

Abstract
This study reports an outbreak of acute febrile respiratory illness caused by human adenovirus B [P14H11F14] in a military training center in China between May and June 2014. In total, 164 military personnel were affected, and two patients were admitted into the intensive care unit of the military regional central hospital. A HAdV-B [P14H11F14] virus was confirmed as the etiological pathogen of this acute outbreak of febrile respiratory illness based on clinical manifestations, epidemiological characteristics, specific molecular detection results, phylogenetic analysis, and serological assays. The virus was isolated by the rhabdomyosarcoma cell culture method, and the complete sequences of the E1A, penton base, hexon, and fiber genes were determined and deposited in the GenBank database. Phylogenetic and sequence homology analyses indicated that the isolated strain is most closely related to some HAdV-55 strains from mainland China. However, this strain appeared to be less virulent than former HAdV-55 strains. According to the chest X-ray results of 31 affected patients, there was no radiological evidence of pneumonia. The most frequent symptoms in these patients were sore throat (95.12 %, 156/164) and tonsillitis (93.29 %, 153/164). During the course of the outbreak, incorrect response measures and some potential risk factors, such as fire training and marching training, may have exacerbated the spread of the infection. This outbreak illustrates the urgent need to improve the epidemiological and etiological surveillance of HAdV infections and to improve the ability of doctors and health officials in basic units of the Chinese army to respond effectively to febrile respiratory illness.
Keywords: Fiber Gene, Hexon, Training Camp, Hexon Gene, Febrile Respiratory Illness
Introduction
Human adenoviruses (HAdVs) are responsible for a broad spectrum of clinical diseases, including febrile respiratory illnesses (FRI), gastroenteritis, pneumonia, and kidney infections [1–4]. These ubiquitous viruses belong to the genus Mastadenovirus in the family Adenoviridae. So far, 68 genotypes of HAdV have been recognized and classified into seven species (A–G) based on their biological and genetic characteristics (http://HAdVwg.gmu.edu/).
Among these genotypes, a re-emerged HAdV-B [P14H11F14] virus has attracted increasing attention in China. This HAdV-B [P14H11F14] virus is an intertypic recombinant of HAdV-B11 and HAdV-B14. It has a HAdV-14 genome chassis, including the HAdV-14 penton gene and fiber gene, but a partial HAdV-11 hexon gene, which encodes the antigenic epitopes of the virus [5]. This virus not only could possess the virulence of HAdV-14 but also could avoid the neutralizing antibody against HAdV-14, which exists much more widely in the population than that against this HAdV-B [P14H11F14] virus [6].
An HAdV-B [P14H11F14] virus was first identified as an atypical HAdV-11 strain in Spain in 1969 [7]. It then appeared in a Turkish military camp in 2004, where hundreds of military trainees fell ill and one man died in this outbreak. In recent years, this acute FRI pathogen has re-emerged several times, once in Singapore in 2005 [8], and twice in China, in Shannxi Province in 2006 [6] and in Beijing in 2011 [9]. In some outbreaks, it has caused severe respiratory disease and death in immunocompetent adults, including military recruits and high school students [5, 10].
In a serological study, this respiratory tract pathogen was classified as “HAdV 14-11 intermediate” [11]. Based on genome typing by restriction enzyme analysis, this virus was designated as HAdV-B11a [12, 13]. When the computational genomic analysis method was used in 2010 by Walsh et al., the results showed that within the context of the molecular evolution of HAdV, this pathogen is a novel adenoviral type and should be designated HAdV-B55 [P14H11F14] [14]. According to a previous study, HAdV-55 has become a major causative agent of acute severe pneumonia in the Chinese population [15].
Here, we report an outbreak of acute FRI in a Chinese military training center in 2014. Over 36 days, from 7 May to 11 June, 164 individuals were affected. Molecular characterization, phylogenetic analysis, and serological assays revealed that HAdV-B [P14H11F14] virus was the etiological agent of this outbreak, and the complete E1A gene, penton base gene, hexon gene, fiber gene of this virus all belonged to the HAdV-55 type. This is the first epidemiological and etiological report of a type-55-like HAdV-B [P14H11F14] virus associated with an FRI outbreak in a Chinese military camp. The clinical characteristics of this outbreak differ from those of previous HAdV-55 outbreaks reported in Chinese civilians and foreign armies. Unlike other HAdV-55 outbreaks, no case of pneumonia developed in any patient.
Materials and methods
Responses to the outbreak
On 16 April 2014, military training commenced at a military training center (MTC) in central Shandong Province, China. About 1600 military personnel were staying at this training center, which consisted of four training camps (about 350 soldiers per camp) and a support team (about 200 support staff, including 23 female soldiers). Of these personnel, about 20 % were new recruits (service period <1 year) and the others were regular soldiers (service period >1 year). According to the training plan, the training schedule included daytime mid-range marches (twice a week) and night fire training (twice a week) between 21 April and 17 May.
