

Abstract
Groundnut bud necrosis virus belongs to the genus Tospovirus, infects a wide range of crop plants and causes severe losses. To understand the role of the nucleocapsid protein in the viral life cycle, the protein was overexpressed in E. coli and purified by Ni-NTA chromatography. The purified N protein was well folded and was predominantly alpha-helical. Deletion analysis revealed that the C-terminal unfolded region of the N protein was involved in RNA binding. Furthermore, the N protein could be phosphorylated in vitro by Nicotiana benthamiana plant sap and by purified recombinant kinases such as protein kinase CK2 and calcium-dependent protein kinase. This is the first report of phoshphorylation of a nucleocapsid protein in the family Bunyaviridae. The possible implications of the present findings for the viral life cycle are discussed.
Electronic supplementary material
The online version of this article (doi:10.1007/s00705-011-1110-0) contains supplementary material, which is available to authorized users.
Keywords: Infectious Bronchitis Virus, Tomato Spotted Wilt Virus, Nucleocapsid Protein, Viral Life Cycle, Rift Valley Fever
Introduction
Groundnut bud necrosis virus (GBNV) is a member of the genus Tospovirus in the family Bunyaviridae. The members of the genus Tospovirus infect a wide range of vegetable, fruit, ornamental and field crops and cause severe economic losses throughout India and elsewhere [30]. Therefore, these viruses are of major economic importance in the world.
The genus tospovirus includes viruses with enveloped quasi-spherical particles of 80-120 nm diameter and tripartite single-stranded RNA genomes with negative or ambisense polarity. The genomes are tightly associated with the nucleocapsid (N) protein and a few copies of the L protein [8]. The three genomic RNA species, large (L), medium (M) and small (S), are of approximate size 8.9, 4.8 and 2.9 kb, respectively. The L RNA codes for RNA-dependent RNA polymerase (RdRP) in the virion complementary sense [1]. The non-structural proteins NSm and NSs are encoded in the virion sense, whereas the precursor for the glycoproteins Gn and Gc and the N protein are encoded in the complementary sense by the M and S RNA, respectively [8, 38].
N proteins of negative-strand RNA viruses play a vital role in the life cycle of the virus and are multifunctional. The major structural component of the virion is the N protein, and it binds to genomic and antigenomic RNAs along their entire length and forms ribonucleoprotein (RNP) complexes. These complexes serve as templates for viral RNA replication and transcription [18, 34]. The N protein has been shown to interact with viral RdRP and display RNA chaperone activity [3, 25]. In addition, it also can replace the cellular eIF4F complex, thereby ensuring efficient translation of viral mRNA [26]. In addition to the viral-encoded proteins, the N protein is also known to interact with host proteins and interfere with different cellular pathways during the life cycle of the virus [24, 29].
The N proteins of hantaviruses have a conserved central domain (residues 136-213) that contains a large cluster of basic residues (15 positive charges) that is involved in RNA binding. However, the number of nucleotides that bind to each N protein is variable [18]. The oligomeric status of the N protein in members of the family Bunyaviridae has not been well studied. This protein is predominantly trimeric in the hantaviruses, and the interaction between subunits has been shown to be mediated by packing of N-terminal coiled coils of adjacent subunits [18]. Analysis of the trimeric N protein by cryo-electron microscopy has confirmed this observation [18].
The N protein of tomato spotted wilt virus (TSWV), the type member of the genus Tospovirus, has been characterized earlier. It binds to single-stranded RNA in a non-sequence-specific manner, but not to double-stranded RNA [34]. However, compared to the N protein of hantaviruses, the TSWV N protein is much smaller, and the RNA-binding domain has not been clearly defined. It has been shown to interact homotypically in vitro and in vivo [37, 38, 42]. The TSWV N protein exists as multimers or aggregates of increasing molecular mass. Analysis of deletion and site-specific mutants of TSWV N protein has revealed that the subunits can interact in a head-to-tail manner, and two conserved phenylalanine residues (F242 and F246) are crucial for this interaction [42]. Apart from its homotypic interaction, the TSWV N protein is known to interact with viral glycoproteins and is proposed to be involved in viral particle assembly in the Golgi [33]. It also interacts with NSm and aids in the cell-to-cell movement of RNP complexes [39]. The three-dimensional structure of the N protein has not been described so far for any member of the genus Tospovirus.
GBNV (also called peanut bud necrosis virus) belongs to serogroup IV of the genus Tospovirus and is the most prevalent virus among the tospoviruses reported from the Indian subcontinent [35]. The complete genomic sequence of GBNV L, M, and S RNA was determined earlier [9, 35, 36]. With a view to understanding the virus life cycle at the molecular level, a strain of GBNV infecting tomato in Karnataka state in India, GBNV- To (K), was purified and characterized. The N gene of GBNV was cloned, overexpressed and purified. The purified recombinant N protein was used for the generation of monoclonal antibodies [15]. Recently, the NSs protein of this virus was characterized and shown to possess ATPase and 5′ phosphatase activities [23]. The N protein of GBNV shares only 29% amino acid identity with that of TSWV [35], the type member of the genus, and therefore, there is an obvious need for specific characterization of the GBNV N protein.
In the present paper, the characterization of the N protein of a serogroup IV virus, GBNV, is described for the first time. The biophysical properties, the domain involved in RNA binding, and in vitro phoshphorylation of the N protein are presented.
Materials and methods
Oligonucleotide primers
The oligonucleotide primers used for amplification of the wild-type N gene and its deletion mutants were custom-made by Sigma Chemicals.
Cloning, expression and purification of full-length and truncated N proteins
The N gene of GBNV- To(K) was cloned into pRSET-A vector at NheI and BamHI sites as described in ref. [15]. To generate N- and C-terminal deletions, pRSET-A plasmid containing the N gene was used as the template, and the region of interest was amplified by PCR using high-fidelity Phusion polymerase and specific sense and antisense primers designed to generate deletions. NheI and BamHI sites were introduced in the sense and antisense primers, respectively for ease of cloning (Table 1). The amplified product was gel-purified (MN gel extraction kit), digested with NheI and BamHI enzymes, and cloned at the same sites in the pRSET-A vector. The clones were confirmed by DNA sequencing.
