

Abstract
Since 2010, porcine epidemic diarrhea has re-emerged with devastating impact on the swine-raising industry in central China. To investigate the epidemic characteristics of PEDV, the complete ORF3 genes of 14 PEDV field strains from central China during 2012 to 2013 were cloned, sequenced and compared with reference strains. Phylogenetic analysis based on the complete ORF3 gene showed that the PEDVs in central China and the reference strains could be divided into three groups: G1, G2, and G3. The 14 PEDV isolates were classified as G1 and showed a close relationship to some Chinese strains isolated previously in central China and differed genetically from recent isolates from southern China, Korean strains (SM98 and DB1865, 2012), the Chinese LZC strain (2007), and the vaccine strain (CV777) being used in China. Our findings suggested that the PEDVs circulating between 2012 and 2013 in central China might have evolved from earlier strains in the local region. To determine the reason for recent vaccination failures, we also studied variations in antigenicity of field strains by analyzing the three neutralizing epitope regions in the S gene. The results showed that the neutralizing epitopes at aa 245-252 were highly conserved, but most of the amino acid changes occurred in the epitope regions aa 7-146 and 271-278. We speculate that the amino acid mutations in the neutralizing epitope regions may be associated with changes in the antigenicity of PEDV and consequently result in vaccination failure. Together, these findings may be useful for understanding the epidemiology of PEDV and may be relevant for designing of new and more efficacious vaccines.
Keywords: Porcine Epidemic Diarrhea Virus, ORF3 Gene, Neutralize Epitope, Korean Strain, Porcine Epidemic Diarrhea Virus Strain
Introduction
Porcine epidemic diarrhea (PED) is an acute, highly contagious viral enteric disease of swine caused by PED virus (PEDV), a member of the genus Alphacoronavirus [1]. PED was first reported in Belgium and the United Kingdom in 1971 [2]. Since then, the disease has been recognized in many European countries, such as Germany, France and Switzerland, and more recently in Korea and Thailand [3–5]. In China, PEDV was first confirmed in 1984 [6], and since 2010, it has caused enteric disease with a devastating impact on the swine-raising industry. This disease is characterized by high morbidity and mortality among preweaning piglets, causing serious economic losses to the swine industry in China [7].
PEDV is an enveloped virus possessing an approximately 28-kb genome. The PEDV genome contains at least seven open reading frames (ORFs), encoding four structural proteins (spike [S], envelope [E], membrane [M], and nucleocapsid [N]) and three non-structural proteins (replicases 1a and 1b and ORF3) [8].
The ORF3 gene is an accessory gene of PEDV and has been demonstrated to be associated with the virulence of PEDV [9]. A region (nt 245 to 295) that is crucial for PEDV pathogenicity, is deleted in all live vaccine strains, and this could be a marker of adaptation to cell culture and attenuation of the virus. Thus, the ORF3 gene could be used as a valuable tool for differentiation of wild- and attenuated-type PEDVs and molecular epidemiology studies of PEDV [10].
The spike protein (S) is the major structural proteins of PEDV and consists of 1383 amino acids. Similar to those of other coronaviruses, the S protein of PEDV consists of three domains, including a large outer domain, a transmembrane domain and a short cytoplasm domain at the carboxyl end. It has been demonstrated to have four neutralizing epitopes (aa 499–638, 748–755, 764–771, and 1,368–1,374) on the surface of the S protein [11–13], which are important for receptor binding and virus entry, the induction of neutralizing antibodies, and host-cell fusion [11, 13, 14]. Therefore, the S protein is also essential for understanding the epidemiological status of PEDV in the field, diversity of PEDV isolates, and the association between genetic mutations and viral antigenicity [5, 15].
Recent studies have shown that the PED caused by PEDV is becoming increasingly serious in China, and current vaccines are ineffective on most swine-raising farms. Molecular epidemiology and virus isolation of PEDV have been undertaken in some regions of China [7, 16–19]. These studies are essential for identifying the epidemic characteristics of PEDV and have the potential to provide information about its pathogenesis. Nevertheless, little is known about the antigenic variability of PEDV prevailing currently in the field. In this study, we aimed to investigate the molecular epidemiology and antigenic variability of PEDV field strains based on analysis of ORF3 and a portion of the S gene encoding three neutralizing epitopes (aa 499–638, 748–755, and 764–771) of the S protein. This study provides new information about the prevalence of PEDV strains currently circulating in China.
