

Abstract
The complete genome sequence of a porcine epidemic diarrhea virus variant, strain SHQP/YM/2013, from China was determined and compared with those of other porcine epidemic diarrhea viruses. The full-length genome was 28,038 nucleotides (nt) in length without the poly (A) tail, and it was similar to that of other reported PEDV strains, with the characteristic gene order 5′-replicase (1a/1b) -S-ORF3-E-M-N-3′. Nucleotide sequence analysis based on individual virus genes indicated a close relationship between the S gene of SHQP/YM/2013 and those of the four Korean field strains from 2008–2009. Its ORF3 gene, however, fell into three groups. Recent prevalent Chinese PEDV field isolates were divided between group 1 and group 3, which suggests that the recent prevalent Chinese PEDV field isolates represent a new genotype that differs from the genotype that includes the vaccine strains. Based on phylogenetic analysis of the M gene, ORF3 gene and S gene, our study demonstrated that prevalent PEDV isolates in China may have originated from Korean strains. This report describes the complete genome sequence of SHQP/YM/2013, and the data will promote a better understanding of the molecular epidemiology and genetic diversity of PEDV field isolates in eastern China.
Keywords: Vaccine Strain, Porcine Epidemic Diarrhea Virus, Field Isolate, ORF3 Gene, Korean Strain
Porcine epidemic diarrhea virus (PEDV) is an enveloped single-stranded, positive-sense RNA virus that is taxonomically classified within the family Coronaviridae, genus Alphacoronavirus [16]. The virus is the causative agent of porcine epidemic diarrhea (PED), which was first reported in Belgium and the United Kingdom in 1978 [16]. PEDV causes a devastating enteric disease that is characterized by watery diarrhea, dehydration and significant mortality in piglets, thereby resulting in tremendous economic losses to the swine industry in Europe and Asia, including Japan, Korea, and China [6, 7, 17, 20].
The PEDV genome is approximately 28 kb in length and contains seven open reading frames (ORFs), which encode replicase 1a and 1b, the spike (S), ORF3, envelope (E), membrane (M), and nucleoprotein (N), arranged in the order 5′-replicase(1a/1b)-S-ORF3-E-M-N-3′ [14]. Two long ORFs (ORF1a and ORF1b) encode the non-structural replicase polyproteins (replicase 1a and 1b), which occupy two thirds of the genome. Genes for the major structural proteins S, E, M, and N are located downstream of the replicase gene. The S glycoprotein makes up the large surface projections of the virion and plays an important role in binding to specific host cell receptor glycoproteins, with subsequent penetration into the cells occurring via membrane fusion. The S glycoprotein also stimulates production of neutralizing antibodies by the host [8]. The M protein is essential for viral envelope formation and release. It not only induces antibodies that neutralize the virus in the presence of complement [18] but also stimulates the production of interferon-α (IFN-α) [13]. In addition, the ORF3 gene is the only accessory gene, and it has been suggested to be an important determinant of virulence in PEDV. The virulence of the virus can be reduced by altering the ORF3 gene through cell culture adaptation, in a manner similar to that for transmissible gastroenteritis virus (TGEV) [19, 23], and its differentiation may be a marker of adaptation to cell culture and virus attenuation [15, 19]. Thus, differentiation of the ORF3 gene could be a valuable tool for molecular epidemiological studies of PEDV [6, 15, 19].
To reveal the characteristics of this virus and determine more precisely the relationships among the PEDV strains currently circulating in China and other PEDV strains, the complete genomic sequence of the SHQP/YM/2013 strain was determined and analyzed.
Intestinal tracts were collected from dead piglets during an outbreak of diarrhea among immunized swine (inactivated transmissible gastroenteritis [TGEV H] and porcine epidemic diarrhea [CV777]) on a breeding farm in Shanghai in 2013. These intestinal samples were confirmed to be positive for PEDV using a commercial real-time reverse transcription polymerase chain reaction (RT-PCR) kit (SuoAo, Beijing, China).
The SHQP/YM/2013 strain was characterized further by complete genome sequencing. Primers were designed to anneal to sites that are highly conserved among PEDV sequences available in the GenBank database, and the complete genome sequence was determined using a primer-walking strategy. Viral RNA extraction, RT-PCR amplification, and cloning were performed according to conventional protocols. The 5′ and 3′ end regions were amplified using 5′ and 3′ full RACE kits (TaKaRa, Dalian, China) according to the manufacturer’s instructions. These plasmids were sequenced by the Invitrogen Company (Life Technologies, Shanghai, China). Each nucleotide was identified from three identical results. Sequences were assembled and analyzed using the DNASTAR software package (DNASTAR Inc., Madison, WI, USA).
