

Abstract
Nariva virus (NarPV) was isolated from forest rodents (Zygodontomys b. brevicauda) in eastern Trinidad in the early 1960s. Initial classification within the family Paramyxoviridae was based mainly on morphological observations including the structure of nucleocapsids and virion surface projections. Here, we report the characterization of the complete genome sequence of NarPV. The genome is 15,276 nucleotides in length, conforming to the rule-of-six, and has a genome organization typical of most members of the family, with six transcriptional units in the order 3′-N–P-M-F–H-L-5′. The gene junctions contain highly conserved gene start and stop signals and a tri-nucleotide intergenic sequence present in most members of the subfamily Paramyxovirinae. Sequence comparison studies indicate that NarPV is most closely related to Mossman virus, which was isolated from wild rats (Rattus leucopus) in Queensland, Australia, in 1970. This study confirmed the classification of NarPV as a member of the subfamily Paramyxovirinae and established the close genome organization and sequence relationship between the two rodent paramyxoviruses isolated almost a decade apart and from two locations separated by more than 15,000 km.
Keywords: Newcastle Disease Virus, Measle Virus, Editing Site, Canine Distemper Virus, cDNA Subtraction
Introduction
Members of the family Paramyxoviridae are pleomorphic enveloped viruses possessing a nonsegmented negative-strand (NNS) RNA genome [11]. They are divided into two subfamilies, Paramyxovirinae and Pneumovirinae. The subfamily Paramyxovirinae is currently classified into five genera: Respirovirus, Morbillivirus, Rubulavirus, Avulavirus and Henipavirus. The subfamily Pneumovirinae consists of two genera: Pneumovirus and Metapneumovirus [10].
Paramyxoviruses are well-known pathogens of humans (measles virus, mumps virus, etc.) and livestock animals (Newcastle disease virus, rinderpest virus, etc.). The emergence of henipaviruses (Hendra and Nipah viruses) further highlighted the broad host range and the severity of the diseases that can be caused by novel paramyxoviruses [4, 24].
Bats are increasingly recognized as an important reservoir of novel viruses, including paramyxoviruses, coronaviruses and, potentially, filoviruses [2]. At least five bat paramyxoviruses have been identified, which include Hendra virus, Nipah virus, Tioman virus, Menangle virus and Mapuera virus. There is anecdotal evidence suggesting that porcine rubulavirus may also originate from bats [19, 26], and Indian bats may carry a paramyxovirus antigenically related to simian virus 41 and parainfluenza virus 2 [15]. While it is not clear why bats seem to be an ideal reservoir host for many different viruses, one suggested possibility is the direct result of their great species diversity and population abundance. If these are the main drivers for virus distribution and evolution, one would expect to find even more viruses in rodents, since they are the most diverse mammalian animals on earth [1].
Sendai virus (SeV), although first isolated from a human specimen, is believed to have rodents as its natural reservoir [5] and represents the first and best characterized paramyxovirus of rodent origin. Since then, at least four other paramyxoviruses, Mossman virus (MosPV), J virus (JPV), Beilong virus (BeiPV) and Nariva virus (NarPV) have been isolated from rodents. The complete genome sequences of the first three rodent viruses were determined previously by our group [7, 12, 13]. Here, we report the full-length genome sequence of NarPV to complete the molecular analysis of all known rodent paramyxoviruses.
NarPV was isolated from forest rodents, Zygodontomys b. brevicauda, trapped in the Nariva swamp in eastern Trinidad in the early 1960s [8, 21]. The virus grew in suckling mouse brain and formed syncytia in Vero and BHK cells. NarPV was identified as a member of the family Paramyxoviridae, mainly based the structure of its nucleocapsids (approximately 20 nm in diameter and mean length 1.8 μm) and its virion morphology, being enveloped, spherical and pleomorphic with surface projections [23]. The virus displayed no serological cross-reactivity with a range of known paramyxoviruses at the time of isolation, including parainfluenza virus types 1-4, mumps virus, Newcastle disease virus and measles virus [8]. NarPV resembles members of the genera Respirovirus and Rubulavirus in that it generates only cytoplasmic inclusion bodies in virus-infected cells [23]. In this respect, it differs from members of the genus Morbillivirus that produce both cytoplasmic and nuclear inclusions in infected cells. However, haemagglutination and cell-binding studies performed on guinea pig and monkey red blood cells suggested that NarPV, like measles virus, does not use sialic acid receptors on red blood cells as do respiroviruses and rubulaviruses [8].
The data presented in this study confirmed the classification of NarPV as a member of the subfamily Paramyxovirinae. Although NarPV could not be classified into any of the existing genera, it is clear that NarPV is most closely related to MosPV, confirming its rodent origin. Analysis of the NarPV H protein also revealed the lack of the consensus NRKSCS sequence motif known to be important for sialic acid binding for the HN proteins of respiroviruses and rubulaviruses [14].
