatrics Dr. von Hauner Childrens Hospital, Ludwig- Maximilians-University, Munich, Germany
6Research Assistant, Medical Genetics Laboratory, Alzahra University Hospital, Isfahan University of Medical Sciences, Isfahan, Iran
7Assistant Prof., Nosocomial Infection Research Center, Isfahan University of Medical Sciences, Isfahan, Iran.
8Scientist, Acquired Immunodeficiency Research Center, Isfahan University of Medical Sciences , Isfahan , Iran
9Associate prof., Integrative Functional Gastroenterology Research Center, Isfahan University of Medical Sciences, Isfahan, Iran
10Prof., Immunology Department, Isfahan University of Medical Sciences, Isfahan, Iran.
Background: Genetic defect in IL12R1 affect cellular immunity, underlie Mendelian Susceptibility to Mycobacterial Disease (MSMD) and Inflammatory bowel disease (IBD) through different pathways.
We present for the first time a patient with IL-12R1 deficiency from a consanguine family with two different phenotypes. Initially diagnosed as Crohn's disease prior to the MSMD diagnosis.
Method and Material:Patient was referred to the clinical Immunology and Allergy clinic at the at Alzahra University Hospital for immunological and genetic evaluation . Blood samples from patient, his family and healthy donor controls were collected upon informed consent.
In this study, we investigated effect of IL12R1 mutation in IL-12/IFN- axis by evaluation of patients whole blood cell response to IL-12 and IFN-, IL-12R1 expression in PBMCs and T cell blasts. Also Whole-exome sequencing has been performed.
Result and Discussion: A 26 years old male from consanguine family , with history of right sub-axillary BCG lymphadenitis, recurrent mouth ulcers , chronic diarrhea in childhood and appendectomy at age of 5 was investigated. Based on his clinical presentation abdominal pain, significant weight loss, chronic and bloody diarrhea , endoscopic and pathological findings treatment for Crohn's disease (CD) was started at the age of seven . Unfortunately, protracted patient's symptoms ends up to resection of his colon and colostomy two years later. He was presented with multi focal osteomyelitis at the age of 13 . Although no bacteria was detected in PCR and tissue culture of the bone biopsy and the patient was not responded to antibacterials , he had a dramatic response to empirical anti mycobacterial treatment and his severe bone pain and lesions were healed. Even though the bone manifestations were completely controlled, he continuously was under treatment for his gastrointestinal symptoms. Genetic analysis was confirmed segregation of homozygous mutation in 3splice site of exon 15 in IL-12R1. Expression of gene was completely abolished in PBMCs of patient and the surface expression of IL12RB1 was not detectable in T cell derived PBMCs of the patient compared to healthy control. Furthermore, did not response to IL12 stimulation since we could not detect increase of INF- after stimulation with Il12 and BCG.
Our patient received BCG vaccination at birth and had BCG lymphadenitis as an infant, CD and mycobacterial multifocal osteomyelitis as a child. Furthermore there are some evidences which indicate the role of atypical mycobacterial infections as a trigger for CD.
Conclusion: We reported for the first time contemporary MSMD and IBD in 26 years old patient, who had impaired IL-12 signaling and abolished IL12 R1 expression in PBMCs and T cell blast. However, mycobacterial osteomyelitis is a typical phenotype of MSMD patients with deficiency in IFN-R1 or STAT, there were no mycobacterial osteomyelitis reported in IL-12R1 deficient patients.
(103) Submission ID#601130
NBAS Compound Heterozygous Variants as a Cause of Recurrent Acute Liver Failure Triggered by Common Childhood Infections
Suthida Kankirawatana, MD1, Anna Hurst, MD2, Janaina Nogueira, MD3, Julie Jones, PhD4, Prescott Atkinson, MD, PhD5
1Assistant Professor, Division of Allergy and Immunology, Department of Pediatrics, Children's of Alabama, UAB
2Assistant Professor, Division of Genetics, Department of Pediatrics, Children's of Alabama, UAB
3Associate Professor, Division of Gastroenterology, Department of Pediatrics, Children's of Alabama, UAB
4 Director, Clinical Genomic Sequencing Program, Greenwood Genetic Center
5Professor, Division of Allergy, Immunology, Department of Pediatrics, Children's of Alabama, UAB
Background: Advanced genetic studies help explain the occurrence of many undiagnosed, rare conditions. Recently, NBAS variants were identified as a causative basis of recurrent liver failure in infants (Infantile Liver Failure Syndrome 2, ILFS2). The NBAS (Neuroblastoma Amplified Sequence) gene encodes a protein involved in Golgi to ER retrograde transport. NBAS functions seem to be broad and loss of function variants in NBAS have been associated with multisystem manifestations.
Case report: A 5y 9m old Chilean male presented to the ER with a three day history of vomiting, diarrhea and one day of fever (38.3° F). On examination he was pale, lethargic, and tachycardic. A chemistry profile revealed markedly elevated liver enzymes, increased bilirubin, and coagulopathy, consistent with the acute hepatic failure (ALT 6291, AST >4000, total bilirubin 3.49 (2.82 DB), GGT 52, and INR of 2.1). He was hospitalized, given Vitamin K, and kept on intravenous fluids, ursodiol, and anti-pyretics. His liver function improved significantly within 5 days of admission (ALT was down to 980, AST 45, total bilirubin 1.62). Work-up of possible etiologies including autoimmunity and infectious hepatitis was negative. Liver sonogram was normal, but liver biopsy was consistent with acute hepatitis with some necrosis. Urine organic acid and plasma amino acid screens were not consistent with any inherited metabolic disorders. His parents recalled two previous episodes of liver failure at ages 3 and 4 years. Both were preceded with a mild febrile illness and non-specific symptoms including fever, coughing, vomiting, diarrhea, lethargy, and decreased PO intake. These subsequently were followed by jaundice and marked elevation of liver enzymes. Flu A and adenovirus were identified as causes of febrile illnesses of the two previous episodes. For this admission, adenovirus was found in the respiratory secretions and a mild EBV viremia was also detected. Genetic evaluation in Chile was reportedly normal. After a literature review we obtained sequencing of NBAS which revealed two variants: c.2827G>T,p.Glu943* and NBAS c.2951T>G, p.IIe984Ser. Both variants have been previously reported in patients with an infantile onset, recurrent liver failure syndrome. His other clinical features include developmental and speech delays, failure to thrive, and facial dysmorphism. He also has a history of recurrent ear infections and has had 3 sets of tympanostomy tubes. Further testing was limited due to the lack of insurance coverage.
Conclusion: NBAS deficiency is a newly described syndrome of recurrent acute liver failure that occurs early in life. Once individuals have survived to adulthood they do not seem to develop liver failure with illness. Typically, liver crisis is triggered by a common childhood febrile illness. The mechanism of disease is thought to be thermal instability of hepatocytes which improves over time in most cases. However, although spontaneous recovery can occur following the crises, each episode can be fatal or result in permanent liver damage required liver transplantation. Increased awareness of this disease will lead to the early establishment of the diagnosis. Appropriate and timely management of fever at the onset of illness can significantly improve outcome in this potentially fatal disease.
(104) Submission ID#601139
Molecular Study in Children with Chronic Granulomatous Disease (CGD) at a Tertiary Care Center in North India
Dharmagat Bhattarai, MD, DM Fellow1, Pandiarajan Vignesh, MD;DM (pediatric clinical immunology and rheumatology)2, Madhubala Sharma, PHD scholar3, Jitendra Shandilya, PHD scholar3, Amit Rawat, MD, PDCC4, Kohsuke Imai, MD, PHD5, Shigeaki Nonoyama, MD, PHD6, Osamu Ohara, PHD7, Yu-lung Lau, MBChB, MD (Hon), FRCPCH, FRCPS8, Surjit Singh, MD; DCH (Lon.); FRCP (Lon.); FRCPCH (Lon.); FAMS9
1DM Fellow, Postgraduate Institute of Medical Education and Research, Chandigarh
2Assisstant professor, department of pediatrics, Postgraduate Institute of Medical Education & Research
3PHD Scholar, Postgraduate Institute of Medical Education and Research, Chandigarh
4Professor, Postgraduate Institute of Medical Education and Research, Chandigarh
5Faculty, Tokyo Medical and Dental University
6Faculty, Tokyo Medical and Dental University
7Immunologist, Kazusa DNA Research Institute
8Chair Professor of Paediatrics Doris Zimmern Professor in Community Child Health, Department of Paediatrics and Adolescent Medicine, The University of Hong Kong
9Head, Department of Pediatrics and Chief, Allergy Immunology Unit, Advanced Pediatrics Centre,Principal Investigator, Indian Council of Medical Research (ICMR) Centre for Advanced Research in Primary Immunodeficiency DiseasesVice-President, Indian Rh, Postgraduate Institute of Medical Education & Research
Background: Chronic granulomatous disease (CGD) results from an inherited, genetically heterogeneous functional defect of phagocytes. CGD results from defects in different components of NADPH oxidase enzyme complex. Mutations in the seven structural genes of this complex (CYBB, CYBA, NCF1, NCF2, NCF4 and Rac1/Rac2 GTPase binding protein gene) cause CGD.
Methods: All children diagnosed to have CGD between 1998 and 2018 on the basis of abnormal result of nitroblue tetrazolium dye reduction test (NBT) and Dihydrorhodamine 123 (DHR) assay were enrolled in the study. Clinical findings, diagnostic tests and outcomes were recorded from scrutiny of case notes. Assessment of carrier state of mother and level of gp91phox, p47phox, p67phox , p22phox by flow cytometry were done as guiding tools for possible genetic defects. Mutation analysis was done by gene scan, Sanger sequencing and next-generation sequencing.
Results: Among 62 patients with CGD, 51 (40 boys; 11 girls) had been tested proven for mutation. Male female ratio was 3.63:1. Twenty five (49.1%) patients had X-linked CGD and 26 (50.9%) had autosomal recessive (AR) forms of CGD. Mean age at initial presentation was 2 years (range 15 days - 10 years), while mean age of diagnosis was 3.5 years (range 15 days - 20 years). Forty nine percent (25 among 51) children were diagnosed to have CYBB gene mutation. Sixteen patients (31%) had NCF1 gene mutation. NCF2 gene defect was detected in 10 patients (19.6%). There was history of consanguineous marriage in 15% of the patients.
All children were receiving cotrimoxazole and itraconazole prophylaxis after being diagnosed with CGD. Thirty three (64.7%) patients recovered completely from their infections. Two patients needed rib resection due to locally invasive Aspergillus pneumonia while two patients lost follow up. Sixteen (32%) children succumbed to their illness despite therapy.
Conclusion: A high proportion of heterogeneity was detected in our cohort of CGD. Assessment of mothers carrier status and flow cytometric evaluation of membrane bound and cytosolic protein component of NADPH oxidase complex can be assessed to get help for determining possible mutation. Genetic testing has diagnostic and prognostic importance for the children with CGD.
References:
1. Gennery A. Recent advances in understanding and treating chronic granulomatous disease. F1000Res. 2017 Aug 11;6:1427.
2. Wolach B, Gavrieli R, de Boer M, van Leeuwen K, Berger-Achituv S, Stauber T, et al. Chronic granulomatous disease: clinical, functional, molecular, and genetic studies. The Israeli experience with 84 patients: research article. Am J Hematol. 2017;92(1):28–36.
(105) Submission ID#601150
Cytomegalovirus Specific Cell-mediated Immunity Status in Women with Pre-eclampsia: A Case-control Study
Roya Sherkat, MD1, Zahra Shahshahan, MD2, Sahar Memar Montazerin, MD3, Maryam Kalateh Jari, MD4, Majid Yaran, PhD5, Maryam Nasirian, PhD6, Somaye Najafi, MSc3
1Associate Prof. ,Head of Acquired Immunodeficiency Research Center, Acquired Immunodeficiency Research Center, Isfahan University of Medical Sciences , Isfahan , Iran
2Associate prof., Obstetrics and Gynecology department, Isfahan University of Medical Sciences, Isfahan, Iran.
3Research Assistant, Acquired Immunodeficiency Research Center, Isfahan University of Medical Sciences , Isfahan , Iran
4Obstetrics and Gynecology Assistant, Obstetrics and Gynecology department, Isfahan University of Medical Sciences, Isfahan, Iran.
5Scientist, Acquired Immunodeficiency Research Center, Isfahan University of Medical Sciences , Isfahan , Iran
6Associate prof., Infectious Diseases and Tropical medicine research center, Isfahan University of Medical Sciences, Isfahan, Iran
Background: Pre-eclampsia, a pregnancy-specific complication, has been shown to be associated with Cytomegalovirus (CMV) infection. CMV specific T-cell response plays the major role in CMV infection or disease .We explored whether a change in CMV-specific cell-mediated immunity (CMI) Is related to the development of pre-eclampsia.
Method: CMV-specific CMI was assessed using CMV-QuantiFERON (QF-CMV) assay in serum from 35 women with pre-eclampsia as well as 35 normal pregnancy controls retrospectively. Participants were matched for gestational age individually. Proportion of reactive results, mean value of Interferon- level produced in mitogen and antigen tubes were compared between the cases and controls via Chi-Square, Wilcoxon rank-sum tests, respectively. Odds ratio (OR) and confidence interval (CI) were calculated as well.
Result: No significant differences observed between demographic characteristics of the case and control groups. The QF-CMV assay turned reactive (QF-CMV [+]) in 22 of 35 of patients (63%) VS. 32 of 35 controls (91.4%) (P = 0.004). Women with pre-eclampsia had lower mean IFN- levels in antigen tube (1.57 ± 1.79) compared with normal pregnancy controls (2.40 ± 2.21) (P = 0.028). There was no statistically significant differences in this value of mitogen tube between cases (3.53 ± 1.67) and controls (3.53 ± 1.67) (P = 0.209). Women with suppressed CMV-CMI were 6.3 times more likely to manifest pre-eclampsia (OR= 6.30, 95% CI: 1.60-24.7). This result even strengthened after adjustment for age, gestational age and gravidity (OR = 12.86, 95% CI: 2.68-61.6).
Conclusion: Our finding support an association between suppressed CMV specific CMI and pre-eclampsia.
(106) Submission ID#601152
Auto-inflammation and Immunodeficiency 2 Genes One Presentation
Amarilla B. Mandola, MD1, Chaim M. Roifman, MD2
1Fellow in Training, Canadian Centre for Primary Immunodeficiency, The Roifman Laboratory, Research Institute Division of Immunology and Allergy, Department of Paediatrics, The Hospital for Sick Children, University of Toronto
2Head of Canadian Centre for Primary Immunodeficiency, Head of the Roifman Laboratory, Canadian Centre for Primary Immunodeficiency, The Roifman Laboratory, Research Institute Division of Immunology and Allergy, Department of Paediatrics, The Hospital for Sick Children, University of Toronto
Introduction: The triad of susceptibility to infections, auto-inflammation, and cancer in a patients personal and family history are always suggestive of an underlying primary immunodeficiency; however, in some cases the diagnosis might be delayed for years. Furthermore, the results of immunological and inflammatory evaluation can also be affected by ongoing immunomodulatory therapy initiated by different specialists upon clinical diagnosis.
Objective: To describe a unique presentation of auto-inflammatory disease with combined immunodeficiency in an adult patient.
Case presentation: We report here the case of a 64 year old male, who had a long history of infections including recurrent sino-pulmonary bacterial infections starting during childhood, osteomyelitis at 7 years of age, recurrent tonsillitis requiring tonsillectomy at 21 years of age, recurrent cellulitis, an episode of prostatitis with septicaemia, as well as recurrent varicella zoster and warts. The patient was also diagnosed with sclerosing mesentheritis, and Reynauds phenomenon, recurrent oral ulcers, arthritis, uveitis, autoimmune thyroiditis, lung fibrosis and suffered repeated episodes of abdominal pain. Furthermore, there is a family history of early childhood death, multiple soft tissue cancers, Crohns disease, and autoimmune thyroiditis.
Upon physical examination, the patient had multiple telangiectasia, baseline erythroderma, and flushing. Immunological evaluation showed lymphopenia with significant reduction in both circulating B and T cells, however, assessment of humoral immunity revealed low IgG and decreased IgM with normal IgA levels. At the time of the evaluation he had been on low dose daily prednisone (7.5mg), colchicine, and methotrexate as immuno-modifying therapy.
Genetic evaluation revealed a heterozygous mutation in NOD2 as well as compound heterozygous mutations in the MEFV gene.
Discussion: Mutations in NOD2 have been described in association with Blau syndrome a multisystem auto-inflammatory syndrome which may explain many of the features experienced by our patient. To our surprise next generation sequencing revealed a second aberration in the MEFV gene which causes Familiar Mediterranean Fever, another multisystem auto-inflammatory disease, which might lead to the phenotype observed in the patient.
Conclusion: This is the first report of genetic lesions in two different genes leading to a severe course of auto inflammation.
(107) Submission ID#601164
Novel CDC42 Mutation Causes Severe Autoinflammatory Syndrome Responsive to IL-1 Inhibition
Yael Gernez, MD, PhD1, Matthew Kirbey, PhD2, Jay M. Balagas, MD3, Claudia I. Macaubas, PhD4, Scott Canna, MD, PhD5, David B. Lewis, MD6, Elizabeth Mellins, MD, PhD7, Rosa Bacchetta, MD8, Katja G. Weinacht, MD, PhD9
1Clinical Assistant Professor, Stanford School of Medicine
2Scientist, Department of Pediatrics, Stem Cell Transplantation, at the Lucile Salter Packard Childrens Hospital, Stanford school of medicine, Stanford, CA, USA
3Physician, Pediatric Hematology/Oncology, 2018 John Muir Health Physician Network Member
4Basic Life Res Scientist, Department of Pediatrics Rheumatology/RK Mellon Institute, Children's Hospital of Pittsburgh of UPMC, Pittsburgh, Pa, USA
5Assistant Professor, Department of Pediatrics Rheumatology/RK Mellon Institute, Children's Hospital of Pittsburgh of UPMC, Pittsburgh, Pa, USA
6Professor, Department of Pediatrics, Allergy, Immunology and Rheumatology at the Lucile Salter Packard Childrens Hospital, Stanford School of Medicine
7MD, PhD, Department of Pediatrics, Human Gene Therapy at the Lucile Salter Packard Childrens Hospital, Stanford school of medicine, Stanford, CA, USA
8ASSOCIATE PROFESSOR, Department of Pediatrics, Stem Cell Transplantation, at the Lucile Salter Packard Childrens Hospital, Stanford school of medicine, Stanford, CA, USA
9Assistant Professor, Division of Stem Cell Transplantation and Regenerative Medicine, Department of Pediatrics, Stanford School of Medicine, Stanford, CA
Monogenic autoinflammatory syndromes (MAIS) are a diverse group of disorders characterized by primary over-activation of the innate immune system. Induction of the inflammasome complex by innate immune sensors and increased production of IL-1b are implicated in the pathogenesis of MAIS. Macrophage activation syndrome (MAS) is a life-threatening illness defined by acute hyper-inflammation and unopposed cytokine release. It is considered an acquired condition secondary to infection, rheumatoid disease or malignancy. The early therapeutic use of IL-1b inhibition has profoundly improved the prognosis MAS. It has recently been shown that increased free IL-18 levels in the blood are causatively linked to the development of MAS. Significant overlap in clinical presentation and laboratory markers between patients with MAIS and MAS led us to explore the role of free IL-18 and therapeutic use of IL-1b inhibition in a patient with CDC42 mutation.
Here, we report the case of an 18 months-old female who presented with hydrops fetalis in utero, and later developed failure-to-thrive, splenomegaly, anemia, thrombocytopenia, arthralgias, rashes, frequent febrile episodes and mild facial dysmorphism along with massive increase in CRP, ESR and ferritin. Whole Exome Sequencing (WES) identified a heterogenous likely pathogenic de novo variant in cell division control protein 42 homolog (CDC42) c.563G>A (p.C188Y). CDC42 encodes a small RHO family GTPase that regulates multiple signaling pathways controlling cell polarity, migration, endocytosis and cell cycle progression. Single allele mutations in the CDC42 gene were recently reported to cause Takenouchi-Kosaki syndrome manifesting with growth retardation, developmental delay, facial dysmorphism, and thrombocytopenia however systemic autoinflammation has not been described. CDC42 closely interacts with the Wiskott-Aldrich Syndrome Protein but little is known about the mechanism underlying immune abnormalities associated with CDC42 mutations.
Our patient had an inflammamosopathy-like syndrome. Because of significant clinical overlap to MAS, we measured IL-6, IL-18, free IL-18 and IL-18 binding protein, all of which were significantly increased. This increase in free IL-18 heightened her risk of developing MAS. Her IL1-b level was normal, but an increase in IL-1b is hardly ever detectable in the serum despite playing a critical role in this type of inflammation. Indeed, chronic IL-1b excess in the tissues promotes systemic inflammation and is associated with chronically elevated CRP and ESR. With this rationale we started the IL-1 receptor antagonist anakinra. Within 48 hours from starting anakinra, the parents observed an increase in appetite, resolution of arthralgias and improved mobility. Over the course of the following weeks, fever, anemia, thrombocytopenia and rash disappeared, the spleen massively decreased in size and the patient started to meet developmental milestones. CRP, ESR eventually normalized while ferritin and free IL-18 are still trending down.
Conclusions: Significant increase in free IL-18 and extremely encouraging clinical response to therapy with anakinra in a patient with novel CDC42 mutation suggests a link between MAS and defects in CDC42. Elucidating the mechanism of inflammasome activation and the drivers of IL-18 increase in MAS and MAIS more broadly may shed light on novel therapeutic targets like the use of human recombinant IL-18 binding protein.
(108) Submission ID#601190
First Three Years Experience in the Immunology Outpatient Clinic of a University Hospital in Cali,Colombia
Andres F. Zea-Vera, MD, PhD1, Vanessa Montoya-Lozano2, Mario A. Chacon-Acevedo, MD3
1Assistant Professor, Universidad del Valle. Hospital Universitario del Valle.
2Nursing Student, Universidad del Valle
3MSc Student, Universidad del Valle. Hospital Universitario del Valle.
Introduction: In August 2015 the Clinical Immunology outpatient clinic was established in the Hospital Universitario del Valle (HUV) in Cali, Colombia. The clinic evaluate an average of 8 to 10 patients weekly (new and follow up patients). Most of the cases are referred in the context of Recurrent Infection syndrome, disseminated mycobacterial disease, hypogammaglobulinemia and severe autoimmune or allergic disease.
Results: The Clinical Immunology outpatient clinic has evaluated 261 patients classified as: Primary Immunodeficiencies (PID)= 64 patients (25%), Secondary Immunodeficiencies= 14 patients (5%), Autoimmunity/Rheumatic disease= 49 patients (19%), Severe/Refractory Allergy= 61 patients (23%) and Infectious diseases with high suspicious of PID in follow up= 24 patients (9%).
According to the IUIS-2017 classification, 64 patients with confirm Inborn Errors of Immunity (PID) were diagnosed: I. Immunodeficiencies affecting cellular and humoral immunity 4(6%), II. CID with associated or syndromic features 10(16%), III. Predominantly Antibody deficiencies 26(41%), V. Congenital defects of phagocyte number, function or both 7(11%), VI. Defects in intrinsic and innate immunity 6(9%), VII. Auto-inflammatory disorders 7(11%), VIII. Complement deficiencies 2(3%), IX. Phenocopies of PID 2(3%).The mean age was 16 years with a Male:Female ratio 34:30. Molecular tests have been done in 14 cases with 8 gene mutation confirmation.
Conclusion: Our Clinical Immunology service constitutes an opportunity for low income people with public health care insurance in the southwest of Colombia. The Universidad del Valle and Hospital Universitario del Valle combine effort has contribute to improve the suspicious, diagnosis and treatment of patients living with Inborn Errors of Immunityr (PID) as well as patients with other immune disorders.
(109) Submission ID#601193
Telomeres in Schimke Immuno-Osseous Dysplasia: Comparing Telomere Length in Individuals with Homozygous and Heterozygous SMARCAL1 Mutations
Elizabeth A. Lippner, MD1, Geraldine Aubert, PhD2, Vasavi Ramachandran, MS3, David B. Lewis, MD4
1Postdoctoral Scholar and Clinical Instructor, Division of Allergy, Immunology, and Rheumatology, Department of Pediatrics, Stanford University School of Medicine
2Director of Clinical Development & Research, Repeat Diagnostics, Inc
3Life Science Research Professional, Division of Allergy, Immunology, and Rheumatology, Department of Pediatrics, Stanford University School of Medicine
4Professor, Department of Pediatrics, Allergy, Immunology and Rheumatology at the Lucile Salter Packard Childrens Hospital, Stanford School of Medicine
BACKGROUND: Schimke Immuno-Osseous Dysplasia (SIOD) is a rare, autosomal recessive disease characterized by spondyloepiphyseal dysplasia, vasculopathy, T-cell immunodeficiency, progressive nephrotic disease, and increased neoplastic risk. SIOD is caused by homozygous mutations in the SMARCAL1 gene. SMARCAL1 encodes a DNA-annealing helicase that functions in gene expression modulation and maintaining genome integrity at stalled DNA replication forks. Recent in vitro studies implicate a role for SMARCAL1 in telomere maintenance; SMARCAL1 is enriched in cells that maintain telomeres via the alternative lengthening of telomeres pathway and SMARCAL1-decifient cells demonstrate telomere instability with replication fork collapse and increased telomere-associated DNA damage.[1,2] Telomere analysis of 4 SIOD patients, including one patient who received a hematopoietic stem cell transplant (HSCT) 20 years prior, as well as 5 heterozygous family members revealed significantly shorter telomeres in SIOD patients compared to heterozygous family members and compared to age-matched, healthy controls.
METHODS: Peripheral blood mononuclear cells were isolated using a Ficoll-Hypaque density gradient, cryopreserved, then sent to Repeat Diagnostics in North Vancouver, BC. Telomere length measurements were performed at a single-cell level using flow-fluorescence in situ hybridization as previously described.[3] Telomere length was measured in total lymphocytes, naive and memory enriched T cells, B cells, and NK cells and compared to reference samples from age-matched, healthy individuals.
RESULTS: Compared to age-matched healthy controls, three SIOD individuals had mean telomere lengths (MTLs) less than the 1st percentile for age across all lymphocyte subsets (total lymphocytes, B cells, NK cells, naïve and memory T cells). In comparison, three unaffected family members had normal MTLs (10th percentile< x <90th percentile) across all subsets, and two unaffected family members had low MTLs (1st< x <10th percentile) in some subsets. The SIOD individual who received a matched-sibling HSCT 20 years prior, had normal MTL in NK cells (10th < x <90th percentile) but low MTLs (1st< x <10th percentile) for all other subsets.
CONCLUSIONS: These data show that SIOD patients have significantly impaired telomere lengths across multiple lymphocyte lineages and support a limiting role for SMARCAL1 deficiency in telomere maintenance. In comparison, unaffected family members, heterozygous for SMARCAL1 mutations, have mean telomere lengths that are normal or slightly low for age. This suggests that abnormally short telomeres are seen in individuals with homozygous but not heterozygous SMARCAL1 mutations. For the individual who received a HSCT, we do not have pre and post-HSCT telomere data, but these results support obtaining pre and post-HSCT telomere length analysis in future cases.
Abnormally short telomeres have been linked to widespread perturbation of gene expression.[4] We hypothesize that SMARCAL1 deficiency, by the effect of stalled forks and shortened telomeres, leads to perturbation in the transcriptome of affected tissues. Shortened telomeres may explain the reduced hematopoietic bone marrow production in SIOD, as bone marrow failure is a cardinal feature of dyskeratosis congenita, a disorder of impaired telomere maintenance. Future studies to investigate the role of telomere maintenance in SIOD include measurement of telomerase activity in polyclonally activated T cells and transcriptome analysis using RNA-Seq.
REFERENCES:
1. Cox KE, Maréchal A, Flynn RL. SMARCAL1 resolves replication stress at ALT telomeres. Cell Reports. 2016; 14:1032-1040.
2. Poole, LA et al. SMARCAL1 maintains telomere integrity during DNA replication. Proc Natl Acad Sci U S A. 2015; 112:14864-14869.
3. Baerlocher GM, Vulto I, de Jong G, Lansdorp PM. Flow Cytometry and FISH to measure the average length of telomeres (flow FISH). Nat Protocols. 2006; 1:2365-2376.
4. Robin JD, Ludlow AT, et al. SORBS2 transcription is activated by telomere position effect-over long distance upon telomere shortening in muscle cells from patients with facioscapulohumeral dystrophy. Genome Res. 2015; 25:1781-1790.
(110) Submission ID#601200
Yellow Fever: Is It Possible to Vaccinate Patients with IgA Deficiency?
Magda Carneiro-Sampaio, MD, PhD1, Mariana Castiglioni, MD2, Nathalia Souza, MD2, Bruna Aquilante, MD3, Mayra Dorna, MD3, Ana Paula Castro, MD3, Antonio Carlos Pastorino, MD, PhD4,
1Full Professor, Department of Pediatrics, Faculdade de Medicina da Universidade de Sao Paulo, Sao Paulo, Brazil
2Allergy Immunology Fellow, Allergy Immunology Department from Child´s Institute from Medicine Faculty from Uiversity of Sao Paulo (Instituto da Criança do Hospital das Clinicas da Faculdade de Medicina da Universidade de São Paulo)
3Allergy Immunologist, Allergy Immunology Department from Child´s Institute from Medicine Faculty from Uiversity of Sao Paulo (Instituto da Criança do Hospital das Clinicas da Faculdade de Medicina da Universidade de São Paulo)
4Allergy Immunologist Professor, Allergy Immunology Department from Child´s Institute from Medicine Faculty from Uiversity of Sao Paulo (Instituto da Criança do Hospital das Clinicas da Faculdade de Medicina da Universidade de São Paulo)
Background: Yellow fever is a potentially fatal disease for which only supportive treatment is available. Vaccination is the primary strategy for prevention of this disease And the vaccine is extremely effective, but there are a few specific populations where it is contraindicated. Regarding IgA deficiency (the most frequent primary immunodeficiency), current recommendations in the literature are controversial. There are no specific studies in this disease, so case series addressing the safety or possible adverse events after vaccination are essential for decision-making during epidemic scenarios, as experienced in Brazil in the last years. In this context, this study aimed to describe adverse events after the use of the yellow fever vaccine in IgA deficient patients.
Method: a retrospective cross-sectional study was conducted including IgA deficient patients followed at a specialized pediatric outpatient clinic between 2017 and 2018. All patients had at least one year of follow-up. Immunoglobulin levels, antibody response to vaccines and lymphocyte subset count were evaluated to exclude other immunodeficiencies or the presence of abnormalities that could contraindicate vaccination. Demographic data, the presence of infections and comorbidities, use of immunosuppressive medication and adverse events after vaccine administration of the vaccine were described.
Results: Thirty-eight patients with IgA deficiency were included in the study and 18 received the vaccine. Vaccinated patients had a mean age at the time of the study of 13.7 years (SD ± 3.5y). Six out of the 18 presented comorbidities: thyroiditis (n=3), type 1 diabetes mellitus (n=1), celiac disease (n=1) and juvenile rheumatoid arthritis (n=1). All patients were atopic and only one had recurrent infections in the last year despite the use of antibiotic prophylaxis. All 18 patients had normal IgG and IgM levels for their age, positive vaccine responses for measles, rubella and mumps, and age-appropriate lymphocyte subset count. After 6 months of observation, no immediate or late adverse events were reported. Among the 20 non-vaccinated patients, only one had a formal contraindication (systemic erythematosus lupus using immunosuppressive therapy). Five out of the 20 non-vaccinated patients reported being afraid of receiving the vaccine, 7 still intended to receive it and for other 7 patients data regarding vaccination was unavailable.
Conclusion: Despite the small number of patients, the absence of adverse events in this case series suggests that immunization with yellow fever vaccine may be safe in IgA deficient patients, excluded other contraindications. More studies are essential to confirm the safety and help the decision-making process regarding the vaccine administration for IgA deficient patients, especially in this yellow fever outbreak scenario.
(111) Submission ID#601201
A Case of Immunodeficiency, Centromeric Instability, and Facial Anomalies Syndrome (ICF) with NK Deficiency and Subsequent EBV-driven Diffuse Large B-cell Lymphoma Treated with Bone Marrow Transplant
Caitlin M. Burk, MD1, Kara E. Coffey, MD2, Emily M. Mace, PhD3, Bret Bostwick, MD4, Ivan K. Chinn, MD5, Zeynep H. Coban-Akdemir, PhD6, Shalini N. Jhangiani, PhD7, James R. Lupski, MD, PhD8, Damara Ortiz, MD9, Jessie L. Barnum, MD10, Steven W. Allen, MD10, Leanna-Marie Robertson, MD2, Jordan S. Orange, MD, PhD11, Hey J. Chong, MD, PhD12
1Pediatric Resident, UPMC Children's Hospital of Pittsburgh, Pittsburgh, PA
2Fellow, Allergy and Immunology, University of Pittsburgh School of Medicine, Pittsburgh, PA
3Assistant Professor, Department of Pediatrics, Columbia University Irving Medical Center, New York, NY
4Assistant Professor, Department of Molecular and Human Genetics, Baylor College of Medicine, Houston, TX
5Assistant Professor, Pediatric Allergy and Immunology, Baylor College of Medicine, Houston, TX
6Postdoctoral Research Fellow, Department of Molecular and Human Genetics, Baylor College of Medicine, Houston, TX
7Project Manager, Department of Molecular and Human Genetics, Baylor College of Medicine, Houston, TX
8Professor, Department of Molecular and Human Genetics, Baylor College of Medicine, Houston, Texas, USA
9Assistant Professor, Pediatric Medical Genetics, University of Pittsburgh School of Medicine, Pittsburgh, PA
10Assistant Professor, Pediatric Hematology and Oncology, University of Pittsburgh School of Medicine, Pittsburgh, PA
11Professor and Chair, Department of Pediatrics, Columbia University Irving Medical Center, New York, NY
12Assistant Professor and Chief, Pediatric Allergy and Immunology, University of Pittsburgh School of Medicine, Pittsburgh, PA
Introduction/Backround: Immunodeficiency, centromeric instability, and facial anomalies syndrome (ICF) is a rare group of autosomal recessive disorders involving the triad of hypogammaglobulinemia, centromeric instability, and facial anomalies. The majority of patients have hypo- or agammaglobulinemia, but T cell defects have also been reported. We present the case of a child with ICF-2 who presented with NK deficiency and ultimately developed an EBV-driven malignancy and was successfully treated with bone marrow transplant.
Methods: Whole exome sequencing and NK cell function via 51-Cr cytotoxicity assay and phenotyping via flow cytometry were performed at Baylor College of Medicine and Texas Childrens Hospital. Centromeric banding studies were performed at University of Pittsburgh Medical Center.
Results: The female patient presented at 3 months of age with CMV pneumonitis and persistent CMV viremia requiring treatment followed by prophylaxis with valgancyclovir. She initially had hypogammaglobulinemia and low T, B, and NK cells; she had normal TRECs, lymphocyte mitogen proliferation responses and Zap 70, MHCI and MHCII expression. The hypogammaglobulinemia and T- and B-cell lymphopenia resolved within 9 months after initial presentation as she clinically improved from her CMV infection. She was found to have NK cell deficiency on three separate commercially tested samples. Whole exome sequencing revealed a homozygous variant in ZBTB24 indicative of ICF-2 syndrome that was confirmed with Sanger sequencing (c.1492_1493del, p.Q498Vfs). Repeat NK cell studies confirmed impaired function, and phenotyping showed an increase in CD56-bright and a decrease in CD16-positive cells, suggesting either impaired transition from immature to mature NK cells or impaired survival of mature cells. Her karyotype and centromeric banding studies were normal, as were centromeric instability studies.
She later developed a memory B-cell defect and presented at 34 months of age with persistent fever, respiratory distress, loss of vaccine titers, hypogammaglobulinemia and low B and T cells. She was found to have EBV viremia and an EBER-positive diffuse large B-cell lymphoma in her right lung. Due to tenuous clinical status, she received rituximab for treatment of EBV prior to definitive lymphoma diagnosis. She was treated with chemotherapy per protocol ANHL1131, group B (pre-phase with COP, courses 1 and 2 with COPADM, and courses 3 and 4 with CYM) and her course was complicated by seizures attributed to methotrexate toxicity. She ultimately underwent reduced intensity conditioning with hydroxyurea, alemtuzumab, fludarabine, mephalan, and thiotepa followed by a CD-34 selected, HLA-matched, unrelated donor peripheral blood stem cell transplant. Her early post-transplant course was complicated by adeno- , EBV, and CMV viremia, all successfully treated with antivirals and a donor lymphocyte infusion. She is now greater than 8 months post-transplant, off immunosuppression with 100% donor engraftment, no evidence of organ toxicity or GVHD, and with excellent immune reconstitution.
Conclusions: This is the first reported case of impaired NK cell function and phenotype and EBV-driven malignancy in a patient with ICF-2. This case expands the phenotype of ICF-2 and suggests that early bone marrow transplant should be considered in these children. It also demonstrates a novel requirement for ZBTB24 in human NK cell maturation and function.
(112) Submission ID#601233
Variant Mutation in PLCG2 Associated with Common Variable Immundeficiency Without Cold Urticaria
Veronica Solivan-Vargas, MD1, Wilfredo Cosme-Blanco, MD/Phd2, Cristina Ramos-Romey, MD3, Sylvette Nazario-Jimenez, MD4
1Resident, Hospital Episcopal San Lucas
2Allergy & Immunology Physician, Veteran Affairs Caribbean Healthcare System
3Assistant Director of Allergy Immunology Program, University of Puerto Rico
4Director of Allergy Immunology Program, University of Puerto Rico
Rationale: Common variable immunodeficiency (CVID) is a disorder that affects the production of immunoglobulins and is associated with development of autoimmunity. Multiple mutations have been described that are associated with CVID, but PLCG2 mutations have only been described in patients with phospholipase C gamma 2 (PLC2) associated antibody deficiency and immune dysregulation (PLAID) and autoinflammatory PLC2 associated antibody deficiency and immune dysregulation (APLAID). We present a case of a 44 y/o male CVID patient with recurrent upper respiratory tract infections, steroid-dependent autoimmune thrombocytopenia, low B cell count, hepatosplenomegaly, and restrictive lung disease. He was found with a variant of unknown significance at the PLCG2 gene. In contrast to PLAID our patient does not exhibit cold urticaria.
Method: Case presentation of a CVID patient followed in our clinics. Patients chart and previous laboratories were reviewed. Sequence analysis and deletion/duplication CVID panel testing was performed using Invitae©
Discussion: Genetic testing has revolutionized the diagnosis of immune deficiencies, but variants of unknown significance are being increasingly reported. In this case, a variant of uncertain significance was identified which replaces threonine for alanine at codon 829 of the PLCG2 protein. This codon is located at the SH3 domain, which is part of a region that provides auto-inhibitory enzymatic functions. PLAID mutations have been identified in SH2 domain, but it has been known that both SH2 and SH3 domains facilitate PLCG association with other proteins. Studies with deletion of PLCG2 gene have shown functional abnormalities in B cells, natural killer cells and mast cells. To our knowledge, there has not been any previous report of a CVID patient with a variant mutation at the SH3 domain of the PLCG2 gene without being diagnosed as PLAID or APLAID. Our patient has immunodeficiency, recurrent upper respiratory tract infections, steroid-dependent recurrent autoimmune thrombocytopenia, rheumatoid arthritis, hepatosplenomegaly, early-osteoporosis and restrictive lung disease. He does not have cold urticaria as seen in PLAID, but exhibits autoimmunity not observed in APLAID.
Conclusion: Conclusion: PLCG2 is an important protein in the pathway of B cell development. A novel mutation in the SH3 domain of the PLCG2 gene may be associated with the CVID phenotype of low B cells and autoimmunity. This could lead to a gain-of-function mutation as seen in PLAID but without early-onset cold urticaria. Functional studies are required to confirm the significance of this mutation.
(113) Submission ID#601257
JAK-dependent and Independent Cytokines Drive the Pathogenicity of HLH : Targets for Combination Therapy
Josée-Anne Joly1, Sara Bourbonnais, MSc2, Alexis Vallée3, Chloé Berthe, MSc2, Hélène Decaluwe, MD, PhD, FRCPC4
1Master student, CHU Sainte-Justine Research Center, Université de Montréal
2Research Assistant, CHU Sainte-Justine Research Center, Université de Montréal
3PhD student, CHU Sainte-Justine Research Center, Université de Montréal
4Pediatric Immunologist and Clinician Scientist, CHU Sainte-Justine and Université de Montréal
Primary (or Familial) Hemophagocytic lymphohistiocytosis (HLH) is a rare, life-threatening hyper-inflammatory disease affecting mainly young children. It is caused by mutations in genes involved in the granule-dependent cytotoxic pathway, and is characterized by extreme inflammation and massive tissue infiltration by activated T cells and macrophages. To this day, hematopoietic stem cell transplantation is the only available curative treatment with a transplant-related mortality of 30%. Thus, the development of new, more efficient anti-inflammatory treatments would be a significant advancement in the treatment of HLH. Here, we hypothesize that combination therapies targeting both JAK-dependent and independent cytokines will be more effective than either one alone to reduce the life-threatening symptoms induced by this pathology.
Using a perforin-deficient mouse model of HLH, we first compared the effect of targeting individual cytokines with blocking antibodies on the progression of the disease. We show that blocking IFNg and IL-18, but not IL-6, significantly reduces the severity of HLH. Targeting the JAK-STAT signalling pathway with ruxolitinib, a specific inhibitor of JAK1 and JAK2, downstream of IFNg and IL-6, but not IL-18, is similarly beneficial. More importantly, combination therapies using ruxolitinib and blocking antibodies to either IFNg or IL-18 show synergistic effects, further mitigating the progression of the disease. These results suggest that JAK-dependent and independent cytokines drive the pathogenicity of HLH in perforin-deficient mice. It further supports that ruxolitinib, although effective in reducing the symptoms of HLH, should be used in combination with anti-IFNg and/or anti-IL-18 antibodies to prevent HLH progression. This is particular relevant since the former were recently approved for the treatment of HLH while the latter (IL-18 binding proteins) are in clinical trials for IL-18-dependent macrophage activation syndromes.
This project was supported by funds from the Fondation de Cancérologie Charles Bruneau and the Canadian Institutes of Health Research.
(114) Submission ID#601260
A Case of Disseminated Pneumocystis Jiroveci in a Non-Human Immunodeficiency Virus Infected Patient
Aminaa E. Siddiqi, MD1, Joshua Sacha, MD2, Rebecca Saenz, MD, Phd1, Anne Liu, MD3, Christian Kunder, MD4, Gulbu Uzel, MD5, Beth Martin, MD6, David B. Lewis, MD7, Yael Gernez-Goldhammer, MD, PhD8
1Fellow Physician, Division of Allergy and Immunology, Department of Pediatrics, Stanford School of Medicine
2Clinician, David Grant USAF Medical Center in Travis AFB, California
1Fellow Physician, Division of Allergy and Immunology, Department of Pediatrics, Stanford School of Medicine
3Clinical Assistant Professor, Division of Allergy and Immunology, Department of Pediatrics, Stanford School of Medicine
4Clinical Assistant Professor, Department of Pathology, Stanford School of Medicine
5Staff Clinician, Laboratory of Clinical Immunology and Microbiology, National institute of Allergy and Infectious Diseases, NIH, Bethesda, MD, USA
6Clinical Assistant Professor, Division of Hematology, Department of Hematology, Stanford School of Medicine
7Professor, Department of Pediatrics, Allergy, Immunology and Rheumatology at the Lucile Salter Packard Childrens Hospital, Stanford School of Medicine
8Clinical Assistant Professor, Division of Allergy and Immunology, Department of Pediatrics, Stanford University School of Medicine
Disseminated Pneumocystis jiroveci (PJP) infection is a well described entity in patients with acquired immunodeficiency syndrome (AIDS). Despite the increased risk of opportunistic lung infection in patients with severe T cell dysfunction (e.g. CD40L deficiency) and/or severe CD4 T cell lymphopenia, we are not aware of any reports of disseminated Pneumocystis jiroveci infection in non- human immunodeficiency virus (HIV) patients with primary immunodeficiency (PID).
We report the first case, to our knowledge, of disseminated PJP in a patient with CVID like/CTLA4 haploinsufficiency. He had been diagnosed with common variable immunodeficiency (CVID) in 2009, approximately eight years prior to being referred to us, and was on intravenous immunoglobulin (IVIG). He was also diagnosed with multilineage Evans syndrome in 2015. His medical history was also significant for potential granulomatous lymphocytic interstitial lung disease (GLILD) (lung biopsy in the remote past with interstitial disease), significant splenomegaly (29.4 cm), severe portal hypertension, nodular liver disease (likely nodular regenerative hyperplasia) complicated by anasarca, history of chronic diarrhea (potential enteropathy), lymphadenopathy s/p biopsy with nodular lymphoid hyperplasia, and a history of multiple pneumonias. In 2017, he had developed disseminated PJP with lung, liver, and bone involvement. The T2 vertebra PJP invasion was confirmed with a bone biopsy; Gomori methenamine silver staining and PCR were performed and concluded PJP. He was treated with Trimethoprim sulfamethoxazole (TMP-SMX) and steroids, then was continued on TMP-SMX prophylaxis. Due to his liver damage and his chronic neutropenia, TMP-SMX was replaced by atovaquone as a secondary prophylaxis for PJP.
His laboratory studies were significant for an absolute neutrophil count of 1.54 K/uL, absolute lymphocyte count of 0.61 K/uL, hemoglobin of 12.7 g/dL, platelets of 78 K/uL, total bilirubin of 2.3 mg/dL, and LDH of 243 U/L. His HIV PCR was negative. His infectious workup showed negative PCR for Epstein Barr virus, Cytomegalovirus, Herpes virus and Hepatitis B and C. His stool PCR was negative for norovirus. Flow cytometry demonstrated almost absent B cells and decreased T cells count; 586 CD3+ T cells, 12 CD20+ cells, 12 CD19+ cells, 181 CD3+CD8+ cells, 392 CD3+CD4+ cells, 15 CD56+CD16+ cells, 19 CD4+CD45RA, and 380 CD4+CD45RO cells. On IVIG, his IgA, IgG and IgM levels were respectively <8 mg/dL; 995 mg/dL, and IgM 48.4 mg/dL. T-cell receptor beta chain repertoire analysis showed an oligoclonal distribution. Severe combined immunodeficiency panel through Ambry genetic testing was negative as was genetic testing for CD40L deficiency. Given his complex clinical history, whole exome sequencing was obtained and detected an autosomal dominant heterozygous missense mutation (C.436G>A) implicated in CTLA-4 haploinsufficiency and previously reported by Schwab et al.
Our patient is currently undergoing therapy with abatacept (CTLA-4 fusion protein), which has been reported to improve GLILD, splenomegaly and enteropathy in patients with CTLA-4 haploinsufficiency. He is improving on this regimen. He has met with the Stem Cell transplant team, but at this point of time, due to his abnormal lung function, his liver damage and his significant splenomegaly, he is not a good candidate.
(115) Submission ID#601261
Deficiency of the Non-classical Inhibitor of NF-kappaB, IkappaBNS, Causes a Novel Primary Immunodeficiency Due to Dysregulated NF-kappaB Signaling
Charlotte Slade, MBBS, FRACP, FRCPA1, Maryam Rashidi, MD, PhD2, Tom Scerri, PhD2, Melanie Bahlo, PhD3, Steven Holt, MBBS, PhD, FRCP, FRACP4, Samantha Chan, MD5, James Vince, PhD6, Philip Hodgkin, PhD7, Jo A Douglass, MBBS, MD, FRACP8, Vanessa L. Bryant, BSc (Hons) PhD9
1Researcher/Clinical Immunologist, Walter & Eliza Hall Institute/Royal Melbourne Hospital
2Postdoctoral scientist, WEHI
3Head, Population Health and Immunity, WEHI
4Head, Department of Nephrology, Royal Melbourne Hospital
5Clinical Immunologist, Royal Melbourne Hospital
6Laboratory Head, WEHI
7Division Head, Immunology, WEHI
8Head, Clinical Immunology & Allergy, Royal Melbourne Hospital
9Head, Immunogenetics Research Unit, Walter & Eliza Hall Institute
Defects in the NF-B signaling pathway are implicated in the pathogenesis of several primary immune deficiencies in humans. The clinical features of these conditions vary significantly, reflecting the complexity of the pathway, and its broad role in innate and adaptive immune responses, and the development and differentiation of lymphoid organs.
Here we report the first case of a human PID caused by a homozygous mutation in NFKBID in a 30 year-old male. He was the second child of consanguineous parents, and was diagnosed with possible CVID at the age of 16, after recurrent episodes of pneumococcal pneumonia. However the clinical features have evolved over time; he developed severe EBV infection at age 18, causing hepatitis and pancreatitis. At the age of 20, he presented with an ANCA-negative systemic vasculitis, manifesting as pulmonary haemorrhage, and acute necrotizing pauci-immune glomerulonephritis. Pulsed methylprednisolone and cyclophosphamide induced an initial remission, however, relapse a year later led to end-stage renal failure. He is now dialysis-dependent, and due to the underlying PID, and chronic CMV viraemia, is not a candidate for renal transplantation.
Genomic DNA was subjected to whole-exome sequencing. Variants were filtered using a model of autosomal-recessive inheritance and functional analysis of primary cells was performed. We identified a novel, homozygous, single-base deletion resulting in a frame-shift, and premature stop in NFKBID. NFKBID encodes IBNS, a non-classical inhibitor of NF-B signaling.
At diagnosis the patient had reduced levels of IgG2, IgA and IgM, elevated IgE, with absent humoral immune responses to pneumococcal polysaccharide vaccine, and an intact response to tetanus. Lymphocyte numbers were initially within normal reference ranges, albeit with an increased proportion of CD4+:CD8+ T cells. However, over time there has been a significant reduction in B cells and CD8+ T cells. CD4+ T cells demonstrated a skewing towards a central memory phenotype (CD45RO+/CCR7+), and CD4 T cell proliferative responses to PHA were comparable to a healthy control. Functional analysis of primary cells from the proband revealed a complete absence of BNS protein expression, dysregulated NF-B signaling, and elevated pro-inflammatory cytokine production. The patient is currently receiving a trial of targeted therapy to modulate the aberrant immune responses.
This novel PID highlights the importance of regulation of NF-B signalling, in orchestrating an appropriate immune response, maintenance of self-tolerance, and protection against viral pathogens.
(116) Submission ID#601278
A Demonstration of the Diagnostic and Clinical Utility of Genomic Sequencing in Primary Immunodeficiency Diseases in Australia
Charlotte Slade, MBBS, FRACP, FRCPA1, Fiona Moghaddas, PhD2, Sebastian Lunke, BSc, PhD, RCPA3, Zornitza Stark, MA BMBCh DM MBioeth FRACP4, Ingrid Winship, MB ChB, MD, FRACP, FACD, FAICD5, Kirsty West6, Alison Trainer7, Samar Ojaimi, MBBS (Hons) PhD, FRACP FRCPA8, Matthew Hunter, MBChB, FRACP9, Yael Prawer10, Katherine Nicholls, MBBS BMedSc FRACP FRCPA11, Mittal Patel, MBBS FRACP11, Pricilla Auyeung, MBBS PhD FRACP FRCPA11, Kymble Spriggs, MBBS, MPH, GDipClinEd, DTMH, MRCP(UK) FRACP11, Jeremy McComish, MBBS FRACP FRCPA11, Gary Unglik, MBBS(Hons) FRACP FRCPA11, Joseph De Luca11, Samantha Chan, MD11, Giulia Valente12, Anna Jarmolowicz13, Laine Hosking14, Ben van Dort15, Theresa Cole, BM MRCPCH (UK) PhD FRACP14, Joanne Smart, BSc MBBS PhD FRACP16, Sharon Choo, MBBS FRACP FRCPA14, Elly Lynch17, Clara Gaff, BSc(Hons) PhD FHGSA (genetic counselling)18, Seth Masters, BSc (Hons) PhD19, Jo A Douglass, MBBS, MD, FRACP20, Vanessa L. Bryant, BSc (Hons) PhD21
1Researcher/Clinical Immunologist, Walter & Eliza Hall Institute/Royal Melbourne Hospital
2Researcher/Clinical Immunologist, Royal Melbourne Hospital
3Head of the Translational Genomics Unit, Victorian Clinical Genetics Service
4Clinical Geneticist, Victorian Clinical Genetics Service
5Head, Clinical Genetics, Royal Melbourne Hospital
6Associate Genetic Counsellor, Royal Melbourne Hospital
7Clinical Geneticist, Royal Melbourne Hospital
8Immunopathologist, Monash Health
9Head, Monash Genetics Clinic, Monash Health
10Associate Genetic Counsellor, Monash Health
11Clinical Immunologist, Royal Melbourne Hospital
12 Associate Genetic Counsellor, Austin Health
13Associate Genetic Counsellor, Royal Children's Hosptial
14Clinical Immunologist, Royal Children's Hosptial
15Immunology Nurse, Royal Children's Hosptial
16Head, Clinical Immunology, Royal Children's Hosptial
17Clinical Project Manager, Melbourne Genomics Health Alliance
18Executive Director, Melbourne Genomics Health Alliance
19Head, Masters Lab, Walter & Eliza Hall Institute
20Head, Clinical Immunology & Allergy, Royal Melbourne Hospital
21Head, Immunogenetics Research Unit, Walter & Eliza Hall Institute
Primary Immunodeficiency diseases (PID) are a heterogeneous group of conditions with variable clinical features that are frequently associated with significant diagnostic delay. Accurate diagnosis has significant therapeutic benefit and may lead to personalized therapies. We established the Immunology Flagship of Melbourne Genomics Health Alliance in Australia to determine the clinical utility of genomic sequencing for diagnosis and management of individuals with suspected and confirmed cases of PID.
198 adults and children with suspected or confirmed PID (n=153), autoinflammatory disease (n=33) and hereditary angioedema (HAE, n=11) were recruited to the Melbourne Genomics Immunology Flagship. Whole-exome sequencing (WES) was performed, with targeted gene analysis. Variant curation and reporting was performed according to the American Council of Medical Genetics guidelines. Overall, WES was diagnostic in 15% (30/198), confirming a preexisting diagnosis in 7% (14/198), and offering a new or more specific diagnosis in 8% (16/198). Variants of uncertain significance were identified in a further 28 patients (14%) in genes known to be associated with their clinical diagnosis, that warrant further functional validation. In the HAE group, diagnosis was confirmed in only 5 patients (45%), suggesting that WES may not be the appropriate technique for genetic diagnosis in this condition. A higher diagnostic rate was observed for autoinflammatory disorders (20%; 8/40) compared to PID (12%; 18/146). Of those who received a diagnosis, immediate changes to patient management and treatment occurred for 17/29 patients (59%), including HSCT for 3 and specific targeted therapy for 11 (38%) individuals.
We have demonstrated the utility of WES for accurate diagnosis of complex immune diseases, with the potential to change diagnoses, guide therapeutic intervention and provide opportunities for genetic counseling. Further longitudinal analysis will determine clinical outcomes and health economic implications of genomic sequencing for diagnosis and management of immunological conditions in Australia.
(117) Submission ID#601297
Ataxia Telangiectasia and Common Variable Immunodeficiency with B-cell Lymphoma in Adolescent
William Rafael. Marquez, Sr., MD1, Lorenzo Benitez, MD2, Lina Jaramillo, MD3
1Misericordia Children Hospital, Bogota. Colombia, Pediatric immunologist
2Fellow pediatric neumology, Bosque University, pediatric neumology
3professor National University, Misericordia Children Hospital, Bogota. Colombia, pediatric pathologist
At birth he had neonatal asphyxia and cerebral palsy. At 4 years old he had presented involuntary movements, left paresis, bilateral horizontal nystagmus. At 8 years of age, he had a right nasal obstruction. It was resected by otorhinolist and informed by biopsy: inflammatory polyp and chronic sinusitis. He has had 3 pneumonias, sinusitis and diarrhea.
At the age of 13 years, the ataxia telangiectasia was confirmed by sequencing with PCR (62 exons, 91711 bp) of the ATM gene: transition G> A, nucleotide position 2250, codon 750, affecting splicing. Alpha fetoprotein 572-606.90 U/ml. Brain MRI, say Cerebellar Atrophy.
He had IgG 685 mg / dl - 734 mg / dl, IgA 0.00 mg / dl, <1 mg / dl, IgM 268 mg / dl - 315 mg / dl, IgE 0.10 - <1 IU / ml. Subclasses of IgG: IgG3: 0.05 G / dl, IgG4: 0.04 gr/dl, low. IgG anti hepatitis B 6,22. No seroconversion. HIV negative TCD3 + lymphocytes: 32,40%, = 553 cells / mm3, LTCD4 +: 23,78% = 413,21 CEL / mm3, LTCD8 +: 7,69% = 133,5 cells / mm3, CD4 / CD8: 3.09. For all of the above, common variable immunodeficiency was diagnosed. He receives human immunoglobulin.
At 16, I arrived at this hospital due to fever, respiratory distress and lymphadenopathy in the neck. CT showed ganglionic conglomerate on right side neck.
Lymph node biopsy: strong tumors with CD20 and BCL2, weak and moderate diffuse PAX-5; Negativity with CD68, CD3 and CD10, and a cell proliferation index with Ki67 of 50%, diagnosis: Diffuse large B cell lymphoma. Treated with rituximab and chemotherapy. Lymphoma completely remitted.
Conclusion: the association Ataxia Telangiectasia and lymphoma is frequent. By contrast, CVID and Ataxia Telangiectasia are extraordinarily rare.
(118) Submission ID#601301
Loss of Donor Chimerism 20 Years After Bone Marrow Transplant for Chronic Granulomatous Disease
Christa S. Zerbe, MD, MS1, Jennifer Treat, PA-C, MSHS2, Samantha Kreuzburg, BA, RN3, Steven Holland, MD4, Harry L. Malech, MD5
1Senior Research Physician, Laboratory of Clinical Immunology and Microbiology, NIAID, NIH
2Physician Assistant, Medical Science & Computing
3Research Nurse Specialist, National Institutes of Health/National Institute of Allergy and Infectious Diseases
4Director, Division of Intramural Research, NIAID
5Chief, Genetic Immunotherapy Section, Laboratory of Clinical Immunology and Microbiology, IDGS, DIR, NIAID, NIH, Bethesda, MD, USA
Introduction: Chronic granulomatous disease (CGD) is a primary immunodeficiency wherein affected patients are susceptible recurrent infections caused by specific bacteria and fungi as a result of defective NADPH activity. Additionally, inflammatory complications involving the bowel and lungs can cause significant morbidity. Currently the only proven permanent cure to CGD remains hematopoietic stem cell transplant.
Case: A 25-year-old patient was diagnosed in infancy with X-linked CGD. At age 5yrs he received a nonmyeloablative peripheral blood stem cell transplant from his 10/10 non-carrier sister as previously reported (NEJM 344:881, 2001). Conditioning was cyclophosphamide (60 mg/kg) on D-6 and D-7; daily fludarabine (25mg/m2) on D-5 through D-1; Antithymocyte globulin at 40mg/kg on D-5 through D-2. Post-transplant immunosuppression consisted of cyclosporine on D-4 through D+100. He received 7.8x106 CD34+ peripheral blood stem cells which were T-cell depleted with 1x105 add back of CD3+ cells on Day 0. After 10 days of neutropenia (ANC <500) there were signs of engraftment. Per protocol, he received donor peripheral-blood lymphocytes containing 2.0x106 CD3+ cells/kg on D+ 30 after transplantation. Since donor T cells constituted less than 60 percent of his circulating CD3+ T cells and he had no graft versus-host disease, he received 1.0¬107 CD3+ cells/kg on D+60. After the discontinuation of cyclosporine, he received a total of three donor-lymphocyte infusions (1.0¬107 CD3+ cells/kg) at 90-day intervals achieving 100% T cell and myeloid engraftment at 26 months post-transplant with no acute nor chronic GvHD. At last follow-up 6 years post-transplant (2004) he had 100% and 98% lymphoid and myeloid peripheral chimerisms, respectively. The patient and family declined further periodic followup. Then, in October 2018 he presented with malaise, cough and fevers. He eventually was found to have a large consolidation and a BAL grew Burkholderia cepacia. His DHR showed 12% activity and peripheral blood myeloid and lymphoid chimerisms were 12% and 60%, respectively.
Discussion: This late graft failure following peripheral blood transplant occurred following a conditioning regimen which is not the current standard for transplant in CGD. In the case series in which this patients transplant is reported (NEJM 2001), another patients myeloid chimerism fell to 15% by 3 years post-transplant, remaining stable at that level of chimerism without any serious infections over regular periodic follow up to the present time. Current regimens typically include busulfan to enhance engraftment and prevent graft failure. This case reinforces the need for prolonged monitoring of primary immune deficiency patients after transplantation.
(119) Submission ID#601303
Atypical Presentation of Complete DiGeorge Syndrome Without Correlating Genetic Defect: Rescued by State Newborn Screening
Aba Al-Kaabi, MD, FAAP1, Lovya George MD, FAAP2, Erin M. Calhoun, B.S., MD Candidate, Class of 20203, Selina Gierer, DO4
1Fellow - Allergy and Clinical Immunology, Univeristy of Kansas Medical Center
2Assistant Professor – Neonatology, Univeristy of Kansas Medical Center
3medical student, Univeristy of Kansas Medical Center
4Assistant Professor - Allergy and Clinical Immunology, Univeristy of Kansas Medical Center
Introduction: With the introduction of severe combined immunodeficiency (SCID) newborn screen (NBS) in the state of Kansas in 2017, a case of complete DiGeorge Syndrome (DGS) was discovered in an infant born to a diabetic mother with atypical features. This is the first DGS case diagnosed after adding the SCID NBS, which emphasizes the need to establish SCID NBS in all 50 states.
Case presentation: The female infant was born via spontaneous vaginal delivery at 39 1/7 weeks to a 31 year old G1 now P1 mother. Maternal history was significant for chronic hypertension, obesity, insulin dependent type 2 diabetes, anxiety, depression, and scoliosis. The infant was noted to have a left sided abdominal wall defect and hernia, imaging identifying left renal agenesis, and was initially suspicious for VATER syndrome. Fortunately, the infant's SCID NBS revealed low T cell receptor excision circles (TRECS). Her initial white blood cell count was 14.1 with an absolute lymphocyte count of 2.679 K/UL. EBV PCR, CMV PCR, and HIV studies were negative. Chest imaging discovered absent thymus, abnormal vertebrae with only 10 ribs on the right and 12 ribs on the left, and abnormally formed thoracic vertebrae (T7). Echocardiogram detected an atrial septal defect measuring 0.32 cm, possible PFO versus secundum ASD. Endocrinology was consulted for management of labile calcium and phosphorus levels. FISH was negative for 22q11.2 deletion. Microarray revealed a variant of unknown significance arr[GRCh37]2p11.2(86285942_86506132)x3. Sequence analysis of combined and severe immune deficiency genes showed a variant of uncertain significance c.544C>A (p.Leu182Met).
Management and Outcome: Additional evaluation included: CD3 67UL (1700-3600UL), CD4 51UL (1700-2800UL), CD8 19UL (800-1200UL), CD45RA 14 cells/uL (1100-5200cells/uL), normal CD 19, and CD 16/56, normal immunoglobulin G level, and normal dihydrorhodamine assay. Skeletal survey, CT abdomen and chest, and HLA typing were performed in preparation for thymic transplant.
Discussion: Patients with complete DGS, a form of SCID found in less than 1 percent of patients with 22qDS, have absent thymus and a T cell count <3 standard deviations below normal for age (typically <50 naïve CD3+ T cells/mm3). In a large series of patients with complete DGS, only 52 percent had an identifiable 22q11.2 deletion [1].
Infants of a diabetic mother have various genetic and syndromic associations including diabetic embryopathy. [2] Despite the importance of immunological aspects in pregnancy, few studies have reported on the cellular immune modifications of diabetic embryopathy. Diabetes during pregnancy may affect the development of the thymus and thus maturation of the immune system in the offspring. [3]
The recent addition of a TREC assay to newborn screening can identify such a subset of infants with atypical presentations. SCID NBS uses an assay for TRECs, a biomarker of T cell development. [4-6] This initial presentation now places the immunologist in the role of "first responder" with regard to diagnosis and management of these patients, who may present with atypical features. Newer genetic and molecular techniques now allow for earlier identification of immune defects in such disorders with life-long clinical concerns. [7]
References:
1. Markert et al. Review of 54 patients with complete DiGeorge anomaly enrolled in protocols for thymus transplantation: outcome of 44 consecutive transplants. Blood. 2007;109(10):4539. Epub 2007 Feb 6.
2. Stiehm et al. Steim’s Immune Deficiencies. Academic Press. 2014. Print.
3. Warncke K et al. Thymus Growth and Fetal Immune Responses in Diabetic Pregnancies. Horm Metab Res. 2017 Nov;49(11):892-898. doi: 10.1055/s-0043-120671. Epub 2017 Nov 14.
4. Kwan et al. Newborn screening for severe combined immunodeficiency in 11 screening programs in the United States. JAMA. 2014 Aug 20;312(7):729-38. doi: 10.1001/jama.2014.9132. Erratum in: JAMA. 2014 Nov 26;312(20):2169.
5. Kwan et al. History and current status of newborn screening for severe combined immunodeficiency. Semin Perinatol. 2015 Apr;39(3):194-205. doi: 10.1053/j.semperi.2015.03.004. Epub 2015 Apr 30. Review.
6. http://primaryimmune.org/idf-advocacy-center/idf-scid-newborn-screening-campaign.
7. Kuo et al. Immune and Genetic Features of the Chromosome 22q11.2 Deletion (DiGeorge Syndrome). Curr Allergy Asthma Rep. 2018 Oct 30;18(12):75. doi: 10.1007/s11882-018-0823-5.
(120) Submission ID#601306
Clinical and Laboratory Features of Thymoma and Immunodeficiency (Good's Syndrome): A Report from the USIDNET Registry and the Mount Sinai Hospital Cohort
Hsi-en Ho, MD1, Ramsay Fuleihan, MD2, A Wesley. Burks, MD3, Shradha Agarwal, MD4, Charlotte Cunningham-Rundles, MD, PhD5
1Fellow - Allergy and Clinical Immunology, Icahn School of Medicine at Mount Sinai
2Professor of Pediatrics, Division of Allergy and Immunology, Northwestern University Feinberg School of Medicine, Chicago, NY
3Professor of Pediatrics, Division of Allergy and Immunology, UNC School of Medicine
4Associate Professor, Division of Clinical Immunology, Icahn School of Medicine at Mount Sinai
5Professor in Medicine, Division of Clinical Immunology, Icahn School of Medicine, Mount Sinai, NY, NY, USA
Introduction/Background: Goods syndrome is a rare cause of combined B- and T-cell immunodeficiency occurring in association with a thymoma. Affected patents are susceptible to bacterial, fungal, viral, and opportunistic infections. An association with autoimmunity has also been reported. Current knowledge of Goods syndrome is primarily limited to case reports and small series.
Objectives: To examine the spectrum of clinical and laboratory features of a major cohort of Goods syndrome patients in the US.
Methods: We conducted a retrospective analysis of patients with Goods syndrome in the USIDNET Registry and the Mount Sinai Hospital (MSH) cohort.