Phylogenetic analysis based on the seven coding regions of the SHQP/YM/2013 strain and the other PEDV strains available in the GenBank database was carried out. Phylogenetic trees were constructed by the neighbor-joining method using MEGA, version 4 [21]. The topology of trees based on the nucleotide sequences was obtained by majority-rule consensus using 1000 bootstrap replicates, shown as percentages, and bootstrap values greater than 60 % were considered statistically significant for grouping. The newly characterized sequence has been deposited in the GenBank database under the accession number KJ196348.
The complete genomic sequence of SHQP/YM/2013, excluding the poly (A) tail, comprises 28,038 nucleotides (nt), with 5′ (292 nt) and 3′ (334 nt) ends containing untranslated regions (UTRs). The entire genome has a GC content of 41.81 %. The genomic organization of SHQP/YM/2013, which is typical of all previously characterized PEDV strains, is summarized as follows: 5′ UTR-replicase (1a/1b)-spike (S)-ORF3-envelope (E)-membrane (M)-nucleoprotein (N)-3′ UTR (Table 1). UTRs were found at both ends: the 5′ UTR was 292 nt long with a relatively high GC content of 44.86 % when compared with the whole genome. The 3′ UTR of 334 nt was present immediately downstream of the N gene of the genome and had 46.41 % GC content. Seven coding regions, respectively, encode four structural proteins (S [nt 20,634 to 24,794], E [nt 25,449 to 25,679], M [nt 25,687 to 26,367], and N [nt 26,379 to 27,704]), two nonstructural proteins (replicase 1a [nt 293 to 12,601] and replicase 1b [nt 12,601 to 20,637]), and the single accessory protein, ORF3 (nt 24,794 to 25,468).
Table 1.
Differences in genome organization between the SHQP/YM/2013 strain and the CV777 strain
| Gene | SHQP/YM/2013 strain | CV777 strain | ||||||
|---|---|---|---|---|---|---|---|---|
| Start | Stop | Size (nt) | Size (aa) | Start | Stop | Size (nt) | Size (aa) | |
| 5′UTR | 1 | 292 | 292 | – | 1 | 296 | 296 | – |
| ORF1a | 293 | 12601 | 12309 | 4103 | 297 | 12605 | 12309 | 4103 |
| ORF1b | 12601 | 20637 | 8037 | 2678 | 12605 | 20641 | 8037 | 2678 |
| S | 20634 | 24794 | 4161 | 1386 | 20638 | 24789 | 4152 | 1383 |
| ORF3 | 24794 | 25468 | 675 | 224 | 24789 | 25463 | 675 | 224 |
| E | 25449 | 25679 | 231 | 76 | 25444 | 25674 | 231 | 76 |
| M | 25687 | 26367 | 681 | 226 | 25682 | 26362 | 681 | 226 |
| N | 26379 | 27704 | 1326 | 441 | 26374 | 27699 | 1326 | 441 |
| 3′UTR | 27705 | 28038 | 334 | – | 27700 | 28033 | 334 | – |
The nucleotide and amino acid (aa) sequence identities of different regions of the SHQP/YM/2013 genome were compared with another 32 PEDV strains (Table 2). The SHQP/YM/2013 genome exhibited identities ranging from 96.6 % to 99.8 % to other strains; the highest level of similarity, with JS-HZ2012, was 99.8 %. The 5′ UTR of SHQP/YM/2013 shared 96.6 %–99.3 % identity with other PEDV strains. The 3′ UTR of SH/YM/2013 showed greater genetic conservation than the 5′ UTR and displayed 97.3 %–99.8 % homology to other PEDV stains.
Table 2.