Table 1.
Oligonucleotide primers used in the study
| Primers | Sequence 5′–3′ | Restriction enzyme site (underlined sequence) |
|---|---|---|
| N sense | GCTAGCCATATGTCTACCGTTAAGCAGCTCAC | NheI |
| N antisense | CAGCGGATCCTTACAATTCCACAGAAGC | BamHI |
| N ∆ 20 sense | GCTAGCCATATGGCAGATGTTGAAATTGAAAC | NheI |
| N ∆ 40 sense | GCTAGCCATATGGACACTAACAAAAGTCTTG | NheI |
| C ∆ 15 anti sense | GGATCCCTACTCGAGATGTTCACCATAATCATC | BamHI |
| C ∆ 37 anti sense | CCGGATCCTTAATATTTCTTGAGTGAGATAGAGC | BamHI |
To express and purify the N protein, E. coli BL21(DE3) pLys S cells were transformed with the pREST-A clone containing the N gene. Cells were cultured in 2 × YT medium, expression was induced by the addition of 0.3 mM IPTG, and the culture was grown for 4-5 h at 30°C or 10-12 h at 18°C. The cell pellet was resuspended in lysis buffer (50 mM Tris-HCl pH 8.0, 300 mM NaCl, 1 % Triton-X 100, 10% glycerol) and sonicated. The overexpressed hexa-histidine-tagged N protein was purified from the soluble fraction by Ni-NTA affinity chromatography [15]. The purity of the protein was checked by SDS-PAGE [19], and the identity of the protein was confirmed by western blot analysis with a monoclonal antibody (A10D10) [15] raised against the N protein. E. coli BL21(DE3) pLysS cells were transformed with the deletion mutant clones of the N gene, and the proteins expressed upon induction with 0.3 mM IPTG were purified by Ni-NTA chromatography as described for the wild-type N protein [15].
Biophysical characterization of purified N protein
Circular dichroism (CD) spectroscopy
The far-UV CD spectrum of purified N protein was recorded using a Jasco-815 spectropolarimeter. The ellipticity was monitored at 25°C from 200 to 250 nm using 0.1 mg/ml of the purified protein in 50 mM Tris-HCl, pH 8.0. A scan speed of 50 nm/min, a 0.2-cm-path-length cuvette, a bandwidth of 1 nm, and a response time of 1 s were used. The spectra were averaged for three scans and corrected with buffer blanks. The percentage of α-helix, β-pleated sheet and random coil present in the N protein was determined using the K2D2 program, which is available online. Thermal denaturation studies were carried out using a Peltier thermocycler; 0.2 mg/ml of the purified protein in 50 mM Tris-HCl, pH 8.0, was heated from 30 to 90°C. The molar ellipticity was monitored at 208 nm as a function of temperature to obtain the thermal melting curve. Tm was determined by plotting a first-derivative graph of molar ellipticity vs temperature. The temperature corresponding to the peak of the profile gave the Tm, the temperature at which 50% of the molecules are denatured.
Fluorescence spectroscopy
Fluorescence measurements were carried out using a Perkin Elmer Life Science LS 55 spectrofluorimeter. The protein (0.1 mg/ml) in 50 mM Tris-HCl buffer, pH 8.0, was excited at 280 nm, and emission was measured from 290 to 450 nm. The excitation and emission band-pass wavelength was 5 nm.
Size-exclusion chromatography
The oligomeric status of the recombinant N protein was analyzed using a Superdex S-200 analytical gel filtration column attached to a Bio-Rad FPLC system. Purified N protein was subjected to size-exclusion chromatography along with molecular mass markers (Bio-Rad).
Agarose gel electrophoresis
Nucleic acid associated with the N protein was analysed on a 0.6% agarose gel in TAE buffer. The gel was viewed by ethidium bromide staining for the presence of nucleic acid and later dried and stained with Coomassie blue for visualizing the protein.
Spectrophotometric analysis
The presence of nucleic acid in the purified wild-type N protein and its truncated forms was monitored by spectrophotometric analysis of A260 / 280 ratios.
RNA extraction and analysis
Total RNA associated with the N protein was extracted using the TRIzol method [5]. To the purified N protein sample an equal volume of TRI Reagent (Sigma) was added, the tube was agitated gently, and the aqueous phase was collected by centrifugation at 12,000 rpm for 10 min. The aqueous phase was then extracted with an equal volume of chloroform. The RNA present in the aqueous phase was precipitated by the addition of ethanol and 3 M sodium acetate (pH 5.5). The RNA pellet was dissolved in DEPC-treated water and analysed on a 2% native agarose gel in 0.5X TBE buffer along with low-range RNA size markers (New England Biolabs).
Bioinformatic analysis
Bioinformatic analysis was carried out to identify the presence of RNA binding sites in the N protein using the RNABindR software available at www.bindr.gdcb.jastate.edu/RNABindR. To predict the folded and unfolded regions in the protein, the software FoldIndex© (http://bip.weizmann.ac.il/fldbin/findex#info), available at the Weizmann Institute of Science, was used. The NetPhos 2.0 server (http://www.cbs.dtu.dk/cgi-bin/nph) was used to predict potential phosphorylation sites present in the protein.
In vitro protein kinase assay
In vitro phosphorylation assays were performed as described previously [16]. Briefly, N protein (as substrate) was incubated with soluble fraction of tobacco (Nicotiana benthamiana) plant sap (as kinase source) in 25 mM HEPES buffer, pH 7.5, 2 mM MnCl2 and 1 μCi of γ32P ATP/GTP in a 20-μl reaction mixture. The reaction was carried out for 30 min at 30°C, followed by SDS-PAGE analysis and autoradiography. Similar in vitro experiments were carried out by using purified recombinant protein kinase CK2 (CK2) from tobacco (the tobacco-CK2-expressing clone was a kind gift from Prof. Kristiina Makinen, University of Helsinki) and by using purified recombinant calcium-dependent protein kinase (CDPK) from chickpea (a kind gift from Prof. C. Jayabaskaran, IISc.), except that the reaction with CDPK was carried out in 50 mM Tris-HCl buffer, pH 7.2, containing 250 μM CaCl2.