Results: We identified 20 patients with thymoma and hypogammaglobulinemia (USIDNET, n=11; MSH, n=9; median age: 60 years; female: 45%), representing data from 151 patient years. The median age at diagnosis of thymoma and hypogammaglobulinemia were 52 years (range 31-85), and 50.5 years (range 28-86), respectively. Two patients were deceased (at age 65 and 70 years, cause unspecified). All patients had low IgG (median 313mg/dL, range 47-699). IgA and IgM were reduced in 90% and 45% of patients, respectively. Low CD19+ B cells (median 0.5/mm^3, range 0-28) were reported in all available records. The absence of CD19+ B cells was observed up to 21 years post-thymectomy. A wide range of additional laboratory abnormalities were identified: low CD4+ T cells (n=5), low CD8+ T cells (n=2), low CD4/CD8 ratio (n=6), low NK cells (n=6), and absent peripheral eosinophils (n=8). The most common sites of infections were lower respiratory (70%), upper respiratory (55%), and gastrointestinal (35%). In addition, sepsis (15%), meningoencephalitis (5%), osteomyelitis (5%), and urinary tract infection (5%) were also observed. Identifiable infectious agents included: bacteria (35%), virus (35%), fungus (25%), parasites (10%), and protozoa (5%), with opportunistic infections recorded in 25% of patients. Opportunistic infections were significantly associated with absolute CD4 lymphopenia (p=0.048, Fishers exact test). Enterovirus was identified as a previously unreported cause of meningoencephalitis in this population. Autoimmune manifestations were reported in 45% of patients, with a higher prevalence of inflammatory colitis (20%) than previously reported. Hashimoto thyroiditis, fibromyositis, and bronchiolitis obliterans organizing pneumonia (n=1 each) were identified as previously unreported autoimmune/inflammatory conditions in this population. A case of alopecia areata was also observed. Additionally, bronchiectasis was recorded in 20% of patients. All patients were initiated on immunoglobulin replacement, with antibiotics prophylaxis in 20%, and immunosuppressive medications employed in 10% of patients post diagnosis of immunodeficiency.
Conclusion: Goods syndrome is a combined immunodeficiency, with a wide range of autoimmunity in a subset of patients. We expanded upon the spectrum of associated infectious and inflammatory complications through a major US cohort. Persistent immune dysregulation was observed up to 2 decades post-thymectomy.
(121) Submission ID#601308
Immune Dysregulation: Diagnosis of Behcets Disease in an Affected Chronic Granulomatous Disease Carrier
Aba Al-Kaabi, MD, FAAP1, Jessica Hobson, MD2, John Martinez, MD3
1Fellow - Allergy and Clinical Immunology, University of Kansas Medical Center
2Assistant Professor - Allergy and Clinical Immunology, University of Kansas Medical Center
3Assistant Professor - Allergy and Clinical Immunology, University of Kansas Medical Center
Introduction: Primary immunodeficiencies (PIDs) constitute a large group of rare disorders that affect the immune systems function. Some PID patients develop autoimmunity in addition to having increased susceptibility to infections due to their impaired immunity [917]. (1)
Case presentation/ Management: A 43 year old Caucasian female with history of bipolar disorder, Factor V Leiden deficiency, anti thrombin 3 deficiency, pulmonary embolism, endometriosis, and seasonal allergies was evaluated for Chronic Granulomatous Disease (CGD) in 2007. The main symptoms were inflammatory breast lesions necessitating 4 surgeries on the right breast, and back, facial, genital, ocular, mouth, and scalp sores. Biopsy with cultures of the wounds was positive for Corynebacterium, coagulase-negative staphylococcus, enterococcus, bacteroides species, and Provatella. Neutrophil oxidative burst was ordered by the infectious disease specialist and showed normal and abnormal neutrophil populations, a finding consistent with CGD carrier. Patient was started on Interferon gamma-1b after failing multiple courses of antibiotics. Her symptoms were well controlled on Interferon gamma-1b 100mcg/0.5ml SQ every other day, Trimethoprim 100mg tab (2tabs in am and 1 tab in pm), cefixime 400mg once daily, and topical mupirocin as needed except for her recurrent genital ulcers. CGD can be rarely associated with oral ulcers however there is a limited literature describing associated genital ulcers.
According to the International Study Group diagnostic criteria published in 1990 (2), the patient was diagnosed by a rheumatologist as having Behcets disease (BD). There are no pathognomonic laboratory tests in BD; as a result, the diagnosis is made clinically. Patient failed a trial of colchicine and was later started on Cyclosporine, which resulted in decrease of her mouth and genital ulcers.
Discussion: BD is a rare disease mostly seen along the Silk Road. The prevalence has been reported as 0.12 (USA) to 370 (in a single village, northern Turkey) for 100 000 inhabitants. (3) CGD is a primary immunodeficiency caused by defects in any of the five subunits of the NADPH oxidase complex responsible for the respiratory burst in phagocytic leukocytes. Patients with CGD are at increased risk of life-threatening infections with catalase-positive bacteria and fungi, and inflammatory complications such as CGD colitis. (4)
Reports of CGD female carriers with discoid lupus erythematosus, photosensitivity rashes, and other autoimmune phenomena have been published [48,49] (4). To the best of our knowledge, this is the first case to report BD in an affected CGD carrier.
The treatment of inflammatory disease in patients with CGD poses a difficult balance between therapeutic immunosuppression and the increased risk of severe infection. (5). High dose intravenous immunoglobulin, and targeted therapies such as CTLA4-Ig for T cell mediated pathologies, Rituximab for B-cell mediated pathologies, and anti-TNF for IBD, may be preferable over the broad immunosuppressive activity of glucocorticoids. In addition, emerging evidence suggests that hematopoietic stem cell transplantation has indication for cases that have been difficult to control using immunosuppression. (1) Given all that, our case emphasizes the need to maintain suspicion for autoimmune disorders / immune dysregulation in patients with PID.
References:
1) Aziz et al. Cellular and molecular mechanisms of immune dysregulation and autoimmunity. Cell Immunol. 2016 Dec;310:14-26. doi: 10.1016/j.cellimm.2016.08.012. Epub 2016 Aug 27.
2) Criteria for diagnosis of Behçet's disease. International Study Group for Behçet's Disease. Lancet. 1990;335(8697):1078.
3) Zouboulis CC (2003) Epidemiology of adamatiades-Behcet's disease. In: Zierhut M, Ohno S (eds) Immunology of Behcet's Disease, pp 1–16. Zeitlinger BV, Lisse, The Netherlands.
4) Arnold, D and Heimall, J. A Review of Chronic Granulomatous Disease. Adv Ther. 2017 Dec;34(12):2543-2557. doi: 10.1007/s12325-017-0636-2. Epub 2017 Nov 22.
5) Thomsen IP et al. A Comprehensive Approach to the Management of Children and Adults with Chronic Granulomatous Disease. J Allergy Clin Immunol Pract. 2016 Nov - Dec;4(6):1082-1088. doi: 10.1016/j.jaip.2016.03.021. Epub 2016 May 10.
(122) Submission ID#601310
Encephalopathy in an Adolescent with CD40-ligand Deficiency
Raquel Rozner, MD1, Elizabeth Feuille, MD2, James Bussel, MD3, Luigi D. Notarangelo, MD, PhD4, Charlotte Cunningham-Rundles, MD, PhD5
1Resident Physician, New York Presbyterian Hospital-Weill Cornell Medical Center
2Attending Physician, New York Presbyterian Hospital- Weill Cornell Medical Center
3Professor in Pediatrics, Department of Hematology and Oncology, Weill Cornell Medicine, NY, USA
4Chief, Laboratory of Clinical Immunology and Microbiology, IDGS, DIR, NIAID, NIH, Bethesda, MD, USA
5Professor in Medicine, Division of Clinical Immunology, Icahn School of Medicine, Mount Sinai, NY, NY, USA
Introduction: CD40-ligand deficiency is an X-linked combined immunodeficiency, characterized by susceptibility to infection, often with associated neutropenia, malignancy, and autoimmunity. Central nervous system (CNS) manifestations are less commonly reported than respiratory or gastrointestinal complications, but are most often attributed to infection. Herein we describe a challenging case of gradual onset episodic memory loss, confusion, and unilateral hemiplegia in a young male with CD40-ligand deficiency.
Case Presentation: The patient is a 13-year-old male with CD40-ligand deficiency on immunoglobulin replacement therapy presenting with recurrent, episodic altered mental status (AMS) and gradual neurocognitive decline. Initial neurologic symptoms began at age 11 years, and included fever, nausea, and eyelid fluttering. Initial comprehensive infectious workup at this time, including blood and urine cultures, Lyme antibody, serum PCR for HSV, CMV, EBV, respiratory viral PCR including atypical viruses, CSF studies including culture, Lyme EIA, PCRs for enterovirus, VZV, EBV, CMV, HSV1/2 were unrevealing. Electroencephalogram (EEG) and MRI displayed generalized slowing and global atrophy, respectively. Definitive diagnosis was not made. The patient continued to decline with worsening developmental delay and memory loss. One year later, at age 12 years, he had a recurrent episode of AMS with repeat negative infectious workup including blood and urine cultures, respiratory virus PCR including atypical viruses, CSF culture including acid fast bacillus and fungi, cryptococcal antigen, viral encephalitis panel by PCR, and serum PCR for EBV and HHV-6. EEG at this time showed left hemispheric epileptogenic potential, consistent with seizure activity.
His presentation, at age 13 years, was notable for right-sided hemiplegia with facial numbness, dysarthria, nausea, and fever. He was found to have anello virus on PCR of CSF, abnormal left temporal region on EEG, and global atrophy with stable, diffuse generalized volume loss on MRI. He was diagnosed with occult anello virus-induced encephalitis with hemiplegic migraine and discharged on valproate.
Discussion: Here we present the first reported case of Anello virus detected by PCR in a CD40-ligand deficient male with neurocognitive manifestations, attributed primarily to hemiplegic migraine. Given the anello virus prevalence and relatively avirulent character, it is presumed to be unlikely culprit for encephalitis; however, the significance of this finding is as yet unknown. This case highlights diagnostic challenges in immunodeficiency: infection may go undetected by standard diagnostic techniques; however, the significance of infections identified with advanced techniques may not yet be understood.
(123) Submission ID#601323
Etanercept Use in Refractory Chronic Henoch-Schönlein Purpura
Sana Habib, MD1, Elif Dokmeci, MD2
1Pediatric Resident, UNM
2Assoc Prof, University of New Mexico
Background: Henoch-Shönlein purpura (HSP) is an IgA-mediated small vessel vasculitis that presents with a tetrad of abdominal pain, arthritis, glomerulonephritis, and purpura. HSP is typically a self-limiting disease of childhood following a viral illness. There is no universal treatment for patients with chronic or recurrent HSP. We report a chronic refractory case of HSP that was successfully treated with a tumor necrosis factor inhibitor (TNFi), Etanercept. Etanercept functions as recombinant protein that consists of a TNF-alpha receptor ligand-binding region that links to the Fc portion of human IgG. It is currently approved for use in 5 diseases: juvenile rheumatoid arthritis, rheumatoid arthritis, ankylosing spondylitis, plaque psoriasis, psoriatic arthritis. TNFi are categorized into two broad categories, recombinant receptors (etanercept) and neutralizing antibodies (ex. infliximab and adalimumab). There have been prior case reports of HSP associated with TNFi agents during the treatment of other autoimmune conditions in the adult population. To our knowledge, there have been 3 prior etanercept related HSP reports, one report associated with adalimumab, and one with infliximab. However, there has been no prior report of etanercept use successfully treating chronic refractory HSP.
Case Presentation: A 16-year-old Native American male with 3 year history of chronic HSP, HLA-B27 positive, and enthesitis related arthritis who was initially treated with steroids, sulfasalazine and methotrexate for symptoms of joint pain and purpura. His IgA level was 545 mg/dL prior to therapy. Despite treatment for one month of steroids, eight months of sulfasalazine exclusively and eight months of methotrexate and sulfasalazine, he continued to have persistent purpura on bilateral extremities without improvement. He was subsequently initiated on Etanercept 50mg weekly and methotrexate was discontinued. Approximately one month later, his rash significantly improved. His rash and joint pain recurs when he misses a dose of Etanercept. Punch biopsies were taken 3 months after initiation of etanercept. The biopsies of a lesion from his left arm showed early leukocytoclastic vasculitis and from his left leg showed weak granular deposition of IgA, IgM and C3 within vessel walls. There is controversy whether this is a true IgA vasculitis. However, we believe that his clinical presentation and the deposition of IgA and C3 within blood vessel walls seen on biopsy correlates with chronic Henoch-Shönlein purpura.
Conclusion: There is no standard treatment of chronic HSP, but there are reports of benefit with NSAID and corticosteroids. Per our literature review, there are no prior reports of Etanercept use in the treatment of chronic HSP. TNF inhibitor, Etanercept should be considered as a treatment for chronic refractory HSP in the pediatric population as it has showed rapid resolution of purpura in this case report. Further studies of Etanercept in the treatment of chronic HSP should be conducted given the controversial literature of anti-TNF ab induced HSP during the treatment of other autoimmune diseases.
(124) Submission ID#601334
Recurrent Sinusitis in Heterozygous Hemochromatosis; Is It a Risk Factor?
Osman Dokmeci, MD1
1Assist Prof, University of New Mexico
BACKGROUND: Hereditary hemochromatosis (HH) is one of the most common inherited disorders in people of northern European descent. HH patients are at risk for a number of infections including invasive fungal sinus infections. Although clinical manifestations of iron overload appear to be quite uncommon in patients who are heterozygous carriers of HFA mutation, we present cases that appear to suggest an increased risk non allergic rhino-sinusitis.
CASE REPORT: We present a 66 year old gentleman with perennial colored rhinorrhea, with facial pressure and tenderness, constant post nasal drip, dry cough and bilateral congestion that had been going on for the past several years. He also had a frequent urge to clear his throat and had frequent episodes of sore throat despite having no history of GERD or LPR.
He reported to have multiple sinus infections every year that would progress to pneumonia and eventually require long courses of oral antibiotics. All started in his 40s intensified in the recent past. He had 3 other siblings; one died in his 40s due to liver complications of HH and had a carrier sister and brother with a hx of sino nasal problems exactly similar to the patients. His exam was remarkable for bilateral narrowed nasal passages and moderate edema of the mucosa. His rhinolaryngoscopy showed significant edema and purulent drainage, most notably from bilateral middle meati.
His skin test was negative. His CBC showed a WBC count of 6.7/ml with 2% eosinophils and his immunoglobulin panel showed an IgA of 236 mg/dl, IgG of 1190 mg/dl and IgE of 31 mg/dl. Patient was placed on Alkalol sinus rinses and Azelastine nasal spray, which he reported to work pretty well. He left for Costa Rica and is expected to return back with his siblings to A&I clinic in the coming months.
DISCUSSION: HH is one of the most common inherited disorders in people of northern European descent with an incidence of 1:200 and carrier rate of 1:10.. Most affected HH patients are homozygous for the mutation designated C282Y at the HFE gene located at the 6th chromosome. Unlike hereditary hemochromatosis, clinical manifestations of iron overload appear to be quite uncommon in patients who are heterozygous carriers. HH patients are at risk for a number of infections with bacteria whose virulence is increased in the presence of excess tissue iron. HH is also a risk factor for acute fulminant FRS . Here the mechanism is postulated to be due to quantitative or qualitative neutrophil defects as this condition is mostly seen in patients with DM, aplastic anemia, and can happen in patients undergoing antineoplastic chemotherapy. No known increased susceptibility for infections through either mechanism is postulated for patients with the heterozygous carrier state. Here we present 3 HH carrier patients who present with recurrent rhinosinusitis with no allergen sensitizations and normal IgE levels. Since most fungal immunity is at the tissue level and is cytokine driven, it can be speculated that increased tissue levels of iron might interfere with mechanisms of innate immunity.
(125) Submission ID#601340
Compound Heterozygous DOCK8 Mutations in a Patient with pre-B Cell Acute Lymphoblastic Leukemia and EBV-associated Diffuse Large B-cell Lymphoma
David K. Buchbinder, MD, MSHS1, Ivan Kirov, MD2, Jeffrey Danielson, MS3, Nirali N. Shah, MD4, Alexandra F. Freeman, MD5, Helen C. Su, MD, PhD6
1Assistant Clinical Professor, Department of Hematology, Children's Hospital of Orange County, Orange, CA, Department of Pediatrics, University of California at Irvine, Orange, CA
2Clinical Professor, Department of Oncology, Children's Hospital of Orange County, Orange, CA, Department of Pediatrics, University of California at Irvine, Orange, CA
3Research Staff Member, Laboratory of Clinical Immunology and Microbiology, NIAID, NIH, Bethesda, MD
4Associate Research Physician, Pediatric Oncology Branch, NCI, NIH, Bethesda, MD
5Director, Primary Immune Deficiency Clinic, Laboratory of Clinical Immunology and Microbiology, NIAID, NIH, Bethesda, MD, USA
6Chief, Human Immunological Diseases Section, Laboratory of Clinical Immunology and Microbiology, NIAID, NIH, Bethesda, MD
Background: Dedicator of cytokinesis 8 (DOCK8) mutations are associated with a combined immunodeficiency disorder marked by atopic features, infectious susceptibility with a striking preponderance of cutaneous viral disease, and a risk for the development of malignancy including lymphoma. Almost all cases can be diagnosed by documentation of the loss of DOCK8 protein expression.
Methods: We describe a 22-year-old male with a diagnosis of pre-B cell acute lymphoblastic leukemia (ALL) followed by Epstein-Barr Virus (EBV) associated diffuse large B cell lymphoma (DLBCL). Compound heterozygous mutations in DOCK8 were documented following the completion of whole exome sequencing (WES). The pathogenicity of the variants was assessed. Flow cytometric quantification of intracellular DOCK8 protein was completed. DOCK8 protein function was assessed by evaluating the morphology of patient lymphocytes when migrating in a 3D collagen matrix.
Results: A concern for a primary immunodeficiency was raised due to a history of recurrent otitis media which began at 12 months of age. By 4 years of age, numerous warts were noted on his fingers; however, they were transient for a duration of only 2 years. No atopic features were appreciated. At 15 years of age, a diagnosis of pre-B cell ALL was made. During ALL therapy, infectious complications were severe including an intestinal perforation, osteomyelitis, and sepsis. At 22 years of age, still in an ongoing remission from his ALL, an incidental finding of a lung nodule led to a diagnosis of EBV-associated DLBCL. During therapy, however, infectious complications were again severe including a soft tissue infection and sepsis. WES was performed and compound heterozygous mutations in DOCK8 (c.1128_1132del and c.4474-1G>C) were documented. Flow cytometric quantification of intracellular DOCK8 protein was normal when compared to a normal control. Nevertheless, additional functional assessment of DOCK8 protein was completed. When migrating through a 3D collagen matrix, 45% of the patient lymphocytes studied demonstrated abnormal elongation (stretch ratio > 8 defined by length/width) compared with 10% of lymphocytes from a normal control. He is being evaluated for hematopoietic stem cell transplant.
Conclusion: Autosomal recessive mutations in DOCK8 are a rare cause of a combined immunodeficiency marked by atopic features, infectious susceptibility with a striking preponderance of cutaneous viral disease, and a risk for the development of malignancy including lymphoma. Here, pre-B cell ALL followed by the development of a subsequent malignant neoplasm (EBV-associated DLBCL) led to the discovery of DOCK8 deficiency. Hence, as our case underscores, for rare instances of high clinical suspicion despite normal DOCK8 protein expression, additional functional testing is crucial to make a definitive diagnosis and plan treatment. Understanding the spectrum of DOCK8 mutants and their phenotypes will improve our understanding of DOCK8 deficiency.
(126) Submission ID#601345
Recalcitrant Abdominal Abscesses in a Patient with Hyperimmunoglobulin E Syndrome
Anh Nguyen, MD/MPH1, Victoria Dimitriades, MD2
1Allergy and Immunology Fellow, Division of Rheumatology, Allergy and Clinical Immunology, Department of Internal Medicine, University of California Davis Health
2Associate Clinical Professor of Pediatrics, Division of Pediatric Allergy, Immunology & Rheumatology, University of California Davis Health
Background: Autosomal dominant hyperimmunoglobulin E syndrome (AD-HIES) is a rare primary immunodeficiency caused by heterozygous loss-of-function mutations in the signal transducer and activator of transcription 3 (STAT3) gene. AD-HIES classically characterized by recurrent cold Staphylococcal abscesses, pneumonia, eczema, and an elevation of IgE level. Other additional clinical manifestations of HIES have been recognized including skeletal dysplasia (scoliosis, pathologic fractures, delayed dental deciduation), pneumatoceles, coronary-artery aneurysms, brain lesions, and Chiari malformations.
Objective: To describe a unique case of abdominal abscesses in a patient with AD-HIES.
Method: A 22-year-old female with known AD-HIES (C.1144 C>T (p.Arg382Trp)) and a complicated history of early Pneumococcal pneumonia and meningococcemia resulting in bilateral amputation below the knees along with loss of several digits, presented for evaluation of skin infection. She had a history of recurrent Staphylococcal skin abscesses and presented with inability to use her prostheses due to pain from inflammation around her amputation sites. She underwent imaging and was found to have bilateral extremity abscesses with an associated osteomyelitis of her L tibia (which was found to be MRSA after incision and drainage). While receiving intravenous antibiotics for her osteomyelitis, she developed intractable abdominal pain. Imaging showed a thick-walled, multi-septated, paranephric abscess as well as several smaller abscesses scattered throughout her abdomen. She underwent multiple drain placements and drainage of retroperitoneal fluid collections via interventional radiology (IR). Purulent fluid from the abdominal abscess drainage grew MRSA. The patient continued to have re-accumulation of abscesses despite multiple drainages. Repeat imaging noted increased paranephric abscesses which were not communicating with drains. Given lack of response to several IR-placed abdominal drains and to 6 weeks of intravenous antibiotics, she had an open surgical washout with minimal improvement. Hospital course was further complicated by development of a left lower lung lobe consolidation and sub-segmental pulmonary embolism necessitating treatment with heparin. Finally, after several weeks of escalating antimicrobial therapy and with additional drain placements, the retroperitoneal abscesses started to recede. Repeat abdominal imaging several months later while asymptomatic revealed slow but continuing resolution of the abscesses.
Conclusion: The present case raises awareness of an unusual location for infection in a patient with AD-HIES. Although the majority of complications of AD-HIES are sinopulmonary and skin infections, recalcitrant intra-abdominal abscesses should be considered in the differential of infections in HIES.
(128) Submission ID#601352
Epidemiology on Primary Immunodeficiencies in Korea: A Systematic Review of Reported Literature and Analysis of Bigdata from National Health Insurance System
Sohee Son, PhD1, Ji-Man Kang, MD, PhD2, Younsoo Hahn, MD, PhD3, Kang Mo Ahn, MD, PhD1, Yae-Jean Kim, MD, PhD1
1Department of Pediatrics, Samsung Medical Center, Sungkyunkwan University School of Medicine, Seoul, Korea
2Department of Pediatrics, Severance Children's Hospital, Yonsei University College of Medicine, Seoul, Korea
3Department of Pediatrics, Chungbuk National University Hospital, Cheongju, Korea
Introduction/Background: The recent epidemiologic studies have revealed that primary immunodeficiencies (PIDs) are more common than previously thought. However, there are very few data on epidemiology of PIDs in Korea.
Objectives: We attempted to estimate the PID epidemiology and disease burden in Korea and provide the background information for PID registry for future.
Methods: To review the previously reported scientific studies, PubMed, KoreanMed, Google Scholar were searched. Any studies on PIDs reported in scientific journal (Korean or International) from January 2001 to November 2018 were searched. Both Korean and English reports were searched. Diagnosis for PID was categorized from group I to group XI according to 2017 IUIS Phenotypic Classification. Study period was divided into two periods: period 1 from 2001 to 2005 and period 2 from 2006 to 2018, because there was a multicenter study to estimate PID epidemiology from 2001 to 2005. In addition, the number of PID patients and the cost for care were estimated among patients who requested reimbursement to Health Insurance Review and Assessment Service (HIRA) Korea for one year in 2017.
Results: A total of 334 PID patients were identified in 75 reports. One hundred and ninety-nine patients (20 reports) and 135 patients (55 reports) were found in period 1 and period 2, respectively. The PIDs were reported in 11 patients for immunodeficiencies affecting cellular and humoral immunity, 23 patients for combined immunodeficiency with associated or syndromic features, 143 patients for predominantly antibody deficiencies, 33 patients for diseases of immune dysregulation, 113 patients for congenital defects of phagocyte, 1 patient for defects in intrinsic and innate immunity, 4 patients for auto-inflammatory disorders, 6 patients for complement deficiencies, and none for phenocopies of PID.
From HIRA reimbursement data, the number of PID patients were 42 for combined immunodeficiency, 486 for predominantly antibody deficiency, 47 for common variable immunodeficiency, 135 for functional defect of neutrophils, 238 for immunodeficiency associated with other major defects, 272 for other immunodeficiencies. A total of 1,220 PID patients were treated for 14,316 days and $3,351,678 was reimbursed in 2017.
Conclusions: We performed a systematic review on published studies for PID in medical journals and national open data system of HIRA to estimate the PID disease burden for the first time in Korea. To obtain more information on true PID epidemiology and disease burden in Korea, a national multicenter study for PID registry is required in the future.
(129) Submission ID#601355
Lentiviral Gene Therapy Corrects Platelet Phenotype and Function in Wiskott-Aldrich Patients
Lucia Sereni, PhD1, Maria Carmina Castiello, PhD2, Dario Di Silvestre, PhD3, Patrizia Della Valle4, Chiara Brombin, PhD5, Francesca Ferrua, MD6, Maria Pia Cicalese, MD, PhD6, Loris Pozzi, MSc4, Maddalena Migliavacca, MD, PhD6, Maria Ester Bernardo, MD6, Claudia Pignata, MD7, Roula Farah, MD8, Lucia Dora Notarangelo, MD9, Nufar Marcus, MD10, Lorella Cattaneo, MD11, Marco Spinelli, MD12, Stefania Giannelli, PhD13, Marita Bosticardo, PhD14, Koen van Rossem, MD, PhD15, Armando D'Angelo, MD16, Alessandro Aiuti, MD, PhD17, Pierluigi Mauri, PhD18
1Junior Postdoctoral Fellow, San Raffaele Telethon Institute for Gene Therapy (SR-Tiget), Division of Regenerative Medicine, Stem Cells and Gene Therapy, San Raffaele Scientific Institute, Milan, Italy
2Senior Postdoctoral Fellow, San Raffaele Telethon Institute for Gene Therapy (SR-Tiget), Division of Regenerative Medicine, Stem Cells and Gene Therapy, San Raffaele Scientific Institute, Milan, Italy
3Researcher, Proteomic and Metabolomic Laboratory, Institute of Biomedical Technologies, National Research Council (ITB-CNR), Segrate (MI), Italy
4Biologist, Coagulation Service & Thrombosis Research Unit, San Raffaele Scientific Institute, Milan, Italy
5Researcher, University Centre for Statistics in the Biomedical Sciences (CUSSB), Vita-Salute San Raffaele University, Milano, Italy 5Vita-Salute San Raffaele University, Milan, Italy
6Clinician, San Raffaele Telethon Institute for Gene Therapy (SR-Tiget), Division of Regenerative Medicine, Stem Cells and Gene Therapy, San Raffaele Scientific Institute, Milan, Italy
7Clinician, Pediatric Section, Dep of Translational Medical Sciences, University of Naples Federico II, Naples, Italy
8Clinician, Department of Pediatrics, Division of HematologyOncology. Saint George Hospital University Medical Centre, Beirut, Lebanon
9Clinician, Pediatric Onco-Haematology and BMT Unit, Children's Hospital, ASST Spedali Civili of Brescia, Brescia, Italy
10Clinician, Department of Pediatrics B, Schneider Children's Medical Center of Israel, Petach Tikva, Israel
11Clinician, A.O. SS. Antonio e Biagio C. Arrigo. Alessandria, Italy
12Clinician, Pediatric Clinic, MBBM Foundation, Maria Letizia Verga Center, Monza, Italy
13Biologist, San Raffaele Telethon Institute for Gene Therapy (SR-Tiget), Division of Regenerative Medicine, Stem Cells and Gene Therapy, San Raffaele Scientific Institute, Milan, Italy
14Staff Scientist, Laboratory of Clinical Immunology and Microbiology, IDGS, DIR, NIAID, NIH, Bethesda, MD, USA
15Clinical Research Director, Rare Diseases Unit, GlaxoSmithKline, Brentford, UK
16Clinician, Head of Unit, Coagulation Service & Thrombosis Research Unit, San Raffaele Scientific Institute, Milan, Italy
17Clinician, Head of Unit, San Raffaele Telethon Institute for Gene Therapy (SR-Tiget), Division of Regenerative Medicine, Stem Cells and Gene Therapy, San Raffaele Scientific Institute, Milan, Italy.
18Staff Scientist, Head of Unit, Proteomic and Metabolomic Laboratory, Institute of Biomedical Technologies, National Research Council (ITB-CNR), Segrate (MI), Italy
Anna Villa, MD
Clinician, Head of Unit, San Raffaele Telethon Institute for Gene Therapy (SR-Tiget), Division of Regenerative Medicine, Stem Cells and Gene Therapy, San Raffaele Scientific Institute, Milan, Italy. 8 Milan Unit, Istituto di Ricerca Genetica e Biomedica, CNR, Milan, Italy
Micro-thrombocytopenia is one of the most serious challenges for Wiskott-Aldrich Syndrome (WAS) and X-linked Thrombocytopenia (XLT) patients. Thrombocytopenia leads to severe, potentially life-threatening, bleeding episodes, which require frequent transfusions and account for 23% of deaths in patients experiencing WAS mutations.
The gold standard treatment for WAS patients is hematopoietic stem cell transplantation (HSCT) from an HLA-identical donor but more recently a number of gene therapy (GT) trials in Europe and USA showed promising results. In particular, it has been shown that WAS patients receiving lentiviral mediated GT, consisting of autologous CD34+ cells transduced with lentiviral vector encoding the human WAS gene under the control of the endogenous promoter, in combination with a reduced intensity conditioning regimen, have a significant increase in platelet (PLT) counts. Even though PLT counts do not reach normal levels, treated patients decreased the severity and frequency of bleedings.
Here, in a cohort of 4 XLT and 16 WAS patients, fifteen treated with GT, the PLT phenotype and function were analyzed by electron microscopy, flow cytometry and proteomic profile. The aim of the project is to assess the presence of PLT defects in WAS untreated patients and the impact of GT treatment on the correction of PLT behavior.
We demonstrate that PLTs of untreated WAS patients have reduced size and abnormal ultrastructure along with hyperactivated phenotype at steady state, showing increased expression of CD62P, activated IIb3 integrin and CD40L; conversely, activation response to agonist and aggregation capacity are both decreased. Analyzing PLT samples isolated from treated patients, we found that GT restores PLT size and ultrastructure very early after treatment and reduces the hyperactivated phenotype proportionally to WAS protein (WASp) expression and follow-up length. PLTs isolated from GT treated patients showed a normal activation response to agonists and restored aggregation capacity in 5 out of 7 analysed patients.
By proteomics, various protein pathways were found downregulated in untreated PLT samples, mainly involving cytoskeletal-rearrangement proteins, integrins, signal transduction molecules, vesicles-transport proteins; additionally, decreased metabolic capacity were observed. These results are in line with the functional defects observed in PLTs in terms of activation and aggregation. Conversely, the expression of protein-pathways found downregulated in untreated patients is comparable to healthy controls in GT-treated PLT samples, reflecting the amelioration of PLT phenotype and function.
Overall, our study highlights the coexistence of multiple defects in the activation and aggregation responses occurring in WAS patient PLTs in absence of WASp. GT was able to normalize the PLT proteomic profile followed by consequent restoration of PLT ultrastructure and phenotype, suggesting GT is responsible for the observed reduction of bleeding episodes in treated patients.
(130) Submission ID#601392
PIK3CD, a Rare Autosomal Dominant Disorder of the Immune System: A Reason for the Use of Next Generation Sequencing
Jacob L. Barish, MD1, Lyda Cuervo-Pardo, MD2, Mario Rodenas, MD, FAAAAI3
1Internal Medicine Resident, University of Florida, Department of Internal Medicine
2Assistant Professor, University of Florida, Division of Rheumatology & Clinical Immunology, Department of Medicine
3Assistant Professor, University of Florida, Division of Rheumatology & Clinical Immunology, Department of Medicine
Introduction: PIK3CD is an autosomal dominant genetic disorder of the immune system that results in persistent activation of PI3K. Signaling through PI3K is essential for immune cell regulation of metabolism, migration, proliferation and differentiation, leading patient to present with lymphadenopathy, immunodeficiency and senescent T cells. The mutated protein causes T cells to over activate and mature too quickly leading to their death, this over activation also blocks the maturation of B cells.
Case presentation: A 51-year-old female with a childhood history of failure to thrive, asthma, chronic rhinitis and Common variable Immunodeficiency on intravenous immunoglobulin replacement, was seen in immunology clinic to establish care. She reported frequent episodes of pneumonia and bronchitis in her childhood. Her family history was significant for family members with leukopenia, but no diagnosed immunodeficiency. Patient had 1 son who did not report symptoms concerning for immunodeficiency. Physical exam was within normal limits with no lymphadenopathy.
Laboratory examinations exhibited normal IgA (185 mg/dL), IgG (800 mg/dL), and IgM (100 mg/dL). While flow cytometry showed normal absolute CD3 687 (570-2400 cells/uL), CD4 (540 cells/uL), NK Cells (151 cells/uL), CD19 (179 cells/uL), CD45RA (160 cells/uL), CD45RO (311 cells/uL), CD2 (757 cells/uL), and HLA-DR (173 cells/uL), nonswitched memory cells (9 cell/uL) and class-switched memory cells: (15 cells/uL). (4-62 cells/uL). Vaccine response was not pursued as patient had been on IVIG. Genetic testing was pursued, and revealed a mutation in PIK3CD gene, specifically a mutation in the c.2320G>A; p.Val774Met variant (rs370932461). This mutation though seen in databases, is not currently reported in medical literature as associated with this condition. Based on these, CT chest was ordered to screen for bronchiectasis, adenopathy and lymphoma. CT showed no cardiopulmonary disease or adenopathy, but did show an incidental adrenal mass which is now being worked up. While the pattern of inheritance of this mutation is autosomal dominant, her son is asymptomatic and testing of her son has not been pursued, though it was advised for her cousins given history of leukopenia. Patient has continued on IgG replacement therapy.
Conclusion: Recent publication by the Clinical Immunology Society suggests consideration for next generation sequencing when it can affect future family planning or it has treatment and prognostic implications. This case highlights all aspects of the importance of genetic testing as part of the diagnosis of CVID, since it can affect progeny, it offers the possibility of treatment with immune modulating agents and has implications on screening, since patients are at increased risk for malignancies.
(131) Submission ID#601396
Rapid Identification of Patients with RAG Mutations Using Valpha 7.2 Antibody
Kerry Dobbs, BSc1, Julie E. Niemela, MS, MLS2, Kenneth Olivier, MD, MPH3, Alexandra F. Freeman, MD4, Jenna Bergerson, MD/MPH5, Gregory M. Constantine, MD6, Sergio D. Rosenzweig, MD/PhD7, Luigi D. Notarangelo, MD, PhD8
1Biologist, Laboratory of Clinical Immunology and Microbiology, NIAID, NIH, Bethesda, MD, USA
2Sequencing Laboratory, Team Leader, Immunology Service, Department of Laboratory Medicine, NIH Clinical Center, Bethesda, MD, USA
3Chief, Pulmonary Branch, NHLBI, NIH, Bethesda, MD, USA
4Director, Primary Immune Deficiency Clinic, Laboratory of Clinical Immunology and Microbiology, NIAID, NIH, Bethesda, MD, USA
5Staff Clinician, Laboratory of Clinical Immunology and Microbiology, NIAID, NIH, Bethesda, MD, USA
6Clinical Fellow, Allergy and Immunology, Laboratory of Clinical Immunology and Microbiology, NIAID, NIH, Bethesda, MD, USA
7Chief, Immunology Service, Department of Laboratory Medicine, NIH Clinical Center, Bethesda, MD, USA
8Chief, Laboratory of Clinical Immunology and Microbiology, IDGS, DIR, NIAID, NIH, Bethesda, MD, USA
Background: Abnormal V(D) J recombination activity in patients with mutations in the Recombination-Activating Genes 1 and 2 (RAG1/2) results in markedly reduced usage of distal V and J genes at the T cell receptor alpha (TRA) locus. Mucosa-associated invariant T (MAIT) cells express a semi-invariant T cell receptor containing the distal TRAV1-2 gene. MAIT cells can be identified by flow cytometry using a mAb directed against Valpha 7.2, which recognizes the product of the TRAV1-2 gene. By performing high throughput sequencing (HTS) of TRA rearrangements and flow cytometry, we have confirmed lack of T cells using distal Valpha genes in patients with known RAG mutations. We now report that flow cytometry with mAb against Valpha 7.2 successfully identified RAG deficiency in two patients with an atypical presentation.
Methods: TRA rearrangements were analyzed by HTS using gDNA from sorted T cell subsets from RAG-mutated patients and healthy donors. Distal Valpha usage was measured in whole blood by flow cytometric analysis with an anti-Valpha 7.2 antibody. RAG mutations were detected by Sanger sequencing. Patients were enrolled in NIAID protocol 18-I-0041.
Results: HTS of TRA rearrangements revealed lack of distal TRAV and TRAJ gene usage in patients with RAG1/2 mutations. The presence of circulating MAIT cells in controls and patients with known RAG1/2 mutations and various clinical phenotypes was analyzed by flow cytometry using mAb against Valpha 7.2. We found a virtual lack of Valpha 7.2 expression in RAG mutated patients (<0.5%) compared to controls (2-8%). We used the Valpha 7.2 assay to test two patients with unknown immunodeficiency manifesting as skin granulomas and autoimmune cytopenia, and found nearly absent expression (0.14% and 0.08%). Targeted sequencing of RAG1/2 revealed that both patients were compound heterozygous for RAG1 mutations: p.R112H/p.C328Y and p.R410W/p.R507Q, respectively.
Conclusions: Patients with mutations in RAG1/2 demonstrate a skewing of their TCRalpha repertoire. The reduction in recombinase activity in these patients does not allow for rearrangements of the most distal Valpha segments. Rapid identification of patients lacking Valpha 7.2+ T cells by flow cytometry may prompt Sanger sequencing and identification of RAG1/2 mutations in a matter of days. This assay represents a simple but powerful tool to reduce the cost and time associated with other analysis methods.
Acknowledgements: Supported by DIR/NIAID/NIH.

(132) Submission ID#601398
Human STAT5 Deficiency Results in an Increase of Follicular T Cells Leading to Expanded Germinal Center B Cells and Autoimmunity
Maria Soledad. Caldirola, MSc, PhD1, María Isabel. Gaillard, MSc2, Norberto Walter Zwirner, PhD3, Liliana Bezrodnik, MD4
1Biochemist, Grupo de Inmunología Hospital de Niños "R.Gutierrez"- IMIPP-CONICET, Buenos Aires-Argentina
2Biochemist, Grupo de Inmunología Hospital de Niños
3Researcher, Instituto de Biología y Medicina Experimental (IBYME-CONICET), Laboratorio de Fisiopatología de la Inmunidad Innata, Buenos Aires, Argentina, Departamento de Química Biológica, Facultad de Ciencias Exactas y Naturales, Universidad de Buenos Aires,
4Director, Centro de Inmunología Clínica Dra.Bezrodnik y equipo
Introduction: The fate of effector T cells is strongly dependent on the expression of Bcl-6 or Blimp-1, which are inhibited reciprocally through a complex signaling pathway. Several studies have shown that Bcl-6 is a key transcription factor for differentiation towards the follicular helper T cells (Tfh) lineage able to collaborate with B lymphocytes (BL). On the contrary, the transcription factor Blimp-1 is highly expressed in T lymphocytes Th1, Th2 and Treg, thus regulating the differentiation towards Tfh. Materials and methods: whole fresh blood and peripheral mononuclear cells from a patient with homozygous mutation in STAT5b were analysed by flow cytometry. Analysis of cTfh (CD4+CD45RA-CXCR5+), cTfh1 (CXCR3+), cTfh17 (CCR6+), cTfh2 (CXCR3-CCR6-), naïve BL (LB IgM+IgD+CD27-), memory (MBL) (LB IgM+ IgD- CD27+), switched (MBL-Sw) (IgD-IgM-) and plasmablast (PBC) (CD27+CD38++) cells was performed. Immunoglobulins were measured in serum. Results: the patient with STAT5b deficiency showed increased values of cTfh (38%) (Healthy donors p10-p90: 7,9-17,8 %) that presented an activated phenotype (ICOS+ and PD-1+) with a skewed to a Th17 profile (CCR6+), consistent with her hipergammaglobulinemia and the marked and sustained increase in the switched MBL and PBC subpopulations in peripheral blood over the years. Discusion: This immunological phenotype described in the patient with STAT5b deficiency could explain in part the pathophysiology of the autoimmune disorders. This patient (as well as the other two patients with mutations in STAT5b previously described by our group), have had chronic hypergammaglobulinemia, autoantibodies and consequently autoimmune processes (psoriasis, hypothyroidism, eczema, alopecia and celiac disease, among others). We believe that the link between this clinical symptomatology and the molecular defect relies in the fact that the absence of STAT5b promotes a greater expression of Bcl-6, which generates a bias towards the production of cTfh cells, that give rise to a greater activation of LB, generation of LBM and plasma cells (dysregulation in the CG), events that manifest as hypergammaglobulinemia and autoimmunity. In summary, we provide promising evidence of the mechanisms that lead to autoimmunity in this type of patients that could also be a consequence of the defect in the regulation of GC, highlighting the crucial role of STAT5b in the humoral immune response and maintenance of the tolerance of the immune system.
(133) Submission ID#601403
The First Year: Experience from Mayo Clinic Laboratories After Clinical Implementation of Nine Primary Immunodeficiency Next Generation Sequencing Tests
Michelle L. Kluge, M.S., LCGC1, Susan A. Lagerstedt, BS2, Laura J. Train, BS3, Linda Hasadsri, MD, Ph.D.4, Ann M. Moyer, MD, Ph.D.4
1Genetic Counselor, Department of Laboratory Medicine and Pathology, Mayo Clinic, Rochester, MN
2Development Technologist Coordinator, Department of Laboratory Medicine and Pathology, Mayo Clinic, Rochester, MN
3Development Technologist II, Department of Laboratory Medicine and Pathology, Mayo Clinic, Rochester, MN
4Laboratory Director, Department of Laboratory Medicine and Pathology, Mayo Clinic, Rochester, MN
Background/Introduction: The term primary immunodeficiencies (PID) encompasses a phenotypically and genetically diverse group of conditions. Genetic testing for these conditions can guide treatment, reduce morbidity and mortality, allow for genetic counseling, and identification of additional at-risk family members. However, this testing can be complicated by a number of factors, including pseudogenes, high homology, methodology limitations, and the heterogeneous nature of PIDs.
Methods: Mayo Clinic Laboratories launched their first set of nine PID next generation sequencing (NGS) tests approximately one year ago. These tests include one single gene assay for GATA2 deficiency and eight targeted next generation sequencing panels for: atypical hemolytic uremic syndrome (aHUS), autoinflammatory disorders, B-cell disorders, monogenic irritable bowel disease (IBD), phagocytic defects, severe combined immunodeficiencies (SCID), and severe or cyclic neutropenia. Herein we summarize our first year of experience with these NGS tests, with a focus on the eight targeted panel tests.
Results: From March 2018 through November 2018 we performed testing for 341 cases. Our highest volume of tests was for the aHUS panel (127/341 cases, 41%). A variant was reported in 76/341 cases (22.29%). These variants included variants of uncertain significance, likely pathogenic variants and pathogenic variants. The indication with the highest percentage of cases where a variant was reported was SCID (9/13 cases, 69.23%). The number of cases that were considered solved, where the genotype likely explains the patients phenotype, varied widely by indication. Twenty cases were found to have a pathogenic or likely pathogenic variant or variants; however 2/20 cases were heterozygotes for an autosomal recessive condition and were not considered solved cases. The panel with the highest percentage of solved cases is our SCID panel (4/13 cases, 30.77%). Conversely, we have yet to solve an autoinflammatory, irritable bowel disease, or telomere defects case; however ~20% of cases in each of those three panels have had a variant of uncertain significance reported. We hypothesize that one of the reasons for the low detection rate for these three panels is inappropriate test orders. We are also actively looking for ways to update all 8 panels to increase detection rates and clinical utility, for example expanding the gene list of our IBD panel, including large deletion/duplication detection, and including NCF1, a difficult gene to capture by NGS, on the phagocytic panel. Finally, we present the molecular findings from a number of interesting cases that were solved using our targeted NGS panels.
Conclusions: The launch of our PID NGS tests in March of 2018 has allowed us to aid patients by confirming diagnoses and providing molecular diagnoses that will enable more accurate genetic counseling and risk assessment. We have also uncovered areas for improvement, both on the clinical side: provider education is important to enable better identification of patients who can benefit from molecular genetic testing for PIDs, and on the laboratory side: introduction of more expanded panels and additional methodologies.
(134) Submission ID#601405
Treatment-resistant Autoimmune Cytopenias as a Sign of Primary Immunodeficiency Disorders (PIDs)
Sara Ciullini Mannurita, MSc1, Enrico Attardi, MD2, Ebe Schiavo, PhD1, Maria Luisa Coniglio, MSc3, Maddalena Bagni, MD2, Marina Vignoli, PhD1, Fabio Tucci, MD4, Marinella Veltroni, MD4, Claudio Favre, MD5, Eleonora Gambineri, MD6
1Research fellow, University of Florence, Dep. Neurosciences, Psychology, Drug Research and Child Health (NEUROFARBA), Florence, Italy
2Haematology trainee, AOU Careggi, Haematology Unit, University of Florence, Florence, Italy
3Biologist, Anna Meyer Children's Hospital, Department of Haematology-Oncology, Florence, Italy
4Medical doctor, Anna Meyer Children's Hospital, Department of Haematology-Oncology, Florence, Italy
5Head of Department, Anna Meyer Children's Hospital, Department of Haematology-Oncology, Florence, Italy
6Associate Professor, Anna Meyer Children's Hospital, Department of Haematology-Oncology - Bone Marrow Transplantation BMT Unit, University of Florence, Dep. Neurosciences, Psychology, Drug Research and Child Health (NEUROFARBA), Florence, Italy
The progressive decrease of red blood cells, platelets or neutrophils via a self-directed immune process is jointly termed as autoimmune cytopenias. While autoimmune cytopenias, including autoimmune hemolytic anemia (AIHA), immune thrombocytopenic purpura (ITP), and autoimmune neutropenia (AN), are a common presentation of autoimmunity in the general population, they are particularly frequent and can appear as the first sign in patients with primary immunodeficiencies (PIDs). Possible causes of cytopenia in PIDs comprise mainly immune dysregulation, bone marrow failure (BMF) and myelodysplasia.
Our goal is to investigate possible immune mediated mechanisms underlying chronic cytopenia in children in order to achieve an early diagnosis and consequently offer timely and appropriate therapy.
We selected 24 patients affected by chronic cytopenia, evaluated with immunophenotyping by flow-cytometry; data were subjected to multivariate analysis by Principal Component Analysis (PCA). Next Generation Sequencing (NGS) analysis of genes frequently implicated in PIDs was performed.
Among the patients, 5 were affected by bone marrow failure, of which 2 were diagnosed with Fanconi Anemia and severe congenital neutropenia; 12 were affected by immune-mediated cytopenia and 7 by idiopathic cytopenia. The immunephenotyping showed a typical pattern of CD8 T cell subpopulations expression in patients compared with healthy donors with an increase of naïve T cells and a reduction of central memory (CM) and effector memory (EM) T cells levels. We observed a decrease in total B cells, B switched and B memory cells and an increase in CD21low cells. PCA showed an overlap between groups, however it revealed a peculiar trend of some single patient, suggesting the pathway involved in immune defect.
Preliminary results from NGS studies revealed genetic variations in genes previously associated with PIDs in 10 out of 11 patients investigated. In particular we identify one patient with a mutation in FAS, one with a mutation in AIRE and one with a mutation in IKAROS. Concerning the remaining patients further studies are ongoing to validate the pathogenicity of the genetic variations.
PCA is a very effective tool to analyze several parameters at the same time, highlighting patients whose phenotype shows the main peculiarities. The presence of specific lymphocyte subpopulation patterns can be important indicators of immune-mediated cytopenias and helpful signs of specific PIDs that should promptly be investigated with genetic analysis.
(135) Submission ID#601434
What We Are Missing with PID Exomes, Including Poorly Covered Exons
Sarah E. Henrickson, MD, PhD1, Manish Butte, MD, PhD2
1Attending Physician, The Children's Hospital of Philadelphia, Divsion of Allergy Immunology
2Division of Allergy/Immunology Chair, Division of Immunology, Allergy, and Rheumatology, Dept. of Pediatrics and Jeffrey Modell Diagnos-tic and Research Center, University of California, Los Angeles
The rapid of discovery of novel, monogenic primary immunodeficiencies has been made possible by the broad availability of clinical whole exome sequencing (WES). However, clinical WES has major shortcomings that should be understood by practicing immunologists. Focusing on the 2017 IUIS list of ~330 monogenic primary immunodeficiency genes, we show here limitations in coverage that could significantly impact clinical interpretation. On the Agilent Whole Exome capture kit, the most common WES platform, there are a number of genes with exons that are poorly covered. Specifically, there are at least 94 genes with less than 100% exonic coverage, 26 with less than 99% coverage and 5 with less than 90% coverage (e.g. IKBKB, NCF1, TACI, UNC93B1 and TBX1). Beyond this challenging technical issue, there are more subtle issues as well. These include the presence of pseudogenes in at least 17 of our genes (e.g. AK2, C1QBP, CD46, CFTR, CR2, MSN, NCF1, NCSTN, IKBKG, NHP2, PMS2, PTEN, RNASEH2C, RPS, SBDS and WAS), which can make accurate sequencing very challenging. Finally, there are many known causative intronic (e.g. BTK, CTLA-4, WASP) and copy number variant mutations (e.g. RAG1 and XIAP) as well as large deletions (e.g. DOCK8) that we cannot expect to be optimally covered using WES. This list of genes requires consideration even with a negative exome and may require additional approaches including Whole Genome Sequencing, Sanger sequencing, CNV arrays and/or long-read NGS sequencing. WES is a powerful genomic diagnostic tool, but to avoid missing key diagnostic insights using these alternative approaches may be critical when certain genes are in the differential diagnosis. Going forward, as PID phenotypes continue to broaden, these issues remain fundamentally important even if these genes are not obviously implicated in a given clinical phenotype.
(136) Submission ID#601465
Recurrent Viral Encephalitis in a Child with Several Variants of Uncertain Significance in Primary Immunodeficiency Genes
Sara Sussman, MD1, Zoya Treyster, MD2, Artemio M. Jongco, III, MD, PhD, MPH3
1Fellow, Department of Pediatrics, Zucker School of Medicine at Hofstra Northwell School of Medicine
2Fellow, Division of Allergy & Immunology, Donald and Barbara Zucker School of Medicine at Hofstra/Northwell
3Assistant Professor of Medicine and Pediatrics, Division of Allergy & Immunology, Donald and Barbara Zucker School of Medicine at Hofstra/Northwell; Center for Health Innovations and Outcomes Research, Feinstein Institute for Medical Research, Manhasset, NY
More physicians are utilizing targeted genetic panels to reach a definitive diagnosis for their patients with immunodeficiency. However, this increase in testing also has led to the discovery of many more variants of uncertain significance (VUS) in the genes tested. These findings can often leave the patient and the physician with more questions than answers. We present a patient with recurrent infections found to have multiple variants of uncertain significance in several genes associated with primary immunodeficiency.
A 13-year-old female who was diagnosed with Crohns disease at age 9 after intestinal perforation and jejunal resection experienced two discrete episodes of Epstein Barr Virus (EBV) meningoencephalitis and septic shock. The first episode was diagnosed when patient had fever and altered mental status and occurred prior to her Crohns disease diagnosis and the second episode was complicated with altered mental status, disseminated intravascular coagulation (DIC) and hypotension requiring PICU admission. Aside from these two major infections, the family denied any other infections requiring antibiotics in the last 5 years and reported a remote history of repeated streptococcal pharyngitis that have not recurred. Immunology was consulted at the time of the second episode of meningoencephalitis and work up was mainly unremarkable with normal immunoglobulins, adequate vaccine response to Hib, tetanus, diphtheria, rubella, measles and pneumococcus (18 out of 22 protective titers). She had normal T cell numbers with slightly decreased natural killer numbers for age. Neutrophil studies showed normal Dihydrorhodamine (DHR) analysis, glucose-6-phosphate dehydrogenase levels and Myeloperoxidase (MPO) stain. Commercial testing of her Toll like Receptors (1-8) showed normal function. Invitae Primary Immunodeficiency Panel demonstrated a heterozygous variant in NOD2 (c2.104C>T; p.Arg702Trp) as well as heterozygous variants of uncertain significance in IL7R (c.662G>T; p.Ser221Ile) and TLR3 (c.889C>G; p.Leu297Val). The patients NOD2 variant is known to be associated with an increased risk for Crohns disease.
Even with our patients presentation with recurrent severe viral infections and IBD, it is not immediately clear how these genetic results explain the pathology. Innate immune defects probably contribute to her presentation and it is currently unclear if and how the combination of multiple genetic variants has left her immunologically vulnerable. We use this case to demonstrate that even when genetic testing does not elucidate a clear-cut diagnosis of primary immunodeficiency, it can still provide helpful insight into a patients underlying immune phenotype.
(137) Submission ID#601470
Quercetin Halts Abnormal IL-1beta and IL-18 Production: A Natural Calm for XIAP Deficiency?
Erika Owsley, B.S.1, Vijaya Chaturvedi, B.S.1, Michael Jordan, MD2, Catherine Terrell, B.S.1, Parinda Mehta, MD3, Rebecca A. Marsh, MD4
1Research Associate, Cincinnati Children's Hospital
2Professor of Pediatrics, Divisions of Immunobiology, and Bone Marrow Transplantation and Immune Deficiency Cincinnati Children's Hospital
3Professor, University of Cincinnati
4Associate Professor, Division of Bone Marrow Transplantation and Immune Deficiency, Cincinnati Childrens Hospital Medical Center
Introduction: XIAP Deficiency is a rare primary immune deficiency characterized by hemophagocytic lymphohistiocytosis, recurrent fever and inflammatory syndromes, inflammatory bowel disease, hypogammaglobulinemia, recurrent infections, and other manifestations. Loss of XIAP results in abnormal TNF receptor signaling and NLRP3 inflammasome actvity which leads to dysregulated production of IL-1beta and IL-18. We hypothesized that suppressing the NLRP3 inflammasome with either targeted deletion or pharmacologic inhibition would suppress abnormal production and secretion of inflammatory IL-1beta and IL-18.
Methods: Bone marrow derived macrophages (BMDMs) from control, XIAP-deficient, and XIAP and NLRP3 double knock-out mice were derived with 1 week of culture in L929-cell conditioned media. BMDMs were stimulated with a variety of TLR agonists or TNF-alpha, with or without a variety of inhibitors including the NLRP3 inhibitor MCC950, the cathepsin B inhibitor CA-074, and quercetin, which is a natural flavonoid (antioxidant) found in many fruits and vegetables, and available as a nutritional supplement. IL-1beta, IL-18, and TNF-alpha were measured in supernatants by ELISA, and cell death was evaluated by flow cytometry using PI exclusion.
Results: As expected, BMDMs from XIAP deficient mice had markedly increased TLR-agonist- or TNF-alpha-induced IL-1beta production compared to normal BMDMs. Genetic deletion of NLRP3 and the pre-treatment of cells with the NLRP3 inhibitor MCC950 greatly reduced abnormal IL-1beta production; residual production of IL-1beta could be inhibited by caspase-8 inhibition. Pre-treatment of cells with the cathepsin B inhibitor CA-074 also decreased cytokine production but was toxic at higher concentrations. Quercetin reliably abrogated IL-1beta, and also IL-18. Quercetin was found to inhibit priming of the NLRP3 inflammasome (decreased upregulation of pro-IL1beta and NLRP3) and also decreased TNF-alpha secretion following TLR agonist stimulation.
Conclusion: Quercetin suppresses the NLRP3 inflammasome and may be a promising therapeutic option for patients with XIAP deficiency. It prevents IL-1beta and IL-18 secretion. It is a particularly appealing option given that it is a naturally occurring antioxidant, has a great safety profile, and is readily available as a nutritional supplement. Human studies are needed.
(139) Submission ID#601493
Single Cell RNAseq Analysis Reveals Profound Abnormalities in the Distribution and Diversity of Thymic Epithelial Cells in Wild-type and Rag1 Mutant Mice
Francesca Pala, PhD1, Andrew Martins, PhD2, John Tsang, PhD3, Luigi D. Notarangelo, MD, PhD4, Marita Bosticardo, PhD5
1Post-Doctoral Fellow, Laboratory of Clinical Immunology and Microbiology, IDGS, DIR, NIAID, NIH, Bethesda, MD, USA
2Research Fellow, LABORATORY OF IMMUNE SYSTEM BIOLOGY, NIAID, NIH
3Investigator, LABORATORY OF IMMUNE SYSTEM BIOLOGY, NIAID, NIH
4Chief, Laboratory of Clinical Immunology and Microbiology, IDGS, DIR, NIAID, NIH, Bethesda, MD, USA
5 Staff Scientist, Laboratory of Clinical Immunology and Microbiology, IDGS, DIR, NIAID, NIH, Bethesda, MD, USA
Background: The thymus contains a heterogeneous population of stromal cells which orchestrate T cell development and selection. Recently, single cell RNA sequencing (scRNAseq) analysis in mice has disclosed an unexpected complexity of thymic stromal cells, and medullary thymic epithelial cells (mTECs) in particular. However, the developmental origin, hierarchy, and function of these subpopulations remain ill-defined. Moreover, although cortical TECs (cTECs) are thought to represent a more homogeneous population, their characterization has been largely restricted to the adult thymus. We have previously shown that impaired lymphostromal cross-talk in the thymus of patients with combined immunodeficiency (and of corresponding mouse models) is associated with abnormalities of thymic architecture and TEC maturation. Here, we sought to compare TEC distribution and gene expression in wild-type (WT) and in mice carrying Rag1 hypomorphic mutations observed in patients with combined immune deficiency and immune dysregulation.
Methods: Multi-color flow cytometry and scRNAseq were used to analyze composition and distribution of cTEC and mTEC subpopulations in WT and Rag1 mutant mice at various weeks of age (NIAID animal protocol: LCIM-6E).
Results: We observed that Rag1 mutant mice have an excess of cTECs, and that their mTEC compartment is predominantly represented by cells with high levels of MHC class II (MHC-II) expression, recapitulating the phenotype of neonatal WT thymi. While MHC-IIhi mTECs are thought to represent a minor fraction of mTECs in adult WT mice and include mature AIRE+ cells, a relative abundance of MHC-IIhi mTECs is observed also at neonatal age, where they are thought to represent immature mTECs. To define more precisely TEC maturation, we performed scRNAseq on sorted CD45- EPCAM+ cells, and identified 8 and 10 distinct clusters of TECs in WT and Rag1 mutant mice, respectively. A large proportion of cells in Rag1 mutant mice could be ascribed to the cTEC compartment, confirming our previous flow cytometry and histopathology results. Furthermore, scRNAseq analysis also disclosed a different distribution of mTEC subsets in WT and Rag1 mutant mice. To address the hypothesis that this difference in cTEC and mTEC abundance and subset distribution may reflect different maturation stages in TEC development in WT and Rag1 mutant mice, we will perform lineage tracing and transplantation experiments, and we will also extend TEC scRNAseq analysis to WT and mutant mice of embryonic and neonatal age. In parallel, to evaluate the contribution of thymocyte maturation in shaping the stromal populations, scRNAseq will be performed on thymocytes.
Conclusions: We have further refined the complexity of TECs, and shown that impaired development of T cells in combined immune deficiency (as exemplified by Rag1 mutant mice) has profound effects on the composition and maturation of TECs and may thus contribute to abnormalities of immune tolerance that are often associated with these conditions.
(140) Submission ID#601506
Multigenicity of the Deficit of the Immune System: Novel Frontiers of Primary Immunodeficiencies (PIDs)
Ebe Schiavo, PhD1, Sara Ciullini Mannurita, MSc1, Maria Luisa Coniglio, MSc2, Enrico Attardi, MD3, Marina Vignoli, PhD4, Laura Luti, MD5, Annalisa Tondo, MD6, Ilaria Fotzi, MD6, Elena Chiocca, MD6, Fabio Tucci, MD6, Marinella Veltroni, MD6, Claudio Favre, MD7, Eleonora Gambineri, MD8
1Research fellow, University of Florence, Dep. Neurosciences, Psychology, Drug Research and Child Health (NEUROFARBA), Florence, Italy.
2Biologist, Anna Meyer Children's Hospital, Department of Haematology-Oncology, Florence, Italy.
3Haematology trainee, AOU Careggi, Haematology Unit, University of Florence, Florence, Italy.
4Research fellow, University of Florence, Dep. Neurosciences, Psychology, Drug Research and Child Health (NEUROFARBA), Florence, Italy.
5Medical doctor, Pediatric Hematology Oncology, Bone Marrow Transplant, S. Chiara Hospital, Pisa, Italy.
6Medical doctor, Anna Meyer Children's Hospital, Department of Haematology-Oncology, Florence, Italy.
7Head of Department, Anna Meyer Children's Hospital, Department of Haematology-Oncology, Florence, Italy.
8Associate Professor, Anna Meyer Children's Hospital, Department of Haematology-Oncology - Bone Marrow Transplantation BMT Unit, University of Florence, Dep. Neurosciences, Psychology, Drug Research and Child Health (NEUROFARBA), Florence, Italy.
The advent of next-generation sequencing (NGS), with the development of whole-exome sequencing (WES) in particular, has allowed the identification of unknown genetic lesions for many diseases and the implementation of specific therapeutic strategies. Primary immunodeficiencies (PIDs) are a group of rare diseases which have benefited from NGS, with the discovery and molecular characterization of previously genetically undefined diseases and the identification of novel molecules involved in the regulation of the immune system.
PIDs are often associated with autoimmune disease due to the dysregulation of the immune system as a whole. The clinical phenotypes are heterogeneous and often overlapping. While a monogenic cause of disease has been identified in a most subsets of patients, the recent application of whole-genome sequencing has found that a polygenic cause is likely.
Our aim is to investigate the genetic background of patients with immunedysregulations and autoimmunity and to evaluate the possible pathogenicity of the identified gene variants through extensive functional studies.
We select 19 patients with sign of immunedysregulation and autoimmunity, extended immunophenotyping and next-generation sequencing (NGS) analysis of 50 genes frequently implicated in PIDs was performed.
In six of them we identify a single gene as responsible of the clinical feature. In particular, we identify two patients with gain of function mutation in STAT3, one patient with a mutation in CTLA4, one patient with an activating PIK3CD mutation, one with a RAG1 mutation and one with a FAS mutation. In most of them variants in multiple genes have been detected. Interestingly, we find that some genes are recurrently mutated in more then one patient such as WAS, DOCK8, CASP10, CASP8, NFATC2 and FCGR3A. Further studies are ongoing to validate the effect of the variations identified.
Our results strongly suggest that the old hypothesis, based on a single gene mutation as a cause of illness, should be revised in favor of the concept that "is the sum that causes the effect" and that a different point of view on PIDs now seems inevitable.
(141) Submission ID#601507
Dose and Clinical Outcomes in Patients with CVID and Bronchiectasis Receiving Immunoglobulin Replacement Therapy in the Home
Allyson Checkley, PhD1, Loretta Kristofek, BSN, RN2, William Bolgar, PharmD3, Luqman Seidu, MD4
1Research and Registry Program Advisor, Coram CVS Specialty Infusion Services
2Clinical Service Liaison, Coram CVS Specialty Infusion Services
3VP Clinical Operations, Coram CVS Specialty Infusion Services
4Physician, Omni Allergy, Immunology, and Asthma
Introduction/Background: Immunoglobulin replacement therapy (IGRT) may be optimized to reduce the severity and incidence of infections and potentially delay or abrogate the development of pulmonary complications of primary immune deficiencies. Pulmonary complications including bronchiectasis are common in common variable immune deficiency (CVID) and contribute significantly to morbidity and mortality in these patients. It remains unclear whether continued obstructive bronchial changes are a result of repeated respiratory infections, associated inflammation and immune dysregulation, or simply lung-damage that is irreversible by the time therapy is initiated. It has also been suggested that under-treatment in addition to the diagnostic delay may contribute to the development of bronchiectasis in patients with PID. Lower serum IgG levels with any given dose of immunoglobulin replacement therapy have been demonstrated in patients with bronchiectasis compared to those PID patients without this complication. In addition, earlier studies have shown that greater doses of Ig (600 mg/kg/ month) may reduce the frequency and duration of infections and help prevent or slow progression of chronic lung disease.
Objective: To evaluate the prevalence of bronchiectasis in a cohort of patients with a diagnosis of CVID and identify associated Ig dosing patterns and clinical outcomes.
Methods: Data were analyzed from the IDEaL (Immunoglobulin, Diagnosis, Evaluation, and key Learnings) Patient Registry. This is a prospective, longitudinal registry study of patients receiving Ig replacement therapy in the home or ambulatory infusion suite with one national home infusion provider. Nursing and pharmacy standard of care forms were collected, and dose, infection rate, and prevalence of bronchiectasis were evaluated in patients with a diagnosis of CVID (ICD-10 codes: D83.9, D83.1)
Results: There were 310 patients in the Registry with CVID, 14 (4.5%) of which bronchiectasis was also observed. Seventy-nine percent (n=246) of the study population was female, and 50% (n=7) of the cases of bronchiectasis were observed in females. The mean age of the patients with concurrent bronchiectasis was 65±15.8 at start of care compared to 57±15.8 in those without this observed bronchial obstruction. Most bronchiectasis patients (n=11) received IGRT subcutaneously every week with a mean dose of 123.8±22.8 mg/kg/wk. The mean dose of Ig in the 3 remaining patients receiving Ig intravenously was 506.8±82.0 mg/kg/month. The average annual rate of infection in IVIG and SCIG patients with bronchiectasis was 1.6±1.0 and 2.2±1.3, respectively, however many were serious bacterial infections. At time of analysis, 7 of the bronchiectasis patients remained active in the registry and 7 had withdrawn. Reasons for withdrawal included stopping IGRT due to the following: patient decision (n=3), physician decision (n=1) insurance change (n=1), and patient expired (n=2).
Conclusions: There were 14 documented cases of bronchiectasis in our cohort of CVID registry patients, and dosing patterns aligned with standard doses despite the presence of bronchial obstruction. Further studies are necessary to assess evolution of lung damage with respect to Ig dosing in patients with CVID.
(142) Submission ID#601511
NGS Reveals Repertoire Restriction of Treg Cells in APDS1 Patients
Ottavia M Delmonte, MD, PhD1, Riccardo Castagnoli, MD2, Stephen Daley, DVM3, Kerry Dobbs, BSc4, Marita Bosticardo, PhD5, Su Hua, PhD6, Gulbu Uzel, MD7, Luigi D. Notarangelo, MD, PhD8
1Staff Clinician, 1 Laboratory of Clinical Immunology and Microbiology, Division of Intramural Research, National Institute of Allergy and Infectious Diseases, National Institutes of Health, Bethesda, MD.
