

Abstract
Human coronavirus OC43 (HCoV-OC43) is a respiratory virus that usually causes a common cold. However, it has the potential to cause severe infection in young children and immunocompromised adults. Both SARS-CoV and MERS-CoV were shown to express proteins with the potential to evade early innate immune responses. However, the ability of HCoV-OC43 to antagonise the intracellular antiviral defences has not yet been investigated. The potential role of the HCoV-OC43 structural (M and N) and accessory proteins (ns2a and ns5a) in the alteration of antiviral gene expression was investigated in this study. HCoV-OC43M, N, ns2a and ns5a proteins were expressed in human embryonic kidney 293 (HEK-293) cells before challenge with Sendai virus. The Human Antiviral Response PCR array was used to profile the antiviral gene expression in HEK-293 cells. Over 30 genes were downregulated in the presence of one of the HCoV-OC43 proteins, e.g. genes representing mitogen-activated protein kinases, toll-like receptors, interferons, interleukins, and signaling transduction proteins. Our findings suggest that similarly to SARS-CoV and MERS-CoV, HCoV-OC43 has the ability to downregulate the transcription of genes critical for the activation of different antiviral signaling pathways. Further studies are needed to confirm the role of HCoV-OC43 structural and accessory proteins in antagonising antiviral gene expression.
Introduction
Human coronavirus OC43 (HCoV-OC43) is taxonomically classified within the Coronaviridae family which also includes severe acute respiratory syndrome (SARS) and Middle East respiratory syndrome (MERS) coronaviruses. HCoV-OC43 belongs to subgroup A of the betacoronaviruses. These are positive-sense single-stranded RNA viruses with a large genome size at 30,780 bases [1]. The viral proteins that contribute to the structure of coronaviruses include the Membrane (M), Nucleocapsid (N), Envelope (E) and Spike (S) glycoprotein. Betacoronaviruses possess a unique protein on the surface alongside the S glycoprotein called Hemagglutinin esterase (HE). Each coronavirus possesses its own set of unique accessory proteins. The accessory proteins of HCoV-OC43 are called ns2a and ns5a [2].
HCoV-OC43 and other common circulating coronaviruses cause 10 to 30% of all common colds, and 5 to 10% of all upper and lower respiratory tract infections [3–6]. Subclinical or very mild infections are common, and can occur throughout the year. However, human coronaviruses other than SARS-CoV and MERS-CoV are responsible for 10 to 20% of hospitalisations of young children and immunocompromised adults with respiratory tract illness [7]. HCoV-OC43 seems to have also the potential for both neurotropism and neuroinvasion that are associated with multiple sclerosis [8, 9]. Moreover, HCoV-OC43 was detected in the cerebrospinal fluid of a child with acute disseminated encephalomyelitis [10]. A case of fatal encephalitis associated with HCoV-OC43 infection was recently reported in an immunodeficient 11-month-old boy [11].
Several studies on human coronavirus interaction with host cells and the innate immune response have been conducted on SARS-CoV and MERS-CoV, because of the high case-fatality rate associated with these infections. In Vero E6 cells expressing SARS-CoV N protein, interleukin 6 (IL6) was activated through nuclear factor kappa B (NF-κB) [12]. On the contrary, the M protein of SARS-CoV suppresses NF-κB activity and contributes to the pathogenesis of the virus [13]. In addition, many proteins in SARS-CoV were shown to have the ability to antagonise interferon (IFN) and counteract the innate immune response. The ORF3b and ORF6 accessory proteins have the potential to antagonise IFN by different mechanisms [14]. SARS-CoV M protein was recently shown to suppress IFN production [15]. The structural and accessory proteins M, ORF4a, ORF4b, and ORF5 of MERS-CoV were shown to be potent IFN antagonists [16, 17]. It is still unknown whether the evasion mechanisms described above have a certain role in the severity of SARS and MERS. It is noteworthy that several people infected with SARS-CoV or MERS-CoV do not develop severe symptoms [18–21].
Most research on SARS and MERS has been done on one specific virus and how it activates or inhibits the innate immune response. None of the studies conducted so far have compared results with those of HCoV-OC43 that is associated with low mortality rate. HCoV-OC43 may have the same ability as SARS-CoV and MERS-CoV to evade innate immunity. Indeed, there are few studies investigating HCoV-OC43 interaction with host cells and the innate immune response. The N protein was shown to cause prolonged activation of NF-κB in N-expressing HEK-293T cells [22]. Furthermore, HCoV-OC43 infection of cell lines derived from human respiratory tract epithelia and hepatocytes, was enhanced by all three types of IFN, due to the interaction with the IFN-inducible transmembrane (IFITM) proteins [23]. Moreover, the acetyl-esterase activity of HE protein strongly enhances the production of infectious virus [24].
While SARS-CoV and MERS-CoV have received much attention from the scientific community, very little research has addressed the effect of other human coronavirus infections on antiviral gene expression. The potential role of the HCoV-OC43 structural and accessory proteins in the alteration of antiviral gene expression in HEK-293 cells was investigated in this study.
Materials and methods
Cell culture
Human embryonic kidney 293 (HEK-293) cells were obtained from American Type Culture Collection (ATCC, Manassas, VA, USA), and were grown in Dulbecco’s minimal essential medium (DMEM) containing GlutaMAX™ (Life Technologies CorporationTM, Grand Island, NY, USA) using 25 cm2 tissue culture flasks. The medium was supplemented with 10% fetal bovine serum (FBS) (Life TechnologiesTM), Fungizone® (250 µg/ml) (Life TechnologiesTM), penicillin G (10,000 U/ml) (Life Technologies) and streptomycin sulfate (10,000 pg/ml) (Life TechnologiesTM). Monolayers of HEK-293 cell culture flasks were incubated at 37 °C in the presence of 5% carbon dioxide (CO2) and 90% humidity.
