

Abstract
We report a child with short stature since birth who was otherwise well, presenting at 2.8 years with progressive granulomatous skin lesions when diagnosed with severe T cell immunodeficiency. When previously investigated for short stature, and at the time of current investigations, she had no radiological skeletal features characteristics for cartilage hair hypoplasia, but we found a disease causing RMRP (RNase mitochondrial RNA processing endoribonuclease) gene mutation. Whilst search for HLA matched unrelated donor for haematopoietic stem cell transplantation (HSCT) was underway, she developed rapidly progressive EBV-related lymphoproliferative disorder requiring laparotomy and small bowel resection, and was treated with anti-B cell monoclonal antibody and eventually curative allogeneic HSCT. Screening for RMRP gene mutations should be part of immunological evaluation of patients with ‘severe and/or combined’ T cell immunodeficiency of unknown origin, especially when associated with short stature and regardless of presence or absence of radiological skeletal features.
Keywords: Cartilage-hair hypoplasia, granulomatous inflammation, RNase mitochondrial RNA processing endoribonuclease, severe T cell immunodeficiency, EBV driven lymphoproliferative disease
Introduction
Cartilage-hair hypoplasia (CHH) (MIM #250250) is a rare autosomal recessive syndrome characterized by clinical features of metaphyseal dysplasia (short-limbed dwarfism) and light-coloured hypoplastic hair, bone marrow failure, defective spermatogenesis, an increased risk of Hirschsprung disease and malignancies, and variable degree of immunodeficiency [1]. CHH is caused by mutations of the untranslated RMRP gene, which encodes for the RNA component of the mitochondrial RNA processing (RMRP) endoribonuclease complex. Disease-causing mutations in RMRP result in disruption of ribosomal processing and cell cycle progression in rapidly dividing cells such as lymphocytes and chondrocytes, thus explaining the pleiotropy of clinical manifestations [2–4].
Despite individual variability, it has been recently shown that functional lymphocyte abnormalities are an integral component of CHH, with defects in thymic generation of T lymphocytes and in peripheral T-cell proliferation, cell cycle control, and activation-induced cell death [5]. In patients with clinical and laboratory features of (severe) T cell and/or combined immunodeficiency (CID) such as failure to thrive, recurrent severe infections (bacterial, fungal, viral), lymphopaenia with absent/low ‘naïve’ T cells and/or T cell function [5, 6], allogeneic haematopoietic stem cell transplantation (HSCT) is the only curative treatment, although the skeletal abnormalities and the growth failure remains unaffected [7].
Here we report progressive granulomatous inflammation as a leading clinical presentation of CHH due to RMRP gene mutation in an otherwise well child with short stature since birth, but no radiological features of skeletal dysplasia, associated with severe T cell immunodeficiency resulting in EBV-related lymphoproliferative disease (LPD), eventually cured by allogeneic HSCT.
Case Report
A 2 year 8 month female, the first child of unrelated Caucasian parents, presented with a 12 months history of initially small painless erythematous lesion affecting the right arm, gradually progressing in size (Fig. 1a–b), and a smaller lesion on the right leg. Except for recent ear infection with fever (38.6 °C), there was no history of infections, bowel symptoms, foreign travel or exposure to mycobacteria (tuberculosis). She was fully vaccinated (except BCG), and had been previously investigated for persistent short stature since birth (height <0.4th percentile with mid-parental height of 50th percentile; weight 2nd percentile) but no underlying reason was found. She was short but proportionate, with fine blond hair, and with normal nails, teeth, body lanugo hair and sweating.
Fig. 1.
