

Abstract
Introduction
LPS-responsive beige-like anchor protein (LRBA) deficiency is a primary immunodeficiency categorized as common variable immunodeficiency associated with autoimmune manifestations and inflammatory bowel diseases; however, the clinical spectrum has been extended. Here, we present our cohort of Turkish LRBA-deficient patients from a single center, demonstrating a diversity of clinical manifestations.
Method
Seven affected individuals from five families were assessed retrospectively in this study.
Results
Of the seven patients with LRBA deficiency, four had homozygous, and two had compound heterozygous mutations. One patient remained disease free until the last follow-up (age 17 years). The most common clinical manifestations of the six symptomatic patients were organomegaly (6/6), autoimmunity (6/6), and chronic diarrhea (5/6). Recurrent infectious episodes were observed in three patients. None of the patients had hypogammaglobulinemia at presentation. B cell subpopulation analysis revealed low numbers of switched-memory B cell numbers in two of the four tested patients. During the disease course, three of the patients died, two of them underwent successful hematopoietic stem cell transplantation (HSCT) from matched sibling donors, and one is under abatacept therapy.
Conclusion
LRBA defects should always be kept in mind as a differential diagnosis for patients with autoimmune disease affecting multiple organs, chronic diarrhea, and organomegalies. In our experience, early HSCT is a life-saving therapeutic strategy.
Keywords: LRBA deficiency, autoimmunity, ALPS, HSCT
Introduction
Primary immunodeficiencies (PIDs) are a heterogeneous group of disorders affecting distinct components of the immune system. Following the definition of agammaglobulinemia by Bruton in the 1950s as one of the first prototypic PIDs [1], these diseases have been the subject of intense studies to elucidate etiology and develop effective treatments. Several new diseases and novel mutations have been identified during the last 20 years so that the number of genetically defined PIDs exceeds 300 [2]. Recently defined genetic defects and clinical presentations reveal that PIDs not only present with recurrent infections but also lymphoproliferation, autoimmunity, allergy, and malignancies [3].
LPS-responsive beige-like anchor protein (LRBA) deficiency is a prototypical PID which presents with a broad spectrum of clinical phenotypes. Mutations in LRBA, which encodes LRBA, have been linked to defective vesicle trafficking including transendocytosis and turnover of CTLA-4 in regulatory T cells (Treg) [4]. A decrease in LRBA protein function leads to the diminished expression of CTLA-4 on the surface of Treg cells [4]. Patients who present with defects in CTLA-4 biology further show autoantibody-mediated cytopenias, lymphoproliferation, hypogammaglobulinemia, organ-specific autoimmunity, and lymphocytic infiltration of non-lymphoid organs [5].
Although LRBA deficiency was first categorized as common variable immunodeficiency (CVID), with autoimmune manifestations and inflammatory bowel diseases [6], the clinical spectrum has been extended with the latest reports of phenotypic variations. In particular, patients with phenotypes resembling autoimmune lymphoproliferative syndrome (ALPS), including autoimmune cytopenias, and lymphoproliferation without any hypogammaglobulinemia have been described [7, 8]. In this study, we present our cohort of Turkish LRBA-deficient patients from a single center, illustrating a wide range of clinical manifestations, with different therapeutic approaches depending on the clinical status of the patients.
Method
Patients
Seven individuals from five families affected with LRBA defect, who were under follow-up in Ankara University Department of Immunology, were assessed retrospectively in this study. Informed consent was taken from all parents according to the Declaration of Helsinki. Complete blood counts, immunoglobulin levels, lymphocyte subsets, lymphocyte activation responses, and genetic aberrations were analyzed as well as the clinical data and histopathological findings of biopsy specimens.
For the two patients who underwent hematopoietic stem cell transplantation (HSCT), posttransplant follow-up data regarding the immunophenotyping and the chimerism analysis were evaluated separately.
Genetic Analyses
Genetic analyses of the patients P1 and P4 were defined previously [9, 10]. All other patients were analyzed with NGS-based gene panel screening, which was further confirmed with Sanger sequencing as described [11].
Protein Analysis
Peripheral blood mononuclear cells (PBMCs) were transformed into EBV B cell lines and expanded. B cells were harvested and lysed in Frackleton buffer (10 mM Tris-HCl, pH 7.5; 50 mM NaCl; 30 mM NaPPi; 50 mM NaF, 1% Triton X-100) supplemented with protease inhibitor cocktail (Sigma-Aldrich). For Western blot, 150 μg of the protein was loaded. The following antibodies were used: anti-LRBA (HPA023597, Sigma-Aldrich) and anti-CAD (11933S, Cell Signaling Technologies). Spectra™ Multicolor High Range Protein Ladder (Thermo Fisher) was used as a size marker.
Immunofluorescence
PBMCs of patients and healthy donors (including shipment controls) were stimulated with antibodies against CD3 (clone OKT3) and CD28 (clone CD28.2, both Thermo Fisher Scientific) for 48 h. Cells were seeded on poly-L lysine-coated coverslips and subsequently fixed in 4% PFA and permeabilized in 0.2% Triton X-100. Antibodies against LRBA (HPA023597, Sigma-Aldrich) and CTLA-4 (550405, BD Biosciences) were used. Secondary staining was performed with goat-anti-mouse-AF488 (A-11029) and goat-anti-rabbit-AF647 (A-21244, both Life Technologies). The nucleus was counterstained with DAPI. Slides were analyzed on a LSM 700 with a 63×/1.4 Plan-Apochromat oil objective. Five different areas on each coverslip were screened for DAPI, LRBA, and CTLA-4 expressing cells, and three representative images were taken.