Nucleotide and amino acid sequence identity (%) of different regions of the SHQP/YM/2013 genome compared with those of the other viruses (values in bold show highest identities)
| Strain | 5′UTR | ORF1a | ORF1b | S | ORF3 | E | M | N | 3′UTR |
|---|---|---|---|---|---|---|---|---|---|
| Virulent DR13 | 98.0(−) | 98.0(98.6) | 98.8(99.6) | 95.1(94.5) | 98.4(99.1) | 97.8(98.7) | 98.2(98.2) | 96.9(96.8) | 98.2(−) |
| Attenuated DR13 | 98.0(−) | 97.9(98.4) | 97.8(99.4) | 94.1(93.2) | 92.8(91.3) | 97.1(95.7) | 97.8(97.4) | 96.1(96.8) | 98.8(−) |
| SM98 | 96.9(−) | 96.9(97.4) | 97.7(99.0) | 93.9(92.7) | 96.8(95.5) | 95.7(94.8) | 97.9(97.4) | 95.2(96.2) | 97.6(−) |
| CV777 | 98.3(−) | 97.1(97.7) | 97.8(99.2) | 94.2(93.6) | 97.0(96.0) | 96.5(97.4) | 98.1(98.2) | 95.4(96.4) | 97.9(−) |
| CH/S | 98.6(−) | 97.9(97.9) | 98.5(99.0) | 94.0(93.5) | 98.4(99.6) | 96.1(96.1) | 97.9(98.2) | 96.0(96.6) | 98.5(−) |
| LZC | 96.6(−) | 97.0(97.4) | 97.7(99.1) | 93.7(92.6) | 95.7(94.2) | 95.7(94.8) | 97.1(96.5) | 95.2(95.9) | 97.3(−) |
| JS2008 | 97.9(−) | 98.0(98.4) | 98.2(99.4) | 94.4(93.5) | 92.8(91.3) | 95.7(93.5) | 97.7(96.9) | 96.1(96.8) | 98.8(−) |
| LC | 98.3(−) | 99.4(99.3) | 99.0(99.7) | 97.9(98.3) | 96.1(96.9) | 98.7(98.7) | 98.5(98.2) | 96.2(96.8) | 99.4(−) |
| GD-1 | 98.3(−) | 98.2(98.8) | 99.0(99.6) | 97.8(97.8) | 95.4(96.0) | 98.3(97.4) | 98.5(98.2) | 96.3(97.1) | 99.1(−) |
| GD-A | 98.3(−) | 98.3(98.8) | 99.0(99.6) | 97.8(98.0) | 95.7(96.0) | 98.7(98.7) | 98.4(97.4) | 96.1(96.8) | 99.4(−) |
| GD-B | 98.3(−) | 99.8(99.8) | 99.8(99.8) | 99.4(98.9) | 99.6(99.6) | 99.1(98.7) | 99.9(99.6) | 99.2(99.1) | 99.8(−) |
| AJ1102 | 99.0(−) | 99.4(99.4) | 99.0(99.7) | 97.9(98.3) | 96.1(96.9) | 98.7(98.7) | 98.5(98.2) | 96.2(97.1) | 99.4(−) |
| SD-M | 98.0(−) | 97.9(98.4) | 97.8(99.4) | 94.0(92.9) | 92.8(91.3) | 96.1(94.8) | 97.9(97.8) | 95.9(96.6) | 98.5(−) |
| BJ-2011-1 | 99.3(−) | 99.7(99.6) | 99.6(99.7) | 99.4(99.1) | 98.8(99.1) | 98.7(98.7) | 99.6(98.7) | 99.3(99.3) | 99.4(−) |
| ZJCZ4 | 98.6(−) | 98.1(98.7) | 99.6(99.8) | 98.4(98.7) | 96.0(96.4) | 98.7(98.7) | 98.2(98.2) | 96.2(97.3) | 99.4(−) |
| CH/FJND-3/2011 | 99.0(−) | 99.3(99.2) | 99.1(99.6) | 97.4(97.1) | 99.1(99.1) | 98.3(98.7) | 99.4(99.6) | 99.0(99.1) | 99.1(−) |
| CH/FJZZ-9/2012 | 98.6(−) | 99.2(99.2) | 98.7(99.3) | 98.0(98.4) | 97.9(99.1) | 97.8(98.7) | 99.4(99.6) | 99.2(99.3) | 98.8(−) |
| CH/ZMDZY/11 | 98.3(−) | 99.4(99.5) | 98.5(99.7) | 98.8(98.6) | 98.8(99.1) | 97.0(96.1) | 99.7(99.1) | 98.6(98.9) | 98.8(−) |
| AH2012 | 99.0(−) | 99.5(99.6) | 99.2(99.3) | 99.3(99.1) | 98.8(99.1) | 98.7(98.7) | 99.6(99.6) | 98.9(98.6) | 98.5(−) |
| JS-HZ2012 | 99.3(−) | 99.9(99.8) | 99.9(99.9) | 99.5(99.3) | 99.6(99.6) | 99.1(98.7) | 99.9(99.6) | 99.2(99.3) | 98.5(−) |
| CH/GDGZ/2012 | 98.3(−) | 98.1(98.5) | 98.8(99.2) | 97.5(97.4) | 95.9(96.4) | 98.3(98.7) | 98.2(98.2) | 94.5(93.2) | 99.1(−) |