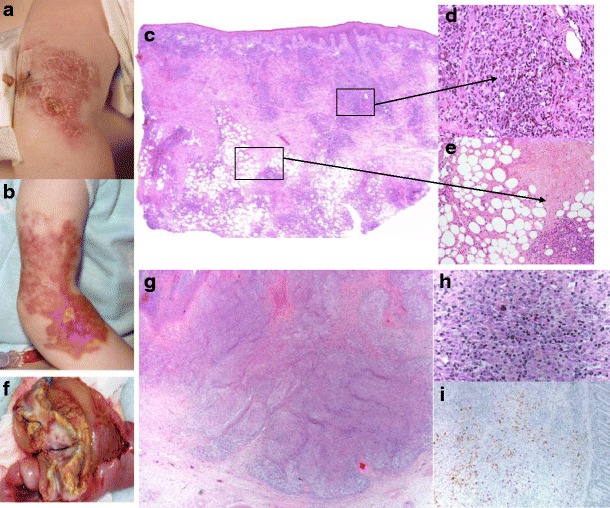
Granulomatous lesions (skin) and EBV lymphoproliferative disorder (small bowel). Right arm, cutaneous granulomatous lesion: a initial; b after 6 months progression. Histopathology, skin biopsy: c Prominent focally diffuse dermal and subcutaneous lympho-histiocytic infiltrate suggesting lymphoma; d Poorly formed granulomas, composed of epithelioid histiocytes; e Focal areas of necrosis in the subcutaneous tissue. Resected 17 cm of small bowel, EBV-LPD: f Lobulated, ulcerated, partially necrotic tumour (4 cm in diameter); g Tumorous mass: small intestine containing diffuse infiltrate/sheets of atypical large centroblastic lymphoid cells amongst which are scattered larger, more pleomorphic forms; background of small reactive lymphocytes, macrophages, scattered eosinophils and plasma cells. h High power: Hodgkin/Reed–Sternberg cells set singly in a mixed inflammatory background with numerous eosinophils. i Immunohistochemical staining: centroblastic and pleomorphic B cells (express CD20, Pax5, CD30 and MUM1), most are cyclin D1-negative but several aggregates of positive cells morphologically suggestive of atypical lymphoid cells are present throughout. EBV-EBER in situ hybridisation shows strong widespread positivity; the pleomorphic cells express EBV-LMP1. The Ki67 proliferation fraction is high (approximately 80 %). Staining for CD2, CD3 and CD5 highlights the reactive T-cell population. In situ hybridisation of immunoglobulin light chains shows the plasma cells to be polytypic. EBV-EBER EBV-encoded RNA, EBV-LMP1 EBV-latent membrane protein 1, CD cluster of differentiation
Initial investigations revealed raised inflammatory markers (ESR 96 mm/h; CRP 131 mg/l), significant neutropaenia (ANC 0.01–0.11 × 10e9/L), non-haemolytic anaemia (Hb 8.6 g/l, negative direct Coombs test, normal serum bilirubin), variable lymphopaenia (3.3–1.95 × 10e9/L), elevated serum immunoglobulin (Ig) M (4.62 g/l; range 0.5–2.0), and negative antinuclear, double stranded-DNA, ENA and anti-neutrophil antibodies. Bacterial, mycobacterial and fungal cultures (blood, skin) were negative, and viral serology non-diagnostic (positive EBV VCA IgM and IgG, CMV IgG, and negative VZV IgG and Parvovirus B19 IgM and IgG). Chest and bone (skeletal survey) radiography and abdominal ultrasound were normal. Bone marrow aspiration and biopsy (trephine) showed somewhat reduced erythroid series, and a reactive picture with significant left shift and good myeloid activity with increased numbers of eosinophils, although with a lack of maturation. The initial skin biopsy (Fig. 1c–e) demonstrated CD3+ lymphocyte infiltrate and a T cell clone by T cell receptor (TCR) gene rearrangement analysis; EBV in-situ hybridization was negative as were mycobacterial PCR and fungal stains.
Further immunological investigations showed normal neutrophil oxidative burst and protective antibody titres to haemophilus influenzae type b (1.0 mg/l; range 1.0–20), tetanus (0.66 IU/ml; range 0.1–10) and pneumococcus (>0.35 mcg/l for 11/13 tested strains). However, she was found to have severe T cell immunodeficiency (Table I, Fig. 2) and viral infections (by PCR, EBV 5.5 × 10e4 and HHV6 2 × 10e3 (copies/ml) in blood, sapovirus in stool, adenovirus in nasal swab, and few EBV-positive cells were detected in the second skin biopsy).
Table I.