Materials and methods
Virus culture and RNA isolation
Vero cells were infected with NarPV or Salem virus (SalPV) [17], which was used as a driver in the cDNA subtraction experiment described below, and incubated at 37°C until the appearance of syncytia. Upon reaching approximately 80% CPE, the medium was replaced with PBS, and cells were collected in PBS. Cells were pelleted by centrifugation at 600 × g for 10 min, and virus in the supernatant was then harvested by ultracentrifugation at 300,000 × g for 30 min. RNA extraction was performed using the RNeasy mini kit (Qiagen) according to the manufacturer’s instructions. After elution from the column in RNase-free water, RNA concentration was determined using a Gene QuantII (Pharmacia). The RNA concentration was adjusted to approximately 1 μg/μl for subsequent applications.
Genome characterization by cDNA subtraction and gap-filling PCR
A Clontech PCR-Select cDNA Subtraction Kit (Clontech, USA) was used to select virus-specific fragments as previously described by our group [3, 7, 13]. Double-stranded cDNA was made using random hexamer oligonucleotide primers and 4 μg each of total RNA as prepared above from pelleted NarPV (tester cDNA) and SalPV (driver cDNA). Digestion, adaptor ligation, hybridization and PCR reactions were then carried out as described in the instructions provided with the kit. Nested PCR products from both subtractions were size-purified on a 1% agarose gel in three fractions (0.1–0.4, 0.4–0.8 and 0.8–2.0 kb) using the QIA-Quick PCR Gel Extraction Kit (Qiagen). The three purified fractions were cloned, and plasmids with insert sizes 100 bp or greater were randomly selected and sequenced.
For filling the gaps that were not covered by fragments obtained from the cDNA subtraction above, random cDNA was synthesized using the Omniscript RT Kit (Qiagen) with random hexamer primers. Virus-specific primers were designed using either cDNA subtraction-derived sequences or consensus sequences of published paramyxovirus genomes and made by a commercial provider (GeneWorks, Australia). The Platinum PCR SuperMix Kit (Invitrogen, USA) was used to perform PCR on the cDNA template synthesized as described above. PCR products were visualized with ethidium bromide on 1–2% agarose gels and purified, using either the QIAquick PCR Purification Kit (Qiagen) or the QIAquick Gel Purification Kit (Qiagen), prior to DNA sequencing.
Characterization of genome termini
The sequences of the 5′ genome and 5′ anti-genome termini were determined using a modified procedure from a previously published method [22]. Virus growth and RNA extraction were conducted as described above. Total RNA (containing both genome and anti-genome RNA) was used for cDNA synthesis using the Thermoscript RT-PCR System kit (Life Technologies, USA) and virus-specific primers located within approximately 100–200 nt of the genome termini. Reverse transcriptase reactions were incubated at 37°C for 60 min followed by RT inactivation at 85°C for 5 min and treatment with Rnase H. The first-strand cDNA was purified using the QIAquick PCR Purification Kit (Qiagen) prior to ligation with the anchor oligonucleotide (5′-GAAGAGAAGGTGGAAATGGCGTTTTGG, 5′-phosphorylated and 3′-blocked) using T4 RNA ligase (New England BioLabs, USA). The ligated product was amplified by PCR using a virus-specific primer, nested with respect to the first primer used for cDNA synthesis, and a 27-nt adaptor primer complementary in sequence to the adaptor. When required, a hemi-nested PCR, using the same adaptor primer and an additional (third) nested virus-specific primer, was also performed. The PCR products obtained were gel purified as described earlier and either sequenced directly or cloned before sequencing, in which case at least six individual clones were sequenced to ensure a reliable consensus sequence.
DNA sequencing
Purified PCR products or plasmid DNA were sequenced using the BigDye® Terminator v1.0 Kit (Applied Biosystems, USA) and an ABI PRISM 377 DNA Sequencer (Applied Biosystems). Every nucleotide in the genome was sequenced with a minimum of threefold redundancy, at least once in each sense and at least once directly from PCR products without cloning.
Sequence analysis
The Clone Manager and Align Plus programs in the Sci Ed Central software package (Scientific and Educational Software, USA) were used for routine sequence data management and analysis. Sequence similarity searches were conducted using the BLAST service at the National Center for Biotechnology Information (NCBI). Phylogenetic trees were constructed using the neighbour-joining algorithm with bootstrap values determined by 1,000 replicates in the MEGA4 software package [20].
