

Abstract
Torque teno sus virus 1 (TTSuV1) is a novel virus that has been found widely distributed in the swine population in recent years. Analysis of codon usage can reveal much about the molecular evolution of TTSuV1. In this study, synonymous codon usage patterns and the key determinants in the coding region of 29 available complete TTSuV1 genome sequences were examined. By calculating the nucleotide content and relative synonymous codon usage (RSCU) of TTSuV1 coding sequences, we found that the preferentially used codons were mostly those ending with A or C nucleotides; less-used codons were mostly codons ending with U or G nucleotides, and these were mainly affected by composition constraints. Although there was a variation in codon usage bias among different TTSuV1 genomes, the codon usage bias and GC content in the TTSuV1 coding region was lower, which was mainly determined by the base composition in the third codon position and the effective number of codons (ENC) value. Moreover, the results of correspondence analysis (COA) indicated that the codon usage patterns of TTSuV1 isolated from different countries varied greatly and had significant differences. In addition, Spearman’s rank correlation analysis and an ENC plot revealed that apart from mutation pressure, which was critical in determining the codon usage pattern, other factors were involved in shaping the evolution of codon usage bias in TTSuV1, such as natural selection. Those results suggested that synonymous codon usage patterns of TTSuV1 genomes were the result of interaction between mutation pressure and natural selection. The information from this study not only provides important insights into the synonymous codon usage pattern of TTSuV1, but also helps to identify the main factors affecting codon usage by this virus.
Keywords: Codon Usage, Synonymous Codon, Codon Usage Bias, Synonymous Codon Usage, Torque Teno Virus
Introduction
It is well known that the genetic code is composed of 61 codons to represent the 20 standard amino acids, and three codons to represent termination signals (UAA, UAG, UGA). Alternative codons encoding the same amino acid are termed “synonymous codons”. In general, each amino acid can be encoded by one (Met, Tyr) or more (up to six) synonymous codons. Previous studies have indicated that synonymous codons are not favored equally within a genome, and members of some species and some genes preferentially use one or several particular synonymous codons. The higher frequency of these preferred codons is called “codon bias”. Genes from different species or the same species have their own bias of synonymous codon usage [1, 2]. However, mutations resulting in synonymous codons tend to occur in the third base position, and the codons can be interchanged without altering the primary sequence of the protein product. Generally, compositional constraints under mutational pressure and translation selection in nature have been found to be the two main factors accounting for codon usage variation among genes [3–5]. In some genes with extremely high A + T or G + C content [3, 6–8], mutation pressure plays an important role in affecting the synonymous codon usage pattern. In addition, synonymous codon usage bias is also related with to the processes of DNA replication and transcription [9], gene structure, protein secondary structures [10–12] and environmental conditions [13].
Torque teno virus (TTV) is a small, circular, non-enveloped, negative-sense single-stranded DNA (ssDNA) virus [14, 15] that belongs to the newly created family Anelloviridae [16], together with torque teno minivirus (TTMV) and torque teno midivirus (TTMDV). TTV was first detected in a human patient with post-transfusion hepatitis of unknown aetiology in 1997 [17] and was then later detected in many other vertebrate animals and domesticated farm animals, such as chickens, pigs, cows and sheep [18–21]. In swine, two genetically distinct viruses, torque teno sus virus 1 (TTSuV1) and 2 (TTSuV2), have been found [22, 23]. Sequence analysis has shown that the TTSuV1 and TTSuV2 genomes are only 44 % homologous [23]. Torque teno sus viruses (TTSuVs) are widely distributed and have been detected in serum, plasma, semen, colostrum, feces and tissues of swine [24–27]. Because a definitive pathogenic role of this virus has not been demonstrated, it is considered a non-virulent commensal inhabitant of vertebrates. Although TTSuV infection in swine has not directly lead to any diseases and is considered non-pathogenic, the virus still plays an important role in co-infection. It is considered to be a symbiotic virus in vertebrates, and it is associated with economically important diseases.
The TTV genome has an extremely wide range of sequence divergence. The TTSuVs genome approximately 2.8 kb in length, including a coding region translated from the negative-sense ssDNA and a short stretch of untranslated region (UTR) with a high GC content [16, 28]. In the coding region, three open reading frames (ORFs), ORF1, ORF2 and ORF3 (also known as ORF1/1 and ORF2/2), have been found, and the three ORFs have many overlapping genes. Previous studies have confirmed that the Rep protein is encoded by the largest open reading frame (ORF1), which encodes mostly highly hydrophilic viral capsid proteins. The functions of ORF2 and ORF3 and their encoded proteins are unknown. Although many genomic analyses have been performed on TTSuVs in recent years, in-depth studies on codon usage patterns and the factors influencing them are lacking. The codon usage pattern may be important for revealing the molecular mechanism and evolutionary process of TTSuVs. In this study, the key genetic determinants of codon usage in TTSuV1 are examined based on the TTSuV1 coding region sequences. To our knowledge, this is the first systemic study to analyze the synonymous codon usage pattern as well as the main factors affecting codon usage in TTSuV1.
Materials and methods
Sequence data
The 29 available complete genome sequences of TTSuV1 were downloaded from the National Center for Biotechnology Information (NCBI) (http://www.ncbi.nlm.nih.gov/Genbank/). Detailed information about these sequences is given in Table 1. The coding-region sequence of each TTSuV1 isolate was analyzed using biosoftware DNAStar 7.0 for Windows.
Table 1.