Immunologic parameters I: a/Peripheral blood lymphocyte markers and b/Mitogen stimulated T cell proliferation
| Age | 2.8 year | 3 year | (NV) |
|---|---|---|---|
| (Post-rituximab) | |||
| a/Lymphocytes (cells/μl) | 3602 | 909 | (2–8000) |
| CD3+T cells (cells/μl) | 1885 | 721 | (900–4500) |
| CD3+/CD8+T cells (cells/μl) | 714 | 250 | (300–1600) |
| CD3+/CD4-/CD45RA+/CD27+ (% T cells) | 4 % | 6 % | |
| CD3+/CD4-/CD45RA+/CD27+ (cells/μl) | 75 | 43 | |
| CD3+/CD4-/CD45RA+/CD27- (% T cells) | 0 | 0 | |
| CD3+/CD4+T cells (cells/μl) | 459 | 243 | (500-2400) |
| CD3+/CD4+/CD45RA+/CD27+ (% T cells) | <1 % | <1 % | |
| HLA-DR+/CD3+T cells (% T cells) | 50 % | 26 % | |
| T cell receptor alpha/beta (% T cells) | 43 % | n/a | |
| T cell receptor gamma/delta (% T cells) | 57 % | n/a | |
| CD19+B cells (cells/μl) | 1064 | 0 | (200–2100) |
| CD19+CD27-IgD+ (% B cells) | 71 % | n/a | |
| CD19+CD27+IgD+ (% B cells) | 22 % | n/a | |
| CD19+CD27+IgD- (% B cells) | 4 % | n/a | |
| CD3-/CD56+/CD16+ NK cells (cells/μl) | 615 | 172 | (100–1000) |
| MHC class I and II expression | Normal | ||
| CD40 expression on B cells | Normal | ||
| b/Mitogen stimulated T cell proliferation | (Control) | ||
| Background (cpm) | 2695 | (2741) | |
| PHA | 6429 | (165591) | |
| Anti-CD3 | 106910 | (142813) | |
| PMA+Ionophore | 67039 | (154643) | |
(CD3+/CD4+/CD45RA+/CD27+)—markers for ‘naïve’ CD4+T cells; (CD3+/CD4-/CD45RA+/CD27+)—markers for ‘naïve’ CD8+ (CD4-)T cells; (CD3+/CD4-/CD45RA+/CD27-)—markers for “effector” CD8+ T cells
Profound T cell deficiency is demonstrated by complete absence of CD4+ naïve T cells and very low numbers of CD8+ ‘naïve’ T cells, so that most of the present T cells are of the memory population. This is confirmed by the functional in vitro tests of T cell proliferation showing strikingly reduced proliferation to PHA, but preserved capacity to proliferate to anti-CD3 and PMA mitogens. No antigen-specific T cell proliferation assays were performed. Severe T cell dysregulation is demonstrated by inverted CD4+/CD8+ ratio, very high percentage of ‘activated’ (CD3+/HLA-DR+) T cells, moderate oligoclonality of T lymphocytes expressing TCR-alpha/beta (as shown in Fig. 2), with unusually high proportion of T lymphocytes expressing TCR-gamma/delta (usually <10 %) (no clonality studies of this cell population were performed). Some of these features may be due to the chronic viral (EBV) infection, although there is a complete lack of the ‘effector’ CD8+ T cell population (CD45RA+/CD27-) which would otherwise be expected to be expanded
CD cluster of differentiation; NK natural killer; HLA human leukocyte antigen; MHC major histocompatibility complex; PHA phytohaemmaglutinin; PMA pokeweed mitogen; cpm counts per minute; IL interleukin
Fig. 2.