Viruses
Human coronavirus OC43 (HCoV-OC43; VR-1558), influenza A virus (H3N2, A/Hong Kong/8/68 strain; VR-544), and Sendai virus (VR-105) were obtained from ATCC.
Amplification of HCoV-OC43 ns2a, ns5a, M, and N genes by RT-PCR
HCoV-OC43 RNA was extracted using the QIAamp® Viral RNA Minikit (QiagenTM GmbH, Hilden, Germany) according to the manufacturer’s instructions. The HCoV-OC43 ns2a, ns5a, M, and N genes were amplified by a two-step reverse transcription-polymerase chain reaction (RT-PCR) using the GeneAmp® RNA Core Kit (Applied BiosystemsTM, Foster City, CA, USA) and 10 pmol of previously described primers [2] on a GeneAmp® PCR System 9700 (Applied BiosystemsTM). The RT reaction conditions were: annealing at 37 °C for 60 minutes, denaturation at 90 °C for 5 minutes, and cooling at 4 °C. Thereafter, the cDNA was amplified by PCR using the following conditions: denaturation at 94 °C for 10 minutes, then 35 cycles of denaturation at 95 °C for 30 seconds, annealing at 60 °C for 30 seconds, and extension at 72 °C for 30 seconds, followed by a final extension step at 72 °C for 7 minutes, with cooling at 4 °C. PCR products were run on agarose gels and the bands that were size-specific for the amplified gene of interest were cut and purified by Wizard® SV Gel and PCR Clean Up System (PromegaTM, Madison, WI, USA) according to the manufacturer’s instructions. Influenza A virus RNA was also isolated and was used to amplify the NS1 gene by RT-PCR using NS1-specific primers [25]. Influenza A NS1 protein was used as positive control for IFN antagonism [14].
Expression of HCoV-OC43 ns5a, ns2a, M and N proteins in HEK-293 cells
The pAcGFP1-N In-Fusion® Ready Vector (ClontechTM, Takara Bio Company, Mountain View, CA, USA) was used for cloning RT-PCR products. The In-Fusion® HD Cloning Kit (ClontechTM) was used to set up the ligation reaction. Competent TOP10 E. coli cells (InvitrogenTM, Carlsbad, CA, USA) were used for transformation. The isolation of vector was achieved by using the PureYield™ Plasmid Miniprep Kit (Promega) according to the manufacturer’s instructions. Confirmation of successful cloning was achieved using restriction digestion and direct sequencing. HEK-293 cells were seeded at 5 x 105 per well of 24-well plates, and transfected with ns2a-pAcGFP1, ns5a-pAcGFP1, M-pAcGFP1, N-pAcGFP1, or NS1-pAcGFP1 using Lipofectamine® 2000 according to the manufacturer’s instructions. Expression of ns2a, ns5a, M, N and NS1 proteins was confirmed by indirect immunofluorescence assay (IFA) using anti-GFP monoclonal antibody (10 μg/ml) (Clone JL-8, ClontechTM). Expression of N and NS1 proteins was further confirmed by IFA using monoclonal antibody against HCoV-OC43N (EMD MilliporeTM, Billerica, MA, USA) and polyclonal antibody against influenza A NS1 protein (EMD MilliporeTM), respectively.
Challenge of HEK-293 cells with Sendai virus
Transfected and mock-transfected HEK-293 cells were grown to confluency in a 96-well plate at 37 °C in the presence of 5% CO2. The media was then removed, and Sendai virus suspension (ATCC) was added to each well at 1 multiplicity of infection (m.o.i). The plates were further incubated at 37°C with 5% CO2 and at 90% humidity.
Antiviral gene expression profile in cells expressing HCoV-OC43 structural and accessory proteins
The Human Antiviral Response PCR array (QiagenTM) was used to profile the antiviral gene expression in HEK-293 cells in two separate experiments. The PCR array monitored the transcriptional activity of different genes involved in the activation of antiviral and pro-inflammatory proteins following amplification of RNA transcripts by real-time RT-PCR. Total RNA from transfected and mock-transfected HEK-293 cells was extracted using an RNeasy® Kit (QiagenTM) with on-column DNase digestion according to the manufacturer’s instructions. The RT2 First Strand Kit (QiagenTM) was used for cDNA synthesis. The RT reaction was then added to RT2 SYBR Green ROX qPCR Mastermix (QiagenTM), and the PCR was carried out in a 96-well plate (QiagenTM) on an ABI 7500 Fast Real-Time PCR system (Applied BiosystemsTM). Cycle threshold (CT) values were exported to an Excel file to create a table of CT values. This table was then uploaded on to the Qiagen data analysis web portal (http://www.qiagen.com/geneglobe). Quality control was performed as the array contains controls to monitor genomic DNA contamination (GDC), first strand synthesis (RTC) and real-time PCR efficiency (PPC). Samples were assigned to controls and test groups. CT values were normalised based on a manual selection of reference genes with glyceraldehyde 3-phosphate dehydrogenase (GAPDH) chosen as the housekeeping reference gene. Fold regulation comparisons were used to profile antiviral gene expression. Fold change/regulation was calculated using delta delta CT method (∆∆CT) where ∆CT is calculated between the gene of interest (GOI) and GAPDH, followed by ∆∆CT calculations (∆CT (Test Group)-∆CT (Control Group)). Fold change is then calculated using 2^ (-∆∆CT) formula. Thereafter, the data analysis web portal was used to provide the scatter plot, the volcano plot, the clustergram, and the heat map. The fold regulation threshold was ≥ 2 for upregulation and ≤ -2 for downregulation.