| CHGD-01 | 96.6(−) | 98.4(98.8) | 98.9(99.4) | 97.8(98.1) | 96.1(96.9) | 98.3(97.4) | 98.2(97.8) | 96.2(96.8) | 98.5(−) |
| 13-019349 | 99.0(−) | 99.2(99.4) | 99.2(99.4) | 99.0(99.0) | 99.0(99.6) | 98.7(98.7) | 99.9(99.6) | 99.1(99.1) | 99.4(−) |
| IA1 | 99.3(−) | 99.2(99.4) | 99.2(99.6) | 99.1(99.4) | 99.0(99.6) | 98.7(98.7) | 99.7(99.6) | 99.1(99.1) | 99.4(−) |
| IA2 | 99.3(−) | 99.2(99.4) | 99.2(99.7) | 99.1(99.3) | 99.0(99.6) | 98.7(98.7) | 99.9(99.6) | 99.0(98.1) | 99.4(−) |
| MN | 99.3(−) | 99.2(99.4) | 99.2(99.7) | 99.1(99.2) | 99.0(99.6) | 98.7(98.7) | 99.9(99.6) | 99.1(99.1) | 98.8(−) |
| USA/Colorado/2013 | 99.0(−) | 99.3(99.5) | 99.2(99.7) | 99.2(99.4) | 98.8(99.1) | 98.7(98.7) | 99.9(99.6) | 99.1(99.1) | 99.4(−) |
| USA/Indiana/17846/2013 | 99.0(−) | 99.2(99.4) | 99.2(99.3) | 99.1(99.4) | 98.7(99.1) | 98.7(98.7) | 99.9(99.6) | 99.1(99.1) | 99.4(−) |
| USA/Iowa/16465/2013 | 99.0(−) | 99.2(99.4) | 99.1(99.3) | 98.8(99.4) | 99.0(99.6) | 98.7(98.7) | 99.9(99.6) | 99.1(99.1) | 99.4(−) |
| USA/Iowa/18984/2013 | 99.3(−) | 99.2(99.4) | 99.2(99.4) | 99.1(99.4) | 99.0(99.6) | 98.7(98.7) | 99.9(99.6) | 99.1(99.1) | 99.4(−) |
| ISU13-19338E-IN-homogenate | 99.3(−) | 99.2(99.4) | 99.2(99.7) | 99.1(99.4) | 98.7(99.1) | 98.7(98.7) | 99.9(99.6) | 99.1(99.1) | 99.4(−) |
| ISU13-22038-IA-homogenate | 99.3(−) | 99.2(99.4) | 99.2(99.7) | 99.1(99.3) | 99.0(99.6) | 98.7(98.7) | 99.9(99.6) | 99.1(99.1) | 99.4(−) |
Two open reading frames, ORFs 1a and 1b, reside in the first two thirds of the SHQP/YM/2013 genome and are translated into non-structural proteins 1a and 1b. Replicase 1a was predicted to encode a protein of 4103 aa, while replicase 1b was predicted to encode a protein of 2678 aa. Nucleotide sequence analysis revealed that there were no deletions or insertions in the replicase gene of any of the PEDV strains. Both the replicase 1a and the replicase 1b proteins were highly conserved, while the replicase 1b protein exhibited the highest amino acid sequence similarity among strains (99.0 %–99.9 %) (Table 2). The frameshift “slippery sequence” UUUAAAC [4] in ORF1b was identified in SHQP/YM/2013, as shown in Fig. 1. The sequences downstream of UUUAAAC were predicted to form a pseudoknot to support the translational frameshift [4, 10, 12]. Based on analysis of the nucleotide sequences of the PEDV ORF1a and ORF1b, all of the PEDV strains could be divided into three groups (Fig. 2A and B). Group 1 comprised one Chinese strain (LZC), one Korean strain (SM98) and one European strain (CV777). Group 2 consisted of vaccine strains (attenuated DR13) and two Chinese PEDV field strains (SD-M and JS2008). Group 3 was made up of virulent DR13, 10 USA strains isolated in 2013 and 16 Chinese strains (except CH/S), which were isolated from China during PED outbreaks in 2011–2013.