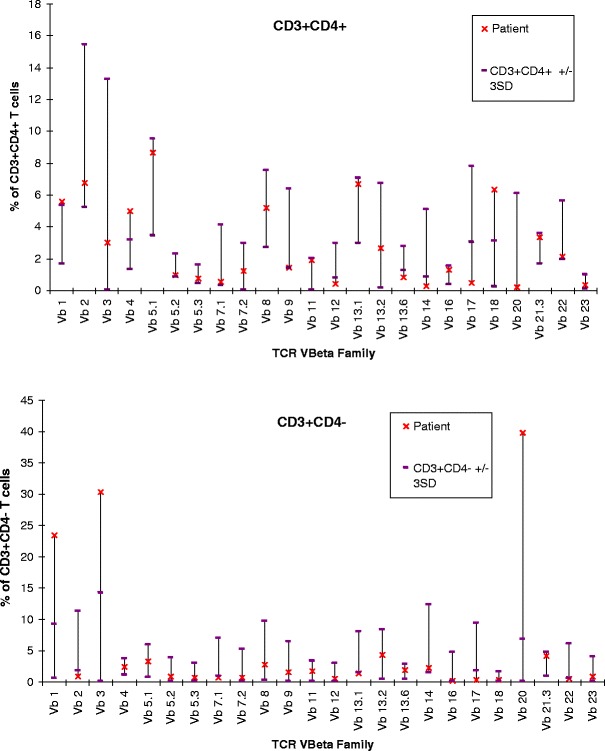
Immunologic parameters II: TCR V-beta family pattern. Skewed usage of TCR V-beta family, with increased number of expansions seen in the CD4+ population, and increased number and size of expansions in the CD4- (CD8+) population. See Table Legend for further explanation. CD cluster of differentiation, TCR T cell receptor
Based on the features of short stature since birth, severe T cell immunodeficiency and unusual granulomatous skin inflammation, in spite of normal bone radiography (Fig. 3), we searched for disease-causing mutations in the RMRP gene. Compound heterozyosity for a 13 bp duplication in the promoter (g.−26_−14 dup TACTACTCTGTGA) inherited from the mother (new mutation) and a nucleotide substitution (g.4C>T) close to the transcription initiation site inherited from the father (previously reported) confirmed the diagnosis (2,3). The severe T cell immunodeficiency and persistent EBV viraemia were indications for HSCT. However, before an unrelated (URD) donor was identified she developed abdominal pain and distension, fever and raised CRP (150 mg/l). Exploratory laparotomy revealed an ileal tumorous mass (Fig. 1f) which was resected and confirmed to be EBV-related diffuse large B-cell lymphoma (Fig. 1g–i). She was treated with anti-B cell monoclonal antibody (rituximab), and in June 2012 received 11.1 × 10e6/kg CD34+ peripheral blood stem cells from a 10/10 HLA matched URD after conditioning with alemtuzumab, treosulfan and fludarabine. Engraftment was uneventful (platelets day + 11; neutrophils day + 15) resulting in 100 % donor chimaerism. Complications post-HSCT included transient grade 2 acute (skin, gut) graft versus host disease (GvHD) at 3 weeks, treated with corticosteroids and TNF-alpha blocking agent (infliximab), autoimmune cytopenias (Coombs positive haemolytic anaemia, thrombocytopaenia and neutropaenia) at 3 months, treated with corticosteroids, immunomodulatory high-dose intravenous Ig (IVIg) and rituximab, and viral reactivation and/or new infections (parainfluenza III, adenovirus, HHV6, rhinovirus, coronavirus, norovirus) of which only persistent HHV6 viraemia (10e4–10e5/ml) and biopsy-proven enterocolitis needed treatment (ganciclovir, valganciclovir, foscarnet). One year post-HSCT she is well with markedly receded granulomatous skin lesions, only on IVIg substitution and azithromycin prophylaxis. Chimaerism remains 100 % donor in myeloid, T and B cell lineages, she has normal numbers of naïve ‘early thymic emigrant’ T cells, in vitro proliferation to PHA, and emerging B cells and serum IgM post-rituximab.
Fig. 3.
Bone radiography (age 2.8 years)
Discussion
The clinical features of CHH are more heterogeneous than usually thought [1, 2], with marked variability of immunological phenotype [5, 6, 8] and radiographic features [9]. Importantly for the diagnostic process, as illustrated by our case, the characteristic skeletal abnormalities should not always be expected to be present, particularly in early childhood [9, 10]. Bone marrow failure is common, as in our patient, with hypoplastic anaemia reported in ~80 %, lymphopaenia in ~60 %, and neutropaenia in ~25 % of patients [11].
