

Abstract
In this study, we demonstrate that infection of HSB-2 cells with human herpesvirus 6 (HHV-6) resulted in the accumulation of infected cells in the G2/M phase of the cell cycle. Analysis of various cell-cycle-regulatory proteins indicated that the levels of cyclins A2, B1, and E1 were increased in HHV-6-infected cells, but there was no difference in cyclin D1 levels between mock-infected and HHV-6-infected cells. Our data also showed that inducing G2/M phase arrest in cells infected by HHV-6 provided favorable conditions for viral replication.
Keywords: Human herpesvirus 6, Cyclin, Cell cycle arrest
Human herpesvirus 6 (HHV-6) is a ubiquitous pathogen of the subfamily Betaherpesvirinae, which includes cytomegalovirus and HHV-7, and it primarily infects CD4+ T cells [32]. Like other herpesviruses, HHV-6 establishes latency after the initial productive infection and thus is never cleared from its host [28]. Two subtypes of HHV-6 have been identified: variants A and B [30]. Although these two variants are similar in sequence and genome organization, they are associated with different pathogenesis. HHV-6B causes exanthema subitum in young children [34]. HHV-6A is associated with several adult diseases. It is a cofactor in AIDS progression[18] and is involved in various neurological disorders, including encephalitis, multiple sclerosis, seizures, and chronic fatigue syndrome; however, the causal link between human disease and virus infection remains to be fully elucidated [1, 6].
Many viruses, including DNA viruses, retroviruses, and RNA viruses, can perturb the cell cycle during their infections [2, 4]. Recently, there has been increasing evidence demonstrating different levels of cell cycle arrest in HHV-6-infected cells. It has been shown that HHV-6A infection induces cell cycle arrests at the G2/M phase in infected cord blood mononuclear cells [5]. Furthermore, recent studies have suggested that HHV-6A and HHV-6B infection can also alter E2F1/Rb pathways and cause cell cycle arrest in the G2/M phase in infected SupT1 T cells [20] and that HHV-6B infection of MOLT 3 cells causes cell cycle arrest at the G1 phase concomitant with p53 phosphorylation and accumulation [25]. In addition, G1/S arrest induced by HHV-6 infection has been observed in the other types of cells, such as epithelial cells and neural cells [7, 26]. Although HHV-6 has been implicated in halting cell cycle progression, the precise mechanisms behind this phenomenon have not yet been fully understood.
The aim of this work was to investigate the effects of HHV-6 on the cell cycle, with emphasis on the patterns of expression of cyclins as the infection progressed. We demonstrate that HHV-6 induced elevated protein levels of cyclin A2, cyclin B1, and cyclin E1 following virus-induced cell cycle arrest. Furthermore, we show here for the first time that the G2/M arrest provides a favorable environment for viral replication.
The GS strain of HHV-6 variant A was propagated in cord blood mononuclear cells (CBMCs), as described previously [33]. HHV-6A viral DNA loads were quantified by SYBR Green real-time PCR. Primers were specific for the HHV-6 late gene U22. The forward primer was 5’-CGCTCGGAAAGGAAACATTA-3’, and the reverse primer was 5’-AAGTGGAACTGCTTGGTGGC-3’. A standard curve was generated by amplification of an HHV-6A U22 DNA sequence incorporated into the pMD™ 19-T vector, designated as pMD19T-U22. A multiplicity of infection (MOI) of 10 virus DNA copies per cell was used for all of the experiments. After infection with HHV-6A, HSB-2 cells showed typical cytopathic effects (CPE), rounding up like balloons, and fusion of infected cells to form giant multinucleated syncytia (Fig. 1A). To further demonstrate HHV-6A infection in HSB-2 cells, expression of a late protein gp60/110, which is essential for viral propagation in infected HSB-2 cells, was analyzed by western blot using an anti-gp60/110 monoclonal antibody (Chemicon). Prominent expression of HHV-6 gp60/110 was detected in HHV-6A-infected HSB-2 cells compared with that in control mock-infected cells, and the levels of this marker increased in a time-dependent manner following HHV-6A infection (Fig. 1B). We also investigated the extent of viral genome replication in HHV-6A-infected HSB-2 cells by real-time PCR at 24, 48, and 72 h postinfection. As shown in Fig. 1C, viral loads in cell lysates from HHV-6A-infected cells increased from 1.17 × 107 copies/106 cells at 24 h and 2.42 × 107 copies/106 cells at 48 h to 5.15 × 107 copies/106 cells at 72 h after infection. These results showed that HHV-6A could efficiently infect HSB-2 cells.