2Research fellow, 1 Laboratory of Clinical Immunology and Microbiology, Division of Intramural Research, National Institute of Allergy and Infectious Diseases, National Institutes of Health, Bethesda, MD.
3Senior Research Fellow, Infection and Immunity Program, Monash Biomedicine Discovery Institute and Department of Biochemistry and Molecular Biology, Monash University, Melbourne, VIC, Australia
4Biologist, Laboratory of Clinical Immunology and Microbiology, NIAID, NIH, Bethesda, MD, USA
5Staff Scientist, Laboratory of Clinical Immunology and Microbiology, IDGS, DIR, NIAID, NIH, Bethesda, MD, USA
6Staff Scientist, Immunopathogenesis Unit, LCIM, NIAID, NIH
7Staff Clinician, Laboratory of Clinical Immunology and Microbiology, National institute of Allergy and Infectious Diseases, NIH, Bethesda, MD, USA
8Chief, Laboratory of Clinical Immunology and Microbiology, IDGS, DIR, NIAID, NIH, Bethesda, MD, USA
Background: Activated phosphoinositide 3-kinase syndrome type 1 (APDS1) is a combined immunodeficiency resulting from gain-of-function (GOF) mutations in PIK3CD, the gene encoding the catalytic subunit of phosphoinositide 3-kinase (PI3K). This form of PID is characterized by recurrent respiratory tract infections, susceptibility to herpes virus infections, impaired antibody responses, lymphoproliferation and autoimmunity. Previous studies showed that patients with APDS1 have B cell defects that contribute to the clinical phenotype. Furthermore, these patients display T cell abnormalities, including increased numbers of memory T cells and T follicular helper cells (Tfh), reduction of naïve T cells and impaired T regulatory cell (Treg) function. Whether these T cell abnormalities are also associated with perturbations of T cell repertoire in unknown.
Objective: We aimed to investigate the effects of increased PI3K signaling on the T-cell repertoire of patients with APDS.
Methods: High throughput sequencing was used to study composition and diversity of T-cell receptor (TRA) and T-cell receptor (TRB) repertoire in sorted Treg, Tfh, conventional CD4+ (Tconv), and CD8+ T cells from 4 patients with PIK3CD GOF mutations and healthy controls.
Results: Treg cells of patients with APDS1 show restriction of TRA and TRB repertoire diversity, and increased clonality. No repertoire restriction was detected in Tfh, Tconv, and CD8+ T cells from the same patients. However, the TRB repertoire of Treg and CD8+ cells was enriched for the presence of hydrophobic amino acids in position 6 and 7 of the CDR3, a biomarker of self-reactivity.
Conclusion: These data demonstrate that the T-cell repertoire of patients with APDS1 is characterized by a molecular signature that may contribute to the increased rate of autoimmunity associated with this condition. Furthermore, our result support the notion that the PI3K pathway is a key regulator of Treg cell development and homeostasis in humans.
(143) Submission ID#601524
Combined Immunodeficiency in a Patient with a Heterozygous TNFRSF13B (TACI) Variant
Amanda V. Grippen Goddard, DO1, Mohini Pathria, MD1, Flavia Hoyte, MD2, Rafeul Alam, MD, PhD3
1Allergy/Immunology Fellow, National Jewish Health
2Associate Professor, National Jewish Health
3Professor, National Jewish Health
Introduction/Background: Monogenetic defects are responsible for approximately 10% of cases of common variable immunodeficiency disorder (CVID). In most cases of CVID, there is a polygenic mode of inheritance. Variants in TNFRSF13B (TACI) are seen in 7-10% of CVID patients, but also in asymptomatic relatives and in about 1-2% of normal individuals who are heterozygotes. This highlights the incomplete penetrance of TACI mutations and suggests that heterozygous mutations increase the risk, but are neither necessary nor sufficient to cause CVID. TACI mutations are known to mainly result in defects in B-cell switching and differentiation; however, some of these patients are also found to have T-cell defects.
Objectives: 1) Illustrate the polygenic nature of CVID. 2) Discuss predominantly T-cell immunodeficiency in a patient with a heterozygous TACI variant.
Results: A 26-year-old, Mexican male, was diagnosed with Tuberculosis at the age of 6, shortly after moving to the United States. He received one year of antimycobacterial therapy. He continued to have frequent, but mild, respiratory infections that resulted in approximately 2 missed school days per month, but did not require any hospitalizations. At the age of 22, he was diagnosed with multi-lobar pneumonia, which progressed to sepsis and hypoxic respiratory failure requiring intubation. One year later, he developed a second episode of pneumonia with sepsis requiring ICU admission. Subsequently, an immunodeficiency evaluation revealed an IgA of 40 mg/dL (70-400), IgG of 667 mg/dL (700-1600), IgM of 31 mg/dL (60-300), protection against 7/14 streptococcal pneumoniae serotypes, and protective Tetanus and Diptheria IgG antibodies (2.4 and 11.5 IU/mL, respectively). Quantitative lymphocytes subsets were significantly abnormal. They showed a CD45 of 0.47 K/mcL (0.82-2.84), CD3 of 300 cells/mcL (550-2202), CD4 of 197 cells/mcL (385-1437), CD8 of 97 cells/mcL (199-846) and CD 19 of 26 cells/mcL (91-409). Immature B cells (CD38lo/CD21lo) were elevated at 13.9% (0.5-8.0). Lymphocyte proliferation in response to PHA, Con A and PWM was 4-10x lower in the patient as compared to a healthy control. He was subsequently started on intravenous immunoglobulin (IVIG) replacement therapy and continued this for 1 year without any infections. Due to an insurance related lapse of IVIG infusion for 8 months, he developed another multi-lobar pneumonia requiring hospitalization. He is currently back on monthly IVIG replacement therapy and doing well. Comprehensive genetic testing for 207 known primary Immunodeficiency genes revealed one likely pathogenic heterozygous variant, c.310T>C (p.Cys104Arg) in TNFRSF13B (TACI), and three additional variants of uncertain significance: c.31G>A (p.Asp11Asn) in RAC2, c.237G>C (p.Gln79His) in TRAF3IP2, and c.4720G>A (p.Ala1574Thr) in VPS13B.
Conclusions: Features that stand out in our patient are 1). Recurrent bacterial infection in the setting of normal antibody response and mildly depressed IgA, IgM and IgG; and 2). Absence of viral and fungal infections despite markedly reduced CD4 and CD8 T cell number and proliferation. Although TACI mutations can result in B and T cell deficiencies, it is unlikely that a heterozygous TACI variant alone can explain this unusual clinical manifestation in our patient. We speculate that epistasis with other immune gene variant(s) contributed to this complex phenotype.
(144) Submission ID#601532
Diagnostic Challenges in Hospitalized Patients with Suspected Inborn Errors of Immunity in a Reference Center in the Southwest of Colombia
Andres F. Zea-Vera, MD, PhD1, Fabio S. Vargas-Cely2, Vanessa Montoya-Lozano3
1Assistant Professor, Universidad del Valle. Hospital Universitario del Valle.
2Medicine Student, Universidad del Valle
3Nursing Student, Universidad del Valle
Introduction: The Hospital Universitario del Valle (located in Cali, Colombia) is a reference center for the low income people of the southwest of Colombia with a population of influence close to 4.5 million people. The low number of clinical Immunologist in Colombia represents a huge challenge for the field.
Results: Since June 2016, sixty one (61) hospitalized patients have been evaluated by the Clinical Immunology service. The most common causes of consultation are recurrent infection syndrome, disseminated tuberculosis, hypergammaglobulinemia, refractory autoimmune disease and early age malignancies. The main diagnoses at admission were complicated pneumonia, meningitis and opportunistic infections in HIV negative patients.
Patients were classified as Primary Immunodeficiencies (10 patients 16%), suspected Immunodeficiencies or patients in follow up after discharge (27 patients 44%) and secondary Immune disorders due to autoimmunity, malignancy, immunosuppressant therapy or chronic infections (24 patients 60%).
According to the IUIS-2017 classification, 10 patients with confirm Inborn Errors of Immunity (PID) were diagnosed: II. CID with associated or syndromic features (4), III. Predominantly Antibody deficiencies (2), I. Immunodeficiencies affecting cellular and humoral immunity (1), VII. Auto-inflammatory disorders (2), IX. Phenocopies of PID (1). Two non related cases of Ataxia-Telangiectasia and one case of Schimke syndrome (SMARCAL1 compound heterozygous mutation) were diagnosed in the last year. We observed a wide range of age (we evaluate adult and pediatric population) with a Male:Female ratio close to 1:1
Conclusions: The Hospital Universitario del Valle - Clinical Immunology inpatient service had increased the opportunity for the subsidiary health care system patients in Colombia. Interestingly our main PID group was combined Immunodeficiency with associated or syndromic features opposite to previous reports. Molecular and functional testing diagnosis is a growing necessity in Colombia. Future characterization of patients with PID is necessary to reduce complications.
(145) Submission ID#601541
Diagnostic Yield of a Next-Generation Sequencing Panel for Primary Immunodeficiencies in a Cohort of Pediatric Patients with Immunohematologic Disorders
Elizabeth A. Varga, MS, LGC1, Kristin Zajo, MA, MS, LGC2, Melissa Rose, DO3, Benjamin Prince, MD, MSCI4
1Genetic Counselor, Co-Director of Personalized Medicine, Division of Hematology/Oncology/BMT and the Institute for Genomic Medicine, Nationwide Childrens Hospital
2Genetic Counselor, Division of Hematology/Oncology/BMT, Nationwide Children's Hospital
3Director, Cytopenia and Bone Marrow Failure Program, Division of Hematology/Oncology/BMT, Nationwide Children's Hospital
4Assistant Professor of Pediatrics, Division of Allergy-Immunology, Nationwide Children's Hospital,
Immunohematologic disorders encompass a broad array of clinical conditions in which hematologic manifestations, usually cytopenias, are caused by aberrant immune responses. These often lead to the development of neutropenia, hemolytic anemia, or thrombocytopenia, either separately or in combination. Common underlying mechanisms include immunodeficiency, immune dysregulation, and systemic autoimmunity. Clinical diagnosis of these disorders is complicated by overlapping phenotypes.
In April 2017, a 207-gene next generation sequencing (NGS) panel inclusive of copy number variation analysis was launched by a commercial laboratory to facilitate clinical diagnosis of primary immunodeficiency (PID), monogenic autoimmunity and autoinflammatory disorders. We assessed the outcomes of genetic testing utilizing this panel on a cohort of pediatric patients with immunohematologic phenotypes evaluated at our tertiary care center during an 18-month period (5/1/17-10/31/18). Eligible subjects were evaluated by at least two of three providers from a multidisciplinary pediatric hematology-immunology team, including a hematology physician, immunology physician and a geneticist or genetic counselor.
Twenty-three patients met inclusion criteria; 20 (87%) were Caucasian, 12 (52%) were male with an average age of 11.7 years. The two most common phenotypic diagnoses included cytopenias, single- or multi-lineage (leukopenia, neutropenia, anemia, thrombocytopenia) primarily attributed to autoimmune causes or hypogammaglobulinemia. Five (22%) were given a definitive genetic diagnosis as a result of panel testing, though in two of these cases, the causative mutations were listed as variants of uncertain significance (VUS). Diagnoses included common variable immunodeficiency due to a pathogenic variant in NFKB2, STAT3 multiorgan autoimmunity due to gain-of-function mutation, and familial cold autoinflammatory syndrome due to a pathogenic mutation in NLRP12. Biallelic DNMT3B VUS were found in a patient whose phenotype and further laboratory studies (including karyotype) were consistent with immunodeficiency-centromeric instability, facial anomalies syndrome. Further, a STAT3 VUS was identified in a patient with multiorgan autoimmunity and his father with hypothyroidism; studies from an outside research laboratory were consistent with gain-of-function with this variant (private communication). An additional three patients had VUS identified that were suspected to be related to their phenotype, prompting eligibility for research studies. Four (17%) patients had increased risk alleles in NOD2, conferring an increased risk of Crohns disease. Three (13%) patients had pathogenic or likely pathogenic carrier findings warranting genetic counseling. In addition, 47 VUS (an average of 2 per patient) thought to be unrelated to phenotype were identified, necessitating further investigation and counseling.
The use of an NGS panel in a cohort of pediatric patients with immunohematologic disorders led to a definitive diagnosis in 22% of previously undiagnosed patients and prompted further research investigation in several more. Genetic testing also led to the identification of clinically significant carrier findings, risk alleles and 47 VUS unrelated to phenotype, necessitating genetic counseling. Our experience illustrates the value of genetic testing for diagnosis of immunohematologic disorders, and the importance of multidisciplinary care, including genetic counseling, for the proper evaluation and management of these patients.
(146) Submission ID#601557
Reduced-intensity, T Cell-replete, Alternative Donor Allogeneic Hematopoietic Cell Transplantation with Post-transplantation Cyclophosphamide Is Safe and Effective for Primary Immune Deficiencies
Orly R. Klein, MD1, Dimana Dimitrova, MD2, Ellen Carroll, RN3, Kenneth R. Cooke, MD4, Stephanie N. Hicks, RN5, Christopher G. Kanakry, MD6, Howard M. Lederman, MD, PhD7, Jennifer Sadler, RN5, Elias T. Zambidis, MD, PhD8, Heather J. Symons, MD, MHS9, Jennifer A. Kanakry, MD10
1Instructor, Oncology and Pediatrics, Johns Hopkins University School of Medicine
2Medical Officer, National Institutes of Health, National Cancer Institute, Experimental Transplantation and Immunology Branch
3Transplant Coordinator, National Institutes of Health, National Cancer Institute, Experimental Transplantation and Immunology Branch
4Professor, Oncology and Pediatrics, Johns Hopkins University School of Medicine
5Research Nurse, National Institutes of Health, National Cancer Institute, Experimental Transplantation and Immunology Branch
6Tenure-track Investigator, Lasker Clinical Research Scholar, National Institutes of Health, National Cancer Institute, Experimental Transplantation and Immunology Branch
7Professor of Pediatrics, Medicine and Pathology, Division of Pediatric Allergy and Immunology at Johns Hopkins University School of Medicine
8Associate Professor, Oncology and Pediatrics, Johns Hopkins University School of Medicine
9Assistant Professor, Oncology and Pediatrics, Johns Hopkins University School of Medicine
10Clinical Head of Transplant, National Institutes of Health, National Cancer Institute, Experimental Transplantation and Immunology Branch
Background: Allogeneic hematopoietic cell transplantation (alloHCT) is curative for primary immune deficiencies (PID). However, many patients lack a fully-matched unaffected sibling, or may have an unknown underlying genetic defect, rendering it undesirable to use related donors. Many PID patients have significant comorbidities at the time they are referred to alloHCT, precluding the use of myeloablative conditioning. The use of alternative donors with reduced-intensity conditioning (RIC) has historically led to increased rates of graft failure, graft-versus-host disease (GVHD), and transplant-related mortality (TRM). Post-transplantation cyclophosphamide (PTCy) as GVHD prophylaxis immunomodulates the graft through the preferential sparing of regulatory T cells and hematopoietic stem cells from its cytotoxic effects, thus allowing for robust donor engraftment that overcomes the HLA barrier while effectively preventing severe acute and chronic GVHD. We report the outcomes of two institutions using a RIC alloHCT regimen with alternative donors and PTCy in patients with PID.
Design: We transplanted 35 PID patients (Table 1) using alternative donors and RIC, either serotherapy-free (n=21) or alemtuzumab-based (n=14). All patients received PTCy for GVHD prophylaxis on days +3 and +4, either alone (n=3), or combined with mycophenolate mofetil and either sirolimus (n=21) or tacrolimus (n=11). Donors included haploidentical family members (n=16), matched unrelated (n=15), and mismatched unrelated (n=4). Stem cell source was T cell-replete bone marrow (n=33) or peripheral blood stem cells (n=2).
Results: The median follow-up is 17 months (range 0.5-8 years). At 17 months, overall survival is 91%, and event-free survival (defined as alive without graft failure) is 83%. The median days of neutrophil and platelet engraftment are 17 (range 14-42) and 28 (range 15-110), respectively. There were 10 patients who developed acute GVHD, grade 1 (n=5) or grade 2 (n=5), and there were no cases of grade 3 or 4 aGVHD. Seven of eight patients treated with systemic corticosteroids responded, and one was corticosteroid-dependent, then responded to second-line therapy. One patient developed skin-only chronic GVHD, which responded to corticosteroids and PUVA light therapy. Five patients developed graft failure, either primary (n=1) or secondary (n=4), and four were successfully re-transplanted and remain engrafted. One patient with secondary graft failure had autologous recovery and has not required a second alloHCT given some durable infection control gained during initial engraftment. There were three deaths prior to day 180 due to infection, and one death at 1.5 years secondary to presumed overdose. In ongoing follow-up of engrafted survivors (n=30), evidence of phenotype reversal has been demonstrated in all patients, with complete or ongoing resolution of some or all of their underlying disease manifestations, including infection, transfusion-dependence, autoimmunity, malignancy, and/or immune dysregulation.
Discussion: We have observed high rates of engraftment, low rates and severity of acute and chronic GVHD, and low TRM in 35 patients with PID transplanted using alternative donors, RIC, and PTCy-based GVHD prophylaxis. RIC alloHCT with PTCy shows promise for curing PID, and its use minimizes toxicity and widely expands the donor pool, thus allowing us to offer this curative therapy to many more patients with PID.
Table 1. Patient and donor characteristics.
|
Patients (n=35) |
|
| Male, n (%) | 25 (71%) |
| Age at time of HCT in years, median (range) | 13 (0.5-54) |
| Diagnosis, n | |
| Unknown primary immunodeficiency | 8 |
| Chronic granulomatous disease | 8 |
| MAGT1 deficiency | 3 |
| PI3KCD gain of function | 3 |
| RAG1/2 deficiency, hypomorphic | 2 |
| XIAP deficiency | 2 |
| STAT3 deficiency | 1 |
| IFNGR1 deficiency | 1 |
| CTLA4 haploinsufficiency | 1 |
| IL10R1 deficiency | 1 |
| NFKB1 haploinsufficiency | 1 |
| NFKBIA gain of function | 1 |
| Wiskott-Aldrich syndrome | 1 |
| ADA2 deficiency | 1 |
| IPEX syndrome | 1 |
| Allograft type, n (%) | |
| T cell-replete bone marrow | 33 (94%) |
| T cell-replete peripheral blood stem cells | 2 (6%) |
| Donor source, n (%) | |
| HLA-haploidentical | 16 (46%) |
| HLA-matched unrelated | 15 (43%) |
| HLA-mismatched unrelated | 4 (11%) |
Abbreviations: HCT, hematopoietic cell transplantation; IPEX, immunodysregulation polyendocrinopathy enteropathy X-linked; HLA, human leukocyte antigen
(147) Submission ID#601564
Generating a CYBB-KO THP-1 Cell Line Model for Studying Auto-inflammation in Chronic Granulomatous Disease
Aissa Benyoucef1, Lorie Marchitto2, Fabien Touzot, MD, PhD3
1Research Assistant, CHU Ste-justine
2Master Student, CHU Ste-Justine
3Department of Pediatrics, CHU Ste-Justine, University of Montreal, Montreal, QC, Canada
Chronic granulomatous disease (CGD) is a primary immune disorder that involves mutations in the nicotinamide adenine dinucleotides (NADPH) oxidase complex (Deffert, Cachat, & Krause, 2014). Two-third of CGD cases are caused by loss-of-function mutations in the CYBB gene that encodes the gp91pox subunit of the NADPH. The increased in patients' life expectancy thanks to progress in diagnosis and management has underlined the burden of inflammatory manifestations occurring independently of infectious agents (Dunogue et al., 2017; Marciano et al., 2018). CGD patients develop inflammatory granulomatous disorders, notably colitis, as a consequence of a dysregulated inflammasome activation. The treatment of inflammatory manifestations remains challenging, as it can be associated with an increased risk of infections. Thus, understanding the pathophysiological mechanism of auto-inflammation in CGD could help improve the therapeutic arsenal for the management of these manifestations. To reveal the precise pathophysiological mechanism of auto-inflammation in CGD, we have developed a cellular model that reproduces the CGD phenotype in phagocytic cell. Through CRISPR-Cas9 gene-editing we generated a THP-1 cell line harboring the previously described mutation c.90_92delCCGinsGGT (p.Tyr30Ter) in the CYBB gene responsible for gp91phox knock-out by early termination of translation. This cell line recapitulates the phenotype of CGD phagocytes: (i) decreased H2O2 production (ii) and enhanced inflammatory responses after PMA stimulation as evidenced by increased IL-1, IL-6 and TNFa secretion levels (Kuijpers & Lutter, 2012). These features were rescued by complementation through lentiviral transduction of a wild type CYBB gene. This new model will help us to investigate the auto-inflammation reported in CGD patients and also to propose new therapeutic targets of inflammatory manifestations in this disorder.
Deffert, C., Cachat, J., & Krause, K. H. (2014). Phagocyte NADPH oxidase, chronic granulomatous disease and mycobacterial infections. Cell Microbiol, 16(8), 1168-1178. doi:10.1111/cmi.12322
Dunogue, B., Pilmis, B., Mahlaoui, N., Elie, C., Coignard-Biehler, H., Amazzough, K., . . . Lortholary, O. (2017). Chronic Granulomatous Disease in Patients Reaching Adulthood: A Nationwide Study in France. Clin Infect Dis, 64(6), 767-775. doi:10.1093/cid/ciw837
Kuijpers, T., & Lutter, R. (2012). Inflammation and repeated infections in CGD: two sides of a coin. Cell Mol Life Sci, 69(1), 7-15. doi:10.1007/s00018-011-0834-z
Marciano, B. E., Zerbe, C. S., Falcone, E. L., Ding, L., DeRavin, S. S., Daub, J., . . . Holland, S. M. (2018). X-linked carriers of chronic granulomatous disease: Illness, lyonization, and stability. J Allergy Clin Immunol, 141(1), 365-371. doi:10.1016/j.jaci.2017.04.035
(148) Submission ID#601588
Genetic Testing Reveals a Homozygous RTEL1 Mutation in a 12 Month Old Female with Pancytopenia, Failure to Thrive and Low Immunoglobulins
Miriam Samstein, MD, PhD1, Idil Ezhuthachan, MD1, Sherry Farzan, MD2, Artemio M. Jongco, III, MD, PhD, MPH3
1Fellow, Northwell Health
2Attending, Northwell Health
3Assistant Professor of Medicine and Pediatrics, Division of Allergy & Immunology, Donald and Barbara Zucker School of Medicine at Hofstra/Northwell; Center for Health Innovations and Outcomes Research, Feinstein Institute for Medical Research, Manhasset, NY
Introduction/background: Dyskeratosis congenita (DC) is a disease of short telomeres characterized by bone marrow failure, immune dysfunction and somatic abnormalities including abnormal skin pigmentation, nail dystrophy and oral leukoplakia. Several genes have been implicated in the pathogenesis of DC including RTEL1 and ACD. Inheritance patterns include autosomal dominant, autosomal recessive and X-linked. Successive generations of affected individuals can be born with progressively shorter telomeres. While classic presentations include skin findings, bone marrow failure can be the first presenting sign. Hoyeraal-Hreidarsson Syndrome is a severe form of DC with symptoms beginning in early childhood and including microcephaly, cerebellar hypoplasia, intrauterine growth retardation (IUGR), severe immunodeficiency and early-onset progressive bone marrow failure.
Objectives: Understand the clinical features of DC and Hoyeraal-Hreidarsson syndrome
Describe a novel mutation leading to a DC like phenotype
Highlight the utility of genetic testing for severely ill patients with immune disorders
Methods: This is a case presentation of a patient with immunodeficiency and bone marrow failure found to have a homozygous variant of uncertain significance (VUS) in RTEL1 on genetic testing with the Invitae 207 gene primary immunodeficiency panel.
Results: The patient is a female born at 38 weeks as the product of a pregnancy complicated by IUGR. Zika testing, head ultrasound and NY state newborn screen were normal. She began to fall off the growth curve and had watery diarrhea at age 4 months despite high calorie formula implementation. At age 9 months she began having low grade fevers and was found to have a hemoglobin of 4 prompting urgent hospital admission for transfusion. Hematology sent testing for bone marrow failure syndromes, with suspicion for Diamond Blackfan anemia. She required PRBC transfusion roughly once every 2-3 weeks. At 11 months of age she became pancytopenic and had an urgent hospital admission. Immunology and GI were first consulted at this time. Due to persistently poor weight gain GI performed a colonoscopy which revealed IBD. Initial immune evaluation revealed hypogammaglobulinemia (IgG = 82 mg/dL, IgA = 7 mg/dL, and IgM = 32 mg/dL), nonprotective titers to HiB, pneumococcus, and diphtheria despite vaccination, and the patient received IVIG. She was also panlymphopenic (CD3 = 1092, CD4 = 837, CD8=253, CD19 = 31, CD16/56=7). Invitae primary immune deficiency panel demonstrated a homozygous variant of uncertain significance in RTEL1 (c.1742T>C p.Leu581Pro) as well as a heterozygous mutation in ACD c.262G>C (p.Ala88Pro), CARD14 (c.1192G>C (p.GLu398Gln), EPG5 (c.4231C>A(p.Leu1411Ile). She was also heterozygous for a pathogenic variant of SLC37A4 (c.1043delCT(p.Leu348Valfs*53).
Conclusions: In appropriately selected patients genetic testing can shed light on previously unidentified immune deficiencies. Although the clinical features of DC were present in this patient, the rarity of Hoyeraal-Hreidarsson syndrome makes it a difficult to make diagnosis and these patients are typically diagnosed with other bone marrow failure syndromes or idiopathic anemia. Prompt evaluation by an immunologist for babies with failure to thrive and bone marrow failure is warranted.
(149) Submission ID#601601
Chronic Rhinosinusitis and Nasal Polyposis in Patients with IRAK-4 Deficiency
Sara Seghezzo, MD1, Lauren Sanchez, MD2, Dylan K. Chan, MD, PhD3, Kristina W. Rosbe, MD4, Morna J. Dorsey, MD, MMSc5
1Clinical Fellow, Department of Pediatrics, Division of Allergy, Immunology, and Bone Marrow Transplant, University of California, San Francisco
2Assistant Clinical Professor, Department of Pediatrics, Division of Allergy, Immunology, and Bone Marrow Transplant, University of California, San Francisco
3Assistant Professor, Clinical Professor, Department of Pediatric Otolaryngology and Pediatrics, University of California San Francisco, San Francisco, CA
4Clinical Professor, Department of Pediatric Otolaryngology and Pediatrics, University of California San Francisco, San Francisco, CA
5Pediatric Immunologist and Allergist, Department of Pediatrics, Division of Allergy, Immunology, and Bone Marrow Transplant, University of California San Francisco, San Francisco, CA
Introduction: Homozygous or compound heterozygous mutations in IRAK4 underlie IRAK-4 deficiency, a rare autosomal recessive immunodeficiency that results in impaired toll-like receptor (TLR) and Interleukin-1 (IL-1) driven responses. Children with IRAK-4 deficiency are predisposed to recurrent and invasive infections secondary to Streptococcus pneumoniae, Staphylococcus aureus and other pyogenic bacteria with high mortality rates in early childhood. The frequency and severity of infections is thought to decrease with age due to the acquisition of humoral immunity and immunologic memory, however due to the rarity of the disease, the natural history of this condition beyond early childhood is not well described.
Objectives: We present three unrelated IRAK-4 deficient patients with persistent chronic rhinosinusitis with nasal polyposis that developed in childhood.
Cases: Patient 1 is a 15 y/o male with compound heterozygous mutations in IRAK4 (p.G75Afs*14/c.717-1G>T) with a history of recurrent S. pneumoniae osteomyelitis (left hip at age 9 and left knee at age 10) and C. septicum sepsis at age 9 following acute bowel perforation. Additionally, he experienced recurrent AOM during infancy and recurrent UTI since age 9. Despite prophylactic antibiotics and IVIG, he has had recurrent polymicrobial (MRSA, S. pneumoniae, H. influenzae, P. aeruginosa, A. fumigatus) rhinosinusitis with nasal polyposis since age 4 refractory to medical management requiring surgical intervention and prolonged courses of IV antibiotics.
Patient 2 is an 11 y/o female with homozygous deletions (exons 10-12) in IRAK4 with a history of ruptured appendicitis complicated by Pseudomonas abscess and bacteremia at age 2, culture-negative sepsis with septic arthritis and osteomyelitis of the right leg at age 3, and septic shock secondary to MSSA bacteremia complicated by rhabdomyolysis and DIC at age 5. She has a history of chronic rhinosinusitis, and despite IVIG and prophylactic antibiotics, she developed polymicrobial (H. influenzae, B. fragilis) rhinosinusitis with associated nasal polyposis pending surgical management.
Patient 3 is a 10 y/o female with homozygous mutations in IRAK4 (Q293X/Q293X on exon 8) with a history of S. pneumoniae meningitis at 3 months, M. catarrhalis epiglottitis and neck cellulitis at 4 months, RSV bronchiolitis at 6 months, Enterococcus bacteremia at 8 months, S. pneumoniae sepsis at age 2 and Streptococcus lymphadenitis at age 9. Despite IVIG and prophylactic antibiotics, she developed recurrent polymicrobial (H. influenzae, B. fragilis, MSSA, V. cholera, P. aeruginosa, A. fumigatus) rhinosinusitis refractory to medical management requiring surgical intervention and IV antibiotics.
Conclusions: In our centers experience, IRAK-4 deficient patients continue to suffer from infectious complications, most prominently recurrent polymicrobial sinus infections beyond early childhood. The consistent presence of sinonasal polyps in these children is unusual, as it is not typically found in uncomplicated pediatric chronic rhinosinusitis. These infections have occurred despite antimicrobial prophylaxis and IVIG, highlighting the role of IRAK-4 in sinopulmonary epithelium. Additionally, the infectious organisms identified in our patient cohort are not commonly associated with IRAK-4 deficiency. Further study of chronic rhinosinusitis and nasal polyposis in a larger cohort of IRAK-4 deficient patients and other innate immunodeficiencies may help identify pathways for targeted treatment of these patients.
(150) Submission ID#601621
Osteomyelitis in Chronic Granulomatous Disease: Experience from a Tertiary Care Centre in North-West India
Johnson Nameirakpam1, Pandiarajan Vignesh, MD;DM (Pediatric Clinical Immunology and Rheumatology)2, Amit Rawat, MD (Pathology) PDCC (Laboratory Immunology) PDCC (Nephropathology) MAMS3, Deepti Suri, MD4, Anju Gupta5, Surjit Singh, MD; DCH (Lon.); FRCP (Lon.); FRCPCH (Lon.); FAMS6
1DM Fellow Pediatric Clinical Immunology and Rheumatology, Allergy and Immunology Unit, Advanced Pediatrics Centre, Postgraduate Institute of Medical Education and Research
2Assisstant professor, department of pediatrics, Postgraduate Institute of Medical Education & Research
3Professor of Pediatric Allergy and Immunology, Paediatric Allergy Immunology Unit, Department of Paediatrics, Advanced Paediatric Centre, Postgraduate Institute of Medical Education & Research
4Consultant, Allergy immunology unit, Department of Pediatrics, Advanced Pediatrics Centre, Postgraduate Institute of Medical Education and Research, Chandigarh, India, Dept. of Pediatrics, Allergy- Immunology Unit, Postgraduate Institute of Medical Education and Research
5Professor, Allergy and Immunology unit, Advanced pediatrics Centre, , Postgraduate Institute of Medical Education and Research, Chandigarh, India
6Head, Department of Pediatrics and Chief, Allergy Immunology Unit, Advanced Pediatrics Centre,Principal Investigator, Indian Council of Medical Research (ICMR) Centre for Advanced Research in Primary Immunodeficiency DiseasesVice-President, Indian Rh, Postgraduate Institute of Medical Education & Research
Introduction: Chronic granulomatous disease (CGD) is an inherited phagocytic defect associated with inability to clear catalase positive organisms. Infections in patients with CGD are severe and recalcitrant. Commonest infections are pulmonary followed by soft tissue infections and suppurative lymphadenitis. Osteomyelitis is an uncommon infection in patients with CGD. It poses several diagnostic and therapeutic challenge. We herein report our experience of osteomyelitis in CGD over the last 10 years.
Material and methods: Review of records was carried out to describe the profile of osteomyelitis in cohort of patients with CGD at Pediatric Immunodeficiency Clinic, Advanced Pediatrics Centre, Postgraduate Institute of Medical Education and Research, Chandigarh, India. The diagnosis of CGD was based on Nitroblue tetrazolium dye reduction test (NBT) and Dihydrorhodamine reduction (DHR) assay.
Results: Of the 63 patients with CGD, 8 (12.7%) had osteomyelitis (6 males and 2 females; age range 1- 10 years). Most patients had their first episode of serious infection in early childhood (mean age: 1.5 years). Stimulation index (SI) of DHR assay ranged from 1 to 4.58. Mutational analysis was done in 5/8 patients (3 X-linked; 2 autosomal recessive). Site of involvement was variable ribs- 4; vertebrae- 2; radius- 1; skull- 2; tibia- 1. Aspergillus fumigatus was the most common isolate (62%; 5/8); others had Aspergillus flavus, Aspergillus terreus and Serratia marcescens each. All 4 patients with rib osteomyelitis had concurrent pneumonia, and fungus was isolated in all of them (Aspergillus fumigatus- 2, Aspergillus flavus- 1, Zygomyces spp.- 1). Antifungals (intravenous amphotericin B) were given for a duration of 4-6 weeks and were followed by oral voriconazole in therapeutic doses for 3 to 6 months in majority of them. Debridement and resection of ribs was required in one patient, while other patients were managed conservatively. Out of 8 patients, 2 (25%) succumbed to pneumonia and respiratory failure.
Conclusion: Osteomyelitis in the context of CGD is usually caused by Aspergillus spp. Involvement of ribs and vertebra usually occurs with the contiguous spread of infection from the lungs. Therapy often requires prolonged duration of anti-microbials, and may require surgical debridement in addition to it.
(151) Submission ID#601626
A 29-year-old Woman with History of Hypogammaglobulinemia and Acute Liver Failure
Beth K. Thielen, MD, PhD1, Timothy Moss, MD, PhD2
1Fellow, University of Minnesota, Departments of Internal Medicine and Pediatrics, Divisions of Infectious Diseases and International Medicine and Pediatric Infectious Diseases and Immunology
2Assistant Professor, University of Minnesota, Department of Pediatrics, Division of Genetics and Metabolism
A 29-year-old woman with a 7-month history of nausea, vomiting, and abdominal pain was admitted to an outside hospital with new onset of jaundice and anasarca. Liver biopsy was thought most consistent with alcoholic steatohepatitis, and she was discharged with counseling on alcohol cessation and medical management of liver disease. She presented to our facility for a second opinion. Over the following days, she developed further rise in direct hyperbilirubinemia up to 19.2 mg/dL, new coagulopathy with an INR 2.06 and hypoalbuminemia to 1.7 mg/dL in the absence of ongoing alcohol consumption. Liver sonography revealed course echotexture and patent vessels. PCRs directed against multiple hepatotropic viruses were negative and copper studies were normal. Due to a history of moderate alcohol consumption, she was started on high-dose corticosteroids due to a presumptive diagnosis of alcoholic hepatitis.
Additional history raised concern for a possible primary immunodeficiency, including idiopathic thrombocytopenic purpura at 11 years of age, multiple episodes of sinusitis treated with antibiotics and sinus surgery, one episode of suspected bacterial pneumonia, and one hospitalization for influenza A during which she developed neutropenia. In her 20s, she developed refractory genital warts, prompting infectious diseases evaluation. Initial immune evaluation had revealed low immunoglobulins (IgA <7 mg/dL, IgG 198 mg/dL, IgM 13 mg/dL) with very low responses to tetanus and diphtheria, despite a recent booster dose, and B and T cell lymphopenia (CD19+ 89 cells/μL, CD3+ 567 cells/μL, CD4+ 345v, CD 8+ 244 cells/μL, CD16/56+ 236 cells/μL); antigen and mitogen proliferation were not assessed. Intravenous immunoglobulin replacement was initiated but discontinued by the patient due to infusion-related adverse effects, and she was lost to follow up until she presented with liver failure. Both parents were deceased from cardiovascular disease in their 40s and she had no siblings. She had limited knowledge of family history but no known immune diseases.
Due to suspicion for genetic etiology of immune disorder and liver disease, we performed next-generation sequencing of a panel of over 200 genes implicated in primary immune deficiencies. Patient was heterozygous for a nucleotide substation (c.1752+1G>A) within a splice site at the exon 16/intron 16 boundary of the NFKB1 gene. During the hospitalization, immunoglobulin replacement and trimethoprim-sulfamethoxazole prophylaxis were initiated. An attempt was made to refer the patient for additional immunological evaluation and transplantation evaluation but unfortunately, she developed worsening liver failure and multiple complications, including extended-spectrum beta-lactamase (ESBL)-producing E. coli bacteremia, hypotension requiring vasopressors and extensive bowel ischemia, and died in the hospital.
In summary, this case highlights both the risk of diagnostic delay in adult patients presenting with a primary immune deficiency and potential for genetic testing to clarify the diagnosis. While the particular genetic change has not been described, other splice site and predicted loss-of-function mutations have been reported as pathogenic in this gene, which have been implicated in autosomal dominant common variable immunodeficiency. This case further expands on the genetic causes and spectrum of disease associated with changes in the NFKB1 gene.
(152) Submission ID#601723
Acquired Immunodeficiency - More Than Meets the BMI (Body Mass Index)
Laura E. Maurer, MD, MPH1, Victor P. Bilan, MD2
1Resident Physician, Yale New Haven Hospital
2Chief Resident for Quality and Safety, West Haven Veterans Affairs Hospital
Introduction: Malnutrition and micronutrient deficiency are underrecognized causes of acquired immunodeficiency in adults, and may occur even in patients with high body mass index (BMI).
Methods: A 46-year-old woman with a medical history significant for one remote urinary tract infection presented to the emergency department after sudden onset of severe right flank pain. The pain was accompanied by urinary frequency and not relieved by ibuprofen; she denied fevers or chills. She was diagnosed with pyelonephritis and discharged on ciprofloxacin, which was later changed to trimethoprim-sulfamethoxazole after her culture grew resistant E. coli. Her pain continued despite treatment, prompting her to return to the hospital three days later.
Upon presentation, she was afebrile with blood pressure of 128/88 mmHg and heart rate of 86 bpm. Her body mass index was 32.4 kg/m^2. Her physical exam was otherwise notable for right costovertebral angle tenderness. Laboratory studies revealed a leukocyte count of 14,300/ul with 83% neutrophils; alkaline phosphatase of 146 units/L and albumin of 2.7 g/dL, but otherwise normal liver function tests; normal lactic acid; and urinalysis with 3,000 WBC/hpf, 40 RBC/hpf, moderate bacteria, and the presence of WBC clumps. CT scan of the abdomen and pelvis demonstrated an obstructing 13 mm right renal stone with hydronephrosis and a right renal abscess contiguous with a right-sided hepatic abscess measuring 7.8 x 6.0 x 7.5 cm. She was treated with ceftriaxone and metronidazole, and underwent imaging-guided drainage of the abscesses. Abscess cultures again grew resistant E. coli. She was discharged from the hospital with drains in place and a plan to continue trimethoprim-sulfamethoxazole until definitive management of her nephrolithiasis with ureteroscopy and lithotripsy.
Discussion: There remained the question of how an ostensibly immunocompetent patient had developed such severe intraabdominal infection with little systemic inflammatory response (e.g. no fever and only mild leukocytosis). A HIV antibody screen was negative. On further interview, she described a 200lb intentional weight loss over the preceding 2 years, accomplished by dietary restriction to less than 600 calories per day. Nutritional assays revealed prealbumin, vitamin C, and vitamin B6 levels below the threshold of detection. She had low-normal B12 and B1. Out of concern for an acquired immunodeficiency resulting from malnutrition with micronutrient deficiency, balanced nutrition was discussed with the patient who agreed to liberalize her diet.
Conclusion: This case illustrates that marked acquired immunosuppression can result from malnourished and undernourished states. This phenomenon is well recognized in young children and in resource-limited settings, but may occur even in the presence of normal or high body mass index. It further demonstrates the importance of routinely taking a thorough dietary history and encouraging healthy eating practices as part of usual clinical anticipatory guidance.
Uploaded File(s)
Uploads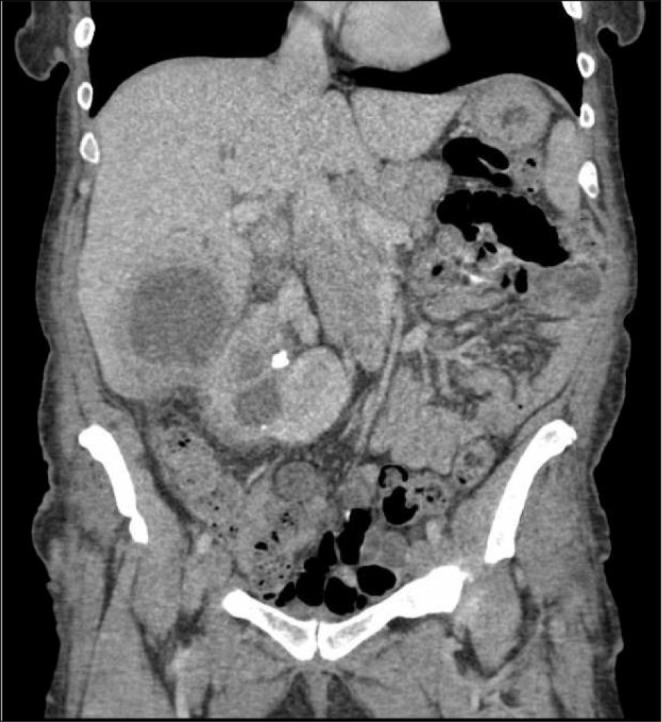

(153) Submission ID#601826
Centralized Sequencing Initiative at NIAID: Year 1
Magdalena A. Walkiewicz, Ph.D.1, Morgan Similuk, ScM2, Celine Hong, Ph.D.3, Leila Jamal, MSc., Ph.D.4, Haley Hullfish, B.S.5, Jia Yan, MSc., Ph.D.6, Patty Littel, B.S.7, Sandhya Xirasagar, Ph.D.8, Adriana Almeida de Jesus, MD9, Elise Ferre, PA-C, MPH10, Raphaela Goldbach-Mansky, MD, Ph.D.11, Michail Lionakis, MD, Sc.D12, Steven M. Holland, MD13
1ABMG certified Clinical Molecular Geneticist, National Institute of Allergy and Infectious Diseases (NIAID)
2Genetic Counselor, Molecular Development of the Immune System Section, Laboratory of Immune System Biology, NIAID, National Institutes of Health, Bethesda, MD, USA
3N/A, NHGRI
4Genetic Counselor, NIAID
5Research Fellow, National Institute of Allergy and Infectious Disease (NIAID)
6Clinical Protocol Coordinator and Genetic Counselor, NIAID
7RN, NIAID
8Health Scientist, NIAID
9Staff Clinician, NIAID
10Physician Assistant, Fungal Pathogenesis Section, Laboratory of Clinical Immunology & Microbiology (LCIM) , National Institute of Allergy & Infectious Diseases (NIAID), NIH
11Clinical Investigator, NIAID
12Chief, Fungal Pathogenesis Section, National Institute of Allergy and Infectious Diseases (NIAID) National Institutes of Health (NIH)
13Director, Division of Intramural Research, Laboratory of Clinical Immunology and Microbiology, National Institute of Allergy and Infectious Diseases, National Institutes of Health
Background: The past decade has brought dozens of new Mendelian disorders of immunity. Yet, the genetic contribution(s) to diverse disorders of the immune system remain largely unelucidated. The majority of research participants referred to the National Institute of Allergy and Infectious Diseases (NIAID) for what may be a Mendelian disorder evade molecular diagnosis. Making progress in this area requires a coordinated, systematic, and transparent approach to clinical genomics research which leverages the unique environment at the National Institutes of Health Clinical Center (NIH CC).
Methods/Design: This study is designed to systematically apply exome sequencing and related technologies with clinical grade interpretation and reporting to NIAID research participants at the NIH CC under a single protocol in order to facilitate research and clinical genetics care across NIAID. We are recruiting approximately 1000 participants per year from approximately 35 intramural clinical investigators. We generate genomic data, collect standardized phenotyping and report clinical interpretation in the medical record, all while providing linked genetic counseling.
Results: To date, we consented 1287 participants, we sent out 1058 samples for exome sequencing and 183 samples underwent copy number variant analysis. We have completed analysis for 359 families (502 individuals) and finalized and resulted 177 cases. Here we present a case series illustrating some of our findings. Case 1: A 10-year-old female was referred to NIAID for neonatal onset multisystem inflammatory disease (NOMID). Developmental delay and mild intellectual disability were appreciated on clinical evaluation. Exome sequencing detected a mosaic novel likely pathogenic variant in NLRP3. Chromosomal Microarray Analysis (CMA) showed a ~5 Mb interstitial deletion of chromosome 12 previously associated with developmental delay and intellectual disability. Case 2: A 10-year-old Ukrainian male was referred to NIAID for the clinical diagnosis of autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy (APECED). Exome sequencing and CMA did not detect pathogenic variants in AIRE, but did find a de novo variant in FAM111B. Defects in FAM111B are associated with poikiloderma with tendon contractures, myopathy, and pulmonary fibrosis (POIKTMP). The clinical features of the patient were consistent with POIKMP. Case 3: A 63-year-old man had a history of brain, liver and kidney nocardiosis, disseminated MAC infection, prostate cancer and lymphoma. Family history was significant for prostate cancer. Exome sequencing showed a heterozygous pathogenic variant in BRCA2, associated with susceptibility to breast-ovarian, male breast, pancreatic and prostate cancer.
Conclusion: This case series illustrates that multiple diagnoses, unexpected diagnoses, secondary genomic findings, and data sharing helped identify variants in candidate genes. Process standardization supports data integrity and efficiency while accommodating the need for investigator flexibility and providing tailored patient care.
(154) Submission ID#601828
T-cell Receptor Repertoire Clonality in Peripheral Blood and Affected Tissue in Activated PI3 Kinase Delta Syndrome (APDS)
Sara Barmettler, MD1, James M. Heather, PhD2, Jocelyn R. Farmer, MD/PhD3, Gabriel Wong, MD/PhD4, Mark Cobbold, MD/PhD5
1Attending Physician, Massachusetts General Hospital
2Research Fellow, Massachusetts General Hospital
3Instructor, Massachusetts General Hospital
4Physician, University of Birmingham
5Associate Professor, Massachusetts General Hospital
Rationale: Activated PI3 kinase delta syndrome (APDS) is a primary immunodeficiency caused by dominant mutations that increase activity of phosphoinositide-3-kinase (PI3K). The catalytic subunit p110 is mainly expressed in cells of the hematopoietic system, primarily lymphocytes and myeloid cells, and mutations affect both B- and T-cells. We sought to further evaluate the role of the T-cell receptor (TCR) repertoire in immune dysregulation and the pathogenesis of autoimmunity and lymphoproliferation in patients with APDS.
Methods: We evaluated the TCR repertoire in the peripheral blood in 3 patients with PIK3CD mutations and compared these to the peripheral TCR repertoire in 26 patients with common variable immunodeficiency (CVID) and 50 healthy controls to investigate the role of the TCR in disease. The TCR repertoire in affected tissue of 2 patients with PIK3CD mutations was also evaluated (tissue included lymph nodes for both patients, in addition to gastrointestinal tract and lung tissue in one patient). A fixed number of TCRs were subsampled (35,000 for blood and 5,000 for tissue) and diversity was calculated using the Gini and Shannon indexes.
Results: Using the Shannon and Gini diversity indexes, the TCR repertoire in patients with PIK3CD mutations had less diversity/increased clonality as compared to healthy controls and those with CVID (Figure 1). For the two APDS patients with biopsy tissue available for analysis, the diversity of the TCRs in tissue was increased as compared to the peripheral blood TCR repertoire (Figure 2).
Conclusions: PI3K plays an important role in the development and function of both B- and T-cells. Patients with APDS were found to have decreased TCR repertoire diversity in the circulating T-cell compartment compared to healthy controls and other CVID patients. The increased TCR diversity in the affected tissues compared to peripheral blood implicates the PI3K/AKT signaling pathway with T-cell trafficking and tissue immune homeostasis, and suggests this pathway may play a role in the development of inflammatory and lymphoproliferative complications in these patients.
Uploaded File(s)
Uploads
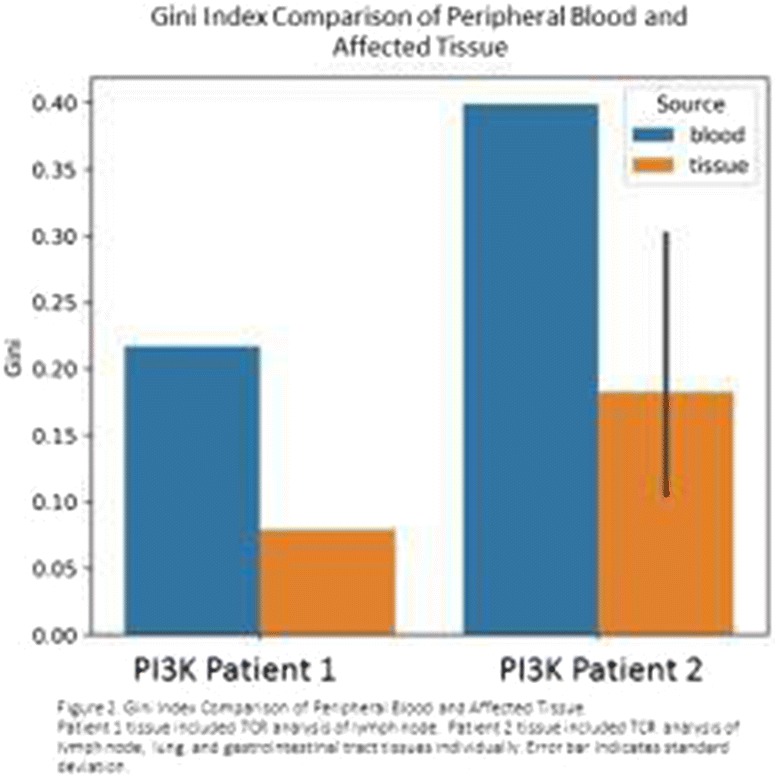
(155) Submission ID#601830
Hyperactivated PI3Kd Promotes Self and Commensal Reactivity at the Expense of Optimal Humoral Immunity
Silvia Preite, PhD1, Jennifer L. Cannons, PhD2, Andrea J. Radtke, PhD3, Ivan Vujkovic-Cvijin, PhD4, Julio Gomez-Rodriguez, PhD2, Stefano Volpi, MD5, Bonnie Huang, PhD6, Cheng Jun7, Nicholas Collins, PhD8, Kerry Dobbs, BSc9, Julie Reilley10, Quan-Zhen Li, MD, PhD11, Stefania Pittaluga, MD, PhD12, Gulbu Uzel, MD13, Luigi D. Notarangelo, MD, PhD14, Yasmine Belkaid, PhD15, Ronald N. Germain, MD, PhD16, Pamela L. Schwartzberg, MD, PhD17
1Postdoctoral fellow, National Human Genome Research Institute. Laboratory of Immune System Biology, NIAID, NIH, Bethesda, MD, USA
2Staff Scientist, National Human Genome Research Institute. Laboratory of Immune System Biology, NIAID, NIH, Bethesda, MD, USA
3Postdoc, Laboratory of Immune System Biology, NIAID, NIH, Bethesda, MD, USA
4Postdoc, Laboratory of Parasitic Diseases, NIAID, NIH, Bethesda, MD, USA
5Researcher, Clinica Pediatrica e Reumatologia, Centro per le Malattie Autoinfiammatorie e Immunodeficienze, Istituto Giannina Gaslini e Università degli Studi di Genova, Genoa, Italy
6Postdoc, National Human Genome Research Institute. Laboratory of Immune System Biology, NIAID, NIH, Bethesda, MD, USA
7Technician, National Human Genome Research Institute, NIH, Bethesda, MD, USA
8Postdoc, Laboratory of Parasitic Diseases, NIAID, NIH
9Biologist, Laboratory of Clinical Immunology and Microbiology, NIAID, NIH, Bethesda, MD, USA
10Biologist, National Human Genome Research Institute. Laboratory of Immune System Biology, NIAID, NIH, Bethesda, MD, USA
11Associate Professor, Microarray Core Facility and Department of Immunology, University of Texas Southwestern Medical Center, Dallas, TX, USA
12Senior Research Physician, Laboratory of Pathology, National Cancer Institute, NIH, Bethesda, MD, USA
13Staff Clinician, Laboratory of Clinical Immunology and Microbiology, National institute of Allergy and Infectious Diseases, NIH, Bethesda, MD, USA
14Chief, Laboratory of Clinical Immunology and Microbiology, IDGS, DIR, NIAID, NIH, Bethesda, MD, USA
15Senior Investigator, Laboratory of Parasitic Diseases. Microbiome Program, NIAID, NIH, Bethesda, MD, USA
16Chief, Laboratory of Immune System Biology. Chief, Lymphocyte Biology Section, Laboratory of Immune System Biology, NIAID, NIH, Bethesda, MD, USA.
17Senior Investigator, National Human Genome Research Institute. Laboratory of Immune System Biology, NIAID, NIH, MD, USA
Gain-of-function mutations in PI3Kd result in a human primary immunodeficiency, named APDS (Activated PI3K-delta syndrome), characterized by lymphopenia, lymphoproliferation, respiratory infections and inefficient responses to vaccination. However, what promotes these immune disturbances at the cellular and molecular level remains unknown. We have recently published a mouse model that recapitulates major features of this disease and used this model and patient samples to probe how hyperactive PI3Kd fosters aberrant humoral immunity. We found that mutant PI3Kd alters the intrinsic function of T and B cells, leading to ICOS-independent increases in T follicular helper (Tfh) and germinal center (GC) B cells, disorganized GCs, and poor class-switched antigen-specific responses to immunization. These phenotypes were associated with increased phosphorylation of AKT and S6 in T and B cells, and lower threshold of activation, with altered regulation of FOXO1 and BCL2 family members. Moreover, B cells showed enhanced responsiveness and proliferation to both antigens and innate stimuli, accompanied by reduced cell death. Strikingly, aberrant responses were accompanied by increased reactivity to gut bacteria, and a broad increase in autoantibodies that were dependent on commensal microbial stimulation, as demonstrated by striking reduction of self-reactivity upon antibiotic treatment in mutant mice. We now have further examined B cell function in these mice and demonstrate that altered FOXO1 plays a major role in disruption of both B and T cell function. We further provide evidence for altered activation of metabolic pathways in B cells, compared to WT cells, that may contribute to the dysregulated B cell reactivity. Our findings suggest that proper PI3Kd regulation is critical for ensuring optimal host-protective humoral immunity despite tonic stimulation from the commensal microbiome.
This research was supported in part by the Intramural Research Program of the NIH, NHGRI and NIAID.
(156) Submission ID#601877
The B-cell Subset Mileu of Autoimmune Cytopenias in Primary Immunodeficiency
Travis Sifers1, Charlotte Cunningham-Rundles, MD, PhD2
1Fellow-in-Training, Mount Sinai School of Medicine, Ichan School of Medicine at Mount Sinai
2Professor in Medicine, Division of Clinical Immunology, Icahn School of Medicine, Mount Sinai, NY, NY, USA
Autoimmune cytopenias are seen in a significant proportion of patients with immunodeficiencies affecting antibody production. Previous B-cell maturation studies using fluorescence-activated cell sorting (FACS) have associated various phenotypes of primary immunodeficiency diseases affecting antibody production with differing levels of B-cell differentiation. In this study we analyzed the peripheral B-cell compartment of 84 patients with a hypogammaglobulinemia and >1% B-cells with and without a history of autoimmune cytopenias. B-cells were isolated from peripheral blood using monoclonal anti-CD19 and these cells were gated to identify the proportion of memory B cell (CD19+CD27+), IgM+ memory B (CD27+IgM+), marginal zone B-cells (IgM+IgD+), isotype-switched memory B-cells (CD27+IgM-IgD-) and transitional cells (IgMhiCD38hi).
PID patients with a history of AIC had decreased proportions of total CD27+ B-cell (11.6% vs 25.6%; p=0.0003) and IgM memory B cells (8.3% vs 18.4%; p = 0.0018). Conversely, the proportion of marginal zone B-cells was increased in this group (82.0% vs 66.5%; p = 0.0043).
Consistent with previous reporting, the proportion of isotype-switched memory B-cells was significantly lower in the AIC group (0.75% vs 2.8%; p = 0.0003). Statistically significant inter-group difference was not seen within the transitional B-cell subset.
Our data suggest that maturation arrest of marginal zone (CD27+IgM+IgD+) B-cells may be implicated in the development of autoimmune cytopenias in humoral immunodeficiency.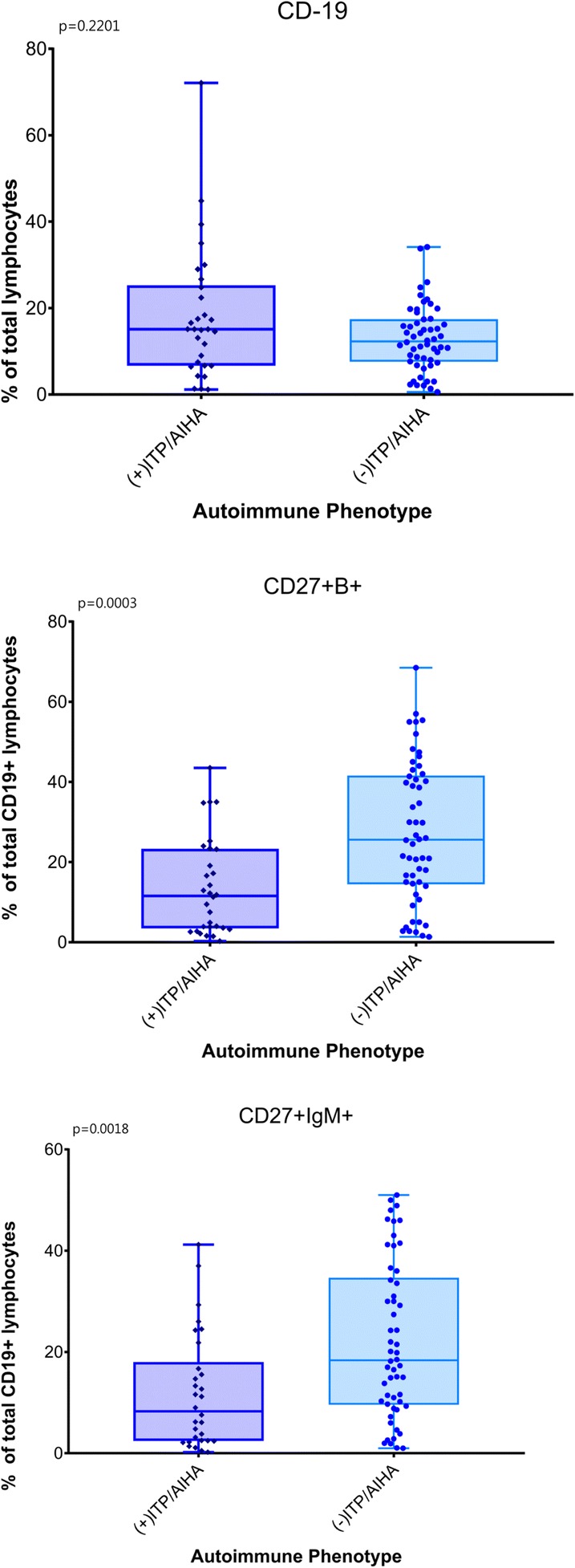
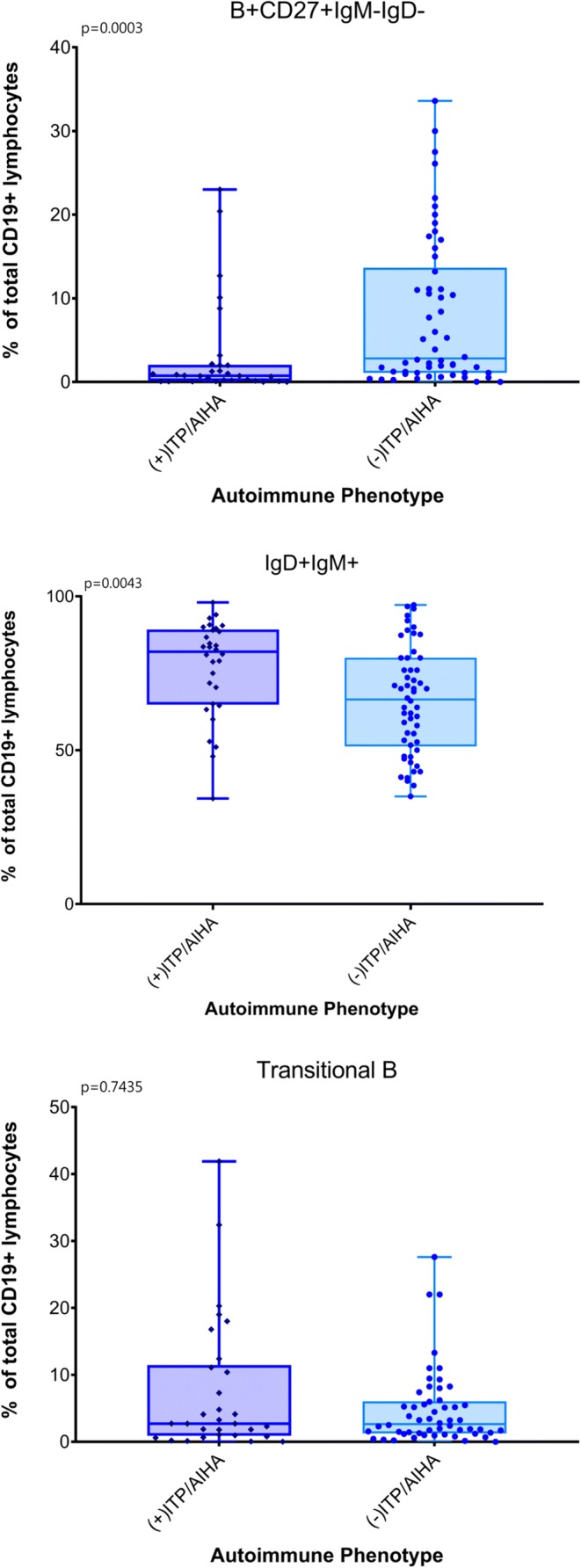
(157) Submission ID#601907
XLA Presenting as HHV-6 Meningitis and Pseudomonas Meningitis/Bacteremia in a Patient Without History of Recurrent Infections
Melissa A. Cardenas-Morales, MD1, Camile Ortega, D.O.2, Jose Calderon, MD3, Paul Martinez, MD4, Vivian Hernandez-Trujillo, MD3
1Allergy and Immunology Fellow, Nicklaus Childrens Hospital
2Allergy and Immunology Fellow, Nicklaus Children's Hospital
3Allergy Immunology Attending, Nicklaus Children's Hospital
4Critical Care Physician, Nicklaus Childrens Hospital
Introduction: X-linked agammaglobinemia (XLA) is a primary immunodeficiency due to the inheritance of a pathologic variant of the Bruton Tyrosine Kinase (BTK) protein. The condition usually manifests within the first 2 years of life. An estimated 60% of patients present with a severe, life-threatening infection. Invasive Pseudomonas aeruginosa infection is uncommon in immunocompetent hosts. Several cases of XLA patients presenting as pseudomonas sepsis, meningitis, and bacteremia have been reported.
While severe viral infections may also be an initial presentation of primary immunodeficiency, an immune evaluation is not always obtained in this scenario. Patients with XLA have an increased susceptibility to severe enterovirus infections, manifesting as chronic meningoencephalitis, which can be fatal.
The following case describes a patient with newly diagnosed XLA presenting as suspected coxsackievirus and confirmed HHV-6 meningitis, Pseudomonas meningitis and bacteremia. This may be the first reported new diagnosis of XLA presenting with both severe bacterial and viral co-infection.
Case Description: A 2 year old, partially vaccinated, Hispanic male with a history of febrile seizures presented to the emergency room with fever, oliguria, watery diarrhea, lethargy, meningismus, ecthyma gangrenosum and lower abdominal pain. Eight days prior to presentation, he was seen by his pediatrician for facial rash and low grade temperature, and was diagnosed with hand-foot-and mouth disease. He worsened on empiric antibiotics. He had no history of sinopulmonary infections. He did not attend daycare. His vaccines were delayed due to parental choice, and he had not received live vaccines (Rotavirus, MMR or VZV). Full sepsis evaluation was performed. CSF demonstrated pleocytosis, and he was started on empiric antibiotics and transferred to PICU. Due to worsening abdominal pain, CT of the abdomen was performed, which was consistent with ruptured appendicitis and septic emboli at the lung bases. CSF PCR panel was positive for HHV-6 and he was started on Gancyclovir. CSF and blood cultures subsequently grew Pseudomonas aeruginosa. Immune evaluation was performed. Serum immunoglobulins were undetectable. In addition to IV antibiotics, he received 500 mg/kg IVIG and lymphocyte subsets revealed profound B cell lymphopenia (0.23 %, 5 cells/uL). BTK protein analysis revealed hemizygous BTK pathogenic variant confirming the diagnosis of X-Linked agammaglobulinemia. The hospital course was further complicated by brain abscesses and pyoventriculitis. He was treated with 3 additional doses of 500 mg/kg IVIG and IV antibiotics. Repeat MRI of the brain nearly 4 weeks after admission demonstrated significant improvement. There was significant clinical recovery. He was discharged home at baseline neurological status. His IgG level upon discharge home was 605 mg/dL with the plan to increase dose to 600 mg/kg per month with close monitoring.
Conclusion: Both severe opportunistic bacterial infections and severe viral infections as the initial presentation of XLA have been well reported in the literature. This case describes the first reported severe Pseudomonas aeruginosa and HHV-6 co-infection in a newly diagnosed XLA patient. This case further highlights the necessity for an increased index of suspicion of primary immunodeficiency in a patient who presents with a severe first infection, despite lack of recurrent infections.
(158) Submission ID#601911
Missense Variants as a Contributing Cause to DOCK8 Immune Deficiency
Haley Hullfish, B.S.1, Morgan Similuk, ScM2, Huie Jing, PhD3, Jeffrey Danielson, MS4, Leila Jamal, MSc., Ph.D.5, Celine Hong, Ph.D.6, Jia Yan, MSc., Ph.D.7, Steven M. Holland, MD8, Alexandra F. Freeman, MD9, Magdalena A. Walkiewicz, Ph.D.10, Helen C. Su, MD, PhD11
1Research Fellow, National Institute of Allergy and Infectious Disease (NIAID)
2Genetic Counselor, Molecular Development of the Immune System Section, Laboratory of Immune System Biology, NIAID, National Institutes of Health, Bethesda, MD, USA
3Research Staff Member, NIAID
4Research Staff Member, Laboratory of Clinical Immunology and Microbiology, NIAID, NIH, Bethesda, MD
5Genetic Counselor, NIAID
6N/A, NHGRI
7Clinical Protocol Coordinator and Genetic Counselor, NIAID
8Director, Division of Intramural Research, Laboratory of Clinical Immunology and Microbiology, National Institute of Allergy and Infectious Diseases, National Institutes of Health
9Director, Primary Immune Deficiency Clinic, Laboratory of Clinical Immunology and Microbiology, NIAID, NIH, Bethesda, MD, USA
10ABMG certified Clinical Molecular Geneticist, National Institute of Allergy and Infectious Diseases (NIAID)
11Chief, Human Immunological Diseases Section, Laboratory of Clinical Immunology and Microbiology, NIAID, NIH, Bethesda, MD
We present two patients with DOCK8 deficiency due to compound heterozygous variants including a copy number loss at chromosome band 9p24.3 spanning approximately .107 Mb with partial deletion of the DOCK8 gene and a novel c.2603C>T (p.Ser868Leu) missense variant [chr9:379933 (GRCh37) NM_203447] in DOCK8. Functional data is presented to support the pathogenicity of the missense change, along with a review of the literature on DOCK8 variants.