Fig. 1.

The putative ribosomal frame shift region. The nucleotide sequence covers the pseudoknot structures of PEDV reference strains. The putative slippery sites are represented by the dotted-line box, the stems are boxed, the loops are indicated by thick dark lines, and the 144 nucleotides (N144) are boxed in gray
Fig. 2.


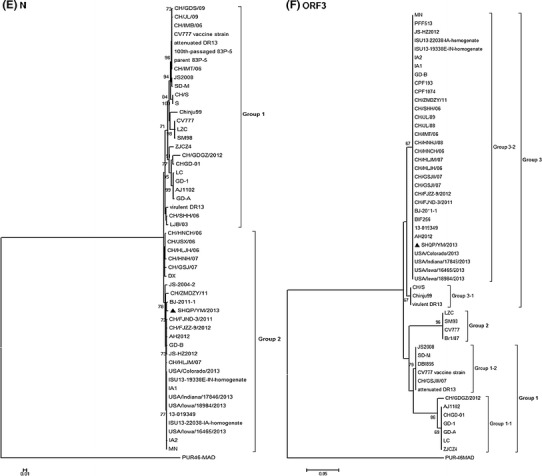
Phylogenetic analysis using the neighbor-joining method based on nucleotide sequences of different genes (A: ORF1a; B: ORF1b; C: S; D: M; E: N; F: ORF3) of PEDVs. Bootstrapping for 1,000 replicates with a value >60 % was performed to determine the percentage reliability of each internal node. PUR46-MAD is an outgroup control. The scale bar indicates nucleotide substitutions per site. The sequence of the SH/QPYM/2013 strain is indicated by the black triangle
The S protein gene was located immediately downstream of ORF1b; it encoded a predicted protein of 1386 amino acids. It contained 4161 nt and was therefore 9 nt longer than that of the PEDV reference strain CV777 (Table 1). Compared with CV777, two insertions (at positions 56–59 and 140) and one deletion (at position 156–157) were observed (Fig. 3). The S gene shared 93.8 %–99.4 % nucleotide sequence identity with those of other PEDV strains (Table 2). Phylogenetic analysis of the S gene nucleotide sequences revealed that all PEDV strains in this study could be separated into three groups (Fig. 2C): Group 1 comprised one Japanese strain (NK) and seven strains from South Korea (KNU-0801, KNU-0901, KNU-0903–KNU-0905, Spk1 and Chinju 99). Group 2 comprised two vaccine strains (the attenuated strains DR13 and CV777 vs), two Europe strains (CV777 and Br1/87), two Korean field strain (SM98 and virulent DR13), three strains from Japan (parent 83p-5, 100th-passaged 83p-5 and MK) and 12 strains from China, including six field strains from 2011. SHQP/YM/2013 belonged to group 3, which also including four Korean field strains (KNU-0802, KNU-0902, CNU-091222-01 and CNU-091222-02) from 2008–2009, 10 USA field isolates from 2013, and 15 Chinese strains, which were isolated during severe PED outbreaks in China during 2011–2013.
Fig. 3.
