

Abstract
Porcine reproductive and respiratory syndrome virus (PRRSV) and porcine epidemic diarrhea virus (PEDV) are porcine nidoviruses that are considered emerging and re-emerging viral pathogens of pigs that pose a significant economic threat to the global pork industry. Although cholesterol is known to affect the replication of a broad range of viruses in vitro, its significance and role in porcine nidovirus infection remains to be elucidated. Therefore, the present study was conducted to determine whether cellular or/and viral cholesterol levels play a role in porcine nidovirus infection. Our results showed that depletion of cellular cholesterol by treating cells with methyl-β-cyclodextrin (MβCD) dose-dependently suppressed the replication of both nidoviruses. Conversely, cholesterol depletion from the viral envelope had no inhibitory effect on porcine nidovirus production. The addition of exogenous cholesterol to MβCD-treated cells moderately restored the infectivity of porcine nidoviruses, indicating that the presence of cholesterol in the target cell membrane is critical for viral replication. The antiviral activity of MβCD on porcine nidovirus infection was found to be predominantly exerted when used as a treatment pre-infection or prior to the viral entry process. Furthermore, pharmacological sequestration of cellular cholesterol efficiently blocked both virus attachment and internalization and, accordingly, markedly affected subsequent post-entry steps of the replication cycle, including viral RNA and protein biosynthesis and progeny virus production. Taken together, our data indicate that cell membrane cholesterol is required for porcine nidovirus entry into cells, and pharmacological drugs that hamper cholesterol-dependent virus entry may have antiviral potential against porcine nidoviruses.
Introduction
Nidovirales is a large order of enveloped positive-sense, single-stranded RNA viruses that consists of the families Arteriviridae, Coronaviridae, Roniviridae, and Mesoniviridae, whose members infect a broad range of hosts including humans and other mammals, birds, fish, insects, and crustaceans [10, 38, 47, 51]. Although the genome sizes and virion morphologies of nidoviruses are strikingly different, the genome organization and replication strategy are comparable across the order. The nidovirus genome is composed of two large open reading frames (ORFs), 1a and 1b, encompassing the 5′-proximal two-thirds of the viral genome that encode non-structural proteins (nsps) and the remaining ORFs located in the 3′-proximal genome part that code for structural proteins [25, 50]. The initial translation from replicase ORF1a and ORF1b yields large polyprotein (pp) precursors, pp1a and pp1ab, via a –1 ribosomal frameshift (RFS), which then undergo autoproteolysis by viral proteases to eventually produce functional nsps, including the viral RNA-dependent RNA polymerase (RdRp) [3, 47, 59]. The membrane-bound RdRp-containing replication complex engages in viral genomic RNA replication and subgenomic (sg) mRNA transcription. The latter finally generates a 3′ co-terminal nested set of sg mRNAs that are used to express nidoviral structural proteins [25, 49, 50].
Porcine reproductive and respiratory syndrome virus (PRRSV) is a pathogenic macrophage-tropic arterivirus of swine that results in reproductive failure in pregnant sows and acute or chronic respiratory illnesses in pigs of all ages. PRRSV primarily replicates in porcine alveolar macrophages (PAMs) and can establish persistent infection in lymphoid tissues of infected pigs that lasts for several months. As a result, PRRSV infection suppresses normal macrophage function and immune responses and is often associated with severe disease outcomes, including increased pre-weaning mortality in growing pigs resulting from secondary bacterial or viral infections, thereby affecting the swine production system [17, 37, 49]. Porcine epidemic diarrhea virus (PEDV) is a pathogenic enterocyte-tropic swine coronavirus that causes acute enteritis with high mortality rates in neonatal piglets. PEDV infection is characterized by severe villous atrophy in the small intestine that results in watery diarrhea followed by fatal dehydration and death in newborn piglets [27, 45]. Although PEDV outbreaks have been reported in Europe and Asia, the most serious epizootics for nearly the past three decades have occurred in Asia. However, since the virus first emerged in the United States in 2013 [52], PEDV has become recognized globally as a highly contagious and deadly virus. These two viruses, PRRSV and PEDV, represent emerging and re-emerging porcine nidoviruses that continue to threaten pork-producing countries around world, leading to huge financial losses to the global swine industry [20, 27].
Lipid rafts, which are enriched in cholesterol, sphingolipids, and associated proteins, are unique liquid-ordered microenvironments in the plasma membrane and are involved in a variety of cellular processes as well as in multiple stages of the virus life cycle. Cholesterol, a major constituent of lipid rafts, maintains the tight packaging of sphingolipids, and several proteins are partitioned into these microdomains. Cholesterol depletion destroys this structural order, leading to disorganization of lipid raft microdomains and dissociation of bound proteins [2, 22]. Therefore, plasma membrane cholesterol plays important roles in the infection processes of various non-enveloped and enveloped viruses [5, 34, 36, 56]. In particular, enveloped virus entry requires cholesterol in either the viral or cellular membrane or both [1, 4, 13, 15, 16, 35, 43, 57, 62]. However, there are few reports on the potential relationship between cholesterol and the replication of porcine nidoviruses, although cellular membrane cholesterol has been shown to be a determinant of PRRSV entry in African monkey kidney MARC-145 cells [21, 55]. In the present study, therefore, we investigated the requirement for cholesterol and its mechanism of action in porcine nidovirus infection. Independent depletion of cholesterol from the plasma membrane of target cells by treatment with methyl-β-cyclodextrin (MβCD) significantly impaired PRRSV and PEDV infection. These inhibitory effects on viral replication were partially reversible by replenishment with exogenous cholesterol. In contrast, porcine nidoviruses were shown to be resistant to pharmacological reduction of the cholesterol content of the viral envelope. Our data indicate that cholesterol-enriched microdomains are essential for PRRSV and PEDV in the cellular membrane, but not in the viral membrane. Further experiments revealed that pharmacological depletion of cellular cholesterol primarily interferes with virus binding and penetration and subsequently influences post-entry stages of the PRRSV and PEDV replication cycle, including viral genomic and sg RNA synthesis, viral protein expression, and virus production. Altogether, our results suggest that cholesterol in the cellular membrane is critical for porcine nidovirus entry and that disruption of the cholesterol-dependent entry process may be an excellent therapeutic option for nidovirus infection in human or veterinary subjects.
Materials and methods
Cells, viruses, reagents, and antibodies
PAM-pCD163 cells [31] were cultured in RPMI 1640 medium (Invitrogen, Carlsbad, CA) supplemented with 10% fetal bovine serum (FBS, Invitrogen), antibiotic-antimycotic solution (100×, Invitrogen), 10 mM HEPES (Invitrogen), 1 mM sodium pyruvate (Invitrogen), and nonessential amino acids (100×, Invitrogen) in the presence of 50 μg of Zeocin (Invitrogen) per ml. Vero cells were cultured in alpha minimum essential medium (α-MEM, Invitrogen) with 10% FBS and antibiotic-antimycotic solution. ST-pAPN cells [39] were cultured in α-MEM with 10% FBS and antibiotic-antimycotic solution in the presence of 200 μg of G418 (Invitrogen) per ml. The cells were maintained at 37 °C in a humidified 5% CO2 incubator. PRRSV strain VR-2332 was propagated in PAM-pCD163 cells as described previously [29]. PEDV strain SM98-1 was kindly provided by the Korean Animal and Plant Quarantine Agency and propagated in Vero cells as described previously [19, 39]. MβCD and water-soluble cholesterol were purchased from Sigma (St. Louis, MO) and dissolved in ethanol and phosphate-buffered saline (PBS), respectively. These compounds were diluted to the desired concentrations in maintenance medium. PRRSV N and PEDV N protein-specific monoclonal antibodies (MAb) were obtained from ChoogAng Vaccine Laboratory (CAVAC; Daejeon, South Korea). Antibodies to porcine CD163 (pCD163) and β-actin were purchased from AbD Serotech (Raleigh, NA) and Santa Cruz Biotechnology (Santa Cruz, CA), respectively. The polyclonal antibody recognizing porcine aminopeptidase N (pAPN) obtained from BALB/c mice immunized with purified pAPN (Sigma) was a gift from Bang-Hun Hyun (Animal and Plant Quarantine Agency, Gimcheon, South Korea).
Cell viability assay
The cytotoxic effects of reagents on PAM-pCD163 and Vero cells were analyzed using a 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay (Sigma) that allows detection of cell viability. Briefly, PAM-pCD163 and Vero cells were grown at 1 × 104 cells/well in 96-well tissue culture plates with MβCD or water-soluble cholesterol treatment for 48 h. After 2 days of incubation, 50 μl of MTT solution (1.1 mg/ml) was added to each well and the samples were incubated for an additional 4 h. The supernatant was then removed from each well, and 150 μl of DMSO was added to dissolve the formazan crystals produced by MTT. The absorbance of the solution was measured at 540 nm using an enzyme-linked immunosorbent assay plate reader. All MTT assays were performed in triplicate.