Complete TTSuV1 genome sequences used in this study
| No. | Accession no. | Name | Country | Year | Length (bp) |
|---|---|---|---|---|---|
| 1 | AB076001 | SD-TTV31 | Japan | 2009 | 2878 |
| 2 | AY823990 | 1p | Brazil | 2010 | 2872 |
| 3 | GQ120664 | swSTHY-TT27 | Canada | 2005 | 2875 |
| 4 | GU188045 | 471819 | Germany | 2008 | 2863 |
| 5 | GU450331 | PTTV1SH0822 | China | 2008 | 2823 |
| 6 | GU456383 | PTTV1a-VA | USA | 2008 | 2878 |
| 7 | GU456384 | PTTV1b-VA | USA | 2008 | 2875 |
| 8 | GU570198 | TTV1_19 N | Spain | 2011 | 2913 |
| 9 | GU570199 | TTV1_20 N | Spain | 2011 | 2913 |
| 10 | GU570200 | TTV1_1914 | Spain | 2011 | 2913 |
| 11 | GU570201 | TTV1_G21 | Spain | 2011 | 2910 |
| 12 | GU570202 | TTV1_G26 | Spain | 2011 | 2910 |
| 13 | HM633242 | TTV1Bj1-1 | China | 2009 | 2897 |
| 14 | HM633243 | TTV1Bj2-1 | China | 2009 | 2910 |
| 15 | HM633244 | TTV1Bj2-2 | China | 2009 | 2876 |
| 16 | HM633245 | TTV1Bj3 | China | 2009 | 2910 |
| 17 | HM633246 | TTV1Bj4-1 | China | 2009 | 2869 |
| 18 | HM633247 | TTV1Bj4-2 | China | 2009 | 2875 |
| 19 | HM633248 | TTV1Bj5 | China | 2009 | 2868 |
| 20 | HM633249 | TTV1Bj6-1 | China | 2009 | 2882 |
| 21 | HM633250 | TTV1Bj7-1 | China | 2009 | 2873 |
| 22 | HM633251 | TTV1Bj10 | China | 2009 | 2914 |
| 23 | HM633252 | TTV1Fj3 | China | 2009 | 2897 |
| 24 | HM633253 | TTV1Gx3-1 | China | 2009 | 2868 |
| 25 | HM633254 | TTV1Hlj5 | China | 2009 | 2878 |
| 26 | HM633255 | TTV1Hlj16 | China | 2009 | 2874 |
| 27 | HM633256 | TTV1Hlj20 | China | 2009 | 2911 |
| 28 | HM633257 | TTV1Hlj21 | China | 2009 | 2875 |
| 29 | HM633258 | TTV1Ln23-1 | China | 2009 | 2875 |
The primary indicators of codon usage analysis
Nucleotide content
“Nucleotide content” refers to the frequency of the nucleotides A, C, U (T), G in the genomic sequence (A %, C %, U %, and T %). The frequency of each nucleotide in the third position of synonymous codons (A3 %, C3 %, U3 % and G3 %) of the TTSuV1 coding region were calculated. The frequency of the occurrence of G and C nucleotides in the genomic sequence, also known as GC content, and the frequency of G and C in the variable third position of codons (GC %) were also calculated. Codons AUG and UGG are the only codons for Met and Trp, respectively, and the termination codons UAA, UAG and UGA do not encode any amino acids. Therefore, these five codons would not exhibit any usage bias and were therefore excluded from this study.
Effective number of codons
The effective number of codons (ENC) is often used to measure the magnitude of codon bias for an individual gene in a way that is independent of the gene length and number of amino acids. The ENC value ranges from 20, for a gene in which only one of the possible synonymous codons is used for each amino acid, to 61, for a gene in which all synonymous codons are used equally [29]. If the ENC value is closer to 20, the codon usage bias is stronger, and the degree to which codons are used non-randomly is higher. It is generally accepted that genes have a significant codon bias when the ENC value is less than or equal to 35. Thus, the ENC value provides an intuitively meaningful estimate of the codon preference in a gene. This method was used in this study to evaluate the degree of codon usage bias in the TTSuV1 coding region.
Relative synonymous codon usage
To investigate the characteristics of synonymous codon usage, the relative synonymous codon usage (RSCU) of TTSuV1 coding region sequences was calculated. The RSCU value represents the ratio between the usage frequency of one codon in gene sample and the expected frequency in the synonymous codon family. It is calculated by dividing the observed codon usage by the expected usage when all codons for the particular amino acid are used equally. RSCU values are calculated according to the previously published equation [30, 31]
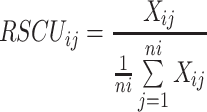 |
where Xij is the observed number of the “ith” codon for the “jth” amino acid. “ni” is the total number of synonymous codons that the “jth” amino acid has. If a codon RSCU value is equal to 1, the codon usage frequency is close to the expected frequency, and there is no codon usage bias for that amino acid; if a codon RSCU value is greater than 1, it has positive codon usage bias and the usage frequency is higher than expected for that codon; otherwise, it is lower than the expected value. A synonymous codon with an RSCU value greater than 1.6 is thought to be over-represented; otherwise, it is regarded as under-represented [32].
Correspondence analysis
Correspondence analysis (COA), also known as principal component analysis (PCA), which is a commonly used multivariate statistical method [33], is used to analyze major trends in codon usage pattern among different TTSuV1 coding sequences. In COA, all genes were plotted in a 59-dimensional vector, and each dimension was represented as a synonymous codon, corresponding to the RSCU values of the 59 codons. The results could be applied for finding out the major factors affecting codon usage bias. We set up a two-dimensional coordinate system made up of the first principal component (f 1’) and second principal component (f 2’). Major trends within this dataset could be determined using measurements of relative inertia, and genes were ordered according to their positions along the axis of major inertia. This method has been used successfully to investigate the variation of RSCU values among all strains [34–36].
Correlation analysis
Correlation analysis of TTSuV1 was used to identify the relationship between the nucleotide composition and the codon usage pattern, which was implemented based on Spearman’s rank correlation analysis.
Analysis tools
The nucleotide content was calculated using the program MEGA5 for Windows. A3 %, C3 %, U3 %, G3 % values, GC content, GC3 %, RSCU, ENC values and COA were calculated using the program CodonW version 1.4 (http://codonw.sourceforge.net). Correlation analysis was carried out by using the multianalysis software SPSS version 11.6.
Results
The characteristics of synonymous codon usage in TTSuV1
The nucleotide content and A3 %, C3 %, U3 %, G3 % values in TTSuV1 coding-region sequences are listed in Table 2. In order to investigate the codon usage pattern of TTSuV1, we calculated various RSCU values of various codons from 29 TTSuV1 coding sequences. The overall RSCU values of 59 codons in the TTSuV1 coding region are listed in Table 3.
Table 2.