The proband is a 14-year-old female with elevated serum IgE, severe atopic dermatitis, mild persistent asthma, food allergies, and seasonal allergic rhinitis. She is currently healthy following haploidentical bone marrow transplant in June 2018. She has a 17-year-old brother with DOCK8 deficiency with the same compound heterozygous variants. The brother had later onset of symptoms and a milder presentation of intermittent asthma and seasonal allergic rhinitis. Each of the parents is heterozygous for one of the two variants.
We evaluated the pathogenicity of the c.2603C>T missense variant with western blots of DOCK8 protein expression, intracellular flow cytometry, and DOCK8 stretch assays. Flow cytometry showed decreased DOCK8 protein expression and stretch assays revealed T cells that were stretched in collagen gels.
Notably, DOCK8 is a large gene containing 47 exons spanning 190 kb and it is relatively common to be a carrier of a rare missense change. In fact, gnomAD has approximately 1500 individuals with rare (<0.002 frequency) missense alleles in DOCK8. Therefore, it is important to demonstrate the potential pathogenicity of any given rare missense change, since few pathogenic missense variants in DOCK8 have been reported. Of the 168 published DOCK8 variants listed in the Human Gene Mutation Database (HGMD) only 13 are missense. The majority are gross deletions, 97 of which were reported in HGMD. The remaining reported DOCK8 variants include 19 nonsense, 15 splicing, 13 small deletions (all frameshifting), 3 small insertions (all frameshifting), 2 small indels, and 5 gross insertions/duplications.
This case demonstrates the relatively infrequent but important contribution of missense changes to pathogenic DOCK8 alleles. Functional validation of missense alleles is critical in the complex evaluation of DOCK8 deficiency.
(159) Submission ID#601984
IgD Class Switched B Cells in Patients with Common Variable Immunodeficiency
Taissa de Matos. Kasahara1, Sudhir Gupta, MD2
1PhD student, State University of Rio de Janeiro and University of Californis Irvine
2Professor, University of California at Irvine, Irvine, CA, USA
Introduction/Background: Common variable immunodeficiency (CVID) is the most frequent form of primary hypogammaglobulinemia with decreased serum IgG and IgA levels and variable levels of IgM in adults. In addition to decreased serum immunoglobulins, 25-30% of CVID patients present autoimmune manifestations. The mechanisms that lead to a breakdown of self-tolerance in CVID are not completely understood. However some differences in B and T cells subsets and autoreactive B and T cells can be detected. Elevated expression of surface IgD and downregulation of IgM receptor are hallmarks of anergic naïve B cells that contain autoreactive receptors in human peripheral blood. Moreover, memory B cells that have class switched to IgD and present an IgD+IgM- phenotype are also highly reactive to self-antigens in healthy individuals. The role of these autoreactive naïve and memory B cells in the immunopathogenesis of CVID has not been evaluated. Here we investigated the frequency of CD27- and CD27+ B cells expressing IgD and IgM in peripheral blood of CVID patients.
Methods: Peripheral blood mononuclear cells (PBMC) from CVID patients (n=29) and health subjects (n=32) were separated by FicollHypaque and incubated with anti-human CD19-PerCP, CD27-FITC, IgD-BV510 and IgM-APC to identify different subsets of B cells by flow cytometry. CD19+CD27-IgD+IgM- and CD19+CD27-IgD+IgM+ B cells were sorted, loaded with CFSE and cultured with CpG and ant-CD40 for 5 days to evaluate the proliferation.
Results: Among the compartment of CD27- B cells, CVID patients showed an increased frequency of IgD+IgM+ cells and a lower frequency of IgD-IgM- cells as compared to control group. No differences were observed in the frequency of IgD+IgM- cells in CD27- B cells between CVID patients and controls. In contrast, in the compartment of CD27+ B cells, CVID patients showed an increased frequency of IgD+IgM-, IgD+IgM+ and IgD-IgM+ cells and a lower frequency of IgD-IgM- cells when compared to health subjects. When the patients were divided in two groups based on autoimmune manifestations, the group with autoimmune disease showed an increased frequency of IgD+IgM+ and IgD-IgM+ cells in CD27- B cells when compared to the control groups. Both patient groups showed an increased frequency of IgD+IgM-, IgD+IgM+ and IgD-IgM+ cells and a lower frequency of IgD-IgM- cells when compared to health subjects. Regarding the proliferation, naïve B cells from CVID patients showed a reduced proliferative capacity in response to in vitro stimulation as compared with naïve B cells from health subjects.
Conclusion: Our results suggest that the increase of CD27+IgD+IgM- B cells can be related to the susceptibility of autoimmunity in CVID patients.
(160) Submission ID#601996
A Case Review of IgG4 Related Disease
Blake A. Thompson, MD1, Lyda Cuervo-Pardo, MD2, Mario Rodenas, MD, FAAAAI2
1Internal Medicine Resident, University of Florida
2Assistant Professor, University of Florida, Division of Rheumatology & Clinical Immunology, Department of Medicine
Introduction: Immunoglobulin G4-related disease (IgG4-RD) is a group of immune-mediated conditions where tissues are affected with dense lymphoplasmacytic infiltrations with a predominance of IgG4-positive plasma cells and storiform fibrosis, usually in the setting of elevated serum concentrations of IgG4. Common presentations include autoimmune pancreatitis, sclerosing cholangitis, retroperitoneal fibrosis, salivary gland disease, and orbital disease, among others. Symptoms of asthma or allergy are present in approximately 40 percent of patients and they typically exhibit a good initial therapeutic response to glucocorticoids.
Case presentation: A 61-year-old female with a history of gastroparesis, cutaneous lupus erythematosus and suspected autoimmune pancreatitis was referred to Allergy/Immunology clinic for evaluation of elevated IgG4.
She reported a 15-year history of recurrent abdominal pain attributed to recurrent pancreatitis based on previous mild lipase elevations. Prior endoscopic ultrasound (EUS) of the pancreas revealed edema. There was concern for gallstone pancreatitis but ERCP followed by cholecystectomy, biliary and pancreatic sphincterotomy had no change in her symptoms. In 2016, she was noted to have a positive ANA and high serum IgG4, per patient (values from OSH records could not be obtained). Symptoms improved with a course of steroids, hence suspicion for autoimmune pancreatitis.
In 2018 she developed a rash on her arms and face. Biopsies of the affected areas revealed cutaneous lupus erythematosus on the arms and a basal cell carcinoma on the face, which was excised. ANA was only 1:80 at that time.
At the visit, she complained of severe allergic rhinitis, joint pains, as well as a malar rash, which responded to intermittent courses of prednisone by prior providers.
Laboratories obtained at initial visit were significant for thrombocytopenia (135 thou/cu mm), positive Lupus anticoagulant (56 sec) and elevated IgG4 (95 mg/dL; normal range 4-86 mg/dL). C3, C4, C1q, ANA, anti-double stranded DNA, anti-Smith antibodies, antiphospholipid panel, UPEP and SPEP were all unremarkable. CT chest and abdomen were also normal.
Given the patient's history of cutaneous lupus erythematosus, Plaquenil was started as a steroid sparing agent. EUS of the pancreas with possible biopsy was ordered in an attempt to obtain a histopathologic diagnosis of IgG4-RD.
Conclusion: This case exhibits the association between elevated IgG4, pancreatitis of unknown origin, allergic rhinitis, and cutaneous lupus erythematosus, highlighting the value of identifying a pathologic connection between seemingly unrelated disorders in patients with elevated IgG4, as they may be manifestations of IgG4-RD. In order to make the diagnosis, histopathologic findings showcasing lymphoplasmacytic tissue infiltration consisting mainly of IgG4-positive plasma cells and small lymphocytes is essential.
The majority of patients respond to glucocorticoids, and while the duration of response is variable, most patients flare during or after glucocorticoids are tapered, as noted in this patient. Rituximab has been shown to be effective in some patients and will be considered in this patient if symptoms persist.
(161) Submission ID#602042
Newborn Infant with Purine Nucleoside Phosphorylase (PNP) Deficiency and Congenital Cytomegalovirus (CMV) Infection
Benjamin Rahoy, DO1, Sachit Patel, MD2, Shirley Delair, MD, MPH3, Michael Hershfield, MD4, Hana Niebur, MD5
1Pediatric Resident, University of Nebraska Medical Center
2Clinical Director, Pediatric Blood and Marrow Transplantation; Assistant Professor, Division of, Hematology/Oncology, University of Nebraska Medical Center
3Associate Professor, Division of Pediatric Infectious Diseases, University of Nebraska Medical Center
4Professor of Medicine, Professor of Biochemistry, Duke University Medical Center
5Assistant Professor, Division of Pediatric Allergy/Immunology, University of Nebraska Medical Center
Rationale: PNP deficiency is an autosomal recessive disorder due to defective purine metabolism leading to Severe Combined Immunodeficiency (SCID) and neurological deterioration. Newborn screening utilizing T-Cell Receptor Excision Circle (TREC) assay can detect affected patients before complications arise. Herein, we describe an infant initially identified by newborn screening with PNP deficiency and congenital CMV, a previously unreported presentation.
Methods: CMV quantitative PCR (qPCR) was performed by Nebraska Medicine, PNP enzyme activity by Duke and genetic sequencing by Invitae.
Results: A small for gestational age (SGA) male infant was reported to have an abnormal TREC assay on day of life (DOL) 7. He was hospitalized for further evaluation. Initial studies revealed profound lymphopenia, normal lymphocyte proliferation to mitogens and no evidence of maternal engraftment. Additionally on DOL 10, he had CMV viremia and viruria; thus with SGA, failed unilateral hearing screen and head ultrasound with bilateral parenchymal calcifications, congenital CMV was suspected. PNP enzyme activity was abnormal. CMV treatment was initiated with ganciclovir on DOL 10. Foscarnet was added on DOL 13. CMV qPCR levels decreased below the limit of detection by DOL 30. Genetic testing found a pathogenic homozygous mutation in PNP (c.286-18G>A). The infant has a 10/10 HLA-matched, unaffected, CMV positive sibling and will proceed to hematopoietic stem cell transplantation.
Conclusions: To our knowledge, this is the first reported case of PNP deficiency identified through newborn screening. This novel case of congenital CMV and PNP deficiency highlights the importance of CMV screening and need for treatment strategies for congenital CMV in SCID.
(162) Submission ID#602066
A Clinical Genomic Research Ecosystem Maximizes Data and Improves Patient Care
Morgan Similuk, ScM1, Leila Jamal, MSc., Ph.D.2, Haley Hullfish, B.S.3, Sandhya Xirasagar, PhD4, Magdalena A. Walkiewicz, Ph.D.5, Steven Holland, MD6
1Genetic Counselor, Molecular Development of the Immune System Section, Laboratory of Immune System Biology, NIAID, National Institutes of Health, Bethesda, MD, USA
2Genetic Counselor, NIAID
3Research fellow, National Institute of Allergy and Infectious Disease (NIAID)
4GRIS program lead, NIAID
5ABMG certified Clinical Molecular Geneticist, National Institute of Allergy and Infectious Diseases (NIAID)
6Scientific Director, NIAID, NIH
Despite a dramatic increase in the use of next generation sequencing over the last decade, the majority of the more than 50 million identified human genomic variants do not have well-established clinical implications. Progress is being made on this complex challenge through multiple approaches, including data sharing. To maximize our understanding of genomic data, platforms that enable effective and responsible data-sharing are essential. This means that genotypic and phenotypic data must be findable, accessible, interoperable, and reusable under conditions that are ethical and transparent.
To highlight innovations in data-sharing and their potential to advance discovery, we present three data-sharing mechanisms. For each platform, we will present a case highlighting its key functionality and discuss opportunities and challenges that may arise as each platform is scaled up.
(1.) Genomic Research Integration System (GRIS) is a collaboration-engendering web application that facilitates the identification of genetic variants associated with rare immunological disorders. Users can access integrated and standardized phenotypic and genomic data that is analyzable within the platform. GRIS enables systematic and automated capturing, and links patient data from disconnected systems and paper-based records. Standardized annotations allow for the comparison of data from different clinical studies. The main goal of this tool is discoverability of other affected individuals enrolled in separate protocols within the NIAID intramural research program. This internal database was used to find a second family with a rare variant in a candidate gene.
(2.) The Genomic Ascertainment Cohort (TGAC) is a resource that aims to improve our understanding of the phenotypic consequence of genetic variation by providing access to aggregate, de-identified genomic data from large NIH intramural and related cohorts. Participants have provided informed consent to be re-contacted for additional phenotyping in the future. The main goal of this tool is to enable further study of the clinical consequence of variants in a large, unbiased cohort of patients ascertained for many indications. This database was used to investigate findings in participants with previously published pathogenic variants in genes associated with primary immune deficiency based on medical record review.
(3.) ClinGen is dedicated to building an authoritative central resource that defines the clinical relevance of genes and variants for precision medicine and research. Through the sharing of genetic and health data, ClinGen seeks to answer whether a given gene is associated with a disease (clinical validity)?; whether a given variant is causative (pathogenicity)?; and whether the information is actionable (clinical utility)? This resource is meant to convene disease- and gene-specific expert groups to curate the medical literature on Mendelian disease to better define gene-disease and variant-disease relationships using many lines of evidence. This resource was used to clarify clinical validity of disease-gene assertions.
Together these efforts help create a clinical research ecosystem that maximizes the value of clinical research data and ultimately improves patient care.
This research was supported by the Intramural Research Program of the NIH, NIAID.
(163) Submission ID#602340
Immunodeficiency in Elderly: Data from the USIDNET Registry
Charmi Patel, MD1, Hannah Wright2, Ramsay Fuleihan, MD3, Charlotte Cunningham-Rundles, MD, PhD4, Daniel Suez, MD5, Artemio M. Jongco, III, MD, PhD, MPH6
1Attending physician, Donald Barbara Zucker School of Medicine at Hofstra/Northwell
2Research Data Analyst, USIDNET Consortium, National Institute of Allergy and Infectious Diseases (NIAID), Towson, MD
3Professor of Pediatrics, Division of Allergy and Immunology, Northwestern University Feinberg School of Medicine, Chicago, NY
4Professor in Medicine, Division of Clinical Immunology, Icahn School of Medicine, Mount Sinai, NY, NY, USA
5President, Allergy, Asthma & Immunology Clinic, PA
6Assistant Professor of Medicine and Pediatrics, Division of Allergy & Immunology, Donald and Barbara Zucker School of Medicine at Hofstra/Northwell; Center for Health Innovations and Outcomes Research, Feinstein Institute for Medical Research, Manhasset, NY
Introduction: According to the Population Reference Bureau, the number of elderly Americans, defined as age 65 and older, is projected to more than double from 46 million to 98 million by 2060, rising from 15% to 24% of the total population. The impact of immunodeficiency in this important segment of the population remains understudied.
Methods: The USIDNET Registry was queried to obtain demographic, clinical data of elderly patients defined as age 65 and older. Descriptive analyses were performed on the data.
Results: 373 participants (7.2%) were eligible out of 5176 total registry participants. The median age of the cohort was 70 years and predominantly female (74.7%) and White (78.0%) with a median BMI of 26.6 ± 6.6.The majority (81.8%) of subjects were living. Humoral deficiencies comprised the majority of diagnoses (94.6%), with Common Variable Immune Deficiency being the most frequent (76.9%). Of the remaining non-humoral diagnoses, immune dysregulation (1.3%) and immunodeficiency with myelodysplasia (1.1%) were the most frequent. The majority (79.1%) of subjects reported having received immunoglobulin replacement therapy (IGRT) at some point, with 51.7% reporting via IV route. Of the 1275 infections that occurred in this cohort, sinopulmonary infections were the most commonly reported, specifically sinusitis (18.5%), pneumonia (13.8%), upper respiratory infection (6.7%), and otitis media (5.5%). In this cohort, 107 autoimmune, 49 cardiovascular, and 11 granulomatous complications were reported . The number of patients with malignancy was 89, with some patients diagnosed with multiple malignant disorders. Of the reported malignancies, the majority (69.9%) were solid tumors.
Conclusions: Compared to the age-matched non-immunodeficiency United States population, this cohort had more females 74.7% (USIDNET) versus 56.0% (US population) and fewer whites 78.0% (USIDNET) vs 86.0% (US population. Humoral immunodeficiencies, specifically CVID, were most common diagnoses, similar to other age groups of immunodeficiency patients. Majority of these patients have received IGRT, with approximately half via IV route. This cohort reported living with a variety of non-infectious complications, including autoimmunity and malignancies. More research which specifically focuses on elderly patients with immunodeficiency is needed.
(164) Submission ID#602465
A Rare Case of Helicobacter Bilis Chronic Complicated Osteomyelitis with Pyomyositis and Cellulitis in a Patient with XLA Agammaglobulinemia : Discussion of Challenges in Diagnosis and Management
Candace Rypien, MD1, Nicola A. Wright, MD2, Luis Murguia-Favela, MD3, Andrea Fong, MD4, Dan Gregson, MD5
1Pediatric Infectious Disease, Alberta Children's Hospital
2Associate Professor, Department of Pediatrics, Alberta Children's Hospital, Calgary, Alberta, Canada
3Clinical Assistant Professor, University of Calgary
4Physician, University of Saskatchewan
5Clinical Microbiologist and Infectious Disease Physician, University of Calgary
X-linked agammaglobulinemia (XLA) is a primary immunodeficiency caused by mutations in the Bruton tyrosine kinase gene which leads to B cell maturation failure and defective antibody production. This puts patients at risk of recurrent sinopulmonary infections, gastrointestinal infections, and recurrent skin infections including infections caused by Helicobacter sp.
Helicobacter sp are gram negative bacilli commonly found in the gastrointestinal tract of various animals. Helicobacter sp. have been linked with gastritis most notably Helicobacter pylori causing gastric ulcers in humans. Helicobacter sp. has been found in rare cases to cause disseminated infections including pyodermic gangrenosum and cellulitis notably in patients with agammaglobulinemia.
Infections caused by Helicobacter bilis are challenging to diagnosis due to difficulties with culturing the pathogen as well as poor guidelines for antimicrobial management.
Case Report:
The patient was diagnosed with X-linked agammaglobulinemia at the age of 16 months with a history of recurrent sinusitis and was started on IVIG q3weeks. Despite regular IVIG, he developed bronchiectasis. At 11 years of age in 2013, he developed a chronic rash around his left knee resembling erythema nodosum. By 2014, he had developed a left knee effusion associated with left sided calf pain.
His knee pain was found to improve during courses of ciprofloxacin to treat recurrent lung infections. Given case report data of H. pylori causing erythema nodosum in patients with agammaglobulinemia, he was treated empirically for an H. pylori infections with no improvement. In 2015 he was found to have progressive cellulitis with pyomyositis of the left leg. A skin biopsy of a calf nodule was found to be culture negative but 16S PCR was positive for H. bilis. He was started on treatment with ertapenem and levofloxacin with subsequent resolution of his rash.
His left ankle pain progressed and by late 2015 and was found to have possible osteomyelitis of the left ankle on MRI. In 2016 he was found to be bacteremic with H bilis. Due to progressive symptoms with significant impact on function and rising inflammatory markers despite 12 months of antimicrobial treatment, doxycycline and flagyl were added leading to clinical improvement and normalization of his inflammatory markers. He was continued on oral doxycycline and flagyl for 12 months for a chronic osteomyelitis.
Discussion:
H. bilis is a slow growing pathogen which is challenging to culture in the laboratory often requiring special agar plates and prolonged incubation. In patients with agammaglobulinemia and associated chronic skin infections or erythema nodosuma, H bilis should be suspected as a possible pathogen. Due to challenges with culturing, 16S PCR or amplification of the 16S ribosomal subunit should be considered to try to identify the pathogen.
There are poorly delineated clinical antimicrobial breakpoints to help guide therapy with minimal evidence. Case reports suggest prolonged therapy with aminoglycosides and penicillin. Other studies have successfully treated patients with a carbapenem, azithromycin and levofloxacin. In the absence of sensitivity data, prolonged treatment (12months) should be considered with a combination of antimicrobials. Patients should be followed closely as recurrent infections are not uncommon.
(165) Submission ID#604074
Clinical Phenotyping of a DOCK8 Deficiency Cohort
Alexandra F. Freeman, MD1, Nirali N. Shah, MD2, Amanda Urban, DNP, CRNP3, Dennis Hickstein, MD4, Helen C. Su, MD, PhD5
1Director, Primary Immune Deficiency Clinic, Laboratory of Clinical Immunology and Microbiology, NIAID, NIH, Bethesda, MD, USA
2Associate Research Physician, Pediatric Oncology Branch, NCI, NIH, Bethesda, MD
3Nurse Practitioner, Clinical Monitoring Research Program Directorate, Frederick National Laboratory for Cancer Research sponsored by the National Cancer Institute
4Senior Investigator, NCI, NIH
5Chief, Human Immunological Diseases Section, Laboratory of Clinical Immunology and Microbiology, NIAID, NIH, Bethesda, MD
Introduction: DOCK8 deficiency is a combined immunodeficiency characterized by eczema, recurrent sinopulmonary infections, viral skin infections, malignancy and early mortality. In recent years, liver disease and vasculopathy have been increasingly recognized as a complication of DOCK8 deficiency. We clinically characterized our cohort of DOCK8 deficient patients, with a specific focus on these newly identified areas of disease involvement.
Methods: Chart reviews were performed on patients seen at NIH with genetic and clinical diagnosis of DOCK8 deficiency. Patients were all enrolled on IRB approved NIAID protocols.
Results: We identified 52 patients from 40 families with DOCK8 deficiency in our NIH cohort, ranging in age from 6-44 years. Of the 40 families, 17 had homozygous mutations. Of the 52 patients, food allergy was diagnosed in 31 (60%), eczema in 49 (94%), and asthma in 30 (58%). Chronic or recurrent viral skin infections were seen in 49/52 (94%). Chronic EBV viremia by PCR positivity was seen in 18/46 patients (39%); only 2 patients were known to be EBV immune without viremia. CMV viremia was infrequent. Sinopulmonary infections were common, with bronchiectasis occurring in 23 /50 (46%) with available imaging. Liver disease was diagnosed in 14 (27%), with 7 having biliary tract abnormalities on imaging and stool positive for Cryptosporidia; most patients with Cryptosporidia were without diarrhea. The incidence of Cryptosporidia is likely under-represented due to more recent availability of sensitive assays for Cryptosporidia detection. Other liver abnormalities included fatty liver, metastatic disease from malignancy and medication related hepatitis. Vasculopathy, predominantly of the aorta and cerebral arteries, was diagnosed in 7, with patients in the last 5 years being prospectively imaged. Autoimmunity was rare (5%) including autoimmune cytopenias and hypothyroidism. 36 of 50 with follow-up are alive (70%) with age range 6-44 years. Of the 36 living patients, 28 (78%) have had a HSCT. Causes of deaths include malignancy (6), infection (1), and HSCT complications (7). Long-term follow-up of patients with HSCT (up to 6 years) has revealed resolution of the infection susceptibility and eczema, no new cancers, and stabilization of vasculopathy.
Conclusions: In addition to the well described manifestations of DOCK8 deficiency including eczema, allergy, recurrent sinopulmonary infections, skin viral infections and malignancy, our cohort revealed a relatively high incidence of liver disease, frequently associated with stool positivity for Cryptosporidia, as well as vasculopathy. Both of these clinical manifestations should be considered during preparation for HSCT as they may affect management through transplant. Autoimmunity has likely been over-estimated in prior descriptions of DOCK8 deficiency. Long-term follow-up after HSCT is needed to determine the prognosis from the vasculopathy, liver disease, and malignancy risk.
(166) Submission ID#604115
Transcriptome Analysis Reveals an Important Role for EXTL3 in Human Hematopoietic Cell Differentiation
Yasuhiro Yamazaki1, Stefano Volpi2, Luigi D. Notarangelo1
1Laboratory of Clinical Immunology and Microbiology, IDGS, DIR, NIAID, NIH, Bethesda, MD, USA
2U.O. Clinica Pediatrica e Reumatologia, IRCCS Istituto Giannina Gaslini, Genova, Italy
Introduction/Background: EXTL3 (Exostosin Like Glycosyltransferase 3) is an exostosin family member which initiates heparan sulfate (HS) chain biosynthesis and elongation. We have reported homozygous EXTL3 hypomorphic mutation (R339W) as a cause of immuno-osseous-dysplasia syndrome. Fourteen patients who have EXTL3 homozygous mutation were reported so far. Eight of them manifested T cell lymphopenia, and 5 presented with severe combined immunodeficiency (SCID) or Omenn syndrome. Using patient-derived induced pluripotent stem cells (iPSCs) as a model, we have previously reported that EXTL3 mutations affect differentiation to thymic epithelial progenitor cells as well as expansion of hematopoietic progenitor cells. Consistent with the latter, previous studies have suggested that mutations in other genes involved in HS biosynthesis affect hematopoietic stem cell (HSC) differentiation. However, the exact mechanisms by which EXTL3 mutations affect hematopoiesis are not known.
Objectives: We tried to clarify gene expression difference in HSCs derived from wild-type, EXTL3 hypomorphic and EXTL3 knock-out (KO) human iPSCs.
Methods: The control BJ iPSC line was engineered by CRISPR/Cas9 gene targeting. EXTL3 KO iPSCs were obtained which carried compound heterozygous EXTL3 mutations (c.1003_1004insT; c.1005_1006insGATATTT). HSC differentiation was induced using the STEMdiff hematopoietic kit (STEMCELL technologies). Bulk RNA from each iPS cells and each differentiated CD34+CD43+CD45+ was analyzed by RNA sequencing.
Results: As compared to control iPSCs, patient-derived cells showed slightly lower capacity to generate CD34+CD43+CD45+ cells. On the other hand, EXTL3 KO cells showed no differentiation into CD34+CD43+CD45+ cells. Gene Set Enrichment Analysis showed enriched expression of genes involved in hematopoietic progenitor cell differentiation, regulation of hemopoiesis, and positive regulation of hemopoiesis in both control and patient-derived CD34+CD43+CD45+ cells compared to parental iPSCs. Moreover, these gene sets were more abundantly enriched in control than in patient-derived CD34+CD43+CD45+ cells. The gene set of Response to type I interferon was significantly enriched in control versus patient-derived CD34+CD43+CD45+ cells.
Conclusions: These results confirm that EXTL3 plays an important role for HSC homeostasis in human cells. Because type 1 interferons play a role in HSC proliferation, the decreased type I interferon signature may account for the reduced number of HSCs that we have previously reported upon in vitro differentiation of EXTL3-mutated versus control-derived iPSCs.
This study was supported by the Division of Intramural Research, NIAID, NIH, under protocol 16-I-N139.
(167) Submission ID#604171
A Case of Autoinflammatory Syndrome with Osteoporosis and Specific Antibody Deficiency
Irina Dawson, MD1, Mark Ballow, MD2, Lori Broderick, MD, PhD3, Jolan Walter, MD, PhD4
1Allergy and Immunology Fellow, Division of Allergy and Immunology, Department of Pediatrics, University of South Florida, St Petersburg, FL
2Associate Professor, Division of Allergy and Immunology, Department of Pediatrics, University of South Florida, St Petersburg, FL
3Assistant Professor, Division of Allergy, Immunology and Rheumatology, Department of Pediatrics, University of California, San Diego, CA
4Associate Professor, Robert A. Good Endowed Chair and Division Chief, Division of Pediatric Allergy & Immunology, Department of Pediatrics, University of South Florida, Johns Hopkins All Children's Hospital, St. Petersburg, FL.
Autoinflammatory syndromes are inherited disorders with an exaggerated inflammatory response with no specific trigger. The clinical phenotypes of variants of autoinflammatory syndromes may overlap.
We report a case of a 13 year old male with prior diagnosis of specific antibody deficiency, Periodic Fever, Aphthous Stomatitis, Pharyngitis, Cervical Adenitis (PFAPA) syndrome, arthralgia and moderate atopic dermatitis.
He was diagnosed at 3 years of age with specific antibody deficiency based on persistently low pneumococcal titers against repeat immunizations. Due to recurrent infections, he was placed on immunoglobulin replacement therapy (IgRT) at 8 years of age. IgRT was discontinued at 13 years of age due to full resolution in infections and patient demonstrated robust response to immunizations. Patient had lifelong history of recurrent fevers (every 5 weeks) associated with pharyngitis and aphthous ulcers consistent with diagnosis of PFAPA. As he became older these episodes became less frequent. Last episode of fever was over a year ago. The father had similar symptoms of recurrent fevers and oral ulcers as a child but currently remains asymptomatic. Paternal grandfather died of kidney disease.
Patient has been generally in good health until recent year with intermittent abdominal pain, arthralgia and several long bone fractures with no history of prior trauma. A bone density scan revealed osteopenia and osteoporosis with a Z score of -2.2 of lumbar spine, -4.0 of left femoral neck, -3.1 of left hip.
Given history of familial autoinflammatory disease, and antibody deficiency genetic testing was obtained which identified a pathogenic heterozygous variant of TACI and MEFV c.2082G>A (p.Met694lle). TACI mutation has been linked to antibody deficiency syndromes. Genetic study for family members is pending.
The MEFV gene is associated with autosomal recessive familial Mediterranean fever (FMF) and has been reported in autosomal dominant FMF as well. FMF is characterized by recurrent episodes of fever associated with serositis, arthralgia, and arthritis. Patients with FMF have elevation in acute phase reactants during attacks with most returning to normal levels during the episode-free periods. Multiple studies have shown that patient with FMF have lower bone mineral density and Z-scores than the general population. Inflammation in FMF is thought to be mediated by several different cytokines (IL-1, IL-2, IL-6, IL-7, IL-8, IL-11, IL-15 and TNF-). These same cytokines play a role in osteoclast activity and bone resorption. It has been suggested chronic inflammation during acute attacks and subclinical inflammation during the disease-free period lead to bone loss and osteoporosis. Regular use of colchicine, the main treatment for FMF, may slow down osteoporosis.
Beside careful monitoring of clinical and laboratory phenotype, genetic evaluation is an important step in distinguishing between overlapping entities and can prevent complication and promote targeted intervention.
(168) Submission ID#604373
When Mosquito Bite Allergy Is Treated with Bone Marrow Transplantation (BMT)
Joseph A. Church, MD1, Ronald M. Ferdman, MD2, Neena Kapoor, MD3
1Professor, Pediatrics, Children's Hospital Los Angeles and Keck School of Medicine of U.S.C.
2Associate Professor, Pediatrics, Children's Hospital Los Angeles and Keck School of Medicine of U.S.C.
3Professor, Pediatrics, Children's Hospital Los Angeles and Keck Schood of Medicine of U.S.C.
A 5 year old previously healthy boy was referred for periodic fever/PFAPA and mosquito bite hypersensitivity. Eight weeks earlier he developed fever to 104F, mouth sores and exudative tonsillitis; a rapid strep screen was negative. One week later he developed moderate cervical lymphadenopathy and had a positive EBV Early Antigen antibody.. One month later he had several severe local reactions to mosquito bites. Each manifested 6-8 cm of erythema and induration with a 1+ cm bullae which left an ulcer after rupture and healed with a hypopigmented scar. The bites were accompanied by fever to 104F for 4 days. One febrile episode was treated with low dose prednisolone for presumed PFAPA, and the fever resolved within hours. His past history was positive for nasal allergy and mild asthma. His parents are not related: mom is of European-Indonesian and dad European-African (Creole ancestry. Testing prior to this visit showed normal IgG, IgA and IgM, elevated IgE (12,000 U/L) and normal CBC. Lymphocyte subsets revealed CD3+ 23% (1538/mcL), CD4+ 17% (1109/mcL), CD8+ 6% (363/mcL), CD19+ 9% (587/mcL), NK cells 67% (4435/mcL). On examination he appeared well with height at 86th%ile and weight at 58th%ile. There was no lymphadenopathy, hepatosplenomegaly or inflammed skin lesions; there was a 1cm round scar on the right plantar surface at the site of a prior mosquito bite. Laboratory studies confirmed NK lymphocytosis 64% (5459/mcL) and elevated IgE (29,600 U/L). Lymphoproliferation to mitogens, CD3/CD28, CMV and HSV were normal, but absent to tetanus and candida antigens. EBV antibodies reflected past infection (VCA-IgG+, VCA-IgM-, EBNA+); quantitative EBV PCR was >5,000,000 copies/mL whole blood. NK cytotoxicity and CD107a expression were decreased. Bone marrow NK analysis suggested conality. The patient was diagnosed with "hypersensitivity to mosquito bites with EBV-associated T-/NK lymphoproliferation." This disorder represents a subset of chronic active EBV (CAEBV) that is rarely seen outside of East Asia. The lack of organomegaly or lymphadenopathy with hyper-IgE and NK lymphocytosis and decreased NK function support the likelihood that NK cells are the target of EBV infection in this patient. This diagnosis may be a precursor to hemophagocytosis, liver necrosis or lymphoma/leukemia, and the only curative treatment is bone marrow transplantation. The patient's sister is a 10/10 HLA match. She is seropositive for past EBV infection, and she has no history of extreme reactions to mosquito bites. Genetic mutations that cause familial hemophagocytic lymphohistiocytosis have not been reported in CAEBV, and to the best of our knowledge familial cases of this disorder have not been identified. The response to BMT in this patient is pending.
(169) Submission ID#604432
Epidemiology of Anti-epileptic Drug Induced Hypogammaglobulinemia in a Tertiary Care Network
ErinMarie Kimbrough, MD1, Keith Sacco, MD1, Ismael Carrillo-Martin, MD2, Natalia Chamorro-Pareja, MD3, Daniela Haehn, MD4, Alexei Gonzalez Estrada, MD5
1Resident, Department of Medicine, Mayo Clinic Jacksonville
2Research Trainee, Department of Medicine,Mayo Clinic Jacksonville
3Research Trainee, Division of Pulmonary, Allergy, and Sleep Medicine, Mayo Clinic Jacksonville
4Research Trainee, Department of Anesthesia and Perioperative Medicine, Mayo Clinic Jacksonville
5Division of Pulmonary, Allergy, and Sleep Medicine, Mayo Clinic Jacksonville
Introduction/Background: A number of case reports have described symptomatic hypogammaglobulinemia following administration of anti-epileptic drugs (AEDs), specifically lamotrigine, carbamazepine, and levetiracetam. The mechanism by which symptomatic hypogammaglobulinemia develops is unclear. We evaluated the prevalence and the clinical significance of hypogammaglobulinemia associated with use of these AEDs.
Objectives: Our aim was to characterize the prevalence of AED-induced hypogammaglobulinemia, identify specific AEDs associated with hypogammaglobulinemia, and characterize the timeline to development of hypogammaglobulinemia after initiation of therapy.
Methods: A retrospective, multicenter, electronic medical record review spanning 18 years identified patients with hypogammaglobulinemia who were on AED therapy (lamotrigine, carbamazepine, or levetiracetam). Patients were excluded if they had a pre-existing primary immunodeficiency (PID), malignancy, protein-losing enteropathy, or significant proteinuria. Patients on chronic immunosuppressive therapy, those without laboratory criteria for hypogammaglobulinemia, or those on one of the AEDs for less than one month were also excluded.
Results: Of the 316 cases reviewed, 5 patients met our inclusion criteria. The median age was 35; 80% were adults, 80% were female, and 80% were white. Lamotrigine was implicated in 3/5 of the cases, carbamazepine in 2/5, and levetiracetam in 1/5. Tetanus and pneumococcal titers were available for 4/5 patients. Of those patients, 3/4 had protective titers to both per report with responses to >70% of the serotypes. Only one patient reported severe, recurrent infections while the remaining four had little to no symptoms. Interestingly, the patient with severe infections did have protective titers. Of the five laboratory proven hypogammaglobulinemia patients, one died of an infection, two have continued on the medication due to refractory seizures responsive only to these medications, and two are currently being tapered off of their AED.
Conclusion: While it appears that AED-induced hypogammaglobulinemia is quite rare, it should be considered in a patient without other secondary causes of hypogammaglobulinemia on AED therapy. Many antiepileptics downregulate NFkB signaling suggestive that patients who develop symptomatic hypogammaglobulinemia may have hypomorphic mutations in the NFkB signaling pathway.
(170) Submission ID#604503
Autoimmune Lymphoproliferative Syndrome with Histopathologic Features of Castleman Disease
Rachelle Lo, MD1, Dita Gratzinger, MD, PhD2, Elizabeth Keiser, MD, MPH3, Kay Chang, MD4, Yael Gernez-Goldhammer, MD, PhD5
1Fellow, Allergy and Immunology, Division of Allergy and Immunology, Department of Pediatrics, Stanford University School of Medicine
2Associate Professor, Department of Pathology, Stanford University School of Medicine
3Fellow,General Surgical Pathology, Department of Pathology, Stanford University School of Medicine
4Professor, Professor of Otolaryngology and Pediatrics, Stanford University, Department of Otolaryngology, Lucile Packard Children's Hospital at Stanford, Division of Pediatric Otolaryngology
5Clinical Assistant Professor, Division of Allergy and Immunology, Department of Pediatrics, Stanford University School of Medicine
Autoimmune Lymphoproliferative Syndrome (ALPS) results from defective apoptosis of lymphocytes mediated through the Fas/Fas ligand (FasL) pathway. The hallmark lab finding is an expansion of T cells that express the alpha/beta T cell receptor, but lack both CD4 and CD8 (double negative T cells) in the setting of normal or elevated lymphocyte counts. Patients present with chronic, nonmalignant, noninfectious lymphadenopathy or splenomegaly. For definitive diagnosis, patients need to have (1) a pathogenic mutation in FAS, FAS ligand or caspase 10 or (2) a defective FAS-induced lymphocyte apoptosis. We describe a probable case of ALPS with heterozygous mutation in FAS c.287A>G(p.His96Arg), a variant that has not been previously reported (his lymphocyte apoptosis assay is pending). Unique to this case is the patients Castleman disease-like features on pathology.
A 15 year-old male referred from hematology clinic presented with an 8 year history of chronic lymphadenopathy, splenomegaly, anemia, and no underlying diagnosis. Malignancy had previously been excluded by bone marrow aspirate and biopsy 8 years prior. However, he had a right sided lymph node that had increased in size for the past 4 months. He was otherwise asymptomatic. A lymph node biopsy 7 years prior was reportedly normal. His exam demonstrated significant bilateral lymphadenopathy, greater on right, with an approximately 8 x 6 cm mobile right neck mass. He had splenomegaly palpated 7 cm down and across to midline. He was therefore admitted for excisional lymph node biopsy to evaluate for possible malignancy and labs were sent to evaluate for ALPS.
Labs were supportive of ALPS. He had elevated T cell receptor alpha beta double negative T cells (TCR a/b DNTCs) in blood (10.5%). B12 level was elevated (>1000 pg/mL). Plasma soluble FASL level was elevated (5517 pg/mL). Interleukin-10 (IL-10) and IL-18 levels were elevated (88 and 909 pg/mL respectively). He had multilineage cytopenias: anemia with Hgb of 9.5 g/dL and neutropenia (absolute neutrophil count of 1380 K/uL). He had hypergammaglobulinemia with an IgG level of 2010 mg/dL. Broad infectious work-up was negative, including HIV, QuantiFERON, Cocci, Bartonella, Toxoplasma, Coxiella burnetii, EBV PCR and, CMV IgM.
Lymph node biopsy showed no evidence of malignancy. Immunostains and flow cytometry showed the presence of expanded TCR a/b DNTCs in the lymph node, consistent with ALPS. Interestingly, lymph node histology showed morphologic features typical of plasma cell variant Castleman disease. Numerous Castleman-like follicles showed typical regressive changes with onion-skinning morphology. Paracortical hyperplasia with sheets of plasma cells was noted. There was negative staining for HHV8 (a well-known cause of plasma cell variant Castleman disease).
The diagnosis of idiopathic multicentric HHV8-negative Castleman disease was excluded by definition in the setting of ALPS, per evidence-based consensus criteria published in 2017. In addition, our patient did not show any symptoms typically associated with it, such as fever, night sweats, weight loss, weakness or fatigue. Should his FAS-induced lymphocyte apoptosis be defective (in 2 separate assays), this would confirm his ALPS-FAS diagnosis and we would start the patient on sirolimus.
(171) Submission ID#604721
SLC46A1 Deficiency Presenting as Hypogammaglobulinemia and Wide Clinical Manifestations
Agostina Llarens, MD1, Carolina Dorfman, MD1, Daniela Di Giovanni, MD2, Andrea Gomez Raccio, MD2, Gisela Seminario, MD2, María Isabel. Gaillard, MSc3, Patricia Carabajal, MD4
1Physician – Immunology trainer, Children's Hospital Ricardo Gutierrez
2Physician – Immunologist, Children's Hospital Ricardo Gutierrez
3Biochemist, Grupo de Inmunología Hospital de Niños
4Head of Immunology Unit, Children' s Hospital Ricardo Gutierrez
Introduction: SLC46A1 gene encodes the proto-couple folate transporter (PCFT), which supports intestinal folate uptake, and participates in folate transport into the central nervous system. SLC46A1 mutations cause PCFT defects, resulting in low folate levels in serum and cerebrospinal fluid. Hereditary folate malabsorption (HFM) is a rare, autosomal recessive disorder with PCFT deficiency resulting in cerebral folate deficiency. Most of the patients present megaloblastic anaemia, moderate pancytopenia in the first few months of life, failure to thrive, diarrhoea and/or later onset neurological symptoms including seizures and developmental delay.
Immunodeficiency in HFM can manifest itself with hypogammaglobulinemia with normal T-cell function. B-cell precursor compartment seems to be particularly vulnerable to folate deficiency in some HFM patients. This immunodeficiency can be restored with specific treatment with folic acid.
Aim: To describe a female patient with a homozygous pathological variation in the SLC46A1 gene.
Results: A 17 months old girl, born of non-consanguineous parents. She started at 3 months old with diarrhoea due to rotavirus, low weight and bicytopenia with normal bone marrow aspiration. She presented low levels of folic acid 1.5ng/ml (NV 3.1-20.5 ng/ml) at first thought due to secondary to malnutrition. Treatment with folic acid supplementation was administrated, improving platelets counts. At 5 months old she presented steatorrhea with severe perianal panniculitis which required surgical treatment. No germs were rescued after a skin biopsy. Moreover, she suffered from a respiratory infection due to Picornavirus with two episodes of pneumothorax which required intensive care. At that moment IVIG treatment was administered due to hypogammaglobulinemia and clinical severity.
Chronic diarrhoea worsened with bloody depositions. Three rectal ulcers were found in the gut biopsy. Bowel inflammatory disease was suspected and mesalazine administration was started with weight improvement.
Furthermore, at 10 months old she presented 3 status epilepticus, with pathological EEG and normal MRI; one of them related to a CMV infection, successfully treated.
In the immunological evaluation IgG and IgA were low with normal IgM and IgD. The protein-antibody response was not evaluated. She presented normal lymphocyte and T cells extended populations, T cells proliferation assay, DHR, Treg cells, complement, CD107a expression, alpha-fetoprotein, without autoantibodies
A molecular panel testing was done by NGS and a homozygous variant in SLC46A1 gene was found, causing impaired intestinal folate absorption.
Conclusion: HFM should be considered in the diagnosis of patients with cytopenias and hypogammaglobulinemia in order to provide specific treatment.
HFM has wide clinical manifestations, not only with megaloblastic anaemia and neurological impairment but also with gastrointestinal and skin manifestations. With folate treatment, clinical and immunological defects can be normalized.
(172) Submission ID#604754
A Case Report of Focal Epithelial Hyperplasia (Hecks Disease) with Elevated Tumor Necrosis Factor Alpha
Zoya Treyster, MD1, Sara Sussman, MD2, David Rosenthal, DO, PhD3
1Fellow, Division of Allergy & Immunology, Donald and Barbara Zucker School of Medicine at Hofstra/Northwell
2Fellow, Department of Pediatrics, Zucker School of Medicine at Hofstra Northwell School of Medicine
3Assistant Professor of Medicine and Pediatrics, Donald and Barbara Zucker School of Medicine at Hofstra/Northwell
Introduction: Multifocal epithelial hyperplasia (MEH), or Hecks Disease, is a rare, benign infection of the mucosa caused by human papilloma virus (HPV). Clinically, MEH manifests as numerous painless, soft, sessile papules or plaques, and typically occurs in the labial, lingual, and buccal mucosa. MEH lesions are usually associated with HPV types 13 and 32, and seen more commonly in patients of Caribbean or Central/South American descent. Prior studies in adults have shown that Tumor Necrosis Factor Alpha (TNF) promotes HPV, and may influence duration of HPV infection.
Case: We present a five-year-old full term male of Haitian descent referred for assessment of multiple flesh colored, papular lesions on the buccal and labial mucosa that had persisted and quantitatively increased over one year, although some lesions regressed. He had no pain or difficulty eating. Medical history significant for one seizure; negative for infection. No family history of infection, immunodeficiency, consanguinity, or miscarriage. Head and neck examination failed to reveal cervical lymphadenopathy, masses, or hypertrophy in the salivary glands. Intraoral examination revealed multiple papular nodules, mostly flat although some were corrugated. The greatest concentration was noted on the lower left labial surface extending to the mucosal vermillion interface, not involving the vermillion or commissure region. Lesions extended into the mandibular vestibule and the left buccal mucosa. No other lesions were noted on extremities, genitalia, or any other visualized mucosal surface. Based on history and exam, he was diagnosed with MEH. White blood cell count, neutrophils, lymphocytes, CD4 and CD8 T cell, B cell, NK cell enumeration, and immunoglobulin panel were normal for age. Tetanus and Streptococcus pneumoniae titers were protective. Cytomegalovirus IgG and IgM were negative. Epstein-Barr Virus IgG was positive, IgM and Early Antigen Ab negative. Serology was significant for elevated TNF (84 pg/mL; reference range <22pg/mL) while interferon gamma and interleukins 1, 2, 4, 5, 6, 8, 10, 12, 13, and 17 were normal, as was IL-2 receptor CD25. One month after the initial visit, lesions were stable and unchanged. Nine-valent HPV vaccination was considered, but not administered.
Conclusions: MEH is a rare but benign disease caused by HPV. Awareness of the disease and its course is important to prevent unnecessary expanded immunodeficiency work-up and possible procedures to eliminate lesions. Although mucosal immunity can be site specific, especially with HPV, our understanding of T-cell cytokine and chemokine responses to HPV in cervical and laryngeal lesions may be instructive. The mechanism which allows HPV persistence in MEH is not characterized, but it likely is due to increased viral persistence and an inability for the host immune response to successfully induce viral latency and successful containment. Elevated TNF levels, with normal levels of IL-2, IL-6, IL-8, IL-10, may correlate with decreased clearance of HPV and prolonged duration of MEH. It remains unclear if viral persistence is the cause of, or the sequela of, increased TNF. Longitudinal monitoring of cytokine (TNF, IL-2, IL-6, IL-8, IL-10) and chemokine (CCL17, CCL18, CCL19, CCL20, CCL21, and CCL22) serum concentrations may be useful biomarkers for disease resolution.
(173) Submission ID#604905
Patient Education with a Self-Efficacy Focus for Adult Autosomal Dominant Hyper IgE Syndrome Patients
Amanda Urban, DNP, CRNP1, Dirk A. Darnell, MA, RN2, Ladan Foruraghi, CRNP3, Alexandra F. Freeman, MD4
1Nurse Practitioner, Clinical Monitoring Research Program Directorate, Frederick National Laboratory for Cancer Research sponsored by the National Cancer Institute
2Nurse Case Manager, Laboratory of Clinical Immunology and Microbiology, NIAID, NIH, Bethesda, MD, USA
3Nurse Practitioner, Laboratory of Clinical Immunology and Microbiology, NIAID, NIH, Bethesda, MD, USA
4Director, Primary Immune Deficiency Clinic, Laboratory of Clinical Immunology and Microbiology, NIAID, NIH, Bethesda, MD, USA
Introduction: Autosomal dominant Hyper IgE (Jobs) Syndrome is a rare primary immunodeficiency characterized by eczema and sinopulmonary infections as well as musculoskeletal and vascular complications. As in all chronic illnesses, patient education is an ongoing need. In the rare disease population, patient education is especially important as patients must be able to explain their unique healthcare concerns in a variety of medical settings. We focused on AD-HIES, due to our relatively large cohort of patients, the frequent lack of classic signs of illness often impairing diagnosis of severe infection, and the diverse non-immunologic clinical features of this disease.
Objectives: We aimed to increase understanding of the clinical manifestations of AD-HIES to promote earlier recognition of symptoms and to increase self-efficacy for symptom management in the adult HIES population.
Methods: Adult patients were asked to participate in a patient education project. Demographic information was collected from participants. They also completed a 12-item multiple choice test about symptom recognition in AD-HIES and PROMIS Self-Efficacy for Managing Symptoms, an 8-item validated survey. Then, patient education handouts that focused on pulmonary symptoms, eczema, bone health, and cardiovascular complications were reviewed with the participant. Six weeks later, participants were asked to repeat the 12-item test and the self-efficacy Survey. The demographic information, test, and self-efficacy were collected anonymously.
Results: 33 participants provided demographic information, completed the test and the Self-Efficacy Survey. Of the 33 participants, 15 were male and 17 were female. Participants ranged in age from 18 to 66 years. 22/33 (67%) reported looking for information about AD-HIES using search engines and most patients (91%) report that they have been given information about AD-HIES from a doctor. 19/33 (58%) participants identified pulmonary symptoms as the symptom that concerns them most and 10/33 (30%) participants identified more than one symptom of concern. 25 participants returned the second test and second survey. The mean test score increased from 9.08 to 10.28 with 23/25 participants achieving a score of 9/12 or higher. The self-efficacy scores were unchanged with a mean score of 50.08 before reviewing the patient education handouts and 50.13 after.
Conclusions: Participant feedback to this project was generally positive. AD-HIES patients are seeking information and an educational intervention can improve their understanding of disease. Self-Efficacy results were mixed and unchanged overall, but suggest that AD-HIES patients manage symptoms as well as other patients with chronic illnesses. Patient education should continue at each encounter. This project can be expanded to include more topics, pediatric patients, and other rare disease populations. Funded by the NCI Contract No. HHSN261200800001E
(174) Submission ID#605066
T and NK Cell Dysfunction Arising from BCL11B Deficiency
Samuel Chiang, PhD1, Sharat Chandra, MD, MRCPCH2, Vijaya Chaturvedi, B.S.3, Erika Owsley, B.S.3, Jack Bleesing, MD, PhD4, Brian Dawson, PhD5, Rebecca A. Marsh, MD6, Miao Sun, PhD7
1Research Associate, Cincinnati Childrens Hospital Medical Center
2Assistant Professor, UC Department of Pediatrics, Division of Bone Marrow Transplantation and Immune Deficiency, Cincinnati Childrens
3Research Associate, Cincinnati Children's Hospital
4Professor, Division of Bone Marrow Transplantation and Immune Deficiency, Cincinnati Childrens
5Director, Clinical Laboratories, Division of Human Genetics
6Associate Professor, Division of Bone Marrow Transplantation and Immune Deficiency, Cincinnati Childrens Hospital Medical Center
7Assistant Director, Clinical Laboratories, Division of Human Genetics
Introduction: BCL11B plays an important role in the development and maintenance of the immune system and the central nervous system. Expression of BCL11B represses NK and myeloid factors while inducing T cell lineage genes in thymocytes at the DN2 stage. Conditional loss of Bcl11b expression in murine thymocytes leads to T cell deficiency while complete knockout of Bcl11b was fatal within a few days of birth. Recently, specific heterozygous BCL11B mutations have been reported in 11 individuals with global development delay. However, only 2 of these cases, both carrying heterozygous missense variants, had low TREC values with 4 other cases having frequent infections. Little is known regarding the impact of BCL11B on human NK and T cell function.
Methods: We identified a novel heterozygous truncating mutation in BCL11B in an infant who was first detected by TREC newborn screening. She subsequently developed severe autoimmune hemolytic anemia at the age of 3 months. We used standard immunoblotting and flow cytometry methods to assess protein expression and the impact of this BCL11B mutant on T cell and NK cell development and function.
Results: The patient has a novel single base-pair deletion in the BCL11B gene, which is predicted to produce a truncated protein with the loss of 3 of 6 zinc finger domains in BCL11B. Immunoblotting of T cell blast lysates revealed a reduced BCL11B expression in the patient consistent with the heterozygous defect in BCL11B but also generated a novel band with a smaller molecular weight that we postulate represents the truncated protein product. While mitogen responses to ConA and PHA were normal, both CD4+ and CD8+ T cell counts were decreased, especially CD4+ naïve and CD4+CD31+ naïve T cells, suggesting reduced thymic output. The function of TH1 cells was skewed with reduced IL-2 production but increased IFN levels after PMA and ionomycin stimulation. Moreover, T regulatory cell counts were below normal range. NK cell counts were normal but these were mostly CD56bright NK cells. Of the few CD56dim NK cells that presented, approximately half did not express CD16, the Fc receptor for ADCC. Perforin was only present in CD16 expressing NK cells. As such, anti-CD16 stimulation understandably led to low but not defective NK cell degranulation. Function after stimulation with K562 cells was normal when controlled for NK cell counts.
Conclusion: We report a novel BCL11B truncating mutation with a leaky SCID phenotype that manifested with T-cell lymphopenia and autoimmunity. Lowered thymic-derived naïve T and regulatory T cells, skewed TH1 cytokine response, and incomplete NK cell development suggests that BCL11B is important for the development and differentiation of multiple lymphocyte lineages.
(175) Submission ID#605266
Refractory Giardia Infection in a Patient with Common Variable Immune Deficiency
Ekta Kakkar, MD1, Laila Woc-Colburn, MD2, Joud Hajjar, MD, MS3
1Allergy and Clinical Immunology Fellow, National Jewish Health and The University of Colorado
2Associate Professor in Infectious Diseases, Baylor College of Medicine
3Assistant Professor, Baylor College of Medicine, 1Texas Childrens Hospital Center for Human Immunobiology and Division of Immunology, Allergy and Rheumatology
Introduction: Chronic diarrhea is one of the most common gastrointestinal complaints in patients with common variable immune deficiency (CVID) and can lead to life-threatening complications such as malabsorption and malnutrition. Chronic diarrhea in CVID could be caused by infections, an inflammatory bowel disease-like picture, as well as malignancy. Giardia lamblia is one of the most common parasites causing diarrhea in CVID (up to 40%), and can be refractory in these patients, leading to villous atrophy, weight loss, and failure to thrive.
Case Report: A 41-year-old female with a history of CVID presents with chronic diarrhea and significant weight loss. Her CVID was diagnosed by hypogammaglobulinemia (low levels of IgG, IgM, and IgA), inadequate responses to protein and polysaccharide-based vaccines, decreased memory B cells (CD19+CD27+ 0.5%), and recurrent sinopulmonary infections. She was started on immune globulin replacement therapy and had significant improvement in her rate of infections. Four years before her presentation to our center, she developed chronic, severe diarrhea. Work up revealed Giardia lamblia infection on endoscopy and colonoscopy. Biopsy showed intraepithelial lymphocytes, villous blunting, and atrophic gastritis with rare plasma cells concerning for non-infectious enteropathy related to her CVID, in addition to the high burden of Giardia organisms. She was initially treated with metronidazole for several weeks. However, her diarrhea did not improve, and she developed significant peripheral neuropathy leading to lower extremity weakness and limited mobility. Her diarrhea persisted and was associated with approximately a 20-pound weight loss. Repeat endoscopy and colonoscopy two years later showed persistent high burden Giardiasis of the small intestine, as well as reactive lymphocytic infiltrates and atrophic gastritis. She was treated with nitazoxanide but continued to have diarrhea, and her stool continued to show trophozoites. Given the significant inflammation and the lack of response to multiple antiparasitic agents, she was referred to our center for further evaluation. She was started on oral budesonide (9 mg daily) and oral immune globulin (20 grams weekly for 12 weeks). With this regimen, she had significant improvement in her diarrhea with a 10-pound weight gain. Repeat colonoscopy showed considerable improvement in inflammation and resolution of her Giardia infection, though her stool antigen continues to be positive.
Conclusions: Persistent diarrhea in our patient is most likely due to a combination of CVID enteropathy and Giardiasis. A prolonged course of metronidazole and later nitazoxanide did not control her diarrhea and led to significant side effects. Switching to an immunomodulatory approach significantly decreased the inflammation in her bowel and may even have helped to reduce the burden of Giardia in the gut. Targeting both underlying bowel inflammation as well as active infection in CVID patients with chronic diarrhea might be needed to control symptoms.
(176) Submission ID#605352
Sphingosine-1-phosphate Lyase Deficiency Identified by Newborn T Cell Receptor Excision Circle Screening for Severe Combined Immunodeficiency
Cullen M. Dutmer, MD, Austin A. Larson, MD, Elena WY. Hsieh, MD
1Assistant Professor, Children's Hospital Colorado - University of Colorado School of Medicine
Introduction: Sphingosine-1-phosphate (S1P) is a lipid chemoattractant that is critical for lymphocyte egress from lymphoid organs. Following a S1P concentration gradient maintained by S1P lyase ubiquitously expressed in tissues, lymphocytes within lymphoid organs are drawn to efferent lymph and blood unless their S1P receptor is internalized or downregulated. Owing to diminished degradation of not only S1P, but also other sphingoid bases, deleterious mutations in SGPL1 (encoding S1P lyase) perturb sphingolipid catabolism in numerous tissues. Correspondingly, human S1P lyase deficiency results in multiorgan dysfunction including kidney, skin, endocrine gland, and neurologic impairment alongside expected lymphopenia. Although severe T cell lymphopenia (<300 cells/microliter) rivaling that of severe combined immunodeficiency (SCID) can be seen in patients with S1P lyase deficiency, no such patients have been identified by newborn screening of T cell receptor excision circle (TREC) counts, which are a surrogate measure of effective T cell production. Herein, we describe an infant boy with an undetectable TREC count at birth who was found to have two novel, biallelic SGPL1 mutations resulting in S1P lyase deficiency.
Case Description: A 1-day-old boy with a preceding history of fetal hydrops is born at a gestational age of 36 weeks and presents with renal failure, anasarca, and respiratory failure. TREC analysis of a dried blood spot obtained at 24 hours of life reveals zero copies/microliter. Subsequent peripheral blood studies show profound lymphopenia, with diminished CD3+ T (129/microliter; 96 CD4+, 27 CD8+), CD19+ B (130/microliter), and CD16/56+ natural killer (124/microliter) cell counts. Recent thymic emigrants are reduced (11.3% of CD4+ T cells are CD45RA+CD31+), as is the ratio of naïve-to-memory CD4+ T cells (63% CD45RA+, 37% CD45RO+). Expedited whole genome sequencing identifies two novel variants in SGPL1 a paternally inherited splice site variant (c.1566+2T>C) predicted to impact a canonical splice donor site, and a maternally inherited missense change (c.854G>A; p.Cys285Tyr) located in a well-established functional domain of S1P. In addition to nephrotic syndrome and lymphopenia, the patient displays evidence of adrenal insufficiency and has increased plasma levels of sphingoid bases and ceramides. Before further analyses could be pursued, the infant dies at 40 days of age due to ongoing complications of renal failure and eventual cardiorespiratory failure.
Summary: We report the first case of S1P lyase deficiency identified by newborn TREC screening for SCID. As SGPL1 is not included in most commercially-available, SCID-tailored gene panels, S1P lyase deficiency would be missed by conventional genetic testing. Therefore, analysis for variants in SGPL1 should be considered in neonates with low-to-undetectable TREC counts, nephrotic syndrome, and other suggestive sequelae.
(177) Submission ID#605673
Case of WHIM Syndrome with Unique CXCR4 Variant
Hassan A. Ahmad, MD1, G. Wendell Richmond, MD2
1Allergy/Immunology Fellow, Rush University Medical Center
2Allergist/Immunologist, Rush University Medical Center
Introduction/Background: WHIM Syndrome (warts, hypogammaglobulinemia, recurrent infections, and myelokathexis) is a rare autosomal dominant primary immunodeficiency. It is caused by a defect in the gene encoding the chemokine receptor CXCR4. This receptor, along with the associated ligand CXCL12, regulates leukocyte migration. We present the case of a 40-year-old female, who presented after she self-identified the signature signs of WHIM syndrome in herself and multiple family members.
Objectives: We present the case of a 40-year-old female who presented with a history of recurrent warts, leukopenia of unknown cause, and recurrent infections as a child. As a child, she experienced multiple ear and sinus infections, along recurrent warts on her upper and lower extremities that have persisted to this day. Furthermore, during a routine examination when she was 14-years-old, she had a complete blood count drawn significant for leukopenia. No further workup was undertaken at that time. When continued leukopenia was noted at the age of 30, referral to a hematologist and a bone marrow biopsy was completed. Bone marrow was significant for myelokathexis with borderline hypercellular marrow for patient age (80% cellularity), and normal cell line quantity. A trial of neupoegen was undertaken, without significant improvement. Her family history is significant for father and brother with both leukopenia and recurrent warts.
Results: Genetic analysis showed a heterozygous pathogenic variant in the CXCR4 gene, C.1012_1015dup (p.Ser339Phe fs*6). Recent complete blood count was significant for a total WBC count of 1.0 K/uL, with a differential consisting of 30% neutrophils and 57% lymphocytes. Lymphocyte subsets were significant for quantitatively low CD3+, CD8+ and CD19+ subsets, with normal numbers of CD4+ and NK cells. Immunoglobulin levels revealed an IgG of 835 mg/dL, IgA of 145 mg/dL, and IgM of 54 mg/dL; IgG anti-diphtheria and tetanus titers were protective, however, none of the 23 S. pneumoniae serotype titers were > 1.3 ug/mL. Mitogen (PHA, ConA and PWM) and antigen (Candida and tetanus) stimulation of lymphocytes were normal for all stimuli.
Conclusions: We present the case of a 40-year-old female with a history of recurrent infections, warts, and myelokathexis. On genetic analysis, she is noted to have a pathogenic mutation of the CXCR4 gene. The substitution of a phenylalanine for a serine decreases one of the seven serine phosphorylation sites in the carboxy tail of the molecule that occurs upon binding to its ligand, CXCL12 (SDF1). Additionally, the variation generates a premature stop condon terminating the remainder of the carboxy terminal amino acids including Ser346-7, known to have a role in carboxy terminial beta-arrestin binding. Failure to generate adequate beta-arrestin binding sites leads to prolonged CXCR4 CXCL12 interaction resulting in myelokathexis.
(178) Submission ID#605697
Low Dose Azithromycin Prophylaxis Reduces Respiratory Exacerbations in Patients Affected by Primary Antibody Deficiencies : A Multicenter, Double-blind, Placebo-controlled Randomized Clinical Trial
Cinzia Milito, MD, PhD1, Federica Pulvirenti, MD, PhD2, Francesco Cinetto, MD, PhD3, Maria Carrabba, MD, PhD4, Giovanna Fabio, MD, PhD4, Andrea Matucci, MD5, Giuseppe Spadaro, MD6, Baldassarre Martire, MD7, Alessandro Plebani, MD, PhD8, Carlo Agostini, MD, Ph9, Stefano Tabolli, MD, PhD10, Isabella Quinti, MD, PhD10
1Department of Molecular Medicine Sapienza University of Rome
2Department of Molecular Medicine Sapienza University of Rome, Rome, Italy
3Clinical Immunology- Padova Univ. Hospital, Dpt. of Medicine-DIMED, Padua, Italy, Padua, Italy
4Fond. IRCCS Ca' Granda Ospedale Maggiore Policlinico, Dpt of Internal Medicine, Milan, Italy, Milan, Italy
5Immunoallergology Unit- Policlinico di Careggi- Dpt. of Biomedicine, Firenze, Italy
6Allergy and Clinical Immunology- Univ. of Naples Federico II-, Dpt. of Translational Medical Sciences, Naples, Italy
7Pediatric Hospital, Bari, Italy
8Pediatrics Clinic and Institute for Molecular Medicine A. Nocivelli- Univ. of Brescia, Dpt. of Clinical and Experimental Sciences-, Brescia, Italy, Brescia, Italy
9Clinical Immunology- Padova Univ. Hospital, Dpt. of Medicine-DIMED, Padua, Italy, Padua, Italy
10Department of Molecular Medicine Sapienza University of Rome, Rome, Italy
Background: Lacking protective antibodies, patients with Primary Antibody Deficiencies (PAD) suffer from frequent respiratory infections leading to chronic pulmonary damage. Macrolides prophylaxis has been proven effective to successfully manage chronic lung diseases as cystic fibrosis, bronchiectasis, COPD. We conducted a trial to evaluate the efficacy and safety of orally low-dose azithromycin prophylaxis when added to the usual care in PAD patients.
Methods: A 3-year, phase II, prospective, multicenter, randomized, double-blind, placebo-controlled trial on PAD patients (age 18-74 years) with chronic infection-related pulmonary disease. Patients received azithromycin 250 mg or placebo once daily three-times a week for 24 months. The primary outcome was the decrease of annual episodes of respiratory exacerbations. Secondary endpoints included: time to the first exacerbation, number of hospitalizations, additional doses of antibiotics, Health Related Quality of Life measures, and safety.
Results: Forty-four patients received azithromycin and 45 patients received placebo. The mean number of exacerbations was 3·6 per patient-year (95%CI 2·5-4·7) in the azithromycin arm, and 5·2 (95%CI 4·1-6·4) in the placebo arm (p=0·02). In the azithromycin group the HR for having an acute exacerbation was 0·5 (95%CI 0,3-0·9, p=0,03) and the HR for hospitalization was 0.5 (95%CI 0,2-1·1) (p=0·04). The rate of additional antibiotic treatment per patient-year was 2·3 (95%CI 2·1-3·4) in the intervention and 3·6 (95%CI 2·9-4·3) in placebo groups (p=0·004). Improvement in HRQofL was observed in intervention group. Azithromycins safety prole was comparable with placebo.
Conclusion: In PAD with respiratory exacerbation, azithromycin prophylaxis led to reduction of exacerbation episodes, of additional courses of antibiotics, and of risk of hospitalization. Given the deleterious effects of respiratory diseases adding azithromycin to PAD treatment should be considered as a valuable option.