Materials and methods
Intestinal and fecal samples were collected from piglets suffering from severe diarrhea from September 2012 to June 2013 in central China. The scraped-off mucosa and the contents of the small intestine were pooled, diluted 1:5 in phosphate-buffered saline (PBS; 0.15 M NaCl, 0.01 M phosphate buffer [pH 7.4]), homogenized by ultrasonication, and clarified by centrifugation for 15 min at 4,800×g after freezing and thawing three times. The supernatants were collected for RT-PCR. PEDV RNA was extracted using TRIzol Reagent (Invitrogen), dissolved in 30 μl of 1 % diethylpyrocarbonate-treated water, and stored at −70 °C.
The complete ORF3 gene was amplified using previously published primers [9], and the size of expected product was 833 bp. The primers for amplifying the partial S gene were designed based on the genome of PEDV CV777 (GenBank no. AF353511), and the size of the final fragments, containing three neutralizing epitopes of PEDV, was 894 bp. Two sets of primers were synthesized by Sangon Biotech, China. The RT-PCR primers are listed in Table 1.
Table 1.
Primers used in this study
| No. | Sense | Sequence | Target gene (fragment size, bp) |
|---|---|---|---|
| 1 | Forward | 5′-TCCTAGACTTCAACCTTACG-3′ | ORF3 gene (833) |
| 2 | Reverse | 5′-GGTGACAAgTGAAGCACAGA-3′ | |
| 3 | Forward | 5′-TTCTGAGTCATGAACAGCCAAT-3′ | Partial S gene (894) |
| 4 | Reverse | 5′-CATACTAAAGTTGGTGGGAAT-3′ |
Synthesis of the first-strand cDNA for the N gene was carried out by reverse transcription using reverse transcription reagents from Promega. The viral RNA (50 μl) was mixed with 2.5 μl of 10 pM antisense primer, incubated at 65 °C for 5 min, and placed on ice for 2 min. After that, 4 μl of 5× RT buffer, 4 μl of 2.5 mM dNTP mixture, 1 μl of RNase inhibitor (40 U/μl), 1 μl of reverse transcriptase M-MLV (200 U/μl), and 2.5 μl H2O were added with gentle mixing. The reaction mixture was incubated for 1 h at 42 °C, and the reaction was terminated by heating for 10 min at 65 °C. The cDNA was either stored at −20 °C or amplified immediately.
For PCR, 2 μl of cDNA was mixed with a reaction mixture containing 2.5 μl of 10× Taq DNA polymerase buffer (Promega, Madison, WI), 3 mM MgCl2, 2.0 μl of dNTPs (2.5 mM), 0.5 μl of each specific primer (10 pmol), 1 μl of Taq DNA polymerase (Promega, Madison, WI) and autoclaved, filtered (0.2 μm) distilled water in a total volume of 25 μl. Amplification was performed as follows: one cycle at 95 °C for 5 min, followed by 30 cycles at 95 °C for 1 min, 50 °C for 1 min, and 72 °C for 1 min, and a final extension at 72 °C for 10 min. The RT-PCR products were visualized by electrophoresis in a 1.5 % agarose gel containing ethidium bromide.
Purified RT-PCR products were identified by electrophoresis in a 1.5 % agarose gel and cloned into pMD® 19-T. The recombinant vector was identified by PCR and enzyme digestion. The positive clones were sent to Sangon Biotech, China, for sequencing. All sequencing reactions were performed in duplicate. The expected sizes of the PCR products were 833 and 894 bp. The former contained the complete ORF3 gene and its flanking sequences, while the latter contained the partial S gene. The nucleotide sequences of the PEDV isolates were deposited in the GenBank database, and the corresponding accession numbers are listed in Table 2.
Table 2.