Nucleotide content of 29 TTSuV1 coding regions (%)
| No. | U | U 3 | C | C 3 | A | A 3 | G | G 3 | GC | GC 3 | ENC |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 19.14 | 25.49 | 22.25 | 30.24 | 32.31 | 39.92 | 26.29 | 30.74 | 50.2 | 47.9 | 57.12 |
| 2 | 19.15 | 26.25 | 21.35 | 34.38 | 33.82 | 36.08 | 25.68 | 32.10 | 48.7 | 51.0 | 57.71 |
| 3 | 19.20 | 26.44 | 21.81 | 33.20 | 33.71 | 38.03 | 25.27 | 31.29 | 48.5 | 49.5 | 55.70 |
| 4 | 19.71 | 25.59 | 22.05 | 34.84 | 32.14 | 34.62 | 26.10 | 32.05 | 49.7 | 52.2 | 57.59 |
| 5 | 18.27 | 25.15 | 22.25 | 33.40 | 34.11 | 37.37 | 25.36 | 32.11 | 49.5 | 50.6 | 56.42 |
| 6 | 19.48 | 24.80 | 22.16 | 30.56 | 32.51 | 40.73 | 25.85 | 29.65 | 49.4 | 47.5 | 56.29 |
| 7 | 18.76 | 25.30 | 21.91 | 33.13 | 34.06 | 39.63 | 25.27 | 30.02 | 48.9 | 49.0 | 54.45 |
| 8 | 19.44 | 28.06 | 22.64 | 32.21 | 32.66 | 38.06 | 25.26 | 28.51 | 49.5 | 47.5 | 57.12 |
| 9 | 19.44 | 28.12 | 22.54 | 32.08 | 32.71 | 38.06 | 25.31 | 28.51 | 49.5 | 47.5 | 57.20 |
| 10 | 19.44 | 28.12 | 22.54 | 32.08 | 32.71 | 38.06 | 25.31 | 28.51 | 49.5 | 47.5 | 57.20 |
| 11 | 18.70 | 26.01 | 23.15 | 30.64 | 31.42 | 36.94 | 26.73 | 32.06 | 51.1 | 49.6 | 58.57 |
| 12 | 18.70 | 25.91 | 23.15 | 30.71 | 31.42 | 37.13 | 26.73 | 31.85 | 51.1 | 49.5 | 58.74 |
| 13 | 19.98 | 25.19 | 22.19 | 32.12 | 34.29 | 40.81 | 23.54 | 29.72 | 47.2 | 48.0 | 54.28 |
| 14 | 18.89 | 26.54 | 22.91 | 30.19 | 31.28 | 36.61 | 26.92 | 31.98 | 51.2 | 49.3 | 58.10 |
| 15 | 18.80 | 25.92 | 21.72 | 33.47 | 33.53 | 36.21 | 25.95 | 31.97 | 49.4 | 50.8 | 54.96 |
| 16 | 18.88 | 26.22 | 22.94 | 32.58 | 31.79 | 36.61 | 26.39 | 31.12 | 50.7 | 49.9 | 57.51 |
| 17 | 19.61 | 25.52 | 22.65 | 34.71 | 31.72 | 34.69 | 26.01 | 31.59 | 50.0 | 52.1 | 57.32 |
| 18 | 18.61 | 24.25 | 22.06 | 33.40 | 33.77 | 38.29 | 25.56 | 31.21 | 49.2 | 50.4 | 54.45 |
| 19 | 19.61 | 25.52 | 22.70 | 34.71 | 31.77 | 34.76 | 25.92 | 31.65 | 50.0 | 52.1 | 57.17 |
| 20 | 19.38 | 25.00 | 22.77 | 30.27 | 31.78 | 39.92 | 26.07 | 28.96 | 50.2 | 47.4 | 55.55 |
| 21 | 19.77 | 26.10 | 22.10 | 31.48 | 31.77 | 35.63 | 26.36 | 31.69 | 50.1 | 50.1 | 55.87 |
| 22 | 19.39 | 27.92 | 22.68 | 32.28 | 32.76 | 37.70 | 25.17 | 28.87 | 49.4 | 47.9 | 56.54 |
| 23 | 20.03 | 24.71 | 22.43 | 32.18 | 34.25 | 40.88 | 23.29 | 29.62 | 47.1 | 48.1 | 54.58 |
| 24 | 19.57 | 25.09 | 22.65 | 35.02 | 31.58 | 34.70 | 26.20 | 31.72 | 50.1 | 52.4 | 57.11 |
| 25 | 18.97 | 26.53 | 22.46 | 34.49 | 33.04 | 35.11 | 25.52 | 31.56 | 50.0 | 51.3 | 56.24 |
| 26 | 18.61 | 24.25 | 22.06 | 33.40 | 33.77 | 38.21 | 25.56 | 31.14 | 49.2 | 50.4 | 54.58 |
| 27 | 18.65 | 26.86 | 22.72 | 32.55 | 32.95 | 37.50 | 25.68 | 31.17 | 50.0 | 49.4 | 57.21 |
| 28 | 19.81 | 25.94 | 22.22 | 32.52 | 31.57 | 35.32 | 26.39 | 31.59 | 50.2 | 50.6 | 57.08 |
| 29 | 18.56 | 24.25 | 22.06 | 33.40 | 33.87 | 38.41 | 25.51 | 30.93 | 49.1 | 50.2 | 54.79 |
| Mean | 19.20 | 25.90 | 22.39 | 32.63 | 32.72 | 37.45 | 25.70 | 30.82 | 49.6 | 49.6 | 56.46 |
Table 3.