Results
Patients
Patient 1
Patient 1 was a female patient who first presented symptoms at the age of 6 months. Her clinical course has been described previously [10]. A missense mutation in LRBA gene (c.A8470C; c.T8471C; p.Ile2824Pr) was identified, which did not result in protein loss. The patient was diagnosed as possible LRBA deficiency, and further functional analysis was planned for definitive LRBA deficiency; however, due to her severe phenotype, she died at the age of 15 years.
Patients 2 and 3
Patient 2 was a 12-year-old girl born to non-consanguineous parents. She presented with recurrent respiratory infections and chronic diarrhea starting at 1 year of age. When she was 6 years old, she developed immune thrombocytopenic purpura (ITP) and autoimmune hemolytic anemia (AIHA) episodes, which were resistant to immunosuppressive therapies. During follow-up, lymphadenopathy and hepatosplenomegaly developed. Her immune work-up revealed lymphopenia and elevated numbers of double negative T cells. She was diagnosed with autoimmune lymphoproliferative syndrome, and intravenous immunoglobulin (IVIG) replacement therapy was initiated together with antibiotic prophylaxis. However, she died as a result of a severe pneumonia episode at the age of 14. In her family history, two deceased siblings had presented a similar clinical phenotype. A newborn brother (patient 3) presented to our clinic with neonatal diabetes. His immune work-up was normal apart from a slight reduction of immunoglobulin G levels. During toddler age, he had an uneventful follow-up apart from two episodes of viral pneumonia due to respiratory syncytial virus (RSV) and adenovirus. However, at the age of 3 years, he developed ITP which was resistant to steroids, IVIG, and plasma exchange therapies, followed by Epstein Barr virus (EBV)-induced lymphoproliferation and splenomegaly. Histopathological evaluation of lymph node biopsy showed reactive lymphocytosis. When he was 4 years old, non-infectious chronic secretory diarrhea started. Colonoscopic biopsies revealed inflammatory bowel disease (IBD)-like mucosal inflammation, which prompted the initiation of total parenteral nutrition (TPN). He had severe nephrotic range proteinuria, for which he underwent a renal biopsy. Lymphocytic infiltration of the renal cortex was observed. During the disease course, lymphocytic interstitial lung disease (ILD) developed. Genetic analysis revealed compound heterozygous mutations affecting LRBA (p.Asp1053fs, p.Ser2659*). Initiation of abatacept therapy (20 mg/kg every 2 weeks) alleviated the lymphoproliferation, but the diarrhea persisted and he died due to persistent thrombocytopenia and subsequent intracranial hemorrhage.
Patient 4
Patient 4 is a previously described female patient. She was successfully transplanted from a matched sibling donor with unknown mutation status following a conditioning regimen with busulfan (7.5 mg/kg), fludarabine (180 mg/m2), and ATG (40 mg/kg) [9].
Patients 5 and 6
Patient 5, a currently 6-year-old male patient born to consanguineous parents, presented at the age of 3 years with therapy-resistant ITP, lymphoproliferation, and severe chronic diarrhea. Complete blood count was normal and immunoglobulins were within reference ranges for age. Furthermore, immune cell subclass analysis including B cell subsets and lymphocyte activation responses were normal. Genetic investigations revealed a stopgain mutation (c.675G > A, p.Trp225*) in LRBA. HSCT was performed from a matched sibling donor, who was heterozygous for the mutation. Non-myeloablative conditioning with busulfan (7.5 mg/kg), fludarabine (180 mg/m2), and anti-thymocyte globulin (ATG) (40 mg/kg) was administered. A total of 7.16 × 106/kg CD34+ stem cells were infused. Neutrophil and thrombocyte engraftment occurred on days 17 and 33, respectively. On day 90 posttransplantation, a CMV infection resulted in an episode of therapy-resistant ITP, which led to chronic hemorrhage due to a complication of catheter insertion. Weekly administration of romiplostim increased thrombocyte counts within 4 weeks. On posttransplant day 270, patient 5 had an episode of autoimmune hemolytic anemia, which was resistant to IVIG replacement therapy. Methylprednisolone (8 mg/kg) was introduced, and sirolimus was initiated due to a second attack of autoimmunity. After hemolysis was under control, steroid doses were rapidly tapered down. He is currently under sirolimus therapy on posttransplant of 1 year, with mixed-donor chimerism. LRBA protein expression was tested 9 months after transplantation and found to be reduced if compared with a healthy control. However, the expression is similar to the LRBA expression of his heterozygous matched sibling donor (Fig. 1a).
Fig. 1.
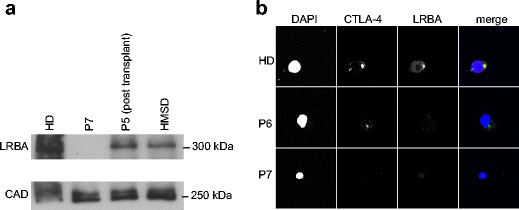
Molecular analysis of patient cells. a Western blot of lysates derived from EBV-transformed B cell lines testing for LRBA expression (size 319 kDa) reveals the absence of protein expression in P7 and a reduction of protein expression in P5 after transplantation which corresponds to the signal of the HMSD (heterozygous matched sibling donor). CAD (size 243 kDa) serves as a loading control. b Immunofluorescence analysis of stimulated PBMCs derived from P6 and P7 confirms the absence of LRBA. Note that CTLA-4 levels were also altered in this assay
Patient 6 is the elder sister of patient 5. During the diagnostic work-up of her brother, her DNA samples were also studied. The same stop codon mutation was detected, although she was completely asymptomatic. Her general immune work-up was normal, with slightly low switched-memory B cells. She was negative for the autoantibodies ANA, ASMA, ANCA, anti-TPO, anti-thyroglobulin, and tissue transglutaminase antibody, as well as in direct Coombs test. Abdominal ultrasound was normal and no deep lymphadenopathy was detected. Vaccination responses were normal. Despite these findings, LRBA expression in PBMCs as assessed by immunofluorescence staining was absent (Fig. 1b) which is in concordance with the identified mutation. Patient 6 is currently under follow-up.