Skin granulomatous inflammation can be caused by infections (mycobacteria in particular), inflammatory conditions (sarcoidosis, inflammatory bowel disease, Blau syndrome etc.), but may also be a feature of primary immunodeficiency disorders associated with marked immune dysregulation such as common variable immunodeficiency, chronic granulomatous disease, combined/T cell immunodeficiency, etc. [reviewed in 12]. Omenn syndrome has been reported in patients with RMRP mutations [13], and a variety of patients classified as ‘short-limbed skeletal dysplasia with combined immunodeficiency’ (MIM 200900) presented with skin features such as ectodermal dysplasia [14] and granulomatous lesions [15]. Moshous et al. described inflammatory granulomas as a new clinical feature in 4 out of a series of 21 patients with CHH [16], and a further case of extensive granulomatous lesions was reported recently in a foetus [17]. Similar to ours, these 5 patients with proven RMRP mutation had severe CD4 T cell lymphopaenia [16, 17]. Even in otherwise clinically ‘mild’ phenotype [10], severe T cell lymphopaenia should not be undermined. Our patient clearly demonstrated one of the hallmarks of severely impaired T cell immunity due to RMRP mutation, the poor handling of the herpes family of viruses, with development of life-threatening EBV-related LPD [1, 5]. The only curative therapy is allogeneic HSCT [7], highlighted by the ‘natural history’ observed during the long-term follow up of a child diagnosed with CHH at the age 8 years, who at the age 32 years presented with relapsing EBV-related anaplastic large cell lymphoma and granulomatous lymphomatoid papulosis, both conditions being part of the spectrum of primary cutaneous CD30+ T cell LPD, and who died 14 years after the primary diagnosis of lymphoma in spite of several courses of chemotherapy [18]. A recent insight in the role of dysregulated long non-coding RNAs in cancer initiation and progression, in the case of RMRP specifically related to leukemia and lymphoma [19] further expands our understanding of the well recognised increased incidence of cancer in patients with CHH, particularly non-Hodgkin lymphoma and basal cell carcinoma [20].
All this evidence strongly suggests that patients with severely impaired T cell immunity due to RMRP mutation should be classified in the group of “severe and/or combined primary immunodeficiencies” [21–24].
Summary
We present a patient with granulomatous lesions and severe T cell immunodeficiency due to RMRP gene mutation, who developed rapidly progressive and life-threatening EBV-related LPD.
Screening for RMRP gene mutations should be part of immunological evaluation of patients with ‘severe and/or combined’ immunodeficiency of unknown origin, especially if associated with short stature and/or granulomatous inflammatory lesions, irrespective of presence or absence of radiographic features characteristic for cartilage hair hypoplasia.
Allogeneic haematopoietic stem cell transplantation is the only curative treatment for immunodeficiency, while growth failure remains unaffected.
Acknowledgments
We are grateful to Drs Ali Al-Sharqi, Poonam Dharmaraj, Helen Campbell and Eileen Baildam, Alder Hey Children’s NHS Foundation Trust, Liverpool and the staff at the Children’s Bone Marrow Transplantation Unit, Great North Children’s Hospital, Royal Victoria Infirmary, Newcastle upon Tyne Hospitals NHS Foundation Trust, for their involvement in management of the patient.
Declaration of Funding
None.
Conflict of Interest
The authors declare that they have no conflict of interest.
Consent
Informed consent has been obtained from the patient’s parents for the case report and medical photographs used in this manuscript. All studies have been performed in accordance with the 1964 Declaration of Helsinki and its later amendments.
Contributor’s Statement
Liza J McCann, Jo McPartland, George Kokai, Chris Bacon, Julian Verbov and Andrew Riordan were involved in direct patient care and/or diagnostic process, provided patient data, and reviewed and approved the manuscript.
Dawn Barge performed and interpreted the immunological investigations results for the manuscript, and reviewed and approved the manuscript.
Eduardo Calonje interpreted the histopathology findings of the skin biopsy, and reviewed and approved the manuscript.
Lisa Strain and David Bourn performed and interpreted the genetic mutation analysis, and reviewed and approved the manuscript.
Michael Wright was involved in direct patient care and diagnostic process, interpreted the skeletal radiography and genetic mutation analysis, and reviewed and approved the manuscript.
Mario Abinun was involved in direct patient care and diagnostic process, and wrote the manuscript.