Immunofluorescence assay (IFA)
PAM-pCD163 and Vero cells grown on microscope coverslips placed in 6-well tissue culture plates were pretreated with MβCD or ethanol for 1 h and mock infected or infected with PRRSV and PEDV, respectively, at a multiplicity of infection (MOI) of 1. Virus-infected cells were then grown in the presence of MβCD or vehicle for 48 h, fixed with 4% paraformaldehyde for 10 min at room temperature (RT) and permeabilized with 0.2% Triton X-100 in PBS at RT for 10 min. The cells were blocked with 1% bovine serum albumin (BSA) in PBS for 30 min at RT and then incubated with N-specific MAb 7 for 2 h. After washing five times in PBS, the cells were incubated for 1 h at RT with a goat anti-mouse secondary antibody conjugated with Alexa Fluor 488 (Molecular Probes, Carlsbad, CA), followed by counterstaining with 4′,6-diamidino-2-phenylindole (DAPI; Sigma). The coverslips were mounted on glass microscope slides in mounting buffer (60% glycerol and 0.1% sodium azide in PBS), and cell staining was visualized using a Leica DM IL LED fluorescence microscope (Leica, Wetzlar, Germany). In addition, PAM-pCD163 and ST-pAPN cells stably expressing pCD163 and pAPN, respectively, were grown in the presence of MβCD or vehicle for 24 h, fixed, and subsequently subjected to IFA with anti-pCD163 or anti-pAPN antibody as described above. Cell staining was analyzed using a confocal laser scanning microscope (Carl Zeiss, Gattingen, Germany).
Fluorescence-activated cell sorting (FACS) analysis
Quantification of virus-infected cells after treatment with MβCD was analyzed by flow cytometry. PAM-pCD163 and Vero cells were pretreated with MβCD, infected with virus, and maintained as described above. Virus-infected cells were trypsinized at 48 h postinfection (hpi) and centrifuged at 250 × g (Hanil Centrifuge FLETA 5) for 5 min. Cell pellets were washed with cold washing buffer (1% BSA and 0.1% sodium azide in PBS), and 106 cells were resuspended in 1% formaldehyde solution in cold wash buffer for fixation at 4 °C in the dark for 30 min, followed by centrifugation and incubation of the pellets in 0.2% Triton X-100 in PBS at 37 °C for 15 min for permeabilization. After centrifugation, the cell pellets were resuspended in a solution of primary anti-N MAb, and the mixture was incubated at 4 °C for 30 min. The cells were washed and allowed to react with an Alexa Fluor 488-conjugated anti-mouse IgG secondary antibody at 4 °C for 30 min in the dark. The stained cells were washed again and analyzed on a FACSAria III flow cytometer (BD Biosciences). Expression of the viral receptor on the cell surface upon cholesterol depletion was also evaluated by FACS analysis as described previously with some modifications [16]. Briefly, PAM-pCD163 and ST-pAPN cells were trypsinized at 48 h post-seeding. The detached cells were fixed and then reacted with the primary antibody or normal mouse IgG1 (Santa Cruz Biotechnology), followed by incubation with secondary antibody as described above. The stained cells were either left untreated or were treated with MβCD at 37 °C for 1 h and analyzed using a flow cytometer.
Virus titration
PAM-pCD163 and Vero cells were infected with PRRSV or PEDV and treated with MβCD or vehicle. The culture supernatants were collected at different time points (6, 12, 24, 36, and 48 hpi) and stored at –80 °C. The PRRSV titer was measured by limiting dilution on PAM-pCD163 cells in duplicate by IFA as described above, and the 50% tissue culture infectious dose (TCID50) per ml was calculated using the Spearman-Kärber method [14]. The PEDV titer was determined by plaque assay using Vero cells as described previously [39] and was expressed as plaque-forming units (PFU) per ml.
Depletion of cholesterol from the virus
Viral stocks were treated with MβCD at various concentrations at 37 °C for 1 h followed by ultracentrifugation to remove the MβCD. The MβCD-treated virus supernatants were purified through a 20% sucrose cushion (wt/vol) prepared in TE buffer [10 mM Tris-HCl (pH 8.0), 1 mM EDTA] by centrifugation at 36,900 rpm for 1 h at 4 °C in a P70AT rotor (model CP100WX; Hitachi, Hitachinaka, Japan). The virion cholesterol content was determined using fluorescence intensity analysis. Briefly, 96-well plate wells were coated with 50 ng of the purified viruses in 50 mM sodium bicarbonate buffer (pH 9.6) and incubated at 4 °C overnight. Plates were washed three times with washing buffer (0.05% Tween-20 in PBS) and blocked with 5% powdered skim milk (BD Biosciences, Belford, MA) in PBS at 37 °C for 2 h. After washing, filipin III (Cayman Chemical, Ann Arbor, MI) was added in triplicate for 1 h in the dark. The plates were washed, and fluorescence intensity was measured with a SPARK 10M multimode microplate reader (TECAN, Männedorf, Switzerland). In parallel, the purified samples were used to infect PAM-pCD163 or Vero cells for 48 h, and the virus-infected cells were independently subjected to FACS analysis and virus titration to determine PRRSV or PEDV infection as described above.
Replenishment of cholesterol in the presence of MβCD
PAM-pCD163 and Vero cells were first preincubated with vehicle or MβCD at various final concentrations for 1 h and then supplemented with or without 100 μg/ml or 40 μg/ml exogenous cholesterol, respectively, and incubated for 1 h. The cells were then inoculated with PRRSV or PEDV as described above. The virus inoculum was removed, and the infected cells were maintained in fresh medium containing MβCD and exogenous cholesterol. At 48 h dpi, the virus-infected cells were harvested and subjected to FACS analysis to assess the infectivity of PRRSV and PEDV as described above. In parallel, the cellular cholesterol content was determined using a Cholesterol Cell-Based Detection Assay Kit (Cayman Chemical) according to the manufacturer’s instructions. Briefly, virus-infected cells were cultivated in the presence of MβCD and exogenous cholesterol for 48 h, fixed with Cell-Based Assay Fixative Solution (Cayman Chemical) for 10 min at RT, and then washed three times for 5 min each with Cell-Based Assay Wash Buffer (Cayman Chemical). The cells were incubated with filipin III for 1 h in the dark. After washing three times in wash buffer, the cells were counterstained with DAPI, and filipin staining was visualized using a fluorescent Leica DM IL LED microscope.
Time course of MβCD treatment
PAM-pCD163 and Vero cells were infected with PRRSV and PEDV, respectively, at an MOI of 1 as described above. At –1, 0, 1, 2, 4, 6, 8, 10, 12, or 24 hpi, MβCD was added to maintain the indicated final concentration over the remainder of the time course experiment. The virus-infected and inhibitor-treated cells were further maintained and trypsinized at 48 hpi, followed by centrifugation. The harvested cells were subjected to FACS analysis to assess the presence of PRRSV or PEDV infection as described above.
Virus binding and internalization assays
Binding and internalization assays were performed as described previously with some modifications [8]. PAM-pCD163 and Vero cells grown in 6-well culture plates were pretreated and infected with PRRSV and PEDV, respectively, at an MOI of 1 at 4 °C for 1 h in the presence of MβCD. Unbound viruses were then removed by washing with PBS, and the cells were either incubated at 4 °C (allowing virus binding only) or 37 °C (permitting virus binding and internalization) in the presence of MβCD for 1 h. In the latter case, the cells were further treated with proteinase K (0.5 mg/ml) at 37 °C for 45 min to remove bound but uninternalized virus particles. The PRRSV-infected cells were then serially diluted in RPMI medium and inoculated onto fresh PAM-pCD163 cell monolayers in 96-well tissue culture plates. At 48 h post-incubation, bound or internalized viruses were titrated by IFA as described above, and the TCID50 was determined. For PEDV, the serially diluted infected cells were inoculated onto uninfected Vero cells, and, after 48 h, viruses were titrated using plaque assay and quantified as PFU per ml.
Quantitative real-time reverse transcription polymerase chain reaction (RT-PCR)
PAM-pCD163 and Vero cells were incubated with MβCD for 1 h prior to infection and then inoculated with PRRSV or PEDV at an MOI of 1 for 1 h at 37 °C. The virus inoculum was subsequently removed, and the infected cells were maintained in fresh medium containing MβCD for 48 h. Total RNA was extracted from lysates of the infected cells at 48 hpi using TRIzol Reagent (Invitrogen) and then treated with DNase I (TaKaRa, Otsu, Japan) according to the manufacturer’s protocols. The concentrations of extracted RNA were measured using a NanoVue spectrophotometer (GE Healthcare, Piscataway, NJ). Quantitative real-time RT-PCR was conducted using a Thermal Cycler Dice Real Time System (TaKaRa) with gene-specific primer sets as described previously [23]. The RNA levels of viral genes were normalized to that of mRNA for the β-actin or glyceraldehyde-3-phosphate dehydrogenase (GAPDH) gene, and relative quantities (RQ) of mRNA accumulation were determined using the 2-ΔΔCt method. To detect alterations in genomic RNA and sg mRNA levels in the presence of MβCD during porcine nidovirus infection, the results obtained from drug-treated cells were compared with those from vehicle-treated cells.