Statistical analysis
Fold change in gene expression from 2 different experiments was summarised as mean ± standard deviation. The difference in mean fold change between two groups was determined by independent-samples t-test. P-value < 0.05 was considered significant. All statistical analyses were performed using IBM SPSS statistics software version 25.0 for windows (IBM Corporation, Armonk, NY, USA).
Results
PCR array profiling of antiviral genes in HEK-293 cells expressing HCoV-OC43 structural and accessory proteins
Figure 1 shows the expression of HCoV-OC43 and influenza proteins in HEK-293 cells. The antiviral gene expression in transfected HEK-293 cells expressing one HCoV-OC43 protein was compared to that in mock- and non-transfected cells. Only the expression of the oncogene FOS was upregulated across all samples (Table 1). Nevertheless, over 30 genes were downregulated in the presence of one of the HCoV-OC43 proteins, e.g. mitogen-activated protein kinases (MAP3K7), toll-like receptors (TLR3 and TLR8), caspases (CASP1), cathepsins (CTSS), interferons (IFNA1), interleukins (IL1B, IL12A, IL15, and IL18), and signaling transduction proteins (MYD88 and MAVS) (Table 1). Similar results were obtained in the presence of the influenza A NS1 protein, a well-known IFN antagonist [14] (Table 1). The mean negative fold change for CARD9 in the presence of the structural protein M or N was larger than that in the presence of the accessory protein ns2a (p < 0.01). In contrast, the expression of the MAP3K7 gene was more downregulated in the presence of ns2a or ns5a protein than in the presence of M or N protein (p < 0.01). Downregulation of CTSS gene expression was greater in the presence of ns2a, ns5a or N protein than in the presence of M protein (p < 0.01). IL1B, CASP1, and TLR3 gene expression was more downregulated in the presence of ns2a or ns5a protein than in the presence of M or N protein (p < 0.01). On the contrary, CD86 was more downregulated in the presence of M protein than in the presence of N, ns2a or ns5a protein (p ≤ 0.01).
Fig. 1.
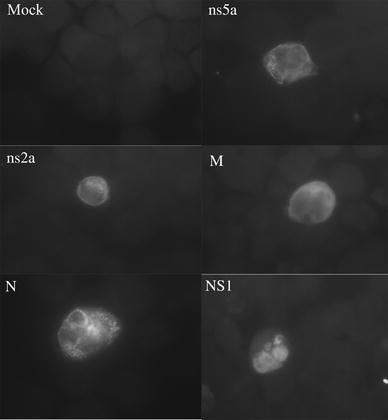
Expression of HCoV-OC43 and influenza A proteins in HEK-293 cells. HEK-293 cells were transfected with the expression vector pAcGFP1 coding for HCoV-OC43 ns2a, ns5a, M or N proteins, or for influenza A NS1 protein. Three days following transfection, the expression of viral proteins was confirmed by indirect immunofluorescence assay using anti-GFP monoclonal antibody
Table 1.
Mean fold change in antiviral gene expression in HEK-293 cells expressing HCoV-OC43 structural or accessory proteins
| ns2a | ns5a | M | N | NS1 (FluA) | |||||
|---|---|---|---|---|---|---|---|---|---|
| Mean | ±SD | Mean | ±SD | Mean | ±SD | Mean | ±SD | ||
|
FOS MAP3K7 TLR3 CTSS CASP1 MEFV IL12A PYCARD IL1B MX1 IFIH1 RIPK1 IFNA1 CD80 CD86 CXCL11 MYD88 FADD MAVS TRIM25 IL15 CYLD TLR8 TNF IL18 TRAF6 IRF3 TBK1 IFNAR1 SUGT1 CARD9 |
+9.94 -83.63 -55.56 -21.91 -16.73 —— 7.18 -10.06 -24.83 -6.97 -7.89 -9.61 -4.99 -4.41 -4.15 -5.84 -7.55 -4.61 -3.81 -4.59 -3.77 -4.98 -2.39 -2.22 -3.16 -4.71 —– -3.35 -3.12 —– -4.81 |
0.34 2.17 0.79 1.64 1.56 —– 1.34 1.24 0.67 0.57 0.23 1.25 2.78 1.23 1.20 0.07 2.22 1.23 1.49 1.93 0.72 0.04 0.16 0.03 0.06 0.45 —– 0.02 0.05 —– 0.45 |
+9.49 -88.75 -32.59 -23.85 -16.52 -14.36 -13.62 -12.64 -9.94 -7.24 -6.99 -6.90 -6.59 -6.38 -6.01 -5.05 -4.79 -4.30 -2.80 -4.06 -3.96 -3.76 -3.46 —– -3.17 -3.09 -2.98 -2.97 -2.93 -2.92 —– |
0.23 2.45 1.23 1.85 1.93 1.52 0.95 0.97 0.25 1.22 1.24 0.75 0.53 0.54 0.12 1.26 1.89 0.03 0.74 0.08 0.19 1.35 1.23 —– 0.04 0.63 0.35 0.69 0.04 0.09 —– |
+3.45 —– -14.68 -7.68 —– -5.21 -6.74 -18.10 —– —– -2.84 -5.21 -3.24 -5.71 -13.61 -2.17 -2.34 —– —– —– —– —– -3.06 -7.27 -3.14 -2.47 -2.19 -2.85 —– —– -16.75 |
0.45 —– 0.63 0.93 —– 2.22 1.23 0.63 —– —– 0.09 2.55 1.24 0.56 0.83 0.11 0.05 —– —– —– —– —– 0.05 0.68 0.91 0.33 0.03 0.45 —– —– 0.93 |