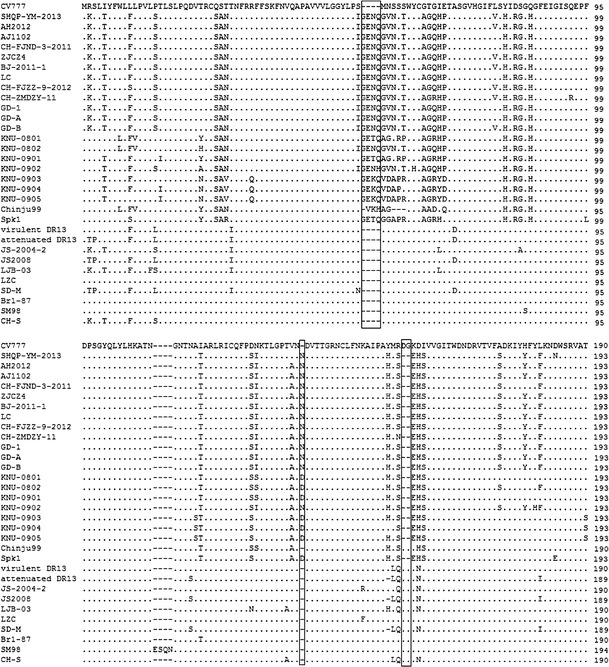
Amino acid sequence alignment of the S glycoprotein genes of the SHQP/YM/2013 and PEDV reference strains. The dashes (−) indicate the deleted sequences. Insertions and deletions in PEDV isolates are boxed
The E gene was 231 nucleotides long, encoding a protein of 76 amino acids. It shared 93.5 %–98.7 % amino acid sequence identity with other PEDV strains. The M gene was 681 nucleotides long and was able to make a protein of 226 amino acids. It had no nucleotide deletions or insertions but did show point mutations. The M gene had a conserved ATAAAC sequence 11 nucleotides upstream of the initiator ATG, as previously recognized in Br1/87 [9]. There was a highly conserved domain of 12 amino acids (SWWSFNPETDAL) located at aa 108 to 119. These residues are almost completely conserved across the entire family Coronaviridae [1]. Sequence analysis of the complete M gene showed that the PEDVs fell into three groups (Fig. 2D). The SHQP/YM/2013 strain belonged to the third group, which comprised three Japanese strains (JMe2, parent 83p-5, 100th-passaged 83p-5), 10 Korean strains, 10 USA field isolates from 2013, and all Chinese isolates (excluding LZC).
The N gene was 1326 nucleotides in length, encoding a polypeptide of 441 amino acids. The amino acid sequences had 93.2 %–99.3 % identity to those of other PEDV strains, and the highest identity (99.3 %) to BJ-2011-1, CH/FJZZ-9/2012 and JS-HZ2012. The N protein of SHQP/YM/2013 had 16 amino acid substitutions compared with CV777. All PEDV strains could be divided into two groups based on their N gene sequences (Fig. 2E). Group 2 comprised 10 USA field isolates from 2013 and 16 Chinese strains, including SHQP/YM/2013, isolated from China during 2004–2013.
The ORF3 gene, which is an accessory gene, is located between the structural genes for S and E. It had a single ORF of 675 nucleotides encoding a protein of 224 amino acids. The ORF3 gene had a conserved sequence (CTAGAC) 46 nucleotides upstream of the initiator ATG, similar to that described above for the M gene. Based on phylogenetic analysis of ORF3, all of the PEDV strains could be divided into three groups (Fig. 2F). One group had two subgroups (1-1 and 1-2). Seven Chinese field isolates from 2011–2012 formed one subgroup (1-1), and three Chinese field isolates (CH/GSJIII/07, JS2008 and SD-M) formed the second subgroup (1-2), together with vaccine strains (the attenuated DR13 and CV777 vs) and the DBI865 Korean field isolate. The second group included the CV777, Br1/87, SM98 and LZC strains. The third group, containing six Korean field isolates, 10 USA field isolates from 2013 and 19 Chinese isolates, had two subgroups (3-1 and 3-2). Two Korean strains (virulent DR13 and Chinju99) and the Chinese field isolate formed one subgroup (G3-1), and 18 Chinese field strains, including SHQP/YM/2013, formed the second subgroup (G3-2) with 10 USA field isolates from 2013 and four Korean field isolates from 2007.
As mentioned above, SHQP/YM/2013 was obtained in 2013 from a piglet with severe diarrhea on a vaccinated (inactivated transmissible gastroenteritis [TGEV H] and porcine epidemic diarrhea [CV777]) farm in eastern China. According to the investigation of the pig farm during the outbreak, about 90 % of piglets presented typical clinical signs of PED, and most of them died, whereas none of the sows immunized with CV777 showed any clinical signs. However, we also detected PEDV in sows’ milk, which suggests vertical transmission of the virus, as reported previously [20].
We have determined the complete genomic sequence of SHQP/YM/2013 and analyzed the phylogenetic relationships among PEDV strains. According to the complete M gene sequence, all Chinese PEDV strains (except LZC) are closely related to group 3, and SHQP/YM/2013 is especially closely related to 10 strains obtained from the USA in 2013 and two Korean field strains isolated in 2007.