In May 2014, clustered cases of acute FRI (febrile respiratory illnesses) appeared in this MTC, which was defined as an individual with a body temperature >37.5 °C and with at least one sign or symptom of acute respiratory tract infection, such as a cough or sore throat. In the early stage of the outbreak, 1–2 cases of FRI were admitted to the MTC hospital every day. This situation did not draw the attention of the MTC managers, and the outbreak was mistaken for severe seasonal influenza by the doctors of the MTC hospital. However, after 22 May, the incidence of disease suddenly increased to more than 10 cases per day, and all patients showed similar symptoms. On 29 May, in an effort to prevent an outbreak of the illness, about 798 soldiers were gathered into several crowded classrooms to attend a health lecture. The MTC managers also appealed to the regional Center for Disease Control and Prevention (CDC) for help. On 30 May, an epidemic survey was conducted by the regional CDC. At the same time, disease control and prevention measures were adopted to control the outbreak, including the cessation of training, isolation of affected individuals, environmental disinfection, and morning temperature screening. No new cases were reported after 11 June.
Specimen collection
To investigate the etiological agent of this outbreak, throat swab specimens (n = 49, from the date of onset on 27 May to 11 June) were collected. Sixty serum samples from FRI patients affected after 7 May were collected on 30 May. On 15 June, 43 paired serum samples from patients in the convalescent phase were also collected. At the same time, another 51 healthy soldiers were randomly selected from all of the training camps as the control group. Their paired sera were collected on 31 May (as the outbreak-phase samples) and 20 June (as the after-outbreak-phase samples). The sera and swab specimens were transferred to the regional CDC laboratory at 4 °C within 24 h and stored at –20 °C and –80 °C, respectively, until further analysis.
Extraction and analysis of viral nucleic acids from throat swabs
Viral nucleic acids were extracted directly from the throat swab specimens using a MiniBEST Viral RNA/DNA Extraction Kit (TaKaRa, Dalian, China). A one-step reverse transcription (RT)–PCR was then performed using an RV13 PCR Detection Kit (Neuro Hemin, Hangzhou, China). This PCR kit can detect 11 types of RNA virus (including influenza type A viruses, influenza type B viruses, human respiratory syncytial viruses A and B, human metapneumovirus, human parainfluenza viruses 1, 2, and 3, human rhinovirus, human coronaviruses 229E and OC43, and human enteroviruses) and two types of DNA virus (human bocavirus 1, 2, 3, 4 and human adenovirus).
The primary identification of HAdV in the positive specimen was performed by PCR using the adenovirus-specific primers described by Allard et al. [16]. The amplified target of this specific PCR was the partial hexon gene sequence (467 bp) in the HAdV genome. The amplicon was sequenced and analyzed using the BLAST program (National Center for Biotechnology Information).
Cell culture and viral isolation
Rhabdomyosarcoma (RD) cells (ATCC CCL-136) were cultured in Dulbecco’s modified Eagle’s medium (DMEM, containing 10 % fetal bovine serum, 100 U/ml penicillin G, 100 μg/ml streptomycin; Gibco, Carlsbad, California, USA) at 37 °C in an closed system with 5 % CO2 incubation.
To isolate the virus, RD cells were inoculated with the patients’ throat swab specimens (300 μl of each specimen) and cultured in DMEM containing 2 % fetal bovine serum, 100 U/ml penicillin G, 100 μg/ml streptomycin (Gibco, Carlsbad, California, USA) at 37 °C in an closed system with 5 % CO2 incubation. The cultures were observed daily for a cytopathic effect (CPE). In 2–7 days, the cultures displaying an adenovirus-like CPE were passaged again to confirm the presence of the virus. The positive isolates were identified by adenovirus-specific PCR as described by Allard et al. [16].
Extraction of viral DNA and gene amplification
Viral DNA was extracted from the HAdV-positive cell cultures using a TaKaRa MiniBEST Viral RNA/DNA Extraction Kit. The complete E1A, penton base, hexon, and fiber genes were amplified and sequenced using specific primer sets designed at the Jinan Junqu CDC (listed in Table 1). The PCR was performed using a Ready-To-Use PCR Kit (Pfu B532073; Sangon, Shanghai, China). The 50-μl PCR reaction mixture contained 25 μl of 2 × HiFi PCR Master, 21 μl of sterile ddH2O, 2 μl of DNA template, and 0.2 μM each primer. The PCR cycling parameters were initial denaturation at 94 °C for 4 min, followed by 35 cycles of denaturation at 94 °C for 30 s, annealing at 51 °C for 50 s, and extension at 72 °C for 80 s, with a final extension step at 72 °C for 10 min. The amplification products were analyzed by 1.5 % agarose gel electrophoresis.
Table 1.