+10.06 -59.54 -31.73 -24.57 -12.03 -13.59 -8.61 -11.48 -6.63 -8.14 -6.05 -8.85 -5.39 -6.04 -5.69 —– -5.15 -5.66 -3.75 -5.20 -3.27 —– -3.28 —– -3.25 -3.76 —– -3.05 -2.93 -3.97 -12.19 |
0.29 1.23 0.83 0.73 0.53 2.22 1.23 2.56 1.59 0.03 0.94 0.35 0.48 0.54 2.34 —– 0.53 0.23 0.05 1.29 0.45 —– 1.24 —– 0.53 0.73 —– 0.23 0.04 0.05 0.02 |
+8.23 -83.27 -25.79 -17.94 -21.32 -9.89 -9.59 -2.60 -2.15 -9.12 -7.10 -6.66 -4.42 —– -4.14 -2.17 -5.18 -4.58 -4.67 -4.57 -4.14 -3.36 -2.39 —– -3.14 -3.50 -3.15 -3.20 -2.93 -2.99 —– |
Results are shown as the mean ± standard deviation (SD) of two independent experiments. Mean ± SD was not available for influenza NS1 because only one experiment was performed. (—–) indicates no significant fold change in one or two experiments
Sendai virus, a well-known inducer of antiviral genes, was used to challenge HEK-293 cells. A total of 22 genes were upregulated in the presence of Sendai virus, whereas 9 genes were downregulated (Table 2). There was strong positive fold change for MAP3K7, FOS, CXCL9, JUN, TLR7, IRF7, IL12B, NOD2 and IL6 in HEK-293 cells. In the presence of ns2a, ns5a, M, N or NS1 protein, the challenge of cells with Sendai virus resulted in the upregulation of only 7 genes and downregulation of 25 genes (Table 3). The expression of OAS2 was upregulated in the presence of HCoV-OC43 ns5a or M protein, or influenza A NS1 protein. A positive fold change was observed for three chemokines (CCL5, CXCL8, CXCL10), two RIG-I-like receptors (IFIH1 and DDX58), one interferon (IFNB1), and one ubiquitin-like protein (ISG15), whereas there was a drastic downregulation of MAP3K7 expression with negative fold change ranging from -484 to -27,496 (Table 3). FOS, CXCL9, JUN, TLR7, IRF7, IL12B and IL6 also showed substantial negative fold changes (Table 3). The expression of MAP3K7, TLR7, IL12B, IL6 and NOD2 genes was more downregulated in the presence of ns5a protein than in the presence of other HCoV-OC43 proteins (p < 0.05), whereas the expression of the CXCL9 gene was more downregulated in the presence of ns2a protein than in the presence of other proteins (p < 0.001). Moreover, the expression of JUN and IRF7 genes was most downregulated in the presence of N protein (p ≤ 0.01). Interestingly, the expression of IRF7 and IFNA1 genes was more downregulated in the presence of ns2a, ns5a or N protein than in the presence of influenza NS1 protein. The expression of NOD2 was more downregulated in the presence of ns2a or ns5a protein than in the presence of influenza NS1 protein, while the expression of PYCARD was more downregulated in the presence of one of the tested HCoV-OC43 proteins than in the presence of influenza NS1 protein. Moreover, FOS gene expression was more downregulated in the presence of ns5a, M or N protein than in the presence of influenza NS1 protein (Table 3).
Table 2.
Mean fold change in antiviral gene expression in HEK-293 cells challenged with Sendai virus
| Upregulated genes | Cells (w/ Sendai) | Downregulated genes | Cells (w/ Sendai) | ||
|---|---|---|---|---|---|
| Mean | ±SD | Mean | ±SD | ||
| MAP3K7 | +276.05 | 8.27 | IFIH1 | -16.21 | 3.45 |
| FOS | +177.10 | 2.13 | CCL5 | -14.95 | 3.43 |
| CXCL9 | +25.64 | 2.71 | CTSS | -13.06 | 1.54 |
| JUN | +24.61 | 2.34 | IL12A | -8.27 | 1.73 |
| TLR7 | +21.87 | 1.87 | TLR3 | -6.53 | 1.35 |
| IRF7 | +15.61 | 2.30 | DDX58 | -5.18 | 2.69 |
| IL12B | +15.54 | 0.89 | RIPK1 | -5.03 | 2.27 |
| NOD2 | +15.26 | 1.74 | CASP1 | -4.01 | 0.37 |
| IL6 | +15.17 | 4.78 | MEFV | -2.29 | 0.01 |
| SPP1 | +4.20 | 0.01 | |||
| NLRP3 | +4.16 | 0.01 | |||
| IFNA2 | +3.25 | 0.62 | |||
| TRADD | +3.04 | 0.45 | |||
| MAVS | +3.02 | 0.75 | |||
| IFNA1 | +2.90 | 0.06 | |||
| CCL3 | +2.81 | 0.01 | |||
| IRAK1 | +2.78 | 0.12 | |||
| CTSB | +2.72 | 0.06 | |||
| PIN1 | +2.50 | 0.05 | |||
| TLR8 | +2.48 | 0.32 | |||
| IRF5 | +2.36 | 0.06 | |||
| MAP2K3 | +2.31 | 0.21 | |||
Results are shown as the mean ± standard deviation (SD) of two independent experiments
Table 3.
