

Abstract
Porcine reproductive and respiratory syndrome virus (PRRSV) infection appears to elicit a weak innate immune response suppressing type 1 interferon (IFN) production. Recent studies have revealed that several nonstructural proteins encoded by the PRRSV genome independently antagonize the type 1 IFN system. The present study sought to identify the structural proteins that possess the immune evasion properties in immortalized porcine alveolar macrophages (PAM). Each structural protein gene was stably expressed in a porcine monocyte-derived macrophage cell line, PAM-pCD163, and tested for its potential to inhibit IFN-β induction. We then focused on the nucleocapsid (N) protein, which has a strong inhibitory effect on dsRNA-induced IFN-β production. Upon dsRNA stimulation, IFN-β production was shown to decrease proportionally with increasing levels of N expression. Furthermore, the PRRSV N protein was found to down-regulate IFN-dependent gene production by dsRNA. Taken together, these results indicate the ability of N to modulate the dsRNA-mediated IFN induction pathways. In addition, the N protein significantly interfered with dsRNA-induced phosphorylation and nuclear translocation of IRF3. Our data suggest that the PRRSV N protein is a responsible component, independent of other nonstructural elements, for evading the IFN response by antagonizing IRF3 activation.
Keywords: Porcine Alveolar Macrophage, ISG15 mRNA, Porcine Alveolar Macrophage Cell, Immune Evasion Property, dsRNA Stimulation
Introduction
Porcine reproductive and respiratory syndrome (PRRS) is an important and re-emerging disease of swine that was first recognized independently in North America in 1987 and in Europe in 1990 [19, 46]. The disease is characterized by reproductive failure in pregnant sows and mild to severe respiratory distress in piglets and growing pigs [47]. Since its emergence, the PRRS situation has become endemic in most pork-producing countries, causing tremendous economic losses in the pig industry worldwide [2, 36]. The causative agent of the disease is PRRS virus (PRRSV), which belongs to the family Arteriviridae in the order Nidovirales [5, 31, 41]. The PRRSV genome is a single-stranded positive-sense RNA containing ten open reading frames (ORFs), designated ORF1a, ORF1b, and ORF2 through 7, including ORF2b and ORF5a [12, 15, 41]. Two large ORFs, 1a and 1b, occupying the 5′ two-thirds of the genome, encode the nonstructural polyproteins pp1a and pp1ab, which are proteolytically cleaved into 14 nonstructural protein (NSP) products: NSP1α, NSP1β, NSP2 through NSP12 including NSP7α and NSP7β, in order from the N-terminus [41, 44]. The remaining ORFs in the 3′-terminal region code for structural GP2, small envelope (E), GP3, GP4, 5a, GP5, membrane (M), and nucleocapsid (N) proteins [12, 15, 32, 51].
The type 1 IFN system is a central feature of the antiviral innate immune response, which is the first defense against viral infection [37, 39]. Viral replication initially generates viral double-stranded RNA (dsRNA), which is then recognized by host-cell receptors. This interaction stimulates host signaling pathways, which leads to the coordinated activation and subsequent nuclear translocation of transcription factors, including interferon regulatory factor 3 (IRF3), nuclear factor kappa B (NF-κB), and activating transcription factor-2 (ATF-2), to induce the expression of type 1 IFN. Once IFN is secreted, it signals via IFN receptors on adjacent cell surfaces, which activates a cell-signaling cascade through the Janus kinase (JAK)-mediated signal transducer and activator of transcription (STAT) pathway. This triggers the transcription of several hundred IFN-stimulated genes (ISGs), enabling target cells to produce an antiviral response to inhibit virus replication.
PRRSV primarily infects cells of the monocyte/macrophage lineage, such as porcine alveolar macrophages (PAMs) [9, 10] and can establish persistent infection, which may last for up to six months in the natural host [1, 8, 48]. Consequently, PRRSV infection appears to suppress normal macrophage function and immune responses, and it is complicated by secondary opportunistic bacterial infections in most cases [11]. Indeed, studies have revealed that lungs of pigs infected with PRRSV are incapable of eliciting type 1 interferon (IFN) responses [4, 29, 33, 45]. The impairment of type 1 IFN production by PRRSV would result in a weak host adaptive immunity, including a delayed or slow development of humoral and cell-mediated immune responses, leading to viral persistence in infected pigs [28, 30, 34, 38].
Viruses have evolved the strategy of expressing proteins to circumvent the IFN response for their survival in the host. PRRSV has been postulated to encode viral products that possess immune evasion properties by regulating IFN activity. Thus, several recent studies have attempted to identify NSPs with innate immune evasion features encoded by the PRRSV genome. Beura et al. [3] first reported that various NSPs, such as NSP1, NSP2, NSP4, and NSP11, are able to suppress dsRNA-induced IFN-β promoter activation. In that study, NSP1α and NSP1β, auto-cleavage products of NSP1, were shown to modulate type 1 IFN induction by antagonizing IRF3 activation. Other studies have independently demonstrated that NSP1 exerts its inhibitory effect on the IFN response in different ways by interfering with other transcription factors such as CREB-binding protein (CBP), NF-κB, and STAT-1 [6, 20]. In addition, the PRRSV NSP2 has been shown to antagonize type I IFN production by inhibiting the NF-κB signaling pathway or IRF3 activation [27, 43]. However, little is known about structural proteins that regulate IFN activity. In the present study, therefore, we aimed to identify the structural proteins of PRRSV that are responsible for mediating IFN down-regulation in immortalized natural target cells. It was showed that several structural proteins inhibit type 1 IFN induction upon dsRNA treatment. Among those viral proteins, the N protein was chosen to investigate the inhibitory mechanism and was subsequently found to block dsRNA-induced IRF3 activation. Our data indicate that the N protein has a novel function related to the immune modulation and evasion strategy during PRRSV infection.