Western blot analysis
PAM-pCD163 and Vero cells were grown in 6-well tissue culture plates for 1 day and were mock infected or infected with PRRSV and PEDV, respectively, at an MOI of 1 in the presence of MβCD. At the indicated times, cells were harvested in 80 μl of lysis buffer (0.5% Triton X-100, 60 mM β-glycerophosphate, 15 mM ρ-nitrophenyl phosphate, 25 mM MOPS, 15 mM MgCl2, 80 mM NaCl, 15 mM EGTA [pH 7.4], 1 mM sodium orthovanadate, 1 μg of E64 per ml, 2 μg of aprotinin per ml, 1 μg of leupeptin per ml, and 1 mM PMSF) and sonicated on ice five times for 1 s each. Homogenates were lysed for 30 min on ice and clarified by centrifugation at 15,800 × g (Eppendorf centrifuge 5415R, Hamburg, Germany) for 30 min at 4 °C. The protein concentrations of the cell lysates were determined by BCA protein assay (Pierce, Rockford, IL). The cell lysates were mixed with 4× NuPAGE sample buffer (Invitrogen) and boiled at 70 °C for 10 min. The proteins were then separated on a NuPAGE 4–12% gradient Bis-Tris gel (Invitrogen) under reducing conditions and electrotransferred onto Immobilon-P (Millipore, Billerica, MA). The membranes were subsequently blocked with 3% powdered skim milk in TBS (10 mM Tris-HCl [pH 8.0], 150 mM NaCl) with 0.05% Tween-20 (TBST) at 4 °C for 2 h and reacted at 4 °C overnight with primary antibodies against PRRSV N, PEDV N, or β-actin. The blots were then incubated with secondary horseradish peroxidase (HRP)-labeled antibody (Santa Cruz Biotechnology) at a dilution of 1:5,000 for 2 h at 4 °C. Proteins were visualized using enhanced chemiluminescence (ECL) reagents (Amersham Biosciences, Piscataway, NJ) according to the manufacturer’s instructions. To quantify the viral proteins produced, band densities of PRRSV N and PEDV N proteins were quantitatively analyzed using a computer densitometer with the Wright Cell Imaging Facility (WCIF) version of the ImageJ software package (http://www.uhnresearch.ca/facilities/wcif/imagej/), based on the density value relative to that of the β-actin gene.
Statistical analysis
All statistical analyses were performed using Student’s t-test, and P-values less than 0.05 were considered statistically significant.
Results
Suppression of porcine nidovirus replication by cellular cholesterol depletion
To investigate whether cholesterol plays a role in viral infection, we used MβCD, which is the most common cholesterol-sequestering agent used for plasma membranes. To examine the effect of MβCD on porcine nidovirus replication, PRRSV and PEDV were selected because they are economically important viral pathogens in the pork industry. Based on MTT assay, none of the doses of MβCD tested in the current study caused detectable levels of PAM-pCD163 or Vero cell death (Fig. 1A). PAM-pCD163 and Vero cells were pretreated with MβCD at concentrations of 0.5 to 2 mM or with ethanol as a vehicle control for 1 h prior to infection. MβCD or vehicle was present throughout infection. Virus production was initially measured by monitoring cytopathic effect (CPE) after infection and then confirmed by immunofluorescence using the respective anti-N protein MAb at 48 hpi (Fig. 1B). In vehicle-treated control cells, visible CPE appeared at 24 hpi (data not shown) and became predominant by 48 hpi, and virus-specific staining was pronounced in many cell clusters, indicating infection and spread of the viruses to neighboring cells. In contrast, MβCD had an obvious inhibitory effect on porcine nidovirus propagation. As shown in Fig. 1B, the cholesterol-sequestering compound dramatically diminished virus-induced CPE (first and fourth panels) and expression of PRRSV and PEDV genes in a dose-dependent manner. Based on the quantification of N protein by flow cytometry, the proportion (%) of virus-infected cells was noticeably reduced after MβCD treatment. A maximum of ~80% inhibition of both viruses was observed in response to 1.5 mM MβCD (Fig. 1C and D). In addition, the effective doses for inhibiting 50% (ED50) of the replication of PRRSV and PEDV were determined to be about 770 μM and 820 μM, respectively. Taken together, these data show that cholesterol depletion of target cells efficiently suppresses the replication of porcine nidoviruses.
Fig. 1.
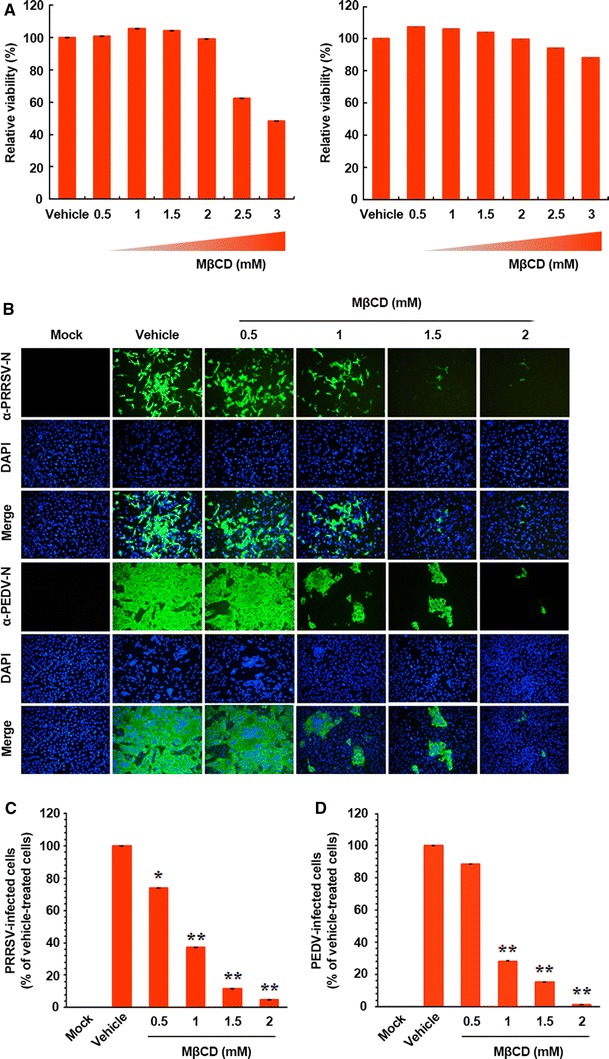
Effects of cellular cholesterol depletion on the replication of porcine nidoviruses. (A) PAM-pCD163 (left) and Vero (right) cells were incubated with various concentrations of MβCD for 48 h prior to the MTT assay, and the cytotoxicity of MβCD was determined by the MTT assay. (B) PAM-pCD163 and Vero cells were preincubated with MβCD at the indicated concentrations for 1 h prior to infection and were mock infected or infected with PRRSV or PEDV at an MOI of 1. Virus-infected cells were further maintained for 48 h in the presence of vehicle or MβCD. For immunostaining, infected cells were fixed at 48 hpi and incubated with MAb against the N protein of PRRSV or PEDV, followed by incubation with Alexa-green-conjugated goat anti-mouse secondary antibody (first and fourth panels). The cells were then counterstained with DAPI (second and fifth panels) and examined using a fluorescent microscope at 200× magnification. (C and D) Viral production in the presence of MβCD was calculated by measuring the percentage of cells expressing N proteins of PRRSV (C) or PEDV (D) by flow cytometry. The values shown are the means of three independent experiments, and error bars represent standard deviations. *, P = 0.001 to 0.05; **, P < 0.001
We then investigated the effects of cholesterol depletion on the envelopes of porcine nidoviruses. Each viral stock was treated with MβCD up to 2 mM prior to inoculation of the respective target cells. The virion cholesterol content after MβCD treatment was measured using filipin III as a fluorescent polyene antibiotic that binds to cholesterol. As shown in Fig. 2A, viral cholesterol levels were significantly reduced in MβCD-treated viruses compared to those in vehicle-treated viruses. However, in contrast to depletion of cellular cholesterol, the removal of cholesterol from virions resulted in no significant reduction in the replication of PRRSV and PEDV, even at the highest concentration used (Fig. 2B). Furthermore, the titers of both PRRSV and PEDV remained unchanged upon treatment of each virus with MβCD (Fig. 2C). Our results indicate that the viral cholesterol content is irrelevant to PRRSV and PEDV infection in vitro.
Fig. 2.