Patient 7
Patient 7 is a 12-year-old male, who presented with autoimmune hemolytic anemia, ITP, hepatosplenomegaly, unexplained urticaria, and angioedema episodes when he was 8 years old. His past medical history was unremarkable for recurrent infections. Initial laboratory investigations demonstrated pancytopenia. Serum immunoglobulins and peripheral blood lymphocyte subsets were within normal ranges. Double-negative T cells were 7.5% of the total lymphocytes. The patient was initially diagnosed with autoimmune lymphoproliferative syndrome. He received steroid and mycophenolate mofetil (MMF) as immunosuppressive treatment. During the follow-up, serum immunoglobulins decreased gradually and IVIG replacement was started. Splenectomy was performed due to steroid-dependent pancytopenia and hypersplenism. Cytopenias alleviated following splenectomy and normal neutrophil and thrombocyte counts were achieved. However, 6 months later, he further developed tachypnea and left axillary lymphadenopathy. A CT scan revealed nodular opacities and lymphocytic interstitial lung disease. The lymph node biopsy showed reactive lymphocytosis. Genetic investigations identified a compound heterozygous mutation in LRBA (c.3028G > A, p.Q1010*; c.7976G > C p.S2659*), which resulted in absent protein expression (Fig. 1a, b). Abatacept was introduced (20 mg/kg every 2 weeks) and the pulmonary lesions resolved markedly (Fig. 2). Matched unrelated donor survey was activated for a planned allogeneic HSCT.
Fig. 2.
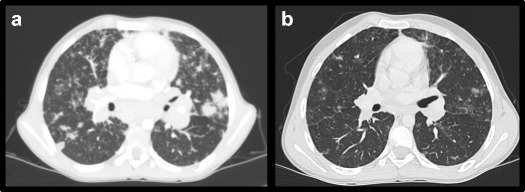
Response of lymphocytic infiltration in lungs to abatacept therapy comparing conditions a before and b after abatacept treatment in P7
Demographic Characteristics
The here-described patients with LRBA mutations have either homozygous or compound heterozygous mutations. The cohort group consists of five different families, three of which were consanguineous. Three of the seven patients were deceased at the time of analysis; two of the living patients underwent HSCT. Average age of disease onset was 2.3 (± 2.9 SD) years. Interestingly, our cohort includes a 17-year-old asymptomatic LRBA mutant patient.
Clinical Phenotype
The most common clinical manifestations of the six symptomatic patients were organomegaly (6/6), autoimmunity (6/6), and chronic diarrhea (5/6) (Table 1). Both splenomegaly and hepatomegaly could be observed in our cohort. Five patients presented organomegalies together with lymphadenopathies. The lymph node biopsy of three patients showed reactive lymphocytosis; no progress to lymphoma was seen in our cohort. Autoimmunity mainly presented as immune-mediated cytopenia. Five patients had recurrent ITP episodes for which IVIG or steroids were used (Table 1). One patient (P3) had resistant ITP, which was occasionally responding to plasma exchange therapy; he died of intracranial hemorrhage during an ITP episode under abatacept therapy. AIHA was observed in three patients. One patient (P7) was splenectomized to control therapy-resistant cytopenias, which was successful in terms of recovery. Other than cytopenias, two of the presented patients had early-onset diabetes, one patient had autoimmune thyroiditis, and one patient had chronic urticaria as autoimmune manifestations.
Table 1.