Materials and methods
Cells, virus, and antibodies
PAM-pCD163 cells [26] were cultured in RPMI 1640 medium (Invitrogen) supplemented with 10% fetal bovine serum (FBS; Invitrogen), 1% antibiotic-antimycotic solution (100×; Invitrogen), 10 mM HEPES (Invitrogen), 1 mM sodium pyruvate (Invitrogen), and nonessential amino acids (100×; Invitrogen) in the presence of 50 μg/ml Zeocin (Invitrogen). The cells were maintained at 37°C with 5% CO2. The PRRSV strain PL97-1 [17] was used to prepare virus stocks in PAM-pCD163 as described previously [26]. The following antibodies were used in the present study: anti-PRRSV N monoclonal antibody (MAb) SDOW17 (Rural Technologies), rabbit anti-IRF3 polyclonal antibody (Santa Cruz Biotechnology), rabbit anti-SP1 polyclonal antibody (Santa Cruz Biotechnology), anti-β-actin MAb (Santa Cruz Biotechnology), anti-α-tubulin (Sigma), anti-phosphorylated IRF3 (Ser396) rabbit MAb (Cell Signaling Technology), and anti-6× histidine tag MAb (IG Therapy).
Construction of expression plasmids
DNA manipulation and cloning were performed according to standard procedures [40]. The E. coli strain DH5α (RBC Bioscience) was used as the host for general cloning. The PRRSV strain PL97-1 was used as a template to extract viral RNA using a Viral RNA Mini Kit (QIAGEN) according to the manufacturer's instructions. RT-PCR was performed to amplify all PRRSV structural protein genes with primer sets, and primer sequences will be provided upon request. The PCR amplicon was initially inserted into a pBudCE4.1 vector (Invitrogen) that contains six repetitive 3′ histidine codons, and each resulting plasmid was verified by nucleotide sequencing. A His-tagged cDNA fragment obtained from the corresponding plasmid vector was then subcloned into a pFB-Neo retroviral vector (Stratagene) using Sal I and EcoR I restriction sites to construct individual PRRSV structural-protein-expressing plasmids, thereby producing C-terminally His-tagged PRRSV structural proteins.
Generation of stable PAM cell lines
A retrovirus gene transfer system (Stratagene) was applied to generate cell lines constitutively expressing each recombinant PRRSV structural protein as described elsewhere [26, 35]. Antibiotic-resistant continuous cell clones were examined by RT-PCR to detect the presence of the full-length structural protein gene, and positive clones expressing the respective PRRSV structural proteins were amplified for subsequent analysis.
Quantitative real-time RT-PCR
PAM-pCD163 cells grown at 3 × 105 cells/well in a 6-well tissue culture plate were infected with PRRSV at a multiplicity of infection (MOI) of 1 for 24 h and transfected with 1 μg polyinosinic:polycytidylic acid (poly[I:C]; Sigma) using X-fect (Clontech) according to the manufacturer's instructions. PAM cells expressing each viral protein were grown in 6-well tissue culture plates to 80% confluency for two days and treated by transfection with 1 μg poly(I:C). At 6 h post-poly(I:C) stimulation, total cellular RNA was extracted using TRIzol Reagent (Invitrogen) and treated with DNase I (TaKaRa) according to the manufacturer's manual. The RNA was used for reverse transcription using a PrimeScript 1st strand cDNA synthesis kit and then for real-time PCR for quantification of porcine IFN-β and ISG15 mRNA copy number on a Thermal Cycler Dice Real Time System using SYBR Green I (TaKaRa). Real-time PCR reactions were performed in a total volume of 20 μl of the reaction mixture containing a template cDNA, forward and reverse primers, and 2× SYBR Premix Ex Taq (TaKaRa). The following primer sets were used in the real-time PCRs: porcine IFN-β forward, 5′-TCGCTCTCCTGATGTGTTTC-3′; porcine IFN-β reverse, 5′-TTCTGACATGCCAAATTGCT-3′; porcine ISG15 forward, 5′-GGGACCTGACGGTGAAGATGC-3′; porcine ISG15 reverse, 5′-GCCAGACGCTGCTGG-3′; porcine β-actin forward, 5′-GACCACCTTCAACTCGATCA-3′; porcine β-actin reverse, 5′-GTGTTGGCGTAGAGGTCCTT-3′. The mRNA levels of the tested genes were normalized to that of porcine β-actin mRNA in all experiments. The porcine IFN-β and ISG15 mRNA levels in virus-infected cells or in viral-protein-expressing cells subjected to poly(I:C) stimulation were expressed as copy numbers relative to unstimulated cells according to a method described previously [42]. Three independent experiments were repeated, and the average of normalized values is presented.
Cell fractionation and western blot analysis
PAM cells were grown in 6-well tissue culture plates to 80% confluency for two days and transfected with 1 μg poly(I:C) for 6 h. The cells were then solubilized in a lysis buffer containing 0.5% Triton X-100, 60 mM β-glycerophosphate, 15 mM ρ-nitrophenyl phosphate, 25 mM MOPS, 15 mM, MgCl2, 80 mM NaCl, 15 mM EGTA (pH 7.4), 1 mM sodium orthovanadate, 1 μg/ml E64, 2 μg/ml aprotinin, 1 μg/ml leupeptin, and 1 mM PMSF for 30 min on ice and clarified by centrifugation at 15,800×g (Eppendorf centrifuge 5415R) for 30 min at 4°C. For cell fractionation, poly(I:C)-transfected PAM cells were fractionated using a Nuclear/Cytosol Fractionation Kit (BioVision) according to the manufacturer's manual. The total protein concentrations in the supernatants were determined using a BCA protein assay (Pierce). Equal amounts of total protein were separated on a NuPAGE 4-12% gradient Bis-Tris gel (Invitrogen) under reducing conditions and electrotransferred onto Immunobilon-P (Millpore). The membranes were blocked with 3% powdered skim milk (BD Biosciences) in TBS (10 mM Tris-HCl [pH 8.0], 150 mM NaCl) with 0.05% Tween-20 (TBST) at 4°C for 2 h and incubated at 4°C overnight with appropriate primary antibodies. The blots were then incubated with horseradish peroxidase (HRP)-labeled goat anti-mouse IgG or anti-rabbit IgG (Santa Cruz Biotechnology) at a dilution of 1:5,000 for 2 h at 4°C. Protein bands were detected using enhanced chemiluminescence (ECL) reagents (Amersham Biosciences) according to the instructions of the manufacturer.