(179) Submission ID#605776
Severe Necrotic Reaction to 23-valent Polysaccharide Pneumococcal Vaccine in a Patient with STAT3 Deficiency
Mervin C. Piñones, MD1, Cecilia Vizcaya, MD2, Guillermo Matamala3, Rodrigo Hoyos, MD4, Arturo Borzutzky, MD4
1Pediatric Immunology And Rheumatology Fellow, Pontificia Universidad Católica de Chile
2Pediatric Infectious Disease Specialist, Pontificia Universidad Catolica de Chile
3Biotechnology, Biotechnologist at Immunology Laboratory, Pontificia Universidad Catolica de Chile
4Pediatric Immunologist, Pontificia Universidad Catolica de Chile
Background: The autosomal-dominant hyper-IgE syndrome (HIES), is a primary immunodeficiency caused by mutations in signal transducer and activator of transcription 3 (STAT3) that leads to defective Th17 immunity. Adverse reactions following 23-valent pneumococcal polysaccharide vaccine (PPSV23) have been reported in 75% of STAT3-HIES patients, including severe local reactions that appear to be specific to this vaccine.
Case report: We present the case of a six-year-old girl, second child of non-consanguineous healthy parents, that developed an extensive inflammatory skin reaction at the vaccination site following a single dose of PPSV23. The vaccine was prescribed due to history of recurrent respiratory tract infections and an incomplete vaccine calendar with no previously administered pneumococcal vaccines. The reaction began after 2 hours with local erythema and edema at vaccination site, expanding in 48 hours to a phlyctenular lesion with no well-defined borders. Within the first 3 weeks, it progressively evolved to a deep necrotic lesion that required surgical debridement. The subsequent skin defect required surgical repair with a split-thickness skin graft from her right thigh as the donor site. The complete wound healing process took about 5 months, leaving a large scar (figure).
The patient had a longstanding history of recurrent infections with multiple hospitalizations including severe neonatal pneumonia that required respiratory support, a colon perforation with secondary peritonitis and septic shock that required a hemicolectomy at 8 months of age, recurrent oral candidiasis, recurrent pneumonias of different lobes, recurrent acute otitis media, a cervical phlegmon, three episodes of dental abscess and multiple kidney abscesses due to Gram-negative bacteria treated with intravenous antibiotics and surgical drainage. Family history is notable for an older sibling that died due to sudden infant death syndrome. The patients mother has large and wide nose suggestive of STAT3-HIES phenotype, but no history of infections. Immunological work up showed mild eosinophilia (850 cells/mm3), elevated IgE (1850 mg/dl), normal IgG, IgA, IgM and lymphocyte subsets (CD3, CD4, CD8, CD16, CD56). Peripheral Th17 cells were markedly decreased (0.6% vs. 3.7% of normal control). Specific pneumococcal antibodies evaluated 1 month after PSV23 revealed 5/10 serotypes in protective levels. High resolution thorax CT showed multilobar bronchiectasis. Echocardiogram and total spine x-rays were normal. STAT3-HIES was suspected with a National Institutes of Health score of 40. A novel heterozygous missense variant in STAT3 affecting the SRC homology 2 (SH2) domain (p.Lys591Glu) was found by next-generation panel sequencing. A variant in the same position (p.Lys591Met) has been previously reported in a HIES patient (ClinVar). Currently, she is on monthly IVIG and prophylactic antibiotics (cotrimoxazole, azithromycin and fluconazole).
Conclusions: The case presented raises awareness on the risk of severe local adverse reactions to PPSV23 in STAT3-HIES patients. The etiology of such reactions is unclear and warrants further study. The benefits and risks of immunizing STAT3-HIES patients with PPSV23 should be weighed carefully by medical providers.
Acknowledgments. Genetic sequencing was kindly provided by Drs. Raif Geha and Janet Chou at the Division of Immunology, Allergy, Rheumatology and Dermatology, Boston Children's Hospital, Harvard Medical School.
(180) Submission ID#605833
DOCK8 Immunodeficiency in a Malay Family from Malaysia: a Family Study
Intan Juliana Abdul Hamid, MD, MMed, PhD1, Nik Khairulddin Nik Yusoff, MBBS, MRCP, FRCPCH, DTM&H2, Mariana Daud, MD, Mmed3, Siti Mardhiana Binti. Mohamad, MD, PhD4, Ilie Fadzilah Hashim, BSc, MSc, PhD5, Zarina Thasneem Zainudeen, BSc, MSc, Dphil6, Bina Menon, MBChB, MRCP, DTM&H7, Elena Sigmund, BSc, MSc, PhD8, Bodo Grimbacher, MD9, Amir Hamzah Abdul Latiff, MBBS, MMed, MRCP, FACAAI, FAAAA10, Lokman Mohd Noh, MBBS, DCH,MRCP,FRCPE,Cert.11
1Paediatric Immunologist, Primary Immunodeficiency Diseases Group, Regenerative Medicine Cluster, Institut Perubatan & Pergigian Termaju, Universiti Sains Malaysia
2Consultant Paediatric Infectious Disease, Department of Paediatrics, Hospital Raja Perempuan Zainab II, Kota Bharu, Kelantan, Malaysia
3Fellowship in Paediatric Respiratory and Sleep Medicine, Consultant Paediatric Respiratory,
Department of Paediatrics, Hospital Raja Perempuan Zainab II, Kota Bharu, Kelantan, Malaysia
4Clinical Scientist, Primary Immunodeficiency Diseases Group, Regenerative Medicine Cluster, Institut Perubatan & Pergigian Termaju, Universiti Sains Malaysia
5Scientist, Primary Immunodeficiency Diseases Group, Regenerative Medicine Cluster, Institut Perubatan & Pergigian Termaju, Universiti Sains Malaysia
6Scientist, Primary Immunodeficiency Diseases Group, Regenerative Medicine Cluster, Institut Perubatan & Pergigian Termaju, Universiti Sains Malaysia
7Consultant Paediatrician, Institut Pediatrik, Hospital Kuala Lumpur, Malaysia
8Scientist, Centre of Chronic Immunodeficiency, Universitatsklinikum Freiburg, Germany
9Scientific Director and Consultant, Centre of Chronic Immunodeficiency, Universitatsklinikum Freiburg, Germany
10Clinical Immunologist and Allergy, Allergy and Immunology Centre, Pantai Hospital, Kuala Lumpur, Malaysia
11Fellowship Immunology, Paediatric Immunologist Consultant, Hospital Kuala Lumpur, Jalan Pahang, 50586 Kuala Lumpur, Wilayah Persekutuan
Abstract (Max 500 words)
Introduction: DOCK8 deficiency is a rare primary immunodeficiency characterized by susceptibility to viral infections, atopic eczema, defective T-cell activation and TH17 differentiation, impaired eosinophil homeostasis and dysregulation of IgE. To date, there are no reported cases from Malaysia.
Objective: We aimed to describe the clinical, immunological profile and mutational analysis of three siblings of consanguineous parents, presented with Hyper-IgE and lymphopenia between the years 1998 and 2012, which were solved by mutational analysis of the second and third siblings.
Methods: Clinical data and investigation results were collated from the medical record. Scoring of the symptoms and physical examination findings using NIH score was performed. T, B, NK lymphocyte subsets and serum IgG, IgA, IgM, total IgE quantification, lymphocyte proliferation test and pneumococcal specific antibody response were performed. Mutational analyses were performed in Freiburg, Germany.
Result: Three siblings presented at different time points over a 20-year span with raised IgE levels, recurrent infections, eczema, hyper-eosinophilia and bronchiectasis. The NIH scores for hyper-IgE syndrome (HIES) ranged from 39 54. We also documented two serious infections in the siblings, which were disseminated Cryptococcus neoformans and Salmonella sp. Immunological results showed T-cell lymphopenia, defective T-cell proliferation, decreased IgM, raised IgE, hyper-eosinophilia and defective pneumococcal antibody responses present but not in all 3 siblings. We identified a large deletion in DOCK8 starting from exon 30-48 in 2 of the siblings from mutational analysis performed. We will proceed with next generation sequencing and DOCK8 protein assay in Malaysia to further characterize the defect.
Conclusion: Our on-going study is the first description of DOCK8 in a family from Malaysia. The diagnosis of DOCK8 should be suspected in cases with raised IgE levels, recurrent infections and lymphopenia, despite no warts infection in the history. This study emphasized the importance of international research collaboration and networking in solving complicated cases.
The following grants are acknowledged: 1. RUI 1.1001/CIPPT/812036 (USM) 2. BMBF 01 EO003 (Freiburg)
The authors would like to thank the Director General of Health of Malaysia for permission to publish this scientific presentation.
Key words
Primary immunodeficiency, DOCK8 deficiency, Idiopathic CD4 lymphopenia, Malaysia, South East Asia
(181) Submission ID#605966
A Family with Hypogammaglobulinemia, ACTH Deficiency, Ectodermal Dysplasia and a Novel NFKB2 Mutation
Linda Geberzahn, MD1, Marc Bienias, MD1, Angela Rösen-Wolff, MD, PhD2, Nicole Toepfner, MD3, Angela Huebner, MD4, Eva-Maria Jacobsen, PhD5, Mingyan Fang, PhD6, MinAe Lee-Kirsch, MD, PhD7, Joachim Roesler, MD8, Catharina Schuetz, MD, MSc9
1Resident, Department of Pediatrics, Medizinische Fakultät Carl Gustav Carus, Technische Universität Dresden, Dresden, Germany
2Senior Scientist, Department of Pediatrics, Medizinische Fakultät Carl Gustav Carus, Technische Universität Dresden, Dresden, Germany
3Resident, Department of Pediatrics, Medizinische Fakultät Carl Gustav Carus, Technische Universität Dresden, Dresden, Germany
4Consultant, Department of Pediatrics, Medizinische Fakultät Carl Gustav Carus, Technische Universität Dresden, Dresden, Germany
5Biologist, Pediatrics, Ulm University Medical Center, Ulm, Germany
6Scientist, 3BGI Genomics, Shenzhen, China
7Senior Scientist, Department of Pediatrics, Medizinische Fakultät Carl Gustav Carus, Technische Universität Dresden, Dresden, Germany
8Senior Physician, Department of Pediatrics, Medizinische Fakultät Carl Gustav Carus, Technische Universität Dresden, Dresden, Germany
9Consultant, Department of Pediatrics, Medizinische Fakultät Carl Gustav Carus, Technische Universität Dresden, Dresden, Germany
The index patient presented at the age of 4 years with increased susceptibility to lower airway and gastrointestinal infections (hospital admissions 5x/year until puberty). She suffered from mumps and varicella disease despite immunization, as well as from recurrent local, partially destructive HSV infections. She was diagnosed with common variable immunodeficiency (CVID) at age 13 and started on immunoglobulin replacement therapy. Following a hypoglycemic seizure at age 20, the patient was diagnosed with isolated ACTH insufficiency with secondary adrenal insufficiency requiring hormone substitution. During and following her first pregnancy at age 25, she suffered from recurrent bronchopneumonias including Pneumocystis jirovecii infection, resulting in bronchiectases documented on chest CT at age 30. Currently, chronic lung disease is severely limiting her quality of life (Table 1). Her daughter was noticed to be hypogammaglobulinemic soon after birth and failed to develop antibody responses to inactivated vaccines. She was started on immunoglobulin replacement therapy. She has not suffered from severe lower airway infections, but developed alopecia totalis at age 10 and nail dystrophy.
Whole exome sequencing revealed a heterozygous c.2553_2554insACCCGAG (p.Lys855ProfsTer33, NM_001077494) mutation in exon 22 of NFKB2 in both mother and daughter. This monoallelic loss-of function frameshift mutation was not found in gnomad, GVS Washington or ClinVar databases. As previously published, a monoallelic mutation in this c-terminal domain leads to impaired phosphorylation and subsequent reduced nuclear translocation of the NFKB2/p52 active form. Pediatricians and internal specialists need to be aware of the combination of hypogammaglobulinemia, ACTH deficiency, immune dysregulation and ectodermal dysplasia which is unusual for CVID - possibly indicating NFKB2 deficiency. This clinical syndrome may overlap with symptoms and signs found in both APECED/AIRE (AR) and EDA-ID/NFKBIA (AD) deficiencies. Besides IG and hormone replacement therapy, curative treatment with hematopoietic stem cell transplantation is a therapeutic option for patients with NFKB2 deficiency, although the experience is limited.
Table 1. Clinical and immunological features of index patient and daughter
| viral infections | bacterial infections | end organ damage immune dysregulation | immunology | |
| mother | mumps and varicella disease (despite immunization), recurrent HSV1 infections (face, index finger) |
tracheobronchitis (H. influenza) P. jirovecii pneumonia |
bronchiectases, COPD, loss of distal phalanx (Dig.II) following HSV infection, ulceration of vocal chords, hypopituitarism, vasculitis of lower legs | IgG 3.44 g/l, IgA < 0.26 g/l, IgM 0.45 g/l, no protective tetanus and diphtheria antibodies following regular immunization, absence of autoantibodies, initally low switched memory B- cells, total loss of peripheral B-cells |
| daughter | recurrent upper and lower respiratory infections, recurrent HSV1 infections periorally | mild restrictive lung disease, alopecia totalis, nail dystrophy |
IgG 2.33 g/l, IgA 0.23 g/l, IgM 0.34 g/l, B-cells 1900/μl, no protective tetanus, measles or HiB antibodies (but positive to rubella) |
(182) Submission ID#605974
Infection Rates and Tolerability in Patients with Primary Immunodeficiency Diseases Treated with Three Different Immunoglobulin Administration Modalities
Richard L. Wasserman, MD, PhD1, Sudhir Gupta, MD2, Mark R. Stein, MD3, Christopher J. Rabbat, PhD4, Werner Engl, PhD5, Heinz Leibl, PhD6, Leman Yel, MD7
1Allergist/immunologist, Allergy Partners of North Texas Research, Dallas, TX, USA
2Professor, University of California at Irvine, Irvine, CA, USA
3Physician, Allergy & immunology, Allergy Section, Good Samaritan Medical Center, West Palm Beach, FL, USA
4Director of Medical Affairs, Shire, Chicago, IL, USA (Affiliation at the time of the study)
5Assoc Dir Biostatistics, Pharmacometrics & Pre-Clinical Biostatistics, Shire, Vienna, Austria
6Sr Medical Director, Global Development Leader, IG, Clinical Research Immunology
Shire, Vienna, Austria
7Sr Medical Director, Global Development Leader, IG, Clinical Research Immunology
Shire, Cambridge, MA, USA
Introduction: The modes of immunoglobulin (IG) administration for primary immunodeficiency diseases (PIDD) differ in pharmacokinetics, infusion parameters, and tolerability. During 3 consecutive clinical studies, a cohort of 30 patients with PIDD experienced all 3 modes of administration with the same IG 10% product in sequence from intravenous (IV) to subcutaneous (SC), then to hyaluronidase-facilitated SC (IGHy), providing a unique opportunity to assess each administration modality within the same patient cohort treated and observed at the same sites. Here we report the rates of infections stratified by IgG trough levels, and the rates of adverse events (AEs) with the 3 modes of IG administration (IVIG, SCIG, IGHy) within this patient cohort.
Design and Methods: This analysis included patients with PIDD aged 4 years who participated in 3 clinical studies: in Study 1 (NCT00546871) patients received IVIG 10% every 34 weeks followed by weekly SCIG 10%; in Study 2 (NCT00814320), patients were treated with IGHy every 34 weeks; in Study 3 (NCT01175313; extension of Study 2), patients continued with the same IGHy dose. To assess a potential association between the administration route at comparable IgG trough levels and the infection rate, IgG trough levels were categorized as 500 <700mg/dL, 700<900mg/dL, 900<1100mg/dL, 1100<1300 mg/dL, 1300 <1500 mg/dL and 1500 mg/dL. Periods where patients had trough levels within these strata were assessed, and the infection frequency was calculated. The time periods for this analysis were 3 months for IVIG and 12 months each for IGHy and SCIG 10% (2.25 years) treatments. In order to account for differences in the frequency of administration, rates of systemic and local AEs were assessed as AEs/patient-year for each mode of therapy.
Results: For IgG trough levels of <1500 mg/dL, the associated annual infection rates were lower or similar for IGHy than SCIG (2.3 vs 3.5 [1300<1500 mg/dL]; 2.6 vs 3.8 [1100<1300 mg/dL]; 3.6 vs 6.2 [900<1100 mg/dL]; 1.4 vs 5.0 [700<900 mg/dL]; 2.0 vs 2.0 [500<700 mg/dL). For IgG trough levels 1500 mg/dL, the annual infection rate (95% CI) appeared to be lower for SCIG versus IGHy treatment (2.6 [1.83.7] vs 4.2 [1.210.3]) with shorter periods of observation and wide 95% confidence intervals. The rates of causally related systemic AEs/patient-year were lowest during IGHy (0.88) versus SCIG (1.93) and IVIG (5.60) treatment; the majority of causally related systemic AEs were mild. The rate of headaches/patient-year, the most common systemic AE, was lowest during IGHy treatment (0.21) versus SCIG (0.45) and IVIG (1.95). The rates of causally related local AEs/patient-year for IGHy, SCIG, and IVIG were 1.57, 0.92, and 0.13, respectively.
Conclusion: Evaluation of the patient cohort in 3 consecutive studies over 2.25 years resulted in lower rates of causally related systemic AEs per patient-year during the IGHy treatment period compared with SCIG. Lower or similar infection rates were found during the IGHy treatment period compared with SCIG treatment at the same IgG trough levels (<1500 mg/dL). This observation warrants further investigation.
(183) Submission ID#606027
Syntaxina11 Mutation with Normal CD107a Surface Expression
Carolina Dorfman, MD1, Agostina Llarens, MD1, Daniela Di Giovanni, MD2, Gisela Seminario, MD3, Andrea Gomez Raccio, MD3, Guadalupe Rodruiguez Broggi, MSc4, Patricia Carabajal, MD5
1Physician – Immunology trainer, Children's Hospital Ricardo Gutierrez
2Physician – Immunologist, Hospital de Niños Ricardo Gutierrez
3Physician – Immunologist, Children's Hospital Ricardo Gutierrez
4Biochemist, Hospital de Niños Ricardo Gutierrez
5Head of Immunology Unit, Children' s Hospital Ricardo Gutierrez
Introduction: Familial hemophagocytic lymphohistiocytosis (FHL) is a severe immune dysregulatory syndrome, inherited in an autosomal recessive way, caused by an impaired T and natural killer (NK) cell cytotoxicity. This results in an uncontrolled T cell and macrophage activation with hypercytokinemia.
Syntaxin 11 (STX11) is one of the known genes of FHL. It is expressed in monocytes, NK cells and cytotoxic T cells and is involved in vesicle priming and membrane fusion.
The clinical manifestations are fever, hepatosplenomegaly and cytopenias. Neurological features usually present later and are associated with poor prognosis. The disease is often triggered by infections, most commonly viral.
Cytopenias, hyperferritinemia, hypertriglyceridemia and hypofibrinogenemia are usually present, along with CD25 soluble in blood and cerebrospinal fluid. Bone marrow may demonstrate hemophagocytosis. Specific functional tests can be done to make an approach to the primary defect. CD107a is present on the membrane of secretory granules within T and NK cells. Absence of CD107a expression on the cell surface after activation, suggests a defect in secretory granule migration, docking, priming or fusion.
The treatment involves the control of infections and immune dysregulation with chemotherapeutic regimens followed by definitive treatment with hematopoietic stem cell transplant (HSCT).
Aim: To describe a female patient with a pathogenic variation in STX11 with normal CD107a expression.
Results: She was a 2 years old female, the 5th daughter of non-consanguineous parents, without relevant personal or family records. She was admitted due to a prolonged febrile syndrome, lymphoproliferation, pancytopenia and hepatitis, with HHV6 rescued in bone marrow and blood. Gancyclovir treatment started with good response. She was admitted one month later with similar clinical symptoms with relapsed HHV6 infection. Furthermore, hemophagocytosis was found in the bone marrow and evaluation of NK cell cytotoxicity demonstrated slightly reduced cytotoxic activity. Functional studies for primary FHL were performed: perforin expression and CD107a surface expression were normal. She fulfilled criteria of FHL, and treatment with gancyclovir and steroids was administered. Despite this treatment, she persisted with activated macrophagic parameters, and started with HLH2014 treatment protocol. She improved the clinical symptoms and laboratory parameters, but persisted with HHV6 low viremia. Three months later, when immunosupression was decreased, she was readmitted with similar clinical manifestations and added neurological symptoms (facial paralysis, abnormal movements and sleep tendency). Cerebral spinal fluid was pathological with HHV6 positive rescue. Immunosupresive treatment was adjusted, but HHV6 copies in blood increased markedly. Foscarnet treatment was administered and immunosupression was suspended for 2 days in order to control viral infection. Unfortunately the patient died 6 days later.
Although specific functional tests were normal, sequencing of STX11 gene by NGS revealed a homozygous variation in c.581_584delTGCC, which is a previously reported mutation responsible for FHL.
Conclusion: Despite the fact that CD107a was normal, the strong clinical and laboratory results must keep the FHL diagnosis in mind and intensive treatment should be early administered; in order to give the patient the opportunity to achieve the curative treatment.
(184) Submission ID#606063
Specific Functional Gammopathy Underlying Infectious Susceptibility in a Patient with Autoimmune-Polyendocrinopathy-Candidiasis-Ectodermal Dystrophy (APECED)
Gregory M. Constantine, MD1, Elise Ferre, PA-C, MPH2, Michail Lionakis, MD, Sc.D.3
1Clinical Fellow, Fungal Pathogenesis Section, Laboratory of Clinical Immunology & Microbiology (LCIM) , National Institute of Allergy & Infectious Diseases (NIAID), NIH
2Physician Assistant, Fungal Pathogenesis Section, Laboratory of Clinical Immunology & Microbiology (LCIM) , National Institute of Allergy & Infectious Diseases (NIAID), NIH
3Senior Investigator, Fungal Pathogenesis Section, Laboratory of Clinical Immunology & Microbiology (LCIM) , National Institute of Allergy & Infectious Diseases (NIAID), NIH
Introduction/Background: Autoimmune-Polyendocrinopathy-Candidiasis-Ectodermal Dystrophy (APECED) is a rare inherited primary immunodeficiency disorder resulting from biallelic mutations in the AIRE gene. Although classically characterized by chronic mucocutaneous candidiasis (CMC), hypoparathyroidism and primary adrenal insufficiency (Addisons Disease), recent evidence has demonstrated broader disease manifestations including: urticarial-like eruption, autoimmune hepatitis, intestinal dysfunction, gastritis, Sjogrens-like syndrome and pneumonitis. Clinical observations have also identified a subset of individuals who suffer from recurrent sinopulmonary infections.
Objectives: To report and characterize the clinical course of a patient with APECED and specific antibody deficiency.
Methods: Retrospective chart review was performed. The patient was enrolled in NIAID IRB-approved protocol 11-I-0187.
Results: The patient is a 13 year-old-girl with APECED caused by homozygous AIRE c.967_979del13, who manifested CMC, hypoparathyroidism, adrenal insufficiency, Sjogrens-like syndrome, autoimmune hepatitis, intestinal dysfunction and autoimmune pneumonitis. She suffered from recurrent sinusitis and severe pneumonias requiring hospitalization and administration of intravenous antibiotics several times per year. At age 9, she presented to our institution with fever and cough, a computed tomography (CT) of the chest revealed bilateral pulmonary infiltrates and bronchiectasis. Bronchoscopy showed mucopurulent secretions in the bilateral lower lobes with culture of the bronchoalveolar lavage fluid growing Streptococcus pneumoniae.
Further evaluation for an underlying disorder such as primary ciliary dyskinesia and cystic fibrosis including exome sequencing and sweat chloride testing was unrevealing. Quantitative immunoglobulins were normal. Despite prior vaccination, specific antibody testing showed negative rubeola IgG and protective levels (> 1.3 mcg/mL) to only 3 of 23 pneumococcal serotypes. Lymphocyte enumeration showed normal B cell subsets. As approximately 15% of APECED patients may experience asplenia, splenic ultrasound was performed confirming the presence of a 7 cm spleen and peripheral blood smear did not reveal Howell-Jolly bodies. Serotyping of the S. pneumoniae isolate confirmed serotype 33F, which is part of the 23-valent vaccine. Follow up vaccine challenge with the 23 valent pneumococcal polysaccharide vaccine showed an inadequate response. Hence, she was started on monthly immunoglobulin replacement and over the following 4 years she has experienced a single methicillin sensitive Staphylococcus aureus pneumonia. She has missed very few school days and other parameters including linear growth have improved, she is now along the fifth percentile for height and along the tenth percentile for weight. Although she continues to experience intermittent cough she remains active participating in sports without limitation.
Conclusions: We report the evaluation, treatment and outcome of a patient with APECED complicated by autoimmune pneumonitis and specific antibody deficiency. As infectious susceptibility of APECED classically pertains to the signature infectious disease, CMC, patients with invasive or recurrent infections should be evaluated for underlying immune deficiency. Investigation should include assessment for asplenia, quantitative immunoglobulins and specific antibodies with response to antigens. In patients with predominate respiratory symptoms, autoimmune pneumonitis should be evaluated given the near 40% prevalence of pneumonitis observed in American APECED patients.
Acknowledgements: Supported by DIR/NIAID/NIH
(185) Submission ID#606159
Two Siblings with a Delayed/Late-Onset Presentation of Combined Immunodeficiency Due to Adenosine Deaminase Deficiency
Tamara C. Pozos, MD, PhD1, Manar Abdalgani, MBBS2, Michael Hershfield, MD3
1Medical Director, Clinical Immunology, Children's Hospital Minnesota
2Clinical Immunologist, Children's Hospital Minnesota
3Professor of Medicine, Professor of Biochemistry, Duke University Medical Center
BACKGROUND: Complete deficiency of adenosine deaminase (ADA) is an autosomally inherited condition that causes one type of SCID secondary to the accumulation of metabolites toxic to lymphocytes. Less severe genetic changes resulting in detectable ADA result in a milder, clinically heterogeneous combined immunodeficiency.
CASE PRESENTATIONS: Patient 1: A 16 year old Somali-American girl presented with progressive pulmonary disease. Past history included recurrent severe pneumonias, eczema and recurrent Staphylococcal infections. Genetic testing for HyperIgE syndrome was negative. She developed autoimmune hypothyroidism at age 7 years and warts covering both hands at age 10 years. On presentation, she had a normal-to-elevated IgG with some therapeutic vaccine titers, normal IgM and IgA. IgE was elevated at 7381 IU/ml. B cells were essentially absent: absolute CD19+ 1cell/ul. NK cells were also low, absolute 39cells/ul. Absolute number of total T cells was normal with decreased CD4+ Tcells. Lymphocyte proliferation responses to mitogens were significantly decreased, and responses to tetanus antigen were absent.
ADA activity was deficient (0.4 nmol/h/mg) with only modestly increased red cell dAXP% (ratio of dAXP/RBC-AXP 6.3%), consistent with delayed/late onset ADA deficiency. After 17 weeks of PEG-ADA injections, dAXP% was zero. B cell counts improved though to normal levels. IgE increased after therapy began, peak 13,000IU/ml, and then slowly decreased. Her cough and dyspnea improved, though pulmonary function tests still show severe airflow obstruction.
Genetic testing revealed a homozygous missense mutation in ADA exon 6, V177M (c.529G>A), a previously identified change shown in vitro to produce 0.1-0.4% of wild type ADA activity (Arredondo-Vega, F. et al, Am J. Hum Genet 1998; 63:1049-59). Patient 1s sister is homozygous for the same mutations.
Patient 2: Though considered by family to be a healthy 11 year old, her past history included a left axillary abscess at age 2 years, eczema, recalcitrant tinea capitus, and 3 warts. She had no pulmonary disease. She had short stature. Labs revealed normal IgG, IgM and IgA. All vaccine titers were strongly therapeutic. IgE was elevated at 1210IU/ml and initially increased with PEG-ADA injections. Like her sister, B cells at presentation were nearly absent with absolute CD19+ 12 cells/ul. T and NK cells were normal. Also like her sister and their mother who is a carrier, Patient 2 had autoimmune hypothyroidism. After 7 weekly PEG-ADA injections, she had rapid decrease in percentage dAXP/total RBC AXP, from 6.3% to 1.0% and absolute B cell number increased to 92.
CONCLUSIONS: We report two siblings with identical genetic changes in ADA and identical presenting ratios of dAXP/total RBC-AXP. Both had dysgammaglobulinemia and B lymphopenia, atopy and autoimmunity on presentation. However, the 16 year old has had significantly more medical complications and has shown a slower improvement with replacement therapy compared to her younger sister.
(186) Submission ID#606161
Isolated Central Nervous System Disease in Familial Hemophagocytic Lymphohistiocytosis a Multicenter Case Series
Annaliesse Blincoe, MBChB FRACP1, Maximilian Heeg, MD2, Patrick Campbell, MD, PhD3, Amer Khojah, MD4, Marisa Klein-Gitelman, MD5, Julie-An Talano, MD6, Claire Booth, MBBs PhD7, Despina Moshous, MD, PhD8, Fabien Touzot, MD, PhD9, Arjan Lankester, MD, PhD10, Jacques Rivière, MD11, Maria Caterina Putti, MD12, Sarah Maier, MD13, Kai Lehmberg, MD14, Itziar Astigarraga, MD15, Steven M. Holland, MD16, Rebecca A. Marsh, MD17, Stephan Ehl, MD18, Elie Haddad, MD, PhD19
1Department of Pediatrics, CHU Sainte-Justine, University of Montreal, Montreal, QC, Canada
2Institute for Immunodeficiency, Center for Chronic Immunodeficiency. Center for Pediatrics and Adolescent Medicine, University of Freiburg, Freiburg, Germany
3Paediatric Haematologist-Oncologist, St. Jude Children's Research Hospital
4Department of Rheumatology, Ann and Robert H Lurie Children's Hopital and Children's Hospital of Chicago, Chicago, IL
5Division Head, Rheumatology, Professor of Pediatrics, Northwestern University Feinberg School of Medicine Chicago, IL
6Associate Professor, Pediatric Haematology and Oncology, Children's Hospital of Wisconsin-Milwaukee Campus, Milwaukee, WI
7Honorary Consultant in Paediatric Immunology, Great Ormond Street Hospital for Children, London, UK
8Paediatric Immunology, Haematology and Rheumatology Unit, Hôpital Necker Enfants Malades, Paris, France
9Department of Pediatrics, CHU Ste-Justine, University of Montreal, Montreal, QC, Canada
10Professor of Pediatrics and Stem Cell Transplantation, University of Leiden Medical Centre, Leiden, Netherlands
11Pediatric Infectious Diseases and Immunology, Vall d'Hebron University Hospital, Barcelona, Spain
12Pediatric Haematologist-Oncologist, Department of Pediatrics, University of Padua Medical School
13Pediatric Haematology and Oncology, University Medical Center Hamburg Eppendorf, Germany
14Paediatric Haematology and Oncology, University Medical Center Hamburg Eppendorf, Germany
15Pediatric Oncology, Hospital Universitario Cruces , Barakaldo, Spain
16Director, Division of Intramural Research, Laboratory of Clinical Immunology and Microbiology, National Institute of Allergy and Infectious Diseases, National Institutes of Health
17Associate Professor, Division of Bone Marrow Transplantation and Immune Deficiency, Cincinnati Childrens Hospital Medical Center
18Institute for Immunodeficiency, Center for Chronic Immunodeficiency. Center for Pediatrics and Adolescent Medicine, University of Freiburg, Freiburg, Germany
19Department of Pediatrics. Department of Microbiology, Infectiology and Immunology, CHU SainteJustine, University of Montreal, Montreal, QC, Canada
Familial hemophagocytic lymphohistiocytosis (FHLH) is a rare, inherited syndrome of immune dysregulation, characterised by genetic predisposition to a systemic hyper-inflammatory disease. Whilst central nervous system (CNS) involvement is well described in FHLH, there have been very few case reports of isolated CNS-HLH in the absence of any systemic features. This clinical presentation may be under-recognised, leading to delays in diagnosis and appropriate treatment. We present 9 new cases together with 11 cases identified in the literature, to describe the clinical presentation, treatment and outcomes.
Patients and Methods: Patients were identified and retrospective data collected through collaborative efforts at North American and European clinical immunology, haematology/oncology and bone marrow transplant centres and through review of previously reported cases in the literature. Inclusion criteria; (1) confirmed diagnosis of FHLH with pathological FHLH gene mutation; (2) any neurological symptoms at time of diagnosis; (3) absence of systemic HLH according to HLH-2004 criteria, in particular absence of bilineage cytopenia and splenomegaly; (4) features of systemic HLH fulfilling HLH-2004 diagnostic criteria no earlier than 3 months after initial neurological presentation.
Results: Nine new patients and 11 patients from the literature were identified. The mean age at time of onset of neurological symptoms was 6.2 years. The most common neurological manifestations were ataxia/gait disturbance (65%) and seizures (50%) with a mean delay from onset of neurological symptoms to confirmed FHLH of 15.8 months. Diffuse multifocal white matter changes and cerebellar involvement were the most common CNS MRI findings at 14/17 (82.4%) and 8/17 (47%) respectively. The mean CSF cell count was 15.9 x 106/L and mean CSF protein 106 mg/dL. Epstein-Barr virus was identified as a trigger in 2 patients, and varicella zoster virus in 1 patient. Seven patients eventually developed systemic HLH manifestations at a mean time of 17.4 months (range 4-28 months) after initial neurological symptoms. Mutations in PRFI were detected in 15 patients (75%), RAB27a mutation in 3 (15%) and UNC13D in 2 (10%). Six patients carried the R225W mutation in perforin.
Nineteen patients received systemic HLH-directed chemo/immunosuppressive therapy. Two patients received intrathecal methotrexate and hydrocortisone. Of the 13 patients treated with HSCT (7 new, 6 literature), 7 patients (53.8%) had improved neurological outcome, 3 patients (23.1%) had stable neurological findings, and one patient had CNS relapse at time of follow-up. There were no cases of neurological improvement in patients who did not receive HSCT. The overall mortality was 35% (2 new and 5 literature patients).
Conclusion: Our study describes the variable clinical presentation and findings in patients with isolated CNS-HLH. We propose that genetic cytotoxicity defects should be considered in any patient with unexplained and progressive neurological symptoms, even in the absence of ANY signs of systemic inflammation. Neurological improvement was observed in patients who received HSCT, emphasising the need for timely institution of treatment in order to improve outcomes. Our study also identified a predominance of PRF1 mutations; however, larger studies would be required to further evaluate the significance of this finding.
(187) Submission ID#606194
Lupus-like Syndrome in a Patient with NOD2-associated Autoinflammatory Disease
Lyda Cuervo-Pardo, MD1, Mario Rodenas, MD, FAAAAI1, Mingjia Li2, Shuhong Han, Ph.D.3, Haoyang Zhuang, Ph.D.3, Westley Reeves, MD4
1Assistant Professor, University of Florida, Division of Rheumatology & Clinical Immunology, Department of Medicine
2Undergraduate Student, Division of Rheumatology, Allergy, & Immunology, University of Florida
3Assistant Professor, Division of Rheumatology, Allergy, & Immunology, University of Florida
4Professor and Chief, University of Florida, Division of Rheumatology & Clinical Immunology, Department of Medicine
Introduction: Autoinflammatory diseases are genetically heterogeneous disorders of innate immunity characterized by recurrent fever, rash, and/or serositis, which generally are considered distinct from autoimmune diseases. We report a case of a patient with lupus-like disease and a mutation of nucleotide-binding oligomerization domain-containing protein 2 (NOD2 R702W, Yao syndrome) suggestive of an overlap between autoinflammatory and autoimmunity processes.
Case Presentation: A 72-year-old man was evaluated for recurrent pleural effusions, morning stiffness, erythematous rashes, and fever up to 103°C. History was notable for Hashimotos thyroiditis and multiple admissions for presumed pneumonia with recurrent bilateral lung infiltrates and pleural effusions. Transbronchial biopsy showed nonspecific pneumonitis and organizing pneumonia. Antinuclear and anti-dsDNA antibodies were positive. He received prednisone for presumed lupus pneumonitis leading to improvement. Prednisone was tapered and hydroxychloroquine was started, but his fevers, pleuritic pain and pleural effusion reoccurred. Genetic testing revealed a NOD2 sequent variant (R702W) associated with autoinflammatory disease. Hydroxychloroquine was stopped and colchicine was added to his regimen, allowing prednisone to be tapered without recurrence of symptoms. Further immunological testing revealed increased signaling through the type I interferon receptor (interferon signature).
Conclusion: Although this patient had several clinical (serositis, arthralgia) and immunological (antinuclear and anti-dsDNA antibodies, interferon signature) manifestations of lupus, his clinical presentation also was consistent with Yao syndrome. In retrospect, he had been having recurrent inflammatory symptoms for many years. Recent studies in both mice and humans suggest that inflammasome activation and IL-1 production are involved in the pathogenesis of lupus. This case provides further support for the idea that lupus and Hashimotos thyroiditis, prototypical autoimmune diseases, may have overlapping autoinflammatory features.
(188) Submission ID#606243
Immunological and Genetic Outcomes of Infants with Positive Newborn Screening for Severe Combined Immunodeficiency (SCID)
Vasudha Mantravadi, MD1, Jeffrey J. Bednarski, MD, PhD2, Michelle A. Ritter, RN3, Megan A. Cooper, MD, PhD4, Maleewan Kitcharoensakkul, MD5
1Resident Physician, Department of Pediatrics, Washington University School of Medicine, St. Louis, Missouri, 63110
2Assistant Professor of Pediatrics, Hematology and Oncology, The Division of Hematology and Oncology, Department of Pediatrics, Washington University School of Medicine, St. Louis, Missouri, 63110
3Research Nurse Coordinator, The Division of Pediatric Rheumatology, Department of Pediatrics, Washington University School of Medicine, St. Louis, Missouri, 63110
4Associate Professor, Rheumatology, The Division of Pediatric Rheumatology, Department of Pediatrics, Washington University School of Medicine, St. Louis, Missouri, 63110
5Assistant Professor of Pediatrics, Rheumatology and Allergy, Immunology and Pulmonary Medicine, The Division of Allergy, Immunology & Pulmonary Medicine, Department of Pediatrics, Washington University School of Medicine, St. Louis, Missouri, 63110
Background: The implementation of severe combined immunodeficiency (SCID) newborn screening by TREC assay has played a pivotal role in identifying these patients early in life. The screen has also led to the identification of infants with other immunologic abnormalities, of which the clinical implications have been unclear and there are limited data on their outcomes.
Objective: To review immunologic and genetic outcomes of infants referred to an immunology service of a tertiary care center with abnormal newborn SCID screens.
Methods: We retrospectively reviewed charts of infants with positive SCID screen from July 2014 to November 2018. We excluded patients who had positive screen at <36 weeks corrected gestational age. We classified outcomes into 3 groups including SCID, non-SCID T-cell lymphopenia (NSCID-TCL) and normal T-cell count. Idiopathic T-cell lymphopenia was defined as NSCID-TCL (CD3+ < 2,500 cells/mcl) with negative chromosome microarray and negative whole exome sequencing/or genetic panel (either GeneDx® SCID panel or Invitae® Primary Immunodeficiency panel).
Results: Of 119 infants, 78% were male, 56% were Caucasian, and 37% were African-American. Fifty-four % and 46% of infants were identified by Illinois and Missouri screens, respectively. The mean age at initial evaluation was 22 days (4-122 days). 69% of infants had a normal T-cell count (N=80) or normal repeat newborn screen (N=2), 25% had NSCID-TCL, including mild (CD3+ 1,500-2,500 cells/mcl, N=20) and moderate (CD3+ 300-1,500 cells/mcl, N=10) TCL, and 6% had SCID (N=5), leaky SCID (N=1) or complete DiGeorge (N=1). Genetic etiologies of NSCID-TCL included 22q11 deletion (N=4), trisomy 21 (N=1), and mutations of TBX1 (N=2), FOXN1 (N=1), and CD3E (N=1). Three of these infants had novel variants at the time of diagnosis. Secondary causes of TCL were identified in 1 infant (thoracic infantile fibrosarcoma). One infant had idiopathic TCL. Eighteen infants with NSCID-TCL were followed clinically without complete genetic testing performed. For SCID, mutations were found in JAK3 (N=2), ADA (N=1), IL2RG (N=1), and RAG1 (N=1). The patient with leaky SCID had negative whole exome sequencing. All patients with SCID and leaky SCID underwent hematopoietic stem cell transplantation at a median age of 5 weeks (3 weeks - 4 months), with successful engraftment in all but 1 patient. Of 19 idiopathic and NSCID-TCL cases followed clinically, 12 had at least one follow-up visit at median age 5 months (2.6 months 2.2 years) and the majority had improved or stable lymphocyte count without serious infections requiring intravenous antibiotics, though 1 had a hospitalization for RSV infection. The MYSM1 patient died after cord blood transplant from unclear etiology. Our study had limitations. Half of infants with NSCID-TCL did not have a complete genetic workup, and only a fifth of patients with NSCID-TCL were inpatients, potentially explaining the relatively low number of infants with secondary lymphopenia.
Conclusions: In our cohort, one-fourth of infants with abnormal SCID screen had NSCID-TCL. Although the majority of NSCID-TCL did well, approximately one-third of them had underlying genetic abnormalities associated with their T-cell lymphopenia.
(189) Submission ID#606320
Nucleus-retained WASP Is Deleterious to T-cell Development
Carole Le Coz, PhD in immunology1, Tanner Robertson2, Caroline Khanna3, Andrew Sy, MD4, David K. Buchbinder, MD, MSHS5, Janis Burkhar, PhD6, Neil Romberg, MD7
1Postdoc, Children's Hospital of Philadelphia
2PhD student, University of Pennsylvania
3Research Assistant, Children's Hospital of Philadelphia
4Fellow, Children's Hospital of Orange County, Orange, CA, Department of Pediatrics, University of California at Irvine, Orange, CA
5Assistant Clinical Professor, Department of Hematology, Children's Hospital of Orange County, Orange, CA, Department of Pediatrics, University of California at Irvine, Orange, CA
6Professor, University of Pennsylvania Perelman School of Medicine
7Assistant Professor, Children's hospital of Philadelphia
Cytosolic Wiskott-Aldrich Syndrome protein (WASp) regulates actin cytoskeleton reorganization but also enters the nucleus to affect gene transcription. Mice and humans without WASp develop normal numbers of naïve T cells but are susceptible to infections by viruses and encapsulated bacteria. Although opportunistic mycobacterial infections are reported in severe combined immune deficiencies (SCID) and more specific Th1-associated monogenic diseases, they have not been previously reported in WASp-deficient patients.
Here we report two unrelated kindreds presenting with opportunistic mycobacterial infections (M. bovis and M. gordonea) that carry nearly identical mutations in the gene encoding WASp (WAS). In one kindred affected males presented as infants with T-B+NK+ SCID, thrombocytopenia and eczema. In the other kindred affected males presented with abundant but dysfunctional T cells. Thrombocytopenia and eczema were present in both groups. In each case mutant WAS encoded truncated WASp lacking its cofilin and the acidic domains (WASp CA). In addition to an inability to normally polymerize actin, primary patient cells retained WASp CA entirely within their nuclei. There it appeared conformationally open and constitutively active even in the absence of stimulation. These data suggest conformationally open WASP retained in the nucleus of T cells alters transcriptional programs leading to new and severe infectious phenotypes.
(190) Submission ID#606328
CVID Plasma Promotes Early Commitment to the Follicular Lineage
Caroline Khanna1, Carole Le Coz2, Neil Romberg, MD3
1Research Assistant, Children's Hospital of Philadelphia
2PhD in Immunology, Postdoc, Children's Hospital of Philadelphia
3Assistant Professor, Children's hospital of Philadelphia
Background: Many CVID patients with undetectable serum IgA concentrations are endotoxemic and possess enlarged pools of circulating T follicular helper (Tfh) cells.
Objective: To determine if endotoxemia and other plasma-soluble factors promote follicular T cell differentiation.
Methods: We cultured healthy donor (HD) naïve CD4+ T cells for 5 days in CVID patient plasma, CVID plasma treated with polymyxin B to neutralize endotoxin, or fetal bovine serum spiked with LPS. After 5 days we measured the frequency of cells expressing the Tfh markers CXCR5 and PD1. We also measured concentrations of cytokines known to promote Tfh differentiation, comparing levels in CVID patient plasma samples that induced expression of Tfh markers with samples that did not induce them.
Results: We found greater frequencies of HD naïve CD4+ T cells expressed Tfh markers when cultured in plasma from IgA deficient CVID patients, than IgA sufficient patients or healthy donors. These differences disappeared upon addition of polymyxin B to patient plasma samples and could be recapitulated by replacing plasma with LPS spiked FBS.
In addition to LPS, patient plasma samples that best promoted Tfh marker expression contained higher concentrations of Activin A but not the traditional Tfh-differentiating cytokines IL-12 and IL-6.
Conclusion: Endotoxin, which circulates in IgA deficient CVID patients, promotes naïve T-cell commitment to the follicular lineage directly and through enhanced release of Activin A.
(191) Submission ID#606365
Infusion Parameters of Patients with Primary Immunodeficiency by Previous Immunoglobulin Routes of Administration Among Enrollees in a Patient Program Initiated on Ig20Gly
Lisa Meckley, PhD1, Yanyu Wu, PhD2, Spiros Tzivelekis, MSc3, Andre Gladiator, PhD4,
1Director, GHEORE, Shire
2Lead, Health Economics and Outcomes Analytics, Shire
3ORE Lead, ORE Immunology & Opthalmolgy, Shire
4Global Medical Lead Immunology - Global Medical Affairs, Shire
Rationale: HCUVP is a patient product-introduction program that provides free-of-charge Cuvitru® (immune globulin subcutaneous [human], 20% solution; Ig20Gly) for the first 4 infusions to eligible patients who have primary immunodeficiency disease (PID). Using data from the ongoing HCUVP, this analysis described the clinical characteristics and infusion parameters of patients initiated on Ig20Gly based on their previous immunoglobulin (IG) routes of administration.
Methods: Patients 2 years of age who had a primary ICD-10 diagnosis code of PID and had no current or prior use of Ig20Gly at the time of program enrollment were eligible for HCUVP. This analysis included data from patients enrolled in HCUVP who received the first Ig20Gly infusion between January 1, 2017 and September 1, 2017. Infusions after October 31, 2017 were excluded. Descriptive statistics were calculated for demographic, clinical, and prescribed infusion characteristics for patients previously treated with intravenous IG (IVIG) and subcutaneous IG (SCIG), respectively.
Results: A total of 420 patients who had previously been treated with IVIG or SCIG completed all 4 infusions and were eligible for this analysis. Prior to enrolling in HCUVP, 268 patients had received IVIG, of whom 46 patients (17%) were < 18 years old, and 152 patients had received SCIG, of whom 17 patients (11%) were < 18 years old. A greater percentage of patients who switched from SCIG infused biweekly (54 of 152, 36%) compared with those who switched from IVIG (64 of 268, 24%). The mean dose administered during the final infusion was higher in patients who previously received SCIG compared with those who previously received IVIG (15.3 g vs 14.5 g, respectively), despite the SCIG group having a lower mean weight (SCIG: 74.1 kg [SD 23.2]; IVIG: 77.2 kg [27.2]) and fewer patients < 18 years. (Both weight and age can influence dosage). The mean number of infusion sites per infusion (SCIG: 2.1; IVIG: 2.0), mean infusion rate per site (SCIG: 41.5 mL/hr/site; IVIG: 42.9 mL/hr/site), and mean infusion volume per site (SCIG: 37.6 mL/site; IVIG 37.1 mL/site) were comparable between both groups. By the third of a total of 4 infusions, 91% and 94% of patients reached their maximum infusion rate when switching from IVIG or SCIG to Ig20Gly, respectively.
Conclusion: Among patients enrolled in an Ig20Gly product-introduction program, those who had previously received SCIG were more likely to infuse biweekly and receive a slightly higher mean dose during the final infusion than patients who had previously received IVIG. However, the number of infusion sites, infusion rates per site, infusion volume per site, and percentage of patients reaching their maximum infusion rate were similar regardless of patients previous routes of IG administration.
Funding: This research was sponsored by Shire.
(192) Submission ID#606375
A Literature Review on Shared Decision-Making (SDM) to Inform the Development of an SDM Tool in Primary Immunodeficiency Diseases
Lisa Meckley, PhD1, Ihor Sehinovych, PharmD2, Spiros Tzivelekis, MSc3
1Director, GHEORE, Shire
2Medical Lead – HyQvia, Shire
3ORE Lead, ORE Immunology & Opthalmolgy, Shire
Background: Shared decision-making (SDM) is an interactive process that allows patients and their physicians to choose treatments that align with patients preferences. For patients with primary immunodeficiency diseases (PIDs) who require immunoglobulin replacement therapy (IGRT), SDM may help individualize IGRT to clinical needs and lifestyles.
Objective: To summarize results of a targeted literature review on SDM models and their impact on clinical outcomes and to introduce a novel SDM tool for PID
Methods: Focused searches for articles in English were conducted in EMBASE and MEDLINE (date range: January 1, 1999 to August 15, 2018). The search targeted the key elements of SDM (defined as the meaningful exchange of information between patient and physician and identification of issues most important to patients) and the impact of SDM on clinical outcomes. Relevant literature was examined for the current state of SDM in PID and used to inform the development of a novel SDM tool.
Results: The search identified 4,730 records with SDM in the title or abstract. A broad range of therapeutic areas (primarily chronic diseases) was represented, and publication frequency had increased with time. Focused searches identified 159 articles that discussed key elements of SDM. Common elements of SDM included recognizing the decision; two-way sharing of knowledge between physician and patient; expression of patient values and preferences; weighing the options; and making and implementing the decision. The impacts of SDM on clinical outcomes were discussed in 59 studies; 15 were reviewed in detail in acute (n = 7), chronic (n = 5), and general/other (n = 3) conditions. Two studies suggested that SDM may improve clinical outcomes in chronic diseases. In 4 studies, patients who participated in SDM used fewer diagnostic tests and medications and underwent fewer intensive tests and treatments for acute illness. Positive effects of SDM were reported in mental health settings, for patients with chronic illness or making longer-term decisions, and in cases in which SDM interventions occurred over multiple sessions. No studies were found that evaluated SDM in the treatment of PIDD. In a survey of US immunologists (n = 15), participants acknowledged the value of SDM; however, in another survey, patient preferences for IGRT were not the same as what physicians perceived of their patients preferences, underscoring a need for SDM in PID.
Conclusions: SDM has been widely studied and increasingly implemented in health care decisions globally; however, its effects on key patient outcomes are not well understood, and there are currently no known SDM applications in PID. The key findings from this review support the applicability of SDM in PID and highlight the need for a novel tool to help patients recognize their own priorities and needs, and to ensure these guide important clinical decisions such as IGRT selection. Based on these findings, an SDM tool and accompanying discussion guide for clinicians is being developed to facilitate SDM in PID.
Funding: This research was sponsored by Shire.
(193) Submission ID#606400
Treatment of Adenosine Deaminase Severe Combined Immunodeficiency with Pegylated Recombinant Adenosine Deaminase. a Clinical Trial of Patients Transitioned from Pegademase to Elapegademase-lvlr
Morna J. Dorsey, MD, MMSc1, Tracy Fausnight, MD2, Heather Lehman, MD, FAAAAI3, Neena Kapoor, MD4, Arye Rubinstein, MD5, Giuseppe Testa, B.S.6, Joseph M. Wiley, MD7,
1Pediatric Immunologist and Allergist, Department of Pediatrics, Division of Allergy, Immunology, and Bone Marrow Transplant, University of California San Francisco, San Francisco, CA
2Associate Professor, Pediatric and Adult Allergy/Immunology, Penn State College of Medicine
3Associate Professor of Pediatrics, Chief Division of Allergy/Immunology and Rheumatology, Children's Hospital of Buffalo
4Professor, Pediatrics, Children's Hospital Los Angeles and Keck Schood of Medicine of U.S.C.
5Professor, Pediatric Allergy/Immunology, Albert Einstein College of Medicine
6VP Product Development, Leadiant Biosciences, Inc.
7VP Medical Affairs, Drug Safety and Pharmacovigilance, Leadiant Biosciences, Inc.
Introduction: Accumulation of intracellular adenosine and deoxyadenosine nucleotides (dAXP) due to adenosine deaminase deficiency results in profound lymphopenia and severe combined immunodeficiency. Left untreated this form of SCID is uniformly fatal. While allogeneic hematopoietic cell transplant (HCT) and autologous gene corrected stem cell therapy (GT) are potential cures for ADA-SCID , initiating enzyme replacement therapy (ERT) immediately upon diagnosis regardless of definitive treatment is standard of care. HCT and GT are not therapeutic options for all ADA-SCID patients and ERT offers immediate therapeutic intervention for these patients leading to partial immune reconstitution, and durable survival in most patients treated. Adagen (pegademase), approved by the FDA in 1990 in the USA, is a pegylated bovine ADA (nADA) with the enzyme harvested from bovine intestines. This unsustainable production process led to the development of a recombinant enzyme source based on the bovine protein sequence and an improved pegylated linker by using succinimidyl carbamate (RevcoviTM- (elapegademase-lvlr).
Methods: A Phase II/III clinical trial was performed at 5 US sites under institutional IRB approval. Eligible ADA-SCID subjects were stable on Adagen and without complicating underlying conditions. Demographics, medical history, lymphocyte counts, immunoglobulin levels, trough plasma ADA activity and RBC dAXP measurements were collected. Patients were treated with Adagen as a single, weekly IM dose adjusted to achieve a trough plasma ADA activity of > 15 mmol/hr/L and RBC dAXP < 0.02 mmol/L (protocol target levels). Once patients had achieved this level (3-9 weeks), a seven-day PK on Adagen was done and the patients were transitioned to Revcovi based on the formula for enzyme equivalent activity of 1mg Revcovi = 150 Units Adagen. After 5 weeks on Revcovi, trough ADA and dAXP were assessed and a seven-day pharmacokinetic study was conducted at week 9. Patients were assessed periodically for clinical and laboratory values and evaluation of the study endpoints was done at week 21. Subjects subsequently continued on Revcovi and were assessed periodically.
Results: Six patients, ages 16-37 entered the trial with initial Adagen dosing at 7.7-42.9U/kg/wk (see Table 1). Adagen dosing was adjusted to target endpoints of ADA trough activity (>15mmol/hr/L) and RBC dAXP (<0.02 mml/l). Patients transitioned to weekly Revcovi using the aforementioned conversion formula at doses of 0.17-0.285 mg/kg/wk. The drug was well tolerated with the most frequent adverse events (AEs) being cough and vomiting. There were no drug attributable serious AEs. Patients achieved trough RBC dAXP < 0.02 mml/L (primary endpoint) in 69/71 measurements through week 21 and trough plasma ADA > 15mmol/hr/L (secondary endpoint) in 68/74 measurements. Total lymphocyte counts increased from a mean of 0.79 x109/L at start of Revcovi to 0.92 x 109/L at week 21.
Conclusion: Revcovi appears safe in ADA-SCID patients, provides adequate detoxification (dAXP<0.02mmol/L) and sustained trough plasma ADA activity at ~ 2X trough activity provided by Adagen with improved lymphocyte counts. Revcovi received FDA approval for treatment of ADA-SCID on 10/5/18.
| Table 1 Study STP-2279-002: Patient Data (Six Patients) | |||||||||||
| Patient ID | Gender/Age/Race b | Start of Adagen Lead In Phase a | Adagen PK Week | Revcovi Tm Treatment Weeks 1 Through 21 d | |||||||
| Weekly Dose (U/kg)b | Trough ADA Activity (mmol/hr/L) | Weekly Dose (U/kg)b | Trough ADA Activity (mmol/hr/L) |
Erythrocyte dAXP (mmol/L) |
Total Lymphocytes (x10 9 /L) | Weekly Dose (mg/kg) b | No. of Trough ADA Activity Values >15 mmol/hr/L |
No. of Erythrocyte dAXP Values < 0.02 mmol/L |
Total Lymphocytes (x10 9 /L) | ||
| 004-001 | Male/19/Other | 28.2 | 20.9 | 28.2 | 14.5 | < 0.02 | 0.57 | 0.188 | 13/14 | 13/14 | 0.73 |
| 005-001 | Male/21/Other | 29.6 | < lloq | 34.5c | 17.6 | < 0.02 | 0.52 | 0.224 | 12/13 | 13/13 | 0.99 |
| 005-002 | Male/37/Black or African American | 7.7 | < lloq | 30c | 22.4 | < 0.02 | 1.71 | 0.2 | 13/13 | 12/13 | 1.76 |
| 006-002 g | Female/30/White | 31.3 | <lloq g | 31.3 | 9.02 | < 0.02 | 0.59 | 0.209 | 10/12 | 11/11 | 0.49 |
| 011-001 | Female/16/White | 42.9 | 11.74 | 42.9 | 16.10 | < 0.02 | 1.17 | 0.285 | 12/13 | 12/12 | 1.39 |
| 012-001e | Male/18/White | 21.6 | 11.55 | 25.9c | 15.44 | < 0.02 | 0.20 | 0.17 | 8/9 | 8/8 | 0.26f |
a Erythrocyte dAXP levels for all patients were below 0.02 mmol/L and testing for total lymphocytes was not conducted at the time point
b Patient demographics and weight were recorded at screening. Dose per kg based on body weight at Screening
c Adagen dose was adjusted during Adagen Lead In Phase
d Data inclusive of pre treatment levels of ADA activity and Erythrocyte dAXP at Treatment Weeks 1 and 21
e Revcovi Treatment data up to Week 15 only
f Total lymphocytes at Treatment Week 9
g Data at start of Lead In unavailable; Week 2 of Adagen Lead In recorded
lloq: lower limit of quantification
(194) Submission ID#606491
Anti-cytokine Antibodies Emerge After Viral Infections and Persist in Patients in Partial RAG Deficiency
Irina Dawson, MD1, Boglarka Ujhazi, MSc2, Krisztian Csomos, PhD3, Roshini S. Abraham, PhD4, John Sleasman, MD5, Taco Kuijpers, MD, PhD6, Benedicte Neven, MD, PhD7, Jennifer Leiding, MD8, Snezhina Mihailova, MD9, Steven M. Holland, MD10, Charles Song, MD11, Vera Goda, MD12, Gergely Krivan, MD13, Sinisa Savic, PhD14, Ravishankar Sargur, MD15, Lauren Henderson, MD16, Waleed Al-Herz, MD17, Mayra Dorna, MD18, Joseph D Hernandez, MD19, Manish Butte, MD, PhD20, Jolan Walter, MD, PhD21
1Allergy and Immunology Fellow, Division of Allergy and Immunology, Department of Pediatrics, University of South Florida
2Scientist, Division of Allergy/Immunology, Department of Pediatrics, Children's Research Institute, University of South Florida, St. Petersburg, FL
3Research Associate, Division of Allergy/Immunology, Department of Pediatrics, Children's Research Institute, University of South Florida, St. Petersburg, FL
4Department of Pathology and Laboratory Medicine, Nationwide Childrens Hospital, Columbus, OH.
5Professor of Pediatrics, Division of Allergy, Immunology, and Pulmonary Medicine, Duke University School of Medicine
6Professor of Pediatrics, Sanquin Research and Landsteiner Laboratory, Department of Blood Cell Research, Amsterdam UMC, University of Amsterdam, Amsterdam, Netherlands
7Professor of Pediatrics, Department for Pediatric Immuno-Hematology and Rheumatology, Necker Hospital, Paris, France
8Associate Professor, University of South Florida
9Immunologist, University Hospital Alexandrovska, Department of Clinical immunology, Sofia, Bulgaria
10Director, Division of Intramural Research, Laboratory of Clinical Immunology and Microbiology, National Institute of Allergy and Infectious Diseases, National Institutes of Health
11Chief of Pediatric Allergy and Immunology, Ronald Reagan UCLA Medical Center, UCLA Mattel Children's Hospital
12Professor, Department of Pediatric Hematology and Stem Cell Transplantation, United Saint Istvan and Saint Laszlo Hospital, Budapest, Hungary
13Professor, Department of Pediatric Hematology and Stem Cell Transplantation, United St. István and St. László Hospital, Budapest, Hungary
14Clinical Associate Professor, Department of Clinical Immunology and Allergy, St. Jamess University Hospital, Leeds, United Kingdom
15Immunologist, Department of Clinical Immunology and Rheumatology, Hannover Medical School, Hannover, Germany
16Professor of Pediatrics, Boston Childrens Hospital, Boston MA
17Associate Professor of Pediatrics, Pediatrics Department, Faculty of Medicine, Kuwait University
18Allergy Immunologist, Allergy Immunology Department from Child´s Institute from Medicine Faculty from Uiversity of Sao Paulo (Instituto da Criança do Hospital das Clinicas da Faculdade de Medicina da Universidade de São Paulo)
19Allergist / Immunologist, Department of Pediatrics, Division of Allergy, Immunology and Rheumatology, Stanford University, Stanford CA
20Division of Allergy/Immunology Chair, Division of Immunology, Allergy, and Rheumatology, Dept. of Pediatrics and Jeffrey Modell Diagnos-tic and Research Center, University of California, Los Angeles
21Associate Professor, Robert A. Good Endowed Chair and Division Chief
Division of Pediatric Allergy & Immunology, Department of Pediatrics, University of South Florida, Johns Hopkins All Children's Hospital, St. Petersburg, FL.
The spectrum of clinical manifestations range from infections to autoimmunity and inflammation among patients with hypomorphic recombination gene 1 and 2 (RAG1/2) pathogenic variants. Auto-antibodies targeting cytokines IFN-alpha, IFN-omega and IL-12 were reported in a large proportion of these patients and their occurrence often coincides with viral infections. We report the time of emergence and relative frequency of anti-cytokine antibodies in children and adults, and their persistence among patients with hypomorphic RAG deficiency.
Antibodies were measured from plasma samples of patients by enzyme linked immunoassay (ELISA).
Our RAG cohort includes 28 patients with RAG1 (n=17, 61%) and RAG2 deficiency (n=11, 39%). Antibodies targeting IFN-alpha (75%) were most common followed by IL-12 and IFN-omega (40% each). Two asymptomatic patients who were detected by newborn screening for SCID and received hematopoietic stem cell transplantation had no detectable anti-cytokine antibodies. In the cohort of young children (ages 11 mo-7 years, n=9), all patients had detectable antibodies to IFN-alpha, prior history of severe viral infection and subsequently developed autoimmune cytopenias. Other anti-cytokine antibodies were less common (IFN-omega 44%, IL-12 33%). Similarly, children between 10-18 yo age (n=9) also had high fraction of anti-IFN-alpha antibodies (89%) with prior history of infections (66%) and continued to have other anti-cytokine antibodies less commonly (IFN-omega 37%, IL-12 62%). In the adult cohort (n=8, ages 25-39 years) the frequency of anti-IFN-alpha anti-cytokine antibodies were lower (62%,) and IL-12 and IFN-omega (50% each) continued to persist. Three adult patients had anti-cytokine (IFN-alpha, IFN-omega and IL-12) antibodies tested at multiple timepoints and elevated titers persisted up to 4 years.
Our data demonstrates that anti-cytokine antibodies, especially those targeting IFN are frequent and emerge early in life in association with viral infections in patients with RAG deficiency. A lower fraction of adult patients have detectable anti-cytokine antibodies, and maintain these over several years. Anti-IFN-alpha may serve as a useful biomarker for identifying partial RAG deficiency among young and adult patients with history of viral infections and autoimmune cytopenias. The role of these antibodies to cytokines is yet to be determined but a specific signature of these antibodies may help to identify an underlying immunodeficiency and initiate early definitive treatment with bone marrow transplantation. Anti-cytokine antibodies appear to be a novel tool in evaluation of auto-immune diseases including RAG deficiency.
(195) Submission ID#606539
Oral Immunoglobulins to Treat Norovirus Gastroenteritis in Patients with Primary and Secondary Immunodeficiency
Matthew Perez, MD1, Stella Hartono, MD, Phd2, Maha N. Syed, MD3, Araceli Elizalde, MD4, James Dunn, Phd5, Claire Bocchini, MD6, Joud Hajjar, MD, MS7
1Medicine Resident, Baylor College of Medicine
2Pediatrics Resident, Baylor College of Medicine
3Research Assistant, Baylor College of Medicine
4Assistant Professor of Pediatrics, Baylor College of Medicine
5Director of Medical Microbiology and Virology, Texas Children's Hospital
6Assistant Professor of Pediatrics Infectious Disease, Baylor College of Medicine
7Assistant Professor, Baylor College of Medicine, 1Texas Childrens Hospital Center for Human Immunobiology and Division of Immunology, Allergy and Rheumatology
Perez, M., Hartono, S., Syed, M., Elizalde, A., Dunn, J., Bocchini, C., Hajjar, J.
Introduction: Norovirus is one of the most common pathogens causing gastroenteritis in immunocompromised patients, often leading to chronic infection, causing villous atrophy, malabsorption, weight loss, organ failure, need for parenteral nutrition, and death. Norovirus treatment in immunocompromised patients is challenging. Oral immunoglobulin (POIG) has been used to treat norovirus gastroenteritis with variable success. Our aim in this study was to determine the outcomes of treating norovirus gastroenteritis in immunocompromised patients
Methods: Electronic medical records were reviewed for patients with norovirus infection confirmed by RT-PCR since January 2012. Our initial cohort was focused on patients with primary immunodeficiency (PID), lung, and liver transplant. Data on demographics, immunological phenotype, treatment with POIG, the number of bowel movements (BM), and virus clearance were collected. Descriptive statistical methods were used to describe treatment outcomes. Further analysis of patients immunophenotype, immunosuppression medications, and co-morbid illnesses is underway.
Results: Twenty-six immunocompromised patients (27 norovirus infection episodes, as one patient had reinfection) were analyzed twelve females, age range 7 months-50 years. Twelve patients had PID diagnosis (3 common variable immunodeficiency, 2 severe combined immunodeficiency, 1 X-linked agammaglobulinemia, 1 Wiskott-Aldrich Syndrome, 1 DiGeorge Syndrome, 1 Hyper-IgM, 1 STAT3 gain-of-function, 1 NEMO and 1 lymphopenia in a patient with Trisomy 21), 13 patients were status-post liver transplant, and two patients were status-post lung transplant. 13 of26 patients were on IG replacement therapy at the time of the norovirus infection. The average number of BM/day in all patients was 8.4 (range 2-20). Eight patients received POIG (250-500 mg/Kg) weekly for a duration from 1-12 weeks. Three of those received additional Nitazoxanide and 2 received Ribavirin. 2/8 patients in the POIG group were receiving Total Parenteral Nutrition (TPN), and 4/19 on no treatment group received TPN. The average number of BM/day in POIG before treatment was 9.5 (range 4-16), and 8.6 (range 2-20) in those who did not receive any treatment. 5 of 8 (62%) on POIG vs. 11 of 19 (57%) in the no treatment group cleared the virus. The average number of weeks to return to baseline BM was 2.6 (range 1-7) in the POIG group vs. 1.5 (range 3 days-5 weeks) in the no treatment group. 2 of 8 on POIG continued to have chronic diarrhea that is still ongoing.
Conclusion: Despite anecdotal reports suggesting successful use of POIG in immunocompromised patients, our data did not show a significant decrease in stool output in patients treated with POIG, compared to no treatment. However, POIG led to a higher rate of virus clearance. A study with larger sample size might be warranted to identify the patients who benefit from POIG in the context of norovirus infection and ensure the appropriate use of IG products, especially given the concerns for the national shortage of IG products.