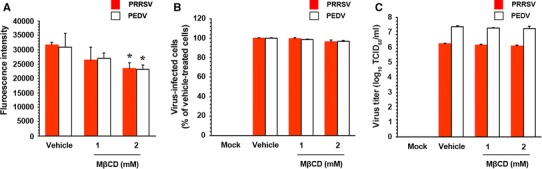
Effects of viral cholesterol depletion on the replication of porcine nidoviruses. (A) PRRSV and PEDV stocks were pretreated with MβCD to remove cholesterol in the viral envelope, followed by ultracentrifugation. Virion cholesterol content was determined using the cholesterol-binding, fluorescing antibiotic filipin III. Fluorescence intensity was measured using a fluorescence microplate reader. (B) PAM-pCD163 and Vero cells were infected with the purified PRRSV and PEDV, respectively. At 48 hpi, the virus-infected cells were fixed, and virus infectivity was determined by measuring the percentage of cells expressing N proteins of PRRSV or PEDV by FACS. (C) Culture supernatants were harvested at the same time point, and the titers of PRRSV and PEDV were measured. The values are representative of three independent experiments, and error bars indicate standard deviations
Effect of depletion and replenishment of cellular cholesterol on porcine nidoviruses
To verify the importance of cellular cholesterol in porcine nidovirus infection, we first examined whether replenishment of exogenous cholesterol restored MβCD-induced inhibition of porcine nidovirus infectivity. To accomplish this, cholesterol-depleted cells were treated with 100 μg or 40 μg of exogenous cholesterol per ml, which is the highest noncytotoxic concentration for PAM-pCD163 or Vero cells, respectively, before virus inoculation. Both MβCD and exogenous cholesterol were supplied throughout the course of infection. The addition of exogenous cholesterol to MβCD-treated and virus-infected cells was found to significantly reverse the antiviral activity of MβCD through depletion of cellular cholesterol. Incubation with MβCD alone greatly reduced PRRSV production to 12% and 5% at 1.5 mM and 2 mM, respectively, whereas supplementation with exogenous cholesterol enhanced virus production to 32% and 23% at the same concentrations of MβCD (Fig. 3A). Likewise, although PEDV infection declined to 48%, 19%, and 2% in the presence of MβCD alone at 1 mM, 1.5 mM, and 2 mM, respectively, virus production increased to 59%, 26%, and 10% at the same concentrations of MβCD when exogenous cholesterol was added (Fig. 3B). To verify these results, we also investigated alterations in cellular cholesterol content in cells treated with MβCD and exogenous cholesterol using a fluorescent filipin III. Cellular cholesterol levels specifically decreased in virus-infected and MβCD-treated cells compared to those in virus-infected and untreated cells. Supplementing exogenous cholesterol distinctly elevated the cholesterol level in virus-infected and MβCD-treated cells (Fig. 4). Altogether, the data reveal that cellular cholesterol content plays a pivotal role in porcine nidovirus infection.
Fig. 3.
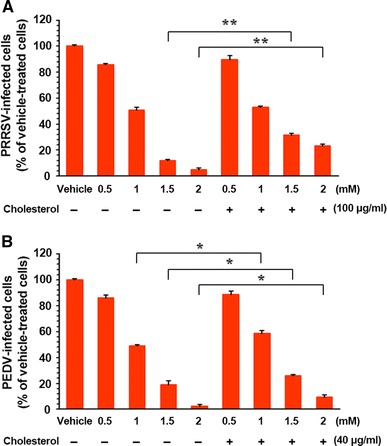
Porcine nidovirus infection efficiency after cholesterol depletion and replenishment in cellular membranes. PAM-pCD163 and Vero cells were preincubated with various concentrations of MβCD with (+) or without (–) exogenous cholesterol and infected with PRRSV and PEDV, respectively, in the presence or absence of MβCD and/or exogenous cholesterol as indicated. At 48 hpi, the virus-infected cells were fixed, and virus infectivity was determined by measuring the percentage of cells expressing N proteins of PRRSV (A) or PEDV (B) by FACS. Virus production in PAM-pCD163 and Vero cells treated with MβCD and cholesterol that was significantly different from that in cells treated with MβCD alone is indicated. The data are presented as the mean values from three independent experiments, and error bars represent standard deviations. *, P = 0.001 to 0.05; **, P < 0.001
Fig. 4.
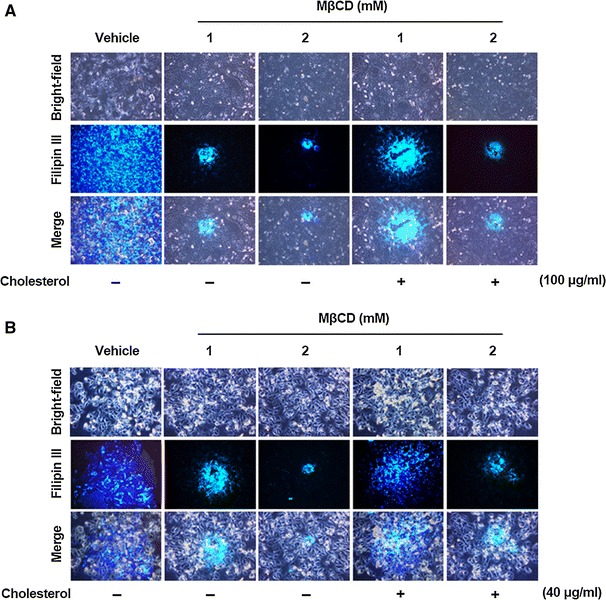
Cholesterol content determination after cholesterol depletion and replenishment in cellular membranes. PAM-pCD163 and Vero cells were preincubated with various concentrations of MβCD with (+) or without (–) exogenous cholesterol and infected with PRRSV (A) and PEDV (B), respectively, in the presence or absence of MβCD and/or exogenous cholesterol as indicated. Virus-specific CPE was observed daily and photographed at 48 hpi using a fluorescent/bright-field microscope at a magnification of 200× (first panels). For immunostaining, infected cells were fixed at 48 hpi and incubated with the cholesterol-binding, fluorescing antibiotic filipin III (second panels). The cells were examined using a fluorescence microscope at 200× magnification
Inhibition of porcine nidovirus replication by cellular cholesterol depletion at the pre-entry stage of viral infection
To determine the point at which MβCD acts during porcine nidovirus infection, PAM-pCD163 and Vero cells were treated with MβCD at various time points postinfection. At 48 hpi, the levels of PRRSV or PEDV replication were measured indirectly by quantifying the cells that expressed viral N protein using flow cytometry (Fig. 5). Treating cells with 2 mM MβCD at –1 and 0 hpi resulted in an approximately 94% and 79% decrease in PRRSV production, respectively, in comparison with control levels (vehicle-treated cells). Strikingly, the addition of MβCD at 1 hpi and thereafter (post-entry periods) had no significant inhibitory effect on PRRSV infectivity compared to the control levels. Similarly, treatment of PEDV-infected cells with 2 mM MβCD up to 0 hpi suppressed viral production by 99–95%, whereas exposure to the compound at 1–24 hpi resulted in no reduction in PEDV infectivity. These data showed that MβCD had to be present pre-infection or at an early stage of viral infection to exert its antiviral effect as a cellular cholesterol depletion reagent. Therefore, there is an important effect of cholesterol in the pre-entry period during porcine nidovirus infection.
Fig. 5.
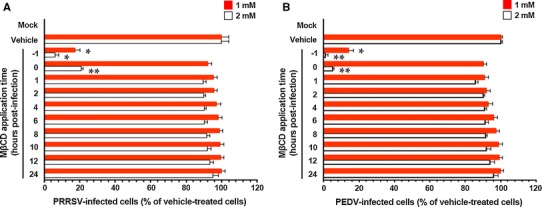
Effects of cellular cholesterol sequestration on porcine nidovirus propagation at early time points postinfection. PAM-pCD163 and Vero cells were pretreated with MβCD and were mock infected or infected with PRRSV (A) and PEDV (B), respectively. At 48 hpi, the virus-infected cells were fixed, and virus infectivity was determined by measuring the percentage of cells expressing N proteins of PRRSV or PEDV by FACS. The data are presented as the mean values of three independent experiments, and error bars represent standard deviations. *, P = 0.001 to 0.05; **, P < 0.001
Influence of porcine nidovirus entry by cellular cholesterol depletion
Next, we sought to pinpoint the step(s) in the replication cycle of porcine nidoviruses that were precisely targeted by pharmacological depletion of cellular cholesterol. To address this, the earliest steps, the two stages of virus entry (virus attachment and penetration), were examined using an internalization assay after treatment with MβCD. PAM-pCD163 and Vero cells were inoculated with PRRSV and PEDV, respectively, at 4 °C for 1 h to allow only virus attachment and were further maintained either at 4 °C or 37 °C to restrict or permit virus internalization, respectively, in the presence of MβCD. The samples incubated at 37 °C were subsequently treated with proteinase K to remove remaining viral particles from the cell surface. Serially diluted infected cells were then subjected to an infectious center assay on uninfected PAM-pCD163 and Vero cell monolayers, and virus titers were measured 2 days later by IFA or plaque assay.
As shown in Fig. 6, the titers of PRRSV and PEDV were reduced in a dose-dependent manner in cells treated with MβCD maintained at 4 °C to permit virus binding but prevent internalization, indicating that cholesterol depletion has an inhibitory effect on virus attachment to these cells. Moreover, production of both viruses was diminished in MβCD-treated cells incubated at 37 °C to allow virus entry to proceed, which suggested that cholesterol sequestration disturbs internalization of PRRSV and PEDV. In addition, we analyzed the amount of pCD163 or pAPN expressed on the cell surface after MβCD treatment and found that the surface expression level of the viral receptor in cells treated with 2 mM MβCD was similar to that of on vehicle-treated cells (Fig. 7). Taken together, these results indicate that pharmacological depletion of cholesterol hinders virus attachment and its subsequent penetration event without altering viral receptor expression and that cellular membrane cholesterol is indispensable for the porcine nidoviral entry process.