PEDV strains used for sequence alignment and phylogenetic analysis
| Reference strain | Accession no. | Origin | Sample strain | Accession no. | Origin |
|---|---|---|---|---|---|
| ZJCZ4 | JX524137.1 (C) | China, 2011 | CH/YF-01 | KF476048 (O) | China, 2012 |
| LC | JX489155.1(C) | China, 2012 | KF484733 (pS) | ||
| CHGD-01 | JX261936.1 (C) | China, 2012 | CH/XY-02 | KF476049 (O) | China, 2012 |
| AJ1102 | JX188454.1 (C) | China, 2012 | KF484734 (pS) | ||
| CH/FJZZ-9/2012 | KC140102.1 (C) | China, 2013 | CH/QX-02 | KF476050 (O) | China, 2012 |
| GD-A | JX112709.1 (C) | China, 2012 | KF484735 (pS) | ||
| DR13 | JQ023161.1 (C) | Korea, 2008 | CH/KF-01 | KF476051 (O) | China, 2012 |
| JS-HZ2012 | KC210147.1(C) | China, 2012 | KF484736 (pS) | ||
| GD-B | JX088695.1 (C) | China, 2012 | CH/XIP-01 | KF476052 (O) | China, 2013 |
| CH/FJND-3/2011 | JQ282909.1 (C) | China, 2011 | KF484737 (pS) | ||
| CH/S | JN547228.1 (C) | China, 1986 | CHLY-09 | KF476053(O) | China, 2013 |
| GD-1 | JX647847.1 (C) | China, 2011 | KF484738 (pS) | ||
| AH2012 | KC210145.1 (C) | China, 2012 | CH/FCH-01 | KF476054 (O) | China, 2013 |
| USA/Colorado/2013 | KF272920.1 (C) | USA, 2013 | KF484739 (pS) | ||
| CH/ZMDZY/11 | KC196276.1 (C) | China, 2011 | CH/LH | KF476055 (O) | China, 2013 |
| BJ-2011-1 | JN825712.1 (C) | China, 2011 | KF484740 (pS) | ||
| CV777 | AF353511.1 (C) | Belgium, 2001 | CH/FH | KF476056 (O) | China, 2013 |
| LZC | EF185992.1(C) | China, 2007 | KF484741 (pS) | ||
| CH/HLJHH-2/2011 | JQ305099.1 (O) | China, 2012 | CH/LSHAN | KF476057 (O) | China, 2013 |
| CH/GDGZ/2012 | KF384500.1 (O) | China, 2012 | KF484742 (pS) | ||
| DX | EU031893.1(O) | China, 2007 | CH/XIP-03 | KF476058 (O) | China, 2013 |
| 13-019349 | KF267450.1 (O) | USA, 2013 | KF484743 (pS) | ||
| SM98 | GU937797.1 (O) | Korea, 2012 | CH/JCH | KF476059 (O) | China, 2013 |
| Chinju99 | EU792474.1 (O) | Korea, 2009 | KF484744 (pS) | ||
| M1595 | HQ537436.1 (O) | Korea, 2012 | CH/ZHZ-04 | KF476060 (O) | China, 2013 |
| DB1865 | HQ537432.1 (O) | Korea, 2012 | KF484745 (pS) | ||
| Br1/87 | Z25483.1 (cS) | Britain,2001 | CH/HEB | KF476061 (O) | China, 2013 |
| SM98 | GU937797.1 (cS) | Korea,2012 | KF484746 (pS) | ||
| MK | AB548624.1 (cS) | Japan, 2013 | |||
| NK | AB548623.1 (cS) | Japan, 2013 | |||
| KH | AB548622.1 (cS) | Japan, 2013 | |||
| KU04RB08 | FJ196224.1 (pS) | Thailand, 2012 | |||
| VN122S8 | HQ883492.1 (pS) | Thailand, 2011 | |||
| TH/NP-65/12 | KC858259.1 (pS) | Thailand, 2013 | |||
| AD01 | KC879280.1 (cS) | Korea, 2013 | |||
| NJ02 | KC876279.1 (cS) | Korea, 2013 |
(C) following the accession number indicates a complete genome sequence of the PEDV reference strain; (O) indicates a complete ORF3 sequence; (pS) indicates a partial S gene sequence; and (cS) indicates a complete S gene sequence
The nucleotide sequences were assembled and proofread using ContigExpress software. Multiple sequence alignments were generated by the Clustal W method using the MegAlign 4.0 program in DNASTAR (DNASTAR Inc. USA) (version 7.0). Phylogenetic trees were constructed by the neighbor-joining method based on nucleotide sequences of ORF3 and the partial S gene, using Molecular Evolutionary Genetics Analysis (MEGA) software (version 5.2.1). To assess the relative support for each clade, bootstrap values were calculated from 1,000 replicate analyses. [20]. In addition, 18 reference strains with complete S gene sequences were chosen for the analysis of the region containing neutralizing epitopes (7-146). B-cell epitope prediction was performed using the BepiPred 1.0b Server (http://www.cbs.dtu.dk/services/BepiPred/) [21]. The reference strains used for phylogenetic analysis and the 14 samples strains are listed in Table 2.