Results and discussion
Bioinformatic analysis
Bioinformatic analysis of the full-length GBNV N protein was carried out to identify the motifs that might have some functional relevance in the viral life cycle. RNA binding is a prerequisite for the encapsidation process. A search for RNA-binding motifs present in the N protein using the RNABindR software did not reveal canonical RNA-binding motifs with optimal and high specificity (data not shown). However, with high-sensitivity prediction by RNABindR (which predicts RNA binding based on positively charged amino acid residues), the segments 1-11, 36-40, 55-114, 186-192, 197-200, 235-237, 245-276 were identified as possible regions of RNA binding (Fig. 1a, underlined regions). Foldindex analysis using the Foldindex © program showed that the C-terminal region, comprising 37 amino acid residues, is unfolded (Fig. 1b). It may be noted that this unfolded region overlaps with one of the predicted RNA-binding segments (amino acids 245-276).
Fig. 1.
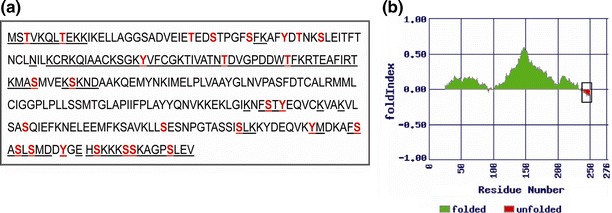
Bioinformatic analysis of GBNV N protein. a Schematic representation of predicted nucleic-acid-binding regions (underlined) and sites of phosphorylation (coloured residues) of the N protein. b Output obtained for the GBNV N protein using the FoldIndex program. The unfolded region in the protein has been boxed
Many plant- and animal-virus-encoded proteins are phosphorylated, and such modifications have regulatory role in the viral life cycle [12, 21, 31]. Prediction of phosphorylation motifs in the N protein using the NetPhos 2 server resulted in the identification of many potential phosphorylation sites in the GBNV N protein (Fig. 1a, coloured residues).
Expression and purification of full-length GBNV-N protein and its truncated forms
Full-length N protein and its truncated forms (N∆20, N∆40, C∆15 and C∆37) used in the present study (Fig. 2a) were expressed in E. coli. Of the different E.coli host strains (BL21(DE3) pLysS, Rosetta, C43(DE3)) tested for expression, BL21(DE3) pLysS cells gave the highest levels of protein expression with all of the constructs, and therefore, this strain was chosen to express the full-length GBNV N protein and its truncated forms. Furthermore, culture conditions and buffers were standardized for obtaining the expressed proteins in the soluble fraction. Culture medium 2 × YT at a growth temperature of 30°C before induction and 18°C after induction (12 h) and 50 mM Tris-HCl (pH 8.0) buffer, containing 300 mM NaCl, 1% Triton X-100 and 10% glycerol were most suitable for this purpose. The recombinant proteins were purified using Ni-NTA affinity chromatography as described in "Materials and methods", and the purity of the proteins was checked by SDS-PAGE analysis (Fig. 2b). Major bands corresponding to the expected monomeric molecular mass of the N protein, 32 kDa; N∆20, 29.8 kDa; N∆40, 27.6 kDa; C∆15, 30.3 kDa; and C∆37, 28.5 kDa were observed along with a minor band at each dimeric position. Western blot analysis (Fig. 2c) with a monoclonal antibody (A10D10) raised against the N protein confirmed the identity of the purified proteins.
Fig. 2.
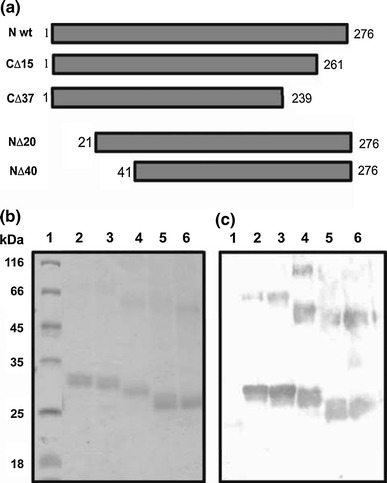
Expression in E. coli and purification of the GBNV N protein and its truncated forms. a Schematic representation of the full-length and deletion mutants of the N protein. Black boxes represent the regions that are present in each protein. b SDS-PAGE analysis of full-length N protein and truncated N proteins purified by Ni-NTA affinity chromatography. Lane 1, molecular mass markers; lanes 2, 3, 4, 5 and 6, full-length, C∆15, N∆20, C∆37 and N∆40 N protein respectively. c Western blot analysis of purified proteins using A10D10 monoclonal antibody to GBNV N protein [14]. Lanes are as marked in panel b
Biophysical characterization of the GBNV N protein
The far-UV CD spectrum of the N protein was typical for a globular protein [11], with molar ellipticity at 208 and 222 nm (Fig. 3a). Analysis of the secondary structure of the N protein using the K2D2 program showed that it is predominantly an alpha-helical protein (84%). The intrinsic fluorescence spectrum showed an emission maximum around 308 to 320 nm upon excitation at 280 nm (Fig. 3b), suggesting that the tryptophan and tyrosine residues are buried [20]. These results demonstrate that the N protein is in a folded conformation. The molar ellipticity of the N protein was monitored as a function of temperature as described in "Materials and methods". The N protein was stable up to 58°C and thereafter started to melt. The midpoint of denaturation, Tm, was found to be 68°C (Fig. 3c).
Fig. 3.