Mean fold change in antiviral gene expression in Sendai-infected HEK-293 cells expressing HCoV-OC43 structural or accessory proteins
| ns2a | ns5a | M | N | NS1 (FluA) | |||||
|---|---|---|---|---|---|---|---|---|---|
| Mean | ±SD | Mean | ±SD | Mean | ±SD | Mean | ±SD | ||
|
CCL5 IFIH1 IFNB1 ISG15 DDX58 CXCL10 OAS2 CXCL8 MAP3K7 FOS CXCL9 JUN TLR7 IRF7 IL12B NOD2 IL6 IL1B PYCARD MYD88 CARD9 IKBKB FADD TRADD MAVS IFNA1 IRAK1 CTSB PIN1 TLR8 IRF5 MAP2K3 TICAM1 |
+53.45 +16.78 +17.76 +8.68 +14.00 +10.20 —– +9.80 -12141.84 -12.68 -104.94 -33.49 -16.23 -59.17 -20.00 -14.32 -12.75 -13.65 -6.51 -3.72 -3.71 -3.12 -4.87 -12.70 -9.74 -18.51 -5.31 —– -5.73 -3.54 -4.87 -5.34 —– |
0.28 0.03 0.77 1.27 0.25 0.39 —– 1.29 0.30 1.25 0.91 2.22 0.01 0.03 0.01 0.01 0.78 0.33 0.56 0.07 0.01 0.19 1.22 1.05 0.07 0.04 1.49 —– 1.26 0.01 0.76 0.10 —– |
+23.24 +8.15 +7.39 +5.84 +4.80 +4.40 +3.23 +3.04 -27496.30 -22.91 -54.81 -51.34 -33.86 -27.66 -41.70 -18.64 -37.00 -30.81 -9.06 -4.96 -3.38 -5.14 -4.80 -7.15 -10.70 -24.28 -3.92 -7.12 -4.34 -7.38 -4.14 -10.28 -3.51 |
0.27 0.59 0.04 0.51 0.59 0.10 0.32 0.51 0.44 0.60 0.62 1.18 0.01 0.72 0.01 0.42 0.15 1.16 2.71 0.01 0.94 0.53 1.49 0.94 1.07 0.20 0.96 0.30 0.19 0.01 0.11 0.27 0.69 |
+30.00 +15.23 +9.16 +6.87 +8.20 +8.03 +4.67 +4.68 -484.06 -17.63 -46.57 -35.35 -19.42 -18.05 -9.86 -5.00 -15.16 -11.86 -4.77 -3.33 -3.85 -3.57 —– -5.20 -6.03 -11.37 -5.30 -4.16 —– -6.31 -5.32 -8.57 -7.15 |
0.22 0.16 0.14 0.70 0.45 0.31 0.63 0.01 7.02 0.91 3.77 1.94 3.91 1.63 1.60 2.60 2.13 0.16 0.34 0.37 1.60 0.07 —– 2.32 1.13 2.32 1.13 0.16 —– 0.49 1.15 0.10 1.74 |
+50.47 +13.60 +22.13 +11.68 +10.98 +10.24 —– +8.88 -16781.69 -29.61 -25.86 -74.12 -14.61 -87.48 -13.24 -5.40 -10.19 -35.01 -7.36 —– -3.34 -7.50 -3.44 -3.91 -9.98 -21.84 -8.89 -4.91 —– -3.18 -5.86 -4.83 —– |
0.07 0.36 0.22 0.17 0.42 0.08 —– 0.71 1.26 2.67 0.63 3.17 0.01 3.49 0.63 1.78 0.51 2.34 1.43 —– 0.01 2.91 1.10 0.83 1.71 1.85 3.91 0.17 —– 0.01 2.59 0.42 —– |
+19.74 +7.47 +2.96 +3.85 +4.07 +2.91 +4.52 +3.61 -31317.90 -14.76 -95.64 -58.19 -53.61 -20.75 -66.03 -7.68 -33.09 -28.98 -3.62 -4.12 -3.21 -4.82 -5.67 -10.01 -10.45 -12.83 -2.47 -4.17 -4.94 -11.68 -2.98 -8.88 -3.51 |
Results are shown as mean ± standard deviation (SD) of two independent experiments. Mean ± SD was not available for influenza NS1 as only one experiment was performed. (—–) indicates no significant fold change in one or two experiments
Discussion
The current study shows for the first time that different genes involved in the activation of antiviral and inflammatory responses, e.g. those representing interleukins, IFNs, MAPKs, PRRs and adaptor proteins, are downregulated in the presence of HCoV-OC43 structural or accessory proteins. Similar antiviral gene expression profiles were observed between influenza NS1 protein, which is known to antagonise type I IFN activity [14, 26–28], and HCoV-OC43 proteins. Interestingly, some genes such as IFNA1 and IRF7 were more downregulated in the presence of HCoV-OC43 proteins than in the presence of influenza NS1 protein.
Accessory proteins are often not essential for virus replication in vitro, but required for optimal replication and virulence in the natural host [29]. HCoV-OC43 ns5a accessory protein is known to be a viroporin involved in virion morphogenesis and pathogenesis [30]. In its absence, there is a reduction of virulence, inflammation and replication in infected mice [30]. Murine coronavirus accessory protein 5a is homologous to ns5a protein, and it has been found to antagonise IFN induction [31]. Our results showed that the expression of PRRs, IFNA1, IRF7, MYD88, and MAVS, all important for the induction of an antiviral response, was downregulated in the presence of HCoV-OC43 accessory proteins ns2a and ns5a. Moreover, the expression of interleukins, chemokines, FOS, JUN, IRFs, and IFNA1 was downregulated in the presence of ns5a protein. FOS and JUN are transcription factors involved in the induction of inflammatory cytokines [32, 33]. Altogether, our results suggest HCoV-OC43 ns2a and ns5a have the potential to induce a shift towards an anti-inflammatory response and blockage of the type I IFN pathway. However, in vivo studies are required to confirm the type of immune response HCoV-OC43 elicits during infection. In the absence of adequate immune responses, the virus will be able to replicate and spread freely.