Fig. 1.

Infection of HSB-2 cells with HHV-6A. A HHV-6A infection, exhibiting typical cytopathic effects in infected HSB-2 cells. The morphological characteristics of HSB-2 cells infected with or without HHV-6A were observed under a light microscope at 72 h postinfection. B HHV-6 gp60/110 expression in HSB-2 cells mock infected or infected with HHV-6A at various time points. Gp60/110 protein expression was determined by western blot analysis using an anti-gp60/110 monoclonal antibody. C Quantitative detection of intracellular HHV-6 DNA at 24, 48 and 72 h postinfection
Cell cycle and nuclear DNA content were determined using propidium iodide (PI) staining and measured by flow cytometry, as described previously [14]. Representative cell cycle profiles in mock- and HHV-6A-infected cells are presented in Fig. 2A. HHV-6A infection markedly decreased the proportion of cells in G1 phase and significantly increased the number of cells in G2/M phase with increasing infection time. Infected cells exhibited an almost sixfold increase in the percentage of G2/M-phase cells compared to that of the control cells at 72 h postinfection. Concomitant with the increase of G2/M-phase cells, there was a threefold decrease in the percentage of HHV-6A-infected cells in the G1 phase compared with mock-infected cells at 72 h postinfection. Collectively, these data revealed that HHV-6A infection promotes cell cycle progression from G1 and S to the G2/M phase and then arrests the infected cells in the G2/M phase.
Fig. 2.
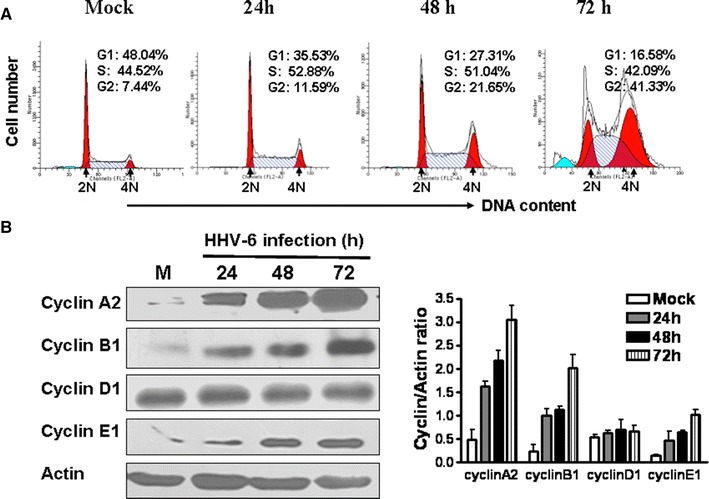
HHV-6A infection induces G2/M phase arrest and significantly increases cyclin A2, cyclin B1 and cyclin E1 expression in HSB-2 cells. A HHV-6A infection markedly decreased the proportion of HSB-2 cells in the G1 phase and increased the proportion in the G2/M-phase with increasing infection time. The relative DNA content of HSB-2 cells at different time points after HHV-6A infection was determined by PI staining and analyzed by a flow cytometry. The 2N (diploid) and 4N (Tetraploid) DNA contents represent the G1 and G2/M phases of the cell cycles, respectively. B Cyclins expression in HSB-2 cells infected with or without HHV-6A. Cell lysates were collected at the indicated time points, and western blot analysis was performed with anti-cyclin A2, anti-cyclin B1, anti-cyclin D1, and cyclin E1 antibodies. Cyclin expression levels were quantitatively analyzed and compared relative to β-actin expression using a densitometer. Results shown at the right are the mean ± SD from three independent experiments
To address the molecular mechanism of HHV-6A-induced cell cycle arrest at the G2/M phase, we examined the expression kinetics of the cell cycle regulatory molecules cyclin A2, B1, D1, and E1 in HHV-6A-infected cells compared to mock-infected cells. At various intervals after infection, HSB-2 cell lysates were collected, and cyclin protein expression was determined by western blot analysis using anti-cyclinA2, anti-cyclinB1, anti-cyclinD1, anti-cyclinE1 and anti-β-actin antibodies (Cell Signaling Technology). As expected, a more drastic increase in cyclin A2, B1, and E1 levels was observed in HHV-6A-infected cells compared with mock-control. At 72 h postinfection, densitometric analysis indicated that cyclin A2, B1 and E1 levels were sixfold, eightfold and sevenfold higher, respectively, in HHV-6A infected cells than that in mock-infected cells. In contrast, there was no obvious change in levels of cyclinD1 in HHV-6A-infected cells compared to mock-infected cells at any time point analyzed (Fig. 2B).