Database accession numbers
The full-length genome sequence of NarPV has been deposited into GenBank under the accession number FJ362497. Accession numbers for other sequences used in this study are listed below. For viruses where full-length genome sequence was not available, individual protein sequences were used and are indicated by the abbreviated gene letter in parentheses following the accession number. The new naming convention for paramyxovirus abbreviations, as proposed in the 8th ICTV report [10], was used in this paper for those viruses which have not been formally classified. Atlantic salmon paramyxovirus (AsaPV) EU156171; avian paramyxovirus type 6 (APMV6) AY029299; Beilong virus (BeiPV) DQ100461; bovine parainfluenza virus 3 (bPIV3) AF178654; canine distemper virus (CDV) AF014953; cetacean morbillivirus (CMV) strain dolphin morbillivirus (DMV) X75961(N), Z47758(P/V/C), Z30087(M), Z30086(F), Z36978(H); fer-de-lance virus (FdlPV) AY141760; Hendra virus (HeV) AF017149; human parainfluenza virus 1 (hPIV1) AF457102; human parainfluenza virus 2 (hPIV2) X57559; human parainfluenza virus 3 (hPIV3) AB012132; human parainfluenza virus 4a (hPIV4a) M32982(N), M55975(P/V), D10241(M), D49821(F), M34033(HN); human parainfluenza virus 4b (hPIV4b) M32983(N), M55976 (P/V) D10242(M), D49822(F), AB006958(HN); J virus (JPV), AY900001; Mapuera virus (MprPV) EF095490; measles virus (MeV) AB016162; Menangle virus (MenPV) AF326114(N,P/V,M,F,HN); Mossman virus (MosPV) AY286409; mumps virus (MuV) AB040874; Newcastle disease virus (NDV) AF077761; Nipah virus (NiV) AF212302; peste-des-petits-ruminants virus (PPRV) X74443(N), AJ298897(P/V/C), Z47977(M), Z37017(F), Z81358(H); phocine distemper virus (PDV) X75717(N), D10371(P/V/C,M,F,H), Y09630(L); parainfluenza virus 5 (PIV5) AF052755; porcine rubulavirus (PorPV) BK005918; rinderpest virus (RPV) Z30697; Salem virus (SalPV) AF237881(N,P/V/C); Sendai virus (SeV) AB005795; Tioman virus (TioPV) AF298895; Tupaia paramyxovirus (TupPV) AF079780.
Results
Characterization of the NarPV genome
Similar to strategies used for characterization of paramyxovirus genomes used in our group, a cDNA subtraction approach was used initially to obtain NarPV-specific sequences. This was then followed by PCR to fill in the “gaps” using specific primers designed from NarPV sequences obtained from the cDNA subtraction or degenerate primers designed using highly conserved consensus sequences of known paramyxoviruses in the subfamily Paramyxovirinae. Finally, the genome terminal sequences were obtained using a modified 5′ RACE technique as described in “Materials and methods”. As shown in Fig. 1a, the NarPV genome organisation is similar to those of most members of the subfamily Paramyxovirinae, containing six transcriptional units in the order 3′-N-P/V/C-M-F-H-L-5′. At 15,276 nt, the NarPV genome is at the lower end of the genome size spectrum known for members of the subfamily Paramyxovirinae, varying from 15,178 nt for porcine rubulavirus [25] to 19,212 nt for Beilong virus [12]. The genome length of NarPV conforms to the rule-of-six [11], a characteristic known to be true for all sequenced members of the Paramyxovirinae.
Fig. 1.
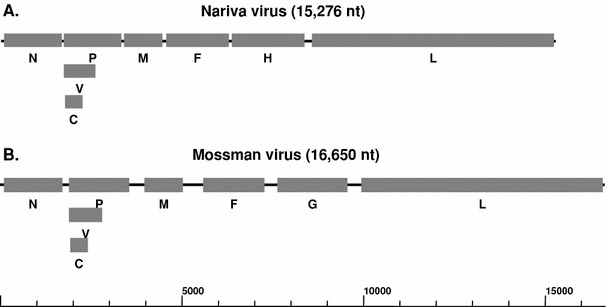
Schematic diagram of NarPV (a) and MosPV (b) genomes. The shaded boxes represent the coding regions for each of the six genes, whereas the lines indicate non-translated regions. The genome size scale is given at the bottom
As with all other members of the Paramyxovirinae, NarPV has a 3′ leader of 55 nt. The 5′ trailer of NarPV is 42 nt long. The first and last 11–13 nt of paramyxovirus genomes are highly conserved and complementary in nature and are critical elements of the genome and anti-genome viral RNA polymerase promoter [11]. This is also true for NarPV (Fig. 2a). When the 3′ leader sequence was aligned with those of other subfamily members (Fig. 2b), it was evident that NarPV’s genome end sequence is most similar to that of MosPV and least similar to those of rubulaviruses or avulaviruses. It is worth noting that the NarPV genome has a C residue at the fifth position from both ends, which is unique among known members of the Paramyxovirinae. This region contains the promoter for replication and transcription and has been shown by reverse genetics to be important for pathogenicity and attenuation of certain paramyxoviruses [11].
Fig. 2.