(196) Submission ID#606540
Emapalumab-lzsg for the Treatment of Pediatric Patients with Primary Hemophagocytic Lymphohistiocytosis
Michael Jordan, MD1, Franco Locatelli, MD, PhD2, Carl Allen, MD, PhD3, Simone Cesaro, MD4, Carmelo Rizzari, MD5, Anupama Rao, MD6, Barbara Degar, MD7, Timothy Garrington, MD8, Julian Sevilla, MD, PhD9, Maria Caterina Putti, MD10, Franca Fagioli, MD11, Martina Ahlmann, MD12, Jose-Luis Dapena, MD13, Alexei Grom, MD14, Fabrizio De Benedetti, MD, PhD15, Walter Giovanni Ferlin, PhD16, Maria Ballabio, MD17, Cristina De Min, MD18
1Professor of Pediatrics, Divisions of Immunobiology, and Bone Marrow Transplantation and Immune Deficiency
Cincinnati Children's Hospital
2Department of Pediatric Hematology and Oncology, Bambino Gesù Childrens Hospital IRCCS
3Associate Professor, Department of Pediatrics, Section of Hematology/Oncology, Baylor College of Medicine
Texas Children's Hospital
4Director, Pediatric Oncology, University Hospital Verona
5Head of the Pediatric Hematology-Oncology Unit, San Gerardo Hospital, University of Milano-Bicocca
6Deputy Chief of Service for Blood, Cells and Cancer, Great Ormond Street Hospital
7Assistant Professor, Dana-Farber Cancer Institute
8Hematology/Oncology - Pediatric, Pediatrics, Children's Hospital Colorado
9Hematología y Hemoterapia, University Children Hospital Niño Jesús
10Pediatric Haematologist-Oncologist, Department of Pediatrics, University of Padua Medical School
11Director, Pediatric Oncology, Regina Margherita-S. Anna Hospital
12Oberärztin, University Children's Hospital Muenster
13Department of Pediatric Hematology and Oncology, University Hospital Vall dHebron
14Research Director, Department of Rheumatology, Cincinnati Children's Hospital Medical Center
15Head of Pediatric Rheumatology, Bambino Gesù Children's Hospital IRCCS
16Department Head, Exploratory Science and Translational Medicine
Novimmune SA
17Head of NI-0501 Clinical Development, Novimmune SA
18Chief Medical Officer, Novimmune SA
Primary hemophagocytic lymphohistiocytosis (pHLH) is a life-threatening, immune regulatory disorder characterized by immune hyperactivation that is driven by high production of interferon (IFN)-. Patients with HLH typically develop fever, splenomegaly, cytopenias and coagulopathy. Until recently, there have been no FDA approved treatments for HLH, and standard dexamethasone/etoposide-based treatment has not evolved significantly in 20+ years. Emapalumab-lzsg (NI-0501) is a fully human, anti-IFN- monoclonal antibody that neutralizes IFN- and which was recently approved (November 2018) by the FDA for the treatment of adult and pediatric (newborn and older) patients with pHLH with refractory, recurrent, or progressive disease or intolerance with conventional HLH therapy. Results of the pivotal trial supporting this approval are presented herein.
Methods: This open-label pivotal study (NCT01818492) includes patients 18 years with a diagnosis of pHLH and active disease. Data presented were from 34 patients, of whom 27 had failed conventional HLH therapy prior to study entry. The initial emapalumab-lzsg dose was 1 mg/kg given intravenously every 3-4 days. Subsequent doses could be increased up to 10 mg/kg based on the evolution of response parameters. Dexamethasone was administered concomitantly at 5 to 10 mg/m2/day and could be tapered during the study. Treatment duration was 8 weeks, with possible shortening to a minimum of 4 weeks, or extension up to the time of allogeneic hematopoietic stem cell transplantation (HSCT). The primary efficacy endpoint was the overall response rate (ORR) at end of treatment, assessed by pre-defined objective parameters, including normalization or at least 50% improvement from baseline of fever, splenomegaly, cytopenias, hyperferritinemia, fibrinogen, D-dimer, central nervous system (CNS) abnormalities, and with no sustained worsening of sCD25 serum levels. The primary analysis used an exact binomial test to evaluate the null hypothesis that ORR be 40% at a one-sided 0.025 significance level. Patients were eligible to enter an extension phase for follow-up after completing the main study (NCT02069899). The data cut-off applied is July 20 2017.
Results: Patient characteristics are summarized in Table 1 and efficacy is summarized in Table 2. Disease at study entry was consistent with the broad spectrum of pHLH abnormalities. Over 30% of patients had signs and/or symptoms of CNS disease. ORR was significantly higher than the pre-specified null hypothesis of 40%, meeting the primary endpoint. The response rate based on investigators clinical judgement was 70.6%. Emapalumab-lzsg infusions were in general well tolerated, with mild to moderate infusion-related reactions reported in 27% of patients. The observed safety events (pre-HSCT conditioning) mostly included HLH manifestations, infections or toxicities due to other administered drugs. Infections caused by pathogens potentially favored by IFN- neutralization occurred in 1 patient during emapalumab-lzsg treatment (disseminated histoplasmosis), and resolved with appropriate treatment. No off-target effects were observed.
Conclusions: Treatment with emapalumab-lzsg was able to control HLH activity with a favorable safety and tolerability profile in a very fragile population. The majority of patients proceeded to HSCT with favorable outcomes. Our results indicate that emapalumab-lzsg should be considered as a new therapeutic option in pHLH thanks to its targeted mode of action.
Table 1. Patient Characteristics
|
All Patients (N=34) |
Patients Failing Conventional Therapy (n=27) |
|
| Age at HLH diagnosis, years (median [range]) | 0.85 [0.03, 13.0] | 1.00 [0.2-13.0] |
| Sex (n [%]) | ||
| Male | 16 [47.1] | 11 [40.7] |
| Female | 18 [52.9] | 16 [59.3] |
| HLH Genetic Confirmation (n [%]) | 27 [79.4] | 22 [81.5] |
| FHL1 | 2 [5.9] | 0 |
| FHL2 | 7 [20.6] | 5 [18.5] |
| FHL3 | 8 [23.5] | 7 [25.9] |
| FHL4 | 1 [2.9] | 1 [3.7] |
| FHL5 | 2 [5.9] | 2 [7.4] |
| Griscelli Syndrome type 2 | 5 [14.7] | 5 [18.5] |
| X-linked Lymphoproliferative Disorder 1 | 1 [2.9] | 1 [3.7] |
| X-linked Lymphoproliferative Disorder 2 | 1 [2.9] | 1 [3.7] |
FHL: Familial HLH.
Table 2. Efficacy
|
All Patients (N=34) |
Patients Failing Conventional Therapy (n=27) | |
| ORR, n/N (%) | 22/34 (65)* | 17/27 (63)** |
| Time to response, days (median [95% CI]) | 8.0 [5.0-10.0] | 8.0 [7.0-14.0] |
| Cumulative duration of response, % days of treatment (median [Q1, Q3]) | 75.7 [33.3-91.2] | 75.4 [30.8-91.2] |
| Proceeded to HSCT, n/N (%) | 22/34 (65) | 19/27 (70) |
| Overall Survival | ||
| Alive at last observations, n/N (%) | 24/34 (70.6) | 20/27 (74.1) |
| 12-month pOS estimate (% [95% CI]) | 69 [50-82] | 73 [52-86] |
| Survival to HSCT, n/N (%)*** | 27/34 (79.4) | 22/27 (81.5) |
| Survival post HSCT, n/N (%) | 20/22 (90.9) | 17/19 (89.5) |
*95% CI: 46 to 80%, P value=0.0031. **95% CI: 42 to 81%, P value=0.0134. ***Patients not receiving HSCT were censored at day last seen or day 180.
CI, confidence interval; Q, quartile; pOS, probability of overall survival.
(197) Submission ID#606549
T Cell Transcriptome in Chromosome 22q11.2 Deletion Syndrome
Nikita Raje, MD1, Marcia A. Chan, PhD2, Nicole M. Gigliotti, BS3, Janelle R. Noel-Macdonnell, PhD4, Daniel P. Heruth, PhD4
1Assistant Professor of Internal Medicine and Pediatrics, University of Missouri Kansas City, Kansas City, Missouri
2Associate Professor, University of Missouri Kansas City
3Genetics Technologist, Children's Mercy
4Assistant Professor of Pediatrics, Children's Mercy, University of Missouri Kansas City
Background: Phenotypic variations of chromosome 22q11.2 deletion syndrome (22qDS) have no clear explanation. T cell lymphopenia in chromosome 22q11.2 deletion syndrome (22qDS) is related to varying degrees of thymic hypoplasia and contributes to the phenotypic heterogeneity. No phenotype correlation with genotype or deletion size is known for lymphopenia. We hypothesized that the T-cell transcriptome is different in 22qDS compared to healthy children and that gene expression in T cells can differentiate patients with low T cells compared to normal T cells.
Methods: Peripheral blood was collected from a convenience sample of participants aged 5-8 years. Standard immune function testing was performed. RNA sequencing was completed on isolated T cells using Illuminas TruSeq technology. Differential gene expression profiles (q<0.05) of T cells between 22qDS and healthy controls were determined with Tuxedo Suite & String Tie pipelines. Bioinformatic analyses were implemented via Ingenuity Pathway Analysis and KEGG to identify enriched pathways. The Spearman correlation between gene expression and clinical variables were calculated using SAS (9.4, Cary, NC) (p-value<0.05).
Results: A total of 360 genes were differentially expressed between T cells of 22qDS patients (n=13) and healthy controls (n=6) (Log 2 fold change range (-2.0747, 15.6724)).When these 360 genes were tested for pathway enrichment, the top 5 pathways in T lymphocytes based on their p value included communication between innate and adaptive immune cells, cross talk between dendritic cells and natural killer cells, allograft rejection signaling, dendritic cell maturation, and B cell receptor signaling. The top 10 biological processes with differential expression included 36 immune response, 31 inflammatory response, 33 apoptotic process, 12 interferon gamma mediated signaling pathway, 14 nucleosome assembly, 16 defense response to virus, 8 lipopolysaccharide mediated signaling pathway, 7 positive regulation of NF-kappa B import into nucleus, 10 type I interferon signaling pathway, and 10 neutrophil chemotaxis genes. We compared gene expression between 22qDS participants with low T cell counts (n=7) and 22qDS participants with normal T cell counts (n=6) and found 94 genes that were differentially expressed (q<0.05) (Log2 fold change range (-4.5445, 5.1297)). We found 29 genes that correlated with T cell counts and subsets (CD3, CD4, CD8, and naïve helper T cells) (R0.8).
Conclusions: T-cell gene expression contributes to phenotypic heterogeneity in chromosome 22q11.2 deletion syndrome. T cell gene expression is different in 22qDS with low T cells compared to normal T cells. Differential gene expression can be used in future to develop biomarkers to differentiate between different phenotypes of 22qDS.
(198) Submission ID#606644
Non-Arteritic Anterior Ischemic Optic Neuropathy in a Patient with XIAP Deficiency: Expanding the Inflammatory Ocular Findings in X-Linked Lymphoproliferative Disorders
Mark Dulchavsky, MS1, Barry Skarf, MD, PhD2, David Frame, PharmD3, Rebecca A. Marsh, MD4, Kelly Walkovich, MD5
1Medical Student, University of Michigan Medical School
2Neuro-Ophthalmologist, Henry Ford Health System
3Clinical Pharmacist, University of Michigan College of Pharmacy
4Associate Professor, Division of Bone Marrow Transplantation and Immune Deficiency, Cincinnati Childrens Hospital Medical Center
5Associate Professor, Pediatric Hematology/Oncology, University of Michigan Medical School
Background: X-linked inhibitor of apoptosis protein (XIAP) deficiency is due to pathologic variants in XIAP/BIRC4, and is most often associated with hemophagocytic lymphohistiocytosis and inflammatory bowel disease. Non-arteritic anterior ischemic optic neuropathy (NAION) is an ophthalmologic condition characterized by idiopathic ischemic infarction of the optic nerve head. It typically presents in older patients with risk factors for vascular disease. In those situations it does not respond to immunomodulatory agents. Although other inflammatory ocular manifestations of XIAP deficiency, such as uveitis, have been documented, this is the first described case of NAION in this disease spectrum.
Case: We report a 51 year-old man who presented with painless, unilateral vision loss in the right eye in 2012, at 44 years of age. Serial visual field testing demonstrated inferior field vision loss. Fundoscopic exam initially revealed a swollen right optic disc. Optical coherence tomography (OCT) demonstrated marked retinal nerve fiber layer thinning in the superior retina, consistent with the known inferior visual field defect, both findings characteristic of NAION. Treatment with IV steroids produced little improvement in vision, but there was no further progression. Two years later the patient began experiencing recurrent high fevers and developed splenomegaly. Elevated transaminases and concern for lymphoproliferative disease prompted a splenectomy and liver biopsy. Both the spleen and liver biopsy were positive for EBV but were negative for malignancy. Bone marrow biopsy was unrevealing. Genetic testing identified a pathogenic variant in XIAP/ BIRC4 (1141C>T), and the patient was treated with high dose oral steroids resulting in an improvement in symptoms. Subsequently, therapy with anakinra was started and steroids were tapered. During the steroid taper, he noticed a change in the vision of his left eye consistent with NAION, as well as worsening of his colitis. There was loss of the inferior visual field and fundoscopic exam was significant for left optic disc swelling. OCT noted superior retinal nerve fiber layer thinning. Oral steroids were restarted with improvement in optic disc swelling, but without improvement or change in vision. As of his most recent exam, the patient has persistent bilateral inferior visual field defects with segmental optic nerve atrophy typical of NAION. He has continued therapy with anakinra, and subsequently tapered off of prednisone; though he remains on a physiologic dose of hydrocortisone.
Conclusions: This case demonstrates an unreported ocular manifestation in a patient with XIAP deficiency, which clinically appeared sensitive to immunomodulation. Our patient is an unusual candidate for NAION due to his young age, the average age of onset being the mid to late 60s, and lack of vascular risk factors. We hypothesize that his hyper-inflammatory condition contributed to irreversible vascular damage in the optic nerve head, resulting in NAION. Therefore, it may be useful to consider the involvement of systemic inflammatory and immune dysregulatory conditions when treating patients with atypical NAION. Additionally, NAION should be considered in patients with XIAP deficiency and sudden unilateral vision loss.
(199) Submission ID#606703
Antibody Responses After Vaccination with Prevnar13® in IgG Subclass Deficient Patients
Leigh Williams, PhD1, Stephen Harding, PhD2, Peter Bergman, MD, PhD3
1Medical Science Liaison, The Binding Site
2Research and Development Director, The Binding Site
3Associate Professor in Clinical Microbiology and Consultant Physician, Karolinska Institutet
Levels of IgG subclasses are often used as markers for vaccine-induced immunity. However, the relationship between IgG and other antibody-classes post-vaccination is not firmly established. Therefore, we set out to investigate the pneumococcal-specific responses of IgG, IgG2, IgA and IgM to Prevnar13® in IgG subclass deficient (IgGScD) patients in this study.
Pneumococcal responses were measured using the VaccZyme Pneumococcal Capsular Polysaccharide IgG, IgG2, IgA and IgM ELISAs (The Binding Site Group, Birmingham, UK) in control (n=10, median age 57 years, range 27-64) and IgGScD patients (n=10, median age 55 years, range 25-65) recruited from the Immunodeficiency Unit at the Karolinska University Hospital. Patients were vaccinated with Prevnar13® (PCV13) and serum samples were collected pre- and 4 weeks post-vaccination.
An increase in concentration of anti-PCV13 IgG, IgG2, IgA and IgM antibodies in response to PCV13 vaccination was observed 4 weeks post vaccination in IgGScD patients (median, 2.5th and 97.5th percentile, p value: PCV13 IgG 83 mg/L, 5-270, p=0.002; PCV13 IgG2 17 mg/L, 1-81, p=0.008; PCV13 IgA 71 U/mL, 10-165, p=0.017; PCV13 IgM 38 U/mL, 13-100, p=0.065). These median concentrations were lower than those observed in control patients (median, 2.5th and 97.5th percentile: PCV13 IgG 215 mg/L, 58-270; PCV13 IgG2 71 mg/L, 14-90; PCV13 IgA 83 U/mL, 26-123; PCV13 IgM 51 U/mL, 39-256). However, percentage changes between pre to post vaccination concentrations of IgG, IgG2 and IgA in response to PCV13 in IgGScD patients were not significantly different to the control patients (PCV13 IgG p=0.595, PCV13 IgG2 p=0.515 and PCV13 IgA P=0.122). Interestingly, pre-vaccination PCV13 IgA concentrations were elevated in IgGScD patients compared to controls (median 45.1 U/mL vs 17.1 U/ml, respectively).
Using the lower 2.5th percentile of the control population at 4 weeks post vaccination (PCV13 IgG 58 mg/L, PCV13 IgG2 14.0 mg/L, PCV13 IgA 26 U/mL and PCV13 IgM 39 U/mL) responders and non-responders of PCV13 IgG, PCV13 IgG2, PCV13 IgA and PCV13 IgM in IgGScD patients were identified. Notably, 40% of IgGScD patients were identified as PCV13 IgG non-responders, 40% PCV13 IgG2, 10% PCV13 IgA and 50% PCV13 IgM non-responders. Post vaccination in IgGScD patients a strong correlation was observed between PCV13 IgA and PCV13 IgM (Pearson correlation r=0.789, p=0.007) and in the control patients a strong correlation was observed between PCV13 IgG and PCV13 IgG2 (Pearson correlation r= 0.921, p=0.001).
In conclusion, concentrations of IgG, IgG2, IgA and IgM in response to PCV13 in IgGScD patients were generally lower compared to the control population. These results support the fact that in addition to IgG and IgG2, measurement of IgA and IgM could also provide useful information for the clinician.
(200) Submission ID#606705
Gain-of-function IKBKB Mutation Causes Human Combined Immune Deficiency
Bahar Miraghazadeh, PhD1, Chelisa Cardinez, MSc2, Kay Tanita, MD3, Elizabeth da Silva, MD4, Akihiro Hoshino, MD, PhD5, Satoshi Okada, MD, PhD6, Rochna Chand, MSc7, Takaki Asano, MD8, Miyuki Tsumura, PhD8, Kenichi Yoshida, PhD9, Hidenori Ohnishi, MD, PhD10, Zenichiro Kato, MD, PhD11, Masahide Yamazaki, MD, PhD12, Yusuke Okuno, MD, PhD13, Satoru Miyano, MD, PhD14, Seiji Kojima, MD/PhD15, Seishi Ogawa, MD, PhD16, Daniel Andrews, PhD17, Matthew Field, PhD18, Gaetan Burgio, MD, PhD19, Tomohiro Morio, MD, PhD20, Carol Vinuesa, MD, PhD21, Hirokazu Kanegane, MD, PhD22, Matthew Cook, MD, PhD23
1Post-doctoral researcher
John Curtin School Medical Research and Translational research Unit
2PhD candidate, John Curtin School Medical Research and Translational research Unit
3Department of Child Health and Development, Graduate School of Medical and Dental Sciences,Tokyo Medical and Dental University, Tokyo
4Department of Immunology Canberra Hospital, Canberra, Australia
5Assistant Professor, Department of Child Health and Development, Graduate School of Medical and Dental Sciences, Tokyo Medical and Dental University, Tokyo
6Associate Professor, Department of Pediatrics, Hiroshima University Graduate School of Biomedical and Health Sciences, Hiroshima, Japan
7Research assistant, Centre for Personalised Immunology and Department of Immunology Canberra Hospital, Canberra, Australia
8Department of Pediatrics, Hiroshima University Graduate School of Biomedical and Health Sciences,Hiroshima, Japan
9Department of Pathology and Tumor Biology, Graduate School of Medicine,Kyoto University, Kyoto, Japan
10Department of Pediatrics, Gifu University Graduate School of Medicine,Gifu, Japan
11Department of Pediatrics, Gifu University Graduate School of Medicine, Gifu, Japan; Structural Medicine, United Graduate School of Drug Discovery and Medical Information Sciences, Gifu University, Gifu, Japan
12Department of Internal Medicine, Keiju Medical Center,Nanao, Japan
13Center for Advanced Medicine and Clinical Research,Nagoya University Hospital, Nagoya, Japan
14Professor, Laboratory of DNA Information Analysis, Human Genome Center, Institute of Medical Science, The University of Tokyo
15Professor, Department of Pediatrics, Nagoya University Graduate School of Medicine, Nagoya, Japan
16Professor, Department of Pathology and Tumor Biology, Graduate School of Medicine, Kyoto University, Kyoto, Japan
17Researcher, Centre for Personalised Immunology, Australian National University, Canberra, Australia, Department of Immunology and Infectious Diseases, John Curtin School of Medical Research, Australian National University, Canberra
18Researcher, Centre for Personalised Immunology, Australian National University, Canberra, Australia, Department of Immunology and Infectious Diseases, John Curtin School of Medical Research, Australian National University, Canberra
19Researcher, Department of Immunology and Infectious Diseases, John Curtin School of Medical Research, Australian National University, Canberra
20Department of Child Health and Development, Graduate School of Medical and Dental Sciences, Tokyo Medical and Dental University
21Professor, Centre for Personalised Immunology, Australian National University, Canberra, Australia,Department of Immunology and Infectious Diseases, John Curtin School of Medical Research, Australian National University, Canberra
22Professor, Department of Child Health and Development, Graduate School of Medical and Dental Sciences, Tokyo Medical and Dental University, Tokyo,
23Professor, Centre for Personalised Immunology, Australian National University, Canberra, Australia; Department of Immunology Canberra Hospital, Canberra, Australia
The importance of de novo mutations in causing severe sporadic immune disease is well described, yet significance of such a variation in less severe and later onset of immune disease is poorly investigated. Whole exome sequencing has been a powerful tool to resolve and explain the genetic basis of novel syndromes in immune related diseases. However, proving causation can be complicated due to low number of the affected individuals. We performed whole exome sequencing in a cohort of patients with non-congenital immune defects, along with detailed cellular biochemical phenotyping. We report and describe a novel non-congenital combined immune deficiency arising from a de novo gain-of-function mutation in IKBKB(c.607G>A). This gene encodes IKK2, and activates canonical NFKB signalling. Cellular and biochemical studies of the proband revealed that IKK2V203I results in enhanced NF-kB signaling, as well as T and B cell functional defects. IKK2V203 is a highly-conserved residue, and to prove causation, we generated a CRISPR/cas9 mouse model that carry the precise orthologous missense mutation. We show that mice and humans carrying this missense mutation exhibits remarkably similar cellular and biochemical phenotypes.
(201) Submission ID#606707
FAS-mediated Apoptosis Assay in Patients with ALPS/ALPS-like Phenotype Carrying CASP10 mutations
Maurizio Miano, MD1, Enrico Cappelli, PhD1, Agnese Pezzulla, MD2, Roberta Venè, MD3, Elena Palmisani, MD4, Alice Grossi, MD5, Paola Terranova, PhD6, Tiziana Lanza, PHD6, Rosario Maggiore, MD6, Filomena Pierri, MD6, Concetta Micalizzi, MD6, Michaela Calvillo, MD6, Giovanna Russo, MD2, Isabella Ceccherini, Phd7, Carlo Dufour, Genoa6, Francesca Fioredda, MD6
1Hematology Unit, IRCCS Istituto Giannina Gaslini
2Pediatric Hematology/Oncology Unit, University of Catania
3Molecular Oncology and Angiogenesis Unit, IRCCS Ospedale Policlinico San Martino
4Hematology Unit, IRCCS Istituto Giannina Gaslini
5Genetic Unit,Genoa, Italy, IRCCS Istituto Giannina Gaslini
6Hematology Unit, IRCCS Istituto Giannina Gaslini
7Genetic Unit, IRCCS Istituto Giannina Gaslini
INTRODUCTION: Autoimmune lymphoproliferative syndrome (ALPS) is a congenital disorder characterized by an impaired FAS-mediated apoptosis that leads to chronic benign lymphoproliferation and autoimmunity. In most cases mutation on FAS gene are responsible of the disease and the phenotype of individuals carrying the same variants can vary from asymptomatic/mild forms to severe disease, due to incomplete penetrance of pathogenic mutations. More rarely, other genes involved in this pathway, such as CASP10, are mutated. Few clinical and molecular data have been reported on patients carrying CASP10 mutation showing that different genetic variations can produce contrasting phenotypic effects. So far, 2 mutations have been recognized as pathogenic (I406L and L258F) and other have been reported with controversial result on their pathogenicity (V410l, Y446C) or are known to be polymorphic variants (L522l)
AIMS: The aim of this study is to evaluate the FAS-mediated apoptosis function in patients with ALPS or ALPS-like symptoms carrying mutations on CASP10 gene.
METHODS: We evaluated FAS-mediated apoptosis pathway in patients presenting with an ALPS/ALPS-like phenotype that were found to carry a CASP10 mutation in our Institution. Molecular findings have been obtained by NGS analysis of a panel of genes involved in the most common immune-dysregulation syndromes and immune-deficiencies. Functional studies have been performed by Western blot analysis of CASP10, CASP8, and PARP proteins after TRIALinduced apoptosis stimulation. Healthy individuals were used as control.
RESULTS: We identified 6 patients with ALPS (2) or ALPS-like (4) phenotype, carrying the I406L (1), V410l (2), Y446C (1) heterozygous CASP10 mutations and the polymorphic variant L522l (2) associated with another polymorphic homozygote variant on CASP8 gene (p.Met1Thr) or with a compound heterozygous mutation on TNFRSF13C (His159Tyr, Pro21Arg). Patients clinical characteristics are shown in Table 1. Western blot analysis showed an impaired activation of CASP10, CASP8, and PARP proteins in all cases compared to healthy control (Fig. 1)
CONCLUSIONS: In our symptomatic patients, the CASP10 mutations whose pathogenicity is controversial (V410l, Y446C) are associated with an impaired CASP10, CASP8, PARP activity -and therefore with apoptosis dysfunction- as in the case of I406L pathogenic mutation. We can speculate that, in addition to the functional impairment of apoptosis, other genetic and epigenetic factors might be crucial for the development of clinical symptoms in CASP10 mutated patients as already described in FAS mutations.
Nonetheless, patients with polymorphic variants (such as L522l) may have an impairment of apoptosis if this is associated with other polymorphisms on other genes involved in the pathway (such as CASP8) or in other immune-dysregulation syndromes, suggesting that the search of other mutations in patients with ALPS/ALPS-like phenotype should be encouraged in patients carrying CASP10 mutation. Further studies on epigenetic factors potentially implicated in the expression of symptoms are needed to fully understand the heterogeneity of clinical phenotype of this disorder.
Table 1: clinical, immunological and genetic characteristics of the patients
| M/F | Age at onset (years) | ALPS/ALPS-like | Symptoms | Genetic mutation | Defective lymphocyte apoptosis | sFAS-L (>200 pg/ml) | IL-10 (>20 pg/ml) | IL-18 (>500 pg/ml) | Vit B12 (n.r. 191 - 663 pg/ml) | IgG (mg/dl) | Family history | DNT (>1.5% of total lymph) | B220+ DNT (>60%) | CD3+CD25+ / CD3+HLADR+ ratio (<1%) | CD27+ (<15%) | |
| P1 | F | 12 | ALPS-like | + Autoimmunity |
- CASP10 - Leu522lle - CASP8 - Met1Thr (Homozygosis) |
No | 0 | 4 | 365 | 482 | 1407 | Neg | 1.7 | 22 | 0.3 | 84.3 |
| P2 | M | 11 | ALPS-like | Trilinear cytopenia + Autoimmunity + LP |
- CASP10 - Leu522lle - TNFRSF13C – His159Tyr - TNFRSF13C – Pro21Arg |
No | 0 | 3 | 660 | 356 | 820 | Neg | 4 | 70.6 | 0.1 | 4.5 |
| P3 | F | 9 | ALPS-like | monolinear cytopenia + LP | - CASP10 – Val410lle | Yes (100%) | 0 | 1,1 | 550 | 1014 | 1076 | Neg | 1.2 | 46 | 1.3 | 0.3 |
| P4 | F | 5 | ALPS P | monolinear cytopenia + LP | - CASP10 – Tyr446Cys | No | 0 | <1 | 1050 | 466 | 1094 | Neg | 2.3 | 92 | 0.2 | 15.6 |
| P5 | M | 3 | No | monolinear cytopenia + RRI + Recurrent skin abscesse | - CASP10 – Val410lle | ND | 0 | <1 | 75 | 699 | 1154 | Neg | 1.1 | 61.7 | 0.9 | 12.4 |
| P6 | M | 2 | ALPS | Asthenia + LP + RI + Arthromyalgia | - CASP10 – I406L | No | 0 | <1 | 500 | 583 | 1062 | Neg | 3.9 | 57 | 0.7 | 23.6 |
F: female M: male;
LP: lymphoproliferation; ND: not done;
RRI: recurrent respiratory infections; RI: recurrent infections;
ALPS P: probable; ALPS D: definitive
The values indicated above are prior to any treatments (Sirolimus, MMF, IVIG)
(202) Submission ID#606722
Cure of a Complicated Patient with X-linked Lymphoproliferative Syndrome Type I and Hepatitis C Cirrhosis, Through Combined Living Related Liver and Hematopoietic Stem Cell Transplantation
Christian A. Wysocki, MD, PhD1, Madhuri Vusirikala, MD2, Nisa Kubiliun, MD3, Rita Lepe, MD4, Nathan Singh, MD5, David Porter, MD6, Abraham Shaked, MD, PhD7,
1Assistant Professor, Director of the Jeffrey Modell Foundation Diagnostic and Research Center, Division of Allergy and Immunology, Departments of Internal Medicine and Pediatrics, UT Southwestern Medical Center/Children's Medical Center Dallas, TX
2Professor, Division of Hematology and Oncology, Department of Internal Medicine, UT Southwestern Medical Center, Dallas, TX
3Assistant Professor, Division of Digestive and Liver Diseases, Department of Internal Medicine, UT Southwestern Medical Center, Dallas, TX
4Physician, Baylor Scott and White, Liver Consultants of Texas, Dallas, TX
5Fellow, Division of Hematology and Oncology, Perelman Center for Advanced Medicine, University of Pennsylvania, Philadelphia, PA
6Jodi Fisher Horowitz Professor in Leukemia Care Excellence, Division of Hematology and Oncology, Perelman Center for Advanced Medicine, University of Pennsylvania, Philadelphia, PA
7Eldridge L. Eliason Professor of Surgery, Director Penn Transplant Institute, Department of Surgery, University of Pennsylvania, Philadelphia, PA
33-year-old man with X-linked lymphoproliferative syndrome type I (XLP1). He contracted hepatitis C virus (HCV) in childhood through blood products, which progressed to cirrhosis with portal hypertension. He is one of 14 affected male family members, all others deceased due to XLP1 including his brother, who had HCV cirrhosis, and developed lymphoma. Our patient achieved sustained virologic response through treatment of HCV with Ledipasvir/Sofosbuvir. Given his high risk of lymphoma, hematopoietic stem cell transplant (HSCT) for cure of XLP1 was desired, but was precluded by cirrhosis and portal hypertension. Liver transplant was precluded by XLP1, due to concerns for infection and PTLD. His fully HLA matched sister volunteered to donate liver and bone marrow, in hopes of addressing both issues. Living related liver transplant was performed at University of Pennsylvania, followed 3 months later by allogeneic HSCT. Posttransplant he had an anastomotic biliary duct stricture requiring stenting, and chronic GVHD affecting skin and oral mucosa, well controlled on rapamycin. He has not had evidence of parenchymal liver disease or PTLD. Donor chimerism is 94%-96% in T, B and myeloid cells, and he has normal SAP protein expression in CD8+T cells and natural killer cells. Furthermore, he was shown to have normal responses to T dependent vaccine 1 year after transplant. Living related liver and HSCT from an HLA matched relative can be considered in patients with genetic immunodeficiency syndromes who suffer from complications of liver failure. In addition, life-long immunosuppression for solid organ grafting may be avoided with this approach if GVHD is successfully managed.
(204) Submission ID#606735
Droplet Digital PCR Analysis of GATA2 Deficiency
Amy P. Hsu1, Steven M. Holland, MD2
1Biologist, NIH
2Director, Division of Intramural Research, Laboratory of Clinical Immunology and Microbiology, National Institute of Allergy and Infectious Diseases, National Institutes of Health
GATA2 deficiency is a bone marrow failure syndrome caused by dominant heterozygous mutations in GATA2. Initial presentation of patients ranges from cytopenias, including loss of B-cells, NK-cells and monocytes, viral and bacterial infections, including non-tuberculous mycobacteria, myelodysplasia and lymphedema. Mutations are found throughout the gene, although missense and deletion mutations affecting the DNA-binding second zinc finger (2nd ZF) comprise more than half of the probands in our cohort (64/119). Premature stop codons, early frame shifts, splice mutations or deletions leading to mRNA instability or a protein missing the 2nd ZF represent 1/3 of our cohort (40/119) while intronic regulatory mutations are found in 10% of probands (12/119). Transcript analysis in several patients demonstrates reduced or loss of expression of one allele leading to haploinsufficiency.
Several patients meet the clinical phenotype of GATA2 deficiency without an identified mutation. In some cases, cDNA analysis of relative allelic expression has demonstrated reduced or absent transcript from one allele leading to haploinsufficiency. Patients still remain however who are suspicious for GATA2 deficiency with no mutation identified and uninformative transcript assays, often due to lack of heterozygosity of SNPs within the cDNA. GATA2 is tightly regulated, with highest expression occurring during early hematopoietic development and in hematopoietic stem cells; expression is lower or absent in fully differentiated cells making detection and functional assays challenging. We sought to evaluate GATA2 target transcripts as surrogates to determine a pattern of dysregulation in patients. Total RNA isolated from cryopreserved peripheral blood mononuclear cells was reverse transcribed to generate cDNA. We selected four known GATA2 transcriptional targets, GATA1, GATA2, TAL1 and ZFPM1 (encoding FOG1) and used droplet digital PCR to quantify transcript levels normalized to the low-expressing gene TBP1. We used samples from 9 individuals with wild-type GATA2 (WT), 5 known GATA2 mutation patients (Mut) and two individuals suspected of GATA2 deficiency but without identified mutation or allelic imbalance (Unk1, Unk2).
Transcript analysis revealed significantly decreased transcript levels of GATA1, GATA2 and TAL1 in Mut PBMCs compared to WT. Most WT samples had higher ZFPM1 transcripts than GATA2 mutated patients however it did not reach statistical significance. Strikingly, we were able to use this analysis for two individuals suspected of GATA2 deficiency. In the first case (Unk1) a 51 yr old female with primary lymphedema, hypogammaglobulinemia, recurrent infections and possible family history of leukemia was referred for GATA2 testing. No mutation was identified however it was noted that she was homozygous across the gene preventing allelic evaluation. The second patient (Unk2), a 24 yr old female, had erethemya nodosa on legs, Mycobacteria kansasii and cytopenias. In each of the targets analyzed, transcript levels from Unk1 were lower than the WT samples and in a similar range as the GATA2 mutation samples while Unk2 had a profile consistent with the WT samples. We propose the use of GATA2 targets as surrogate markers in cases where a mutation is not identified and allelic expression analysis is uninformative.
(205) Submission ID#606757
Atopic Complications Associated with Elevated IgE in a Subset of Common Variable Immunodeficiency
Nancy Yang, BS1, Jolan Walter, MD, PhD2, Jocelyn R. Farmer, MD/PhD3, Sara Barmettler, MD4
1Clinical Research Coordinator, Massachusetts General Hospital
2Associate Professor, Robert A. Good Endowed Chair and Division Chief, Division of Pediatric Allergy & Immunology, Department of Pediatrics, University of South Florida, Johns Hopkins All Children's Hospital, St. Petersburg, FL.
3Instructor, Massachusetts General Hospital
4Attending Physician, Massachusetts General Hospital
RATIONALE: Atopic manifestations in primary immunodeficiencies, such as common variable immunodeficiency (CVID), are often under-reported and under-recognized. We sought to further understand and evaluate the prevalence, type, and association with serum immunoglobulin E (IgE) for CVID patients with atopic manifestations.
METHODS: We performed a retrospective analysis of CVID patients with atopic manifestations in the Partners HealthCare CVID cohort. We evaluated baseline patient characteristics, atopic diagnoses, and serum IgE levels.
RESULTS: In the Partners CVID cohort, the average age was 52 years old (±17) and 64% female. 92/175 (52.6%) of patients had a diagnosis of asthma, with the majority of these diagnosed by an allergist (65%) or pulmonologist (16%). Eczema/atopic dermatitis was diagnosed in 47/175 patients (26%), by either an allergist (53%) or a dermatologist (8%). Allergic rhinitis was diagnosed in 50/175 (28.5%) with positive skin prick testing in 52% of these patients. Food allergy was diagnosed in 5 patients (2.9%). The median cohort serum IgE was 7.5 IU/mL. The median serum IgE was higher in patients with 2 or more atopic complications compared to those with one or less atopic condition (9 vs. 5 IU/mL), which was statistically significant (p=0.01).
CONCLUSIONS: We report higher rates of atopy than previously described in other CVID cohorts. Consistent with previous reports, we find a low median cohort serum IgE level in CVID patients compared to the general population. However, we identify a subset of patients with a predisposition towards atopy and higher IgE levels within the broader characterization of CVID, and these patients may have a more specific molecular diagnosis that leads to elevated IgE and atopic conditions. Whole exome sequencing is underway to further evaluate this hypothesis.
(206) Submission ID#606773
Abnormal B Cell Function in WHIM Syndrome
Marton Keszei, PhD1, Ashley Lynn Devonshire, MD2, Maryssa Ellison, BSc3, Krisztian Csomos, PhD4, Boglarka Ujhazi, MSc5, Melanie Makhija, MD6, Jenna Bergerson, MD/MPH7, Amer Khojah, MD8, Jolan Walter, MD, PhD9
1Research Associate, Division of Allergy/Immunology, Department of Pediatrics, Children's Research Institute, University of South Florida, St. Petersburg, FL; Department of Microbiology, Tumor and Cell Biology, Karolinska Institutet, Sweden
2Division of Allergy/Immunology, Ann & Robert H. Lurie Children's Hospital of Chicago
3Research Support Specialist, Division of Allergy/Immunology, Department of Pediatrics, Children's Research Institute, University of South Florida, St. Petersburg, FL
4Research Associate, Division of Allergy/Immunology, Department of Pediatrics, Children's Research Institute, University of South Florida, St. Petersburg, FL
5Scientist, Division of Allergy/Immunology, Department of Pediatrics, Children's Research Institute, University of South Florida, St. Petersburg, FL
6Attending, Northwestern University
7Staff Clinician, Laboratory of Clinical Immunology and Microbiology, NIAID, NIH, Bethesda, MD, USA
8Department of Rheumatology, Ann and Robert H Lurie Children's Hopital and Children's Hospital of Chicago, Chicago, IL
9Associate Professor, Robert A. Good Endowed Chair and Division Chief
Division of Pediatric Allergy & Immunology, Department of Pediatrics, University of South Florida, Johns Hopkins All Children's Hospital, St. Petersburg, FL.
WHIM (warts, hypogammaglobulinemia, infections, and myelokathexis) syndrome is a primary immunodeficiency with autosomal dominant inheritance. In most patients, the genetic cause of the disease is a gain-of-function variant in C-X-C chemokine receptor type 4 (CXCR4) that results in arrest of neutrophil migration from the bone marrow. Most patients develop hypogammaglobulinemia and early waning of antibody response with vaccination. However, the exact origin of aberrant humoral immunity in WHIM syndrome patients is yet to be clarified.
Here we describe a 4-year-old Iraqi female with a heterozygous CXCR4 p.Ser338Ter variant, which is presented with haemophilus influenzae meningitis, history of tetralogy of Fallot, early onset intermittent neutropenia, lymphopenia, recurrent bacterial and viral infections. Immunologic evaluation revealed hypogammaglobulinemia, elevated IgM level and a lack of protective vaccine titers after tetanus and Prevnar vaccinations. A bone marrow biopsy was consistent with myelokathexis.
Immune phenotyping, functional studies and apoptosis assays were performed on peripheral blood cells by flow cytometry in our WHIM patient and controls. Although we found that all lymphocyte compartments were reduced, naïve CD4 T helper cells and switched memory B cells were predominantly affected. Spontaneous apoptosis was most pronounced in B rather than T cell compartments in WHIM patients. In addition, naïve B cells easily activated and died upon activation in vitro. CXCL12, a ligand of CXCR4, induced elevated T helper cell migration and increased actin polymerization in p.Ser338Ter mutant cells.
We conclude that intrinsic B cell abnormalities, such as increased rate of apoptosis and altered activation, might be responsible for defective antibody response in WHIM patients.
(207) Submission ID#606778
Human PLCG2 Haploinsufficiency Results in NK Cell Immunodeficiency and Herpesvirus Susceptibility
Joshua B. Alinger1,
1MSTP, St. Louis Children's Hospital
Although most individuals effectively control herpesvirus infections, some suffer from unusually severe and/or recurrent infections requiring anti-viral prophylaxis. A subset of these patients possesses defects in NK cells, innate lymphocytes which recognize and lyse herpesvirus-infected cells; however, the exact genetic etiologies are rarely diagnosed. PLCG2 encodes a signaling protein in NK cell and B cell receptor-mediated signaling. Dominant-negative or gain-of-function mutations in PLCG2 cause cold urticaria, antibody deficiency, or autoinflammation. However, loss-of-function mutations and PLCG2 haploinsufficiency have never been reported in human disease. We examined 2 families with autosomal dominant NK cell immunodeficiency with mass cytometry and whole-exome sequencing to identify the cause of disease. We identified two novel heterozygous loss-of-function mutations inPLCG2 that impaired NK cell function, including calcium flux, granule movement, and target killing. Although expression of mutant PLCG2 protein in vitro was normal, phosphorylation of both mutants was diminished. In contrast to PLAID and APLAID, B cell function remained intact. Plcg2+/- mice, as well as targeted CRISPR knock-in mice, also displayed impaired NK cell function with preserved B cell function, phenocopying human PLCG2 haploinsufficiency. We report the first known cases of PLCG2 haploinsufficiency, a clinically and mechanistically distinct syndrome from previously reported mutations. Therefore, these families represent a novel disease, highlighting a role for PLCG2 haploinsufficiency in herpesvirus-susceptible patients and expanding the spectrum of PLCG2-related disease.
(208) Submission ID#606865
HSCT Utilizing Related Carrier Donor for CD40 Ligand Deficiency
Shanmuganathan Chandrakasan, MD1, Sharat Chandra, MD, MRCPCH2, Lisa Kobrynski, MD, MPH3, Suhag H. Parikh, MD4
1Asst. Professor, Children's Healthcare of Atlanta, Emory University
2Assistant Professor, UC Department of Pediatrics, Division of Bone Marrow Transplantation and Immune Deficiency, Cincinnati Childrens
3Associate Professor of Allergy, Department of Pediatrics, Emory University of School of Medicine
4Associate Professor, Duke University Medical Center
Background: HSCT is the only known curative option currently for CD40L deficiency, an X-linked disorder. In CD40L deficiency and other X-linked immune deficiencies, there is an ongoing debate regarding the use of a carrier female sibling or mother as HSCT donor. Skewed lyonization despite complete donor chimerism has raised concerns for incomplete disease control post-HSCT. No data exist regarding the efficacy of related female carrier as HSCT donor for CD40L deficiency. We herein report outcomes of three patients with CD40L deficiency who underwent HSCT using a related female carrier donor.
Method: Retrospective review of patients who received HSCT from carrier female related donor at three separate institutions.
Results: Three patients with CD40L deficiency underwent HSCT between 2016- 2018. Patient 1 had recurrent episodes of Pneumocystis jiroveci pneumonia (PJP) despite being on Bactrim and immunoglobulin replacement. Patient 2 presented with PJP and severe neutropenia. Patient 3 presented with acute respiratory failure from severe respiratory viral infections, CMV and had severe neutropenia requiring G-CSF treatment. Age at the time of HSCT ranged from 0.5-15 yrs. All three underwent reduced toxicity HSCT with busulfan and fludarabine-based preparatory regimens. Two of them received matched sibling bone marrow HSCT and one received TCR and CD19 depleted mobilized maternal PBSC haploidentical HSCT. Donor CD40L expression varied from 37% - 67% on activated CD4 cells. Immunoglobulin profile and lymphocyte subset were done in two of donors, they were within normal range for age, and none had significant infection history. No history of intermittent neutropenia or oral ulcers noted in donor and the absolute neutrophil count of the donor varied between 2500 6520 /L. Donor age ranged from 3.2 yrs 48 years.
CD34 dose ranged from 6.1 x 106 - 23.1 x 106 cells/kg and CD3 dose ranged from 1 x 105 22.1 x 107 CD3+ cells/kg. GVHD prophylaxis consisted of CSA/MMF (n=2) and TCR-a/b depletion and no CSA (n=1). Neutrophil engraftment ranged from 11- 18 days and platelet engraftment ranged from 13 28 days. None of the patients developed acute or chronic GVHD. All three patients maintain full donor myeloid chimerism at the latest testing (9 months 18 months); T cell chimerism was 100% in one and mixed in two patients (91% at nine months, 80% at 12 months). All three patients had excellent T cell immune reconstitution; Two patients came off immunoglobulin replacement 5 -11 months post HSCT, whereas the 3rd patient is IVIG dependent, though IgA level was 25 mg/dL at nine months post-transplantation. Latest evaluation, 9 18 months post-HSCT, revealed 27% - 63% CD40L expressing activated CD4 T cells, which correlated with donor CD40L expression and T-cell chimerism.
Conclusion: Our data suggest that HSCT utilizing X-linked carrier appears to be safe and results in durable engraftment with excellent humoral and cellular immune reconstitution in patients with CD40L deficiency. Longer follow-up and data from a larger cohort is needed to make a definitive determination of safety and efficacy of utilizing female carrier as HSCT donors in this disease.
(209) Submission ID#606875
A Novel Germline IKAROS C-terminal Mutation in a Patient with Burkitt Lymphoma, Lymphoproliferation and Cytopenias
Hye Sun Kuehn, PhD1, Julie E. Niemela, MS, MLS2, Tala Shahin, Mres3, Annalisa Tondo, MD4, Sara Ciullini Mannurita, MSc5, Raul Jimenez Heredia, MSc6, Kaan Boztug, MD7, Eleonora Gambineri, MD8, Sergio D. Rosenzweig, MD/PhD9,
1Staff Scientist, Immunology Service, Department of Laboratory Medicine, Clinical Center, NIH, USA
2Sequencing Laboratory, Team Leader, Immunology Service, Department of Laboratory Medicine, NIH Clinical Center, Bethesda, MD, USA
3Pre-doctoral fellow, Ludwig Boltzmann Institute for Rare and Undiagnosed Diseases, Vienna, Austria
4Medical doctor, Anna Meyer Children's Hospital, Department of Haematology-Oncology, Florence, Italy.
5Senior research fellow, University of Florence, Dep.NEUROFARBA section of Child's Health, Florence, Italy
6Technical Assistant, Ludwig Boltzmann Institute for Rare and Undiagnosed Diseases, Vienna, Austria
7Director, Ludwig Boltzmann Institute for Rare and Undiagnosed Diseases, Vienna, Austria
8Associate Professor, Anna Meyer Children's Hospital, Department of Haematology-Oncology - Bone Marrow Transplantation BMT Unit, University of Florence, Dep. Neurosciences, Psychology, Drug Research and Child Health (NEUROFARBA), Florence, Italy
9Chief, Immunology Service, Department of Laboratory Medicine, NIH Clinical Center, Bethesda, MD, USA
Background: IKAROS belongs to a hematopoietic-specific zinc-finger (ZF) family of transcription factors. After dimerizing and DNA binding to pericentric-heterochromatin (PC-HC) regions, IKAROS is described as a central regulator of lymphocyte differentiation. Somatic mutations/deletions affecting IKAROS N-terminal ZF have been identified in B-Acute lymphoblastic leukemia (ALL) patients, and germline N-terminal mutations were reported in CVID patients with progressive lack of B cells, hypogammaglobinemia, autoimmune diseases and B-ALL.
Methods: We performed targeted sequencing panel for known inborn errors of immunity disease-causing genes in a previously healthy male pediatric patient with Burkitt lymphoma, followed by benign lymphoproliferation, thrombocytopenia and neutropenia. B-cells and immunoglobulin levels were normal. IKAROS DNA-binding, nuclear localization and protein binding were evaluated by EMSA, fluorescence microscopy and immunoprecipitation. Protein modeling was also performed.
Results: A novel heterozygous germline mutation in IKAROS C-terminal ZF6 dimerization domain (p.R502L) was detected in this patient. This mutant showed normal PC-HC localization but DNA-binding was markedly reduced in terms of IKAROS dimerization and multimerization. Moreover, reduced WT-mutant binding was also detected. Mutant/WT cotransfection experiments suggest a haploinsufficient defect. Geometry based docking of wildtype IKAROS predicted that R502 is within the homodimer interface and may abolish cation-pi interactions and destabilize the IKAROS-ZF6 dimerization domain.
Conclusion: A novel germline IKAROS C-terminal mutation affecting homodimerization/multimerization and resulting in reduced DNA binding to its DNA consensus site was detected in a patient with Burkitt lymphoma, benign lymphoproliferation and cytopenias. Further studies are warranted to formally establish the casual connection between this genotype and phenotype.
(210) Submission ID#606894
Profiling Serum Antibody Specificities in Healthy Toddlers Reveals a Subgroup with Strong IgG Responses to Autoantigens and Infectious Agents
Patricia Pichilingue-Reto, MD1, Prithvi Raj, PhD2, Igor Dozmorov, PhD3, Quan-Zhen Li, MD, PhD4, Edward Wakeland, PhD5, Nancy Kelly, MD6, Maria Teresa de la Morena, MD7, Nicolai S. van Oers, PhD8
1Research Fellow, UT Southwestern Medical Center
2Assistant Professor, UT Southwestern Medical Center
3Associate Professor, UT Southwestern Medical Center
4Associate Professor, Microarray Core Facility and Department of Immunology, University of Texas Southwestern Medical Center, Dallas, TX, USA
5Professor, UT Southwestern Medical Center
6Professor, Children's Health Dallas
7Professor, Seattle Children's Hospital
8Associate Professor, Ut Southwestern Medical Center
Introduction/Background: The immune system of a newborn is relatively immature at birth, with antibodies protecting against infections primarily maternal in origin. In their first two years of life, an infant/toddler begins making antibodies in response to infections, environmental exposures, and vaccinations. However, the specificity of these antibodies towards self-antigens, infections, and vaccinations and how they vary among individuals is poorly characterized.
Objectives: The goal of the study is to characterize the serum IgG and IgM antibody specificities in healthy toddlers at 1 and 2 years of age towards a panel of autoantigens, infectious agents, and vaccine antigens. DNA sequencing is used to identify genetic polymorphisms that may explain differences in immune responses in the cohort.
Methods: Blood samples are obtained from a prospective cohort of 1000 healthy 1- and 2-year old toddlers, with the 2-year-old group including repeat screens. The blood is obtained as part of standard of care wellness checks at Childrens Health Pediatric Group in Dallas, Texas. Serum profiling is done with an antigen array that reveals IgG and IgM responses to autoantigens, pathogens, vaccine antigens, and allergens. Low, intermediate and high responders are defined on the SD below and above the mean fluorescence intensity of antibody reactivity complied for the entire cohort. Clinical information for each healthy toddler is compiled. In addition, a targeted DNA sequencing is performed to identify genetic polymorphisms associated with immune response modifiers.
Results: Serum profiling of 160 samples to date reveals a stratification of the healthy toddlers into low, intermediate, and high IgG response groups. Sixteen % are high responders, with their IgGs recognizing many self-antigens and infectious agents. Longitudinal follow-up of several subjects suggests that the low, intermediate, and high serum responses are relatively stable over time. Interestingly, 26% and 9% of the cohort had moderate and high anti-nuclear antibody (ANA) titers, respectively. Comparing clinical data reveals a significant correlation with the high IgG responders and a family history of asthma and maternal gestational diabetes. Targeted DNA sequencing in the high responder group revealed a strong genetic association signal at the HLA locus, with genetic polymorphisms at this locus associated with high ANA and IgG titers to many antigens. The serum profiling is ongoing, with more healthy toddlers currently being screened.
Conclusions: Healthy toddlers can be stratified into 3 groups based on their IgG antibody reactivity: Low, intermediate and high. Genetic sequencing reveals polymorphisms in the high group that may reveal autoimmune potential. Findings from our study may support implementation of a new wellness screen to identify toddlers at risk for immune system abnormalities later in life.
(211) Submission ID#606898
Late Adaptive Immune Dysfunction 34 Years After Unconditioned Allogeneic Stem Cell Transplant for T-B-NK+ SCID with Novel RAG1 Mutations
Lauren E. Franzblau, MD1, Christian A. Wysocki, MD, PhD2
1Resident Physician, Department of Internal Medicine, UT Southwestern Medical Center, Dallas, TX
2Assistant Professor, Director of the Jeffrey Modell Foundation Diagnostic and Research Center, Division of Allergy and Immunology, Departments of Internal Medicine and Pediatrics, UT Southwestern Medical Center/Children's Medical Center Dallas, TX
36 year old male presented reporting a history of severe combined immunodeficiency (SCID), status post unconditioned sibling-donor allogeneic hematopoietic stem cell transplant at 18 months of age. Since then he has been dependent on monthly intravenous immunoglobulin. He denied opportunistic infections but reported 4-5 lifetime pneumonias and one episode of staphylococcal bacteremia. His problems in recent years included chronic productive cough, chronic sinusitis, asthma, and eczema. Initial workup revealed very low T cells which were >95% memory, absent B cells, and quantitatively normal NK cells. TRECs were absent. TCR V beta repertoire analysis indicated limited diversity. Chimerism studies demonstrated 96% donor T cells, and 100% host NK and myeloid cells. Chest CT revealed bronchiectasis and prominent bilateral tree-in-bud opacities. Spirometry showed severe fixed obstruction. Bronchoalveolar lavage samples grew Mycobacterium abscessus.
We pursued genetic diagnosis, which identified bi-allelic frameshift mutations in the RAG1 gene which had not been previously described: c.967delG (p.V323SfsX22) and c.1048_1075del128insAAAAGAGTG (p.V350KfsX47).
Taken together, his presentation suggested significant immune dysfunction had evolved since transplant leading to extensive pulmonary nontuberculous mycobacterial infection and possible bronchiolitis obliterans. He therefore will undergo a subsequent unconditioned CD34+ stem cell boost from his sister, the original donor, once he completes Mycobacterium abscessus treatment. This case highlights the potential long-term immune dysfunction which may evolve after unconditioned allogeneic stem cell transplant for SCID, in which full engraftment in all myeloid and lymphoid compartments is not expected. It also highlights the importance of guideline-driven follow-up of these patients to monitor for said dysfunction, to prevent serious infection and long-term sequelae.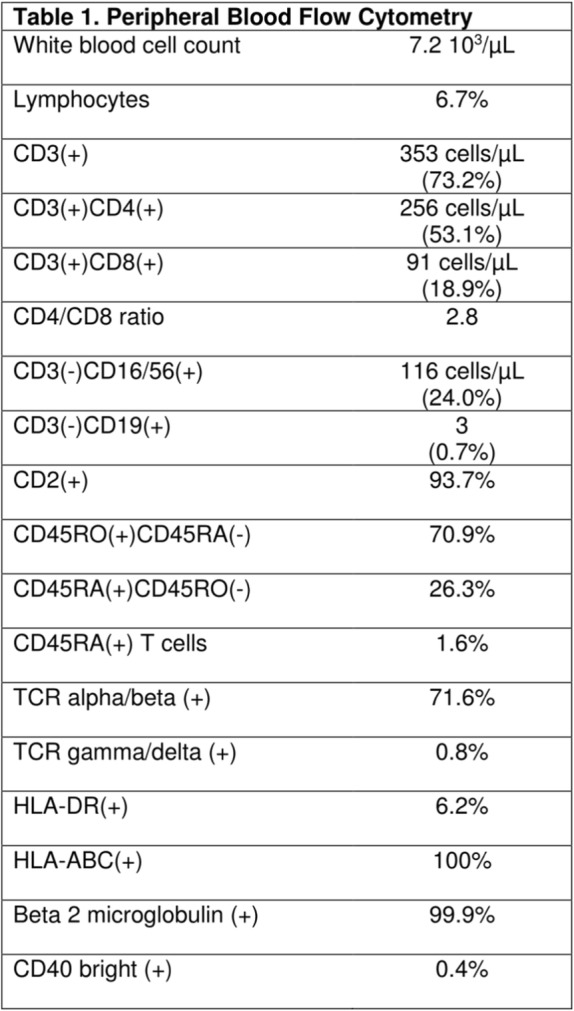
(212) Submission ID#606899
Defective B Cell Fitness Impairs Mutation Away from Self and Sustains Red Blood Cell Reactivity in Hypomorphic RAG Deficiency
Krisztian Csomos, PhD1, Boglarka Ujhazi, MSc2, Kevin Wu3, Rachel Cruz3, Matthew Stowell3, Marton Keszei, PhD4, Waleed Al-Herz, MD5, Joseph D Hernandez, MD6, Sinisa Savic, PhD7, Ravishankar Sargur, MD8, Snezhina Mihailova, MD9, Svetlana O. Sharapova, PhD10, Manish Butte, MD, PhD11, Jolan Walter, MD, PhD12
1Research Associate, Division of Allergy/Immunology, Department of Pediatrics, Children's Research Institute, University of South Florida, St. Petersburg, FL
2Scientist, Division of Allergy/Immunology, Department of Pediatrics, Children's Research Institute, University of South Florida, St. Petersburg, FL
3Student, Division of Allergy/Immunology, Department of Pediatrics, Children's Research Institute, University of South Florida, St. Petersburg, FL
4Research Associate, Division of Allergy/Immunology, Department of Pediatrics, Children's Research Institute, University of South Florida, St. Petersburg, FL; Department of Microbiology, Tumor and Cell Biology, Karolinska Institutet, Sweden
5Associate Professor of Pediatrics, Pediatrics Department, Faculty of Medicine, Kuwait University
6Allergist / Immunologist, Department of Pediatrics, Division of Allergy, Immunology and Rheumatology, Stanford University, Stanford CA
7Clinical Associate Professor, Department of Clinical Immunology and Allergy, St. Jamess University Hospital, Leeds, United Kingdom
8Immunologist, Department of Clinical Immunology and Rheumatology, Hannover Medical School, Hannover, Germany
9Immunologist, University Hospital Alexandrovska, Department of Clinical immunology, Sofia, Bulgaria
10Leading researcher, Research Department, Belarusian Research Center for Pediatric Oncology, Hematology and Immunology
11Division of Allergy/Immunology Chair, Division of Immunology, Allergy, and Rheumatology, Dept. of Pediatrics and Jeffrey Modell Diagnos-tic and Research Center, University of California, Los Angeles
12Associate Professor, Robert A. Good Endowed Chair and Division Chief, Division of Pediatric Allergy & Immunology, Department of Pediatrics, University of South Florida, Johns Hopkins All Children's Hospital, St. Petersburg, FL.
Somatic hypermutation (SHM) in the B cell receptor (BCR) heavy (IGH) and light chain genes promotes affinity maturation and also mutation away from self-reactivity, therefore serves as an important peripheral tolerance checkpoint. As an example, unmutated BCR IGHV4-34 genes give rise to antibodies that bind to I/i antigen on red blood cells (RBC) and may elicit cold agglutinin disease (CAD), a variant of autoimmune hemolytic anemia (AIHA). In case of healthy individuals, frequent SHMs in the I/i binding site of BCR IGHV4-34 genes decrease RBC reactivity and CAD.
Patients with primary immunodeficiencies (PID) paradoxically develop autoimmune diseases, including autoimmune cytopenias, especially AIHA. It is unclear if impaired SHM of BCR, in particular mutation away from i/I binding, is relevant in the development of RBC reactivity and consequently AIHA in a PID background.
Our studies focus on PID patients with hypomorphic recombination activating gene (RAG1 and 2), combined immunodeficiency phenotype and history of autoimmunity, in particular AIHA (RAG CID/AI). We detected increased frequency of unmutated IGHV4-34 BCR in memory B cell repertoires of RAG-CID/AI patients as well as elevated titer of unmutated IGHV4-34 antibodies in the patients' plasma. Lower level of SHM likely reflect abnormal germinal center (GC) reaction. As RAG1 and 2 heterotetramer primarily shapes the pre-immune T and B cell repertoire, we studied the interaction of follicular helper T cells (Tfh) and naive B cells via in vitro co-culture experiment. Interestingly, Tfh cells from RAG CID/AI patients exhibited highly activated phenotype with increased expression of CD40L and IL-21 compared to healthy controls and were able to initiate exaggerated response (class switching and SHM) of healthy donor naive B cells. On the contrary, in vitro activated naive B cells from RAG CID/AI patients showed impaired proliferation, class switching and decreased level of SHM with diminished induction of genes involved T cell co-stimulation (CD40, IL-21R) and SHM (AICDA, repair enzymes) compared to healthy donor naive B cells indicating intrinsic defect in patient B cells. Furthermore, B cells from RAG CID/AI patients also showed increased apoptosis and accumulation of Gamma-H2AX foci at steady state indicating reduced cellular fitness.
These findings suggest that the development of AIHA is a multifactorial process in partial RAG deficiency. Our studies highlight that impaired germinal center reaction is an important tolerance checkpoint with the inability of patient's B cells to respond to hyperactive Tfh cells and introduce proper level of SHM. Hence, we propose that B cell fitness is compromised which impairs proper GC interaction, SHM, including mutation away from self and sustains RBC reactivity in hypomorphic RAG deficiency.
(213) Submission ID#606901
Novel Compound Heterozygous Mutations in Forkhead Box N1 (FOXN1) Cause a Severe Immunodeficiency Without Alopecia or Nail Dystrophy
Qiumei Du, PhD1, Larry Huynh, BSc2, Fatma Coskun, BSc3, Igor Dozmorov, PhD4, Prithvi Raj, PhD5, Shaheen Khan, PhD5, Christian A. Wysocki, MD, PhD6, M. Louise Markert, MD, PhD7, Maria Teresa de la Morena, MD8, Nicolai S. van Oers, PhD4
1Instructor, UT Southwestern Medical Center
2Medical Student, UT Southwestern Medical Center
3Graduate Student, UT Southwestern Medical Center
4Associate Professor, UT Southwestern Medical Center
5Assistant Professor, UT Southwestern Medical Center
6Assistant Professor, Director of the Jeffrey Modell Foundation Diagnostic and Research Center, Division of Allergy and Immunology, Departments of Internal Medicine and Pediatrics, UT Southwestern Medical Center/Children's Medical Center Dallas, TX
7Professor, Duke University
8Professor, Department of Pediatrics, Division of Immunology, University of Washington and Seattle Childrens Hospital
Introduction/Background: The Forkhead Box N1 (FOXN1) transcription factor is an essential regulator of T cell development, affecting the differentiation and expansion of thymic epithelial cells (TECs). Autosomal recessive mutations in FOXN1 cause a T-B+NK+ lymphocyte phenotype due to a thymic aplasia in conjunction with alopecia universalis and nail plate dystrophy resulting from keratinocyte dysregulation. This is a classic nude/SCID (OMIM # 600838) phenotype. We report on the identification of two independent patients, identified through newborn screening with absent TRECs and with a T-NK+B+ SCID phenotype who presented with a T cell lymphopenia who had compound heterozygous mutations in FOXN1. Notably, these individuals had normal hair and nail beds.
Objectives: To determine whether distinct compound heterozygous mutations in FOXN1 cause a novel T-NK+B+ phenotype in the absence of a classic nude presentation.
Methods: Mice were generated by CRISPR/Cas technology to genocopy the FOXN1 compound heterozygous mutations identified in one of the human patients. Thymopoiesis and hair follicle extrusion was analyzed in the various heterozygous and homozygous mutant mice. Gene expression analyses of the hypoplastic and normal- sized thymii and the developing skin were performed. In addition, a structure-function analysis was performed with luciferase reporter assays using 9 distinct and previously unreported FOXN1 mutations uncovered in patients who presented with low TRECs.
Results: Mice harboring compound heterozygous mutations in Foxn1 that match the human patient phenocopy the T-B+NK+ SCID phenotype with normal hair and nails. A functional characterization of the diverse Foxn1 mutations suggests that the severity of the block in thymopoiesis depends on whether the mutations affect the DNA binding or transactivation domains of Foxn1. A 5-amino acid segment at the end of the DNA binding domain appears to be essential for TEC development. However, this segment is not required for normal keratinocyte functions in the skin and nail plate. Gene expression comparisons are revealing key targets of Foxn1 that suggest a dichotomy in its function in the thymus versus the skin.
Conclusions: Novel compound heterozygous mutations in FOXN1 are causal to a T-NK+B+ phenotype with normal hair shaft extrusion and nail plate extension. This differs from the classic nude/SCID (OMIM # 600838) reported for individuals with autosomal recessive mutations in FOXN1.