Manipulation of cell cycle progression is an important strategy exploited by many viruses to create conductive cellular conditions for viral replication. Our data indicated that HHV-6A infection caused an increase in the population of G2/M-phase cells. Therefore, we investigated whether this status of the cell cycle was advantageous for the virus by comparing virus DNA replication and protein expression. Asynchronously growing HSB-2 cells were treated with either DMSO or 0.5 μg/ml nocodazole (Sigma-Aldrich) for 24 h. In asynchronous HSB-2 cells approximately 46 % of cells were in the G0/G1 phase, 48 % in the S phase, and 6 % in the G2/M phase, whereas using nocodazole treatment, approximately 48 % of cells were synchronized in the G2/M phase (Fig. 3A). Then, asynchronously dividing cells and G2/M-phase-synchronized cells were infected with HHV-6A at an MOI of 10 in fresh medium, and virus production was measured by real-time PCR and western blot analysis at 72 h postinfection. As shown in Fig. 3B and C, both the amounts of U22 gene (2-fold) and the expression levels of gp 60/110 protein (2.5-fold) were higher in G2/M-phase-synchronized cells than in asynchronously replicating cells.
Fig. 3.

G2/M arrest promotes HHV-6A replication. A Asynchronously growing HSB-2 cells were treated with DMSO or 0.5 μg/ml nocodazole for 24 h, and the cell cycle profiles were then analyzed by flow cytometric analysis. The 2N (diploid) and 4N (Tetraploid) DNA contents represent the G1 and G2/M phases of the cell cycles, respectively. B and C Synchronously growing HSB-2 cells and asynchronously growing control cells were infected with HHV-6A at an MOI of 10, followed by incubation for 72 h. The amount of viral U22 gene was determined by real-time PCR (B). Viral protein accumulation was analyzed by western blot with anti-HHV-6 gp60/110 antibody, and the expression level was quantitatively analyzed and compared to β-actin expression using a densitometer (C). The data shown are the mean ± SD from three independent experiments. **P < 0.01 compared with the control
Manipulation of the cell cycle in infected cells is a common strategy used by many viruses to regulate their infection. Our previous studies showed that HHV-6A can infect human embryonic fibroblasts and induce G2/M arrest and cell death [13]. We also found that HHV-6A infection inhibits cell cycle progression in HSB-2 cells, resulting from the inhibition of Cdc2-cyclin B1 kinase activity, which is involved in the activation of p53/p21 and Chk1/Chk2/Cdc25C pathways, as well as elevated Wee1 expression [14]. In this study, we demonstrated that HHV-6A infection promotes cell cycle arrest in the G2/M phase and alters the expression of key cell-cycle-regulatory proteins cyclins in HSB-2 cells. A manifest accumulation of cyclins A2, B1, and E1 was observed in HHV-6A-infected cells. However, the levels of cyclin D1 were not obviously changed in infected cells compared to mock-infected cells. Cyclin B1 is an important regulator during the normal cell cycle progression. It accumulates in the S and G2 phases to form a mitosis-promoting factor (MPF) with Cdc2 and then is ubiquitinated and degraded by the anaphase-promoting complex (APC) after the cells pass through mitosis [22, 27]. It is noteworthy that the levels of cyclin B1 remain high in HHV-6A-infected cells, suggesting that an absence of degradation through the ubiquitin pathway might contribute to the cell cycle arrest observed in infected cells. Cyclin E1 has been shown to be rate limiting for S-phase entry, and when overexpressed, to accelerate the G1/S transition [23]. In our study, changes in cyclin D1 expression were not detected after infection, suggesting that the majority of the infected cells do not return to G1 phase after infection, as would be expected with normal cycling and division. In support of our findings, Jault et al. [11] reported HCMV-induced G2/M arrest in cycling cells and found that high levels of cyclin E were induced, with cyclin A appearing only at late time points. De Bolle and coworkers [5] also reported that cyclin B1, cyclin A, and to a slight extent, cyclin E accumulated after HHV-6A infection in cord blood mononuclear cells. Recently, Morita et al. [21] also reported that B19-virus-infected UT7/Epo-S1 cells displayed accumulation of cyclin A, cyclin B1, and phosphorylated cdc2, resulting in cell cycle arrest at the G2 phase; however, the Cdc2-cyclin B1 kinase activity remained high in B19 virus-infected cells.