Alignment of genome terminal sequences. See “Materials and methods” for full virus names and database accession numbers. a Alignment of the 3′ leader and 5′ trailer sequences of NarPV. The 3′ leader is represented as anti-genome written in the 5′ → 3′ sense, and the 5′ trailer is represented as genome written in the 5′ → 3′ sense. The conserved residues are shaded. b Alignment of 3′ leader sequences of selected members of the subfamily Paramyxovirinae. The asterisk indicates the unique 5th C residue present only in the NarPV genome
Overall comparison of deduced sizes and amino acid sequences of all proteins indicated that NarPV is most closely related to MosPV (Table 1). However, the MosPV genome is significantly larger (16,650 nt) than that of NarPV due to the longer untranslated regions (UTRs) located at the 3′ end of most genes (Fig. 1b and Table 1). The NarPV genome has a coding capacity of approximately 94%, which is higher than the average of 92% for members of the subfamily Paramyxovirinae [25], whereas the presence of longer UTRs has resulted in a lower coding capacity of 87% for the MosPV genome.
Table 1.
Molecular features of NarPV and MosPV genes and their deduced proteins
| Gene | mRNA features (nt) | ORF and deduced protein | ||||||
|---|---|---|---|---|---|---|---|---|
| Total length | 5′ UTR | 3′ UTR | Size (aa) | MW (kDa) | pI | Sequence identity (%) | ||
| N | NarPV | 1,659 | 46 | 53 | 519 | 58.2 | 5.26 | 59 |
| MosPV | 1,797 | 52 | 158 | 528 | 58.6 | 4.83 | ||
| P/V/C (P)a | NarPV | 1,661 | 34 | 76 | 516 | 54.4 | 5.28 | 38 |
| MosPV | 2,093 | 46 | 418 | 542 | 58.4 | 5.60 | ||
| P/V/C (V)a | NarPV | 1,662 | – | – | 278 | 29.6 | 4.51 | 39 |
| MosPV | 2,094 | – | – | 295 | 32.1 | 4.90 | ||
| P/V/C (C)a | NarPV | 1,661-2b | – | – | 149 | 17.3 | 10.62 | 49 |
| MosPV | 2,093-4b | – | – | 152 | 17.8 | 8.83 | ||
| M | NarPV | 1,149 | 27 | 99 | 340 | 38.0 | 9.55 | 70 |
| MosPV | 1,419 | 33 | 363 | 340 | 38.3 | 9.16 | ||
| F | NarPV | 1,797 | 32 | 64 | 566 | 62.4 | 5.43 | 47 |
| MosPV | 2,229 | 221 | 349 | 552 | 59.9 | 6.00 | ||
| H/G | NarPV | 2,211 | 41 | 196 | 657 | 72.4 | 7.51 | 29 |
| MosPV | 2,313 | 38 | 376 | 632 | 70.2 | 6.61 | ||
| L | NarPV | 6,684 | 32 | 46 | 2,201 | 250.8 | 6.90 | 65 |
| MosPV | 6,678 | 32 | 28 | 2,205 | 249.7 | 6.54 | ||
aFor the P/V/C gene of NarPV, there are two different types of mRNA made via RNA editing, each encoding a different protein product, i.e., P (non-edited mRNA) and V (+1G)
bThe putative C protein can be made from either of the two possible mRNA species
Transcription of individual genes of Paramyxovirinae members is carried out by a stop-and-reinitiation mechanism controlled by conserved sequences at the gene borders [11]. Members of genera Respirovirus, Morbillivirus and Henipavirus have a conserved trinucleotide intergenic region (IGR), whereas viruses in the genera Rubulavirus and Avulavirus have IGR of variable length and sequence. The gene start, stop and IGR sequences of NarPV are listed in Table 2 (shown as cDNA sequence in the antigenome 5′→3′ sense). The sequence comparison suggests that the transcriptional regulatory elements of NarPV are most similar to those of the MosPV genome.
Table 2.
Sequence of intergenic regions (IGR) and transcriptional start and stop signals of NarPV
| Virus genes | Gene stop | IGR | Gene start |
|---|---|---|---|
| NarPV | |||
| /N | AGGAGTAAAG | ||
| N/P | TTACAAAAAA | CTT | AGGACCAAGG |
| P/M | TTATAAAAAA | CTT | AGGACTAACG |
| M/F | CAATAAAAAA | CTT | AGGACTAACG |
| F/H | CTATAAAAAA | CTT | AGGACCAAGG |
| H/L | TTATAAAAAA | CTT | AGGATCCAGG |
| L/ | TTAAAGAAAA | CTT | |
| Consensus | |||
| Sequences | |||
| NarPV | ywAhArAAAA | CTT | AGGAbymAvG |
| MosPV | ywAhrAAAAA | CKT | AGGrbnmArG |
The coding strategy of the multi-cistronic P gene of Paramyxovirinae members is an important genomic feature used for classification. Similar to most subfamily members, the non-edited mRNA of the NarPV P gene codes for the P protein, whereas the mRNA with a single G-insertion at the RNA editing site codes for the V protein. Both mRNA species have the coding capacity for a C protein, which is coded in an alternative reading frame. However, insertion of two G’s results in an mRNA containing a stop codon immediately after the editing site, suggesting that NarPV does not code for a W-like protein present in certain subfamily members such as henipaviruses [11]. The NarPV P gene has the editing site sequence 5′-ACTAAAAGGGGCA-3′, which is identical to that in the MosPV genome [13].