Fig. 6.
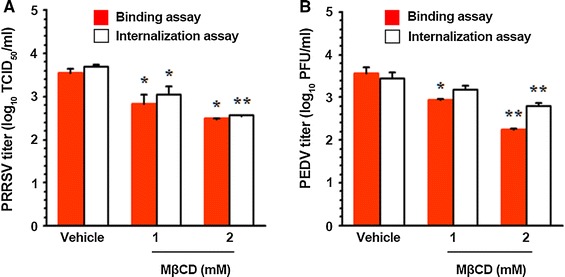
Effects of cellular cholesterol depletion on virus entry. PAM-pCD163 and Vero cells were infected with PRRSV or PEDV at an MOI of 1 at 4 °C for 1 h, respectively. After washing with cold PBS, infected cells were maintained in the presence or absence of MβCD, either at 4 °C (binding) or 37 °C (internalization), for an additional hour. The virus-infected cells maintained at 37 °C were further treated with proteinase K at 37 °C. The infected cells were then serially diluted and plated onto fresh target cells. At 2 days post-incubation, internalized viruses were titrated by IFA and plaque assay for PRRSV (A) and PEDV (B), respectively. The results are expressed as the mean values from three independent experiments performed in triplicate, and error bars represent standard deviations. *, P = 0.001 to 0.05; **, P < 0.001
Fig. 7.
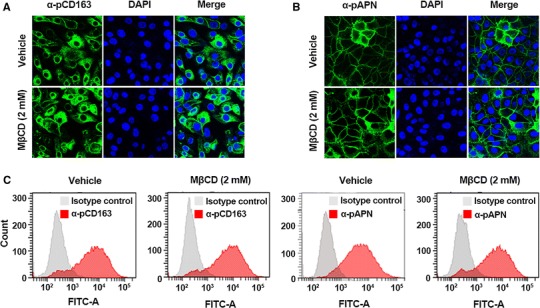
Effects of cellular cholesterol depletion on cell-surface expression of pCD163 or pAPN. (A and B) PAM-pCD163 and ST-pAPN cells cultured in the presence of vehicle or MβCD were fixed at 24 h post-seeding and labeled with antibody against pCD163 or pAPN. The cells were then counterstained with DAPI, and surface expression of pCD163 and pAPN was examined using a confocal microscope at 400× magnification. (C) PAM-pCD163 and ST-pAPN cells were grown for 48 h, and one million cells were harvested and incubated with anti-pCD163 or anti-pAPN antibody (red histogram) or an isotype control (white histogram). The stained cells were then treated with vehicle or MβCD and analyzed by flow cytometry
Impairment of nidoviral biosynthesis and progeny production by cellular cholesterol depletion
Like other positive-sense RNA viruses, following virus entry, the nidovirus genome is released into the cytoplasm and promptly serves as a template for translation of viral proteins by hijacking the host translational machinery. Early nidoviral translation produces the replicase polyproteins that are proteolytically processed into nsps, which subsequently drive de novo synthesis of nidoviral RNA. Therefore, we focused on the post-entry phases of the viral life cycle to investigate the functional mechanisms of sequestration of cellular cholesterol in PEDV infection. Because nidoviral infection generates genomic and sg RNA species, we first tested whether the removal of cellular cholesterol specifically affected genome replication and sg mRNA transcription. For this purpose, relative levels of both genomic RNA and sg mRNA were assessed by quantitative real-time strand-specific RT-PCR in the presence or absence of MβCD following porcine nidovirus infection. As shown in Fig. 8A, MβCD almost completely inhibited the synthesis of PRRSV genomic RNA and sg mRNA at a concentration of 2 mM when compared to untreated infected cells. Furthermore, an analogous effect of MβCD on genome replication and sg mRNA transcription of PEDV was observed. Little PEDV genomic RNA and sg mRNA was detected in cells treated with 1.5 mM MβCD (Fig. 8B). The decreases in viral RNA levels caused by MβCD did not reflect nonspecific inhibition of transcription because the internal control (β-actin or GAPDH) mRNA level remained unchanged in all samples (data not shown). Taken together, these results indicated that treatment with MβCD subsequently suppressed synthesis of nidoviral genomic RNA and sg mRNA.
Fig. 8.
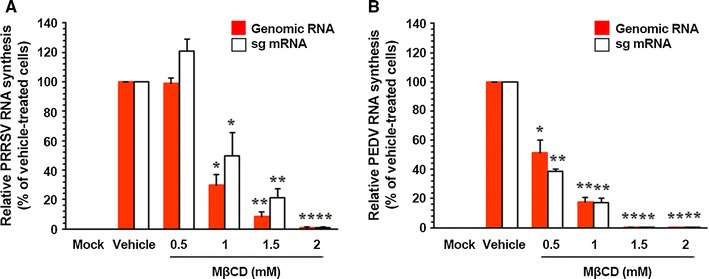
Interference with viral RNA synthesis by cellular cholesterol depletion. PAM-pCD163 and Vero cells pretreated with MβCD were mock infected or infected with PRRSV (A) and PEDV (B) for 1 h, respectively, and then incubated with either vehicle or MβCD. Total cellular RNA was extracted at 48 hpi, and strand-specific viral genomic RNAs (red bars) and sg mRNAs (white bars) of PRRSV and PEDV were amplified by quantitative real-time RT-PCR. Viral positive-sense genomic RNA and sg mRNA levels were normalized to those of porcine β-actin or monkey GAPDH mRNA, and relative quantities (RQ) of accumulated mRNA were determined. The results obtained with MβCD-treated samples were compared with those obtained with vehicle-treated samples. The values presented are the means from three independent experiments, and the error bars denote standard deviations. *, P = 0.001 to 0.05; **, P < 0.001
Since nidoviral structural proteins are translated from their respective sg mRNA transcripts late in the infectious cycle, it is conceivable that suppression of viral protein expression is a consequence of cascade-like inhibition of viral RNA synthesis. Thus, we examined whether viral protein translation was affected by depleting cholesterol from cell plasma membranes. To accomplish this, PAM-pCD163 and Vero cells were exposed to MβCD for 1 h prior to infection, and the compound was allowed to remain in the culture medium during infection and subsequent incubation. The expression levels of PRRSV and PEDV N proteins in the presence or absence of MβCD were evaluated at 48 hpi by western blot analysis. Pharmacological depletion of cellular cholesterol had a detrimental effect on nidoviral protein translation (Fig. 9). Densitometric analysis of the western blots revealed that the intracellular expression of both N proteins was dramatically reduced by MβCD, with a maximum of more than 95% inhibition at the highest concentration (Fig. 9). These data suggested that the sequestration effect of cellular cholesterol on viral protein expression is attributable to its specific preceding actions on viral RNA biosynthesis during nidoviral replication.
Fig. 9.
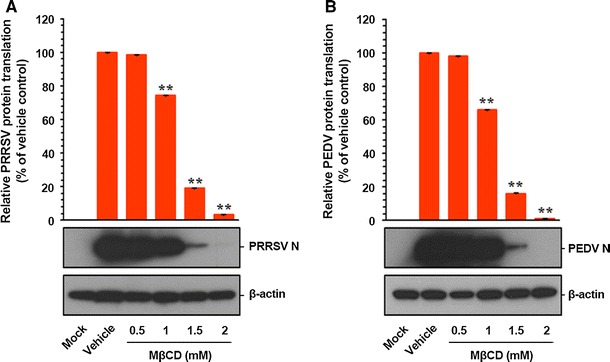
Inhibition of viral protein translation by cellular cholesterol depletion. MβCD-treated PAM-pCD163 and Vero cells were mock infected or infected with PRRSV (A) and PEDV (B) respectively, for 1 h, and were further cultivated in the presence or absence of MβCD. At 48 hpi, cell lysates were prepared, resolved by SDS-PAGE, transferred to a nitrocellulose membrane, and immunoblotted using antibodies that recognize the PRRSV N protein or PEDV N protein. The blot was also reacted with mouse MAb against β-actin to verify equal protein loading. Viral protein expression was quantitatively estimated by densitometry and expressed as the density value relative to that of the β-actin gene, and MβCD-treated sample results were compared to those of the vehicle control. The values shown are the means from three independent experiments, and the error bars denote standard deviations. **, P < 0.001
In addition, virus yields were determined during pharmacological depletion of cellular cholesterol to investigate whether endogenous cholesterol is necessary for production of infectious viral progeny. After infection, viral supernatants were collected at 48 hpi, and viral titers were measured. As illustrated in Fig. 10A, the presence of MβCD suppressed the growth of viral progeny in a dose-dependent manner. The peak viral titer was determined to be 106.05 TCID50/ml and 107.10 PFU/ml in the vehicle-treated control for PRRSV and PEDV, respectively. However, the addition of 2 mM MβCD reduced titers of PRRSV and PEDV to 103.82 TCID50/ml and 105.08 PFU/ml, respectively (representing a more than 2-log reduction compared to control levels). Examination of the growth kinetics also indicated that porcine nidovirus replication was markedly delayed when the cells were treated with MβCD at its optimal concentrations for each virus (Fig. 10B). These findings confirmed that cellular cholesterol content is integral in optimal progeny virus production from host cells.