Indirect immunofluorescence
PAM cells were grown on microscope coverslips placed in 6-well tissue culture plates and induced by transfection with 1 μg poly(I:C). At 48 h post-transfection, cells were fixed with 4% paraformaldehyde for 10 min at room temperature (RT) and permeabilized with 0.2% Triton X-100 in PBS at RT for 10 min. The cells were blocked using 1% bovine serum albumin (BSA) in PBS for 30 min at RT and then co-stained with anti-IRF3 polyclonal antibody and anti-N SDOW17 MAb or anti-His tag antibody for 2 h. After washing five times in PBS, the cells were incubated for 1 h at RT with a mixture of Alexa Fluor 488–conjugated anti-rabbit and Alexa Fluor 594–conjugated anti-mouse secondary antibodies (Invitrogen). The cells were finally counterstained with 4′,6-diamidino-2-phenylindole (DAPI; Sigma) and visualized using a fluorescent microscope (Leica DM IL LED).
Results
Modulation of type 1 IFN induction by PRRSV
Although several research groups have reported the ability of PRRSV to suppress type 1 IFN production, they used primary PAM cells or Marc-145 cells as target cells to determine IFN response upon PRRSV infection [3, 20, 25, 33]. In this study, therefore, we first sought to revisit whether such a feature of PRRSV is reproducible in our continuous PAM cell model [26]. For this experiment, a well-characterized PRRSV strain, PL97-1 [7, 17], corresponding to the type 2 North American genotype, was used to infect immortalized PAM-pCD163 cells to measure IFN-β mRNA levels by quantitative RT-PCR. As shown in Fig. 1, very little IFN-β mRNA was detected in the cells infected with PRRSV. In contrast, PAM-pCD163 cells transfected with poly(I:C), a positive control for type 1 IFN production, showed efficient induction of IFN-β mRNA. However, PRRSV infection significantly suppressed poly(I:C)-mediated IFN-β mRNA production in PAM-pCD163 cells, confirming the regulatory property of PRRSV on type I IFN induction in our cell system.
Fig. 1.
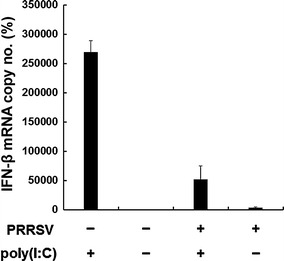
Suppression of IFN-β production in immortalized PAM cells infected with PRRSV. PAM-pCD163 cells were mock-infected or infected with PRRSV (PL97-1) at an MOI of 1. At 24 h postinfection, cells were transfected with poly(I:C) for 6 h. Total cellular RNA was extracted and reverse transcribed, and real-time PCR was performed for the quantitative measurement of porcine IFN-β mRNA. mRNA copy numbers were calculated after normalization with the β-actin copy number and are expressed as a percentage relative to the untreated control. These data are representative of the mean values from three independent experiments, and error bars denote standard deviations
Suppression of type 1 IFN production by N
As with other viruses, PRRSV has been known to code for proteins that antagonize the IFN response by independently inhibiting its related transcription factors. To date, several NSPs of PRRSV have been identified as being responsible for the poor induction of type 1 IFN by PRRSV. The present study was further expanded to the PRRSV structural proteins in order to investigate their role in the modulation of IFN in the natural target cells. To address this issue, sublines of PAM cells were first established to stably express each recombinant structural protein of PRRSV under the control of a retroviral LTR promoter. Cell clones expressing the corresponding viral proteins were initially collected and subjected to RT-PCR and western blotting to verify expression of individual viral genes at the mRNA and protein level, respectively (Fig. 2A). Each of the PAM cell clones constitutively expressing the highest levels of the respective structural protein was chosen for subsequent studies. In addition, a stable PAM cell line expressing the PRRSV NSP1, which alone strongly suppresses type 1 IFN induction, was generated to be used as a positive control.
Fig. 2.
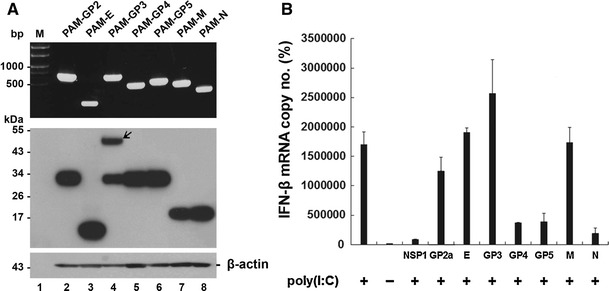
Involvement of specific PRRSV structural proteins in inhibition of IFN-β induction. (A) Established stable PAM cell lines were grown independently for 48 h, and total RNA was extracted from cells. Each viral gene was amplified by RT-PCR and visualized on a 0.8% agarose gel (top panel). Cell lysates were prepared from cells and subjected to western blot using an anti-His tag antibody to determine the expression level of each PRRSV protein in stable cell lines (middle panel). The blot was also reacted with anti-β-actin antibody to confirm equal protein loading (bottom panel). Lane 1 (M), molecular weight marker; lane 2, PAM-GP2; lane 3, PAM-E, lane 4, PAM-GP3, lane 5, PAM-GP4; lane 6, PAM-GP5; lane 7, PAM-M; lane 8, PAM-N. (B) PAM cells stably expressing each structural protein of PRRSV were stimulated with poly (I:C) as described in “Materials and methods”. Total RNA isolated from cells was subjected to real-time RT-PCR, and the porcine IFN-β mRNA levels in each stable cell line were expressed as copy numbers relative to the unstimulated control. These data are representative of the mean values from three independent experiments, and error bars represent standard deviations
Each of these cell lines was treated by transfection with poly(I:C), followed by real-time RT-PCR to quantitatively measure IFN-β mRNA. As expected, NSP1 was found to dramatically down-regulate poly(I:C)-induced IFN-β mRNA production in continuous PAM cells (Fig. 2B). Furthermore, IFN-β mRNA induction by poly(I:C) was suppressed to various degrees by GP4, GP5, and N in PAM-pCD163 cells (Fig. 2B). Of these structural proteins, the N protein was found to have the strongest inhibitory effect on the IFN-β response. To confirm this observation, we used three different PAM cell lines showing low-, moderate-, or high-level expression of N for the same assay. As shown in Fig. 3A, poly(I:C)-stimulated IFN induction was gradually inhibited in proportion to increasing expression levels of N (bottom panel).