Molecular features of deduced NarPV structural proteins
The N proteins of NarPV and MosPV share a 59% sequence identity. As for other paramyxovirus N proteins, the most variable region is located in the C-terminus. The sequence identity increases to 71% when the N-terminal 400-aa regions are compared. The region containing the highly conserved Paramyxovirinae N-protein motif F-X4-Y-X3-Φ-S-Φ-A-M-G (X = any aa; Φ = aromatic aa) has identical sequence between NarPV and MosPV, both having the sequence FAPGNYPLLWSYAMG.
The P and V proteins of NarPV are slightly acidic in nature, with pI values of 5.28 and 4.51, respectively. The sequence identities between these two proteins of NarPV and MosPV are much lower than that observed for their N proteins (Table 1). It is interesting to note that the difference in size of the two P proteins is due to a 26-aa deletion in the NarPV P protein (or a 26-aa insertion in the MosPV P protein), present in the region upstream of the P/V editing site (Fig. 3a). Since the “deletion” area was located close to the stop codon of the NarPV C ORF, the conservation of these proteins was largely unchanged as this resulted in only a 3-aa truncation in comparison to the MosPV C protein (Fig. 3b). It is also interesting to note that, in overlapping coding regions, there are significantly higher amino acid sequence identities between the C proteins and V proteins of the two viruses than the P proteins. For the C and P common region, the percentage sequence identify is 51% for C and 39% for P. For the V and P common region, it is 61% for V and 36% for P.
Fig. 3.
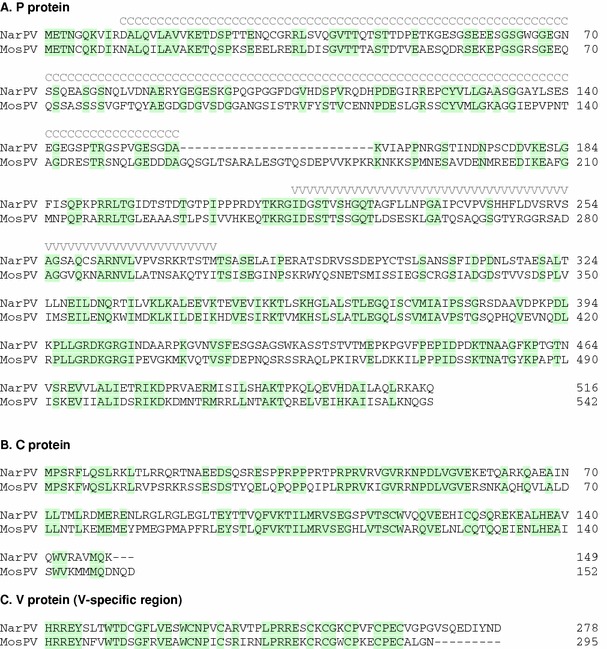
Alignment of deduced amino acid sequences for proteins encoded by the P/V/C genes of NarPV and MosPV. a Alignment of the full-length P protein sequences with identical residues shaded. The overlapping coding regions for the C proteins and V-specific polypeptide are indicated by the light grey letters C and V above. b Alignment of the full-length C protein sequences. c Alignment of the V-specific protein sequences
The M protein of NarPV is 340 aa in length. The M proteins of NarPV and MosPV have the highest sequence identity among all the deduced proteins and a very similar pI, indicating the basic nature of the proteins (Table 1). M is also the only protein that has an identical size between the two viruses.
The F protein of NarPV, like the F proteins of other paramyxoviruses, is predicted to be a type I integral membrane protein. The predicted cleavage site of the NarPV F protein, SGRNK, is dibasic and does not conform to the consensus sequence motif for cleavage by furin, R-X-K/R-R [6]. It is interesting to note that all of the rodent paramyxovirus F proteins characterized so far have the mono- or dibasic cleavage site instead of the more common multi-basic furin cleavage site observed in most other paramyxoviruses. Their cleavage sites resemble those of the bat-derived henipaviruses and the fish-derived AsaPV (see Fig. 4). The predicted cleavage site of the NarPV F protein is immediately followed by a highly conserved 25-aa putative hydrophobic fusion peptide as for all known paramyxovirus F proteins. The MprPV F protein contains 6 potential sites for N-linked glycosylation. Unusually, 4 are located in the F2 region, and 3 of the 4 are also conserved in the MosPV F2 subunit. In contrast, only 2 are located within the F1 subunit, and none of them were conserved in the MosPV F1 protein.
Fig. 4.