Clinical features of LRBA patients
| P1 | P2 | P3 | P4 | P5 | P6 | P7 | |
|---|---|---|---|---|---|---|---|
| Age of genetic diagnosis (year) | 15 | ND | 6 | 13 | 6 | 16 | 12 |
| Age at initial clinical admission (year) | 13 | 12 | 3 months | 11 | 5 | 15 | 12 |
| Gender | F | F | M | F | M | F | M |
| Consanguinity | + | – | – | + | + | + | – |
| Age onset (year) | 1 | 1 | Neonate | 1 | 3 | Asymptomatic | 8 |
| Duration of follow-up (year) | 2 | 4 | 5 | 5 | 2 | 2 | 3 |
| Final outcome | Deceased at age 16 | Deceased at age 14 | Deceased at age 4 | Alive | Alive | Alive | Alive |
| Clinical findings |
Enteropathy Anemia and thrombocytopenia Organomegaly Autoimmune thyroiditis Type I DM Demyelinated PNP Anal fissure/skin tags Intestinal pseudoobstruction |
Enteropathy Lymphadenopathy ITP AIHA Recurrent infections |
Type 1 DM Enteropathy Lymphadenopathy ITP Organomegaly ILD Recurrent infections (brother of P2) |
Enteropathy AIHA Atopic dermatitis Lymphadenopathy Growth failure Bronchiectasis Tubulopathy |
Enteropathy ITP Organomegaly Lymphadenopathy Recurrent infections |
Asymptomatic (sister of P5) |
Pancytopenia AIHA Lymphadenopathy Organomegaly ITP Chronic urticaria LIP |
| Infectious agents |
Acinetobacter Enterococcus Serratia marcescens Candida krusei |
ND |
Coronavirus OC43 Adenovirus EBV RSV Staphylococcus aureus Candida glabrata |
Coronavirus RSV Adenovirus |
Coronavirus CMV |
No documented infectious agent | CMV |
| Mutation | c.A8470C; c.T8471C; p.Ile2824Pr | ND |
c.3196del, c.7976G > C pAsp1053fs, p.Ser2659* (compound heterozygous) |
c.5505delT (p.Ile1836*) | c.675G > A p.Trp225* | c.675G > A p.Trp225* |
c.3028G > A; p.Q1010* c.7976G > C; p.S2659* compound heterozygous |
| Immunosuppressive agents |
Cyclosporine A Methylprednisolone |
Mycophenolate mofetil | None | Methylprednisolone | Mycophenolate mofetil | None |
Methyprednisolone Mycophenolate mofetil |
| IVIG replacement | – | + | + | + | + | – | + |
| HSCT | MSD | MSD (heterozygous for LRBA mutation) | Under donor survey for MUD HSCT | ||||
| Conditioning regimen |
Fludarabine Busulfan ATG |
Fludarabine Busulfan ATG |
|||||
| CD34 infusion | 8 × 106/kg | 7.2 × 106/kg | |||||
| Outcome | Died of sepsis | Died of pneumonia |
5 doses of abatacept Died of intracranial hemorrhage while planning MUD HSCT |
Uneventful at posttransplant 30 months |
-ITP secondary to CMV on posttransplant day 90, controlled by romiplostim -AIHA on posttransplant day 270 responsive to steroids, after which sirolimus is initiated -Uneventful in posttransplant 1 year under sirolimus and romiplostim |
Under follow-up | Abatacept for ILD |
Diarrhea was protracted and resistant to immunosuppressive agents such as steroids, MMF, and cyclosporine A (CsA). No specific infectious pathogens could be isolated. Therapy-resistant diarrhea caused malnutrition and growth failure in three patients and led us to administer TPN for caloric compensation. The colonoscopic examination of the patients with diarrhea revealed IBD-like mucosal pattern. Intraepithelial lymphocytosis similar to celiac disease was observed in the duodenal biopsies.
Infectious Profile
All of the patients had at least one documented infectious episode during the follow-up. Recurrent respiratory tract infections (> 8 times/year) resulting in hospitalization were present in three patients at initial presentation. Viral pathogens were the most common infectious agents, out of which coronavirus was the most frequent. Further viruses included CMV, RSV, and adenovirus, which were detected in patients regardless of the therapeutic scheme. Severe bacterial infections caused by Acinetobacter baumanii and Staphylococcus aureus were documented in two patients who were under immunosuppressive therapy with methylprednisolone and cyclosporine A (Table 1). Candida glabrata and Candida krusei were also isolated from those two patients. No parasitic infection was documented.
Immunologic Phenotype
In addition to the above-mentioned autoimmune-mediated cytopenia, ITP, and AIHA, two patients were lymphopenic on initial admission. Neutropenia was not observed in our patients.
Despite normal T cell counts in all patients, two of them presented reduced activation responses to phytohemagglutinin and anti-CD3 stimulation (Table 2). However, both patients were under chronic immunosuppressive therapy when the analysis was performed. Double-negative T cells (DNT) were elevated in four of our patients (Table 2) who were previously diagnosed with ALPS.
Table 2.
Immunological parameters of LRBA patients
| P1 | P2 | P3 | P4 | P5 | P6 | P7 | |
|---|---|---|---|---|---|---|---|
| Hb (g/dL) | 7.1 | 14.6 | 9.6 | 11.6 | 14.7 | 15.4 | 14.1 |
| Total lymphocyte count (/mm3) | 1000 | 846 | 5000 | 5400 | 2600 | 5390 | 3820 |
| Total neutrophil count (/mm3) | 3600 | 6100 | 2700 | 1900 | 11,500 | 3220 | 5000 |
| Platelet count (/mm3) | 91,000 | 232,000 | 576,000 | 164,000 | 238,000 | 165,000 | 348,000 |
| IgA (mg/dL) |
310 67–433 |