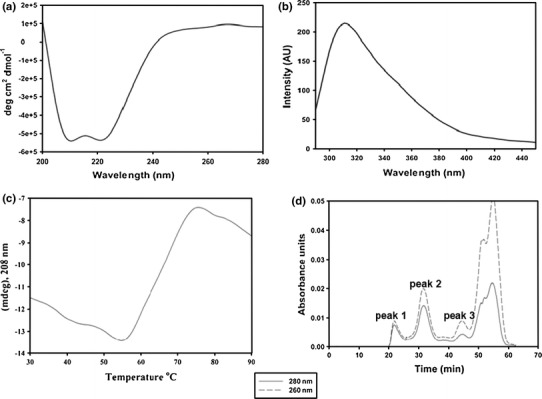
Biophysical characterization of GBNV N protein. a CD spectrum of N protein. The far-UV CD spectrum for the N protein (0.1 mg/ml) was recorded using a Jasco-815 spectropolarimeter. The molar ellipticity was calculated using a subunit mass of 32 kDa. b Fluorescence spectrum of N protein. The fluorescence spectrum for the N protein (0.1 mg/ml) was recorded using a Perkin Elmer LS 55 luminescence spectrometer after excitation at 280 nm. c Thermal melting profile of N protein. The molar ellipticity (y-axis) was monitored at 208 nm as a function of temperature (x-axis) and plotted as shown. d Elution profile of N protein on a Superdex S-200 analytical gel filteration column. Peak 1 represents the void fraction (›600 kDa). Peaks 2 and 3 correspond to 128-kDa (tetramer) and 32-kDa (monomer) proteins, respectively
Oligomerization or multimerization of the N protein is an important step in the assembly of the nucleocapsid. In order to investigate the oligomeric state of the N protein, size-exclusion chromatography was carried out on a Superdex S-200 column as described in “Materials and methods”. The protein eluted in three peaks (Fig. 3d). Peak 1 corresponded to the void fraction (greater than 600 kDa), peak 2 corresponded to 128 kDa (tetramer), and peak 3 corresponded to a 32-kDa (monomer) protein, as estimated using proteins of known molecular mass prior to loading the sample. There was an additional broad peak that eluted late in the run and had a high absorbance at 260 nm. However, when this fraction was analysed by western blot analysis, it did not show the presence of an N-protein-specific band. These results suggest that the purified N protein existed as a heterogeneous mixture of monomers, tetramers and higher-order oligomers. Furthermore, the absorbance at 260 nm was higher than that at 280 nm, indicating that the N protein might be associated with nucleic acids. To confirm the presence of nucleic acid, the protein was further analysed on a 0.6% agarose gel. Agarose gel analysis showed two bands, one at the well and another that entered the gel (Fig. 4a). Both of the bands showed ethidium bromide staining and Coomassie blue staining, confirming that N protein is associated with nucleic acids (Fig. 4a). The bound nucleic acid was degraded specifically by RNase A treatment (Supplementary Fig. 1), suggesting that the N protein is present in a ribonucleoprotein (RNP) complex. Such an association of N protein with RNA has also been reported for Bunyamvera virus [27] and Rift Valley fever virus [32]. Several attempts were made to purify N protein devoid of nucleic acid by using various salts, detergents and nucleases in the buffers throughout the purification steps. However, none of them were successful.
Fig. 4.
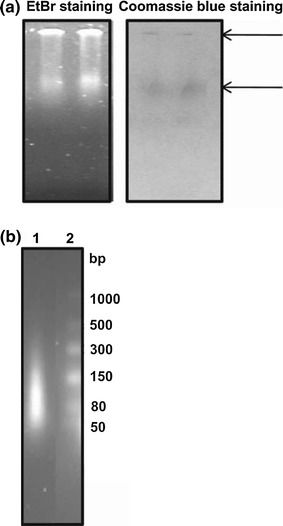
RNA binding properties of recombinant N protein. a Purified N protein was run on a 0.6 % agarose gel and stained with ethidium bromide and Coomassie blue. Lanes 1 and 2 show the N protein (50 μg) in duplicate. b Analysis of RNA species associated with N protein on a 2% native agarose gel. Lane 1: RNA from N protein; lane 2: RNA size markers
Mapping the RNA-binding domain of the GBNV N protein
The interaction of the N protein with the RNA would be essential for encapsidation of viral genomes, for replication, transcription and cell-to-cell movement. As no conserved RNA-binding motifs have been identified for tospoviruses, to map the domain of the N protein that specifically interacts with RNA, the presence of nucleic acid in the purified wild-type N protein and its truncated forms was initially monitored by spectrophotometric analysis of A260 / 280 ratios. For wild-type N protein, the ratio ranged between 1.8 and 1.9, and for the N∆20, N∆40 and C∆15 N proteins, it ranged between 1.2 and 1.7, suggesting the presence of nucleic acid. However, for C∆37, it was 0.7 to 0.8, indicating that the protein was not associated with nucleic acid. Furthermore, the amount of RNA bound to the full-length N protein and its truncated forms was quantitated after extraction of the RNA with TRI Reagent (Table 2). Full-length N protein contained 19.5 ± 0.5 μg of RNA in 100 μg of the protein, whereas the amount of RNA present was reduced by approximately 3-fold upon deletion of 20 amino acids from the N-terminus or 15 amino acids from the C-terminus. When 40 amino acid residues from the N terminus were removed, the amount of RNA associated with the protein was reduced by approximately 6-fold. As expected, no RNA was detected with the C∆37 N protein. Thus, the capacity of the N protein to bind nucleic acid was completely lost upon removal of the unfolded region of the N protein comprising 37 residues from the C-terminus. This suggested that the C-terminal unfolded region (which also contains 46% of the residues predicted to interact with RNA) might play a major role in RNA binding. The location of an RNA-binding domain in the N proteins of negative-strand RNA viruses is variable. Xu et al. [44] mapped a specific RNA-binding domain to a central conserved region comprising residues 175 to 217 of the hantavirus N protein. Multiple regions of RNA binding have been observed for the TSWV N protein [34] and for the influenza virus N protein [7]. It may be noted that even in GBNV, apart from the C-terminal region, the deletion of the N-terminal 40 amino acid residues also affected the protein-nucleic acid interaction. It is possible that the loss of protein-nucleic acid interaction in this mutant is because of the altered structure of the mutant protein. Involvement of disordered regions in RNA binding has also been reported for the N protein of SARS corona virus, a positive-strand RNA virus [4]. Bioinformatic analysis shows that the C-terminal unfolded region is conserved across N proteins of serogroup IV tospoviruses and might be involved in genome encapsidation.