Sequence alignment of the NarPV F cleavage site and fusion peptide with selected members of the Paramyxovirinae. A gap has been introduced at the predicted cleavage site. Amino acid residue numbers for each protein are shown to the left of each sequence. Conserved residues are shaded. See “Materials and methods” for full virus names and database accession numbers
Like other members of the Paramyxovirinae, the NarPV H protein is predicted to be a type II integral membrane protein with a hydrophobic domain located at the N-terminal region (aa 41–68) functioning both as the signal sequence and the transmembrane anchor. For certain paramyxoviruses, the attachment protein can be made in a soluble form by using an alternative in-frame ATG codon within the signal/anchor sequence [18]. For NarPV H protein, an in-frame ATG codon was present at aa 56, which has the potential to produce a soluble H protein if translation initiates from this ATG codon. The sequence surrounding this ATG codon (TGCATGT) does not have a strong conservation to the Kozak consensus sequence (ACCATGG). This is also true for the first ATG codon of the predicted H ORF, which has the sequence AAAATGG. It is therefore possible that both ATG codons are used in vivo, albeit probably with different efficiencies. The 6-aa motif (NRKSCS) present in the predicted neuraminidase active site found in respirovirus and rubulavirus HN proteins [14] is absent in the NarPV H protein. It had the sequence AYDGCA at the corresponding site, with only the C residue conserved. The MosPV G protein has the sequence VFDGCS in this region. There are three potential N-linked glycosylation sites predicted for the NarPV H protein at N70, N177 and N575. Although MosPV G protein also has three predicted N-linked glycosylation sites at N155, N175 and N319, only one site at N177 for NarPV and N175 for MosPV is located at the same location in the protein. For NarPV H protein, the N70 site is located very close to the transmembrane domain and is therefore unlikely to be glycosylated in vivo.
The L proteins of NarPV and MosPV share more than 60% sequence identity. The six strongly conserved linear domains identified within the L proteins of nonsegmented negative-strand (NNS) RNA viruses by Poch et al. [16] can also be identified within these two L proteins. It is interesting to note that in the most conserved domain III, both L proteins contained the GDNE sequence motif, which has only been found in HeV, NiV and TupPV, and is different from the highly conserved GDNQ motif found in all other known viruses in the order Mononegavirales [25].
Phylogenetic analysis
Phylogenetic trees were generated by neighbour-joining methods from sequence alignments of deduced aa sequences of the six major proteins of all known members of the subfamily Paramyxovirinae. A representative tree based on the N protein aa sequences is shown in Fig. 5, which clearly shows the close relationship between NarPV and MosPV and indicates that these two viruses, together with JPV, BeiPV, TupPV and SalPV, cannot be placed in any of the five existing genera in the subfamily. Phylogenetic trees based on other proteins give similar results (data not shown).
Fig. 5.
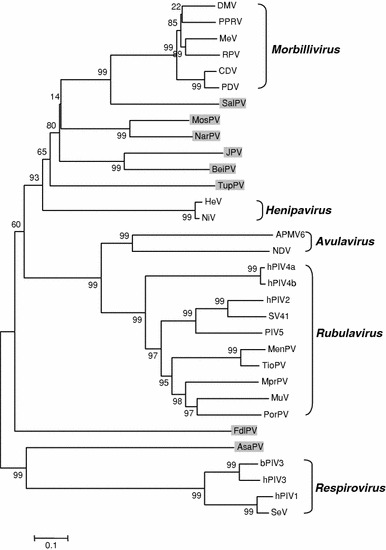
Phylogenetic trees based on complete protein sequences of the N proteins of viruses in the subfamily Paramyxovirinae. Numbers at nodes indicate levels of bootstrap support calculated from 1,000 trees. Branch lengths represent relative genetic distances. Full virus names and database accession numbers are provided in “Materials and methods”
Discussion
The discovery of HeV about 15 years ago opened a new chapter in the genetic diversity of paramyxoviruses. It not only challenged the notion that paramyxoviruses have relatively uniform genome sizes, it also revealed several genetic features not observed in previously known paramyxoviruses, such as the lack of both haemagglutination and neuraminidase activities in the attachment protein, the lack of a multi-basic cleavage site in the F protein and the replacement of the highly conserved GDNQ sequence by GDNE in the L protein.
Interestingly, many of the newly discovered paramyxoviruses seemed to contain genome features that keep pushing the “boundaries” of known paramyxoviruses in terms of genome size, genome organization and gene sequences. This is best demonstrated by genomes of the JPV and BeiPV. They are much larger than most other paramyxovirus genomes, contain several novel genes and have an exceptionally large gene for the attachment protein [7, 12]. Another interesting example is FdlPV [9]. At 15,378 nt, the FdlPV genome is relatively small among the known members of the subfamily Paramyxovirinae but contains a seventh gene, U (for unknown), positioned between the N and P/V genes.
Although rodents represent the most diverse mammals on earth, SeV remained the only known paramyxovirus of rodent origin until the recent characterization of MosPV, JPV and BeiPV by our group [7, 12, 13]. In this context, we decided to determine the genome structure and sequence of NarPV, another unknown rodent paramyxovirus. It is obvious that SeV and the rest of rodent viruses characterized to date do not share a common ancestor virus. In contrast, JPV and BeiPV are closely related both in genome organization and size and in gene sequences. In this paper, we have shown that NarPV and MosPV also share many genetic features, not only confirming their rodent origin, but also suggesting that these two viruses evolved from a common progenitor virus.