RSCU values of codons in the TTSuV1 coding region
| Amino acid | Codon | RSCU | Amino acid | Codon | RSCU |
|---|---|---|---|---|---|
| Phe | UUU | 0.76 | Tyr | UAU | 1.10 |
| UUC | 1.24 | UAC | 0.90 | ||
| Leu | UUA | 1.38 | Ala | GCU | 1.20 |
| UUG | 1.21 | GCC | 1.15 | ||
| CUU | 0.49 | GCA | 1.15 | ||
| CUC | 0.71 | GCG | 0.50 | ||
| CUA | 0.99 | His | CAU | 0.60 | |
| CUG | 1.22 | CAC | 1.40 | ||
| Ile | AUU | 0.57 | Gln | CAA | 0.90 |
| AUC | 1.02 | CAG | 1.10 | ||
| AUA | 1.41 | Asn | AAU | 1.01 | |
| Val | GUU | 0.62 | AAC | 0.99 | |
| GUC | 0.40 | Lys | AAA | 1.22 | |
| GUA | 1.28 | AAG | 0.78 | ||
| GUG | 1.71 | Asp | GAU | 0.74 | |
| Ser | UCU | 0.75 | GAC | 1.26 | |
| UCC | 0.72 | Glu | GAA | 1.08 | |
| UCA | 1.49 | GAG | 0.92 | ||
| UCG | 0.56 | Cys | UGU | 0.55 | |
| AGU | 1.39 | UGC | 1.45 | ||
| AGC | 1.10 | Arg | CGU | 0.51 | |
| Pro | CCU | 1.21 | CGC | 0.93 | |
| CCC | 0.72 | CGA | 0.63 | ||
| CCA | 1.11 | CGG | 0.55 | ||
| CCG | 0.96 | AGA | 1.34 | ||
| Thr | ACU | 1.01 | AGG | 2.05 | |
| ACC | 1.23 | Gly | GGU | 0.74 | |
| ACA | 1.21 | GGC | 0.99 | ||
| ACG | 0.55 | GGA | 1.31 | ||
| GGG | 0.95 |
The preferentially used codon for each amino acid is displayed in bold
As shown in Table 2, we found that A % and A3 % were the highest, with average values of 32.72 % and 37.45 %. U % and U3 % were the lowest, with average values of 19.20 % and 25.90 %. G % (25.70 %) was higher than C % (22.39 %), while G3 % (30.82 %) was lower than C3 % (32.63 %), indicating that the TTSuV1 coding region might preferentially use A-ended synonymous codons over U-ended synonymous codons. In addition, compared with other vertebrate DNA viruses, the TTSuV1 genome had a lower and stable GC %, ranging from 47.2 % to 51.2 %, with an average value of 49.6 %. The GC3 % was also lower and stable, ranging from 47.5 % to 52.4 %, with an average value of 49.6 %. The average value of GC3 % was similar to that of GC %, indicating that GC3 % was mainly affected by GC %. Hence, the fact that the GC content was lower in TTSuV1 coding sequences and that the codons ending in C were used in a biased way suggested that the content of G or C at the third position of the codon had an effect on the synonymous codons usage pattern. Apart from this, we can also see in Table 2 that the ENC values of the TTSuV1 coding sequences had less fluctuation, ranging from 54.45 to 58.74, with an average value of 56.46, which indicates that the codon bias of the TTSuV1 genome is stable. In general, the lower ENC value indicates a higher codon usage preference and a higher gene expression level as well. The results show that the codon usage bias of TTSuV1 is lower, and the codon usage pattern is mainly affected by base composition.
As shown in Table 3, among the 18 preferred synonymous codons, 11 ended in A or C and seven ended in G or U, and therefore, those A-ended or C-ended codons were prone to be used in the TTSuV1 coding region. In addition, the lowest preferentially used synonymous codons in the TTSuV1 coding region were G-ended and U-ended, with four G-ended and four U-ended codons among the nine under-represented synonymous codons, such as CUU for Leu and UCG for Ser, for which the RSCU values ranged from 0.40 to 0.57. These results suggest that compositional limitation often play an integral role in the codon usage pattern of TTSuV1.
Genetic relationship based on synonymous codon usage in TTSuV1
To study the codon usage variation of different TTSuV1 coding genes, CodonW analysis of the ENC values, and GC and GC3 content of TTSuV1 coding sequences was done (Table 2). The ENC values were calculated for each TTSuV1 strain, and an ENC plot was made by plotting ENC values against the G + C content at the third position of the synonymous codon (GC3 %; Fig. 1).
Fig. 1.
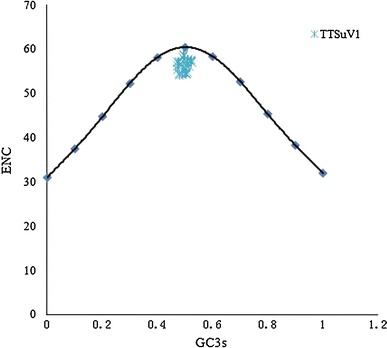
Distribution of the ENC and G + C content at the synonymous third codon position (GC3). The curve indicates the expected codon usage if G + C compositional constrainted alone account for codon usage bias
Figure 1 shows that all points aggregate below the expected curve, which suggests that, apart from mutation pressure, which influences the codon usage pattern in TTSuV1, the codon usage pattern is also influenced by other factors, such as natural selection.
Correspondence analysis of codon usage
To investigate the variation of the RSCU value in the TTSuV1 coding region, COA was implemented for these TTSuV1 coding sequences, examined as a single dataset based on the RSCU value of each gene (Fig. 2). The COA of TTSuV1 coding sequences detected the first principal component (f 1’), which could account for 12.18 % of the total synonymous codon usage variation, and the second principal component (f 2’) accounted for 20.88 % of the total variation. When considering the geographical factors that potentially influenced TTSuV1 evolution, there was an obvious geographical distribution. For example, it was observed that most of the genes from different geographical areas were spread equally over both sides of the two axes, and deviation from the cluster, except one strain isolated from Brazil, concentrated on the first axis. The four strains isolated from Spain were equally spread over both sides of the first axis and partially overlapped with each other. It was obvious that strains from China were distributed over two independent areas and had a significant difference, demonstrating that different strains from the same geographical area showed great differences in their codon usage. In addition, the overall codon usage pattern of TTSuV1 isolated from Spain, the USA and Germany was different from those from Brazil and Canada, and the TTSuV1 isolates from Spain, the USA and Germany had an amount of genetic diversity that was similar to that of strains isolated from China.
Fig. 2.
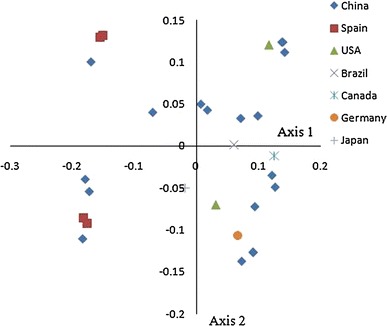
The genetic characteristic of TTSuV1 isolated from different countries
These results indicate that the geographical diversity of the TTSuV1 coding sequences might be a limiting factor affecting the codon usage of the whole TTSuV1 genome, even reflecting the characteristics of TTSuV1 evolution to some degree. In this case, the codon usage variation might be one of factors that drives TTSuV1 evolution.