The M protein is indispensable in virion assembly between the Golgi apparatus and endoplasmic reticulum, and is the most abundant protein among the coronavirus proteins [34]. SARS-CoV M protein suppresses the activation of NF-κB [13]. It can also halt IFN production by blocking the formation of the TRAF3 complex [15]. MERS-CoV M protein is also a potent IFN antagonist, targeting the TRAF3-TBK1 interaction to inhibit phosphorylation of IRF3 [17, 35]. Moreover, TRAF6, TRADD and FADD are key players in the NF-κB and apoptosis pathways [36]. Our results showed that the expression of TRAF6 was downregulated in the presence of M or other tested HCoV-OC43 proteins, but this downregulation could not be confirmed after stimulation with Sendai virus. In contrast, the downregulation of TRADD was only observed after challenge with Sendai virus. However, the expression of FADD was not downregulated in the presence of HCoV-OC43M protein before or after challenge with Sendai virus. Furthermore, the expression of MAVS, MYD88, IRFs and IFNA1 that are essential in the induction of the type I IFN signaling pathway, was downregulated in the presence of M protein. This suggests that HCoV-OC43M protein has similar downregulatory effects on the induction of type I IFN and NF-κB as SARS-CoV and MERS-CoV M protein. Further studies are needed to confirm the effects of HCoV-OC43M protein on the type I IFN and NF-κB signaling pathways.
The function of the N protein is to ensure the replication and transcription of viral RNA while disrupting the cell cycle [37, 38]. Indeed, SARS-CoV N protein inhibits the progression of the cell cycle and activates the proinflammatory factor cyclooxygenase-2 [39]. Moreover, it was shown that SARS-CoV N protein inhibits IFN-β expression [14], while it activates IL-6 expression [13]. HCoV-OC43N protein potentiates NF-κB by binding its inhibitor, microRNA 9 [22]. Our study did not show significant fold changes in NF-κB expression in the presence of N protein; however there was a significant downregulation in expression of PRRs (NOD2 and TLR7), adaptor proteins (MYD88, MAVS, CARD9) and signal mediators (IKBKB, IRAK1, TBK1, RIPK1, TRADD, TRAF), all critical in the induction of NF-κB signaling pathway. Moreover, our results showed that expression of MAPKs (MAP3K7 and MAP2K3) was downregulated in the presence of N protein, which is in line with a previous study showing that murine coronavirus N protein inhibits signaling of the AP-1 complex [40], which is composed of FOS and JUN oncogenes, whose expression was also downregulated in our investigation - in the presence of HCoV-OC43 ns2a, ns5a, N or M proteins after challenge with Sendai virus.
MAP3K7, also called Transforming growth factor-b (TGF-b)-activated kinase 1 (TAK1), was the most downregulated gene in the presence of any HCoV-OC43 protein tested after challenging cells with Sendai virus. TAK1 is a protein kinase known to be a critical mediator of the inflammatory response as it is activated by lipopolysaccharide (LPS) and other TLR/NLR agonists, TNF-α and IL-1 [41]. Binding of IL-1 and LPS to their receptors activates TAK1 through a common pathway whereby the kinases IRAK1 and IRAK4 recruit TRAF6. TNF-α activates TAK1 via a similar mechanism, which is mediated by RIP1 and TRAF2. Activation of TRAF2 and TRAF6 leads to the generation of K63-linked polyubiquitin chains, which bind to the adaptor proteins TAB2 or TAB3. TAK1 is bound constitutively to the accessory protein TAB1 and to the homologs TAB2 or TAB3. Once activated, TAK1 transduces the signal to NF-κB, c-Jun N-terminal kinase (JNK), and p38 via phosphorylation of IKK, mitogen-activated protein kinase kinase 4/7 (MKK4/7), and MKK3/6, respectively. Ultimately, NF-κB and other transcription factors downstream of p38 and JNK are activated, resulting in the transcription of genes important for inflammatory and immune responses. Interestingly, Protein kinase R (PKR) interacts with TRAF6, TAK1, and TAB2 as a scaffold protein to mediate TLR3-dependent NF-κB and MAPK activation [42, 43]. The strong downregulation of TAK1 expression observed in our study could be due to: 1) the inhibition of TAK1 expression by direct interaction of HCoV-OC43 protein with TAK1 preventing binding of other mediators like TRAF2/6 or TAB2/3; 2) the upregulation of expression of negative regulators of TAK1 like PP2C or PP6 phosphatases, and deubiquitinases such as ubiquitin-specific peptidase 4 (USP4) [41].
In certain cell lines, IFITM proteins were shown to facilitate HCoV-OC43 entry into host cells [23]. Our study showed that all tested HCoV-OC43 proteins were able to downregulate the expression of IFNA1, suggesting resistance to IFN-α induction. The interaction between HCoV-OC43 proteins and the host cell requires further investigation to understand the processes that could contribute to overall resistance to the antiviral state. Having all these HCoV-OC43 proteins with potential inhibitory effects expressed in close proximity in infected cells indicates a high replicative capacity of the virus.
In conclusion, our findings suggest that similarly to SARS-CoV and MERS-CoV, HCoV-OC43 has the ability to downregulate the transcription of genes critical for the activation of different antiviral signaling pathways. While SARS-CoV and MERS-CoV were shown to induce proinflammatory cytokines in vitro [44, 45], HCoV-OC43 accessory and structural proteins have the potential to inhibit inflammatory response in HEK-293 cells. Further studies are in progress to confirm the role of HCoV-OC43 structural and accessory proteins in antagonising antiviral gene expression, and to elucidate the nature of the immune response and the level of inflammation seen during HCoV-OC43 infection.
Compliance with ethical standards
Funding
This work was supported by Kuwait University Research Administration Grant no. YM 04/15.