ND |
59 7–123 |
327 67–433 |
64 70–303 |
107 100–447 |
114 70–303 |
| IgG (mg/dL) |
1540 835–2094 |
ND |
785 304–1231 |
1140 835–2094 |
928 764–2124 |
958 913–1184 |
1260 764–2124 |
| IgM (mg/dL) |
277 47–484 |
ND |
133 32–203 |
46 47–484 |
171 69–387 |
172 88–322 |
106 69–387 |
| IgE (kU/mL) | 15.2 | ND | < 0.2 | 3.8 | 39.4 | 3.86 | 5 |
| CD3 + CD16–56- [%(/mm3)] |
85 (850) 55–79 (1000–2200) |
84 (711) 55–79 (1000–2200) |
66 (3300) 55–79 (1900–5900) |
67 (3618) 55–79 (1000–2200) |
72 (1872) 55–79 (1400–3700) |
69 (3719) 55–79 (1000–2200) |
72 (2750) 55–79 (1400–3700) |
| CD3-CD16 + 56+ [%(/mm3)] |
12 (120) 5–28 (70–480) |
13 (110) 5–28 (70–480) |
8 (400) 5–28 (160–950) |
18 (972) 5–28 (70–480) |
3 (78) 5–28 (130–720) |
13 (701) 5–28 (70–480) |
4 (153) 5–28 (130–720) |
| CD3 + CD4+ [%(/mm3)] |
64 (640) 26–49 (530–1300) |
21 (178) 26–49 (530–1300) |
30 (1500) 26–49 (1300–4300) |
43 (2322) 26–49 (530–1300) |
36 (988) 26–49 (700–2200) |
40 (2156) 26–49 (530–1300) |
42 (1604) 26–49 (700–2200) |
| CD3 + CD8 + [%(/mm3)] |
20 (200) 9–35 (60–310) |
60 (508) 9–35 (60–310) |
30 (1500) 9–35 (620–2000) |
24 (1296) 9–35 (60–310) |
30 (780) 9–35 (490–1300) |
27 (1455) 9–35 (60–310) |
26 (993) 9–35 (490–1300) |
| CD19+ [%(/mm3)] |
4 (40) 11–31 (110–570) |
12 (101) 11–31 (110–570) |
22 (1100) 11–31 (720–2600) |
13 (702) 11–31 (110–570) |
25 (650) 11–31 (390–1400) |
14 (754) 11–31 (110–570) |
16 (611) 11–31 (390–1400) |
| CD20 + [%(/mm3)] |
4 (40) 11–29 (110–570) |
13 (111) 11–29 (110–570) |
20 (1000) 11–29 (720–2600) |
13 (702) 11–29 (110–570) |
24 (624) 11–29 (390–1400) |
14 (754) 11–29 (110–570) |
16 (611) 11–29 (390–1400) |
| CD4 + CD45RO+ [%(/mm3)] |
43 (430) 8–42 (240–700) |
20 (1000) 8–42 (240–700) |
11 (594) 8–42 (240–700) |
2 (52) 8–42 (220–660) |
26 (1401) 8–42 (240–700) |
31 (1184) 8–42 (220–660) |
|
| CD4 + CD45RA+ [%(/mm3)] |
32 (320) 20–41 (230–770) |
13 (650) 20–41 (430–1500) |
3 (162) 20–41 (230–770) |
8 (208) 20–41 (430–1500) |
20 (1078) 20–41 (230–770) |
13 (497) 20–41 (430–1500) |
|
| CD4 + CD45RA+CD31+ (Trec) (%) | 24 | 21 | 18 | 19 | 40 | 11 | |
| DNT | 0.6 | > 2.5 | 3.8 | 1 | 3.5 | 1.98 | 5.3 |
| Lymphocyte activation (PHA) | |||||||
| CD3 + CD25+ (%) | 78 | 24 | 80 | 83 | 89 | 91 | 8 |
| CD3 + CD69+ (%) | 76 | 20 | 82 | 41 | 88 | 93 | 16 |
| Lymphocyte activation (anti-CD3) | |||||||
| CD4 + CD25+ (%) | 57 | 20 | 38 | 49 | 50 | 81 | 9 |
| CD4 + CD69+ (%) | 51 | 72 | 41 | 19 | 52 | 73 | 12 |
| B cell subgroups | |||||||
| CD19 + IgM-27 + IgD+ (switched memory) (%) | ND | ND | ND | 12 | 6.7 | 2.2 | 0.7 |
| CD19 + IgM + 27 + IgD+ (marginal zone) (%) | 9.8 | 26 | 7.4 | 2.4 | |||
| CD19+ IgM + 27- IgD+ (naive B) (%) | 86.3 | 70 | 84.3 | 90.5 | |||
| CD19 + CD38highCD21low (activated) (%) | 2.6 | 12.2 | 0.64 | 64.9 | |||
| CD19 + CD39highIgMhigh (transitional B) (%) | 6.1 | 0 | 0.4 | 3.3 | |||
Total B cell numbers were only altered in one patient. However, this patient was under immunosuppresive treatment at the time of analysis (Table 2). B cell subpopulation analysis revealed low numbers of switched-memory B cell numbers in two of the four tested patients. None of our patients had hypogammaglobulinemia on admission; yet, one patient developed it during follow-up combined with low switched-memory B cells.
Genetic and Functional Evaluation
Homozygous mutations were identified in four patients; compound heterozygous mutations in three. Most of the here-described mutations segregated perfectly with the disease as parents of compound heterozygous patients carried only one of the two variants, and parents from homozygous patients were heterozygous for the mutation. One exception was the asymptomatic patient 6, who did not show any signs of immune dysregulation but was a carrier of a homozygous nonsense mutation. Some of the here-described mutations have not been described before (Fig. 3). Novel as well as known mutations are distributed along the whole protein lengths. No specific genotype-phenotype correlation was observed.
Fig. 3.
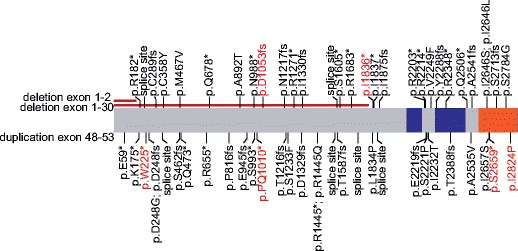
Locations of identified (red) and published (black) missense, non-sense, splice-site, or frameshift mutations affect the whole protein length. No specific cluster can be observed (blue, BEACH domain; range, WD40 domain)
Protein expression analysis was performed on cells derived from patient 7 which revealed a complete absence of LRBA, as well as on material derived from patient 5 after transplantation (Fig. 1a). Patient 5 was transplanted by a heterozygous matched sibling donor (HMSD). Both individuals show a reduced LRBA expression in comparison to healthy controls (Fig. 1a). The absence of LRBA protein in patient 7 was further confirmed by immunofluorescence analysis of patient-derived peripheral blood mononuclear cells which had been stimulated with antibodies against CD3 and CD28 (Fig. 1b). In concordance with the genetic data, analysis of the asymptomatic sibling (P6) also revealed absent LRBA (Fig. 1b).