The most highly variable regions were located in the S and ORF3 genes. As in other coronaviruses, the S and ORF3 genes have been hypothesized to be important in the virulence and pathogenesis of PEDV infections [3, 8, 19, 22]. The S protein is known to play pivotal roles in interacting with cellular receptors to mediate viral entry and in inducing neutralizing antibodies in the natural host [2, 5, 11]. The same amino acid insertions and deletion of the S gene were observed among prevailing PEDV strains (except SD-M, JS2008, CH/BJSY/2011 and CH/JL/2011) in China and two Korean (KNU-0802 and KNU-0902) strains isolated in South Korea during 2008–2009. Our study showed that all of the PEDV strains fell into three groups. Recent prevalent Chinese PEDV field isolates were divided into two different groups (group 2 and group 3), which shows that two different genotypes are prevalent in China. The prevalent PEDV isolates in China, including SHQP/YM/2013, are most closely related to 10 USA strains from 2013 and four Korean field strains from 2008–2009.
The complete ORF3 gene sequence and phylogenetic analysis showed that all of the Chinese PEDV strains fell into three groups, and again the recent prevalent Chinese PEDV field isolates were divided into two different groups (group 1 and group 3). Eighteen Chinese field strains, including SHQP/YM/2013, formed the second subgroup (G3-2) with 10 USA field isolates from 2013 and four Korean field isolates from 2007. These strains are genetically different from the CV777 and attenuated DR13 vaccine strains (G1-2), which have been used for prevention of PEDV infection in China. Therefore, recent prevalent Chinese PEDV field isolates represent a new genotype that differs from the genotype that includes the vaccine strains. Based on phylogenetic analysis of the M gene, S gene and ORF3 gene, our study has demonstrated that the prevalent PEDV isolates in China may have originated from Korean strains.
Since October 2010, outbreaks of PED characterized by high morbidity (approaching 100 %) and mortality (80 % to 100 %) in newborn piglets have emerged in China [20]. Although the bi-combined commercial vaccines (inactivated transmissible gastroenteritis [TGEV H] and porcine epidemic diarrhea [CV777]) against TGEV and PEDV infection are used on swine farms, PED still occurs in immunized pig herds in China. This shows that these vaccines are no longer able to confer protection. Therefore, it is necessary to investigate further the molecular biology, as well as the mechanisms of immunogenicity and pathogenesis, of PEDV. There is an urgent need to develop more effective vaccines to prevent outbreaks of PEDV-induced diarrhea.
Acknowledgments
This research was supported by grant no. 2013: 5-5 from the Key Programs for Agricultural Science and Technology Development of Shanghai.
Conflict of interest
The authors declare that they have no conflicts of interest.
Footnotes
De-Quan Yang and Fei-Fei Ge contributed equally to this work and are co-first authors.
Contributor Information
De-Quan Yang, Email: dequan_yang@126.com.
Fei-Fei Ge, Email: qyfgff2369@yahoo.com.
Jin-Ping Zhou, Email: shzjpvet@163.com.
References
- 1.Arndt AL, Larson BJ, Hogue BG. A conserved domain in the coronavirus membrane protein tail is important for virus assembly. J Virol. 2010;84:11418–11428. doi: 10.1128/JVI.01131-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bosch BJ, Van Der Zee R, de Haan CA, Rottier PJ. The coronavirus spike protein is a class I virus fusion protein: structural and functional characterization of the fusion core complex. J Virol. 2003;77:8801–8811. doi: 10.1128/JVI.77.16.8801-8811.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bosch BJ, de Haan CAM, Smits SL, Rottier PJM. Spike protein assembly into the coronavirion: Exploring the limits of its sequence requirements. Virology. 2005;334:306–318. doi: 10.1016/j.virol.2005.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brierley I, Digard P, Inglis SC. Characterization of an efficient coronavirus ribosomal frameshifting signal: requirement for an RNA pseudoknot. Cell. 1989;57:537–547. doi: 10.1016/0092-8674(89)90124-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chang SH, Bae JL, Kang TJ, Kim J, Chung GH, Lim CW, Laude H, Yang MS, Jang YS. Identification of the epitope region capable of inducing neutralizing antibodies against the porcine epidemic diarrhea virus. Mol Cells. 2002;14:295–299. [PubMed] [Google Scholar]