Table 2.
Amount of RNA present (in μg) per 100 μg of the N protein and its truncated forms
| N protein wild type | 19.5 ± 0.5 |
| N ∆ 20 | 6.6 ± 0.3 |
| N ∆ 40 | 3.3 ± 0.2 |
| C ∆ 15 | 6.2 ± 0.2 |
| C ∆ 37 | 0.0 |
To determine the nature of the RNA and the stoichiometry of the N-RNA association, we next analysed the extracted nucleic acid from the full-length N protein RNP complex on a 2% native agarose gel along with RNA size markers of low range. The majority of the RNAs migrated within the 50-80-nucleotide size range and some RNA species migrated at around 80-150 nucleotides (Fig. 4b). Taking into account that the majority of the N protein exists as a tetramer (Fig. 3d), and taking the average length of the RNA species as 50 nucleotides, 12-13 nucleotides might bind per N monomer. N/RNA stoichiometry has been shown to vary between 6 and 24 nt in negative-strand RNA viruses [2, 10, 27, 43]. Studies carried out by Mohl and Barr [27] with Bunyamwera virus have shown that there is no obligatory requirement of a specific sequence or structure for RNA encapsidation.
Phosphorylation of the GBNV N protein
Bioinformatic analysis using the Net Phos 2 server revealed multiple putative phosphorylation sites in the N protein: at 16 serine, 6 threonine and 5 tyrosine residues in the GBNV N protein (Fig. 1a) and also by multiple kinases (Supplementary Fig. 2). In vitro phosphorylation of the purified N protein was carried out as described in "Materials and methods". As shown in Fig. 5a, lane 1, the N protein was phosphorylated by the kinase(s) present in the soluble fraction of N. benthamiana plant sap. Two negative controls were run in every experiment. In one of these, γ32P ATP was added to the purified N protein in the absence of plant sap, which did not result in labeling of the N protein (Fig. 5a, lane 2), suggesting that the N protein by itself does not bind to radiolabeled ATP. In the other control, the plant sap alone was incubated with γ32P ATP, and no specific bands were observed, suggesting that none of the host proteins present in the sap were phosphorylated (Fig. 5a, lane 3). The gel was stained with Coomassie blue to estimate the amount of protein loaded and the molecular mass of the protein (Fig. 5a, lane 4).
Fig. 5.

In vitro phosphorylation of GBNV N protein. a Phosphorylation of the N protein (5 μg) was carried out by mixing γ32P ATP (1 μCi) and the soluble fraction of tobacco plant sap in 25 mM HEPES buffer, pH 7.2, containing 2 mM MnCl2, incubation for 30 min at 30°C, and analysis by SDS-PAGE. The left panel shows an autoradiogram, and the right panel shows the same gel stained with Coomassie blue. Lane 1, N protein with plant sap; lane 2, N protein alone without plant sap (negative control); lane 3, plant sap alone without N protein (negative control), lane 4: molecular mass markers; lanes 5 and 6, reaction in the presence of 2 mM EGTA and 2 mM EDTA, respectively. b Effect of metal ions on phosphorylation of the N protein. The phosphorylation reaction was carried out without any metal ions (lane 1) or with varying concentrations (0.5, 1 and 2 mM) of MnCl2 (lanes 2, 3, and 4, respectively). Similarly, the reaction was carried out separately with 0.5, 1 and 2 mM MgCl2 (lanes 5-7) and CaCl2 (lanes 8-10). Lane 11, molecular mass markers. The left panel shows an autoradiogram, and the right panel shows the same gel stained with Coomassie blue. c The phosphorylation reaction was carried out with 1 μCi of γ 32P GTP as the phosphoryl group donor instead of labeled ATP. Lane 1, plant sap alone; lane 2, N protein alone; lane 3, reaction carried out in the absence of any metal ions; lanes 4, 5 and 6, reaction carried out in the presence of 2 mM MnCl2, 2 mM MgCl2 and 2 mM CaCl2, respectively; lane 7, molecular mass markers. d Phosphorylation of the N protein (5 μg) was carried out with 200 ng of purified recombinant tobacco CK2 instead of plant sap as described in the legend to Fig. 5 a. Lane 1, CK2 alone without N protein; lane 2, N protein alone without CK2; lane 3, N protein with CK2; lane 4, molecular mass markers; lane 5, BSA (Calbiochem) with CK2 (negative control); lane 6, histone H1 (Sigma) with CK2 (positive control); lane 7, histone H1 (Sigma) without CK2. e Phosphorylation reaction carried out with 100 ng of purified recombinant chickpea CDPK instead of plant sap. Lane 1, CDPK alone without N protein (negative control); lane 2, N protein alone without CDPK; lane 3, N protein with CDPK; lane 4, molecular mass markers; lane 5, histone H1 (Sigma) with CDPK (positive control); lane 6, BSA (Calbiochem) with CDPK (negative control)