Effect of mutational bias on the codon usage variation in TTSuV1
Mutation pressure and natural selection have been considered the two key factors affecting codon usage patterns. In order to investigate whether the primary factor influencing the codon usage pattern in the TTSuV1 coding region was mutation pressure or natural selection, we compared the correlation between A %, U %, G %, C %, GC % and U3 %, G3 %, C3 %, GC3 % by correlation analysis (Table 4). There are three different degrees of correlation, including positive correlation, negative correlation, and non-correlation, among nucleotides [37]. The analysis results showed that A3 % had marked negative correlation with G % and GC % (p < 0.01). U3 %, G3 % C3 % and GC3 % had non-significant corrrelation with A %, U %, G %, C % and GC % (p > 0.05), except that G3 % and C3 % had significant correlation with G % (p < 0.01) and C % (0.01 < p < 0.05), respectively. GC3 % had non-significant correlation with A %, U %, G %, C % and GC % (p > 0.05). The results indicate that nucleotide constraints could influence the codon usage pattern, but they are not the only factor. The codon usage pattern of the TTSuV1 coding region was affected by natural selection, apart from mutation pressure.
Table 4.
Correlation analysis between A, U, C, and G content and A3, U3, C3, and G3 content in TTSuV1 coding sequences
| A3 % | U3 % | G3 % | C3 % | GC3 % | |
|---|---|---|---|---|---|
| A % | 0.529** | −0.263NS | −0.280NS | 0.260NS | −0.097NS |
| U % | −0.145NS | 0.110NS | −0.217NS | −0.40NS | −0.051NS |
| G % | −0.523** | −0.005NS | 0.581** | −0.156NS | 0.326NS |
| C % | −0.141NS | 0.286NS | −0.078NS | −0.369* | −0.230NS |
| GC % | −0.472** | 0.259NS | 0.318NS | −0.304NS | 0.082NS |
The value in this table is the R-value of correlation analysis
NS, non-significant (p > 0.05)
* 0.01 < p < 0.05
** p < 0.01
In addition, the correlation between the G + C content at the first and second codon positions (GC1 % and GC2 %) was compared with that at the synonymous third codon position (GC3 %) in the TTSuV1 coding sequences. A non-significant correlation between GC1 % and GC2 %, GC3 % (r = 0.056, p > 0.05; r = 0.248, p > 0.05), GC2 % and GC3 % (r = −0.147, P > 0.05) was found by using the Spearman’s rank correlation analysis method, implying that the nucleotide content at the first and second codon positions were different from those at the third codon position, and the GC content was highly conserved. During the shaping of the synonymous codon usage pattern of the TTSuV1 coding region, mutation pressure from base composition was not the only factor in shaping genetic diversity of this virus. It might be affected by other factors, such as translational selection in nature, since the effects were present at all codon positions.
Discussion
During protein biosynthesis, synonymous codons are not used randomly, and different genes from different species or from the same one always have an obvious codon usage bias, using one or several particular synonymous codons. Previous studies have mostly focused on higher organisms and many microorganisms with large genomes, and there are few studies on viruses with small genomes. Relatively, there have been more reports on codon usage in genomes from viruses that are important for human health, such as SARS, human immunodeficiency virus, influenza virus A and hepatitis viruses. TTSuVs is an emerging zoonotic DNA virus that has seriously threatened the development of the pig-raising industry and has brought about great economic losses worldwide. The virus is commonly present in the host’s serum, plasma, semen, milk, feces and other tissues [23–26, 38–41]. Although TTSuVs infection occurs in all ages, genders, and varieties of pigs [39, 41–44], a definitive pathogenic role of the virus has not been demonstrated. A recent study has shown that TTSuVs is associated with many economically important diseases, such as postweaning multisystemic wasting syndrome (PMWS) [45]. Krakowka et al. found that TTSuV1 promoted both porcine circovirus type 2 (PCV2) and porcine reproductive and respiratory syndrome virus (PRRSV) infection in gnotobiotic piglets. In addition, TTSuV1 co-infection with PRRSV has been correlated with the development of porcine dermatitis and nephropathy syndrome (PNDS) in gnotobiotic piglets [46].
The codon usage pattern is a genetic characteristic of various organisms. Because C %, U %, C3 % and U3 % play an important role in the formation of the different optimal codons ending with any nucleotide, the codon usage pattern of the TTSuV1 coding region is likely to be influenced by composition constraints. Using nucleotide content and RSCU value analysis, we found that the TTSuV1 coding region was GC3 poor (average value, 49.6 %), and most of the preferentially used codons were A-ended or C-ended codons, and G-ended or U-ended codons were of lower preference. Previous studies have shown that many viruses, including foot-and-mouth disease virus, influenza A virus subtype H5N1, severe acute respiratory syndrome coronavirus, and human bocavirus, preferentially use C-ended or G-ended codons [8, 47, 48]. The overall nucleotide content has an effect on the nucleotide content at the third codon position in TTSuV1, and the over-represented synonymous codons depending on the RSCU value matched with the results deduced from nucleotide content. This evidence indicates that the overall composition constraints affect the nucleotide content at the third codon position and the codon usage pattern of TTSuV1; that is to say, mutation pressure plays an important role in influencing the TTSuV1 codon usage pattern.
The ENC values calculated for the TTSuV1 coding region indicated that a significantly lower codon usage bias existed in TTSuV1. As a case in point, the ENC values varied from 54.45 to 58.74, with a stable diversification. The average ENC value of 56.46 among TTSuV1 coding genes can be compared to those calculated for other viruses, such as H5N1 virus, severe acute respiratory syndrome coronavirus, porcine adenovirus and nucleopolyhedroviruses, for which mean values of 50.91, 48.99, 38.97 and 38.80, respectively, have been reported [13, 48–50]. Therefore, taken together with published data on codon usage bias in other viruses, we conclude that the codon usage bias in TTSuV1 gene is lower and might be affected by natural selection. The low codon bias of TTSuV1 might result from an increase its own replication efficiency in order to adapt to the replication system of its hosts.