Fig. 10.
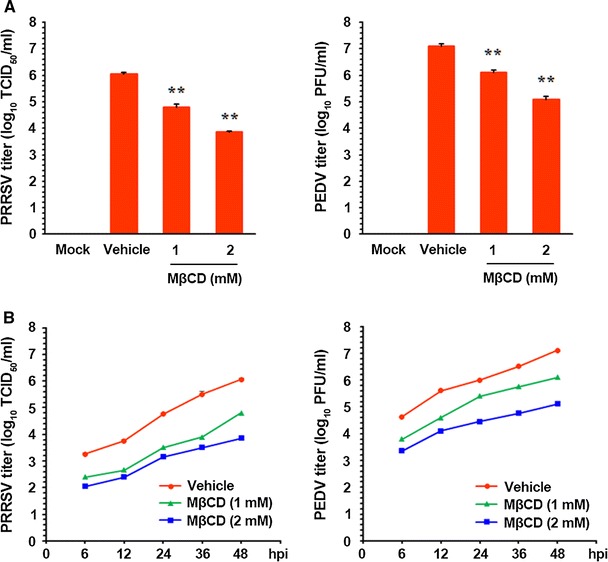
Suppression of viral progeny production by cellular cholesterol depletion. (A) PAM-pCD163 and Vero cells were pretreated with MβCD for 1 h and were mock infected or infected with virus at an MOI of 1. MβCD was present in the medium throughout the infection. At 48 hpi, virus-containing culture supernatants were collected, and the titers of PRRSV (left panel) and PEDV (right panel) were determined. (B) Growth kinetics of PRRSV (left panel) and PEDV (right panel) after treatment with MβCD, using the same experimental conditions as in panel A. At the indicated time points postinfection, culture supernatants were harvested, and virus titers were measured. The results are expressed as the mean values of two independent experiments performed in triplicate, and the error bars represent standard deviations. **, P < 0.001
Discussion
The order Nidovirales is a monophyletic group of enveloped, positive-strand RNA viruses with human and various animal hosts that produce a 3′ co-terminal nested set of sg mRNAs during infection. This order unites the four distantly related families Arteriviridae, Coronaviridae, Roniviridae, and Mesoniviridae, based on a number of common properties such as genome organization, predicted proteomes, and synthesis of genomic and sg viral RNAs, and it also separates them into large- (coronaviruses, toroviruses, and roniviruses), intermediate- (mesoniviruses), and small-genome (arteriviruses) nidoviruses to stress the clear genome size differences [18, 25, 26, 50]. Research on porcine nidoviruses is necessary not only for developing strategies to control these viruses in pig populations but also for understanding the molecular biology of human or veterinary-important nidoviruses. Despite extensive attention and research investment, two porcine nidoviruses, PRRSV and PEDV, continue to plague pig-producing countries, causing a significant economic impact on the swine industry worldwide. This is partially attributable to the lack of efficient vaccines that can confer full protection against nidoviral infections and the lack of antiviral agents to treat these infections. Although cholesterol is required for optimal infectivity of diverse non-enveloped and enveloped viruses [48, 56], its contribution to and specific function in porcine nidovirus replication are currently unknown. The present study showed that pharmacological sequestration of cholesterol in the cellular membrane but not the viral envelope exerts an efficient antiviral effect against PRRSV and PEDV in vitro. This effect could be counteracted by the addition of exogenous cholesterol, indicating the importance of membrane cholesterol for porcine nidovirus infection. Depletion of cellular cholesterol using the drug MβCD primarily affected the virus attachment and internalization stages, significantly affecting post-entry steps in the replication of porcine nidovirus. Altogether, our data indicate that cellular membrane cholesterol plays a critical role in entry of PRRSV and PEDV into target cells.
Because viruses are obligate intracellular parasites, they have evolved elaborate relationships with their host cells and developed the ability to modulate lipid composition, lipid synthesis, and host cell signaling pathways [6]. In particular, cholesterol-rich microdomains appear to be important in the entry of various viruses. Indeed, many viruses have been shown to exploit cholesterol, which is present in either the viral envelope [22, 54], cellular membrane [4, 36], or both [40, 48], for maximal virus entry. Furthermore, accumulating evidence indicates that cholesterol is an essential component in the life cycle of several nidoviruses. The depletion of cellular cholesterol inhibits the entry of coronaviruses, including mouse hepatitis virus [12], severe acute respiratory syndrome coronavirus (SARS-CoV) [32], human coronavirus 229E [42], and avian infectious bronchitis virus [22] as well as arteriviruses, including equine arteritis virus [41] and PRRSV [21, 55]. On the other hand, transmissible gastroenteritis virus (TGEV) and canine coronavirus require cholesterol both in the target cell membrane and in the viral envelope [43, 44]. The current study revealed that PRRSV and PEDV are sensitive to the depletion of cholesterol only in the cell membrane. Although cholesterol depletion has been shown to inhibit PRRSV entry into African green monkey kidney-derived MARC-145 cells, whether plasma membrane cholesterol is involved in either virus attachment or penetration or both is unknown [21, 55]. In the present study, we used a continuous PRRSV-permissive PAM cell line that is considered the primary cell target for PRRSV in the natural host, making it a good system for studying virus-host interactions [31]. In a previous study, Yin et al. [61] suggested that cholesterol is critical for a post-adsorption step in the entry of TGEV, another porcine alphacoronavirus. Porcine CD163 and APN have been shown to confer permissiveness of non-susceptible cell lines to PRRSV and PEDV, respectively, and have been identified as key molecules in the entry of porcine nidoviruses [9, 31, 39]. A previous report showed that cholesterol depletion did not alter CD163 expression in MARC-145 cells [21]. Likewise, the present study indicated that pharmacological depletion of cellular cholesterol had no effect on the levels of pCD163 and pAPN expression in porcine cells. Recent studies have indicated that APN is not a functional cellular receptor for PEDV, suggesting that the presence of the authentic virus receptor is essential for viral entry [33, 46]. Therefore, it is still possible that cellular cholesterol is quantitatively related to a hitherto unidentified receptor for PEDV. Based on our results, nevertheless, we propose that cellular cholesterol is important in both the binding and internalization stages of PRRSV and PEDV entry into susceptible cell lines. These findings are striking in that these two viruses are known to use different cell entry mechanisms for the initiation of virus infection: PRRSV enters PAM cells via receptor-mediated endocytosis followed by pH-dependent fusion between viral and endosomal membranes [60], whereas PEDV enters target cells via virus-receptor interactions, followed by direct pH-independent fusion of the viral and plasma membranes [27].
Because cholesterol is an essential lipid component of cell membranes, its depletion has the potential to inhibit virus entry via several mechanisms. Cholesterol is a critical structural component of lipid rafts, together with sphingolipids. These lipids influence viral infection by regulating viral and/or cellular membranes and thus can function by preferentially partitioning into specific membrane microdomains [6]. Cholesterol may affect virus entry by modifying interactions between virus particles and host cell membranes, and lipid recognition by certain viral constituents may be essential for virus entry [58]. In the case of SARS-CoV, cholesterol in the plasma membrane plays an important role in interactions of the viral spike protein and cellular receptor angiotensin-converting enzyme 2 for optimal infection [16]. By analogy, it is feasible that cellular cholesterol depletion might disturb binding of PRRSV and PEDV to specific cellular receptors. Secondly, the level of cholesterol is important for maintaining biological membrane fluidity, and its removal could reduce the potential for lateral diffusion in the membrane [7]. This reduction in membrane fluidity could have an influence on the entry of PRRSV and PEDV. Thirdly, the lipid environment, including the cholesterol level, is known to contribute to the charge of ion channels formed in cellular membranes [11]. Considering this issue, ion channel alterations in response to the lack of cellular cholesterol may affect the entry process of porcine nidoviruses. Lastly, cholesterol removal has been shown to result in the inhibition of cellular signaling pathways [53]. We previously found that PRRSV and PEDV activate specific intracellular signaling networks such as mitogen-activated protein kinase (MAPK) cascade pathways to favor replication of these viruses, but these signaling pathways are irrelevant to virus internalization [24, 28–30]. Based on these previous data, cellular cholesterol does not appear to act through MAPK signaling pathways. Although our analysis did not provide clear evidence of the mechanism by which cholesterol promotes porcine nidovirus entry, we assume that the mechanism may differ among viruses and that cholesterol-dependent virus entry might be dependent on more than one of the aforementioned pathways simultaneously.