It is worth noting that although JPV/BeiPV and NarPV/MosPV represent two quite different groups of rodent paramyxoviruses in terms of genome size and organization, their common proteins do share more sequence identity than any other known paramyxoviruses, as shown by the phylogenetic tree in Fig. 5.
The only common feature we could identify among all known rodent paramyxoviruses was the lack of the multi-basic cleavage site present in most other paramyxoviruses. The only other paramyxoviruses with a monobasic cleavage site are henipaviruses of bat origin and the only known paramyxovirus of fish origin, AsaPV (Fig. 4). The biological significance of this observation is not known at the present time.
In conclusion, the characterization of the NarPV complete genome sequence in this study further highlights the great genetic diversity present among the paramyxoviruses of wildlife origin, ranging through bats, rodents, reptiles and fish. Further studies are required to determine whether these rodent viruses have the potential to infect and cause diseases in humans and livestock animals. The taxonomic status of these viruses is yet to be determined. Due to their major differences in genome size, genome organization and deduced amino acid sequences, it is most likely that these viruses will be classified into new genera within the subfamily Paramyxovirinae.
Acknowledgments
We thank Dr. N. Karabatsos for supplying the Nariva virus stock, Kaylene Selleck and Eric Hansson for technical assistance, Tony Pye for providing the automated sequencing service, and Drs. Glenn Marsh and Jackie Pallister for critical review of the manuscript.
Footnotes
L. S. Lambeth, M. Yu, and D. E. Anderson contributed equally to this paper.
References
- 1.Adkins RM, Gelke EL, Rowe D, Honeycutt RL. Molecular phylogeny and divergence time estimates for major rodent groups: evidence from multiple genes. Mol Biol Evol. 2001;18:777–791. doi: 10.1093/oxfordjournals.molbev.a003860. [DOI] [PubMed] [Google Scholar]
- 2.Calisher CH, Childs JE, Field HE, Holmes KV, Schountz T. Bats: important reservoir hosts of emerging viruses. Clin Microbiol Rev. 2006;19:531–545. doi: 10.1128/CMR.00017-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chua KB, Wang LF, Lam SK, Crameri G, Yu M, Wise T, Boyle D, Hyatt AD, Eaton BT. Tioman virus, a novel paramyxovirus isolated from fruit bats in Malaysia. Virology. 2001;283:215–229. doi: 10.1006/viro.2000.0882. [DOI] [PubMed] [Google Scholar]
- 4.Eaton BT, Broder CC, Middleton D, Wang LF. Hendra and Nipah viruses: different and dangerous. Nat Rev Microbiol. 2006;4:23–35. doi: 10.1038/nrmicro1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Faisca P, Desmecht D. Sendai virus, the mouse parainfluenza type 1: a longstanding pathogen that remains up-to-date. Res Vet Sci. 2007;82:115–125. doi: 10.1016/j.rvsc.2006.03.009. [DOI] [PubMed] [Google Scholar]
- 6.Hosaka M, Nagahama M, Kim WS, Watanabe T, Hatsuzawa K, Ikemizu J, Murakami K, Nakayama K. Arg-X-Lys/Arg-Arg motif as a signal for precursor cleavage catalyzed by furin within the constitutive secretory pathway. J Biol Chem. 1991;266:12127–12130. [PubMed] [Google Scholar]
- 7.Jack PJM, Boyle DB, Eaton BT, Wang LF (2005) The complete genome sequence of J-virus reveals a unique genome structure in the family Paramyxoviridae. J Virol (in press) [DOI] [PMC free article] [PubMed]
- 8.Karabatsos N, Buckley SM, Ardoin P. Nariva virus: further studies, with particular reference to its hemadsorption and hemagglutinating properties. Proc Soc Exp Biol Med. 1969;130:888–892. doi: 10.3181/00379727-130-33680. [DOI] [PubMed] [Google Scholar]
- 9.Kurath G, Batts WN, Ahne W, Winton JR. Complete genome sequence of Fer-de-Lance virus reveals a novel gene in reptilian paramyxoviruses. J Virol. 2004;78:2045–2056. doi: 10.1128/JVI.78.4.2045-2056.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lamb RA, Collins PL, Kolakofsky D, Melero JA, Nagai Y, Oldstone MBA, Pringle CR, Rima BK. Family paramyxoviridae. In: Fauquet CM, Mayo J, Maniloff J, Desselberger U, Ball LA, editors. Virus taxonomy: 8th report of the International Committee on Taxonomy of Viruses. San Diego: Elsevier; 2005. pp. 655–668. [Google Scholar]