Primers used to amplify and sequence the entire E1A, penton base, hexon, and fiber genes of HAdV-B [P14H11F14] strain SD77001
| Primer | Sequence (5’ - 3’) | Size (bp) |
|---|---|---|
| E1A_1f | TGCTATGAAGACGGGTTTCC | 748 |
| E1A_1r | CGTCCGAAGCGTTCTCTAAC | |
| E1A_2f | ATGGCAGGGTGGAGTATTTG | 700 |
| E1A_2r | AACACTGGCAGCCTTCACTC | |
| Penton_1f | TCGCTTGGGTGGTATGTTGTA | 751 |
| Penton_1r | CACCAATGTCACTTTCAAGCA | |
| Penton_2f | GTGATGGTGTCCAGAAAACCT | 807 |
| Penton_2r | ATCTGAGGTGGTGAGCAATGT | |
| Penton_3f | GCTAACGCTGGAGAGGTCAG | 790 |
| Penton_3r | CACGGGATGTTGGGTAGAAC | |
| Hexon_1f | TCACAGCAGCAGAGGAAAAA | 701 |
| Hexon_1r | TTTCGGTCTTTCCATCAAGG | |
| Hexon_2f | TCAGTGGATTGCAGAAGGTG | 996 |
| Hexon_2r | CTTCGCCATAGATTGGCTTG | |
| Hexon_3f | ATGCAGTGGTTGACTTGCAG | 907 |
| Hexon_3r | TTGGCAGGAATGGGATAGAG | |
| Hexon_4f | CAAAAACCTGCTGCTTCTCC | 908 |
| Hexon_4r | GCCACATGGTTCTGTCACAC | |
| Hexon_5f | CAGATGCTCGCCAACTACAA | 845 |
| Hexon_5r | TCATCCGAGAATCCAAAAGG | |
| Fiber_1f | TTAACCCCGCTAACAACCAC | 899 |
| Fiber_1r | GAAGGGGGAGGCAAAATAAC | |
| Fiber_2f | GGCCAAAGAGCTCAGAGATG | 804 |
| Fiber_2r | ATCCGGCTCCTAGGGATAAA |
Primers were designed according to human adenovirus 55 strain QS-DLL (GenBank accession: FJ643676)
Sequence determination and phylogenetic analysis
The amplified products were sequenced in both directions by the dye terminator method (BigDye Terminator v3.1 Cycle Sequencing Kit; Applied Biosystems) using an ABI Prism 3100 Genetic Analyzer (Applied Biosystems). The nucleotide and predicted amino acid sequences were analyzed using the BioEdit program (http://www.mbio.ncsu.edu/BioEdit/bioedit.html). Nucleotide sequence homologies were inferred from the identity scores determined using the BLASTn program (National Center for Biotechnology Information, Bethesda, MD, USA). Sequence alignments were constructed with ClustalX version 2.1 [17], and phylogenetic analyses were performed with the MEGA 5 program [18]. The reference sequences were retrieved from GenBank (www.ncbi.nlm.nih.gov/GenBank). The reliability of the phylogenetic trees was examined with 1000 bootstrap pseudoreplicates.
HAdV-specific antibody detection using enzyme-linked immunosorbent assays (ELISAs)
All serum specimens from patients (including 43 specimens collected in the acute phase and 60 specimens collected in the convalescent phase) in this outbreak and from healthy controls (including the paired specimens collected during the outbreak phase and after the outbreak phase from 51 healthy soldiers) were examined using an ELISA classic HAdV IgA kit and HAdV IgG kit (Institute Virion/Serion GmbH, Würzburg, Germany). These two kits allowed the detection of HAdV immunoglobulin A (IgA) and immunoglobulin G (IgG) directed against all HAdV serotypes that are pathogenic to humans. According to the instruction of the manufacturer, the sensitivity of the SERION ELISA classic Adenovirus IgA/IgG is 96.9 %, and the specificity is 97.8 % (Instruction of the SERION ELISA classic Adenovirus IgA/IgG KIT V128.14).
Results
Outbreak description
On 7 May 2014, a 19-year-old new recruit from camp 3 developed an acute FRI, with a body temperature of 38.9 °C. Until 11 June, 1–17 cases of FRI per day were reported over an almost 5-week period, and 164 soldiers were admitted to the MTC hospital (10.25 %, 164/1600 soldiers). The outbreak peaked three times in 36 days. The intervals between the peaks were about 6–8 days. All of the patients were male. No symptomatic cases were detected among female soldiers. The mean age of the patients was 20.24 ± 2.16 years (median, 20 years; range, 17–32 years). The distribution of daily cases is shown in Figure 1. Patients were reported in all four training camps. The patient distribution among the training units was as follows: camp 1, 67 (19.14 %, 67/350); camp 2, 24 (6.86 %, 24/350); camp 3, 49 (14.00 %, 49/350); camp 4, 16 (4.57 %, 16/350); support team, 8 (4.00 %, 8/200). The incidence rate among the new recruits was 7.81 % (25/320), and the incidence rate among the regular soldiers (service >1 year) was 10.86 % (139/1280). According to the statistical analysis, these rates did not differ significantly (P > 0.05, χ2 = 2.583; SPSS 19). This result indicates that the pathogen responsible for this outbreak may have been an unusual or newly emergent pathogen in this area.