In conclusion, our findings indicate that optimal infectivity of porcine nidoviruses requires cholesterol in the plasma membrane and that this is critical for the entry of PRRSV and PEDV. However, cholesterol depletion resulted in a reduction, but not abolishment, of virus infectivity, indicating that virus entry may still occur with lower levels of cholesterol but that increased cholesterol content makes this process more efficient. Future work should address the question of whether cholesterol facilitates PRRSV and PEDV entry through interactions between viral attachment proteins and cellular receptors and/or by affecting membrane fluidity. The current study indicates that cellular cholesterol is a key player in the early stages of porcine nidovirus infection, including attachment and penetration. Impeding porcine nidovirus entry is a viable antiviral strategy because it likely acts on extracellular targets, thereby limiting cell damage, and these viruses might be used as surrogate models for testing antiviral agents against human nidoviruses. Although further studies based on in vivo assessments are needed to evaluate the efficacy and safety of the cholesterol-depleting agent MβCD, the results presented here indicate that molecules or drugs that interfere with cholesterol function in virus entry should be considered candidates for antiviral approaches to porcine nidoviral diseases.
Compliance with ethical standards
Funding
This research was supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education (NRF-2015R1D1A1A09057406).
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval
This article does not contain any studies with animals performed by any of the authors.
References
- 1.Ahn A, Gibbons DL, Kielian M. The fusion peptide of Semliki Forest virus associates with sterol-rich membrane domains. J Virol. 2002;76:3267–3275. doi: 10.1128/JVI.76.7.3267-3275.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Barman S, Nayak DP. Lipid raft disruption by cholesterol depletion enhances influenza A virus budding from MDCK cells. J Virol. 2007;81:12169–12178. doi: 10.1128/JVI.00835-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bautista EM, Faaberg KS, Mickelson D, McGruder ED. Functional properties of the predicted helicase of porcine reproductive and respiratory syndrome virus. Virology. 2002;298:258–270. doi: 10.1006/viro.2002.1495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bavari S, Bosio CM, Wiegand E, Ruthel G, Will AB, Geisbert TW, Hevey M, Schmaljohn C, Schmaljohn A, Aman MJ. Lipid raft microdomains: a gateway for compartmentalized trafficking of Ebola and Marburg viruses. J Exp Med. 2002;195:593–602. doi: 10.1084/jem.20011500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bender FC, Whitbeck JC, Ponce de Leon M, Lou H, Eisenberg RJ, Cohen GH. Specific association of glycoprotein B with lipid rafts during herpes simplex virus entry. J Virol. 2003;77:9542–9552. doi: 10.1128/JVI.77.17.9542-9552.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Blaising J, Pécheur EI. Lipids: a key for hepatitis C virus entry and a potential target for antiviral strategies. Biochimie. 2013;95:96–102. doi: 10.1016/j.biochi.2012.07.016. [DOI] [PubMed] [Google Scholar]
- 7.Burger K, Gimpl G, Fahrenholz F. Regulation of receptor function by cholesterol. Cell Mol Life Sci. 2000;57:1577–1592. doi: 10.1007/PL00000643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cai Y, Liu Y, Zhang X. Suppression of coronavirus replication by inhibition of the MEK signaling pathway. J Virol. 2007;81:446–456. doi: 10.1128/JVI.01705-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Calvert JG, Slade DE, Shields SL, Jolie R, Mannan RM, Ankenbauer RG, Welch SK. CD163 expression confers susceptibility to porcine reproductive and respiratory syndrome viruses. J Virol. 2007;81:7371–7379. doi: 10.1128/JVI.00513-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cavanagh D. Nidovirales: a new order comprising Coronaviridae and Arteriviridae. Arch Virol. 1997;142:629–633. [PubMed] [Google Scholar]
- 11.Chang HM, Reitstetter R, Gruener R. Lipid-ion channel interactions: increasing phospholipid headgroup size but not ordering acyl chains alters reconstituted channel behavior. J Membr Biol. 1995;145:13–19. doi: 10.1007/BF00233303. [DOI] [PubMed] [Google Scholar]
- 12.Choi KS, Aizaki H, Lai MM. Murine coronavirus requires lipid rafts for virus entry and cell-cell fusion but not for virus release. J Virol. 2005;79:9862–9871. doi: 10.1128/JVI.79.15.9862-9871.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Desplanques AS, Nauwynck HJ, Vercauteren D, Geens T, Favoreel HW. Plasma membrane cholesterol is required for efficient pseudorabies virus entry. Virology. 2008;376:339–345. doi: 10.1016/j.virol.2008.03.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Finney DJ. The median lethal dose and its estimation. Arch Toxicol. 1985;56:215–218. doi: 10.1007/BF00295156. [DOI] [PubMed] [Google Scholar]
- 15.Funk A, Mhamdi M, Hohenberg H, Heeren J, Reimer R, Lambert C, Prange R, Sirma H. Duck hepatitis B virus requires cholesterol for endosomal escape during virus entry. J Virol. 2008;82:10532–10542. doi: 10.1128/JVI.00422-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Glende J, Schwegmann-Wessels C, Al-Falah M, Pfefferle S, Qu X, Deng H, Drosten C, Naim HY, Herrler G. Importance of cholesterol-rich membrane microdomains in the interaction of the S protein of SARS-coronavirus with the cellular receptor angiotensin-converting enzyme 2. Virology. 2008;381:215–221. doi: 10.1016/j.virol.2008.08.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gómez-Laguna J, Salguero FJ, Pallarés FJ, Carrasco L. Immunopathogenesis of porcine reproductive and respiratory syndrome in the respiratory tract of pigs. Vet J. 2013;195:148–155. doi: 10.1016/j.tvjl.2012.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gorbalenya AE, Enjuanes L, Ziebuhr J, Snijder EJ. Nidovirales: evolving the largest RNA virus genome. Virus Res. 2006;117:17–37. doi: 10.1016/j.virusres.2006.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hofmann M, Wyler R. Propagation of the virus of porcine epidemic diarrhea in cell culture. J Clin Microbiol. 1988;26:2235–2239. doi: 10.1128/jcm.26.11.2235-2239.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Holtkamp DJ, Kliebenstein JB, Neumann EJ, Zimmerman JJ, Rotto HF, Yoder TK, Wang C, Yeske PE, Mowrer CL, Haley CA. Assessment of the economic impact of porcine reproductive and respiratory syndrome virus on United States pork producers. J Swine Health Prod. 2013;21:72–84. [Google Scholar]
- 21.Huang L, Zhang YP, Yu YL, Sun MX, Li C, Chen PY, Mao X. Role of lipid rafts in porcine reproductive and respiratory syndrome virus infection in MARC-145 cells. Biochem Biophys Res Commun. 2011;414:545–550. doi: 10.1016/j.bbrc.2011.09.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Imhoff H, von Messling V, Herrler G, Haas L. Canine distemper virus infection requires cholesterol in the viral envelope. J Virol. 2007;81:4158–4165. doi: 10.1128/JVI.02647-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kim Y, Lee C. Ribavirin efficiently suppresses porcine nidovirus replication. Virus Res. 2013;171:44–53. doi: 10.1016/j.virusres.2012.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kim Y, Lee C. Extracellular signal-regulated kinase (ERK) activation is required for porcine epidemic diarrhea virus replication. Virology. 2015;484:181–193. doi: 10.1016/j.virol.2015.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lai MM, Perlman S, Anderson LJ. Coronaviridae. In: Howley PM, Griffin DE, Lamb RA, Martin MA, Roizman B, Straus SE, Knipe DM, editors. Fields virology. Philadelphia: Lippincott Williams & Wilkins; 2007. pp. 1305–1335. [Google Scholar]
- 26.Lauber C, Ziebuhr J, Junglen S, Drosten C, Zirkel F, Nga PT, Morita K, Snijder EJ, Gorbalenya AE. Mesoniviridae: a proposed new family in the order Nidovirales formed by a single species of mosquito-borne viruses. Arch Virol. 2012;157:1623–1628. doi: 10.1007/s00705-012-1295-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lee C. Porcine epidemic diarrhea virus: an emerging and re-emerging epizootic swine virus. Virol J. 2015;22:193. doi: 10.1186/s12985-015-0421-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lee C, Kim Y, Jeon JH. JNK and p38 mitogen-activated protein kinase pathways contribute to porcine epidemic diarrhea virus infection. Virus Res. 2016;222:1–12. doi: 10.1016/j.virusres.2016.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lee YJ, Lee C. Porcine reproductive and respiratory syndrome virus replication is suppressed by inhibition of the extracellular signal-regulated kinase (ERK) signaling pathway. Virus Res. 2010;152:50–58. doi: 10.1016/j.virusres.2010.06.002. [DOI] [PubMed] [Google Scholar]