- 11.Lamb RA, Parks GD. Paramyxoviridae: the viruses and their replication. In: Knipe DM, Griffin DE, Lamb RA, Straus SE, Howley PM, Martin MA, Roizman B, editors. Fields virology. Philadelphia: Lippincott; 2007. pp. 1449–1496. [Google Scholar]
- 12.Li Z, Yu M, Zhang H, Magoffin DE, Jack PJ, Hyatt A, Wang HY, Wang LF. Beilong virus, a novel paramyxovirus with the largest genome of non-segmented negative-stranded RNA viruses. Virology. 2006;346:219–228. doi: 10.1016/j.virol.2005.10.039. [DOI] [PubMed] [Google Scholar]
- 13.Miller PJ, Boyle DB, Eaton BT, Wang LF. Full-length genome sequence of Mossman virus, a novel paramyxovirus isolated from rodents in Australia. Virology. 2003;317:330–344. doi: 10.1016/j.virol.2003.08.013. [DOI] [PubMed] [Google Scholar]
- 14.Mirza AM, Deng R, Iorio RM. Site-directed mutagenesis of a conserved hexapeptide in the paramyxovirus hemagglutinin-neuraminidase glycoprotein: effects on antigenic structure and function. J Virol. 1994;68:5093–5099. doi: 10.1128/jvi.68.8.5093-5099.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pavri KM, Singh KR, Hollinger FB. Isolation of a new parainfluenza virus from a frugivorous bat, Rousettus leschenaulti, collected at Poona, India. Am J Trop Med Hyg. 1971;20:125–130. doi: 10.4269/ajtmh.1971.20.125. [DOI] [PubMed] [Google Scholar]
- 16.Poch O, Blumberg BM, Bougueleret L, Tordo N. Sequence comparison of five polymerases (L proteins) of unsegmented negative-strand RNA viruses: theoretical assignment of functional domains. J Gen Virol. 1990;71(Pt 5):1153–1162. doi: 10.1099/0022-1317-71-5-1153. [DOI] [PubMed] [Google Scholar]
- 17.Renshaw RW, Glaser AL, Van Campen H, Weiland F, Dubovi EJ. Identification and phylogenetic comparison of Salem virus, a novel paramyxovirus of horses. Virology. 2000;270:417–429. doi: 10.1006/viro.2000.0305. [DOI] [PubMed] [Google Scholar]
- 18.Roberts SR, Lichtenstein D, Ball LA, Wertz GW. The membrane-associated and secreted forms of the respiratory syncytial virus attachment glycoprotein G are synthesized from alternative initiation codons. J Virol. 1994;68:4538–4546. doi: 10.1128/jvi.68.7.4538-4546.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Salas-Rojas M, Sanchez-Hernandez C, Romero-Almaraz Md Mde L, Schnell GD, Schmid RK, Aguilar-Setien A. Prevalence of rabies and LPM paramyxovirus antibody in non-hematophagous bats captured in the Central Pacific coast of Mexico. Trans R Soc Trop Med Hyg. 2004;98:577–584. doi: 10.1016/j.trstmh.2003.10.019. [DOI] [PubMed] [Google Scholar]
- 20.Tamura K, Dudley J, Nei M, Kumar S. MEGA4: molecular evolutionary genetics analysis (MEGA) software version 4.0. Mol Biol Evol. 2007;24:1596–1599. doi: 10.1093/molbev/msm092. [DOI] [PubMed] [Google Scholar]
- 21.Tikasingh ES, Jonkers AH, Spence L, Aitken TH. Nariva virus, a hitherto undescribed agent isolated from the Trinidadian rat, Zygodontomys b. brevicauda (J. A. Allen & Chapman) Am J Trop Med Hyg. 1966;15:235–238. doi: 10.4269/ajtmh.1966.15.235. [DOI] [PubMed] [Google Scholar]
- 22.Tillett D, Burns BP, Neilan BA. Optimized rapid amplification of cDNA ends (RACE) for mapping bacterial mRNA transcripts. Biotechniques. 2000;28:443–452. doi: 10.2144/00283st01. [DOI] [PubMed] [Google Scholar]
- 23.Walder R. Electron microscopic evidence of Nariva virus structure. J Gen Virol. 1971;11:123–128. doi: 10.1099/0022-1317-11-2-123. [DOI] [PubMed] [Google Scholar]
- 24.Wang L-F, Eaton BT. Emerging paramyxoviruses. Infect Dis Rev. 2001;3:52–60. [Google Scholar]
- 25.Wang L-F, Chua KB, Yu M, Eaton BT. Genome diversity of emerging paramyxoviruses. Curr Genomics. 2003;4:263–272. doi: 10.2174/1389202033490385. [DOI] [Google Scholar]
- 26.Wang L-F, Hansson E, Yu M, Chua KB, Mathe N, Crameri G, Rima BK, Moreno-Lopez J, Eaton BT. Full-length genome sequence and genetic relationship of two paramyxoviruses isolated from bat and pigs in the Americans. Arch Virol. 2007;152:1259–1271. doi: 10.1007/s00705-007-0959-4. [DOI] [PMC free article] [PubMed] [Google Scholar]