Fig. 3.
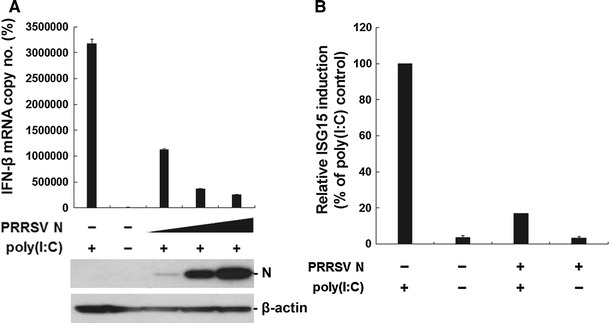
Inhibitory effects on the induction of IFN-β and ISG15 by N. (A) Three different stable PAM cell lines with low-, moderate-, or high-level N expression were stimulated with poly(I:C) for 6 h and used for real-time RT-PCR for the determination of IFN-β mRNA copy number as described earlier. The bottom panels indicate increasing levels of N expression in each stable cell line, and β-actin was used as a protein loading control. (B) Total RNA was isolated from poly(I:C)-treated PAM cells in the presence or absence of N and analyzed for the quantitative detection of ISG15 mRNA by real-time RT-PCR. The level of ISG15 mRNA in the absence of N upon poly(I:C) stimulation was set at 100%, and that of the N protein was normalized accordingly. These values are representative of the average of three independent experiments, and error bars denote standard deviations
Upon virus infection, the biological activities of type 1 IFNs are initiated by binding to their receptors, and they up-regulate expression of many ISGs, which, in combination, specify the antiviral state by sensitizing cells to limit viral replication [37]. Of all the genes regulated by the IFN response, ISG15 is one of the most inducible genes [14]. Therefore, to further verify the IFN-antagonistic activity of the N protein, it was tested whether N is capable of modulating ISG15 expression induced by dsRNA by quantitative RT-PCR. While ISG15 was efficiently induced in response to poly(I:C), the PRRSV N protein significantly impaired poly(I:C)-mediated ISG15 mRNA production in PAM-pCD163 cells (Fig. 3B). Together, our results demonstrate that N is a viral component responsible for IFN down-regulation by PRRSV.
Inhibition of IRF3 phosphorylation and nuclear translocation of N
IRF3 is one of key players of the signaling cascade leading to type 1 IFN induction [18]. This transcription factor is ubiquitously present in the cytoplasm but, upon receiving appropriate extra stimuli, including virus infection, its C-terminus is phosphorylated by an upstream kinase. This event causes a conformational change leading to dimerization and the exposure of a nuclear-localization signal (NLS). Once IRF3 is translocated into the nucleus, it assembles on the IFN-β promoter with other transcription factors, leading to the recruitment of transcriptional coactivators such as CBP/p300, and ultimately up-regulation of IFN mRNA transcription [37]. Various viruses have evolved to disarm IRF3 activation to escape the host IFN defense. Furthermore, PRRSV NSPs are known to possess the ability to interfere with the IRF3 pathway for active IFN antagonism. Thus, essential steps of the IFR3 activation process, such as phosphorylation and nuclear translocation were studied to define the mechanism by which N inhibits type 1 IFN induction. We first determined whether IRF phosphorylation can be impaired by N. As a positive control, poly(I:C) stimulation induced the phosphorylation of IRF3 in PAM cells (Fig. 4A, lane 1). In contrast, dsRNA-mediated IRF3 phosphorylation was undetectable in N-expressing PAM cells (lane 3).
Fig. 4.