The phosphorylation reaction was inhibited to some extent by the addition of metal chelators such as EGTA (2 mM) and EDTA (2 mM) (Fig. 5a, lanes 5 and 6, respectively), suggesting a requirement for divalent metal ions for the reaction. Furthermore, when the reaction was carried out in the absence of MnCl2, the phosphorylation was abolished, confirming the requirement for metal ions (Fig. 5b, lane 1). Therefore, the specificity of the metal ion required for the phosphorylation of the N protein was tested by the addition of increasing concentrations of MnCl2 (Fig. 5b, lanes 2-4), MgCl2 (Fig. 5b, lanes 5-7), and CaCl2 (Fig. 5b, lanes 8-10). As is apparent from the figure, the plant protein kinase that phosphorylates the N protein shows a preference for Mn2+ over Mg2+, and no detectable band could be seen with Ca2+. The Coomassie-stained gel confirmed that the effect observed was not due to variation in the amount of N protein used in the reaction. Similar observations have been made for a plant protein kinase that phosphorylates PVA coat protein and TMV movement protein [16]. Protein kinases that belong to the CK2 class have a unique ability to utilize both ATP and GTP as phosphoryl group donors [28]. The soluble fraction of N. benthamiana plant sap that was used for phosphorylation of the N protein was tested for its ability to utilize GTP instead of ATP. As shown in Fig. 5c, the protein kinase present in the fraction could utilize GTP as phoshphoryl group donor equally well (Fig. 5c, lane 4), and even in this case, Mn2+ was the preferred metal ion (Fig. 5c, compare lane 4 with 5 and 6). Based on these data, we speculated that the protein kinase present in tobacco plant sap that phosphorylates the N protein could be a CK2-like kinase. Heparin, an inhibitor of CK2 [14], reduced the phosphorylation of the N protein by 30% but failed to abolish phosphorylation of the protein completely (Supplementary Fig. 3), suggesting that apart from CK2, there may be other kinases present in the tobacco plant sap that also phosphorylate the protein. To test whether the N protein acts as a substrate for more than one kinase, the phosphorylation reaction was carried out with purified recombinant kinases, viz., tobacco CK2 or chickpea CDPK. It was observed that the N protein could be phosphorylated by both of these kinases. As shown in Fig. 5d, the N protein could be phosphorylated by purified CK2 (Fig. 5d, lane 3), whereas BSA used as negative control was not phosphorylated (Fig. 5d, lane 5), and histone H1 (positive control) was phosphorylated (Fig. 5d, lane 6). In addition to the phosphorylated histone H1 protein (35 kDa), two more bands were observed, suggesting the presence of contaminant proteins that are also phosphorylated. When a similar experiment was performed with purified CDPK, the N protein as well as the histone H1 protein were phosphorylated (Fig. 5e, lanes 3 and 5, respectively) and BSA used as a negative control was not phosphorylated (Fig. 5e, lane 6). It has been demonstrated that CDPKs are components of the defence pathways that are induced upon pathogen attack [22]. It is possible that phosphorylation of the N protein by CDPK is an early response to viral infection. Furthermore, the N protein can be phosphorylated by more than one kinase. Phosphorylation of a viral protein by multiple kinases is not uncommon. Surjit et al. [41] reported that the nucleocapsid protein of SARS corona virus acts as a substrate for cyclin-dependent kinase, glycogen synthase kinase, mitogen-activated kinase and casein kinase 2. Human influenza virus NS1 protein gets phosphorylated by members of the cyclin-dependent kinase and extracellular signal-regulated kinase families [13].
Protein phosphorylation and dephosphorylation is a significant regulatory mechanism in a wide range of cellular processes (reviewed in ref. [6]). In some viruses, like rubella virus and potato virus A, phosphorylation of the capsid or coat protein is known to regulate its RNA-binding activity and has an impact on virus replication and the infection process [16, 17, 21]. Phosphorylation of SARS coronavirus N protein modulates its multimerization, translation inhibitory activity and subcellular localization [31, 41]. In infectious bronchitis virus, phosphorylation of the N protein is involved in distinguishing viral RNA from non viral RNAs [40]. In yet another case, in measles virus, phosphorylation of the nucleoprotein is important for activation of transcription of viral mRNA and/or replication of the genome in vivo [12]. It is possible that phosphorylation of the GBNV N protein regulates its self-association and helps it to distinguish viral RNA from non-viral RNAs, which is an essential process in encapsidation. Phosphorylation may also regulate the subcellular localization of the protein and may act as a regulatory switch between the processes of transcription and translation.
The results presented in this paper clearly demonstrate that the GBNV N protein, like the N proteins of other negative-strand RNA viruses, has an intrinsic ability to bind to RNA. Such an association with RNA would be essential for encapsidation of viral genomes. Furthermore, the N protein interacts with the RNA via a conserved RNA-binding domain present in the C-terminal unfolded region of the protein. Such specific interactions and the regulation of the N protein by phosphorylation would be necessary for eliciting its multiple functions at different stages of the viral life cycle, such as replication, assembly and movement. Future studies in this direction could lead to a better understanding of the role of the N protein in the life cycle of GBNV at the molecular level.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgments
We thank the Department of Biotechnology (DBT) and Department of Science and Technology, New Delhi, India for financial support. ASB acknowledges DBT for a postdoctoral fellowship. We are grateful to Prof. Kristiina Makinen, University of Helsinki, for providing the tobacco CK2 clone. We thank Mr. Ajay and Prof. C. Jayabaskaran, IISc, for a gift of purified recombinant chickpea CDPK.