Therapeutic Approach and Outcome
All of the patients presented with severe immune dysregulations; three of the patients died during the disease course. Two of the patients underwent HSCT from matched sibling donors. For both of them, reduced-intensity conditioning regime consisting of fludarabin (180 mg/m2), ATG (40 mg/kg), and busulfan (7.5 mg/kg) was used (Table 1). As previously published, patient P4 had an uneventful posttransplant follow-up [9]. Currently, 30 months after HSCT, she has no diarrhea and autoimmunity episodes. Patient 5 had also an uneventful period initially, but he encountered ITP and AIHA episodes following HSCT. Currently, 1 year after HSCT, he is under sirolimus and romiplostim with mixed-donor chimerism. Patient 7 is also planned for HSCT, but the survey for matched unrelated donor still continues.
Although in the presented patient cohort the only curative treatment was transplantation, patients received multiple immunosuppressive treatments. All of our patients received chronic or intermittent steroids, to which the symptoms were unresponsive. As steroid-sparing agents, previously MMF was used in two patients and CsA was used in one patient. We used abatacept in two patients with a protocol of 20 mg/kg every 2 weeks. Patient 3 had five doses of abatacept treatment, during which ITP persisted with a fatal outcome, but the lymphoproliferation alleviated. Patient 7 is currently on abatacept therapy for lymphocytic interstitial pneumonia. Lymphocytic infiltration in the lungs resolved after the third dose (Fig. 2).
Discussion
In the current study, we present a single-center experience of LRBA deficiency with seven affected individuals from five families.
As of today, more than 50 patients with LRBA deficiency have been reported, and the numbers keep increasing [12–15]. Although, in 2012 the disease was classified as a CVID-like disorder, recent reports of the extended disease phenotype revealed that the prominent clinical features in LRBA deficiency is due to immune dysregulation presenting mostly as enteropathy, autoimmune hemolytic anemia, and ITP [12, 13, 16]. In concordance, organomegaly, enteropathy, and autoimmune cytopenias were the most common features in our cohort. Most of our patients presented with an ALPS-like phenotype (organomegalies, lymphoproliferation, elevated DNTs) and were under follow-up with ALPS diagnosis before genetic analysis. Previously, other LRBA-deficient patients have been reported who suffer from an ALPS-like phenotype and it was recommended that LRBA defect should be considered in patients who are negative for mutations in the ALPS genes such as FAS, FASL, and CASP10 [7].
The presented patient cohort is the first analysis of a group of Turkish patients with LRBA mutations. Previously, two reports from separate groups, although they included overlapping cases, reviewed phenotypic manifestations of patients bearing LRBA mutations [12, 13]. Major phenotypes were grouped into the categories autoimmunity, enteropathy, and CVID-like immune deficiency [12]. Although the patients present characteristics of autoimmunity and enteropathy phenotypes, CVID-like immune deficiency was not frequent in our cohort (Table 3). In the studied patient group, hypogammaglobulinemia was not a prominent disease feature; ALPS-like disease, with or without diarrhea, was the most common initial presentation. We could not illustrate a specific genotype-phenotype relationship for this phenomenon observed in our cohort.
Table 3.
Main clinical and laboratory characteristics of LRBA deficiency
| Current study (7 patients) n (%) | Alkhairy et al.a (31 patients) n (%) | Gamez-Diaz et al.a (22 patients) n (%) | |
|---|---|---|---|
| Autoimmunity | 6 (86%) | 19 (61%) | 20 (95%) |
| ITP | 5 (71%) | 9 (29%) | 11 (50%) |
| AIHA | 3 (43%) | 12 (39%) | 12 (57%) |
| Diabetes | 2 (28%) | 2 (6%) | 5 (24%) |
| Thyroiditis | 1 (14%) | 3 (10%) | – |
| Organomegaly | 6 (86%) | 19 (61%) | 18 (86%) |
| Chronic diarrhea | 5 (71%) | 19 (61%) | 13 (59%) |
| Recurrent infections | 3 (43%) | 19 (61%) | 15 (71%) |
| Parenchymal lung abnormalities | 3 (43%) | 12 (39%) | 11 (50%) |
| Failure to thrive | 3 (43%) | 13 (42%) | 5 (24%) |
| Hypogammaglobulinemia | 1 (14%) | 18 (58%) | 12 (57%) |
| Low B cell counts | – | 14 (45%) | 11/20 (55%) |
| Low switched-memory B cells | 2/4 (50%) | 13/14 (93%) | 12/14 (85%) |
| Elevated DNT | 4/7 (57%) | NA | 6/16 (37%) |
aPartially overlapping cases are present
Chronic diarrhea was the most troublesome disease symptom for the presented patients, causing prolonged hospitalization and TPN administration for caloric compensation. Diarrhea usually was not the initial symptom but developed during the disease course. No pathogens could be identified as an underlying cause. Furthermore, diarrhea was unresponsive to standard immunosuppressive therapies such as steroids and MMF. Colonoscopic evaluation revealed IBD-like mucosal inflammation. Intraepithelial lymphocyte infiltration of the duodenum in a celiac-like pattern was a common histopathological finding; however, none of the patients responded to a gluten-free diet in concordance with previously published data [9, 17]. In the described patient cohort, diarrhea could only be controlled by HSCT.