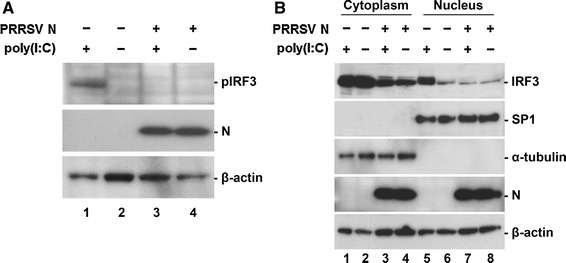
Inhibition of dsRNA-mediated IRF3 phosphorylation and nuclear translocation by N. (A) Phosphorylation of IRF3 in N-gene-expressing cells. PAM cells in the presence or absence of N were induced by poly(I:C). At 6 h post-induction, cell lysates were collected, resolved by SDS-PAGE, transferred to a nitrocellulose membrane, and immunoblotted using phosphor-IRF3 (Ser396) antibody (top panel) and His-tag antibody (middle panel). (B) Nuclear and cytoplasmic fractionation of PAM cells expressing N. Each nuclear and cytosolic fraction was prepared from PAM cells under the indicated conditions and subjected to western blot analysis with an antibody specific for IRF3 (top panel), SP1 as a nuclear protein marker (second panel), α-tubulin as a cytosolic protein marker (third panel), or the PRRSV N protein (fourth panel). All blots were also reacted with β-actin antibody to verify equal protein loading (bottom panel)
Next, the nuclear translocation of IRF3 in PAM cells stably expressing N was investigated by cell fractionation assay. As expected, poly(I:C) treatment was able to cause translocation of IRF3 to the nucleus (Fig. 4B, top panel, lane 5). However, in the presence of N, the level of IRF3 in the nucleus was markedly reduced to the basal level (lane 7). This observation was further confirmed by visualization of subcellular localization of IRF3. As shown in Fig. 5, IRF3 is normally located in the cytoplasm with nuclear diffusion in PAM cell lines without dsRNA stimulation (top, second, fourth, and sixth panels), whereas, in poly(I:C)-treated PAM-pCD163 and E-expressing PAM cells, the majority of IRF3 was translocated to the nucleus (top, first and third panels). This nuclear translocation of IRF3 induced by poly(I:C) was blocked in the presence of N (top, fifth panel), exhibiting a staining pattern similar to that of the unstimulated PAM cell lines (top, second, fourth, and sixth panels). These data indicate that the PRRSV N protein modulates IRF3 activation by inhibiting its phosphorylation and nuclear translocation.
Fig. 5.
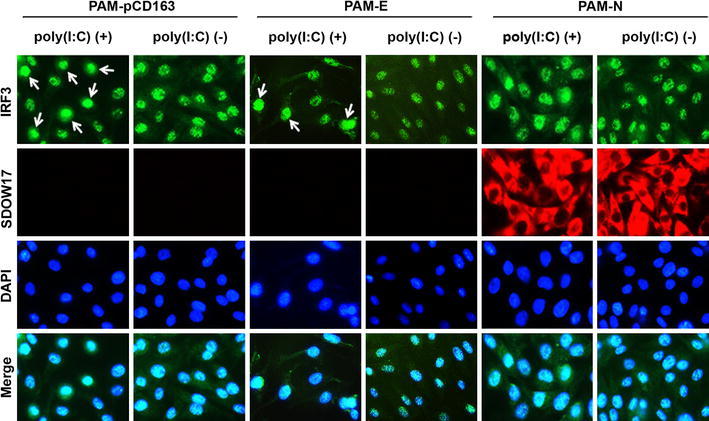
Subcellular localization of IRF3 in the presence of N. PAM-pCD163 and PAM cells stably expressing N or E were grown for two days and stimulated with poly (I:C) for 6 h. Cells were fixed with 4% paraformaldehyde and co-stained with anti-IRF3 and PRRSV N or His-tag antibodies, followed by incubation with a mixture of Alexa Fluor 488 (green)-conjugated goat anti-rabbit and 594 (red)-conjugated goat anti-mouse secondary antibodies (top and second panels). The cells were then counterstained with DAPI (third panels) and examined using a fluorescent microscope at 400× magnification. Arrows indicate nuclear localization of IRF3 upon poly(I:C) stimulation (color figure online)
Discussion
Mammalian hosts have evolved a broad range of cellular sensors to ensure effective protection from viral infection. Although host cells produce multiple cytokines and chemokines for antiviral defense, type 1 IFNs are the principal secreted cytokines that elicit distinct antiviral effects. In turn, viruses have adopted a number of strategies to evade this key system of host immune responses. Many viruses encode components in their genome that subvert the IFN response, and these viral IFN antagonists specifically inhibit pathways involved in either IFN induction or signaling [13, 22]. PRRSV circumvents the innate immune response by suppressing type 1 IFN induction. Recent studies have revealed that this property is mediated independently by several NSPs that individually target signaling pathways to activate various transcription factors responsible for type 1 IFN production [3, 6, 20, 27, 43]. However, the structural proteins with innate immune evasion features encoded by PRRSV remain undetermined, and the molecular mechanisms underlying the IFN suppression are unknown.
In the present study, immortalized PAM cell lines constitutively expressing each structural protein of PRRSV were first established and used as tools for studying dsRNA-induced IFN signaling pathways. The advantages of using this continuous PAM system are that, apart from currently known PRRSV-permissive cells used in other studies, it could facilitate the unlimited supply of PAM and mimic the virus-host interactions in the natural host. GP4, GP5, and N proteins stably expressed in PAM cells are able to significantly suppress upregulation of IFN-β mRNA induced by dsRNA. To investigate the mechanism of impairment of the IFN response by the PRRSV structural protein, we focused on N, which had the strongest inhibitory effect. The N protein was also found to inhibit ISG15 induction by poly(I:C). Although all factors of the IFN system are virtually targeted by a variety of viral IFN antagonistic proteins, IRF3 is one of the ideal components of viruses at the level of the activation process for modulating the IFN response. After dsRNA stimulation in N-expressing PAM cells, our results show that N targets the IRF3 activation pathway, leading to modification of its phosphorylation and subsequent nuclear translocation. Thus, the PRRSV N protein seems to prevent the activation of IRF3, which, in turn, blocks the full assembly of an enhanceosome complex on the IFN promoter, leading to inhibition of IFN mRNA transcription.