It is well established that synonymous codon usage in various organisms often reflects a balance between mutation pressure and natural selection. As for some RNA viruses, previous studies have reported that the major factor in shaping the codon usage pattern appears to be mutation pressure rather than natural selection [51]. Shacklton et al. revealed that codon usage bias was strongly correlated with the overall genomic GC content in DNA virus, suggesting that genome-wide mutational pressure rather than natural selection is the main determinant of codon usage bias [35]. However, Naya et al. found that there was no evidence indicating that the genome composition shaped the codon usage bias in high-GC-content genes [52]. In this study, except for mutation pressure, natural selection also played an important role in determining the codon usage bias of TTSuV1, which was supported by the non-correlation (p > 0.05) between GC1 %, GC2 % with GC3 %, and the results deduced from the ENC plot, in which almost all genes were below the expected curve. In addition, according to the analysis of the codon usage patterns of TTSuV1 isolates from different countries, we found that geographic factors were related to codon usage patterns in TTSuV1. It is interesting to note that TTSuV1 isolates from different countries, and even from the same country, had significant differences in their genetic characteristics. This might suggest that natural selection is a factor that shapes the overall codon usage pattern of this virus and acts as a major force to drive TTSuV1 evolution.
In previous studies, it has been reported that mutation pressure and natural selection are major factors in shaping codon usage patterns in many genomes. As for TTSuV1, the results suggest that there has been an interaction of mutation pressure with natural selection in the process of TTSuV1 evolution, although ENC values for the whole TTSuV1 coding region indicate that mutation pressure is one of the factors influencing the codon usage pattern. If synonymous codon usage bias is affected only by mutation pressure, then the frequency of nucleotides A and T should be equal to that of C and G at the synonymous codon third position. However, our study showed that the frequency of A and T was not balanced with C and G at these positions, indicating that other factors such as natural selection could also be the main determinants for synonymous codon usage bias. This characteristic of TTSuV1 conferred adaptive advantages, which resulted in a highly efficient dissemination of the virus by different modes of transmission.
Although the analysis of synonymous codon usage patterns and the main factors affecting it that we have proposed here do not entirely reflect the genetic variation of TTSuV1, our results have contributed to a better understanding of the evolutionary relationship among TTSuV1 strains, especially the roles played by mutation pressure and natural selection. Additionally, such information might provide a genetic background for future studies on the genetic and molecular aspects of TTSuV1.
Acknowledgments
This study was supported by the Jiangsu Agriculture Science and Technology Innovation Fund (CX(12)3078), the key Technology R&D Program of Jiangsu (BE2009388), the Project Funded by the Priority Academic Program Development of Jiangsu Higher Education Institutions, Natural Science Foundation of Jiangsu Province (BK2012083), Natural Science Foundation of the Higher Education Institutions of Jiangsu Province, China (10kjd230003), and Doctoral Research Foundation of Jinling Institution of Technology (40610047).
References
- 1.Grantham R, Gautier C, Gouy M. Codon frequencies in 119 individual genes confirm consistent choices of degenerate bases according to genome type. Nucleic Acids Res. 1980;8(9):1893–1912. doi: 10.1093/nar/8.9.1893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nakamura T, Suyama A, Wada A. Two types of linkage between codon usage and gene-expression levels. FEBS Lett. 1991;289(1):123–125. doi: 10.1016/0014-5793(91)80923-Q. [DOI] [PubMed] [Google Scholar]
- 3.Sharp PM, Stenico M, Peden JF, Lloyd AT. Codon usage: mutational bias, translational selection, or both? Biochem Soc Trans. 1993;21(4):835–841. doi: 10.1042/bst0210835. [DOI] [PubMed] [Google Scholar]
- 4.Shields DC, Sharp PM, Higgins DG, Wright F. “Silent” sites in Drosophila genes are not neutral: evidence of selection among synonymous codons. Mol Biol Evol. 1988;5(6):704–716. doi: 10.1093/oxfordjournals.molbev.a040525. [DOI] [PubMed] [Google Scholar]
- 5.Stenico M, Lloyd AT, Sharp PM. Codon usage in Caenorhabditis elegans: delineation of translational selection and mutational biases. Nucleic Acids Res. 1994;22(13):2437–2446. doi: 10.1093/nar/22.13.2437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhao S, Zhang Q, Chen Z, Zhao Y, Zhong J. The factors shaping synonymous codon usage in the genome of Burkholderia mallei. J Genet Genomics. 2007;34(4):362–372. doi: 10.1016/S1673-8527(07)60039-3. [DOI] [PubMed] [Google Scholar]
- 7.Karlin S, Mrazek J. What drives codon choices in human genes? J Mol Biol. 1996;262(4):459–472. doi: 10.1006/jmbi.1996.0528. [DOI] [PubMed] [Google Scholar]