Results
Fourteen complete ORF3 genes were amplified by RT-PCR, and the nucleotide sequences were 833 nt in length (GenBank accession nos. KF476048-KF476061), including an open reading frame encoding 225 amino acids. These isolates shared 96.7-100.0 % nucleotide and 94.0-100.0 % amino acid sequence identity with each other and had 96.9-99.8 % and 94.5-99.3 % similarity, respectively, to the reference strains. These analyses indicated that the ORF3 gene sequences determined in this study were relatively conserved. Phylogenetic analysis based on the nucleotide sequence of ORF3 indicated that all of the PEDV strains examined, including the 14 sample strains and the reference strains, could be divided into three groups (1-3). In general, all 14 samples from this study clustered in group G1, together with most strains from China, including CH/ZMDZY/11, BJ-2011-1, JS-HZ2012, GD-B, CH/FJZZ-9/2012, CH/FJND-3/2011, and CH/S, and also two American strains (USA/Colorado/2013 and 13-019349) and three Korean strains (DR13, M1595, and Chinju99). G2 included strains CH/HLJHH-2/2011, DB1865, DX, ZJCZ4, CH/GDGZ/2012, GD-A, GD-1, LC, CHGD-01, and AJ1102. G3 included SM98 (Korean strain), LZC, and CV777 (the vaccine strain). Furthermore, CH/LY-09, CH/FCH-01, CH/HEB, and CH/LH were clustered in the same subgroup with CH/ZMDZY/11, and CH/YF-01 and CH/KF-01 were located in the same subgroup with AH2012. Both CH/ZMDZY/11 and AH2012 were collected previously from Henan (2011) and Anhui (2012) provinces of central China. Notably, CH/XY-02, CH/LSHAN, CH/XIP-03, CH/XIP-01, and CH/QX-02 formed a unique subgroup in the phylogenetic tree constructed based on ORF3 nucleotide sequences (Fig. 1).
Fig. 1.
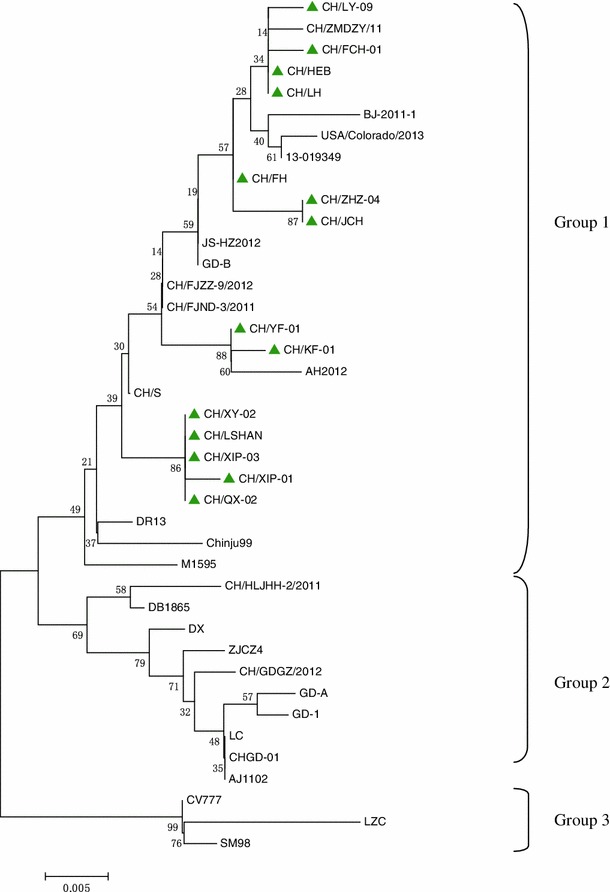
Phylogenetic analysis of the nucleotide sequences of ORF3 genes of PEDV isolates from this study and PEDV reference strains. The tree was constructed using the neighbor-joining method in MEGA 5.2.1 software. Numbers above branches indicate bootstrap values calculated from 1,000 bootstrap replicates; only values greater than 50 are shown. The scale bar represents nucleotide substitutions per site. “▲” indicates the 14 isolates from this study