Abbreviations
- GBNV
Groundnut bud necrosis virus
- TSWV
Tomato spotted wilt virus
- RNP
Ribonucleoprotein
- N protein
Nucleocapsid protein
- CK2
Protein kinase CK2
- CDPK
Calcium-dependent protein kinase
References
- 1.Adkins S, Quadt R, Choi TJ, Ahlquist P, German T. An RNA-dependent RNA polymerase activity associated with virions of tomato spotted wilt virus, a plant- and insect-infecting bunyavirus. Virology. 1995;207:308–311. doi: 10.1006/viro.1995.1083. [DOI] [PubMed] [Google Scholar]
- 2.Albertini AA, Wernimont AK, Muziol T, Ravelli RB, Clapier CR, Schoehn G, Weissenhorn W, Ruigrok RW. Crystal structure of the rabies virus nucleoprotein-RNA complex. Science. 2006;313:360–363. doi: 10.1126/science.1125280. [DOI] [PubMed] [Google Scholar]
- 3.Blakqori G, Kochs G, Haller O, Weber F. Functional L polymerase of La Crosse virus allows in vivo reconstitution of recombinant nucleocapsids. J Gen Virol. 2003;84:1207–1214. doi: 10.1099/vir.0.18876-0. [DOI] [PubMed] [Google Scholar]
- 4.Chang CK, Hsu YL, Chang YH, Chao FA, Wu MC, Huang YS, Hu CK, Huang TH. Multiple nucleic acid binding sites and intrinsic disorder of severe acute respiratory syndrome coronavirus nucleocapsid protein: implications for ribonucleocapsid protein packaging. J Virol. 2009;83:2255–2264. doi: 10.1128/JVI.02001-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chomczynski P, Sacchi N. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal Biochem. 1987;162:156–159. doi: 10.1016/0003-2697(87)90021-2. [DOI] [PubMed] [Google Scholar]
- 6.Cohen P. The regulation of protein function by multisite phosphorylation—a 25 year update. Trends Biochem Sci. 2000;25:596–601. doi: 10.1016/S0968-0004(00)01712-6. [DOI] [PubMed] [Google Scholar]
- 7.Elton D, Medcalf L, Bishop K, Harrison D, Digard P. Identification of amino acid residues of influenza virus nucleoprotein essential for RNA binding. J Virol. 1999;73:7357–7367. doi: 10.1128/jvi.73.9.7357-7367.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fauquet CM, Mayo MA, Maniloff J, Desselberges U, Ball LA. Virus taxonomy eighth report of the international committee on taxonomy of viruses. San Diego: Academic Press; 2005. [Google Scholar]
- 9.Gowda S, Satyanarayana T, Naidu RA, Mushegian A, Dawson WO, Reddy DVR. Characterization of the large (L) RNA of peanut bud necrosis tospovirus. Arch Virol. 1998;143:2381–2390. doi: 10.1007/s007050050468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Green TJ, Zhang X, Wertz GW, Luo M. Structure of the vesicular stomatitis virus nucleoprotein-RNA complex. Science. 2006;313:357–360. doi: 10.1126/science.1126953. [DOI] [PubMed] [Google Scholar]
- 11.Greenfield NJ. Using circular dichroism spectra to estimate protein secondary structure. Nat Protoc. 2006;1:2876–2890. doi: 10.1038/nprot.2006.202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hagiwara K, Sato H, Inoue Y, Watanabe A, Yoneda M, Ikeda F, Fujita K, Fukuda H, Takamura C, Kozuka-Hata H, Oyama M, Sugano S, Ohmi S, Kai C. Phosphorylation of measles virus nucleoprotein upregulates the transcriptional activity of minigenomic RNA. Proteomics. 2008;8:1871–1879. doi: 10.1002/pmic.200701051. [DOI] [PubMed] [Google Scholar]
- 13.Hale BG, Knebel A, Botting CH, Galloway CS, Precious BL, Jackson D, Elliott RM, Randall RE. CDK/ERK-mediated phosphorylation of the human influenza A virus NS1 protein at threonine-215. Virology. 2009;383:6–11. doi: 10.1016/j.virol.2008.10.002. [DOI] [PubMed] [Google Scholar]
- 14.Hathaway GM, Lubben TH, Traugh JA. Inhibition of casein kinase II by heparin. J Biol Chem. 1980;255:8038–8041. [PubMed] [Google Scholar]
- 15.Hemalatha V, Gangatirkar P, Karande AA, Krishnareddy M, Savithri HS. Monoclonal antibodies to the recombinant nucleocapsid protein of a groundnut bud necrosis virus infecting tomato in Karnataka and their use in profiling the epitopes of Indian tospovirus isolates. Current science. 2008;95:952–957. [Google Scholar]
- 16.Ivanov KI, Puustinen P, Merits A, Saarma M, Makinen K. Phosphorylation down-regulates the RNA binding function of the coat protein of potato virus A. J Biol Chem. 2001;276:13530–13540. doi: 10.1074/jbc.M009551200. [DOI] [PubMed] [Google Scholar]
- 17.Ivanov KI, Puustinen P, Gabrenaite R, Vihinen H, Ronnstrand L, Valmu L, Kalkkinen N, Makinen K. Phosphorylation of the potyvirus capsid protein by protein kinase CK2 and its relevance for virus infection. Plant Cell. 2003;15:2124–2139. doi: 10.1105/tpc.012567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kaukinen P, Vaheri A, Plyusnin A. Hantavirus nucleocapsid protein: a multifunctional molecule with both housekeeping and ambassadorial duties. Arch Virol. 2005;150:1693–1713. doi: 10.1007/s00705-005-0555-4. [DOI] [PubMed] [Google Scholar]
- 19.Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 20.Lakowicz JR, Eftink MR. Intrinsic fluorescence of proteins. In: Geddes CD, Lakowicz JR, editors. Topics in fluorescence spectroscopy. US: Springer; 2002. pp. 1–15. [Google Scholar]
- 21.Law LM, Everitt JC, Beatch MD, Holmes CF, Hobman TC. Phosphorylation of rubella virus capsid regulates its RNA binding activity and virus replication. J Virol. 2003;77:1764–1771. doi: 10.1128/JVI.77.3.1764-1771.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lee J, Rudd JJ. Calcium-dependent protein kinases: versatile plant signalling components necessary for pathogen defence. Trends Plant Sci. 2002;7:97–98. doi: 10.1016/S1360-1385(02)02229-X. [DOI] [PubMed] [Google Scholar]