Abbreviations
- CHH
Cartilage-hair hypoplasia
- RNA
Ribonucleic acid
- RMRP
RNA component of the mitochondrial RNA processing
- CID
Combined immunodeficiency
- HSCT
Haematopoietic stem cell transplantation
- EBV
Epstein–Barr virus
- LPD
Lymphoproliferative disorder
- BCG
Bacillus Calmette-Guerin
- ESR
Erythrocyte sedimentation ratio
- CRP
C reactive protein
- Ig
Immunoglobulin
- DNA
Deoxyribonucleic acid
- ENA
Extractible nuclear antigen
- VCA
Virus capsid antigen
- CMV
Cytomegalovirus
- VZV
varicella zoster virus
- CD
Cluster of differentiation
- TCR
T cell receptor
- PCR
Polymerase chain reaction
- HHV
Human herpes virus
- HLA
Human leukocyte antigen
- URD
Unrelated donor
- GvHD
Graft versus host disease
- TNF
Tumour necrosis factor
References
- 1.Abinun M, Kaitila I, Casanova JL. Immunodeficiencies with associated manifestations of skin, hair, teeth, and skeleton. In: Ochs HD, Smith CIE, Puck JM, editors. Primary immunodeficiency diseases. A molecular and genetic approach. (2nd ed). Oxford University Press; 2007. pp. 513–24
- 2.Thiel CT, Mortier G, Kaitila I, Reis A, Rauch A. Type and level of RMRP functional impairment predicts phenotype in the cartilage hair hypoplasia–anauxetic dysplasia spectrum. Am J Hum Genet. 2007;81:519–529. doi: 10.1086/521034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Thiel CT, Rauch A. The molecular basis of the cartilage-hair hypoplasia–anauxetic dysplasia spectrum. Best Pract Res Clin Endocrinol Metab. 2011;25:131–142. doi: 10.1016/j.beem.2010.08.004. [DOI] [PubMed] [Google Scholar]
- 4.Rogler LE, Kosmyna B, Moskowitz D, Bebawee R, Rahimzadeh J, Kutchko K, et al. Small RNAs derived from lncRNA RNase MRP have gene-silencing activity relevant to human cartilage hair hypoplasia. Hum Mol Genet. 2013 doi: 10.1093/hmg/ddt427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.De la Fuente MA, Recher M, Rider NL, Strauss KA, Morton DH, Adair M, et al. Reduced thymic output, cell cycle abnormalities, and increased apoptosis of T lymphocytes in patients with cartilage-hair hypoplasia. J Allergy Clin Immunol. 2011;128(1):139–146. doi: 10.1016/j.jaci.2011.03.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kavadas FD, Giliani S, Gu Y, Mazzolari E, Bates A, Pegoiani E, et al. Variability of clinical and laboratory features among patients with ribonuclease mitochondrial RNA processing endoribonuclease gene mutations. J Allergy Clin Immunol. 2008;122:1178–1184. doi: 10.1016/j.jaci.2008.07.036. [DOI] [PubMed] [Google Scholar]
- 7.Bordon V, Gennery AR, Slatter MA, Vandecruys E, Laureys G, Veys P, et al. Clinical and immunologic outcome of patients with cartilage hair hypoplasia after hematopoietic stem cell transplantation. Blood. 2010;116(1):27–35. doi: 10.1182/blood-2010-01-259168. [DOI] [PubMed] [Google Scholar]
- 8.Notarangelo LD, Roifman CM, Giliani S. Cartilage-hair hypoplasia: molecular basis and heterogeneity of the immunological phenotype. Curr Opin Allergy Clin Immunol. 2008;8:534–539. doi: 10.1097/ACI.0b013e328310fe7d. [DOI] [PubMed] [Google Scholar]
- 9.Kwan A, Manning MA, Zollars LK, Hoyme HE. Marked variability in the radiographic features of cartilage-hair hypoplasia: case report and review of the literature. Am J Med Genet A. 2012;158A(11):2911–2916. doi: 10.1002/ajmg.a.35604. [DOI] [PubMed] [Google Scholar]
- 10.Türkkanı-Asal G, Alanay Y, Turul-Özgür T, Zenker M, Thiel C, Rauch A, et al. Mild clinical phenotype and subtle radiographic findings in an infant with cartilage-hair hypoplasia. Turk J Pediatr. 2009;51:493–496. [PubMed] [Google Scholar]