Most of the known IFN antagonistic proteins are nonstructural proteins of viruses that limit IFN production or specifically inhibit IFN signaling by infected cells by several factors of the IFN pathway [37]. There are only a few reports of viral structural proteins that function as IFN antagonists. These include the core protein of hepatitis C virus (HCV) and the N protein of severe acute respiratory syndrome coronavirus (SARS-CoV) [16, 21]. However, these viral structural proteins were found to inhibit the IFN response by blocking the NF-κB pathway, the other target element of the IFN system. To our knowledge, this is the first instance of a structural N protein directly targeting the IRF3 activation pathway to down-regulate type 1 IFN induction. The N protein of PRRSV is a multifunctional serine phosphoprotein [41, 49]. As the sole structural component of the viral capsid, N associates with itself, providing the pivotal basis for nucleocapsid assembly and virus infectivity [23, 50]. Since the entire life cycle of PRRSV occurs in the cytoplasm of infected cells, the N protein is distributed predominantly in the cytoplasmic and perinuclear regions. However, the PRRSV N protein is specifically observed in the nucleus and nucleolus of infected cells, suggesting a non-structural function(s) in these cellular compartments. Although N protein nuclear localization is dispensable for virus cytopathology and replication, it appears to play an important role in viral attenuation and pathogenesis [24]. Thus, it has been postulated that nuclear translocation of N may have effects on the nuclear function of the host cell in order to favor virus replication and modulate host antiviral responses. The present study supports this hypothesis, providing strong evidence that the N protein inhibits the host innate immune response independently of other nonstructural elements of PRRSV. It would be of further interest to evaluate whether nuclear localization of the N protein is essential for its ability to modulate the IFN response by inhibiting IRF3 activation, and this issue is currently under investigation.
Combining our findings with those from other reports, it appears that the pathogenesis of PRRSV is very complex, with multiple viral proteins contributing to evasion strategies to escape the host innate immune response by targeting different steps in IFN induction. Once the PRRSV genome is released after the uncoating process in virus-infected cells, individual NSPs are initially produced, and among these, five NSPs, including NSP1α, NSP1β, NSP2, NSP4, and NSP11, independently exert their own functions in counteracting the host type 1 response by various mechanisms. Later, while the newly translated N proteins are accumulating in the cytoplasm and function as the principal structural component for nucleocapsid assembly, other N protein populations being shuttled between the cytoplasm and the nucleus appear to play non-structural roles in participating in antagonizing type 1 IFN induction. Consequently, the poor type 1 IFN production mediated by non-structural and structural elements of PRRSV causes the inadequate adaptive immune response and likely leads to viral persistence and continuous virus shedding in the natural host.
Acknowledgments
This research was supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF), funded by the Ministry of Education, Science and Technology (2010-0002318).
References
- 1.Albina E, Madec F, Cariolet R, Torrison J. Immune response and persistence of the porcine reproductive and respiratory syndrome virus in infected pigs and farm units. Vet Rec. 1994;134:567–573. doi: 10.1136/vr.134.22.567. [DOI] [PubMed] [Google Scholar]
- 2.Albina E. Epidemiology of porcine reproductive and respiratory syndrome (PRRS): an overview. Vet Microbiol. 1997;55:309–316. doi: 10.1016/S0378-1135(96)01322-3. [DOI] [PubMed] [Google Scholar]
- 3.Beura LK, Sarkar SN, Kwon B, Subramaniam S, Jones C, Pattnaik AK, Osorio FA. Porcine reproductive and respiratory syndrome virus nonstructural protein 1beta modulates host innate immune response by antagonizing IRF3 activation. J Virol. 2010;84:1574–1584. doi: 10.1128/JVI.01326-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Buddaert W, Van Reeth K, Pensaert M. In vivo and in vitro interferon (IFN) studies with the porcine reproductive and respiratory syndrome virus (PRRSV) Adv Exp Med Biol. 1998;440:461–467. doi: 10.1007/978-1-4615-5331-1_59. [DOI] [PubMed] [Google Scholar]
- 5.Cavanagh D. Nidovirales: a new order comprising Coronaviridae and Arteriviridae. Arch Virol. 1997;142:629–633. [PubMed] [Google Scholar]
- 6.Chen Z, Lawson S, Sun Z, Zhou X, Guan X, Christopher-Hennings J, Knudsen D, Nelson E, Fang Y. Identification of two auto-cleavage products of nonstructural protein (nsp1) in porcine reproductive and respiratory syndrome virus infected cells: nsp1 functions as interferon antagonist. Virology. 2009;398:87–97. doi: 10.1016/j.virol.2009.11.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Choi YJ, Yun SI, Kang SY, Lee YM. Identification of 5′ and 3′ cis-acting elements of the porcine reproductive and respiratory syndrome virus: acquisition of novel 5′ AU-rich sequences restored replication of a 5′-proximal 7-nucleotide deletion mutant. J Virol. 2006;80:723–736. doi: 10.1128/JVI.80.2.723-736.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Christopher-Hennings J, Nelson EA, Hines RJ, Nelson JK, Swenson SL, Zimmerman JJ, Chase CL, Yaeger MJ, Benfield DA. Persistence of porcine reproductive and respiratory syndrome virus in serum and semen of adult boars. J Vet Diagn Invest. 1995;7:456–464. doi: 10.1177/104063879500700406. [DOI] [PubMed] [Google Scholar]