Fig. 1.
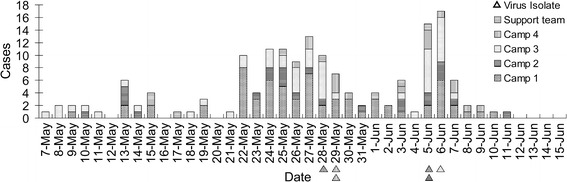
Case distribution in an outbreak of FRI caused by a type-55-like HAdV-B [P14H11F14] virus in a military training center in Shandong Province, China, from 7 May to 15 June. The triangular markers represent six virus isolates, indicating their onset time distribution and unit distribution
Clinical characteristics of the patients
The clinical characteristics of the patients are summarized in Table 2. Among the 164 hospitalized patients, all presented with fever, and most had a sore throat or tonsillitis. Other symptoms reported by the patients were headache, fatigue, cough, sputum, nasal discharge, and diarrhea. Two patients were admitted to the intensive care unit (ICU) of the military regional central hospital because of the prolonged fever (fever more than 5 days, peak temperature over than 39 °C. Patient 1 was diagnosed with acute suppurative tonsillitis, with a peak temperature of 40.0 °C, elevated neutrophilic granulocytes (NE %, 73.4 %, reference value range, 50-70 %), and decreased lympholeukocytes (LYM %, 17.9 %,) reference value range, 20-40 %). Patient 2 was diagnosed with upper respiratory tract infection, with a peak temperature of 39.5 °C, decreased leukocyte counts (WBC, 1.7 × 109/L, reference value range 4.0-10.0 × 109/L)). Other patients were observed and treated at the MTC hospital. On average, the FRI lasted for 4–7 days and all of the patients recovered well. X-ray examinations were conducted of 31 patients (including the two patients admitted to the ICU and 29 patients in the MTC hospital), but no radiological evidence of pneumonia was confirmed. Therefore, no case of pneumonia developed in this FRI outbreak.
Table 2.
Symptoms and characteristics of 164 patients in the military outbreak of FRI, 2014
| Symptom | No. (%) of patients n = 164 |
|---|---|
| Fever | 164 (100) |
| Median peak temperature, °C (range) | 38.5 (37.5 – 40.0) |
| Median duration, days (range) | 3 (2 – 9) |
| Sore throat | 156 (95.12%) |
| Tonsillitis | 153 (93.29%) |
| Headache | 63 (38.41%) |
| Fatigue | 60 (36.59%) |
| Cough | 34 (20.73%) |
| Sputum | 15 (9.15%) |
| Nasal discharge | 7 (4.27%) |
| Diarrhea | 3 (1.83%) |
Detection and identification of the pathogenic agent
A series of serological tests and routine blood cultures were performed to detect Streptococcus, Coxiella, Rickettsia, Mycoplasma, Chlamydia, Legionella, Brucella, and typhoid fever, but all were negative. A diagnostic one-step RT-PCR was performed on all throat swab samples using an RV13 PCR Detection Kit (Neuro Hemin, Hangzhou, China). Of the 49 patient swab specimens, 45 specimens were positive for HAdV DNA, and all 49 specimens were negative for 11other viruses. Then, all 49 specimens were re-tested by HAdV-specific PCR with primers specific for a 467-bp fragment of the partial hexon gene of HAdV. DNA from these 45 HAdV-positive specimens was successfully amplified. The amplicons were sequenced and analyzed using the BLAST program. According to the BLAST analysis results, all 45 amplicon sequences had a high level of sequence similarity, (99 %) to the 2006 China HAdV-B55 strain QS-DLL (Shanxi, China, 2006; GenBank accession number FJ643676). The RV13 detection results and the sequence analysis results indicated that in these 49 throat swab specimens, 91.84 % (45 of 49) were positive for HAdV. A 467-bp HAdV-B55-like hexon gene fragment could be detected in every HAdV-positive specimen.
Virus isolation and amplification of the major structural protein genes
To isolate the virus, RD cells were inoculated with patient throat swab specimens (n = 20). The characteristic adenovirus-like CPE was observed in six cultures from six throat swab specimens, and all these cultures were confirmed to be HAdV positive by HAdV-specific PCR. According to the BLAST analysis, the amplicon sequences (partial hexon gene fragment, 467 bp) of the six HAdV isolates were 100 % identical to each other and to their sequencing results before virus isolation. One of the six HAdV isolates (strain SD77001) was selected for further genetic study. The complete penton base, hexon, fiber, and E1A genes were amplified and sequenced using the specific primer sets listed in Table 1. The sequence data were deposited in GenBank. (GenBank accession numbers: SD77001 strain complete hexon gene sequence: KR912178; penton base gene sequence: KT253552; fiber gene sequence: KT253553; E1A gene sequence: KT253554).