- 8.Zhong J, Li Y, Zhao S, Liu S, Zhang Z. Mutation pressure shapes codon usage in the GC-Rich genome of foot-and-mouth disease virus. Virus Genes. 2007;35(3):767–776. doi: 10.1007/s11262-007-0159-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.McInerney JO. Replicational and transcriptional selection on codon usage in Borrelia burgdorferi. Proc Natl Acad Sci USA. 1998;95(18):10698–10703. doi: 10.1073/pnas.95.18.10698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Griswold KE, Mahmood NA, Iverson BL, Georgiou G. Effects of codon usage versus putative 5’-mRNA structure on the expression of Fusarium solani cutinase in the Escherichia coli cytoplasm. Protein Expr Purif. 2003;27(1):134–142. doi: 10.1016/S1046-5928(02)00578-8. [DOI] [PubMed] [Google Scholar]
- 11.Kahali B, Basak S, Ghosh TC. Reinvestigating the codon and amino acid usage of S. cerevisiae genome: a new insight from protein secondary structure analysis. Biochem Biophys Res Commun. 2007;354(3):693–699. doi: 10.1016/j.bbrc.2007.01.038. [DOI] [PubMed] [Google Scholar]
- 12.Ma J, Zhou T, Gu W, Sun X, Lu Z. Cluster analysis of the codon use frequency of MHC genes from different species. Biosystems. 2002;65(2–3):199–207. doi: 10.1016/S0303-2647(02)00016-3. [DOI] [PubMed] [Google Scholar]
- 13.Levin DB, Whittome B. Codon usage in nucleopolyhedroviruses. J Gen Virol. 2000;81(9):2313–2325. doi: 10.1099/0022-1317-81-9-2313. [DOI] [PubMed] [Google Scholar]
- 14.Zhang Z, Wang Y, Fan H, Lu C. Natural infection with torque teno sus virus 1 (TTSuV1) suppresses the immune response to porcine reproductive and respiratory syndrome virus (PRRSV) vaccination. Arch Virol. 2012 doi: 10.1007/s00705-012-1249-3. [DOI] [PubMed] [Google Scholar]
- 15.Kekarainen T, Segales J. Torque teno virus infection in the pig and its potential role as a model of human infection. Vet J. 2009;180(2):163–168. doi: 10.1016/j.tvjl.2007.12.005. [DOI] [PubMed] [Google Scholar]
- 16.Biagini P. Classification of TTV and related viruses (anelloviruses) Curr Top Microbiol Immunol. 2009;331:21–33. doi: 10.1007/978-3-540-70972-5_2. [DOI] [PubMed] [Google Scholar]
- 17.Nishizawa T, Okamoto H, Konishi K, Yoshizawa H, Miyakawa Y, Mayumi M. A novel DNA virus (TTV) associated with elevated transaminase levels in posttransfusion hepatitis of unknown etiology. Biochem Biophys Res Commun. 1997;241(1):92–97. doi: 10.1006/bbrc.1997.7765. [DOI] [PubMed] [Google Scholar]
- 18.Biagini P, Uch R, Belhouchet M, Attoui H, Cantaloube JF, Brisbarre N, de Micco P. Circular genomes related to anelloviruses identified in human and animal samples by using a combined rolling-circle amplification/sequence-independent single primer amplification approach. J Gen Virol. 2007;88(10):2696–2701. doi: 10.1099/vir.0.83071-0. [DOI] [PubMed] [Google Scholar]
- 19.Okamoto H, Nishizawa T, Takahashi M, Tawara A, Peng Y, Kishimoto J, Wang Y. Genomic and evolutionary characterization of TT virus (TTV) in tupaias and comparison with species-specific TTVs in humans and non-human primates. J Gen Virol. 2001;82(9):2041–2050. doi: 10.1099/0022-1317-82-9-2041. [DOI] [PubMed] [Google Scholar]
- 20.Inami T, Obara T, Moriyama M, Arakawa Y, Abe K. Full-length nucleotide sequence of a simian TT virus isolate obtained from a chimpanzee: evidence for a new TT virus-like species. Virology. 2000;277(2):330–335. doi: 10.1006/viro.2000.0621. [DOI] [PubMed] [Google Scholar]
- 21.Leary TP, Erker JC, Chalmers ML, Desai SM, Mushahwar IK. Improved detection systems for TT virus reveal high prevalence in humans, non-human primates and farm animals. J Gen Virol. 1999;80(8):2115–2120. doi: 10.1099/0022-1317-80-8-2115. [DOI] [PubMed] [Google Scholar]
- 22.Okamoto H, Takahashi M, Nishizawa T, Tawara A, Fukai K, Muramatsu U, Naito Y, Yoshikawa A. Genomic characterization of TT viruses (TTVs) in pigs, cats and dogs and their relatedness with species-specific TTVs in primates and tupaias. J Gen Virol. 2002;83(6):1291–1297. doi: 10.1099/0022-1317-83-6-1291. [DOI] [PubMed] [Google Scholar]
- 23.Niel C, Diniz-Mendes L, Devalle S. Rolling-circle amplification of Torque teno virus (TTV) complete genomes from human and swine sera and identification of a novel swine TTV genogroup. J Gen Virol. 2005;86(5):1343–1347. doi: 10.1099/vir.0.80794-0. [DOI] [PubMed] [Google Scholar]
- 24.Aramouni M, Segales J, Cortey M, Kekarainen T. Age-related tissue distribution of swine Torque teno sus virus 1 and 2. Vet Microbiol. 2010;146(3–4):350–353. doi: 10.1016/j.vetmic.2010.05.036. [DOI] [PubMed] [Google Scholar]
- 25.Martinez L, Kekarainen T, Sibila M, Ruiz-Fons F, Vidal D, Gortazar C, Segales J. Torque teno virus (TTV) is highly prevalent in the European wild boar (Sus scrofa) Vet Microbiol. 2006;118(3–4):223–229. doi: 10.1016/j.vetmic.2006.07.022. [DOI] [PubMed] [Google Scholar]
- 26.Bigarre L, Beven V, de Boisseson C, Grasland B, Rose N, Biagini P, Jestin A. Pig anelloviruses are highly prevalent in swine herds in France. J Gen Virol. 2005;86(3):631–635. doi: 10.1099/vir.0.80573-0. [DOI] [PubMed] [Google Scholar]
- 27.Zhang Z, Wang Y, Fan H, Lu C. Natural infection with torque teno sus virus 1 (TTSuV1) suppresses the immune response to porcine reproductive and respiratory syndrome virus (PRRSV) vaccination. Arch Virol. 2012;157(5):927–933. doi: 10.1007/s00705-012-1249-3. [DOI] [PubMed] [Google Scholar]
- 28.Bendinelli M, Pistello M, Maggi F, Fornai C, Freer G, Vatteroni ML. Molecular properties, biology, and clinical implications of TT virus, a recently identified widespread infectious agent of humans. Clin Microbiol Rev. 2001;14(1):98–113. doi: 10.1128/CMR.14.1.98-113.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wright F. The ‘effective number of codons’ used in a gene. Gene. 1990;87(1):23–29. doi: 10.1016/0378-1119(90)90491-9. [DOI] [PubMed] [Google Scholar]