- 9.Duan X, Nauwynck HJ, Pensaert MB. Effects of origin and state of differentiation and activation of monocytes/macrophages on their susceptibility to porcine reproductive and respiratory syndrome virus (PRRSV) Arch Virol. 1997;142:2483–2497. doi: 10.1007/s007050050256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Duan X, Nauwynck HJ, Pensaert MB. Virus quantification and identification of cellular targets in the lungs and lymphoid tissues of pigs at different time intervals after inoculation with porcine reproductive and respiratory syndrome virus (PRRSV) Vet Microbiol. 1997;156:9–19. doi: 10.1016/S0378-1135(96)01347-8. [DOI] [PubMed] [Google Scholar]
- 11.Feng W, Laster SM, Tompkins M, Brown T, Xu JS, Altier C, Gomez W, Benfield D, McCaw MB. In utero infection by porcine reproductive and respiratory syndrome virus is sufficient to increase susceptibility of piglets to challenge by Streptococcus suis type II. J Virol. 2001;75:4889–4895. doi: 10.1128/JVI.75.10.4889-4895.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Firth AE, Zevenhoven-Dobbe JC, Wills NM, Go YY, Balasuriya U, Atkins JF, Snijder EJ, Posthuma CC. Discovery of a small arterivirus gene that overlaps the GP5 coding sequence and is important for virus production. J Gen Virol. 2011;92:1097–1106. doi: 10.1099/vir.0.029264-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.García-Sastre A, Biron CA. Type 1 interferons and the virus-host relationship: a lesson in détente. Science. 2006;312:879–889. doi: 10.1126/science.1125676. [DOI] [PubMed] [Google Scholar]
- 14.Harty RN, Pitha PM, Okumura A. Antiviral activity of innate immune protein ISG15. J Innate Immun. 2009;1:397–404. doi: 10.1159/000226245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Johnson CR, Griggs TF, Gnanandarajah JS, Murtaugh MP. Novel structural protein in porcine reproductive and respiratory syndrome virus encoded in an alternative open reading frame 5 present in all arteriviruses. J Gen Virol. 2011;92:1107–1116. doi: 10.1099/vir.0.030213-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Joo M, Hahn YS, Kwon M, Sadikot RT, Blackwell TS, Christman JW. Hepatitis C virus core protein suppresses NF-kappaB activation and cyclooxygenase-2 expression by direct interaction with IkappaB kinase beta. J Virol. 2005;79:7648–7657. doi: 10.1128/JVI.79.12.7648-7657.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kang SY, Yun SI, Park HS, Park CK, Choi HS, Lee YM. Molecular characterization of PL97–1, the first Korean isolate of the porcine reproductive and respiratory syndrome virus. Virus Res. 2004;104:165–179. doi: 10.1016/j.virusres.2004.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kawai T, Akira S. Innate immune recognition of viral infection. Nat Immunol. 2006;7:131–137. doi: 10.1038/ni1303. [DOI] [PubMed] [Google Scholar]
- 19.Keffaber KK. Reproductive failure of unknown etiology. Am Assoc Swine Pract Newslett. 1989;1:1–9. [Google Scholar]
- 20.Kim O, Sun Y, Lai FW, Song C, Yoo D. Modulation of type I interferon induction by porcine reproductive and respiratory syndrome virus and degradation of CREB-binding protein by non-structural protein 1 in MARC-145 and HeLa cells. Virology. 2010;402:315–326. doi: 10.1016/j.virol.2010.03.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kopecky-Bromberg SA, Martínez-Sobrido L, Frieman M, Baric RA, Palese P. Severe acute respiratory syndrome coronavirus open reading frame (ORF) 3b, ORF 6, and nucleocapsid proteins function as interferon antagonists. J Virol. 2007;81:548–557. doi: 10.1128/JVI.01782-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Koyama S, Ishii KJ, Coban C, Akira S. Innate immune response to viral infection. Cytokine. 2008;43:336–341. doi: 10.1016/j.cyto.2008.07.009. [DOI] [PubMed] [Google Scholar]
- 23.Lee C, Calvert JG, Welch SK, Yoo D. A DNA-launched reverse genetics system for porcine reproductive and respiratory syndrome virus reveals that homodimerization of the nucleocapsid protein is essential for virus infectivity. Virology. 2005;331:47–62. doi: 10.1016/j.virol.2004.10.026. [DOI] [PubMed] [Google Scholar]
- 24.Lee C, Hodgins D, Calvert JG, Welch SK, Jolie R, Yoo D. Mutations within the nuclear localization signal of the porcine reproductive and respiratory syndrome virus nucleocapsid protein attenuate virus replication. Virology. 2006;346:238–250. doi: 10.1016/j.virol.2005.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lee SM, Schommer SK, Kleiboeker SB. Porcine reproductive and respiratory syndrome virus field isolates differ in in vitro interferon phenotypes. Vet Immunol Immunopathol. 2004;102:217–231. doi: 10.1016/j.vetimm.2004.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lee YJ, Park CK, Nam E, Kim SH, Lee OS, Lee DS, Lee C. Generation of a porcine alveolar macrophage cell line for the growth of porcine reproductive and respiratory syndrome virus. J Virol Methods. 2010;163:410–415. doi: 10.1016/j.jviromet.2009.11.003. [DOI] [PubMed] [Google Scholar]
- 27.Li H, Zheng Z, Zhou P, Zhang B, Shi Z, Hu Q, Wang H. The cysteine protease domain of porcine reproductive and respiratory syndrome virus non-structural protein 2 antagonizes interferon regulatory factor 3 activation. J Gen Virol. 2010;91:2947–2958. doi: 10.1099/vir.0.025205-0. [DOI] [PubMed] [Google Scholar]
- 28.Lopez OJ, Osorio FA. Role of neutralizing antibodies in PRRSV protective immunity. Vet Immunol Immunopathol. 2004;102:155–163. doi: 10.1016/j.vetimm.2004.09.005. [DOI] [PubMed] [Google Scholar]
- 29.Luo R, Xiao S, Jiang Y, Jin H, Wang D, Liu M, Chen H, Fang L. Porcine reproductive and respiratory syndrome virus (PRRSV) suppresses interferon-beta production by interfering with the RIG-I signaling pathway. Mol Immunol. 2008;45:2839–2846. doi: 10.1016/j.molimm.2008.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Meier WA, Galeota J, Osorio FA, Husmann RJ, Schnitzlein WM, Zuckermann FA. Gradual development of the interferon-gamma response of swine to porcine reproductive and respiratory syndrome virus infection or vaccination. Virology. 2003;309:18–31. doi: 10.1016/S0042-6822(03)00009-6. [DOI] [PubMed] [Google Scholar]