Molecular and phylogenetic analysis of the HAdV strain
To investigate the genetic relationships between isolate SD77001 and other HAdV strains, phylogenetic trees were constructed by the maximum-likelihood method with 1000 bootstrap pseudoreplicates using the MEGA 5 program [18]. The phylogenetic analyses were based on the complete hexon, penton base, fiber, and E1A gene sequences of strain SD77001 and their counterparts in 23 other HAdV strains representing HAdV species A–G. In these analyses, HAdV isolate SD77001 clustered with the HAdV-55 strains (Fig. 2). In the phylogenetic analysis based on the complete HAdV hexon gene sequence (Fig. 2a), the hexon gene sequence of SD77001 clustered with those of the HAdV-55 strains and HAdV-11 strains. The strains most similar to SD77001 were HAdV-55 strain QS_DLL_China and ak36_Argentina (BLAST score, 5241; identity, 99 %), followed by Singapore HAdV-11a strain SGN1222 and Taiwanese HAdV-11a strain 760 (BLAST score, 5236; identity, 99 %). On the phylogenetic tree based on the entire penton base gene sequence (Fig. 2b), isolate SD77001 clustered with all the HAdV-55 strains and two HAdV-14 strains: de-Wit and GZ01. The penton base sequence of almost every HAdV-55 strain was 100 % identical to that of SD77001, including that of Singapore strain SGN1222, Chinese strains QS-DLL and CQ-814, Egyptian strain AK37, and Argentinian strain ak36 (BLAST score, 3092; identity, 100 %). On the fiber sequence phylogenetic tree (Fig. 2c), SD77001 clustered with the HAdV-55 strains and HAdV-14 strains. According to the BLASTn analysis, the fiber gene sequence of SD77001 was 100 % identical to those of HAdV-B55 strain 760_Taiwan, strain QS-DLL_China, and strain South-Dakota_USA (BLAST score, 1807; identity, 100 %), and 99 % identical to HAdV-B55 strains 273_Spain, ak36_Argentina, SGN1222_Singapore, and ak37_Egypt (BLAST score, 1801; identity, 99 %). In the phylogenetic tree based on E1A (Fig. 2d), the E1A sequence of isolate SD77001 clustered with those of the HAdV-55 and HAdV-14 strains. Most of the HAdV-55 strains were 100 % identical to SD77001 in the E1A gene sequence. To determine the genetic relationships between SD77001 and the other HAdV-B2 strains, a phylogenetic tree was constructed based on the complete hexon gene sequences of isolate SD77001 and reference strains of HAdV subspecies B2 (Fig. 2e). On this tree, isolate SD77001 clustered with the HAdV-55 strains and HAdV-11 strains. Four main clades were formed in the HAdV-55 (11a) cluster. The first clade was composed of Argentinian strain ak36, some Chinese HAdV-55 strains, and SD77001. The second clade contained Taiwanese strains. The third clade included the HAdV-11a strains from Egypt, Singapore, and the USA. The fourth clade contained the strain isolated in Spain in 1969. To summarize this phylogenetic analysis, SD77001 shared greatest identity with the HAdV-55 strains. This isolate has the HAdV-55 hexon gene, HAdV-55 penton base gene, HAdV-55 fiber gene, and HAdV-55 E1A gene. However, in the presenand t study, we did not determine the complete genome seequence of SD77001, thus the designation HAdV-B55 can not be used directly. As a reference, HAdV-55 was designated as HAdV-55 [P14H11F14]. Therefore, in accordance with the taxonomic nomenclature of HAdV (http://HAdVwg.gmu.edu/), isolate SD77001 should be designated “adenovirus B human/CHN/SD77001/2014[P14H11F14]”.
Fig. 2.