(214) Submission ID#606903
Neutralizing anti-IL-6-autoantibodies Are a Risk Factor for Pyogenic Bacterial Infections
Stephanie Heller1, Uwe Kölsch, MD2, Christoph Tersch, PhD3, Alexej Knaus, PhD4, Lindsey Rosen, PhD5, Rainer Döffinger, PhD6, Laura Perez, PhD7, Charlotte Schaefer8, Michael Kirschfink, MD, PhD9, Mark van der Linden, MD10, Marten Jäger, PhD11, Jacinta Bustamante, MD, PhD12, Peter Kühnen, MD. PhD13, Michail Lionakis, MD, Sc.D.14, Sarah Browne, MD15, Nadine Unterwalder2, Heiko Krude, MD, PhD16, Peter Krawitz, MD, PhD17, Ansgar Schulz, MD, PhD18, Christoph Bührer, MD, PhD19, Hans-Dieter Volk, MD, PhD17, Steven Holland, MD. PhD20, Jean-Laurent Casanova, MD, PhD21, Anne Puel, PhD22, Stefan Rose-John, PhD23, Ulf Reimer, PhD24, Christian Meisel, MD2, Horst von Bernuth, MD, PhD25
1PhD Student, Department of Pediatric Pulmonology, Immunology and Intensive Care Medicine, Charité University Medicine, Berlin, Germany
2Senior Scientist, Labor Berlin, Immunologie, Berlin, Germany
3Researcher, JPT Peptide Technologies GmbH, Berlin, Germany
4Researcher, Berlin-Brandenburg Center for Regenerative Therapies, Berlin, Germany
5Researcher, Laboratory of Clinical Infectious Diseases (LCID), National Institutes of Health, National Institutes of Allergy and Infectious Diseases, NIH/NIAID, Bethesda, Maryland
6Senior investigator, Department of Clinical Biochemistry and Immunology, Addenbrookes Hospital, Cambridge, UK
7Researcher, Service of Immunology and Rheumatology, Garrahan National Pediatric Hospital, Buenos Aires, Argentina
8Student, Kinderklinik der Charité, Universitätsmedizin Charité Berlin
9Senior Investigator, Institute of Immunology, University Heidelberg, Heidelberg, Germany
10Senior Investigator, German National Reference Center for Streptococci, Department of Medical Microbiology, University Hospital RWTH Aachen, Aachen, Germany
11Statistician, Berlin-Brandenburg Center for Regenerative Therapies, Berlin, Germany
12Senior Investigator, Laboratory of Human Genetics of Infectious diseases, Necker Branch, INSERM UMR 1163, Imagine Institute, Necker hospital for Sick children, Paris, France
13Clinical Investigator, Department of Pediatric Endocrinology, Charité University Medicine, Berlin, Germany
14Senior Investigator, Fungal Pathogenesis Section, Laboratory of Clinical Immunology & Microbiology (LCIM) , National Institute of Allergy & Infectious Diseases (NIAID), NIH
15Senior Clinical Advisor, Laboratory of Clinical Infectious Diseases (LCID), National Institutes of Health, National Institutes of Allergy and Infectious Diseases, NIH/NIAID, Bethesda, Maryland, USA
16Senior Investigator, Department of Pediatric Endocrinology, Charité University Medicine, Berlin, Germany
17Senior Investigator, Berlin-Brandenburg Center for Regenerative Therapies, Berlin, Germany
18Senior Investigator, Department of Pediatrics, University Medical Center Ulm, Ulm, Germany
19Head of Department, Department of Neonatology, Charité University Medicine, Berlin, Germany
20Senior Investigator, Laboratory of Clinical Infectious Diseases (LCID), National Institutes of Health, National Institutes of Allergy and Infectious Diseases, NIH/NIAID, Bethesda, Maryland, USA
21Senior Investigator, St. Giles Laboratory of Human Genetics of Infectious Diseases, Rockefeller Branch, The Rockefeller University, New York, USA
22Senior Investigator, Laboratory of Human Genetics of Infectious Diseases, Necker Branch, INSERM U1163, Necker Enfants Malades Hospital, Paris, France
23Senior Investigator, Institute of Biochemistry, University of Kiel, Kiel, Germany
24Senior Scientist, JPT Peptide Technologies GmbH, Berlin, Germany
25Senior Investigator, Department of Pediatric Pulmonology, Immunology and Intensive Care Medicine, Charité University Medicine, Berlin, Germany
Neutralizing autoantibodies (autoAbs) against cytokines increase the susceptibility for selected infections (e.g. anti-IFN-autoAbs for non-tuberculous mycobacteria and non-typhoid salmonella, anti-IL-17-AutoAbs for mucocutaneous candidiasis and anti-GM-CSF-auotAbs for infections by Cryptococcus, Nocardiae and Aspergillus spp). However, the role of anti-IL-6-AutoAbs is less clear. IL-6 is a key mediator of the acute-phase response and released early in bacterial infections. Patients with impaired signaling or affected production of IL-6 are at increased risk for severe bacterial infections. Only three patients with high-titer and neutralizing anti-IL-6-AutoAbs who suffered from severe infections caused by S. aureus, S. intermedius and E. coli have been described so far. To investigate the prevalence of anti-IL-6-AutoAbs in patients with bacterial infections, we investigated a cohort of 350 patients and identified three further patients, all previously healthy, with neutralizing auotAbs against IL-6 who hardly developed an acute-phase response. The first patient suffered from life-threatening pneumonia caused by S. pneumonia, the second patient developed a submandibular abscess and septic arthritis caused by S. pyogenes and the third patient suffered from life-threatening pneumonia caused by S. aureus. We also discovered neutralizing anti-IL-6-AutoAbs in two adults among a cohort of patients with autoimmune diseases (n = 564), in one adolescent among a cohort of obese individuals (n = 455) as well as in three mothers of neonates with impaired IL-6 signaling. So far none of the later individuals developed a severe bacterial infection. This suggests that naturally occurring and neutralizing anti-IL-6-AutoAbs are a risk factor for severe bacterial infections yet with incomplete penetrance.
(215) Submission ID#606931
Persistent Transaminitis in COPA Syndrome
Silpa S. Thaivalappil, MD, MPH1, Andrea Garrod, MD2, Robin LeGallo, MD3, Stephen Borowitz, MD4, Levi Watkin, PhD5, Monica Lawrence, MD6
1Resident, UVA Pediatrics
2Assistant Professor of Pediatrics, Department of Pediatric Pulmonology, UVA
3Assistant Professor of Pathology, Department of Pathology, UVA
4Professor of Pediatrics, Department of Pediatric Gastroenterology, UVA
5Postdoctoral Associate, Department of Allergy and Immunology, Baylor College of Medicine
6Assistant Professor of Medicine and Pediatrics, Department of Allergy and Immunology, UVA
Introduction: COPA syndrome is a recently described monogenic immunodysregulatory syndrome. The COP protein, encoded for by the COPA gene, is expressed in all cell types and is involved in trafficking from the Golgi complex to the endoplasmic reticulum (1). The most common clinical features of COPA syndrome are interstitial lung disease, pulmonary cysts or follicular bronchiolitis, pulmonary hemorrhage, arthritis, glomerular disease, and autoantibody development (2, 3). Atypical features of COPA syndrome identified thus far include: extrapulmonary cysts in the liver and kidney, renal and neuroendocrine malignancies, autoimmune neurological disorders such as neuromyelitis optica, and infections, such as meningitis (4).
Clinical Case: We present a case of a 2 year-old male with COPA syndrome (de novo heterozygous mutation in Exon 9, c.715G>C; p.Ala239Pro) manifesting as lymphocytic interstitial pneumonitis, peripheral blood B-cell lymphocytosis, mediastinal lymphadenopathy and persistent transaminitis (ALT and AST 100-400 U/L, nl AST<35 U/L, ALT <55U/L) with normal bilirubin, alkaline phosphatase and PT/INR. The transaminitis was noted prior to diagnosis of COPA syndrome, and has persisted despite seven months of therapy with pulse dose steroids, two cycles of rituximab and maintenance therapy with hydroxychloroquine and prednisone. He has had a normal CK and aldolase excluding muscle injury as a source of his transaminitis. A congenital cholestasis panel was normal. Markers of autoimmune liver disease including ANA, anti-liver kidney microsomal antibody and anti-smooth muscle were negative. Serum ceruloplasmin and alpha-1-antitrypsin level were normal and celiac serologies, were negative. Liver ultrasound was normal. A liver biopsy did not demonstrate inflammatory changes, hepatocyte necrosis, mononuclear cell infiltrates or fibrosis. Nonspecific biopsy findings included occasional intraparenchymal neutrophils. It is unclear if these scattered neutrophils and the transaminitis are due to an early as yet unidentified autoimmune process, perhaps in response to hepatocellular stress exacerbated by the COPA mutation.
Discussion: Liver involvement has not been reported in COPA syndrome. We describe a child with COPA syndrome who has had chronic transaminitis with no clear alternative cause. If the phenotypic spectrum of COPA syndrome involves the liver, it may limit immunomodulatory options for the treatment of this disease.
References:
(1) Watkin, L. B., Jessen, B., Wiszniewski, W., Vece, T. J., Jan, M., Sha, Y., ... & Forbes, L. R. (2015). COPA mutations impair ER-Golgi transport and cause hereditary autoimmune-mediated lung disease and arthritis. Nature genetics, 47(6), 654.
(2) Vece, T. J., Watkin, L. B., Nicholas, S. K., Canter, D., Braun, M. C., Guillerman, R. P., ... & Forbes, L. R. (2016). Copa syndrome: a novel autosomal dominant immune dysregulatory disease. Journal of clinical immunology, 36(4), 377-387.
(3) Tsui, J. L., Estrada, O. A., Deng, Z., Wang, K. M., Law, C. S., Elicker, B. M., ... & Helfgott, S. M. (2018). Analysis of pulmonary features and treatment approaches in the COPA syndrome. ERJ open research, 4(2), 00017-2018.
(4) Taveira-DaSilva, A. M., Markello, T. C., Kleiner, D. E., Jones, A. M., Groden, C., Macnamara, E., ... & Moss, J. (2018). Expanding the phenotype of COPA syndrome: a kindred with typical and atypical features. Journal of medical genetics, jmedgenet-2018.
(216) Submission ID#606932
The Forest and the Trees: Machine Learning to Classify Cases of Suspected Inborn Errors of Immunity Using Decision Tree and Random Forest Algorithms
Saul O. Lugo Reyes, MD, MS1, Elisa Hierro, MD2, Ana Belen Ramírez López, MD2, Elma Fuentes Lara, MD3, Samuel Rocha, MS4, Chiharu Murata, MS5, Alfredo Mendez Barrera, MS4
1Researcher, Immunodeficiencies Research Unit, National Institute of Pediatrics, Mexico City
2Social service intern, Immunodeficiencies Research Unit, National Institute of Pediatrics
3Pediatrics Resident, Pediatrics Hospital, 21st Century National Medical Center, Mexican Institute of Social Security
4Researcher, Data Science department, Mexican Autonomous Institute of Technology
5Researcher, Department of Research Methodology, National Institute of Pediatrics
BACKGROUND: Inborn errors of immunity constitute a heterogeneous group of over 400 individually rare congenital diseases that involve genes coding for proteins of the immune system, and which result in increased susceptibility to infection, inflammation, autoimmunity, allergy and cancer. The complexity of the diagnostic task, and the intrinsic biases and limitations of the human mind, can be aided by computational tools. Among the available machine learning approaches, decision tree algorithms select the best node to split based on entropy and information gain; random forests build hundreds or thousands of decision trees randomly (bootstrapping), to improve accuracy and reduce overfitting.
AIM: To implement a machine learning-assisted clinical decision support system for the diagnosis of inborn errors of immunity (IEI).
METHODS: With a local database of patients with suspected IEI, we built a decision tree using c4.5 DTC, and a Random Forest on Python 3 (Jupyter Notebook, SciKit, MathPlotLib, Pandas, Numpy). The database was obtained by conducting an electronic search on MedSys of patients with the term immunodeficiency in their electronic medical records, and then hand-picking cases in which an IEI had been confirmed or ruled out. It consisted of 234 patients, of which 201 had been diagnosed with IEI. We first split the dataset randomly into training (70%) and testing (30%) sets. The decision tree was tasked with classifying correctly PID or NOT. After running the algorithm in the training set, we evaluated in the testing set. The random forest classified all cases by majority vote into nine groups (0 to 8), according to the IUIS PID group. Next, we repeated the process on a larger scale with a dataset of 2,400 patients from USIDNET. Accuracy was assessed by out-of-bag (OOB) error estimates.
RESULTS: Accuracy was greater than 95% for the local dataset (PID/Not, 9 groups), and for the USIDNET dataset (9 groups). We provide a list of decision nodes and a diagnostic route with those questions that achieved a greater information gain and less entropy. This might help clinicians direct their interrogation and diagnostic approach of suspected IEI patients.
DISCUSSION: We built two classification models. Decision trees lend themselves more easily to learning and deriving rules of thumb from their sequences. Random forests are more robust and better suited for categoric (as opposed to binary) classification. We next want to develop a chatbot that will ask relevant questions in optimal sequence, and extract undiagnosed patients with suspected IEI, based on statistical red flags.
(217) Submission ID#606934
Evaluation of Novel STAT1 Mutations: Phosphorylation, Luciferase Assay or Both?
Alexander Vargas Hernández1, Sarah E. Henrickson, MD, PhD2, Jennifer Heimall, MD3, Monica Lawrence, MD4, Neha Seth5, Christian A. Wysocki, MD, PhD6, Lisa R. Forbes, MD5
1PostDoc Associate, PhD, Department of Pediatrics, Baylor College of Medicine. Houston, TX, USA.
2Attending Physician, The Children's Hospital of Philadelphia, Divsion of Allergy Immunology
3Assistant Professor, The Children's Hospital of Philadelphia
4Assistant Professor of Medicine and Pediatrics, Department of Allergy and Immunology, UVA
5Assistant Professor, Department of Pediatrics, Baylor College of Medicine, Houston, TX, USA.
6Assistant Professor, Director of the Jeffrey Modell Foundation Diagnostic and Research Center, Division of Allergy and Immunology, Departments of Internal Medicine and Pediatrics, UT Southwestern Medical Center/Children's Medical Center Dallas, TX
Submission Text
Alexander Vargas-Hernández1, 2, Sara Henrickson3, 4, Jennifer Heimall3, 4, Monica G. Lawrence5, Neha Seth1, Christian A. Wysocki6, Lisa R. Forbes1, 2
1Department of Pediatrics, Baylor College of Medicine, Houston, TX, USA.
2Texas Childrens Hospital, Center for Human Immunobiology, Department of Allergy, Immunology, and Rheumatology, Houston, TX, USA.
3Division of Allergy and Immunology, Childrens Hospital of Philadelphia, Philadelphia, PA, USA
4Department of Pediatrics, Perelman School of Medicine at University of Pennsylvannia, Philadelphia, PA, USA
5Division of Asthma, Allergy & Immunology University of Virginia, Charlottesville, VA
6Division of Allergy and Immunology, Departments of Internal Medicine and Pediatrics, UT Southwestern Medical Center/Childrens Medical Center Dallas, TX.
Background: In humans, biallelic STAT1 lost-of-function (LOF) mutations lead to a very low or complete absence of the wild-type (WT) protein. Whereas, heterozygous mutations can lead to partial loss of function. These patients are susceptible to mycobacteria and herpes virus infections. On other hand, heterozygous gain-of-function (GOF) mutations in the STAT1 gene result in a hyperphosphorylated state where patients develop recurrent or persistent chronic mucocutaneous candidiasis (CMC), other cutaneous mycosis, bacterial infections, disseminated dimorphic fungal infections, viral infections and autoimmune disease.
Methods: In this study, we evaluated 4 novel STAT1 mutations, three GOF and one LOF. In vitro, PBMCs from these patients were stimulated with IFN- and IFN- for 30, 60, and 120 minutes and levels of phospho-STAT1 were measured by flow cytometry. The STAT1 phosphorylation and activity (firefly and Renilla luciferase activities) were evaluated in U3A-STAT1 deficient cells transfected with a reporter plasmid (for luciferase), WT or mutant-STAT1 plasmids.
Results: We observed higher levels of STAT1 phosphorylation after two hours of stimulation from three GOF mutations compared to WT. However, a LOF mutation showed absent STAT1 activation at baseline and in response to IFN- and IFN-. Luciferase reporter assay confirmed gain of function and loss of function STAT1 activity observed by flow cytometry.
Conclusions: Using flow cytometry followed by a luciferase assay, we confirmed four novel STAT1 mutations. Measuring phosphorylation of STAT1 by flow cytometry is sufficient to determine whether the STAT1 mutation is disease causing. This assay can be translated to a clinically accessible test for STAT1 related disease.
(218) Submission ID#606967
Failing to Make Ends Meet: The Broad Clinical Spectrum of DNA Ligase IV Deficiency Case Series and Review of the Literature
Aidé Tamara Staines Boone, MD1, Ivan K. Chinn, MD2, Carmen Alaez-Versón, PhD3, Marco A. Yamazaki-Nakashimada, MD4, Karol Carrillo-Sánchez, PhD5, Maria de la Luz García-Cruz, MD6, M. Cecilia Poli, MD, PhD7, Edith González Serrano, MD8, Edgar A. Medina Torres, PhD9, David Muzquiz Zermeño, MD10, Lisa R. Forbes, MD7, Francisco J. Espinosa-Rosales, MD, PhD9, Sara E. Espinosa-Padilla, MD, PhD11, Jordan S. Orange, MD, PhD12, Saul O. Lugo Reyes, MD, MS13
1Attending immunologist, Immunology Department, Hospital de Especialidades UMAE 25, IMSS
2Assistant Professor, Pediatric Allergy and Immunology, Baylor College of Medicine, Houston, TX
3Head, Genomic Diagnostic Laboratory, National Institute for Genomic Medicine
4Attending immunologist, Clinical Immunology Department, National Institute of Pediatrics
5Researcher, Genomic Diagnostic Laboratory, National Institute for Genomic Medicine (INMEGEN)
6Attending immunologist, Otolaryngology department, National Institute of Respiratory Diseases
7Assistant Professor, Department of Pediatrics, Baylor College of Medicine, Houston, TX, USA
8Researcher, National Institute of Pediatrics
9Researcher, Immunodeficiencies Research Unit, National Institute of Pediatrics
10Attending immunologist, Immunology Department, Hospital de Especialidades, UMAE 25 IMSS
11Head, Immunodeficiencies Research Unit, National Institute of Pediatrics
12Professor and Chair, Department of Pediatrics, Columbia University Irving Medical Center, New York, NY
13Researcher, Immunodeficiencies Research Unit, National Institute of Pediatrics, Mexico City
DNA repair defects are inborn errors of immunity that result in increased apoptosis and oncogenesis. DNA Ligase 4-deficient patients suffer from a wide range of clinical manifestations since early in life, including: microcephaly, dysmorphic facial features, growth failure, developmental delay, mental retardation; hip dysplasia, and other skeletal malformations; as well as a severe combined immunodeficiency, radiosensitivity and progressive bone marrow failure; or, they may present later in life with hematological neoplasias that respond catastrophically to chemo- and radiotherapy; or, they could be asymptomatic. We describe the clinical, laboratory and genetic features of five Mexican patients with LIG4 deficiency, together with a review of 36 other patients available in PubMed Medline. Four out of five of our patients are dead from lymphoma or bone marrow failure, with severe infection and massive bleeding; the fifth patient is asymptomatic despite a persistent CD4+ lymphopenia. Most patients reported in the literature are microcephalic females with growth failure, sinopulmonary infections, hypogammaglobulinemia, very low B-cells, and radiosensitivity; while bone marrow failure and malignancy may develop at a later age. Dysmorphic facial features, congenital hip dysplasia, chronic liver disease, gradual pancytopenia, lymphoma or leukemia, thrombocytopenia and gastrointestinal bleeding have been reported as well. Most mutations are compound heterozygous, and all of them are hypomorphic, with two common truncating mutations accounting for the majority of patients. Stem-cell transplantation after reduced intensity conditioning regimes may be curative.
(219) Submission ID#606973
The Clinical and Genetic Spectrum of RAG Deficiency Including a c.256_257delAA Founder Variant in Slavic Countries
Svetlana O. Sharapova, PhD1, Magorzata Skomska-Pawliszak, MD2, Yulia Rodina, MD3, Beata Wolska-Kunierz, MD, PhD2, Nel Dbrowska-Leonik, MD2, Olga Pashchenko, MD, PhD4, Boena Mikou, MD, PhD5, Srdjan Pasic, MD, PhD6, Tomas Freiberger, MD, PhD7, Renata Formánková, MD, PhD8, Tomá Milota, MD9, Anna Szaarska, MD, PhD10, Maciej Siedlar, MD, PhD11, Tadej Avin, MD, PhD12, Gaper Markelj, MD13, Peter inár, MD, PhD14, Krzysztof Kalwak, MD, PhD15, Teresa Jackowska, MD, PhD16, Sylwia Kotan, MD, PhD17, Katarzyna Drabko, MD, PhD18, Alenka Gagro, MD, PhD19, Elisaveta Naumova, MD, PhD20, Magorzata Pac, MD, PhD2, Katarzyna Bbol-Pokora, MD, PhD21, Dzmitry Varabyou, PhD22, Barbara Barendregt, PhD23, Elena Raykina, MD, PhD24, Tatiana Varlamova, MD25, Anna Pavlova, MD25, Irina Mersiyanova, MD25, Hana Grombirikova, MD, PhD26, Marua Debeljak, PhD13, Anastasia Bondarenko, MD, PhD27, Larysa Kostyuchenko, MD, PhD28, Marina Guseva, MD29, Luigi D. Notarangelo, MD, PhD30, Jolan Walter, MD, PhD31, Irina Kondratenko, MD, PhD32, Anna edivá, MD, PhD33, Mirjam van der Burg, PhD34, Natalia Kuzmenko, MD, PhD25, Ewa Bernatowska, MD, PhD35, Olga Aleinikova, MD, PhD36
1Leading researcher, Research Department, Belarusian Research Center for Pediatric Oncology, Hematology and Immunology
2Doctor, Department of Immunology, Childrens Memorial Health Institute
3Allergist-Immunologist, Department of Immunology, National Medical and Research Center of Pediatric Hematology, Oncology and Immunology named after Dmitry Rogachev
4Doctor, Immunology Department, Pirogov Russian National Research Medical University
5Doctor, Department of Pediatrics, Rheumatology, Immunology and Metabolic Bone Diseases, Medical University of Bialystok
6Doctor, Pediatric Immunology, Mother and Child Health Institute, Medical Faculty, University of Belgrade
7Head of laboratory, Molecular Genetics Lab, Center for Cardiovascular Surgery and Transplantation, Masaryk University, CEITEC and Medical Faculty
8Doctor, Department of Pediatric Hematology and Oncology, University Hospital Motol; 2nd Faculty of Medicine, Charles University, Prague, Czech Republic.
9Doctor, Department of Immunology, University Hospital Motol; 2nd Medical School, Charles University, Prague, Czech Republic.
10Doctor, Department of Clinical Immunology, Institute of Pediatrics, Jagiellonian University Medical College
11Head of department, Department of Clinical Immunology, Institute of Pediatrics, Jagiellonian University Medical College; University Childrens Hospital
12Head of department, Department of Allergology, Rheumatology and Clinical Immunology, University Children's Hospital, University Medical Centre Ljubljana, Ljubljana, Slovenia; Faculty of Medicine, University of Ljubljana
13Doctor, Department of Allergology, Rheumatology and Clinical Immunology, University Children's Hospital, University Medical Centre Ljubljana, Ljubljana, Slovenia; Faculty of Medicine, University of Ljubljana
14Doctor, 1st Pediatric Department, Comenius University, Faculty of Medicine Bratislava, Children University Hospital Bratislava
15Head of department, Department of Pediatric Hematology/Oncology and BMT, Wroclaw Medical University
16Doctor, Department of Pediatrics, Medical Center of Postgraduate Education
17Doctor, Department of Pediatrics, Hematology and Oncology Nicolaus Copernicus University in Toru; Collegium Medicum in Bydgoszcz
18Doctor, Department of Pediatric Hematology, Oncology and Transplantology, Medical University of Lublin
19Doctor, Department of Pediatrics, Children's Hospital Zagreb, School of Medicine, University of Zagreb
20Head of department, Department of Clinical Immunology, University Hospital Alexandrovska
21Doctor, Department of Pediatrics, Oncology, Hematology and Diabetology Medical University of ód
22Leading researcher, Department of Geography, Belarusian State University
23Post Doc, Department of Immunology, Erasmus MC, University Medical Center Rotterdam
24Head of laboratory, Department of Immunology, National Medical and Research Center of Pediatric Hematology, Oncology and Immunology named after Dmitry Rogachev
25Doctor, Department of Immunology, National Medical and Research Center of Pediatric Hematology, Oncology and Immunology named after Dmitry Rogachev
26Doctor, Molecular Genetics Lab, Center for Cardiovascular Surgery and Transplantation, Masaryk University, CEITEC and Medical Faculty
27Associate Professor, Department of Pediatric Infectious Diseases and Pediatric Immunology, Shupyk National Medical Academy for Postgraduate Education
28Head of department, Clinical Immunology department, West-Ukrainian Specialized Children's Medical Center
29Doctor, Consulting Center of Pediatric Medical Academy
30Chief, Laboratory of Clinical Immunology and Microbiology, IDGS, DIR, NIAID, NIH, Bethesda, MD, USA
31Chief, University of South Florida at Johns Hopkins All Children's Hospital, Saint Petersburg; Massachusetts General Hospital for Children
32Head of department, Department of Clinical Immunology, Russian Clinical Childrens Hospital
33Head of department, Department of Immunology, 2nd Medical School, Charles University Prague and University Hospital Motol
34Head of laboratory, Department of Immunology, Erasmus MC, University Medical Center Rotterdam, Rotterdam; Department of Pediatrics, Laboratory for Immunology at the LUMC, Leiden
35Head of department, Department of Immunology, Childrens Memorial Health Institute
36Director, Research Department, Belarusian Research Center for Pediatric Oncology, Hematology and Immunology
BACKGROUND: Variants in recombination-activating genes (RAG) are common genetic causes of autosomal recessive forms combined immunodeficiencies (CID) ranging from severe combined immunodeficiency (SCID), Omenn syndrome (OS), atypical SCID (AS) and CID with granulomas and/or autoimmunity (CID-G/AI). The clinical and immunological presentation is broad, ranging from severe infections secondary to near absence of T and B lymphocytes and hypogammaglobulinemia to the occurrence of autoimmunity with late manifestations with partly preserved immune subsets and near normal immunoglobulin levels and broad spectrum of autoantibodies.
OBJECTIVE: We aim to estimate the incidence, clinical presentation, genetic variability and treatment outcome with geographic distribution of patients with the RAG defects in populations inhabiting South, West and East Slavic countries. Due to shared ancestry, we also investigated our cohort for founder variants in RAG1 and RAG2 genes.
METHODS: Demographic, clinical and laboratory data were collected from RAG deficient patients of Slavic origin via chart review, retrospectively.
RESULTS. Based on the clinical and immunologic phenotype, our cohort of 80 patients from 66 families represented a wide spectrum of RAG deficiencies, including SCID (n=19), OS (n=36), AS (n=21) and CID-G/AI (n=4). Sixty-six (82.5%) patients carried RAG1 and 14 patients (17.5%) carried RAG2 biallelic variants. We estimate that the minimal annual incidence of RAG deficiency in Slavic countries varies between 1 in 180,000 300,000 live birth and it may vary secondary to health care disparities in these regions. In our cohort, 70% of the patients carried RAG1 p.K86Vfs*33 (c.256_257delAA), either in homozygous (n=17, 26%) or compound heterozygous (n=29, 44%) form. The majority (77%) of patients with homozygous RAG1 p.K86Vfs*33 originated from Vistula watershed area in Central and Eastern Poland, and compound heterozygote cases distributed among all Slavic countries except Bulgaria. Clinical and immunological presentation of homozygous RAG1 p.K86Vfs*33 cases was highly diverse suggestive of strong influence of other genetic and/or epigenetic factors in shaping the final phenotype. Survival of RAG deficient patients without hematopoietic stem cell transplant (HSCT) (n=3, 8.8%) is poor and dramatically improved in the last decade with access to HSCT and tailored conditioning regimens.
CONCLUSION: We propose that RAG1 p.K86Vfs*33 is a founder variant originating from the Vistula watershed region in Poland, which may explain a high proportion of homozygous cases from Central and Eastern Poland and the presence of the variant in all Slavs. Our studies in cases with RAG1 founder variants confirm that clinical and immunological phenotype only partially depend on the underlying genetic defect. HSCT is becoming available for RAG deficient patients in Eastern Europe with improving outcome.
(220) Submission ID#606997
Acute Central Nervous System GvHD After Liver Transplantation
Valérie Massey, MD1, Hugo Chapdelaine, MD, FRCPC2
1Fellow-in-training, Allergy and Clinical Immunology, Université de Montréal
2Clinical Immunologist, Centre Hospitalier Universitaire de Montréal (CHUM)
Background: Acute GvHD following solid organ transplantation is a rare complication. Intestinal and liver transplantation have the greatest risk of GvHD among solid organs due to high number of donor lymphocytes in these organs. Prevalence of acute GvHD after liver transplantation is estimated to be around 0,1-2% and has a poor prognosis(1). Chronic neurological GvHD is a rare form of GvHD with three subtypes described: cerebral vasculitis, demyelinating disease and immune mediated encephalitis. Acute neurological GvHD has no clear definition and is still considered a controversial entity.
Case presentation: A 63 year-old male underwent cadaveric liver transplantation for alcoholic cirrhosis and hepatocellular carcinoma. The donor was a 70 year-old man who died from anoxic brain injury. The receiver was induced with basiliximab and then put on prednisone, azathioprine and tacrolimus. He was readmitted 10 weeks later for myalgia, headache, fever and neutropenia. Clinical state initially improved with empiric antibiotics. He then developed a skin eruption, colitis and DIC. The latter was thought to be tacrolimus-induced. He was switched to cyclosporine. Skin and rectosigmoid biopsies were compatible with acute GvHD. He received basiliximab and IVIG and developed a refractory convulsive state. CSF analysis showed elevated proteins and slight pleocytosis. Cerebral MRI showed non-specific white matter lesions and conventional angiography was normal. Chimerism on peripheral blood was 0% but was 45% donor on CSF. With the presence of chimerism on CSF, evidence of cutaneous and digestive GvHD and no infectious cause, neurological GvHD was considered the most likely diagnosis. Brain biopsy showed non specific change including neuropil spongiosis, microglial activation and reactive gliosis; but no signs of vasculitis or demyelinating disease. He was treated with ATG, high-dose systemic corticosteroids, cyclosporine, IVIG and intrathecal methotrexate and corticosteroids. CSF pleocytosis, proteins and chimerism improved with treatment (45% to 2% donor). No improvement was noted regarding his neurological state and he developed pancytopenia. He was then transfer to palliative care and died shortly after (4 month and a half after liver transplant).
Discussion: To our knowledge, there is only one prior case published of neurological GvHD following liver transplantation (2). Both patients were old, had hepatocellular carcinoma and had at least one HLA match. Age >50 year, hepatocellular carcinoma and shared HLA antigen are known risk factors for GvHD following liver transplantation (1). Our patient had only one HLA match with the donor. This case is intriguing as there was a great discrepancy between blood and CSF chimerism. Acute neurological GvHD following transplantation is a real complication. It must be taken into consideration in patients with neurological involvement after transplant, even solid organ transplantations.
References
1. Murali AR, Chandra S, Stewart Z, et al. Graft Versus Host Disease After Liver Transplantation in Adults: A Case series, Review of Literature, and an Approach to Management. Transplantation. 2016;100(12):2661-2670.
2. Pahari H, Nagai S, Skorupski S, Salgia R. Graftversushost disease of the central nervous system after liver transplantation: A rare complication. Am J Transplant. 2018;18:25912594.
(221) Submission ID#607032
Hyper IgM2 Diagnosed in a Brazilian Boy
Maine Luellah Demaret Bardou, MD1, Daniele Pontarolli, MD1, Marina Henriques, MD1, Anete Grumach, MD PhD2
1MD, Reference Center on Rare Diseases, Faculdade de Medicina ABC
2MD Phd, Reference Center on Rare Diseases, Faculdade de Medicina ABC
Introduction: Hyper-IgM syndrome are rare. Although no data are available on the frequency of activation-induced cytidine deaminase (AID) deficiency, this disorder is estimated to affect less than 1:1,000,000 individuals. By the year 2012, 110 cases worldwide (1) with such mutation have been described. We describe a patient with hyper IgM by mutation in the AICDA gene.
Case report: MVV, 5-year-old boy, born to consanguineous parents, was referred with recurrent pneumonia, which started shortly after discontinuation of breastfeeding at 6 months old. Repetitive otitis evolved with bilateral tympanic and partial hearing loss. He was submitted to adenoidectomy without improvement. Immunological evaluation showed normal numbers of B and T cells with CD3+ (1290/mm3, 65%), CD4+ (547/mm3, 28%), and CD8+ (259/mm3, 13%). Immunoglobulin concentrations were: IgG = 138mg/dl (p97). Treatment with Intravenous immunoglobulin and prophylactic antibiotic was initiated and he had no infections during the follow up except for one episode of sinusitis. At 10 years of age, molecular evaluation was performed and a mutation in homozygosity in the AICDA gene (OMIM * 605257) at position chr12: 8.757.821 was found, confirming the clinical suspicion.
Conclusion: The role of AID in the immunoglobulin class-switch recombination (CSR) and somatic hypermutation (SHM) have not been fully elucidated. Summarizing within the SHM and CSR processes, AICDA mutation can induce DNA lesions in directed sequences in the S and V regions required for DNA cleavage. Recurrent infections and consanguinity raised the suspicion of inborn errors of immunity in this patient. The literature described late diagnosis as in the second or even the third decade of life. It was suggested that high levels of IgM antibodies may provide effective defense, at least, against some infectious agents. It is important to emphasize that the impossibility to obtain genetic diagnosis did not prevent to introduce therapy.
* AICDA: activation induced cytidine deaminase gene
(222) Submission ID#607035
CARD9Δ11 Gene Dosage: From Mono-allelic Protection to IBD, to Bi-allelic Increased Fungal Infection Susceptibility
Goel S1, Kuehn HS1, Chinen J2, Yamanaka D3, Walkiewicz M4, Lionakis M3 and Rosenzweig S1.
1Department of Laboratory Medicine, Clinical Centre
2Immunology, Allergy and Rheumatology Division, Department of Pediatrics, Baylor College of Medicine, Texas Children’s Hospital, Houston,Texas, USA
3Laboratory of Clinical Immunology and Microbiology, Fungal Pathogenesis Section, National Institute of Allergy and Infectious Diseases,
4Department of Intramural Research, National Institute of Allergy and Infectious Diseases (NIAID), National Institute of Health, Bethesda Maryland, USA
CARD9 deficiency is an autosomal recessive primary immunodeficiency known to underlay increased fungal infection susceptibility mostly presenting as invasive CNS candida infections (in infancy or adulthood) and dermatophyte infections. More recently, a rare CARD9 variant (c.1434+1 G>C, leading to exon 11 skipping, CARD9del11) showed a significant protective association towards inflammatory bowel disease (IBD) when present in heterozygosity. At the NIH we studied an 8-year-old male patient (P1) born to a non-consanguineous marriage who presented as an infant with recurrent/severe thrush, candida esophagitis, and an episode of tinea pedis; P1 also has mild hypogammaglobinemia (IgG ~500mg/dL at age 8y). P1s gDNA was tested by whole exome sequencing and showed a CARD9 c.1434+1 G>C mutation in homozygous state. Segregation analysis and Sanger confirmation determined that both parents and P1s elder brother carried the same variant in heterozygosity, while his asymptomatic younger brother (P2) was also homozygous. As previously described, this variant caused CARD9 exon 11 deletion as determined in P1 and P2s PBMCs by cDNA sequencing and by a lower molecular weight CARD9 protein by immunoblot evaluation. P1 and P2s PBMCs, as well as the heterozygous parents cells, showed a defective cytokine generation (TNF-, IL-1, IL-6 and GM-CSF) in response to heat killed candida (HKC), but not to LPS. While patients PBMCs failed to induce phospho-Erk and phospho-p-38 upon HKC-stimulation but presented an intact response to PMA+ionomycin; the parents cells responded normally to both stimuli. Moreover, T-cell activation and proliferation was affected in response to HKC but not to PHA in both patients, whereas the parents exhibited normal results under the same conditions. When HEK293 cells were transiently transfected with WT or CARD9del11 vectors together with a TRIM62 plasmid (E3-ubiquitin ligase, naturally associated to CARD9), we confirmed that CARD9del11 failed to bind TRIM62 by immunoprecipitation. Furthermore, MALT1, BCL10 and TRIM62 were only co-precipitated by WT CARD9, but no by CARD9del11, strongly suggesting TRIM62 is an integral part of the CARD9/BCL10/MALT1 -CBM- complex.
In summary, herein we demonstrate that the CARD9del11 allele fails to bind TRIM62, and in turn is unable to conform a complete/functional CBM complex. Our data also show that CARD9del11 acts in a dominant negative fashion in terms of cytokine generation (previously reported), but one WT allele seems sufficient to generate normal levels of HKC-induced p-Erk and p-P-38, as well as T-cell proliferation. While decreased cytokine generation associated with CARD9del11 in heterozygosity has been described to be sufficient to protect towards IBD, other defective pathways are affected in homozygosity and likely necessary to confer increased susceptibility to fungal infections. Altogether these results suggest that CARD9del11 acts through a gene dosage mechanism that can dissect pathways that associate IBD protection and fungal infection susceptibility. Further work is warranted to explore CARD9del11 role, if any, in B-cell and T-cell biology.
(223) Submission ID#607037
Subcutaneous Fat Loss in a Patient with CTLA4 Haploinsufficiency and in a Patient Treated with PD-1 Inhibition: Implication for Immune Dysregulation Resulting in Acquired Generalized Lipodystrophy
Kelly Walkovich, MD1, David Frame, PharmD2, Sarita Nestelroad, RN3, Matthew Logsdon, MD4, Mark Hannibal, MD/PhD5, James A. Connelly, MD6, Kenneth Grossmann, MD/PhD7, Megan Othus, PhD8, Gina Ney, MD/PhD9, David Selewski, MD10, Yevgeniya Kashcheyeva, MD/PhD11, Muhammet Ozer, MD12, Rebecca J. Brown, MD13, Elad Sharon, MD14, Elif A. Oral, MD15
1Associate Professor, Pediatric Hematology/Oncology, University of Michigan Medical School
2Clinical Pharmacist, University of Michigan College of Pharmacy
3Clinical Research Nurse, Penrose Cancer Center
4Hematology/Oncology Physician, Rocky Mountain Cancer Research Center
5Associate Professor, Pediatric Genetics, University of Michigan Medical School
6Assistant Professor Hematology/Oncology/Bone Marrow Transplant, Vanderbilt University Medical Center
7Oncology Physician, Moffitt Cancer Center
8Assistant Professor, Biostatistics, University of Washington
9Lecturer, Pediatric Hematology/Oncology, University of Michigan Medical School
10Assistant Professor, University of Michigan Medical School
11Clinical Fellow, Endocrinology, National Institute of Diabetes and Digestive and Kidney Disease
12Endocrinology Physician, University of Michigan Medical School
13Lasker Tenure Track Investigator, National Institute of Diabetes and Digestive and Kidney Disease
14Senior Investigator, National Cancer Institute
15Professor, Endocrinology, University of Michigan Medical School
Background: Acquired generalized lipodystrophy (AGL) syndromes are a heterogeneous group of diseases characterized by selective dysfunction and loss of adipose tissue after birth. This causes ectopic lipid deposition and deficiency of the adipokine leptin, which promotes metabolic dysfunction through impaired glucose handling resulting in insulin-resistant diabetes mellitus, dyslipidemia and steatohepatitis. While the metabolic effects of altered adipokine secretion are known, the molecular mechanism is less clear. Many AGL cases are suspected to have an autoimmune etiology. Effector and regulatory T cells, dendritic cells and macrophages reside in normal adipose tissue. T cells within adipose tissue highly express PD-1 and regulatory T cells express CTLA4, which limits immune activation in the adipose tissue under normal circumstances. Thus, inhibition of these immune checkpoints may hypothetically cause immune activation, leading to adipocyte dysfunction and autoimmune destruction. We have encountered two cases that raise clinical concern for this process.
Patient Cases: Patient 1 is a 16-year-old female who presented with failure to thrive at 6 months. She was diagnosed with insulin-resistant type 1 diabetes and hypertriglyceridemia at ages 2 and 4 years with progressive subcutaneous fat loss and low leptin levels culminating in a diagnosis of AGL. Her childhood clinical course was complicated by hypertrophic cardiomyopathy, hepatomegaly, autoimmune hemolytic anemia with massive splenomegaly and severe chronic diarrhea secondary to autoimmune enteropathy. She presented at 14 years with acute liver failure, thrombotic microangiopathy, nephrotic syndrome and progressive kidney insufficiency. Evaluation for her multi-faceted autoimmune presentation identified a familial heterozygous pathogenic variant in the CTLA4 gene (c.4_5insGTTGG,p.Ala2GlyfsTer14). Despite aggressive immune therapies, including CTLA4-Ig (abatacept), her kidney disease and enteropathy have progressed.
Patient 2 is a 55-year-old male diagnosed with localized malignant melanoma of the right neck in July 2014. He underwent excisional biopsy and regional lymph node dissection with negative margins. He relapsed in November 2017 and underwent a modified radical neck dissection with 1 lymph node positive for disease and received external beam radiation from January-February 2017. Additionally, he was started on anti-PD-1 therapy with the humanized antibody drug pembrolizumab in April 2017 but discontinued the drug in February 2018 in the setting of toxicities including hypothyroidism. Subsequently, he developed up to 7.5% weight loss with progressive loss of subcutaneous fat first in his face, then generalized to the rest of his body. In the ensuing months, imaging with PET-CT demonstrated loss of subcutaneous fat concurrent with elevations in ALT and triglyceride levels plus a low leptin level consistent with AGL.
Conclusion: These cases raise concern that inhibition of the immune checkpoints CTLA4 and PD-1 may facilitate the development of AGL. We hypothesize that these defects significantly increase T cell autoimmune activity in the adipose tissue and/or alter T cell metabolism resulting in AGL. Disorders of immune dysregulation should be considered in the etiology of AGL. Similarly, patients with either genetic or pharmacologic inhibition of immune checkpoints should be monitored for the development of AGL with careful physical exam and periodic monitoring of glucose and triglyceride levels.
(224) Submission ID#607046
A Case of Lymphopenia in Rosai Dorfman Disease
Lisa Liang1
Medical Resident, Memorial University
Background: Rosai-Dorfman Disease (RDD; also known as Sinus Histiocytosis with Massive Lymphadenopathy) is a rare non-Langerhans cell histiocytosis. It is characterized by proliferation and accumulation of activated histiocytes in affected tissues. Classically, RDD presents with bilateral, non-tender, and often markedly enlarged cervical lymphadenopathy.
Case presentation: A 2-year-old female presented with a 6-week history of asymptomatic, persistent and bilaterally enlarged cervical lymph nodes. She was otherwise healthy with no significant past medical history. Operative excision biopsy of the largest lymph node confirmed the diagnosis of RDD. Three months following diagnosis, routine bloodwork revealed that she had developed lymphopenia (lymphocyte count 1.4 x 109/L). Between 1-year and 2-and-a-half-years post-diagnosis, the patient was hospitalized and treated with intravenous antibiotics for 2 presumed episodes of osteomyelitis and 2 presumed episodes of lymphadenitis. Given the recurrent presumed infections and persistent lymphopenia, the patient was referred to Immunology for evaluation. She received a full immunologic work-up. Lymphocyte immunophenotyping revealed low CD4 (288 cells/mm3) and low CD8 (228 cells/mm3) counts. The rest of her immunologic work-up was within normal limits. Approximately 3-and-a-half-years post-diagnosis, the decision was made to initiate treatment for RDD. She was started on a 6-week tapering course of prednisone therapy. Within 2-weeks of starting corticosteroid therapy, the lymphadenopathy had diminished, and by 6-weeks, the lymphopenia completely resolved. At her most recent clinic visit, she had been free of serious infections for more than 3-years, and her lymphocyte counts had remained stable and within normal limits for over one year.
Discussion: In the literature, immune system dysfunction has been reported in RDD, with both auto-antibodies and cellular immunodeficiency implicated. In this patient, the persistent lymphopenia and recurrent episodes of presumed infections appeared consistent with an immunodeficiency. Given the known association of RDD with immunologic dysfunction, this was certainly a reasonable assumption; however, when these issues resolved following corticosteroid therapy, we questioned whether her clinical presentation could instead represent a manifestation of her underlying RDD. This case highlights the diagnostic challenge of differentiating between an infection and an RDD exacerbation. The episodes of presumed infections were considered probable but not confirmed with microbiologic or histopathologic specimens. The mechanism underlying lymphopenia in RDD is not clear but may involve decreased production, increased destruction, or sequestration of lymphocytes. To our knowledge, this has not been specifically studied in RDD in the past, however lymphopenia has been linked to lymphocyte maldistribution in other diseases. For example, studies have shown that experimentally altering either the surface of the lymphocyte or the environment through which the lymphocyte travels through can cause sequestration of lymphocytes in various lymphoid organs including lymph nodes.
Conclusion: We describe the case patient with RDD that developed persistent lymphopenia, and multiple episodes of presumed infections resulting in hospitalization and intravenous antibiotic therapy. Her lymphopenia resolved and she had sustained remission of RDD following treatment with corticosteroids. We hypothesize that lymphocyte sequestration in enlarged lymph nodes may have resulted in lymphopenia. This, combined with recurrent RDD exacerbations that clinically resemble infections created a presentation that mimicked an immunodeficiency.
(225) Submission ID#607048
Expanding Phenotypes: A Complex Case of an Adult Patient with DNA Repair Defect and Immunodeficiency
Claudia L. Gaefke, MD1, Melissa Crenshaw, MD2, Roshini S. Abraham, PhD3, Jolan Walter, MD, PhD4
1Division of Allergy and Immunology, University of South Florida, Tampa, FL.
2Division of Clinical Genetics, Department of Pediatrics, Johns Hopkins All Children's Hospital, St. Petersburg, FL.
3Department of Pathology and Laboratory Medicine, Nationwide Childrens Hospital, Columbus, OH.
4Associate Professor, Robert A. Good Endowed Chair and Division Chief, Division of Pediatric Allergy & Immunology, Department of Pediatrics, University of South Florida, Johns Hopkins All Children's Hospital, St. Petersburg, FL.
Background: There is an expanding spectrum of immunodeficiency phenotypes linked to DNA repair defects, and some patients may not be diagnosed until adulthood. The most well recognized genetic defect linked to DNA repair is in the gene, Ataxia Telangiectasia Mutated (ATM), which causes ataxia telangiectasia, characterized by combined immunodeficiency, neurodegeneration, radiation sensitivity, and ocular telangiectasias. However, there are several other DNA repair defects associated with immunodeficiency, including some syndromic and severe combined immunodeficiency (SCID) disorders.
Objective: We present the case of an adult patient with prolonged history of recurrent infections, facial abnormalities, and autoimmunity who was found to have radiosensitivity suggestive of a DNA repair defect.
Methods: Retrospective chart review, immunodeficiency evaluation, flow-based radiosensitivity assay, gene sequencing.
Results: A 68-year-old female was referred to our clinic due to a complex history of recurrent infections and immune dysregulation.
The patient had a lifelong history of sinopulmonary infections and pan-hypogammaglobulinemia with low vaccine responses, leading to a diagnosis of Common Variable Immunodeficiency (CVID), necessitating treatment with immunoglobulin replacement. Clinical features were also notable for congenital dysmorphia (strabismus, thin and angular face, high arched palate, nasal septal defect, small mouth, missing dentition, clinodactyly, severe equinovarus, and scoliosis). She was subsequently diagnosed with autoimmune features of vasculitides requiring trial of cyclophosphamide, azathioprine, rituximab and belimumab, which was later discontinued due to neutropenia and worsening sinopulmonary and skin infections despite immunoglobulin replacement. In the course of our evaluation she was revealed to have severe B cell lymphopenia (1%), CD4 naïve T cell lymphopenia, persistent IgA and IgM deficiency one-year post Rituximab therapy, and elevated alpha fetoprotein (AFP). Radiosensitivity assay revealed decreased ATM phosphorylation and elevated levels of H2AX 24-hours after low-dose (2Gy) radiation in her lymphocyte subsets (T, B and NK cells).
Due to the evidence of radiosensitivity and elevated AFP levels, there was concern for an ATM or other genetic defects in a DNA repair pathway. Therefore, a targeted (primary immunodeficiency genes) panel was pursued for genetic testing (207 genes, Invitae, San Francisco). The evaluation did not identify a variant in the ATM gene but rather a variant of uncertain significance was identified in the CHD7 gene, in exon 38, c.8440G>A (p.Gly2814Arg), which may be mosaic. This variant has not been reported in population databases. CHD7 is typically associated with CHARGE syndrome, and while this patient has some dysmorphic features, she is not typical for CHARGE syndrome. Currently, studies on copy number variation (CNV) and deep intronic variants in ATM are pending.
Conclusion: DNA repair defects may occur in adult patients with a primary diagnosis of CVID. Our patient exhibits some phenotypic features of both a CHD7 variant, and ATM leading to possible abnormal DNA damage responses (DDR). The exact cause of the immune deficiency in our case remains presently unsolved. This case highlights the relevance of both functional studies and genetic evaluation of complex cases of immune dysregulation, for improving our understanding of the phenotypic variability in these immunological disorders.
(227) Submission ID#607059
Fertility, Pregnancy, and Progeny Outcomes in a Large Academic Cohort of Patients with Antibody Deficiencies
Carolyn H. Baloh, MD1, Rebecca Buckley, MD2, Patricia Lugar, MD, MS3
1Allergy Immunology Fellow, 3nd year, Duke University Medical Center
2Sidbury Professor of Pediatrics, in the School of Medicine; Professor of Immunology, Duke University
3Assistant Professor, Allergy and Immunology, Duke Health
Background: Womens health issues in patients with immunodeficiency are largely underrepresented in the literature. There are no studies assessing for fertility issues in patients with antibody deficiencies, and there are few sizable studies examining pregnancy and outcomes on progeny in the same cohort. The two largest studies of pregnancy in antibody deficiency, an IDF survey and a study of the Czech population, provide conflicting data about the safety of pregnancy for these patients. Immunoglobulin replacement has been shown to be safe and beneficial in pregnancy for patients with CVID, however, dosing strategies are unguided. We sought to further understand these and other issues associated with fertility and pregnancy in a large cohort of patients with antibody deficiencies.
Methods: We performed a retrospective chart review of over 100 patients with ICD9 and/or ICD10 codes of CVID or another antibody deficiency from January 2005 to December 2018. Inclusion criteria also comprised of having reached at least 16 years of age, the beginning of child bearing years. Data collected included disease characteristics, comorbidities, laboratory values, and outcomes. This was followed by a phone survey to elucidate data regarding fertility, pregnancy, delivery complications, and outcomes of children. This study was IRB approved.
Results: The current age of women included ranged from 16 to 88 years of age, currently being in childbearing years to being post-menopausal. Forty percent of the women had been pregnant, delivering an average of 2 babies per woman who had been pregnant. Fertility issues were not a prominent factor for women who never became pregnant. A majority of women who had babies (64%) did not receive a diagnosis of antibody deficiency until after their child bearing years. Recurrent upper respiratory tract infections, bacterial sinusitis, and urinary tract infections during pregnancy were common even in those not yet diagnosed with antibody deficiency. Immunoglobulin levels and dosing of intravenous and/or subcutaneous replacement were recorded for a subset of patients with recent pregnancies. The data re-enforced that increases in dosing are needed in the third trimester. Cord blood IgG levels were also recorded for baby and were the same or higher than the mothers most recent IgG prior to delivery. It was rare for children of our patients to be diagnosed with antibody deficiency or a related condition, although CVID, hypogammaglobulinemia, combined immunodeficiency, lymphoma, rheumatoid arthritis, and other diagnoses were found.
Conclusion: This is the largest report of outcomes before, during, and after pregnancy for patients with antibody deficiencies in the United States. This report highlights the importance of closely monitoring women during pregnancy for recurrent infections regardless of whether a diagnosis of antibody deficiency is present. It also highlights that close monitoring of IgG levels during pregnancy is necessary for women with antibody deficiencies.
(228) Submission ID#607064
Clinical and Laboratory Manifestations of Autoinflammatory Diseases: The Results from the First Iranian Registry
Sahar Memar Montazerin, MD1, Roya Sherkat, MD2, Ali Mosayebian, Phd1, Mohammad Shahrooei, Phd3, Vida Homayouni, Phd1, Somaye Najafi, MSc1, Mahdieh Behnam, MSc4, Aryana Zamanifar, DDS5
1Research Assistant, Acquired Immunodeficiency Research Center, Isfahan University of Medical Sciences , Isfahan , Iran
2Head of A. Immunodeficiency Research Center, Acquired Immunodeficiency Research Center, Isfahan University of Medical Sciences , Isfahan , Iran
3Lab director, Department of Microbiology and Immunology, Laboratory of Clinical Bacteriology and Mycology, KU Leuven, Leuven, Belgium AND Specialized Immunology Laboratory of Dr. Shahrooei, Sina Medical Complex, Ahvaz, Iran.
4Research Assistant, Medical Genetics Laboratory, Alzahra University Hospital, Isfahan University of Medical Sciences, Isfahan, Iran
5Research Assistant, Dental school, Isfahan University of Medical Sciences , Isfahan , Iran
Backgrounds: Autoinflammatory diseases (AIDs) are a group of disorders with an inborn error of innate immunity, characterized by recurrent episodes of fever and inflammatory attacks. The spectrum of AIDs is expanding, but no data on clinical presentation and symptom variability exist for the Iranian population for timely precise diagnosis. This study aims at establishing the first Autoinflammatory registry of an Iranian population focusing on the clinical and laboratory features that may help clinicians toward a better understanding and diagnosis of these disorders.
Methods: Clinical and laboratory characteristics of patients who clinically and or genetically diagnosed with AIDs collected. We used the updated version of classification criteria from the Eurofever Registry for the clinical diagnosis.
Results: In our retrospective study, clinical and laboratory characteristics of the participants collected. Mean age of disease onset, disease course manifestation, the mean duration of episodes, atypical symptoms, laboratory and imaging studies as well as complications, and response to treatment also reviewed.
Data resulted in 26 patients of whom 16 were male. Their age ranged from 2 to 68 years. 5 out of 26 were genetically diagnosed. Familial Mediterranean Fever (FMF) was the most common clinically and genetically approved diagnosis. There were also patients suspected of NLRP12 and NOD2 mutations. Age at disease onset differed variably and ranged from the neonatal period to adulthood. Fever was present in all the participants and the duration of episodes was 1-10 days. The frequency of attacks was between 3 to more than 12 per year. Some of the common clinical manifestations were as follows: myalgia or fatigue (77%), arthralgia and arthritis (70%), abdominal pain (65%), Aphthous stomatitis (38%), chest pain (34%), chronic gastrointestinal symptoms (38%), skin lesion ranging from urticarial rash and severe nodular acne to pyoderma gangrenosum (50%), exudative and or erythematous pharyngitis (46%), consanguineous parents (42%), symptoms of a type of allergy (84%), lymphadenopathy (27%), splenomegaly (27%), Increased acute phase reactant (54%), elevated liver function test (19%). 10 out of 26 of the individuals reported positive family history and in one of the cases, a patient carrying the homozygous mutation in the MEFV gene has shown no clinical manifestation.
Conclusion: This study highlights the most common manifestations of AIDs in the population of Iranian origin and can be used as evidence-based clinical criteria for their diagnosis.
(229) Submission ID#607079
Absolute Neutrophil Counts in Pediatric Duffy Null (FyA-/FyB-) Patients: Assessing Expected Neutrophil Counts in Benign Ethnic Neutropenia
Lauren E. Merz, BA1, Shih-Hon Li, MD/PhD2, Thomas F. Michniacki, MD3, David Frame, PharmD4, James A. Connelly, MD5, Kelly Walkovich, MD6
1Medical Student, University of Michigan Medical School
2Assistant Professor, University of Michigan, Department of Pathology
3Pediatric Hematology/Oncology Fellow, University of Michigan
4Assistance Professor, University of Michigan Department of Clinical Pharmacy
5Assistant Professor Hematology/Oncology/Bone Marrow Transplant, Vanderbilt University Medical Center
6Associate Professor, Pediatric Hematology/Oncology, University of Michigan Medical School
Background: The term benign ethnic neutropenia (BEN) is used to describe patients of African/Arabic descent with absolute neutrophil counts (ANCs) less than 1500 cells/uL in the absence of other causes. Historically, race has been used to support the diagnosis of BEN, but self-reported race is notoriously imprecise. The Duffy null phenotype (Fya -/Fyb-) is a known molecular cause of BEN and may be a more reliable marker of BEN than self-reported race. In addition, although the ANC is known to be lower in patients with BEN, the lower limit of ANCs is poorly described. It is important to differentiate patients with BEN from primary immunodeficiency diseases (PIDD) and to recognize their expected ANC values.
Methods: Eligible subjects included patients less than 21 years seen at the University of Michigan between January 2010-July 2018. Duffy null (Fya -/Fyb-) patients were identified using Electronic Medical Record Search Engine (EMERSE) software and search terms Duffy and FYAB. 105 potential subjects were identified; 67 patients met inclusion criteria including Duffy null status and the absence of other conditions or medications, potentially impacting ANCs. 251 unique healthy ANC values were recorded from the 67 Duffy null patients. Age and sex matched controls were identified using EMERSE software with search terms tonsillectomy, department of anesthesiology and absolute neutrophil count. Subjects with conditions or medications that might impact the ANC or of African/Arabic descent were excluded from the control group. Asian and Caucasian patients included as controls were presumed to be Duffy null given that <1% of these populations are expected to be Duffy null. 363 control subjects were identified; 134 met inclusion and exclusion criteria. Statistical analysis was performed using two-sided two-sample t-test, ANOVA and one-sample t-test.
Results: The median age of the Duffy null cases was 4.78 years (IQR: 1.68-11.48) with 61.2% (n=41) male and all of African or Arabic descent. Mean ANC for Duffy null patients was 1190 cells/uL (n=251, SD= 650) while mean ANC for controls was 4300 cells/uL (n=134; SD=1600) with a mean difference between controls and Duffy null cases of 3100 cells/uL (95% CI: 2950-3380; p=0.0001). The ANC levels between Duffy null individuals and controls were evaluated by five age categories (p=0.0001 for all age categories). However, there was no difference in ANC levels between Duffy null cases at different age categories (ANOVA, p=0.14196). 54 (21.5%) Duffy null CBCs had ANC levels in the non-neutropenic range (>1500 cells/uL), 99 (39.4%) CBCs had mild neutropenia (1001-1500 cells/uL), 70 (27.9%) CBCs had moderate neutropenia (500-999 cells/uL), and 28 (11.2%) CBCs had severe neutropenia (<500 cells/uL).
Conclusions: Although neutropenia can be associated with PIDDs and is often a sign of a compromised immune system, Duffy null patients have a wide range of values that are often much lower than previously appreciated. The degree of neutropenia related to Duffy null phenotype appears to persists throughout childhood and young adulthood. In the context of patients of African/Arabic descent presenting with asymptomatic neutropenia, Duffy null status should be assessed, and BEN should be considered in the differential.
(230) Submission ID#607081
Conventional Treatment versus Stem Cell Transplantation Outcome in 105 Patients with Chronic Granulomatous Disease
Cinzia Dedieu1, Michael H. Albert, MD, PhD2, Nizar Mahlaoui, MD3, Fabian Hauck, MD, PhD4, Christian Hedrich, MD, PhD5, Ulrich Baumann, MD, PhD6, Klaus Warnatz, MD,PhD7, Joachim Rösler, MD, PhD8, Stephan Ehl, MD9, Johannes Schulte, MD, PhD10, Alain Fischer, MD, PhD11, Stephane Blanche, MD3, Jörn Kühl, MD12, Horst von Bernuth, MD, PhD13
1Clinical Investigator, Department of Pediatric Pulmonology, Immunology and Intensive Care Medicine, Charité University Medicine, Berlin, Germany
2Senior Clinician, Department of Pediatric Immunology and Stem Cell Transplantation, Dr. von Hauner Children's Hospital, Ludwig-Maximilians-Universität, Munich, Germany
3Senior Clinician, Pediatric Hematology-Immunology Unit, AP-HP, Necker Hospital, Paris, France
4Senior Investigator, Department of Pediatric Immunology and Stem Cell Transplantation, Dr. von Hauner Children's Hospital, Ludwig-Maximilians-Universität, Munich, Germany
5Senior Investigator, Department of Pediatrics, University Hospital Dresden, Dresden, Germany
6Senior clinician, (1) Department of Pediatric Pulmonology, Allergy and Neonatology, Hannover Medical School, Hannover, Germany
7Senior Investigator, Center for Chronic Immunodeficiency (CCI), Medical CenterUniversity of Freiburg, Faculty of Medicine, University of Freiburg, Freiburg, Germany
8Senior Clinician, Department of Pediatrics, University Hospital Dresden, Dresden, Germany
9Institute for Immunodeficiency, Center for Chronic Immunodeficiency. Center for Pediatrics and Adolescent Medicine, University of Freiburg, Freiburg, Germany
10Senior Investor, Department of Pediatric Hematology, Oncology and Stem Cell Transplantation, Universitätsmedizin Charité Berlin University Hospital Center, Berlin, Germany
11Department Head of Pediatric Hematology-Immunology Unit, AP-HP, Necker Hospital, Paris, France, Pediatric Hematology-Immunology Unit, AP-HP, Necker Hospital, Paris, France
12Senior Clinician, Department of Pediatric Hematology, Oncology and Stem Cell Transplantation, Universitätsmedizin Charité Berlin University Hospital Center, Berlin, Germany
13Senior Investigator, Department of Pediatric Pulmonology, Immunology and Intensive Care Medicine, Charité University Medicine, Berlin, Germany
Patients with chronic granulomatous disease (CGD) are at risk for recurring infections and non-infectious inflammation, reduced quality of life and life expectancy. Conventional treatment with life-long anti-bacterial and anti-fungal prophylaxis prolongs lifespan but does not eliminate the lifelong risk of infection and inflammation. Allogenic stem cell transplantation is currently the only curative option for this disease. Although SCT with reduced intensity conditioning has improved treatment-related mortality and efficacy, it remains a matter of debate whether all patients with CGD benefit from SCT, whether pre-existing infections and non-infectious inflammation are risk factors and at what age SCT should be performed.
We compared patients with CGD on conventional treatment with those after stem cell transplantation for their prognosis and evaluated potential risk factors for stem cell transplantation outcome followed up in six European centers. Frequency of infections, inflammatory complications, hospitalizations, operations and immunomodulative/immunosuppressive therapy, height and weight were compared in patients on conventional treatment /before stem cell transplantation versus patients after SCT. Correlation between transplantation outcome and patient characteristics or medical history was tested. 105 patients were recruited, 55 on CT, 50 after stem cell transplantation. Before/without transplantation 98% of patients suffered from at least one infection, 84,8% from inflammatory complications.
Patients on conventional treatment developed infection/inflammation/hospitalization/surgery at a median of 2,28 (range [0,29-21,82], IQR 2,79) per year, versus 9 (range [1-72], IQR 8,5) in the first year after stem cell transplantation but 0 (range [0-15], IQR 0,53) after the first year post stem cell transplantation. There was a significant decrease of all complications after stem cell transplantation (p < .05). Growth improved significantly after stem cell transplantation (z-score weight -1,692 versus -0,846 (p.017), z-score height -1,906 versus -1,064 (p.029)). Nevertheless, complications post stem cell transplantation are frequent: 88% of patients had at least one infection, 8% had severe acute GvHD, 12% chronic GvHD, 16% had graft rejection, 12% died. Preexisting active mold infection increased the risk for complications after stem cell transplantation.
In summary infections and non-infectious inflammation are common in patients with CGD on conventional treatment, their growth is significantly impaired. Stem cell transplantation, if successful, significantly reduces the risk for infections and non-infectious inflammation. However, treatment related mortality of stem cell transplantation in patients with CGD remains considerable.
(231) Submission ID#607084
Quantitation of T Cell Repertoire Diversity Following Treatment for SCID
Ottavia M Delmonte, MD, PhD1, Riccardo Castagnoli, MD2, Morton J. Cowan, MD3, Christopher C. Dvorak, MD3, Donald B. Kohn, MD, MS, BS4, Harry L. Malech, MD5, Luigi D. Notarangelo, MD, PhD6, Jennifer Puck, MD7
1Staff Clinician, 1 Laboratory of Clinical Immunology and Microbiology, Division of Intramural Research, National Institute of Allergy and Infectious Diseases, National Institutes of Health, Bethesda, MD.
2Research Fellow, 1 Laboratory of Clinical Immunology and Microbiology, Division of Intramural Research, National Institute of Allergy and Infectious Diseases, National Institutes of Health, Bethesda, MD.
3Professor, Department of Pediatrics, Division of Allergy, Immunology, and Bone Marrow Transplant, University of California San Francisco
4Professor of Microbiology, Immunology and Molecular Genetics (MIMG) and Pediatrics, University of California, Los Angeles
5Chief, Genetic Immunotherapy Section, Laboratory of Clinical Immunology and Microbiology, IDGS, DIR, NIAID, NIH, Bethesda, MD, USA
6Chief, Laboratory of Clinical Immunology and Microbiology, IDGS, DIR, NIAID, NIH, Bethesda, MD, USA
7Pediatric Immunologist, Department of Pediatrics, Division of Allergy, Immunology, and Bone Marrow Transplant, University of California San Francisco, San Francisco, CA
Introduction: Development of a diverse T cell repertoire is essential for full immune recovery following definitive treatment for severe combined immunodeficiency (SCID), whether by allogeneic hematopoietic cell transplantation (HCT); autologous gene therapy (GT); or, in the case of adenosine deaminase deficiency, enzyme replacement therapy (ERT). However, the time course and depth of diversity of T cell receptor rearrangements have been difficult to measure directly, necessitating estimates from total and naïve T cell counts and from spectratyping, in which T cell receptor (TCR) beta chain diversity is estimated by the length distributions of cDNA amplicons between a series of TCR beta chain variable (V-beta) segments that have productively recombined with the TCR beta-chain constant region. Analysis of the actual sequences of rearranged TCRs could indicate more precisely the status of the T cell compartment of these patients, and might reveal oligoclonal expansion of dysregulated T cells, T cell insufficiency, or T cell exhaustion.
Objectives: We wished to ascertain whether deep sequencing of individual TCR V-beta rearrangements in peripheral blood could be performed sequentially following diagnosis and treatment of SCID to differentiate satisfactory immune reconstitution from incomplete or skewed repertoire development that might require further cellular therapies.
Methods: Equal amounts of total RNA were obtained from peripheral blood of controls and SCID patients pre-HCT and at 100 d, 6 and 12 mo, and yearly post-treatment(s). cDNA was used as template to semi-quantitatively amplify rearrangements at the TCR-beta locus (TRB). Raw sequences were filtered to remove PCR errors, and resulting FASTQ files were converted into FASTA format (Seqtk software, GitHub, Inc), filtered for productive rearrangement, and analyzed for V, D, and J gene composition and length (IMGT HighV-QUEST software). The VDJ statistics file (PAST program) was used to calculate a Shannon entropy (H) index to measure repertoire diversity, taking into account both abundance and richness of the overall repertoire; and a Gini-Simpson index of unevenness, measuring inequality in the relative representation of species in a given sample. Graphical representations of repertoire diversity were generated by hierarchical tree maps of the TRB repertoires (iRepertoire software): each dot represents a unique sequence and the dot size corresponds to frequency of that sequence in the total sample.
Results: TCR V-beta sequence analysis of 3 SCID patients (Image) showed (top) baseline poor diversity due to pre-treatment ADA deficiency followed by improvement to normal complexity (Shannon H >7.0) after receiving PEG-ADA and autologous lentivirus gene therapy at age 3 m; (middle) increasing diversity in XSCID after maternal T-depleted unconditioned HCT, although B cells did not recover; and (bottom) failure of initial unconditioned maternal T-depleted HCT in another XSCID patient at 12 m, followed by autologous lentivirus gene therapy with subsequent improvement (Shannon H increasing from 3.8 to 6) 12 months later.
Conclusions: TCR V-beta diversity sequence analysis provided a detailed assessment of repertoire diversity in response to cellular therapies for SCID. This method could become a useful predictive tool to measure successful T cell immune reconstitution, both as early as 100 d and in the years following treatment.
Funding: JMP, MJC and DBK were supported by NIAID, and ORDR (NCATS), NIH: U54-AI082973; DBK was supported by NIAID U01-AI100801; and RC, HLM, ODM, and LDN were supported by the Division of Intramural research, NIAID, NIH.
(232) Submission ID#607086
Expanding the Phenotypic Spectrum for STIM1-related Disorders: A Case Report
Anjali S. Sura, MD1, Joseph Jacher, MS2, Erin Neil, DO3, Kelly Walkovich, MD4
1Pediatric Rheumatology Fellow, University of Michigan
2Genetic Counselor, University of Michigan
3Pediatric Neurology, University of Michigan
4Associate Professor, Pediatric Hematology/Oncology, University of Michigan Medical School
Background: The STIM1 (stromal interaction molecule 1) protein, encoded by the STIM1 gene, is involved in calcium regulation in the endoplasmic and sarcoplasmic reticulum. Pathogenic variants in this gene are associated with three different disorders.
Homozygous loss-of-function (LOF) pathogenic variants in STIM1 have been reported to cause autoimmune cytopenias, lymphoproliferation, enamel defects, anhydrosis, and iris hypoplasia. The first described cases had frequent mortality in early childhood due to recurrent life-threatening infections and development of Kaposi sarcoma (1), while recently discovered cases have had more prolonged survival, though still with recurrent serious infections (2).