- 31.Meulenberg JJ, Hulst MM, de Meijer EJ, Moonen PJM, den Besten A, De Kluyver EP, Wensvoort G, Moormann RJM. Lelystad virus, the causative agent of porcine epidemic abortion and respiratory syndrome (PEARS), is related to LDV and EAV. Virology. 1993;192:62–72. doi: 10.1006/viro.1993.1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Meulenberg JJ, Petersen-den Besten A, De Kluyver EP, Moormann RJ, Schaaper WM, Wensvoort G. Characterization of proteins encoded by ORFs 2 to 7 of Lelystad virus. Virology. 1995;206:155–163. doi: 10.1016/S0042-6822(95)80030-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Miller LC, Laegreid WW, Bono JL, Chitko-McKown CG, Fox JM. Interferon type I response in porcine reproductive and respiratory syndrome virus-infected MARC-145 cells. Arch Virol. 2004;149:2453–2463. doi: 10.1007/s00705-004-0377-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Murtaugh MP, Xiao Z, Zuckermann F. Immunological responses of swine to porcine reproductive and respiratory syndrome virus infection. Viral Immunol. 2002;15:533–547. doi: 10.1089/088282402320914485. [DOI] [PubMed] [Google Scholar]
- 35.Nam E, Lee C. Contribution of the porcine aminopeptidase N (CD13) receptor density to porcine epidemic diarrhea virus infection. Vet Microbiol. 2010;144:41–50. doi: 10.1016/j.vetmic.2009.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nuemann EJ, Kliebenstein JB, Johnson CD, Mabry JW, Bush EJ, Seitzinger AH, Green AL, Zimmerman JJ. Assessment of the economic impact of porcine reproductive and respiratory syndrome on swine production in the United States. J Am Vet Med Assoc. 2005;227:385–392. doi: 10.2460/javma.2005.227.385. [DOI] [PubMed] [Google Scholar]
- 37.Randall RE, Goodbourn S. Interferons and viruses: an interplay between induction, signalling, antiviral responses and virus countermeasures. J Gen Virol. 2008;89:1–47. doi: 10.1099/vir.0.83391-0. [DOI] [PubMed] [Google Scholar]
- 38.Royaee AR, Husmann RJ, Dawson HD, Calzada-Nova G, Schnitzlein WM, Zuckermann FA, Lunney JK. Deciphering the involvement of innate immune factors in the development of the host response to PRRSV vaccination. Vet Immunol Immunopathol. 2004;102:199–216. doi: 10.1016/j.vetimm.2004.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sadler AJ, Williams BR. Interferon-inducible antiviral effectors. Nat Rev Immunol. 2008;8:559–568. doi: 10.1038/nri2314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sambrook J, Russell DW. Molecular cloning: a laboratory manual, 3rd edn. New York: Cold Spring Harbor Laboratory; 2001. [Google Scholar]
- 41.Snijder EJ, Meulenberg JJ. The molecular biology of arteriviruses. J Gen Virol. 1998;79:961–979. doi: 10.1099/0022-1317-79-5-961. [DOI] [PubMed] [Google Scholar]
- 42.Stordeur P, Poulin LF, Craciun L, Zhou L, Schandené L, de Lavareille A, Goriely S, Goldman M. Cytokine mRNA quantification by real-time PCR. J Immunol Methods. 2002;259:55–64. doi: 10.1016/S0022-1759(01)00489-6. [DOI] [PubMed] [Google Scholar]
- 43.Sun Z, Chen Z, Lawson SR, Fang Y. The cysteine protease domain of porcine reproductive and respiratory syndrome virus nonstructural protein 2 possesses deubiquitinating and interferon antagonism functions. J Virol. 2010;84:7832–7846. doi: 10.1128/JVI.00217-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.van Aken D, Snijder EJ, Gorbalenya AE. Mutagenesis analysis of the nsp4 main proteinase reveals determinants of arterivirus replicase polyprotein autoprocessing. J Virol. 2006;80:3428–3437. doi: 10.1128/JVI.80.7.3428-3437.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.van Reeth K, Labarque G, Nauwynck H, Pensaert M. Differential production of proinflammatory cytokines in the pig lung during different respiratory virus infections: correlations with pathogenicity. Res Vet Sci. 1999;67:47–52. doi: 10.1053/rvsc.1998.0277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wensvoort G, Tepstra C, Pol JMA, ter Laak EA, Bloemraad M, de Kluyver EP, Kragten C, van Buiten L, den Besten A, Wagenaar F, Broekhuijsen JM, Moonen PLJM, Zestra T, de Boer EA, Tibben HJ, de Jong MF, van Veld P, Groenland GJR, van Gennep JA, Voets MT, Verheijden JHM, Braamskamp J. Mystery swine disease in the Netherlands: the isolation of Lelystad virus. Vet Q. 1991;13:121–130. doi: 10.1080/01652176.1991.9694296. [DOI] [PubMed] [Google Scholar]
- 47.Wensvoort G. Lelystad virus and the porcine epidemic abortion and respiratory syndrome. Vet Res. 1993;24:117–124. [PubMed] [Google Scholar]
- 48.Wills RW, Doster AR, Galeota JA, Sur JH, Osorio FA. Duration of infection and proportion of pigs persistently infected with porcine reproductive and respiratory syndrome virus. J Clin Microbiol. 2003;41:58–62. doi: 10.1128/JCM.41.1.58-62.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wootton SK, Rowland RR, Yoo D. Phosphorylation of the porcine reproductive and respiratory syndrome virus nucleocapsid protein. J Virol. 2002;76:10569–10576. doi: 10.1128/JVI.76.20.10569-10576.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wootton SK, Yoo D. Homo-oligomerization of the porcine reproductive and respiratory syndrome virus nucleocapsid protein and the role of disulfide linkages. J Virol. 2003;77:4546–4557. doi: 10.1128/JVI.77.8.4546-4557.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wu WH, Fang Y, Farwell R, Steffen-Bien M, Rowland RR, Christopher-Hennings J, Nelson EA. A 10-kDa structural protein of porcine reproductive and respiratory syndrome virus encoded by ORF2b. Virology. 2001;287:183–191. doi: 10.1006/viro.2001.1034. [DOI] [PubMed] [Google Scholar]