For successful propagation, viruses may manipulate cell cycle progression to create a more conducive environment for replication [24, 29, 31]. A recent study suggested that influenza A virus may create favorable conditions in infected cells for viral protein accumulation and virus production by inducing a G0/G1 phase arrest in infected cells [10]. EBV could block the host response and actively promote an S-phase-like environment for viral lytic replication [12]. It was also discovered that viral protein expression and progeny virus production were greater in G2/M-phase-arrested cells. For example, it was reported that avian reovirus (ARV) p17 protein facilitates virus replication through initiation of G2/M arrest and host cellular translation shutoff [3]. HIV infection is also favored in the G2/M phase. Groschel and Bushman [9] observed a three- to fivefold increase in HIV transduction compared to other stages of the cell cycle. In addition, the avian coronavirus infectious bronchitis virus (IBV) induces a G2/M phase arrest in infected cells to promote viral replication [8]. In this study, we found that the efficiency of HHV-6A infection is greater in G2/M-synchronized cells than in asynchronously replicating cells. Therefore, we inferred that HHV-6 induces G2/M phase arrest to provide conditions for progeny virus production. The benefits for virus output may be explained by several hypotheses, such as increasing the efficiency of transcription, translation and virus assembly. For example, Lin and Lamb [16] proposed that enveloped RNA viruses could arrest the cell cycle before mitosis to prevent disruption of the Golgi apparatus and endoplasmic reticulum (ER), favoring viruses whose assembly occurs in these structures. It is known that coronaviruses such as IBV also utilize the Golgi apparatus and ER for protein processing and assembly [15, 17, 19].
The studies presented here, coupled with previous work from our lab and others, demonstrate that HHV-6 infection causes dramatic changes in the expression of host-cell regulatory proteins, leading to the cell cycle arrest. Clarification of the molecular mechanisms by which HHV-6 disrupts the cell cycle machinery will not only be important in the study of the cell cycle changes that occur in HHV-6 replication but will also be crucial for better understanding of the processes by which HHV-6 causes disease and for development of novel vaccines and/or therapeutic agents to inhibit HHV-6 infection.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (No. 30972784, No. 81273235 and No. 81201520). We thank Analytical and Testing Center, Nanjing Medical University, for technical assistance.
References
- 1.Cameron B, Flamand L, Juwana H, Middeldorp J, Naing Z, Rawlinson W, Ablashi D, Lloyd A. Serological and virological investigation of the role of the herpesviruses EBV, CMV and HHV-6 in post-infective fatigue syndrome. J Med Virol. 2010;82:1684–1688. doi: 10.1002/jmv.21873. [DOI] [PubMed] [Google Scholar]
- 2.Chaurushiya MS, Weitzman MD. Viral manipulation of DNA repair and cell cycle checkpoints. DNA Repair. 2009;8:1166–1176. doi: 10.1016/j.dnarep.2009.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chulu JL, Huang WR, Wang L, Shih WL, Liu HJ. Avian reovirus nonstructural protein p17-induced G(2)/M cell cycle arrest and host cellular protein translation shutoff involve activation of p53-dependent pathways. J Virol. 2010;84:7683–7694. doi: 10.1128/JVI.02604-09. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 4.Davy C, Doorbar J. G2/M cell cycle arrest in the life cycle of viruses. Virology. 2007;368:219–226. doi: 10.1016/j.virol.2007.05.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.De Bolle L, Hatse S, Verbeken E, De Clercq E, Naesens L. Human herpesvirus 6 infection arrests cord blood mononuclear cells in G(2) phase of the cell cycle. FEBS Lett. 2004;560:25–29. doi: 10.1016/S0014-5793(04)00035-3. [DOI] [PubMed] [Google Scholar]