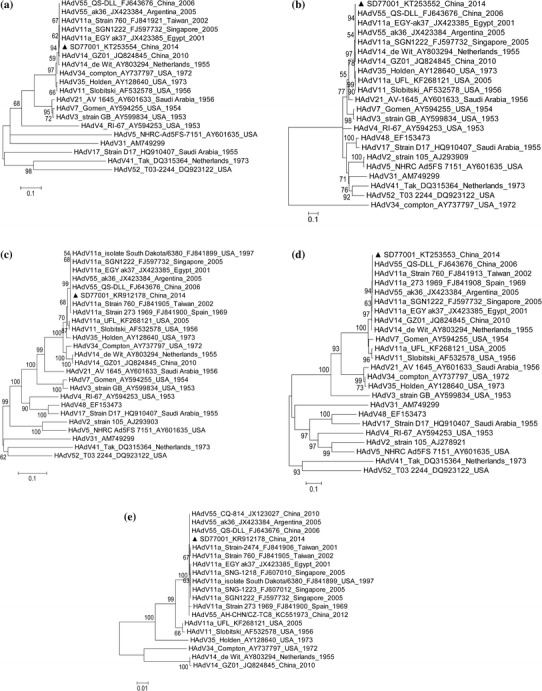
Phylogenetic analysis based on the complete hexon, penton base, fiber, and E1A gene sequences of strain SD77001 and their counterparts in 23 other HAdV strains representing HAdV species A–G. Phylogenetic analysis was conducted by using the neighbor-joining method and 1000 bootstrap pseudoreplicates using the MEGA 5 program. HAdV-B [P14H11F14] isolate SD77001 (this outbreak) is indicated by a black triangle. Reference strains are labeled with their GenBank accession numbers, countries of collection, and dates of collection. (a) Phylogenetic tree based on the complete E1A gene. (b) Phylogenetic tree based on the complete penton base gene. (c) Phylogenetic tree based on the complete hexon gene. (d) Phylogenetic tree based on the complete fiber gene. (e) Phylogenetic tree based on the complete hexon gene of HAdV-B [P14H11F14] strain SD77001 and reference strains of HAdV subspecies B2
HAdV-specific serological assay
To confirm the etiological agent of this outbreak, HAdV-specific serological assays were performed. All serum specimens from patients and healthy controls were examined using a classic ELISA HAdV IgA kit and IgG kit (Institute Virion/Serion GmbH). The detection results are summarized in Table 3. The statistical analysis revealed that in the acute phase (within 4 days of onset), the proportion of IgA-positive samples was significantly higher in the patient group than in the control group, but the proportion of IgG-positive samples did not differ significantly between the patient and control groups (Table 3). In the convalescent phase (>12 days after onset), the proportion of IgA-positive samples was significantly higher in the patient group than in the control group, and the proportion of IgG-positive samples was also significantly higher in the patient group than in the control group. These results confirm that HAdV was associated with this outbreak. On the other, in hand, the healthy group, there were significant differences in the proportions of IgA-positive samples (11.8 % vs 52.9 %, P–IgA < 0.01, χ2 = 19.76) and in the proportions of IgG-positive samples (13.7 % vs 58.8 %, PIgG < 0.01, χ2 = 22.44) between the outbreak phase and the after-outbreak phase (same period of acute phase and convalescent phase). This implies the presence of undetected infections in this military camp, which may have played an important role in the transmission of HAdV.
Table 3.
HAdV-specific serological assay and statistical analysis
| Phase | Patients | Healthy controls | Chi-square testb | |
|---|---|---|---|---|
| Acute phase | IgA |
27.9 % (12 of 43) |
11.8 % (6 of 51) |
P < 0.05 χ2 = 3.93 |
| IgG |
18.6 % (8 of 43) |
13.7 % (7 of 51) |
P = 0.52 χ2 = 0.41 |
|
| CONVa phase | IgA |
85.0 % (51 of 60) |
52.9 % (27 of 51) |
P < 0.01 χ2 = 13.56 |
| IgG |
93.3 % (56 of 60) |
58.8 % (30 of 51) |
P < 0.01 χ2 = 18.81 |
aCONV, convalescent phase
bChi-square tests were conducted using SPSS 19
In conclusion, the regional CDC officials concluded that a type-55-like human adenovirus B human/CHN/SD77001/2014/[P14H11F14] virus was the etiological pathogen responsible for this acute FRI outbreak based on the clinical manifestations in the patients, the epidemiological characteristics of the outbreak, the HAdV-DNA-specific PCR results, isolation of the virus, sequencing and alignment of the PCR amplicons, homology and phylogenetic analyses based on the complete E1A, penton base, hexon, and fiber gene sequences, and serological assays specific for HAdV IgA/IgG.
Discussion
This report describes an acute FRI outbreak caused by a type-55-like HAdV-B [P14H11F14] virus. This virus shared the most sequence similarity with the HAdV-55 in the complete E1A gene, penton base gene, hexon gene, and fiber gene. HAdV-55 infections are often associated with severe respiratory tract diseases and pneumonia, including the outbreaks in Turkey in 2004 and in China in 2006 and 2011. However, in the outbreak presented here, although the disease spread very fast, no pneumonia or other severe disease developed in any of the 164 infected patients. This suggests that the HAdV strain associated with this outbreak was less virulent than previously reported HAdV-B [P14H11F14] strains. Genomic variance or recombination may be responsible for this difference.
Recombination is an important feature of the genetic evolution of HAdV. According to our analysis, the E1A, penton base, and hexon genes of isolate SD77001 are most closely related to those of Chinese mainland strain QS-DLL_2006 and Argentinean strain ak36_2005. However, its fiber gene is 100 % identical to those of USA strain South Dakota/6380_1997, Taiwanese strain 760_2002, and Chinese strain QS-DLL_2006 but differs slightly from that of the Argentinean strain. These characteristics may be the features of regional variants of the same genotype but these variations may have affected the infectivity or virulence of the new recombinant. Further research, including complete genome sequencing and analysis, is required to identify any other relevant genetic variations that are responsible for the milder clinical manifestations of this HAdV-B [P14H11F14] strain.