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
References
- 1.Cavanagh D. Nidovirales: a new order comprising Coronaviridae and Arteriviridae. Arch Virol. 1997;142:629–633. [PubMed] [Google Scholar]
- 2.Vijgen L, Keyaerts E, Lemey P, et al. Circulation of genetically distinct contemporary human coronavirus OC43 strains. Virology. 2005;337:85–92. doi: 10.1016/j.virol.2005.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Larson HE, Reed SE, Tyrrell DA. Isolation of rhinoviruses and coronaviruses from 38 colds in adults. J Med Virol. 1980;5:221–229. doi: 10.1002/jmv.1890050306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lepiller Q, Barth H, Lefebvre F, et al. High incidence but low burden of coronaviruses and preferential associations between respiratory viruses. J Clin Microbiol. 2013;51:3039–3046. doi: 10.1128/JCM.01078-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Razuri H, Malecki M, Tinoco Y, et al. Human coronavirus-associated influenza-like illness in the community setting in Peru. Am J Trop Med Hyg. 2015;93:1038–1040. doi: 10.4269/ajtmh.15-0271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Talbot HK, Shepherd BE, Crowe JEJ, et al. The pediatric burden of human coronaviruses evaluated for twenty years. Pediatr Infect Dis J. 2009;28:682–687. doi: 10.1097/INF.0b013e31819d0d27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Geller C, Varbanov M, Duval RE. Human coronaviruses: insights into environmental resistance and its influence on the development of new antiseptic strategies. Viruses. 2012;4:3044–3068. doi: 10.3390/v4113044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Arbour N, Cote G, Lachance C, et al. Acute and persistent infection of human neural cell lines by human coronavirus OC43. J Virol. 1999;73:3338–3350. doi: 10.1128/jvi.73.4.3338-3350.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Arbour N, Day R, Newcombe J, Talbot PJ. Neuroinvasion by human respiratory coronaviruses. J Virol. 2000;74:8913–8921. doi: 10.1128/JVI.74.19.8913-8921.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yeh EA, Collins A, Cohen ME, et al. Detection of coronavirus in the central nervous system of a child with acute disseminated encephalomyelitis. Pediatrics. 2004;113:e73–e76. doi: 10.1542/peds.113.1.e73. [DOI] [PubMed] [Google Scholar]
- 11.Morfopoulou S, Brown JR, Davies EG, et al. Human coronavirus OC43 associated with fatal encephalitis. N Engl J Med. 2016;375:497–498. doi: 10.1056/NEJMc1509458. [DOI] [PubMed] [Google Scholar]
- 12.Ye J, Zhang B, Xu J, et al. Molecular pathology in the lungs of severe acute respiratory syndrome patients. Am J Pathol. 2007;170:538–545. doi: 10.2353/ajpath.2007.060469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fang X, Gao J, Zheng H, et al. The membrane protein of SARS-CoV suppresses NF-kappaB activation. J Med Virol. 2007;79:1431–1439. doi: 10.1002/jmv.20953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kopecky-Bromberg SA, Martinez-Sobrido L, Frieman M, et al. Severe acute respiratory syndrome coronavirus open reading frame (ORF) 3b, ORF 6, and nucleocapsid proteins function as interferon antagonists. J Virol. 2007;81:548–557. doi: 10.1128/JVI.01782-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Siu K-L, Chan C-P, Kok K-H, et al. Suppression of innate antiviral response by severe acute respiratory syndrome coronavirus M protein is mediated through the first transmembrane domain. Cell Mol Immunol. 2014;11:141–149. doi: 10.1038/cmi.2013.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Niemeyer D, Zillinger T, Muth D, et al. Middle East respiratory syndrome coronavirus accessory protein 4a is a type I interferon antagonist. J Virol. 2013;87:12489–12495. doi: 10.1128/JVI.01845-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yang Y, Zhang L, Geng H, et al. The structural and accessory proteins M, ORF 4a, ORF 4b, and ORF 5 of Middle East respiratory syndrome coronavirus (MERS-CoV) are potent interferon antagonists. Protein Cell. 2013;4:951–961. doi: 10.1007/s13238-013-3096-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Assiri A, McGeer A, Perl TM, et al. Hospital outbreak of Middle East respiratory syndrome coronavirus. N Engl J Med. 2013;369:407–416. doi: 10.1056/NEJMoa1306742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Assiri A, Al-Tawfiq JA, Al-Rabeeah AA, et al. Epidemiological, demographic, and clinical characteristics of 47 cases of Middle East respiratory syndrome coronavirus disease from Saudi Arabia: a descriptive study. Lancet Infect Dis. 2013;13:752–761. doi: 10.1016/S1473-3099(13)70204-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lee HKK, Tso EYK, Chau TN, et al. Asymptomatic severe acute respiratory syndrome-associated coronavirus infection. Emerg Infect Dis. 2003;9:1491–1492. doi: 10.3201/eid0911.030401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li G, Zhao Z, Chen L, Zhou Y. Mild severe acute respiratory syndrome. Emerg Infect Dis. 2003;9:1182–1183. doi: 10.3201/eid0909.030461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lai FW, Stephenson KB, Mahony J, Lichty BD. Human coronavirus OC43 nucleocapsid protein binds microRNA 9 and potentiates NF-κB activation. J Virol. 2014;88:54–65. doi: 10.1128/JVI.02678-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhao X, Guo F, Liu F, et al. Interferon induction of IFITM proteins promotes infection by human coronavirus OC43. Proc Natl Acad Sci USA. 2014;111:1–6. doi: 10.1073/iti0114111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Desforges M, Desjardins J, Zhang C, Talbot PJ. The acetyl-esterase activity of the hemagglutinin-esterase protein of human coronavirus OC43 strongly enhances the production of infectious. Virus. 2013;87:3097–3107. doi: 10.1128/JVI.02699-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lee HK, Tang JWT, Kong DHL, Koay ESC. Simplified large-scale sanger genome sequencing for influenza A/H3N2 Virus. PLoS One. 2013 doi: 10.1371/journal.pone.0064785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hatada E, Fukuda R. Binding of influenza A virus NS1 protein to dsRNA in vitro. J Gen Virol. 1992;73(Pt 12):3325–3329. doi: 10.1099/0022-1317-73-12-3325. [DOI] [PubMed] [Google Scholar]