- 6.De Bolle L, Naesens L, De Clercq E. Update on human herpesvirus 6 biology, clinical features, and therapy. Clin Microbiol Rev. 2005;18:217–245. doi: 10.1128/CMR.18.1.217-245.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dietrich J, Blumberg BM, Roshal M, Baker JV, Hurley SD, Mayer-Proschel M, Mock DJ. Infection with an endemic human herpesvirus disrupts critical glial precursor cell properties. J Neurosci. 2004;24:4875–4883. doi: 10.1523/JNEUROSCI.5584-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dove B, Brooks G, Bicknell K, Wurm T, Hiscox JA. Cell cycle perturbations induced by infection with the coronavirus infectious bronchitis virus and their effect on virus replication. J Virol. 2006;80:4147–4156. doi: 10.1128/JVI.80.8.4147-4156.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Groschel B, Bushman F. Cell cycle arrest in G2/M promotes early steps of infection by human immunodeficiency virus. J Virol. 2005;79:5695–5704. doi: 10.1128/JVI.79.9.5695-5704.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.He Y, Xu K, Keiner B, Zhou J, Czudai V, Li T, Chen Z, Liu J, Klenk HD, Shu YL, Sun B (2010) Influenza A virus replication Induces Cell Cycle Arrest in G0/G1 Phase. J Virol [DOI] [PMC free article] [PubMed]
- 11.Jault FM, Jault JM, Ruchti F, Fortunato EA, Clark C, Corbeil J, Richman DD, Spector DH. Cytomegalovirus infection induces high levels of cyclins, phosphorylated Rb, and p53, leading to cell cycle arrest. J Virol. 1995;69:6697–6704. doi: 10.1128/jvi.69.11.6697-6704.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kudoh A, Fujita M, Zhang L, Shirata N, Daikoku T, Sugaya Y, Isomura H, Nishiyama Y, Tsurumi T. Epstein-Barr virus lytic replication elicits ATM checkpoint signal transduction while providing an S-phase-like cellular environment. J Biol Chem. 2005;280:8156–8163. doi: 10.1074/jbc.M411405200. [DOI] [PubMed] [Google Scholar]
- 13.Li L, Gu B, Zhou F, Chi J, Wang F, Liu G, Ding C, Xie F, Qing J, Guo Y, Yao K. Human herpesvirus 6A infects human embryonic fibroblasts and induces G2/M arrest and cell death. J Med Virol. 2012;84:657–663. doi: 10.1002/jmv.23226. [DOI] [PubMed] [Google Scholar]
- 14.Li L, Gu B, Zhou F, Chi J, Wang F, Peng G, Xie F, Qing J, Feng D, Lu S, Yao K. Human herpesvirus 6 suppresses T cell proliferation through induction of cell cycle arrest in infected cells in the G2/M phase. J Virol. 2011;85:6774–6783. doi: 10.1128/JVI.02577-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lim KP, Liu DX. The missing link in coronavirus assembly. Retention of the avian coronavirus infectious bronchitis virus envelope protein in the pre-Golgi compartments and physical interaction between the envelope and membrane proteins. J Biol Chem. 2001;276:17515–17523. doi: 10.1074/jbc.M009731200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lin GY, Lamb RA. The paramyxovirus simian virus 5V protein slows progression of the cell cycle. J Virol. 2000;74:9152–9166. doi: 10.1128/JVI.74.19.9152-9166.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lontok E, Corse E, Machamer CE. Intracellular targeting signals contribute to localization of coronavirus spike proteins near the virus assembly site. J Virol. 2004;78:5913–5922. doi: 10.1128/JVI.78.11.5913-5922.