Many seroprevalence studies among military recruits have shown that lack of pre-existing immunity to some particular serotypes of pathogen is a critical factor determining susceptibility to infection. From an epidemiological perspective, outbreaks of respiratory illness in a military camp usually show higher infection rate among new recruits than among regular soldiers. However, in the present outbreak, there was no significant difference in the incidence rates of the new recruits and regular soldiers. This phenomenon suggests that the main pathogen of this outbreak is an unfamiliar pathogen to these troops, or even in this whole area. Therefore, both the recruits and regular soldiers lacked pre-existing immunity to this pathogen.
Some earlier statistical analyses have shown that marching training and firing training are significantly associated with the onset of pneumonia and FRI, especially in HAdV outbreaks [19]. During a respiratory illness outbreak in an MTC in the USA, HAdV was detected on rifle surfaces [20]. During the early period of the present outbreak, the main components of the training schedule were mid-range marches and night firing training, which may have contributed to the increased patient numbers or exacerbated their symptoms. Based on this experience, appropriate measures must be identified and implemented to prevent outbreaks of respiratory illnesses in military training units, such as postponing firing training until after the peak stage of the epidemic and disinfecting the training rifles when they are transferred from one trainee to another.
This FRI outbreak also illustrates the pressing need to improve the FRI epidemic response of the doctors and health officials in basic army units. In the acute FRI outbreak reported here, about 25 new cases appeared with the same symptoms in the 15 days after 7 May. However, this situation did not alert the MTC managers or health professionals. No correct diagnostic procedures were implemented, no epidemiological investigation was commenced, and the supervisory CDC received no information about the new cases. When a large wave of new cases (n = 79) appeared between 22 May and 30 May, in an effort to control the outbreak, the MTC healthy managers gathered about 798 soldiers into several crowded classrooms for health education, which may have contributed greatly to the transmission of the disease, and 7 days later, the highest incidence peak of the entire outbreak was observed (Fig. 1). Had the health officials dealt with the outbreak correctly from the very beginning, the spread of this FRI may have been restricted to a much smaller scale.
Because outbreaks of HAdV-55 infections have been frequent in recent years, HAdV-55-associated FRI pose a serious threat to the Chinese army. Therefore, improved epidemiological and virological surveillance of HAdV and many other dangerous pathogens by the Chinese military health system is urgently required. Some simple and accurate diagnostic methods that distinguish HAdV-55 infections from other similar diseases must be established. Moreover, in view of the U.S. army’s experience with an HAdV vaccination program, the development and application of HAdV vaccines may be an excellent way to protect the health of Chinese soldiers.
The present study has several limitations. First, although 91.84 % (45 of 49) of throat swab specimens were positive when using an RV13 kit and HAdV-specific PCR and the amplicons from the 45 PCR-positive samples were found to have high sequence similarities to the HAdV-B55 strains, the remaining 115 FRI cases were still not detected by the specific PCR and sequencing analysis. Therefore, it was still difficult to ensure this HAdV-B55-like virus was the only pathogenic agent responsible for this outbreak. It is possible that some other pathogens were involved in this outbreak. In some previously published descriptions of outbreaks of HAdV infection among military trainees, the co-circulation of two or more types of HAdV had already been reported. Second, the members of the healthy control group were randomly selected from the asymptomatic soldiers in the same MTC on 30 May. At the same time, the disease prevention and control measures had been implemented. None of the healthy controls showed any respiratory tract disease symptoms during the outbreak period. However, these healthy controls had not been screened by laboratory detection methods such as viral nucleic acid detection. Considering the results of the IgA and IgG detection in the healthy control group, there may have been asymptomatic infection cases hiding in the healthy control group. Many “healthy control group” members had already been infected by this HAdV virus. In a future outbreak study, the healthy controls should be selected from a camp where the virus was not present and be screened using laboratory detection methods to differentiate the asymptomatic infection cases.
Acknowledgments
This study was Sponsored by the China Postdoctoral Science Foundation (2012M521933). The sponsors had no role in the study design, data collection, decision to publish, or preparation of the original manuscript.
Compliance with ethical standards
This study was approved by the Review Board of the Jinan Junqu Center for Disease Control and Prevention. Informed consent was received from all participants before the study.
Conflict of interest
The authors declare that they have no competing interests.