We recorded a severe disease course with an average age onset of 2.3 years, which was slightly younger than reported in previous studies [13]. However, one of the patients, who is negative for LRBA protein expressions (Fig. 1b) and carries the same stop codon mutation as her diseased brother, is asymptomatic at the age of 17 years. Currently, her immune work-up is normal, apart from a slightly reduced number of switched-memory B cells. She has no autoimmune diseases documented. Asymptomatic patients have been described previously [12, 13]. However, disease onset varies between patients. The latest age of onset so far is 17 years [13]. This asymptomatic individual of our cohort is currently under close follow-up without any treatment.
The disease course of the patients was severe and often fatal without appropriate intervention. Three out of seven patients died as a result of enteropathy and complications of the immunosuppressive treatment. LRBA patients need early interdisciplinary care; however, due to the lack of genotype-phenotype correlation and clinical heterogeneity of disease presentation, there is no standard therapeutic approach to these patients yet.
Recently, abatacept, a CTLA4-fusion protein, has been introduced as a promising agent for improvement of clinical symptoms in LRBA-deficient patients [4]. However, so far, long-term effects, safety, and cost-effectiveness of this therapy have not been analyzed. We used abatacept treatment for two patients. Patient 7, for whom we applied abatacept in ILD, responded well after three doses. His lymphocytic infiltrates disappeared and respiratory functions improved. Patient 3 received abatacept to treat resistant thrombocytopenia, lymphoproliferation, and enteropathy. Although lymphoproliferation alleviated, diarrhea persisted and thrombocytopenia deteriorated which resulted in the death of the patient due to intracranial hemorrhage. There is increasing evidence that persistent abatacept infusions correct the autoimmune phenotype in LRBA patients; however, not all symptoms are responsive [4]. Until now, five patients have been reported to receive abatacept. In three of those cases, the major complaint was respiratory dysfunction resulting from lung infiltration, which responded completely to abatacept. According to our experience with patients, lymphoproliferative symptoms alleviate, whereas enteropathy and cytopenia persist following abatacept. To date, we cannot give clear advice whether or not to treat with abatacept as more data is needed for the precise therapeutic indications and the effectiveness of abatacept therapy.
Given the severe complications observed in the patients including the high mortality rate, HSCT might be a promising strategy. We preferred HSCT with reduced-intensity conditioning regimen consisting of fludarabin, ATG, and busulfan. Targeted area-under-the-concentration versus time curve (AUC) for busulfan dosing was aimed as published in the protocol for chronic granulomatous disease [18]. The two patients who underwent HSCT using reduced-intensity conditioning responded well. Diarrhea which was the major problem for both patients, ceased in days following the stem cell infusion. P4 was the first LRBA-deficient patient who underwent HSCT with this regime. Currently, she is in posttransplant year 3 with an uneventful survey. The second patient, P5, had one ITP and autoimmune hemolytic anemia episode during the first year post HSCT, which could be controlled by administration of romiplostim and sirolimus, respectively. As both of the presented patients were under cumulative burden of chronic inflammation in combination with organ damages, reduced-intensity conditioning was the only option for HSCT.
By now, 12 LRBA-deficient patients have been reported to be treated with HSCT. Overall survival is 67% (8/12). Of note, 50% of the patients had complete or good partial remission [19]. However, posttransplant autoimmune manifestations are not infrequent in LRBA patients [9, 13, 15]. As speculated before, the autoimmune manifestations seen in these patients may be related to donors who were heterozygous carriers of the disease allele [15, 20]. As of today, there is no controlled study of whether HSCT or abatacept is the preferable treatment option in LRBA deficiency. Given the chronic requirement of CTLA-4 replacement, abatacept treatment might be a bridging strategy before HSCT.
Conclusion
Autosomal recessive disorders are relatively common in Turkey due to high frequency of consanguineous marriages. LRBA defects should always be kept in mind as a differential diagnosis for patients with autoimmune disease affecting multiple organs, chronic diarrhea, and organomegalies. In our experience, early HSCT is a life-saving therapeutic strategy.
Compliance with Ethical Standards
Conflict of Interest
The authors declare that they have no conflict of interest.