Heterozygous gain-of-function (GOF) pathogenic variants in STIM1 have been associated with both tubular aggregate myopathy (TAM) and Stormorken syndrome. TAM is a clinically heterogeneous progressive muscle disorder with a variable age of onset. Muscle biopsy characteristically demonstrates tubular aggregates, with type II muscle fiber atrophy (3). Stormorken syndrome has a phenotype that includes miosis, thrombocytopenia, intellectual disability, mild hypocalcemia, muscle fatigue, asplenia, and ichthyosis (4). The thrombocytopenia has not been reported to be immune-mediated; rather it is due to abnormal platelet calcium regulation (5).
We report a patient with STIM1 pathogenic variant presenting with TAM and immune-mediated thrombocytopenia, along with lymphoproliferative features, arthritis, and a mild immune deficiency.
Case: The patient is a 16-year-old with a history of congenital thrombocytopenia (platelets ranging 60,000-100,000) who presented with acute arthritis of bilateral hand joints after exposure to cold temperatures, which resolved with naproxen. He had back pain without muscle weakness, and preceding sore throat and general fatigue. Labs were significant for leukocytosis and elevations in his inflammatory markers and creatine kinase. MRI of his lower extremities was negative for inflammatory myositis, but did demonstrate bilateral hip and knee effusions, and significant inguinal lymphadenopathy and hyperintense linear signal changes in the mid- and distal femurs with patchy red marrow signal. Abdominal ultrasound could not identify a definite spleen. Bone marrow biopsy was negative for malignancy but significant for toxic granulation of neutrophils, evident of inflammation. Alpha-beta double negative T cells were not elevated. Interferon-gamma was mildly elevated. Flow cytometry demonstrated normal T, B, and NK cell absolute counts. Circulating antibodies against platelets (both IgG and IgA) were detected. On lymphocyte antigen and mitogen proliferation testing, he did not exhibit any proliferation when stimulated with tetanus toxoid even though he had been fully vaccinated against tetanus. Muscle biopsy demonstrated large vacuoles consistent with TAM on both light and electron microscopies. Invitaes Primary Immunodeficiency Panel identified a pathogenic variant in STIM1 (c.910C>T; p.Arg304Trp), consistent with a diagnosis of autosomal dominant STIM1-related conditions, including Stormorken syndrome (6).
Conclusion: This patient expands the phenotypic spectrum of STIM1 related disease. Based on previous evidence, GOF pathogenic variants in STIM1 are associated with TAM and Stormorken syndrome, while LOF pathogenic variants in STIM1 are associated with immune deficiency. However, our patient with a STIM1 GOF pathogenic variant has features of lymphoproliferation and immune dysregulation in addition to TAM. STIM1 GOF pathogenic variants should be considered in the differential of patients with immune thrombocytopenia and lymphoproliferation.
References:
Picard C, McCarl CA, Papolos A, Khalil S, Lüthy K, Hivroz C, et al. STIM1 mutation associated with a syndrome of immunodeficiency and autoimmunity. N Engl J Med (2009) 360(19):1971-80.
Schaballie H, Rodriguez R, Martin E, Moens L, Frans G, Lenoir C, et al. A novel hypomorphic mutation in STIM1 results in a late-onset immunodeficiency. J Allergy Clin Immunol. (2015) 136(3):816-819.
Böhm J, Chevessier F, Koch C, Peche GA, Mora M, Morandi L, et al. Clinical, histological and genetic characterisation of patients with tubular aggregate myopathy caused by mutations in STIM1. J Med Genet (2014) 51(12):824-33.
Morin G, Bruechle NO, Singh AR, Knopp C, Jedraszak G, Elbracht M, et al. Gain-of-Function Mutation in STIM1 (P.R304W) Is Associated with Stormorken Syndrome. Hum Mutat (2014) 35(10):1221-32.
Böhm J, Laporte J. Gain-of-function mutations in STIM1 and ORAI1 causing tubular aggregate myopathy and Stormorken syndrome. Cell Calcium (2018) 76:1-9.
Borsani O, Piga D, Costa S, Govoni A, Magri F, Artoni A, et al. Stormorken Syndrome Caused by a p.R304W STIM1 Mutation: The First Italian Patient and a Review of the Literature. Front Neurol (2018) 9:859.
(233) Submission ID#607089
CARD11 (caspase Recruitment Domain-containing Member 11) Defect: When the Deck Is Stacked
Laura E. Maurer, MD, MPH1, Christina Price, MD2, Joel P. Brooks, DO3
1Resident Physician, Yale New Haven Hospital
2Assistant Professor of Medicine, Immunology, Yale New Haven Hospital
3Allergy and Clinical Immunology Fellow, Yale New Haven Hospital
Introduction / Background: CARD11 is critical for protein binding upstream of NF-kB (nuclear factor kappa B) and mTORC1 (mammalian target of rapamycin complex 1) the signaling pathway involved in T-cell activation and inflammatory response. Prior testing of CARD11 mutations demonstrated variable T-cell dysfunction. In vitro studies have demonstrated reduced interferon gamma cytokine production, interference of T-cell receptor (TCR) signaling, and Th2 phenotype skew in T-cells with CARD11 defects. While homozygous mutation causes severe combined immunodeficiency deficiency, heterozygous CARD11 defect is associated with atopy by way of inappropriate Th2 skewing. Heterozygote atopy is characterized by eosinophilia, elevated IgE, and severe dermatitis. Despite multiple studies demonstrating in vivo consequences of CARD11 on T-cell function, little is known of the clinical significance. Moreover, few studies have demonstrated the impact of CARD11 mutations on B-cell maturation and development, despite the recognized TCR and interleukin 2 signaling deficits.
Objectives: This case demonstrates a CARD11 defect that evolved from atopy to combined immunodeficiency requiring intravenous immunoglobulin therapy. It highlights the poorly understood effect of CARD11 mutation on T-cell function, and the downstream impact on B-cell quality.
Methods: 53-year-old male, with past medical history of T-cell lymphoma and no evidence of disease status post autologous stem cell transplant, was found to have CARD11 E57D missense mutation by genetic testing. Consistent with previous literature regarding heterozygous CARD11 defects, the patient suffered from frequent asthma exacerbations, aeroallergen sensitivity, and eczema. Lab work was consistently positive for elevated IgE and eosinophilia. Family history was positive for a son born with congenital molluscum, and multiple other children with recurrent infections. One child was also identified with CARD11 mutation.
The patient had flow cytometry demonstrating 4% of circulating cells with atypical immunophenotyped CD3+ T-cells, and positive gene rearrangement studies. His qualitative immunoglobulin levels were significant for consistently low IgM, but normal quantity IgG. In the patients adulthood, he had recurrent bronchitis and pneumonia requiring hospitalization and intravenous antibiotics. Given his recurrent infections, the patient underwent immunodeficiency evaluation. Despite previous infection with herpes zoster, the patient did not have protective titers. Additionally, the patient had received the pneumococcal conjugate vaccine once, and the pneumococcal polysaccharide vaccine four times. The most recent vaccination was one year prior to evaluation. Despite repeated vaccinations, titers were unprotective. Consequently, the patient was diagnosed with combined immunodeficiency, and initiated on intravenous immunoglobulin therapy.
Results: In summary, CARD11 defect is a cause of atopy, observed to become less severe with age. Studies of CARD11 heterozygote mutations have demonstrated in vitro deficiencies in T-cell activation, likely secondary to skewed or decreased inflammatory cytokine production and TCR activation. Our patient demonstrates that the variable T-cell dysfunction seen in vitro can have significant clinical implications evidenced by his inadequate vaccine response, and recurrent infections. His combined immunodeficiency poses a connection between CARD11 defects and, not only T-cell, but also B-cell function.
Conclusions: Further studies are needed to determine deficits in T-cell and B-cell function in the setting of CARD11 defect, as this case suggests the clinical implications span further than atopy.
(234) Submission ID#607091
Functional Determination of All Possible Disease-associated Variants of a Region in CARD11 Using Saturation Genome Editing
Richard James, PhD1, Eric Allenspach, MD, PhD2, Iana Meitlis, BS3, Troy Torgerson, MD, PhD4, Debbie Nickerson, PhD5, Michael Bamshad, MD6, David Hagin, MD, PhD7, Jeffrey Stinson, PhD8, Andrew Snow, PhD9, Lea Starita, PhD10, David Rawlings, MD1
1PI, Seattle Children's Research Institute
2Assistant Professor, Department of Immunology, Seattle Children's Hospital
3Research Scientist, Seattle Children's Research Institute
4Principal Investigator, Seattle Children's Hospital
5PI, University of Washington, Department of Genome Sciences
6Division Chief and Professor, University of Washington Division of Clinical Genetics
7Acting Director, Tel-Aviv Sourasky Medical Center
8Post-doctoral Research Associate, National Institute of Standards and Technology
9PI, Uniform Services University
10PI, University of Washington department of Genome Sciences
Genetic variants in the scaffold gene CARD11 cause disorders of the immune system. The clinical course and treatment depends on whether the CARD11 variant causes gain- or loss-of-function. However, lymphocyte immunophenotyping and proliferation assays in cells expressing CARD11 variants don't easily distinguish between gain- and loss-of-function. To address this challenge in variant interpretation, we used multiplexed genome editing in a lymphoma B cell line (TMD8) to generate cell populations expressing all possible single-nucleotide variants in the N-terminal 140 amino acids of CARD11. To assess function in each variant, we tracked its relative abundance over multiple conditions using DNA sequencing. Since CARD11 is required for survival of TMD8 lymphoma B-cells, cells expressing clinically identified gain-of-function variants grew faster relative to cells expressing other variants, even in the presence of upstream pathway inhibitors. Upon evaluation of the relative abundance of each variant in genomic DNA and mRNA, we found that clinically identified loss-of-function variants were depleted in mRNA, which could be attributed to alterations in splicing or to nonsense-mediated decay. To address the impact of splicing, we modeled a newly-identified splice donor mutation (c.358+1G>A) found in two patients from one family diagnosed with combined immune deficiency, autoimmunity and atopy that was also observed in our screen. We show that the variant causes deletion of exon four and that CARD11 missing exon four exerts a dominant-negative effect leading to decreased NF-kB signaling and cell growth. These experiments demonstrate the utility of multiplexed functional assays for determining variant effect in clinically-relevant genes, which will improve diagnosis and treatment in patients.
(235) Submission ID#607098
Genotype-phenotype Correlation in Human RAG1 Deficiency
Enrica Calzoni, MD1, Ezekiel Bello, Post Baccalaureate2, Tomoki Kawai, MD3, Yasuhiro Yamazaki, MD, PhD2, Luigi D. Notarangelo, MD, PhD4
1Graduate Student, Laboratory of Clinical Immunology and Microbiology, IDGS, DIR, NIAID, NIH, Bethesda, MD, USA
2Fellow, NIH, NIAID, LCIM
3Special Volunteer, NIH, NIAID, LCIM
4Chief, Laboratory of Clinical Immunology and Microbiology, IDGS, DIR, NIAID, NIH, Bethesda, MD, USA
Background: The recombination-activating genes (RAG) 1 and 2 play a critical role in the development of T and B cell by initiating the V(D) J recombination process that controls expression of T receptor and immunoglobulin genes and their broad repertoire. Mutations in the RAG1 and RAG2 genes in humans cause a wide spectrum of phenotypes, ranging from severe combined immunodeficiency (SCID) with lack of T and B cells to Omenn syndrome (OS), atypical SCID (AS) and combined immunodeficiency with granulomas and/or autoimmunity (CID-G/AI). Here, we sought to investigate the molecular basis for phenotypic diversity presented in patients with various RAG1 mutations.
Methods: We have recently described a novel flow-cytometry-based assay in which mouse Rag1-/- pro-B cells containing an inverted GFP cassette flanked by recombination signal sequences (RSS) are transduced with a retroviral vector expressing either wild-type or mutant human RAG1 (hRAG1). The green fluorescent protein expression directly relates to the activity of RAG proteins, representing a quick and powerful tool to correlate between defective activity of hRAG1 mutant and severity of the clinical phenotype. The genetic variants of hRAG1 analyzed in this study were affecting the various domains of the protein: RING, zinc finger RING type domain (amino acids 168-283); NBR (amino acids 387-461); HBR (amino acids 531-763) and the core domain (amino acids 385-1011). Using this sensitive assay, we tested the recombination activity of 27 human RAG1 variants that have been reported in patients.
Results: We have demonstrated correlation between the recombination activity of the mutants and the in vivo clinical phenotype of patients. In particular, similarly low levels of recombination activity were observed in patients with SCID and OS, whereas patients with AS and especially those with CID-G/AI carried mutations that retained significant residual levels of activity.
Conclusions: These data provide a framework to better understand the phenotypic heterogeneity of RAG deficiency.
(236) Submission ID#607101
A Case of Burkholderia Cepacia in a Child with Takayasu Arteritis
Michelle M. Clark, MD1, Gretchen A. Harmon, MD2
1Fellow, Nemours/Alfred I. duPont Hospital for Children
2Attending, Nemours/Alfred I. duPont Hospital for Children
Background: Burkholderia cepacia is a catalase positive organism that leads to opportunistic infections typically associated with the underlying conditions cystic fibrosis and chronic granulomatous disease (CGD). Here we report a case of a child with B. cepacia lymphadenitis, ultimately diagnosed with Takayasu arteritis. Takayasu arteritis is a large vessel vasculitis which may have a nonspecific clinical presentation in childhood possibly leading to difficulty in diagnosis.
Case: A 16-month-old female presented with two weeks of fever, respiratory distress, and lymphadenopathy, and was treated with IVIG for presumed atypical Kawasaki disease. Imaging studies performed due to worsening respiratory distress revealed retropharyngeal abscess with bilateral cervical lymphadenopathy, culture-positive for Prevotella oralis and melaninogenica, with improvement following incision and drainage and antibiotic therapy. Recurrence of fever and respiratory distress prompted CT imaging of her neck significant for worsening lymphadenopathy. Cultures from lymph node biopsy grew B. cepacia. Following treatment, she was readmitted with respiratory distress requiring chronic steroid treatment and found to have Candida albicans on bronchoalveloar lavage and necrotizing granulomatous inflammation on lung biopsy.
An immunologic evaluation was notable for two normal DHR assays. CGD genetic panel was negative for pathogenic variants in CYBB (p91), NCF1 (p47), CYBA (p22), NCF2 (p67). Testing was also notably negative for HIV PCR, Bartonella PCR, Cryptococcal antigen, Histoplasma antigen, BAL AFB stain and mycobacterial cultures, CMV PCR, EBV PCR, ANCA, serial blood cultures, and sweat test. Lymphocyte subsets were normal for age. Mitogen stimulation test, myeloperoxidase antibody IgG, serine protease3 IgG, C4 level, LAD panel, and cytokine panel were normal. Autoimmune lymphoproliferative disorders (ALPS) panel was negative. Whole exome sequencing demonstrated heterozygous mutations in CFI and JAK3, not considered to be clinically relevant given the patients clinical picture and laboratory evaluation. The patient was then lost to follow-up for over a year.
At the age of 3 years, the patient presented with fever and back pain. Imaging revealed severe large vessel vasculitis involving the aorta and subclavian, vertebral, mesenteric, and renal arteries. She also had evidence of cardio-embolic strokes on brain MRI. She had had no significant interval infections, and her immunologic evaluation remained unrevealing. In the context of her new vasculitis, evaluation for deficiency of ADA2 (DADA2) was negative. She was ultimately diagnosed with Takayasu arteritis and has begun therapy with systemic corticosteroids, aspirin, and etanercept.
Conclusions: We describe a case of B. cepacia infection in a child without identified immunodeficiency, ultimately diagnosed with a large vessel vasculitis. The presence of B. cepacia infection warrants a thorough investigation. Burkholderia has been previously associated with giant cell arteritis, another type of large vessel vasculitis, though causation has not been established. To our knowledge B. cepacia infection has not been associated with Takayasu arteritis.
(237) Submission ID#607105
Plasma Metabolomic Signatures in Patients with Chronic Granulomatous Disease
Christopher Santaralas, BS1, Valentine Gignon Jadoul, BS1, Jacqueline Squire, MD2, John Cannon, PhD3, Jessica Trotter, BS4, Susan Aja, BS5, Neil Goldenberg, MD, PhD6, David Graham, PhD7, Jennifer Leiding, MD8
1Research Technician, University of South Florida
2Allergy-Immunology Fellow, University of South Florida
3Research Scientist, University of South Florida
4Clinical Research Assistant, University of South Florida
5Research Technician, Johns Hopkins All Childrens Hospital
6Professor, Johns Hopkins School of Medicine
7Associate Professor, Johns Hopkins School of Medicine
8Associate Professor, University of South Florida
Plasma Metabolomic Signatures in Patients with Chronic Granulomatous Disease
Christopher Santaralas, Valentine Jadoul, Jacqueline Squire, John Cannon, Jessica Trotter, Susan Aja, Neil Goldenberg, David Graham, Jennifer Leiding
Background: Chronic granulomatous disease (CGD) is a primary phagocytic immunodeficiency secondary to mutations in any of the components of NADPH oxidase. In addition to infection susceptibility, patients with CGD can develop auto-inflammatory disease that is difficult to manage. Metabolomics is the systematic study of small molecule biomarkers of the clinical phenotype of disease. We sought to investigate plasma metabolic profiles in CGD as we hypothesized that unique signatures may differentiate patients with CGD.
Methods: Plasma collected from 15 subjects with CGD (9 X-linked, 4 p47phox-deficient, 2 p22phox-deficient) and 2 X-linked CGD carriers was analyzed using a targeted multiplex assay by liquid chromatography mass spectrometry (LC-MS) and simultaneously a profiling assay by LCMS. Sufficient signal was present for 34 metabolites. X-linked CGD and p47phox-deficient groups were sufficiently sized for multivariate and univariate analyses in MetaboAnalyst. Twelve patients had a single time point of plasma metabolomics analysis and three had multiple time points, including one in whom both pre- and post-hematopoetic cell transplantation time points were assessed. Post-hoc comparisons were also performed for those with, versus without, clinical comorbidities of auto-inflammation.
Results: Plasma from patients with X-linked and p47phox deficient CGD had a differential metabolomic signature at baseline. Many metabolites as measured by ion intensity were present at high levels, particularly homocysteine, kyneurine, tryptophan, citric acid, carnitine, methionine, and adenosine. Increased values of metabolites reduced to that of normal (compared to post HCT). Homocysteine levels were elevated among patients with (mean 1.5x105), versus without (mean 6.8x104), clinical comorbidities of auto-inflammation (i.e., colitis, lupus). Baseline samples showed elevated kynurenine among all CGD patients, relative to historical normal controls (unmatched, separate analysis). Patients with colitis had elevated citric acid levels that were higher among patients with (mean 2.1x106), versus without (mean 4.5x105), colitis irrespective of genotype.
Conclusions: Preliminary data with a small patient subset suggest that patients with CGD have metabolomic signature distinguishable by phenotype. Citric acid cycle metabolites are elevated in Crohns disease and ulcerative colitis. Based on our data, citric acid may too act as a biomarker for inflammatory bowel disease in CGD. Analyzing a larger number of samples, across time points, will likely describe a metabolomics profile for CGD and identify biomarkers for auto-inflammation in CGD.
(238) Submission ID#607107
The Role of Glycosylation Modification on Resistance to Viral Infections
Yan Su1, Cristiane J. Nunes-Santos, MD2, Hye Sun Kuehn, PhD1, Kevin Spurgers, PhD3, Kelly Warfield, PhD4, Sergio D. Rosenzweig, MD/PhD5
1Staff Scientist, Immunology Service, Department of Laboratory Medicine, Clinical Center, NIH, USA
2Post-Doctoral Research Fellow, Immunology Service, Department of Laboratory Medicine, Clinical Center, NIH, USA
3Senior Project Manager, Anti-Infectives R&D, Emergent BioSolutions
4Vice President, Anti-Infectives R&D, Emergent BioSolutions
5Chief, Immunology Service, Department of Laboratory Medicine, NIH Clinical Center, Bethesda, MD, USA
Background: Congenital disorder of glycosylation type IIb (CDG-IIb, CDG-MOGS) is caused by genetic deficiency of mannosyl-oligosaccharide glucosidase (MOGS, also known as -glucosidase I) the first enzyme in the N-linked glycosylation trimming pathway. This rare primary immunodeficiency presents with dysmorphisms, severe neurological complications, hypogammaglobulinemia and a unique characteristic of decreased susceptibility to enveloped viral infections. Objective: to investigate the role of impaired host N-linked glycosylation on viral susceptibility to Ebola virus. Methods: To mimic the condition observed on CDG-IIb patients, we tested in vitro three proprietary iminosugars (EmergentBioSolutions©), UV4B, UV001, and UV00128, which act as competitive inhibitors of -glucosidase I and II. Their ability to inhibit the trimming of N-glycans was compared to known N-glycans modifiers as castanospermine, tunicamycin, as well as the bacterial enzyme peptide-N-glycosidase F (PNGase-F). Ebola virus envelope protein GP1 was chosen as a prototype glycoprotein, as it is heavily glycosylated with 15 N-glycosylation sites. HEK 293T cells were seeded at 1x10^5 cells/well in 12 well plate. After 18h, cells were transfected with pFlag-Ebolavirus GP1 by coupling with Effectene®. After 24h, cells were treated with the inhibitors and harvested 24h after treatment. Trimming of N-glycans was evaluated via molecular weight assessment by western-blot. Results: All three inhibitors had comparable effectiveness in inhibiting trimming of N-glycans from Ebola GP1 glycoprotein compared to castanospermine. A greater molecular weight shift was seen with tunicamycin and PNGase F as expected. Conclusions: Chemical inhibition of the N-linked glycosylation pathway was successfully achieved using three new MOGS inhibitors. This approach merits further investigation on potential applications on antiviral therapies.
(239) Submission ID#607112
Clinical Description of Rosacea in a Family with STAT 1 GOF Mutation
Lizbeth Blancas Galicia, MD1, Marcos Suárez-Gutierrez, MD2, Anne Puel, PhD3, Melanie Migaud, Msc4, Sara E. Espinosa-Padilla, MD, PhD5
1Reseacher, National Institute of Pediatrics
2Dr, National Institute of Pediatrics
3Senior Investigator, Laboratory of Human Genetics of Infectious Diseases, Necker Branch, INSERM U1163, Necker Enfants Malades Hospital, Paris, France
4Investigator, Laboratory of Human Genetics of Infectious Diseases, Necker Branch, INSERM U1163, Necker Enfants Malades Hospital, Paris, France
5Head, Immunodeficiencies Research Unit, National Institute of Pediatrics
STAT1 GOF mutations are associated with infections, autoimmunity and inflammatory manifestations; the rosacea is one of the manifestations described in this disease, however, the etiology rosacea is not clearly established. The characteristics of rosacea are not described in STAT GOF in the different clinical series. We describe the different characteristics of rosacea in a family with 8 affected members with STAT1 GOF.
A family with eight members with STAT1 GOF mutation were diagnosed through a first affected member affected with tuberculosis and onychomycosis. Seven members more had a clinical history of mycobacterial, viral and fungus infections and autoimmunity disease, in all the seven, was documented the same mutation STAT1GOF. In six of these adults patients, we documented rosacea, it started after adolescence, it was localized in the face and/or eyes, was progressive and not ameliorated with medical treatment and caused nose deformity.
Rosacea has been described previously as a unique manifestation, and the etiology is not clear, an autoimmune hypothesis has been proposed. The fact that is present in patients with STAT1 GOF could suggest that have effectively an autoimmune component. Physicians face the patients with rosacea must look for other manifestation presents in STAT1 GOF mutations. Genetic studies in rosacea patients could evidence an new gene defect.
(240) Submission ID#607113
Heterozygous Variants in FOXN1 in Infants with Abnormal Newborn Screening for SCID
Lauren Sanchez, MD1, Morna J. Dorsey, MD, MMSc2, Mica Muskat, NP3, Jennifer Puck, MD4
1Assistant Clinical Professor, Department of Pediatrics, Division of Allergy, Immunology, and Bone Marrow Transplant, University of California, San Francisco
2Pediatric Immunologist and Allergist, Department of Pediatrics, Division of Allergy, Immunology, and Bone Marrow Transplant, University of California San Francisco, San Francisco, CA
3Nurse Practitioner, Department of Pediatrics, Division of Allergy, Immunology, and Bone Marrow Transplant, University of California, San Francisco
4Pediatric Immunologist, Department of Pediatrics, Division of Allergy, Immunology, and Bone Marrow Transplant, University of California San Francisco, San Francisco, CA
Introduction: Homozygous mutations causing loss of function of the transcription factor Forkhead-box N1 (FOXN1) underlie autosomal recessive severe combined immunodeficiency with congenital alopecia and nail dystrophy (nude SCID). Affected humans, like the scid mouse, have small or absent thymus, absent or severely diminished T cells, alopecia, and nail dystrophy. Infants with nude SCID have had neonatal lymphopenia and severe, life-threatening infections. Studies of heterozygous carriers of FOXN1 mutations are limited, some having been reported with no phenotype or mild disease manifestations, such as nail dystrophy without lymphopenia or recurrent infections.
Objective: We describe six infants, including two brothers, with T-cell lymphopenia (TCL) following abnormal California newborn screens (NBS) for SCID. Each had a single heterozygous variant in FOXN1.
Case Reports: Six infants (3 female, 3 male) were referred for evaluation after abnormal California NBS for SCID (Table 1), with T-cell receptor excision circle (TREC) counts from undetectable to 12 (normal >18). All infants were well at the time of initial evaluation. Five infants with absolute CD3 T cell counts >400 cells/uL and CD4 T cell counts >250 cells/uL began evaluation as outpatients on home isolation. Patient 5, with undetectable TRECs, CD3 T cell count 78, and CD4 T cell count 65 was urgently admitted for inpatient evaluation and management and immediately started on antimicrobial prophylaxis. Patient 5 further evaluation was significant for lymphocyte proliferation to mitogens that was initially normal but waned with time, prompting treatment with a paternal haploidentical hematopoietic cell transplant at 6 months of age. Patients 3 and 5 developed neutropenia within 6 weeks of birth treated with granulocyte colony stimulating factor (GCSF). Patient 3 remains well on GCSF but has had persistent growth failure under continued evaluation. Patients 1, 2, 4 and 6 remain stable off antimicrobial prophylaxis, but with persistent moderate TCL.
As part of an immune evaluation, Patients 1 and 3-6 had gene panel testing revealing heterozygous variants in FOXN1. Only the variant of Patient 1 (presumed shared by Patient 2, his brother) was predicted to be pathogenic; Patient 1 had dystrophic nails and sparse hair most evident after 2 years of age, features shared by his mother and his brother, Patient 2. The other patients lack the clinical features of the previously described phenotype of nude SCID. Their heterozygous FOXN1 variants are of unknown significance; the functional role of these variants in the patients clinical phenotype is unknown.
Conclusion: Six infants with abnormal NBS for SCID had lymphopenia and heterozygous variants in FOXN1. For these infants, variation exists in level of TCL and presence of hair and nail findings. Heterozygous variants of unknown significance in FOXN1 have been uncovered in others, including infants with abnormal NBS for SCID, highlighting the need for functional studies to address the possible role of each heterozygous FOXN1 variant in congenital lymphopenia and neutropenia. More work is needed before attributing TCL to a novel FOXN1 variant of unknown significance in the absence of family history, abnormal hair or nails, or functional evidence.
| Patient 1 (M) | Patient 2 (M) | Patient 3 (F) | Patient 4 (F) | Patient 5 (F) | Patient 6 (M) | |
| Variants Found | FOXN1 c. 1445_1449delinsCCA; p.R482PfsX46 | Brother of Patient 1 with presumed FOXN1 mutation. |
FOXN1 c.79C>T; p.L27F Another gene being investigated (VUS) |
FOXN1 c.950A>C; p.N317T. Additional single VUS in LYST, STX11, and TMC8, TREX1. |
FOXN1 c.965A>G; p.N322S Additional single VUS in DOCK8, IL10RA. |
FOXN1 c.1315delC; p. L439CfsX11 DCLRE1C c. 536G>A; p.R179Q |
| FOXN1 variant prediction | Pathogenic | Under genetic evaluation | VUS | VUS | VUS | |
| Parental Carrier of FOXN1(current age) | Mother, presumed due to phenotype | Father (35) | Unknown carrier | Unknown carrier | Mother (29) | |
| Family History and Phenotype | Maternal thyroiditis; sparse hair, absent eyebrows and brittle, tented nails. Multiple maternal relatives and brother with similar features. | Autoimmune hyperthyroidism. Normal adult WBC, ANC, and T cells at evaluation. | No significant medical history in mother; paternal history is unknown and unavailable. | No significant medical history in mother or father. | Maternal unverified frequent childhood infections and low blood counts; normal adult WBC, ANC, and T cells. | |
| Ancestry | Hispanic (Mexican) | Asian (Chinese) | Asian (Southeast Asian) | Caucasian | Hispanic (Mexican) | |
| Patient Clinical Phenotype | Lymphopenia; sparse, brittle hair, toenail dystrophy | Lymphopenia | Lymphopenia, neutropenia responsive to GCSF, growth failure, abnormal facies, delayed speech | Lymphopenia | Lymphopenia, asymptomatic transaminitis | Lymphopenia, neutropenia responsive to GCSF |
| NBS TRECs (normal >18 copies/uL) | 1 | 9 | Undetectable | 10 | 12 | Undetectable |
| CBC (at referral) | ||||||
| WBC (K/uL) | 5.2 | 5.2 | 5.3 | 7.9 | 4.0 | 6.9 |
| ANC (cells/uL) | 2200 | 1700 | 2279 | 3100 | 1200 | 2600 |
| ALC (cells/uL) | 1200 | 2000 | 1500 | 2400 | 2100 | 1300 |
| Lymphocyte Subsets (at referral) | ||||||
| CD3 | 432 | 759 | 600 | 1224 | 1007 | 78 |
| CD3+CD4+ | 276 | 506 | 330 | 792 | 782 | 65 |
| CD3+CD8+ | 158 | 239 | 256 | 408 | 203 | <20 |
| CD19+ | 276 | 308 | 75 | 600 | 727 | 364 |
| CD56+ | 336 | 730 | 690 | 432 | 362 | 741 |
| CD4+45+RA | 47 | 341 | 33 | 198 | 551 | <20 |
| CD4+45+RO | <20 | 170 | 38 | 208 | 246 | 43 |
| CD8+45+RA | 117 | 234 | 156 | 65 | 191 | <20 |
| CD8+45+RO | <20 | 7 | 18 | <20 | <20 | <20 |
| Immunoglobulins (at referral) | ||||||
| IgG | 747 | 740 | 710 | 907 | 516 | 1360 |
| IgA | <7 | <7 | <7 | 9 | 8 | <7 |
| IgM | 23 | 26 | 64 | 72 | 45 | 41 |
| Lymphocyte proliferation to PHA | Normal | Normal | Normal | Normal | Normal | Normal, then diminished by 1.5 months of age |
| Spectratyping | Not done | Not done | Acceptable TCR Vb diversity for age | Not done | Not done | Abnormal, restricted pattern of TCR Vb diversity |
| Thymus shadow | Unknown | Unknown | Normal | Unknown | Unknown | Diminished |
| Infections | Urinary tract infection, otitis media | None | Thrush, treated with oral fluconazole only | None | None | None pre HCT* |
| Treatment | Home isolation, no prophylaxis | Home isolation, no prophylaxis | Home isolation and fungal prophylaxis until ~12 mo; remains on PCP prophylaxis, GCSF | Home isolation, no prophylaxis | Home isolation, no prophylaxis | Hospital isolation. GCSF, paternal haploidentical HCT |
| Outcome | Alive at 36 months with no major infections | Alive at 7 months with no major infections | Alive at 18 months with no major infections | Alive at 16 months with no major infections | Alive at 10 months with no major infections | Alive >12 months post HCT; delayed T-cell reconstitution, AIHA** |
| Most recent ALC | 1200 cells/uL | 2080 cells/uL | 1020 cells/uL | 5060 cells/uL | 2300 cells/uL | On immunosuppression |
(241) Submission ID#607114
Siblings with Copy Number Gain in ATM Results in Variable Clinical Phenotypes and Defects in ATM, SMC1, and H2AX Phosphorylation
Jasmeen Dara, MD1, Jennifer Puck, MD1, Matthew J Smith, BS2, Roshini S Abraham, PhD3
1Division of Allergy, Immunology, Blood and Marrow Transplantation, UCSF Benioff Children’s Hospital, San Francisco, CA
2Hematology Research, Mayo Clinic, Rochester, MN
3Department of Pathology and Laboratory Medicine, Nationwide Children’s Hospital, Columbus, OH
Rationale: Ataxia telangiectasia is a disorder with variable phenotypes characterized by cerebellar degeneration, immunodeficiency, chromosomal instability, radiosensitivity, and cancer predisposition which may correspond to the degree of ATM protein expression and/or radiosensitivity. We used in vitro cytometric assessment of ATM, SMC1 and H2AX phosphorylation to assess DNA damage in response to radiation and found that two siblings with the same copy number gain in ATM have variable clinical neurologic and immunologic phenotypes.
Methods: Chart review and radiosensitivity assays using cytometric assessment of pATM, pSMC1, and H2AX expression after irradiation with 2Gy.
Results: Patient A is a 6 month old male identified after having low TRECs on newborn screening, then found to have lymphopenia and elevated IgM. He has diffuse café au lait macules and no neurologic symptoms. His 9 year old sister, Patient B, was being followed by neurology for several years for ataxia. She has selective IgA deficiency, normal lymphocyte counts, lymphocyte proliferative responses, gammaglobulins, and vaccine specific antibodies. Both patients have a 4 copy number gains in ATM (exons 48-61). Mother and father both have 3 copy number gains in ATM and are healthy without neurologic symptoms or recurrent infections. Both Patient A and B have normal ATM protein expression. Phosphorylated ATM, SMC1, and H2AX was assessed in lymphocyte subsets (T, B, and NK cells) after low-dose irradiation to induce DNA double-stranded breaks (DSBs). These parameters were assessed at 1 hour post-irradiation when they are expected to be maximal and at 24 hour post-irradiation, when under conditions of normal and effective DNA repair, the phosphorylation state returns to baseline. Patient A had abnormal pATM and pSMC1 but normal H2AX expression 1 hour and 24 hours after irradiation of T, B, and NK cells. Patient B had normal pATM, pSMC1, and H2AX expression in T cells but abnormal pATM and pSMC1 expression in B and NK cells 1 hour after irradiation. Patient B, however, had abnormal ATM phosphorylation at 24 hours after irradiation of T, B, and NK cells.
Conclusions: Our results indicate that a unique copy number gain in ATM within a family can correspond to different clinical and immunologic phenotypes as well as variable degree of radiosensitivity. The persistence of H2AX at 24 hours post-irradiation and impaired phosphorylation of ATM and SMC1 at 1 hour post-irradiation demonstrates defects in DNA DSB repair, and this is variably altered in different lymphocyte subsets. Correlation between ATM phosphorylation in lymphocytes with outcomes may be an area for future studies and particularly important in counseling patients regarding outcomes.
(242) Submission ID#607115
Patients with Chronic Granulomatous Disease Have Distinct Intestinal Microbiome and Metabolomic Signatures
Emilia L. Falcone, MD, PhD1, Yu Han, PhD2, Drew R. Jones, PhD3, Christa S. Zerbe, MD, MS4, Samantha Kreuzburg, BA, RN5, Theo Heller, MD6, Suk See De Ravin, MD, PhD7, Harry L. Malech, MD8, Clay Deming, PhD9, Julia A. Segre, PhD10, Steven M. Holland, MD11
1Director, Microbiome and Mucosal Defense Research Unit; Assistant Professor, IRCM-Montreal Clinical Research Institute
2Research Associate, Immunopathogenesis Section, Laboratory of Clinical Infectious Diseases, National Institute of Allergy and Infectious Diseases, National Institutes of Health, Bethesda, MD, USA.
3Director Metabolomics Core Resource Laboratory and Assistant Professor Biochemistry/Molecular Pharmacology NYU Langone Health
4Senior Research Physician, Laboratory of Clinical Immunology and Microbiology, NIAID, NIH
5Research Nurse Specialist, Laboratory of Clinical Immunology and Microbiology, NIAID, NIH
6Senior Investigator, Translational Hepatology Section, Liver Diseases Branch, NIDDK, NIH
7Clinician, Laboratory of Clinical Immunology and Microbiology, IDGS, DIR, NIAID, NIH, Bethesda, MD, USA
8Chief, Genetic Immunotherapy Section, Laboratory of Clinical Immunology and Microbiology, IDGS, DIR, NIAID, NIH, Bethesda, MD, USA
9Biologist, Translational and Functional Genomics Branch, NHGRI, NIH
10Senior Investigator, Translational and Functional Genomics Branch, NHGRI, NIH
11Director, Division of Intramural Research, Laboratory of Clinical Immunology and Microbiology, National Institute of Allergy and Infectious Diseases, National Institutes of Health
Background and aims: Chronic granulomatous disease (CGD) is characterized by recurrent infections and inflammatory dysregulation, especially in the gut. Almost 50% of patients with CGD have CGD-associated inflammatory bowel disease (CGD-IBD), yet its pathophysiology remains poorly understood. We characterized the intestinal microbiome and metabolome in patients with CGD to determine if intestinal microbiome and metabolomic signatures could distinguish subpopulations of patients with CGD while using the metabolome to add a functional dimension to observed microbiome signatures.
Methods: Clinical metadata and fecal samples were collected cross-sectionally from healthy volunteers (HV; n=16) and patients with CGD (n=77). Metabolomic profiling and 16S rRNA (V4) sequencing was performed on fecal samples (total samples: 108; reads/sample: 15,254 to 191,415; median: 60,816).
Results: Samples from patients with CGD had distinct intestinal microbiome signatures and metabolomic profiles depending on genotype, presence of CGD-IBD and specific interventions (e.g. treatment with an elemental diet). Notably, samples from patients with active CGD-IBD (compared to samples from patients without a history of CGD-IBD) had significantly different alpha- and beta-diversities, and were enriched for Enterococcus spp. (8.5 vs. 1.5%), Serratia spp. (8.6 vs. 3.9%) and Raoultella spp. (6.1 vs. 0.6%), while being depleted of Bacteroides spp. (9.3 vs. 23.6%). Metabolomic profiles from CGD patient samples pointed toward an aberrant metabolism of toxic ammonia waste by the intestinal microbiota compared to HV. Interestingly, use of an elemental diet to treat a patient with CGD-IBD induced long-term changes in the alpha- and beta-diversities of the patients intestinal microbiota, stabilized the intestinal metabolome, and allowed his microbial and metabolic profiles to resemble those of patients without CGD-IBD.
Conclusions: Intestinal microbiome and metabolomic signatures can distinguish subpopulations of patients CGD based on genotype, presence of intestinal inflammation and certain treatment interventions.
(243) Submission ID#607118
Dosing Ruxolitinib for the Treatment of Immunodysregulation in STAT1-GOF
Valentine Gignon Jadoul, BS1, Gretchen Vaughn, ARNP2, Benjamin Oshrine, MD3, Carla Duff, CPNP-PC APRN MSN CCRP IgCN4, Jennifer Leiding, MD5
1Research Technician, University of South Florida
2Nurse Practitioner, Johns Hopkins All Childrens Hospital
3Assistant Professor, Johns Hopkins All Childrens Hospital
4Nurse Practitioner, University of South Florida
5Associate Professor, University of South Florida
Signal Transducer and Activator of Transcription 1 Gain of Function (STAT1-GOF) is a primary immunodeysregulatory disease in which a subset of patients have features of autoimmunity and autoinflammation. Enteropathy with growth failure and nutrient wasting is a more common feature of immunodysregulation. Ruxolitinib is a Janus kinase-STAT inhibitor that has been shown effective for the treatment of immunodysregulatory features in STAT1-GOF.
Our patient is a 13 year old male with STAT1-GOF (c.983A>G p.H328R) with severe total parenteral dependent enteropathy that led to growth failure (weight 28.5kg). Treatment with ruxolitinib led to resolution of diarrhea, return of normal diet, and catch up growth. A dose of 12.5mg twice daily was initially started but was decreased to 12.5mg every morning and 10 mg every evening due to elevated transaminases and thrombocytopenia. Over the following year the patient thrived gaining 7.5kg with normal every other day stools. Despite weight gain, he remained stable on the same dose of ruxolitinib. As he outgrew his dose, he developed an increased frequency of upper respiratory infections (Parainfluenza, Coronavirus, Rhinovirus). One year after initiation of ruxolitinib, he again developed profuse watery diarrhea that was Norovirus positive (weight 36kg, BSA 0.9). He was placed on bowel rest and ruxolitinib was dose escalated with a goal of 15mg/m2/day. When he reached 15mg twice daily, enteropathy completely resolved but liver function tests began to rise. He gained weight and began thriving after 2 weeks of therapy. Six months later, enteropathy is controlled, and transaminases have remained elevated (ALT 88 IU/L, AST 73 IU/ml) but stable.
The appropriate dose and pharmacokinetics for ruxolitinib for the treatment of immunodysregulatory symptoms in pediatric patients has not been thoroughly studied. The dose used was extrapolated from data on the use of ruxolitinib in pediatric myelofibrosis. A dose of 15mg/m2/day appears to provide the most benefit with tolerable adverse effects. This dose should be maintained in order to prevent recurrence of disease related manifestations.
(245) Submission ID#607120
Ex Vivo Generation and Single-Cell Analysis of Human Monoclonal Antibodies from Dengue Virus Infected Patients
Pragati Sharma, M.Sc.1, Harekrushna Panda, PhD2, Anmol Chandele, PhD3, MuraliKrishan Kaja, PhD4
1Graduate Student, International Center for Genetic Engineering and Biotechnology, New Delhi, India
2Research Scientist, International Center for Genetic Engineering and Biotechnology, New Delhi, India
3Assistant Professor, International Center for Genetic Engineering and Biotechnology, New Delhi, India
4Associate Professor, Department of Pediatrics, Emory University School of Medicine/ICGEB-EVC, New Delhi
Antibodies have been implicated in both protection and pathology of dengue virus infections. However, much of this data is gathered from serum/plasma responses that is a cumulative of historical and ongoing infection. To precisely understand the role of antibodies with respect to the ongoing dengue virus infection, we employed the cutting edge approach of generating of human monoclonal antibodies from individual plasmablasts from peripheral blood of dengue patients that allows us to probe for answers at a single cell level.
This method involves ex vivo single cell sorting of plasmablasts from peripheral blood of well-characterized dengue infected patient followed by single cell molecular cloning of immunoglobulin heavy- and light- variable regions into expression vectors containing the defined constant region followed by transient co-transfection of HEK 293A cells with the heavy and light chain expression vectors made from genes arising from the same cell.
Thus far, using this powerful technology, for the first time in India, we have made 140 number of human monoclonals, of which 80 are specific to dengue and 14 neutralize dengue virus at various concentrations. All the neutralizing antibodies are dengue-envelope specific and bind the highly conserved fusion loop of the dengue virus envelope.
Together, with the ongoing comprehensive analysis of the B cell repertoire and somatic hypermutations, these studies provide a detailed understanding of the dengue-specific plasmablast cell response at a single cell level and create a platform for testing these antibodies for basic research, diagnostic, prophylatic and as well as therapeutic applications.
(246) Submission ID#607123
Longitudinal Follow up of EBV-driven Lymphoproliferative Disease and Combined Immunodeficiency in RASGRP1 Deficiency: Successful Treatment and Allogenic Matched Unrelated Bone Marrow Transplant
Alice S. Chau, MD1, Jay Patel, MD2, Suzanne Skoda-Smith, MD3, Adam Lamble, MD4, Aleksandra Petrovic, MD5, K. Scott Baker, MD MS4, Lauri Burroughs, MD4, Eric Allenspach, MD, PhD6, Troy Torgerson, MD, PhD7
1Fellow, University of Washington
2Attending in Allergy and Immunology, Kaiser Permanente, Downy Medical Center
3Associate Professor, Immunology Seattle Children's Hospital
4Associate Professor, Hematology-Oncology, Seattle Children's Hospital
5Associate Professor, Immunology, Hematology-Oncology, Seattle Children's Hospital
6Assistant Professor, Department of Immunology, Seattle Children's Hospital
7Principal Investigator, Seattle Children's Hospital
Introduction: RAS guanyl-releasing protein 1 (RASGRP1) is a guanine-exchange factor that phosphorylates RAS-GDP to RAS-GTP, activating Ras and therefore is integral to lymphocyte development. RASGRP1 deficiency was described in 2016 in patients with a combined immunodeficiency and predisposition to EBV-driven lymphoproliferative disease. In addition to our patient, nine other patients with deleterious RASGRP1 mutations have been described. Patients had recurrent bacterial and viral infections, autoimmunity, and malignancy. There are however no published reports providing details of the successful treatment of EBV lymphoproliferative disease followed by allogeneic hematopoietic cell transplant of RASGRP1 deficiency. Here, we describe a five-year-old male with compound heterozygous mutations who presented with recurrent sinopulmonary infections and EBV-driven lymphoproliferative disease. He was treated with chemotherapy followed by allogenic matched unrelated bone marrow transplant (BMT).
Methods: Retrospective chart and laboratory review.
Results: A two-year-old male was referred to Seattle Childrens Immunology clinic for recurrent otitis media and pneumonia. Initial laboratory evaluation showed elevated IgG (2290 mg/dL), normal CD19 B-cell count, elevated immature/transitional B cells, and normal antibody responses to tetanus, diphtheria, Hib, Pneumovax, and varicella vaccines. However, the T-cell compartment was markedly abnormal with CD4 T-cell lymphopenia (361/mm3), elevated CD8 T-cells (2074/mm3), elevated T-cells (43%), and absent proliferation to mitogens (PHA and anti-CD3) and antigen (tetanus). He was diagnosed with a combined immunodeficiency and was initiated on prophylactic TMP/SMX and azithromycin. Whole exome sequencing identified compound heterozygous mutations in RASGRP1 with a canonical splice donor mutation on one allele (c.1428+1G>A) and a nonsense mutation (c.1780C>T) on the other.
At 3 ½, he developed urinary outlet obstruction and was found to have two large pelvic masses, pulmonary nodules, and diffuse lymphadenopathy. Cervical lymph node and retroperitoneal mass biopsies demonstrated a clonal EBV-driven B cell lymphoproliferative disease with bone marrow and CSF involvement. He was successfully treated with four cycles of prednisone, cyclophosphamide, and rituximab. He also received intrathecal methotrexate twice, which was transitioned to rituximab due to CSF EBV persistence for four treatments, three that followed transplant. EBV PCRs following chemotherapy demonstrated clearance.
Once in remission, the patient underwent conditioning with busulfan and cyclophosphamide and received a HLA-matched unrelated bone marrow graft. He developed grade IIB hyperacute skin GVHD on day +8 with 100% BSA and grade IIA gut GVHD. He was treated with extracorporeal photopheresis and prednisone. Peripheral blood donor chimerisms on day +80 showed full donor engraftment with CD3 98% donor, CD33 100% donor, and CD56 100% donor. CD19 chimerism was not performed, but bone marrow biopsy revealed 100% donor chimerism. Treatment for GVHD was successful with resolution of symptoms. The patient is now 11 months post-transplant and continues to do well.
Conclusions: Like 6/9 of the previously reported RASGRP1 deficient patients, our patient developed profound T cell deficiency complicated by EBV-driven lymphoproliferative disease. Only one other patient has been reported to have survived allogeneic transplant but there are no details about the transplant regimen used. BMT appears to correct the underlying immune defects associated with RASGRP1 deficiency and susceptibility to EBV-driven lymphoproliferation.
(247) Submission ID#607125
Procedure Related Airway Spasms in GOF PI3K Patients with Airway Nodular Hyperplasia
Anahita Agharahimi1, Ashleigh Sun, RN, MSN2, Gulbu Uzel, MD3
1Nurse Practitioner, NIH
2Research Nurse, National Institutes of Health
3Staff Clinician, Laboratory of Clinical Immunology and Microbiology, National institute of Allergy and Infectious Diseases, NIH, Bethesda, MD, USA
Intro/Background: Heterozygous gain-of-function mutations in the kinase domain of PIK3CD, the gene encoding the phosphatidylinositol-3-OH kinase (PI3K) catalytic subunit p110, is an autosomal dominant immune deficiency clinically characterized by recurrent sinopulmonary infections, lymphoproliferation, autoimmune cytopenias, hepatosplenomegaly, EBV and/or CMV viremia, EBV associated lymphoma, and nodular lymphoid mucosal hyperplasia of the gut and respiratory tracts.
Objectives: To present data on PI3K patients who have undergone sedated bronchoscopic procedures that have been complicated by laryngeal spasms related to airway nodular hyperplasia.
Methods: Review of clinical and procedural data of 30 PI3K patients, aged 5- 68 years old who have airway nodular hyperplasia at the National Institutes of Health.
Results: Three patients experienced laryngospasm and one experienced pneumomediastinum up to 24 hours post bronchoscopy procedure which required methylprednisolone for airway inflammation management.
Conclusions: Gain-of-Function PI3K mutations lead to immune deficiency clinically characterized by nodular lymphoid mucosal hyperplasia, which may predispose to airway compromise when procedures involving the airway are performed. Recognizing and preventing complications leading to laryngeal spasm is vital for patient safety. Prophylactic steroids and/or epinephrine pre-procedure may serve to prevent airway compromise in this population.
(248) Submission ID#607128
Evaluation of Persistent Hypogammaglobulinemia Post-rituximab in Patients Undergoing HSCT for XLP-1 Reveals Preserved Ability of B-cells to Class-switch and Differentiate to Plasmablasts In-vitro
Shanmuganathan Chandrakasan, M.D1, Rebecca A. Marsh, MD2, Sharat Chandra, MD, MRCPCH3, Kiran Patel, MD4, Jack Bleesing, MD, PhD5
1Asst. Professor, Children's Healthcare of Atlanta, Emory University
2Associate Professor, Division of Bone Marrow Transplantation and Immune Deficiency, Cincinnati Childrens Hospital Medical Center
3Assistant Professor, UC Department of Pediatrics, Division of Bone Marrow Transplantation and Immune Deficiency, Cincinnati Childrens
4Assistant Professor of Allergy, Department of Pediatrics, Emory University School of Medicine
5Professor, Division of Bone Marrow Transplantation and Immune Deficiency, Cincinnati Childrens
Background: X-linked lymphoproliferative disease type 1 (XLP-1) is a rare immune defect characterized by fulminant Epstein-Barr virus (EBV) hemophagocytic lymphohistiocytosis, lymphoma, dysgammaglobulinemia, aplastic anemia, and vasculitis. Allogeneic HSCT is the only curative option for XLP-1. Due to the underlying predisposition for EBV infection, rituximab has been used pre and post-HSCT (for EBV reactivation and immune cytopenia) in this disorder. We have observed a high incidence of poor B-cell immune reconstitution post-HSCT needing long-term immunoglobulin (Ig) replacement in this disorder. The biology of persistent post-rituximab hypogammaglobulinemia despite complete donor chimerism, the absence of GVHD and discontinuation of immunosuppression is poorly understood. Herein, we report T and B-cell immune profile in patients who are Ig dependent post-HSCT for XLP-1. Additionally, we explored the ability of patients B-cells to undergo class-switching and plasmablast differentiation in-vitro.
Results: 17 XLP-1 patients underwent HSCT at two institutions. At a median follow up of 1406 days (785-2517 days post-HSCT) 13 were surviving. Six of the 13 (46.2%) surviving patients remain dependent on Ig replacement despite robust donor chimerism of 99-100% and no active GVHD. All but two received rituximab pre-HSCT. Of the patients who are independent of Ig replacement, only one (14.2%) received rituximab post-HSCT, whereas 5/6 of the Ig dependent patients received rituximab post-HSCT. T cell immune profiling revealed that the absolute numbers of lymphocyte subsets, CD4+ naïve T cells, and CD4+ recent thymic emigrants were not statistically different between Ig independent and dependent patients (Figure 1). However, there was a marked decrease in the number of total B cells, the percentage of memory B cells (CD27+ B cells), and class-switched memory B cells (CD27+ IgD- IgM- cells) in Ig dependent patients (Figure 1). T follicular helper (Tfh) cell populations (CD4+CD45RA-CXCR5+PD1+) were evaluated in four patients and the frequency was similar to healthy controls (4.5+/-1.2 vs. 3.9+/-1.4%). The ability of the patients naïve B cells to class-switch was assessed following exposure to IL-21, anti-CD40 antibody, and anti-human IgM, and revealed normal B cell class-switching and differentiation to plasmablasts (Figure 1). Additionally, T cell ability to provide B cell help was assessed by co-incubating naïve B cells with activated CD4+ T cells. This revealed comparable B cell class switching to that of healthy controls.
Conclusion: The high incidence of poor long-term functional B cell reconstitution following allogeneic HSCT for XLP-1 could be related to the use of rituximab in the post-HSCT setting rather than pre-HSCT. Normal Tfh numbers and function, and ability of B-cells to class-switch in-vitro suggest that persistent hypogammaglobulinemia is these patients is unlikely from a B or T-cell intrinsic defect. The possibility of rituximab induced acquired lymph nodal stromal defect in these patients is being explored. Further studies are needed to understand the biology of persistent hypogammaglobulinemia in XLP-1. Additionally, due to the high incidence of persistent hypogammaglobulinemia, exposure of rituximab should be limited post-HSCT.
(249) Submission ID#600694
Application of Targeted Proteomics in the Diagnosis and Screening of Primary Immunodeficiency Disorders
Christopher J. Collins, Ph.D.1, Irene Chang, MD2, Fan Yi, PhD1, Remwilyn Dayuha, BA3, Jeffrey Whiteaker, PhD4, Amanda Paulovich, MD, PhD5, Hans Ochs, MD6, Troy Torgerson, MD, PhD6, Sihoun Hahn, MD, PhD6
1Research Scientist, Seattle Children's Hospital
2Senior Fellow, Seattle Children's Hospital
3Research Technician, Seattle Children's Hospital
4Research Scientist, Fred Hutchinson Cancer Research Center
5Director, Fred Hutchinson Cancer Research Center
6Principal Investigator, Seattle Children's Hospital
Background: Tandem mass spectrometry (MS/MS) has emerged as a primary platform for many clinical and newborn screening laboratories. The application of MS/MS mainly focuses on the quantification of accumulated small metabolites in plasma resulting from various metabolic defects. However, many disorders do not yield such metabolic markers and would benefit from the direct quantification of intracellular target proteins. Unfortunately, the extremely low (e.g., pmol/L range) protein concentrations in blood cells limit their detection via MS/MS. In recent years, peptide immunoaffinity enrichment coupled to selected reaction monitoring (immuno-SRM) has emerged as a promising technique for the quantification of low abundance proteins in complex matrices, including dried blood spots (DBS). Our lab has demonstrated that immuno-SRM methods are able to reliably distinguish affected patients from the normal controls for Wilson disease (WD), Wiskott-Aldrich Syndrome (WAS), severe combined immunodeficiency (SCID), and X-linked agammaglobulinemia (XLA) (J. Proteome Res., 2017 and Front. Immunol., in press). These results demonstrate the utilization of immuno-SRM as a sensitive platform for multiplexed quantification of signature peptides in the low pmol/L range.
Methods: Several candidate peptides for each protein were selected based on uniqueness using in Silico BLAST tools and LC-MS/MS response. Monoclonal antibodies (mAbs) were then generated for peptide enrichment from DBS. Blood from normal controls, WD, XLA, SCID, and WAS patients was spotted onto filter paper, dried, and stored at -20 °C until use. Proteins were extracted from DBS, digested with trypsin, and enriched using mAbs bound to magnetic beads. The enriched peptides were then eluted and analyzed using SRM mode with a Waters Xevo TQ-XS.
Results/Conclusions: To date, immuno-SRM methods have been generated for WD, WAS, SCID, XLA, and Cystinosis. Preliminary data shows immuno-SRM methods are able to reliably quantify target proteins using signature peptides and accurately distinguish affected patients from normal controls. Analysis of signature peptides found statistically significant reduction or absence of peptide levels in affected patients compared to control groups in each case (WAS and BTK: p = 0.0001, SCID: p = 0.05). Intra and inter-assay precision ranged from 11 - 22% and 11 - 43%, respectively, and the multiplexed assay showed a broad linear range (1.39 2000 fmol peptide). In a blinded sample set of 42 PIDD patients and 40 normal controls, immuno-SRM-predicted diagnoses showed excellent agreement with clinical or genetic diagnoses. Every molecularly-confirmed case of WAS and BTK was also diagnosed by immuno-SRM analysis. In addition, 62 randomly selected samples provided by the NBS laboratory of Washington State were tested and peptide concentrations were found to be within normal ranges. Efforts are underway to validate and incorporate peptide biomarkers for Adenosine Deaminase deficiency, DOCK8 deficiency, and Ataxia Telangiectasia, as well as general markers for NK cells and platelets into a single multiplexed assay. In addition, SCID, WAS and XLA samples continue to be run while we focus on reducing assay costs, time, and necessary sample input. Our data herein provides proof of concept for the immuno-SRM workflow to be extended to various other genetic diseases as potential multiplexed newborn screening methods.
(250) Submission ID#617782
The Bona-Fide Effect Of Long-Term Glucocorticoids On T Cells: The Endogenous Cushing Syndrome Model And The Role Of IL-21
SuJin Hwang, Ph.D.1, Christina Tatsi, MD, PhD2, Maya Lodish, MD, MHSc3, Hye Sun Kuehn, PhD4, Magdalena A. Walkiewicz, Ph.D.5, Steven M. Holland, MD6, Constantine Stratakis, MD7, Sergio D. Rosenzweig, MD/PhD8
1Research Assoiciate, Immunology Service, Department of Laboratory Medicine, NIH Clinical Center, NIH
2Clinical Fellow in Pediatric Endocrinology, Section of Endocrinology and Genetics, NICHD, NIH
3Professor, University of California San Francisco
4Staff Scientist, Immunology Service, Department of Laboratory Medicine, Clinical Center, NIH, USA
5ABMG certified Clinical Molecular Geneticist, National Institute of Allergy and Infectious Diseases (NIAID)
6Director, Division of Intramural Research, Laboratory of Clinical Immunology and Microbiology, National Institute of Allergy and Infectious Diseases, National Institutes of Health
7Scientific Director, NIH/NICHD
8Chief, Immunology Service, Department of Laboratory Medicine, NIH Clinical Center, Bethesda, MD, USA
Abstract
Background: The long-term effects of glucocorticoids (GCs) on the immune system have been extensively studied in patients with different underlying conditions (e.g, malignancies or autoimmune conditions), as well as in healthy volunteers receiving short-term courses of these drugs. Although these approaches provided highly relevant data, neither of them answered the unbiased/bona-fide effect of long-term GCs use on the immune system. Endogenous Cushing syndrome (ECS) may be caused by pituitary or ectopic ACTH-producing adenomas, or by tumors or hyperplasia of the adrenal cortex. Patients with ECS present with different GCsdependent manifestations, including those affecting the immune system as neutrophilia and lymphopenia. When tumors are removed, most of the effects of GCs tend to progressively regress.
Methods: Paired samples from 15 patients with ECS due to ACTH-producing adenomas (age range 7-16y, 8 females) were studied before (ECS-Pre) and 6-12 months after tumor removal (ECS-Post). Extended lymphocyte phenotypes and apoptosis in different cell subsets were evaluated by flow cytometry. Cytokine production (ELISA) and responses, as well as their effects on cell proliferation and viability, were evaluated using Cell Trace Violet and Annexin-V staining.
Results: Among multiple immunophenotypic changes, ECS-Pre patients showed significantly reduced naïve T cells and recent thymic emigrants (RTE) as well as increased apoptosis in T cells when compared to themselves (ECS-Post) or age matched healthy controls. Moreover, significantly increased exhausted CD8 T cells were observed in ECS-Pre patients. Interestingly, ECS-Post patients showed full cellularity recovery of T cells and RTE with increased proliferation and reduced apoptosis, in addition to correction of most of the other changes evidenced. Significantly lower IL-21 plasma levels were also detected in ECS-Pre when compared to ECSPost patients. To determine the role of IL-21 in an ECS-resembling condition, healthy control PBMCs were treated with GCs in-vitro and the effect of IL-21 and other cytokines was tested. A significant reduction in apoptosis was observed in the IL-21-treated cells that almost completely countered the pro-apoptotic effects of GCs; IL-21 was also significantly more efficient than IL-2, IL-7, IFN-alpha and IFN-gamma in rescuing cells from apoptosis. IL-21-specific upregulation of BCL2 and BCL6 expression was evidenced in these cells.
Conclusions: Chronic use of high dose GCs is a relatively common medical situation frequently associated with T cell lymphophenia and increased susceptibility to opportunistic infections. By studying ECS-Pre and ECS-Post patients we were able to describe the bona-fide effect of GCs on the immune system in general, and T lymphocytes in particular. Decreased lymphocyte/thymic output, as well as increased apoptotic T-cell death underlies lymphopenia in ECS/chronic GCs-exposed patients. Under such conditions, IL-21 was significantly decreased in plasma and our in-vitro studies showed that IL-21 replenishment was able to increase BCL2 (anti-apoptotic molecule) and BCL6 expression, and efficiently counteract the apoptotic effects of GCs. Recombinant IL-21 has been explored as a co-adjuvant treatment for multiple human cancers and may offer a treatment option for lymphopenia and its complications in patients with ECS/ chronic GCs exposure. Further studies are warranted to evaluate this therapeutic option.
(251) Submission ID#619776
F-BAR domain only protein 1 (FCHO1) deficiency is a novel cause of combined immune deficiency in humans
Enrica Calzoni, MD1, Craig D. Platt, MD, PhD2, Sevgi Keles, MD3, Hye Sun Kuehn, PhD4, Yu Zhang, PhD5, Julia Pazmandi, MSc6, Gaetana Lanzi, PhD7, Azzedine Tahiat, PhD8, Hasibe Artac, MD9, Jasmin Dmytrus, Msc10, Ismail Reisli, MD11, Dilara Uygun, MD12, Bertrand Boisson, PhD13, Sergio D. Rosenzweig, MD/PhD14, Helen C. Su, MD, PhD15, Silvia Giliani, PhD16, Michael J. Lenardo, MD17, Raif S. Geha, MD18, Kaan Boztug, MD19, Janet Chou, MD20, Luigi D. Notarangelo, MD, PhD21
1Graduate Student, Laboratory of Clinical Immunology and Microbiology, IDGS, DIR, NIAID, NIH, Bethesda, MD, USA
2Attending Physician, Division of Immunology, Boston Childrens Hospital, Harvard Medical School, Boston, MA 02115
3Attending Physician, Division of Pediatric Immunology and Allergy, Meram Medical Faculty, Necmettin Erbakan University, Konya, Turkey
4Staff Scientist, Immunology Service, Department of Laboratory Medicine, Clinical Center, NIH, USA
5Staff Scientist, Laboratory of Clinical Immunology and Microbiology, National Institute of Allergy and Infectious Diseases, National Institutes of Health, Bethesda, MD 20892, USA
6Graduate Student, Ludwig Boltzmann Institute for Rare and Undiagnosed Diseases, Vienna, Austria
7Staff Scientist, A. Nocicelli Institute for Molecular Medicine, Department of Molecular and Translational Medicine, University of Brescia, Brescia, Italy
8Staff Scientist, Laboratory of Medical Biology, Rouiba Hospital, Algiers, Algeria
9Attending Physician, Pediatric Immunology and Allergy, Selcuk University Medical Faculty, Konya, Turkey
10Graduate Student, Ludwig Boltzmann Institute for Rare and Undiagnosed Diseases, Vienna, Austria
11Professor, Division of Pediatric Immunology and Allergy, Meram Medical Faculty, Necmettin Erbakan University, Konya, Turkey
12Attending Physician, Department of Immunology-Allergy, Akdeniz University School of Medicine, Antalya, Turkey
13Assistant Professor, St. Giles Laboratory of Human Genetics of Infectious Diseases, Rockefeller Branch, The Rockefeller University, New York, NY 10065
14Chief, Immunology Service, Department of Laboratory Medicine, NIH Clinical Center, Bethesda, MD, USA
15Chief, Human Immunological Diseases Section, Laboratory of Clinical Immunology and Microbiology, NIAID, NIH, Bethesda, MD
16Associate Professor, A. Nocicelli Institute for Molecular Medicine, Department of Molecular and Translational Medicine, University of Brescia, Brescia, Italy
17Senior Investigator, Molecular Development of the Immune System Section, Laboratory of Immune System Biology, NIAID, National Institutes of Health, Bethesda, MD, USA
18Chief, Division of Immunology, Boston Childrens Hospital, Harvard Medical School, Boston, MA 02115
19Director, Ludwig Boltzmann Institute for Rare and Undiagnosed Diseases, Vienna, Austria
20Associate Professor, Division of Immunology, Boston Childrens Hospital, Harvard Medical School, Boston, MA 02115
21Chief, Laboratory of Clinical Immunology and Microbiology, IDGS, DIR, NIAID, NIH, Bethesda, MD, USA
Abstract
Clathrin-mediated endocytosis (CME) is the major endocytic pathway by which eukaryotic cells internalize cell-surface cargo proteins and extracellular molecules, thereby allowing for a broad range of biological processes, including cell signaling, nutrient and growth factor uptake, and cell fate and differentiation1. The FBAR domain only proteins 1 and 2 (FCHO1/FCHO2) are involved in the initiation of clathrin coat pit formation. Whether FCHO1 and FCHO2 are functionally redundant or have distinct functions is unclear. We report here the first cases of a severe immunodeficiency due to a genetic defect affecting CME. By using whole exome sequencing and genomic analysis of a targeted PID gene panel, we have identified biallelic loss-of-function FCHO1 mutations in five patients from unrelated families of Italian (P1), Turkish (P2, P3, and P5) and Algerian (P4) origin with severe T cell lymphopenia manifesting as recurrent and severe infections of bacterial, mycobacterial, viral and fungal origin. P3 developed EBV-associated diffuse large B cell lymphoma. Three patients (P3-P5) died in childhood, whereas P1 and P2 are alive with full donor chimerism at 13 and 1.5 years after allogeneic hematopoietic stem cell transplantation, respectively and have cleared pre-transplant infections. Patients P2, P3, and P4 carried homozygous frameshift mutations predicted to cause premature termination. Western-blotting analysis of HA- or FLAG-tagged FCHO1 constructs showed expression of truncated products in P2 and P3, whereas no protein was detected in P4, presumably due to mRNA decay. P1 and P5 carried homozygous splice-site mutations at the invariant -1 and +1 positions, respectively, leading to skipping of exon 6 in P1's FCHO1 cDNA. qPCR analysis demonstrated differential expression of the FCHO1 and FCHO2 genes, with the former being predominantly expressed in lymphoid cells, whereas FCHO2 was more abundantly expressed in fibroblasts and K562 cells.
Analysis of T cell activation in P2 (the only patient for whom pre-transplant PBMC were available) revealed reduced T cell proliferation. While TCR internalization in response to CD3 cross-linking was normal (consistent with recent evidence that TCR internalization occurs through a clathrin-independent pathway), chase experiments demonstrated that transferrin internalization was abolished in activated T cells from P2. We had previously reported that a missense mutation in TFRC, encoding transferrin receptor 1, impairs transferrin internalization and intracellular iron delivery, causing a combined immunodeficiency with defective T cell proliferation.
Our data identify the first form of severe immunodeficiency due to defects of clathrin-mediated endocytosis, and provide additional evidence in support of the critical role played by iron cellular metabolism in T cell function and homeostasis.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.