- 27.Bergmann M, Garcia-Sastre A, Carnero E, et al. Influenza virus NS1 protein counteracts PKR-mediated inhibition of replication. J Virol. 2000;74:6203–6206. doi: 10.1128/JVI.74.13.6203-6206.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lu Y, Wambach M, Katze MG, Krug RM. Binding of the influenza virus NS1 protein to double-stranded RNA inhibits the activation of the protein kinase that phosphorylates the elF-2 translation initiation factor. Virology. 1995;214:222–228. doi: 10.1006/viro.1995.9937. [DOI] [PubMed] [Google Scholar]
- 29.McBride R, Fielding BC. The role of severe acute respiratory syndrome (SARS)-coronavirus accessory proteins in virus pathogenesis. Viruses. 2012;4:2902–2923. doi: 10.3390/v4112902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang R, Wang K, Ping X, et al. The ns12.9 accessory protein of human coronavirus OC43 is a viroporin involved in virion morphogenesis and pathogenesis. J Virol. 2015;89:11383–11395. doi: 10.1128/JVI.01986-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Koetzner CA, Kuo L, Goebel SJ, et al. Accessory protein 5a is a major antagonist of the antiviral action of interferon against murine coronavirus. J Virol. 2010;84:8262–8274. doi: 10.1128/JVI.00385-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Das KC, Muniyappa H. c-Jun-NH2 terminal kinase (JNK)-mediates AP-1 activation by thioredoxin: phosphorylation of cJun, JunB, and Fra-1. Mol Cell Biochem. 2010;337:53–63. doi: 10.1007/s11010-009-0285-0. [DOI] [PubMed] [Google Scholar]
- 33.Krishna M, Narang H. The complexity of mitogen-activated protein kinases (MAPKs) made simple. Cell Mol Life Sci. 2008;65:3525–3544. doi: 10.1007/s00018-008-8170-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Locker JK, Rose JK, Horzinek MC, Rottier PJ. Membrane assembly of the triple-spanning coronavirus M protein. Individual transmembrane domains show preferred orientation. J Biol Chem. 1992;267:21911–21918. doi: 10.1016/S0021-9258(19)36699-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lui P-Y, Wong L-YR, Fung C-L, et al. Middle East respiratory syndrome coronavirus M protein suppresses type I interferon expression through the inhibition of TBK1-dependent phosphorylation of IRF3. Emerg Microbes Infect. 2016;5:e39. doi: 10.1038/emi.2016.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mak TW, Yeh W-C. Signaling for survival and apoptosis in the immune system. Arthritis Res. 2002;4:S243. doi: 10.1186/ar569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Parker MM, Masters PS. Sequence comparison of the N genes of five strains of the coronavirus mouse hepatitis virus suggests a three domain structure for the nucleocapsid protein. Virology. 1990;179:463–468. doi: 10.1016/0042-6822(90)90316-J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kuo L, Masters PS. Genetic evidence for a structural interaction between the carboxy termini of the membrane and nucleocapsid proteins of mouse hepatitis virus. J Virol. 2002;76:4987–4999. doi: 10.1128/JVI.76.10.4987-4999.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yan X, Hao Q, Mu Y, et al. Nucleocapsid protein of SARS-CoV activates the expression of cyclooxygenase-2 by binding directly to regulatory elements for nuclear factor-kappa B and CCAAT/enhancer binding protein. Int J Biochem Cell Biol. 2006;38:1417–1428. doi: 10.1016/j.biocel.2006.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 40.Perlman S, Netland J. Coronaviruses post-SARS: update on replication and pathogenesis. Nat Rev Microbiol. 2009;7:439–450. doi: 10.1038/nrmicro2147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dai L, Aye Thu C, Liu X-Y, et al. TAK1, more than just innate immunity. IUBMB Life. 2012;64:825–834. doi: 10.1002/iub.1078. [DOI] [PubMed] [Google Scholar]
- 42.Jiang Z, Zamanian-Daryoush M, Nie H, et al. Poly(I-C)-induced Toll-like receptor 3 (TLR3)-mediated activation of NFkappa B and MAP kinase is through an interleukin-1 receptor-associated kinase (IRAK)-independent pathway employing the signaling components TLR3-TRAF6-TAK1-TAB2-PKR. J Biol Chem. 2003;278:16713–16719. doi: 10.1074/jbc.M300562200. [DOI] [PubMed] [Google Scholar]
- 43.Ogolla PS, Portillo J-AC, White CL, et al. The protein kinase double-stranded RNA-dependent (PKR) enhances protection against disease cause by a non-viral pathogen. PLoS Pathog. 2013;9:e1003557. doi: 10.1371/journal.ppat.1003557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cheung CY, Poon LLM, Ng IHY, et al. Cytokine responses in severe acute respiratory syndrome coronavirus-infected macrophages in vitro: possible relevance to pathogenesis. J Virol. 2005;79:7819–7826. doi: 10.1128/JVI.79.12.7819-7826.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lau SKP, Lau CCY, Chan K-H, et al. Delayed induction of proinflammatory cytokines and suppression of innate antiviral response by the novel Middle East respiratory syndrome coronavirus: implications for pathogenesis and treatment. J Gen Virol. 2013;94:2679–2690. doi: 10.1099/vir.0.055533-0. [DOI] [PubMed] [Google Scholar]