- 30.Sharp PM, Li WH. An evolutionary perspective on synonymous codon usage in unicellular organisms. J Mol Evol. 1986;24(1–2):28–38. doi: 10.1007/BF02099948. [DOI] [PubMed] [Google Scholar]
- 31.Sharp PM, Li WH. Codon usage in regulatory genes in Escherichia coli does not reflect selection for ‘rare’ codons. Nucleic Acids Res. 1986;14(19):7737–7749. doi: 10.1093/nar/14.19.7737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wong EH, Smith DK, Rabadan R, Peiris M, Poon LL. Codon usage bias and the evolution of influenza A viruses. Codon usage biases of Influenza virus. BMC Evol Biol. 2010;10:253. doi: 10.1186/1471-2148-10-253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhou JH, Zhang J, Chen HT, Ma LN, Liu YS. Analysis of synonymous codon usage in foot-and-mouth disease virus. Vet Res Commun. 2010;34(4):393–404. doi: 10.1007/s11259-010-9359-4. [DOI] [PubMed] [Google Scholar]
- 34.Sau K, Gupta SK, Sau S, Mandal SC, Ghosh TC. Factors influencing synonymous codon and amino acid usage biases in Mimivirus. Biosystems. 2006;85(2):107–113. doi: 10.1016/j.biosystems.2005.12.004. [DOI] [PubMed] [Google Scholar]
- 35.Shackelton LA, Parrish CR, Holmes EC. Evolutionary basis of codon usage and nucleotide composition bias in vertebrate DNA viruses. J Mol Evol. 2006;62(5):551–563. doi: 10.1007/s00239-005-0221-1. [DOI] [PubMed] [Google Scholar]
- 36.Zhao KN, Liu WJ, Frazer IH. Codon usage bias and A + T content variation in human papillomavirus genomes. Virus Res. 2003;98(2):95–104. doi: 10.1016/j.virusres.2003.08.019. [DOI] [PubMed] [Google Scholar]
- 37.Charles H, Calevro F, Vinuelas J, Fayard JM, Rahbe Y. Codon usage bias and tRNA over-expression in Buchnera aphidicola after aromatic amino acid nutritional stress on its host Acyrthosiphon pisum. Nucleic Acids Res. 2006;34(16):4583–4592. doi: 10.1093/nar/gkl597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.McKeown NE, Fenaux M, Halbur PG, Meng XJ. Molecular characterization of porcine TT virus, an orphan virus, in pigs from six different countries. Vet Microbiol. 2004;104(1–2):113–117. doi: 10.1016/j.vetmic.2004.08.013. [DOI] [PubMed] [Google Scholar]
- 39.Kekarainen T, Lopez-Soria S, Segales J. Detection of swine Torque teno virus genogroups 1 and 2 in boar sera and semen. Theriogenology. 2007;68(7):966–971. doi: 10.1016/j.theriogenology.2007.07.010. [DOI] [PubMed] [Google Scholar]
- 40.Martinez-Guino L, Kekarainen T, Segales J. Evidence of Torque teno virus (TTV) vertical transmission in swine. Theriogenology. 2009;71(9):1390–1395. doi: 10.1016/j.theriogenology.2009.01.010. [DOI] [PubMed] [Google Scholar]
- 41.Brassard J, Gagne MJ, Lamoureux L, Inglis GD, Leblanc D, Houde A. Molecular detection of bovine and porcine Torque teno virus in plasma and feces. Vet Microbiol. 2008;126(1–3):271–276. doi: 10.1016/j.vetmic.2007.07.014. [DOI] [PubMed] [Google Scholar]
- 42.Martelli F, Caprioli A, Di Bartolo I, Cibin V, Pezzotti G, Ruggeri FM, Ostanello F. Detection of swine torque teno virus in Italian pig herds. J Vet Med B Infect Dis Vet Public Health. 2006;53(5):234–238. doi: 10.1111/j.1439-0450.2006.00949.x. [DOI] [PubMed] [Google Scholar]
- 43.Sibila M, Martinez-Guino L, Huerta E, Mora M, Grau-Roma L, Kekarainen T, Segales J. Torque teno virus (TTV) infection in sows and suckling piglets. Vet Microbiol. 2009;137(3–4):354–358. doi: 10.1016/j.vetmic.2009.01.008. [DOI] [PubMed] [Google Scholar]
- 44.Zhu CX, Cui L, Shan TL, Luo XN, Liu ZJ, Yuan CL, Lan DL, Zhao W, Liu ZW, Hua XG. Porcine torque teno virus infections in China. J Clin Virol. 2010;48(4):296–298. doi: 10.1016/j.jcv.2010.04.012. [DOI] [PubMed] [Google Scholar]
- 45.Aramouni M, Segales J, Sibila M, Martin-Valls GE, Nieto D, Kekarainen T. Torque teno sus virus 1 and 2 viral loads in postweaning multisystemic wasting syndrome (PMWS) and porcine dermatitis and nephropathy syndrome (PDNS) affected pigs. Vet Microbiol. 2011;153(3–4):377–381. doi: 10.1016/j.vetmic.2011.05.046. [DOI] [PubMed] [Google Scholar]
- 46.Krakowka S, Ellis JA. Evaluation of the effects of porcine genogroup 1 torque teno virus in gnotobiotic swine. Am J Vet Res. 2008;69(12):1623–1629. doi: 10.2460/ajvr.69.12.1623. [DOI] [PubMed] [Google Scholar]
- 47.Zhao S, Zhang Q, Liu X, Wang X, Zhang H, Wu Y, Jiang F. Analysis of synonymous codon usage in 11 human bocavirus isolates. Biosystems. 2008;92(3):207–214. doi: 10.1016/j.biosystems.2008.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhou T, Gu W, Ma J, Sun X, Lu Z. Analysis of synonymous codon usage in H5N1 virus and other influenza A viruses. Biosystems. 2005;81(1):77–86. doi: 10.1016/j.biosystems.2005.03.002. [DOI] [PubMed] [Google Scholar]
- 49.Das S, Paul S, Dutta C. Synonymous codon usage in adenoviruses: influence of mutation, selection and protein hydropathy. Virus Res. 2006;117(2):227–236. doi: 10.1016/j.virusres.2005.10.007. [DOI] [PubMed] [Google Scholar]
- 50.Gu W, Zhou T, Ma J, Sun X, Lu Z. Analysis of synonymous codon usage in SARS Coronavirus and other viruses in the Nidovirales. Virus Res. 2004;101(2):155–161. doi: 10.1016/j.virusres.2004.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jenkins GM, Holmes EC. The extent of codon usage bias in human RNA viruses and its evolutionary origin. Virus Res. 2003;92(1):1–7. doi: 10.1016/S0168-1702(02)00309-X. [DOI] [PubMed] [Google Scholar]
- 52.Naya H, Romero H, Carels N, Zavala A, Musto H. Translational selection shapes codon usage in the GC-rich genome of Chlamydomonas reinhardtii. FEBS Lett. 2001;501(2–3):127–130. doi: 10.1016/S0014-5793(01)02644-8. [DOI] [PubMed] [Google Scholar]