- 6.Chen J, Wang C, Shi H, Qiu H, Liu S, Chen X, Zhang Z, Feng L. Molecular epidemiology of porcine epidemic diarrhea virus in China. Arch Virol. 2010;155:1471–1476. doi: 10.1007/s00705-010-0720-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Debouck P, Pensaert M, Coussement W. The pathogenesis of an enteric infection in pigs, experimentally induced by the coronavirus-like agent, CV777. Vet Microbiol. 1981;6:157–165. doi: 10.1016/0378-1135(81)90007-9. [DOI] [Google Scholar]
- 8.Duarte M, Laude H. Sequence of the spike protein of the porcine epidemic diarrhoea virus. J Gen Virol. 1994;75:1195–1200. doi: 10.1099/0022-1317-75-5-1195. [DOI] [PubMed] [Google Scholar]
- 9.Duarte M, Tobler K, Bridgen A, Rasschaert D, Ackermann M, Laude H. Sequence analysis of the porcine epidemic diarrhea virus genome between the nucleocapsid and spike protein genes reveals a polymorphic ORF. Virology. 1994;198:466–476. doi: 10.1006/viro.1994.1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Eleouet JF, Rasschaert D, Lambert P. Complete sequence (20 kilobases) of the polyprotein-encoding gene 1 of transmissible gastroenteritis virus. Virology. 1995;206:817–822. doi: 10.1006/viro.1995.1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Godet M, Grosclaude J, Delmas B, Laude H. Major receptor-binding and neutralization determinants are located within the same domain of the transmissible gastroenteritis virus (coronavirus) spike protein. J Virol. 1994;68:8008–8016. doi: 10.1128/jvi.68.12.8008-8016.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kocherhans R, Bridgen A, Ackermann M, Tobler K. Completion of the porcine epidemic diarrhoea coronavirus (PEDV) genome sequence. Virus Genes. 2001;23:137–144. doi: 10.1023/A:1011831902219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Laude H, Gelfi J, Lavenant L, Charley B. Single amino acid changes in the viral glycoprotein M affect induction of alpha interferon by the coronavirus transmissible gastroenteritis virus. J Virol. 1992;66:743–749. doi: 10.1128/jvi.66.2.743-749.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Murphy FA, Gibbs EP, Horzinek MC, Studdert MJ. Veterinary virology. 3. San Diego: Academic Press; 1999. pp. 496–501. [Google Scholar]
- 15.Park SJ, Moon HJ, Luo Y, Kim HK, Kim EM, Yang JS, Song DS, Kang BK, Lee CS, Park BK. Cloning and further sequence analysis of the ORF3 gene of wild- and attenuated-type porcine epidemic diarrhea viruses. Virus Genes. 2008;36:95–104. doi: 10.1007/s11262-007-0164-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pensaert MB, De Bouck P. A new coronavirus-like particle associated with diarrhea in swine. Arch Virol. 1978;58:243–247. doi: 10.1007/BF01317606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pensaert MB, Yeo SG. Porcine epidemic diarrhea. In: Straw BE, Zimmerman JJ, D’Allaire S, Taylor DJ, editors. Diseases of Swine. 9. Ames: Wiley-Blackwell; 2006. pp. 367–372. [Google Scholar]
- 18.Saif LJ. Coronavirus immunogens. Vet Microbiol. 1993;37:285–297. doi: 10.1016/0378-1135(93)90030-B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Song DS, Yang JS, Oh JS, Han JH, Park BK. Differentiation of a Vero cell adapted porcine epidemic diarrhea virus from Korean field strains by restriction fragment length polymorphism analysis of ORF3. Vaccine. 2003;21:1833–1842. doi: 10.1016/S0264-410X(03)00027-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sun RQ, Cai RJ, Chen YQ, Liang PS, Chen DK, Song CX. Outbreak of porcine epidemic diarrhea in suckling piglets, China. Emerg Infect Dis. 2012;18:161–163. doi: 10.3201/eid1801.111259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tamura K, Dudley J, Nei M, Kumar S. MEGA4: Molecular Evolutionary Genetics Analysis (MEGA) software version 4.0. Mol Biol Evol. 2007;24:1596–1599. doi: 10.1093/molbev/msm092. [DOI] [PubMed] [Google Scholar]
- 22.Vaughn EM, Halbur PG, Paul PS. Sequence comparison of porcine respiratory coronavirus isolates reveals heterogeneity in the S, 3, and 3-1 Genes. J Virol. 1995;69:3176–3184. doi: 10.1128/jvi.69.5.3176-3184.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Woods RD. Efficacy of a transmissible gastroenteritis coronavirus with an altered ORF-3 gene. Can J Vet Res. 2001;65:28–32. [PMC free article] [PubMed] [Google Scholar]