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lusso P, Crowley RW, Malnati MS, Di Serio C, Ponzoni M, Biancotto A, Markham PD, Gallo RC. Human herpesvirus 6A accelerates AIDS progression in macaques. Proc Natl Acad Sci USA. 2007;104:5067–5072. doi: 10.1073/pnas.0700929104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Machamer CE, Mentone SA, Rose JK, Farquhar MG. The E1 glycoprotein of an avian coronavirus is targeted to the cis Golgi complex. Proc Natl Acad Sci USA. 1990;87:6944–6948. doi: 10.1073/pnas.87.18.6944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mlechkovich G, Frenkel N. Human herpesvirus 6A (HHV-6A) and HHV-6B alter E2F1/Rb pathways and E2F1 localization and cause cell cycle arrest in infected T cells. J Virol. 2007;81:13499–13508. doi: 10.1128/JVI.01496-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Morita E, Tada K, Chisaka H, Asao H, Sato H, Yaegashi N, Sugamura K. Human parvovirus B19 induces cell cycle arrest at G(2) phase with accumulation of mitotic cyclins. J Virol. 2001;75:7555–7563. doi: 10.1128/JVI.75.16.7555-7563.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nurse P. Universal control mechanism regulating onset of M-phase. Nature. 1990;344:503–508. doi: 10.1038/344503a0. [DOI] [PubMed] [Google Scholar]
- 23.Ohtsubo M, Roberts JM. Cyclin-dependent regulation of G1 in mammalian fibroblasts. Science (New York) 1993;259:1908–1912. doi: 10.1126/science.8384376. [DOI] [PubMed] [Google Scholar]
- 24.Op De Beeck A, Caillet-Fauquet P. Viruses and the cell cycle. Prog Cell Cycle Res. 1997;3:1–19. doi: 10.1007/978-1-4615-5371-7_1. [DOI] [PubMed] [Google Scholar]
- 25.Oster B, Bundgaard B, Hollsberg P. Human herpesvirus 6B induces cell cycle arrest concomitant with p53 phosphorylation and accumulation in T cells. J Virol. 2005;79:1961–1965. doi: 10.1128/JVI.79.3.1961-1965.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Oster B, Kaspersen MD, Kofod-Olsen E, Bundgaard B, Hollsberg P. Human herpesvirus 6B inhibits cell proliferation by a p53-independent pathway. J Clin Virol. 2006;37(Suppl 1):S63–S68. doi: 10.1016/S1386-6532(06)70014-2. [DOI] [PubMed] [Google Scholar]
- 27.Page AM, Hieter P. The anaphase-promoting complex: new subunits and regulators. Annu Rev Biochem. 1999;68:583–609. doi: 10.1146/annurev.biochem.68.1.583. [DOI] [PubMed] [Google Scholar]
- 28.Sandhoff T, Kleim JP, Schneweis KE. Latent human herpesvirus-6 DNA is sparsely distributed in peripheral blood lymphocytes of healthy adults and patients with lymphocytic disorders. Med Microbiol Immunol. 1991;180:127–134. doi: 10.1007/BF00206116. [DOI] [PubMed] [Google Scholar]
- 29.Schang LM. The cell cycle, cyclin-dependent kinases, and viral infections: new horizons and unexpected connections. Prog Cell Cycle Res. 2003;5:103–124. [PubMed] [Google Scholar]
- 30.Schirmer EC, Wyatt LS, Yamanishi K, Rodriguez WJ, Frenkel N. Differentiation between two distinct classes of viruses now classified as human herpesvirus 6. Proc Natl Acad Sci USA. 1991;88:5922–5926. doi: 10.1073/pnas.88.13.5922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Swanton C, Jones N. Strategies in subversion: de-regulation of the mammalian cell cycle by viral gene products. Int J Exp Pathol. 2001;82:3–13. doi: 10.1046/j.1365-2613.2001.00165.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Takahashi K, Sonoda S, Higashi K, Kondo T, Takahashi H, Takahashi M, Yamanishi K. Predominant CD4 T-lymphocyte tropism of human herpesvirus 6-related virus. J Virol. 1989;63:3161–3163. doi: 10.1128/jvi.63.7.3161-3163.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang F, Yao K, Yin QZ, Zhou F, Ding CL, Peng GY, Xu J, Chen Y, Feng DJ, Ma CL, Xu WR. Human herpesvirus-6-specific interleukin 10-producing CD4+ T cells suppress the CD4+ T-cell response in infected individuals. Microbiol Immunol. 2006;50:787–803. doi: 10.1111/j.1348-0421.2006.tb03855.x. [DOI] [PubMed] [Google Scholar]
- 34.Yamanishi K, Okuno T, Shiraki K, Takahashi M, Kondo T, Asano Y, Kurata T. Identification of human herpesvirus-6 as a causal agent for exanthem subitum. Lancet. 1988;1:1065–1067. doi: 10.1016/S0140-6736(88)91893-4. [DOI] [PubMed] [Google Scholar]