- 23.Lokesh B, Rashmi PR, Amruta BS, Srisathiyanarayanan D, Murthy MR, Savithri HS. NSs encoded by groundnut bud necrosis virus is a bifunctional enzyme. PLoS One. 2010;5:e9757. doi: 10.1371/journal.pone.0009757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Longhi S. Nucleocapsid structure and function. Curr Top Microbiol Immunol. 2009;329:103–128. doi: 10.1007/978-3-540-70523-9_6. [DOI] [PubMed] [Google Scholar]
- 25.Mir MA, Panganiban AT. The bunyavirus nucleocapsid protein is an RNA chaperone: possible roles in viral RNA panhandle formation and genome replication. RNA. 2006;12:272–282. doi: 10.1261/rna.2101906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mir MA, Panganiban AT. A protein that replaces the entire cellular eIF4F complex. EMBO J. 2008;27:3129–3139. doi: 10.1038/emboj.2008.228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mohl BP, Barr JN. Investigating the specificity and stoichiometry of RNA binding by the nucleocapsid protein of Bunyamwera virus. RNA. 2009;15:391–399. doi: 10.1261/rna.1367209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Niefind K, Putter M, Guerra B, Issinger OG, Schomburg D. GTP plus water mimic ATP in the active site of protein kinase CK2. Nat Struct Biol. 1999;6:1100–1103. doi: 10.1038/70033. [DOI] [PubMed] [Google Scholar]
- 29.Ontiveros SJ, Li Q, Jonsson CB (2010) Modulation of apoptosis and immune signaling pathways by the Hantaan virus nucleocapsid protein. Virology. doi:10.1016/j.virol.2010.1002.1018 [DOI] [PMC free article] [PubMed]
- 30.Pappu HR, Jones RA, Jain RK. Global status of tospovirus epidemics in diverse cropping systems: successes achieved and challenges ahead. Virus Res. 2009;141:219–236. doi: 10.1016/j.virusres.2009.01.009. [DOI] [PubMed] [Google Scholar]
- 31.Peng TY, Lee KR, Tarn WY. Phosphorylation of the arginine/serine dipeptide-rich motif of the severe acute respiratory syndrome coronavirus nucleocapsid protein modulates its multimerization, translation inhibitory activity and cellular localization. FEBS J. 2008;275:4152–4163. doi: 10.1111/j.1742-4658.2008.06564.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Raymond DD, Piper ME, Gerrard SR, Smith JL. Structure of the Rift Valley fever virus nucleocapsid protein reveals another architecture for RNA encapsidation. Proc Natl Acad Sci USA. 2010;107:11769–11774. doi: 10.1073/pnas.1001760107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ribeiro D, Borst JW, Goldbach R, Kormelink R. Tomato spotted wilt virus nucleocapsid protein interacts with both viral glycoproteins Gn and Gc in planta. Virology. 2009;383:121–130. doi: 10.1016/j.virol.2008.09.028. [DOI] [PubMed] [Google Scholar]
- 34.Richmond KE, Chenault K, Sherwood JL, German TL. Characterization of the nucleic acid binding properties of tomato spotted wilt virus nucleocapsid protein. Virology. 1998;248:6–11. doi: 10.1006/viro.1998.9223. [DOI] [PubMed] [Google Scholar]
- 35.Satyanarayana T, Mitchell SE, Reddy DV, Brown S, Kresovich S, Jarret R, Naidu RA, Demski JW. Peanut bud necrosis tospovirus S RNA: complete nucleotide sequence, genome organization and homology to other tospoviruses. Arch Virol. 1996;141:85–98. doi: 10.1007/BF01718590. [DOI] [PubMed] [Google Scholar]
- 36.Satyanarayana T, Mitchell SE, Reddy DV, Kresovich S, Jarret R, Naidu RA, Gowda S, Demski JW. The complete nucleotide sequence and genome organization of the M RNA segment of peanut bud necrosis tospovirus and comparison with other tospoviruses. J Gen Virol. 1996;77:2347–2352. doi: 10.1099/0022-1317-77-9-2347. [DOI] [PubMed] [Google Scholar]
- 37.Snippe M, Borst JW, Goldbach R, Kormelink R. The use of fluorescence microscopy to visualise homotypic interactions of tomato spotted wilt virus nucleocapsid protein in living cells. J Virol Methods. 2005;125:15–22. doi: 10.1016/j.jviromet.2004.11.028. [DOI] [PubMed] [Google Scholar]
- 38.Snippe M, Goldbach R, Kormelink R. Tomato spotted wilt virus particle assembly and the prospects of fluorescence microscopy to study protein-protein interactions involved. Adv Virus Res. 2005;65:63–120. doi: 10.1016/S0065-3527(05)65003-8. [DOI] [PubMed] [Google Scholar]
- 39.Soellick T, Uhrig JF, Bucher GL, Kellmann JW, Schreier PH. The movement protein NSm of tomato spotted wilt tospovirus (TSWV): RNA binding, interaction with the TSWV N protein, and identification of interacting plant proteins. Proc Natl Acad Sci USA. 2000;97:2373–2378. doi: 10.1073/pnas.030548397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Spencer KA, Dee M, Britton P, Hiscox JA. Role of phosphorylation clusters in the biology of the coronavirus infectious bronchitis virus nucleocapsid protein. Virology. 2008;370:373–381. doi: 10.1016/j.virol.2007.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Surjit M, Lal SK. The SARS-CoV nucleocapsid protein: a protein with multifarious activities. Infect Genet Evol. 2008;8:397–405. doi: 10.1016/j.meegid.2007.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Uhrig JF, Soellick TR, Minke CJ, Philipp C, Kellmann JW, Schreier PH. Homotypic interaction and multimerization of nucleocapsid protein of tomato spotted wilt tospovirus: identification and characterization of two interacting domains. Proc Natl Acad Sci USA. 1999;96:55–60. doi: 10.1073/pnas.96.1.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Vulliemoz D, Roux L. “Rule of six”: how does the Sendai virus RNA polymerase keep count? J Virol. 2001;75:4506–4518. doi: 10.1128/JVI.75.10.4506-4518.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Xu X, Severson W, Villegas N, Schmaljohn CS, Jonsson CB. The RNA binding domain of the hantaan virus N protein maps to a central, conserved region. J Virol. 2002;76:3301–3308. doi: 10.1128/JVI.76.7.3301-3308.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.