- 11.Makitie O, Rajanite J, Kaitila I. Anaemia and macrocytosis—unrecognized features in cartilage-hair hypoplasia. Acta Paediatr. 1992;81:1026–1029. doi: 10.1111/j.1651-2227.1992.tb12168.x. [DOI] [PubMed] [Google Scholar]
- 12.Petersen HJ, Smith AM. The role of the innate immune system in granulomatous disorders. Front Immunol. 2013;4:120. doi: 10.3389/fimmu.2013.00120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Roifman CM, Gu Y, Cohen A. Mutations in the RNA component of RNase mitochondrial RNA processing might cause Omenn syndrome. J Allergy Clin Immunol. 2006;117:897–903. doi: 10.1016/j.jaci.2006.01.003. [DOI] [PubMed] [Google Scholar]
- 14.Gatti RA, Platt N, Pomerance HH, Hong R, Langer LO, Kay HEM, et al. Hereditary lymphopenic agammaglobulinemia associated with a distinctive form of short-limbed dwarfism and ectodermal dysplasia. J Pediatr. 1969;75(4):675–684. doi: 10.1016/S0022-3476(69)80465-8. [DOI] [PubMed] [Google Scholar]
- 15.Gotoff SP, Esterly NB, Gottbrath E, Liebner EJ, Lajvardi SR. Granulomatous reaction in an infant with combined immunodeficiency and short-limbed dwarfism. J Pediatr. 1972;80(6):1010–1017. doi: 10.1016/S0022-3476(72)80015-5. [DOI] [PubMed] [Google Scholar]
- 16.Moshous D, Meyts I, Fraitag S, Janssen CEI, Debre M, Suarez F, et al. Granulomatous inflammation in cartilage-hair hypoplasia: risks and benefits of anti-TNF-a mAbs. J Allergy Clin Immunol. 2011;128:847–853. doi: 10.1016/j.jaci.2011.05.024. [DOI] [PubMed] [Google Scholar]
- 17.Crahes M, Saugier-Veber P, Patrier S, Aziz M, Pirot N, Brasseur-Daudruy M, et al. Foetal presentation of cartilage hair hypoplasia with extensive granulomatous inflammation. Eur J Med Genet. 2013;56(7):365–370. doi: 10.1016/j.ejmg.2013.04.004. [DOI] [PubMed] [Google Scholar]
- 18.Taskinen M, Jeskanen L, Karjalainen-Lindsberg ML, Mäkitie A, Mäkitie O, Ranki A. Combating cancer predisposition in association with idiopathic immune deficiency: a recurrent nodal and cutaneous T-cell lymphoproliferative disease in a patient with cartilage-hair hypoplasia. Clin Lymphoma Myeloma Leuk. 2013;13(1):73–76. doi: 10.1016/j.clml.2012.06.005. [DOI] [PubMed] [Google Scholar]
- 19.Shi X, Sun M, Liu H, Yao Y, Song Y. Long non-coding RNAs: a new frontier in the study of human diseases. Cancer Lett. 2013;339(2):159–166. doi: 10.1016/j.canlet.2013.06.013. [DOI] [PubMed] [Google Scholar]
- 20.Makite O, Pukkala E, Teppo L, Kaitila I. Increased incidence of cancer in patients with cartilage-hair hypoplasia. J Pediatr. 1999;134:315–318. doi: 10.1016/S0022-3476(99)70456-7. [DOI] [PubMed] [Google Scholar]
- 21.Liston A, Enders A, Siggs OM. Unravelling the association of partial T cell immunodeficiency and immune regulation. Nat Rev Immunol. 2008;8:545–558. doi: 10.1038/nri2336. [DOI] [PubMed] [Google Scholar]
- 22.Al-Herz W, Bousfiha A, Casanova JL, Chapel H, Conley ME, Cunningham-Rundles C, et al. Primary immunodeficiency diseases: an update on the classification from the International Union of Immunological Societies Expert Committee for Primary Immunodeficiency. Front Immunol. 2011;2:54. doi: 10.3389/fimmu.2011.00054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Roifman CM, Somech R, Kavadas F, Pires L, Nahum A, Dalal I, et al. Defining combined immunodeficiecny. J Allergy Clin Immunol. 2012;130:177–183. doi: 10.1016/j.jaci.2012.04.029. [DOI] [PubMed] [Google Scholar]
- 24.Maggina P, Gennery AR. Classification of primary immunodeficiency: need for a revised approach. J Allergy Clin Immunol. 2013;131(2):292–294. doi: 10.1016/j.jaci.2012.10.008. [DOI] [PubMed] [Google Scholar]