References
- 1.Bruton OC. Agammaglobulinemia. Pediatrics. 1952;9(6):722–728. [PubMed] [Google Scholar]
- 2.Bousfiha A, Jeddane L, Al-Herz W, Ailal F, Casanova JL, Chatila T, et al. The 2015 IUIS phenotypic classification for primary immunodeficiencies. J Clin Immunol. 2015;35(8):727–738. doi: 10.1007/s10875-015-0198-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gennery AR. The evolving landscape of primary immunodeficiencies. J Clin Immunol. 2016;36(4):339–340. doi: 10.1007/s10875-016-0273-6. [DOI] [PubMed] [Google Scholar]
- 4.Lo B, Zhang K, Lu W, Zheng L, Zhang Q, Kanellopoulou C, et al. AUTOIMMUNE DISEASE. Patients with LRBA deficiency show CTLA4 loss and immune dysregulation responsive to abatacept therapy. Science. 2015;349(6246):436–440. doi: 10.1126/science.aaa1663. [DOI] [PubMed] [Google Scholar]
- 5.Lo B, Fritz JM, Su HC, Uzel G, Jordan MB, Lenardo MJ. CHAI and LATAIE: new genetic diseases of CTLA-4 checkpoint insufficiency. Blood. 2016;128(8):1037–1042. doi: 10.1182/blood-2016-04-712612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lopez-Herrera G, Tampella G, Pan-Hammarstrom Q, Herholz P, Trujillo-Vargas CM, Phadwal K, et al. Deleterious mutations in LRBA are associated with a syndrome of immune deficiency and autoimmunity. Am J Hum Genet. 2012;90(6):986–1001. doi: 10.1016/j.ajhg.2012.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Revel-Vilk S, Fischer U, Keller B, Nabhani S, Gamez-Diaz L, Rensing-Ehl A, et al. Autoimmune lymphoproliferative syndrome-like disease in patients with LRBA mutation. Clin Immunol. 2015;159(1):84–92. doi: 10.1016/j.clim.2015.04.007. [DOI] [PubMed] [Google Scholar]
- 8.Burns SO, Zenner HL, Plagnol V, Curtis J, Mok K, Eisenhut M, et al. LRBA gene deletion in a patient presenting with autoimmunity without hypogammaglobulinemia. J Allergy Clin Immunol. 2012;130(6):1428–1432. doi: 10.1016/j.jaci.2012.07.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sari S, Dogu F, Hwa V, Haskologlu S, Dauber A, Rosenfeld R, et al. A successful HSCT in a girl with novel LRBA mutation with refractory celiac disease. J Clin Immunol. 2016;36(1):8–11. doi: 10.1007/s10875-015-0220-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Serwas NK, Kansu A, Santos-Valente E, Kuloglu Z, Demir A, Yaman A, et al. Atypical manifestation of LRBA deficiency with predominant IBD-like phenotype. Inflamm Bowel Dis. 2015;21(1):40–47. doi: 10.1097/MIB.0000000000000266. [DOI] [PubMed] [Google Scholar]
- 11.Erman B, Bilic I, Hirschmugl T, Salzer E, Cagdas D, Esenboga S, et al. Combined immunodeficiency with CD4 lymphopenia and sclerosing cholangitis caused by a novel loss-of-function mutation affecting IL21R. Haematologica. 2015;100(6):e216–e219. doi: 10.3324/haematol.2014.120980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Alkhairy OK, Abolhassani H, Rezaei N, Fang M, Andersen KK, Chavoshzadeh Z, et al. Spectrum of phenotypes associated with mutations in LRBA. J Clin Immunol. 2016;36(1):33–45. doi: 10.1007/s10875-015-0224-7. [DOI] [PubMed] [Google Scholar]
- 13.Gamez-Diaz L, August D, Stepensky P, Revel-Vilk S, Seidel MG, Noriko M, et al. The extended phenotype of LPS-responsive beige-like anchor protein (LRBA) deficiency. J Allergy Clin Immunol. 2016;137(1):223–230. doi: 10.1016/j.jaci.2015.09.025. [DOI] [PubMed] [Google Scholar]
- 14.Schreiner F, Plamper M, Dueker G, Schoenberger S, Gamez-Diaz L, Grimbacher B, et al. Infancy-onset T1DM, short stature, and severe immunodysregulation in two siblings with a homozygous LRBA mutation. J Clin Endocrinol Metab. 2016;101(3):898–904. doi: 10.1210/jc.2015-3382. [DOI] [PubMed] [Google Scholar]
- 15.Tesi B, Priftakis P, Lindgren F, Chiang SC, Kartalis N, Lofstedt A, et al. Successful hematopoietic stem cell transplantation in a patient with LPS-responsive beige-like anchor (LRBA) gene mutation. J Clin Immunol. 2016;36(5):480–489. doi: 10.1007/s10875-016-0289-y. [DOI] [PubMed] [Google Scholar]
- 16.Levy E, Stolzenberg MC, Bruneau J, Breton S, Neven B, Sauvion S, et al. LRBA deficiency with autoimmunity and early onset chronic erosive polyarthritis. Clin Immunol. 2016;168:88–93. doi: 10.1016/j.clim.2016.03.006. [DOI] [PubMed] [Google Scholar]
- 17.Alangari A, Alsultan A, Adly N, Massaad MJ, Kiani IS, Aljebreen A, et al. LPS-responsive beige-like anchor (LRBA) gene mutation in a family with inflammatory bowel disease and combined immunodeficiency. J Allergy Clin Immunol. 2012;130(2):481–488. doi: 10.1016/j.jaci.2012.05.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gungor T, Teira P, Slatter M, Stussi G, Stepensky P, Moshous D, et al. Reduced-intensity conditioning and HLA-matched haemopoietic stem-cell transplantation in patients with chronic granulomatous disease: a prospective multicentre study. Lancet. 2014;383(9915):436–448. doi: 10.1016/S0140-6736(13)62069-3. [DOI] [PubMed] [Google Scholar]
- 19.Seidel MG, Bohm K, Dogu F, Worth A, Thrasher A, Florkin B, et al. Treatment of severe forms of LPS-responsive beige-like anchor protein (LRBA) deficiency by allogeneic hematopoietic stem cell transplantation. J Allergy Clin Immunol. 2017. [DOI] [PubMed]
- 20.Seidel MG, Hirschmugl T, Gamez-Diaz L, Schwinger W, Serwas N, Deutschmann A, et al. Long-term remission after allogeneic hematopoietic stem cell transplantation in LPS-responsive beige-like anchor (LRBA) deficiency. J Allergy Clin Immunol. 2015;135(5):1384–90 e1-8. doi: 10.1016/j.jaci.2014.10.048. [DOI] [PMC free article] [PubMed] [Google Scholar]