Footnotes
Y. Dongliu, Y. Guoliang, X. Haocheng and Q. Shuaijia contributed equally to this paper.
References
- 1.Schmitz H, Wigand R, Heinrich W. Worldwide epidemiology of human adenovirus infections. Am J Epidemiol. 1983;117:455–466. doi: 10.1093/oxfordjournals.aje.a113563. [DOI] [PubMed] [Google Scholar]
- 2.Thomas L. Adenovirus infections in immunocompetent and immunocompromised patients. Clin Microbiol Rev. 2014;27:441–462. doi: 10.1128/CMR.00116-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Heim A, Ebnet C, Harste G, Pring Akerblom P. Rapid and quantitative detection of human adenovirus DNA by real-time PCR. J Med Virol. 2003;70:228–239. doi: 10.1002/jmv.10382. [DOI] [PubMed] [Google Scholar]
- 4.Wo Y, Lu QB, Huang DD, Li XK, Guo CT, Wang HY, Zhang XA, Liu W, Cao WC. Epidemical features of HAdV-3 and HAdV-7 in pediatric pneumonia in Chongqing, China. Arch Virol. 2015;160:633–638. doi: 10.1007/s00705-014-2308-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chmielewicz B, Benzler J, Pauli G, Krause G, Bergmann F, Schweiger B. Respiratory disease caused by a species B2 adenovirus in a military camp in Turkey. J Med Virol. 2005;77:232–237. doi: 10.1002/jmv.20441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yang Z, Zhu Z, Tang L, Wang L, Tan X, et al. Genomic analyses of recombinant adenovirus type 11a in China. J Clin Microbiol. 2009;47:3082–3090. doi: 10.1128/JCM.00282-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hierholzer JC, Pumarola A, Rodriguez-Torres A, Beltran M. Occurrence of respiratory illness due to an atypical strain of adenovirus type 11 during a large outbreak in Spanish military recruits. Am J Epidemiol. 1974;99:434–442. doi: 10.1093/oxfordjournals.aje.a121632. [DOI] [PubMed] [Google Scholar]
- 8.Kajon AE, et al. Outbreak of febrile respiratory illness associated with adenovirus 11a infection in a Singapore military training camp. J Clin Microbiol. 2010;48:1438–1441. doi: 10.1128/JCM.01928-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gu L, Liu Z, Li X, Qu J, Guan W, Liu Y, Song S, Yu X, Cao B, et al. Severe community-acquired pneumonia caused by adenovirus type 11 in immunocompetent adults in Beijing. J Clin Virol. 2012;54:295–301. doi: 10.1016/j.jcv.2012.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhu Z, Zhang Y, Xu S, Yu P, Tian X, Wang L, Liu Z, et al. Outbreak of Acute Respiratory Disease in China Caused by B2 Species of Adenovirus Type 11. J Clin Microbiol. 2009;47:697–703. doi: 10.1128/JCM.01769-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hierholzer JC, Pumarola A. Antigenic characterization of intermediate adenovirus 14-11 strains associated with upper respiratory illness in a military camp. Infect Immun. 1976;13:354–359. doi: 10.1128/iai.13.2.354-359.1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li QG, Hambraeus J, Wadell G. Genetic relationship between thirteen genome types of adenovirus 11, 34, and 35 with different tropisms. Intervirology. 1991;32:338–350. doi: 10.1159/000150218. [DOI] [PubMed] [Google Scholar]
- 13.Kajon AE, Lu X, Erdman DD, Louie J, Schnurr D, George KS, Koopmans MP, Allibhai T, Metzgar D. Molecular epidemiology and brief history of emerging adenovirus 14-associated respiratory disease in the United States. J Infect Dis. 2010;202:93–103. doi: 10.1086/653083. [DOI] [PubMed] [Google Scholar]
- 14.Walsh MP, Seto J, Jones MS, Chodosh J, Xu W, Seto D. Computational analysis identifies human adenovirus type 55 as a re-emergent acute respiratory disease pathogen. J Clin Microbiol. 2010;48:991–993. doi: 10.1128/JCM.01694-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cao B, Huang GH, Pu ZH, Qu JX, Yu XM, Zhu Z, Dong JP, Gao Y, Zhang YX, Li XH, Liu JH, Wang H, Xu Q, Li H, Xu W, Wang C. Emergence of community-acquired adenovirus type 55 as a cause of community-onset pneumonia. Chest. 2014;145:79–86. doi: 10.1378/chest.13-1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Allard A, Girones R, Juto P, Wadell G. Polymerase chain reaction for detection of adenoviruses in stool samples. J Clin Microbiol. 1990;28:2659–2667. doi: 10.1128/jcm.28.12.2659-2667.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R, Thompson JD, Gibson TJ, Higgins DG. Clustal W and Clustal X version 2.0. Bioinformatics. 2007;23:2947–2948. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
- 18.Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol. 2011;28:2731–2739. doi: 10.1093/molbev/msr121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hwang SM, Park DE, Yang YI, Park SJ, Lee HK, Kim MJ, Chun BC. Outbreak of febrile respiratory illness caused by adenovirus at a South Korean military training facility: clinical and radiological characteristics of adenovirus pneumonia. Jpn J Infect Dis. 2013;66:359–365. doi: 10.7883/yoken.66.359. [DOI] [PubMed] [Google Scholar]
- 20.Russell KL, Broderick MP, Franklin SE, et al. Transmission dynamics and prospective environmental sampling of adenovirus in a military recruit setting. J Infect Dis. 2006;194:877–885. doi: 10.1086/507426. [DOI] [PMC free article] [PubMed] [Google Scholar]