- 30.Lee YJ, Lee C. Stress-activated protein kinases are involved in porcine reproductive and respiratory syndrome virus infection and modulate virus-induced cytokine production. Virology. 2012;427:80–89. doi: 10.1016/j.virol.2012.02.017. [DOI] [PubMed] [Google Scholar]
- 31.Lee YJ, Park CK, Nam E, Kim SH, Lee OS, Lee DS, Lee C. Generation of a porcine alveolar macrophage cell line for the growth of porcine reproductive and respiratory syndrome virus. J Virol Methods. 2010;163:410–415. doi: 10.1016/j.jviromet.2009.11.003. [DOI] [PubMed] [Google Scholar]
- 32.Li GM, Li YG, Yamate M, Li SM, Ikuta K. Lipid rafts play an important role in the early stage of severe acute respiratory syndrome-coronavirus life cycle. Microbes Infect. 2007;9:96–102. doi: 10.1016/j.micinf.2006.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li W, Luo R, He Q, van Kuppeveld FJM, Rottier PJM, Bosch BJ. Aminopeptidase N is not required for porcine epidemic diarrhea virus cell entry. Virus Res. 2017;235:6–13. doi: 10.1016/j.virusres.2017.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liao Z, Cimakasky LM, Hampton R, Nguyen DH, Hildreth JE. Lipid rafts and HIV pathogenesis: host membrane cholesterol is required for infection by HIV type 1. AIDS Res Hum Retroviruses. 2001;17:1009–1019. doi: 10.1089/088922201300343690. [DOI] [PubMed] [Google Scholar]
- 35.Liao Z, Graham DR, Hildreth JE. Lipid rafts and HIV pathogenesis: virion-associated cholesterol is required for fusion and infection of susceptible cells. AIDS Res Hum Retroviruses. 2003;19:675–687. doi: 10.1089/088922203322280900. [DOI] [PubMed] [Google Scholar]
- 36.Lu X, Xiong Y, Silver J. Asymmetric requirement for cholesterol in receptor-bearing but not envelope-bearing membranes for fusion mediated by ecotropic murine leukemia virus. J Virol. 2002;76:6701–6709. doi: 10.1128/JVI.76.13.6701-6709.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lunney JK, Fritz ER, Reecy JM, Kuhar D, Prucnal E, Molina R, Christopher-Hennings J, Zimmerman J, Rowland RR. Interleukin-8, interleukin-1beta, and interferon-gamma levels are linked to PRRS virus clearance. Viral Immunol. 2010;23:127–134. doi: 10.1089/vim.2009.0087. [DOI] [PubMed] [Google Scholar]
- 38.Mayo MA. A summary of taxonomic changes recently approved by the ICTV. Arch Virol. 2002;147:1655–1656. doi: 10.1007/s007050200039. [DOI] [PubMed] [Google Scholar]
- 39.Nam E, Lee C. Contribution of the porcine aminopeptidase N (CD13) receptor density to porcine epidemic diarrhea virus infection. Vet Microbiol. 2010;144:41–50. doi: 10.1016/j.vetmic.2009.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nayak DP, Hui EK. The role of lipid microdomains in virus biology. Subcell Biochem. 2004;37:443–491. doi: 10.1007/978-1-4757-5806-1_14. [DOI] [PubMed] [Google Scholar]
- 41.Nitschke M, Korte T, Tielesch C, Ter-Avetisyan G, Tünnemann G, Cardoso MC, Veit M, Herrmann A. Equine arteritis virus is delivered to an acidic compartment of host cells via clathrin-dependent endocytosis. Virology. 2008;377:248–254. doi: 10.1016/j.virol.2008.04.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nomura R, Kiyota A, Suzaki E, Kataoka K, Ohe Y, Miyamoto K, Senda T, Fujimoto T. Human coronavirus 229E binds to CD13 in rafts and enters the cell through caveolae. J Virol. 2004;78:8701–8708. doi: 10.1128/JVI.78.16.8701-8708.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pratelli A, Colao V. Role of the lipid rafts in the life cycle of canine coronavirus. J Gen Virol. 2015;96:331–337. doi: 10.1099/vir.0.070870-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ren X, Glende J, Yin J, Schwegmann-Wessels C, Herrler G. Importance of cholesterol for infection of cells by transmissible gastroenteritis virus. Virus Res. 2008;137:220–224. doi: 10.1016/j.virusres.2008.07.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Saif LJ, Pensaert MB, Sestack K, Yeo SG, Jung K. Coronaviruses. In: Straw BE, Zimmerman JJ, Karriker LA, Ramirez A, Schwartz KJ, Stevenson GW, editors. Diseases of swine. Ames: Wiley-Blackwell; 2012. pp. 501–524. [Google Scholar]
- 46.Shirato K, Maejima M, Islam MT, Miyazaki A, Kawase M, Matsuyama S, Taguchi F. Porcine aminopeptidase N is not a cellular receptor of porcine epidemic diarrhea virus, but promotes its infectivity via aminopeptidase activity. J Gen Virol. 2016;97:2528–2539. doi: 10.1099/jgv.0.000563. [DOI] [PubMed] [Google Scholar]
- 47.Siddell S, Snijder EJ. Nidoviruses. In: Gallagher T, Snijder EJ, Perlman S, editors. Introduction to nidoviruses. Washinton, DC: ASM Press; 2008. pp. 1–13. [Google Scholar]
- 48.Smith AE, Helenius A. How viruses enter animal cells. Science. 2004;304:237–242. doi: 10.1126/science.1094823. [DOI] [PubMed] [Google Scholar]
- 49.Snijder EJ, Kikkert M, Fang Y. Arterivirus molecular biology and pathogenesis. J Gen Virol. 2013;94:2141–2163. doi: 10.1099/vir.0.056341-0. [DOI] [PubMed] [Google Scholar]
- 50.Snijder EJ, Spaan WJM. Arteriviridae. In: Knipe DM, Howley PM, Griffin DE, Lamb RA, Martin MA, Roizman B, Straus SE, editors. Fields virology. Philadelphia: Lippincott Williams & Wilkins; 2007. pp. 1337–1361. [Google Scholar]
- 51.Spaan WJM, Brian D, Cavanagh D, de Groot RJ, Enjuanes L, Gorbalenya AE, Holmes KV, Masters PS, Rottier PJ, Taguchi F, Talbot P. Family Coronaviridae. In: Fauquet CM, Mayo MA, Maniloff J, Desselberg U, Ball LA, editors. Virus taxonomy: eighth report of the international committee on taxonomy of viruses. London: Elsevier Academic Press; 2005. pp. 947–964. [Google Scholar]
- 52.Stevenson GW, Hoang H, Schwartz KJ, Burrough ER, Sun D, Madson D, Cooper VL, Pillatzki A, Gauger P, Schmitt BJ, Koster LG, Killian ML, Yoon KJ. Emergence of Porcine epidemic diarrhea virus in the United States: clinical signs, lesions, and viral genomic sequences. J Vet Diagn Invest. 2013;25:649–654. doi: 10.1177/1040638713501675. [DOI] [PubMed] [Google Scholar]
- 53.Stulnig TM, Berger M, Sigmund T, Stockinger H, Horejsí V, Waldhäusl W. Signal transduction via glycosyl phosphatidylinositol-anchored proteins in T cells is inhibited by lowering cellular cholesterol. J Biol Chem. 1997;272:19242–19247. doi: 10.1074/jbc.272.31.19242. [DOI] [PubMed] [Google Scholar]
- 54.Sun X, Whittaker GR. Role for influenza virus envelope cholesterol in virus entry and infection. J Virol. 2003;77:12543–12551. doi: 10.1128/JVI.77.23.12543-12551.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sun Y, Xiao S, Wang D, Luo R, Li B, Chen H, Fang L. Cellular membrane cholesterol is required for porcine reproductive and respiratory syndrome virus entry and release in MARC-145 cells. Sci China Life Sci. 2011;54:1011–1018. doi: 10.1007/s11427-011-4236-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Suzuki T, Suzuki Y. Virus infection and lipid rafts. Biol Pharm Bull. 2006;29:1538–1541. doi: 10.1248/bpb.29.1538. [DOI] [PubMed] [Google Scholar]
- 57.Takano T, Satomi Y, Oyama Y, Doki T, Hohdatsu T. Differential effect of cholesterol on type I and II feline coronavirus infection. Arch Virol. 2016;161:125–133. doi: 10.1007/s00705-015-2655-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Teissier E, Pécheur EI. Lipids as modulators of membrane fusion mediated by viral fusion proteins. Eur Biophys J. 2007;36:887–899. doi: 10.1007/s00249-007-0201-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.van Aken D, Zevenhoven-Dobbe J, Gorbalenya AE, Snijder EJ. Proteolytic maturation of replicase polyprotein pp1a by the nsp4 main proteinase is essential for equine arteritis virus replication and includes internal cleavage of nsp7. J Gen Virol. 2006;87:3473–3482. doi: 10.1099/vir.0.82269-0. [DOI] [PubMed] [Google Scholar]
- 60.Van Breedam W, Delputte PL, Van Gorp H, Misinzo G, Vanderheijden N, Duan X, Nauwynck HJ. Porcine reproductive and respiratory syndrome virus entry into the porcine macrophage. J Gen Virol. 2010;91:1659–1667. doi: 10.1099/vir.0.020503-0. [DOI] [PubMed] [Google Scholar]
- 61.Yin J, Glende J, Schwegmann-Wessels C, Enjuanes L, Herrler G, Ren X. Cholesterol is important for a post-adsorption step in the entry process of transmissible gastroenteritis virus. Antivir Res. 2010;88:311–316. doi: 10.1016/j.antiviral.2010.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhu L, Ding X, Tao J, Wang J, Zhao X, Zhu G. Critical role of cholesterol in bovine herpesvirus type 1 infection of MDBK cells. Vet Microbiol. 2010;144:51–57. doi: 10.1016/j.vetmic.2009.12.031. [DOI] [PMC free article] [PubMed] [Google Scholar]