Fourteen partial S genes amplified from the clinical samples (GenBank accession nos. KF484733-KF484746) had a size of 894 nt, encoding a 298-amino-acid polypeptide, corresponding to nt 1477–2370 of the complete S gene of the CV777 strain, containing three neutralizing epitopes (499–638, 748–755, 764–771), with 97.5-100.0 % nucleotide and 95.7-100.0 % deduced amino acid sequence similarity to each other, and 94.7-99.9 % nucleotide and 93.6-99.7 % amino acid sequence identity to the reference strains. A phylogenetic tree based on the partial S nucleotide sequence showed that the isolates could be clustered into three groups. All 14 isolates belonged to G1, together with most Chinese strains and six foreign strains, including two American strains (USA/Colorado/2013 and 13-019349), three strains from Thailand (VN12258, KU04RB08, and TH/NP-65/12) and one strain from Japan (MK). The 14 samples in G1 were clearly separated into two different subgroups: CH/FCH-01, CH/LH, CH/LY-09, CH/JCH, CH/ZHZ-04, and CH/HEB formed a unique subgroup and differed clearly from the other PEDV isolates, and the unique subgroup was adjacent to MK, a strain from Korea. In addition, G2 included two Korean strains (AD01 and NJ02). G3 included one European strains (Br1/7), four Korean strains (DR13, KH, NK, and SM98), two early domestic strains (LZC and CH/S), and the vaccine strain CV777). These strains differed genetically from the 14 sample strains because of their distant location in phylogenetic tree (Fig. 2).
Fig. 2.
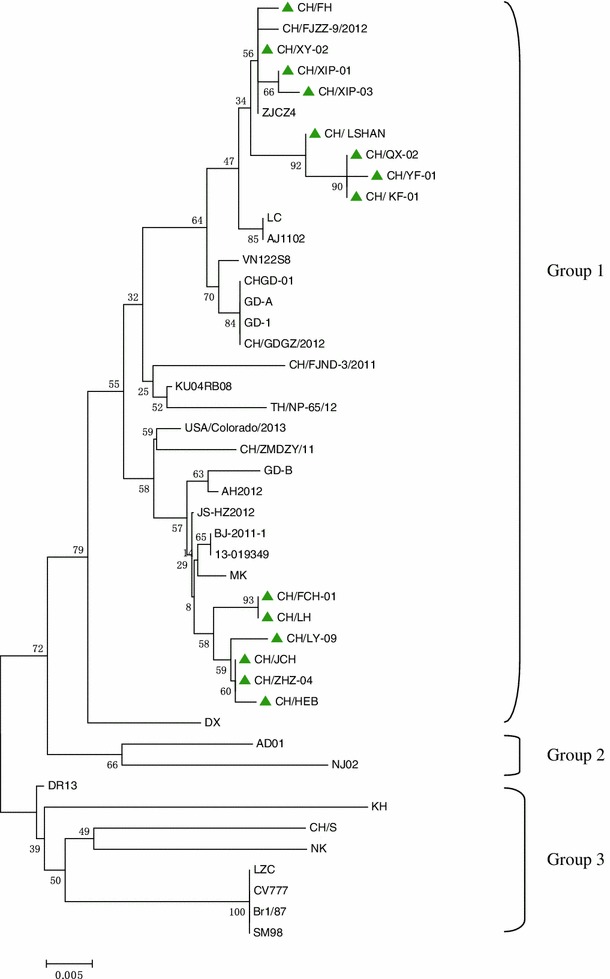
Phylogenetic analysis of the nucleotide sequences of partial S genes of PEDV isolates from this study and PEDV reference strains. The tree was constructed using the neighbor-joining method in MEGA 5.2.1 software. Numbers above branches indicate bootstrap values calculated from 1,000 bootstrap replicates; only values greater than 50 are shown. The scale bar represents nucleotide substitutions per site. “▲” indicates the 14 isolates from this study
In addition, the deduced amino acid sequences from the partial S gene analysis showed that there was no amino acid change in the epitope at aa 245-252 in any of the isolates. However, when compared with the reference strains, some variants were observed in the epitope regions at aa 7-146 and 271-278, including T → I at aa 11 (CH/LH), T → A at aa 11 (CH/LSHAN), S → A at aa 14 (CH/YF-01, CH/QX-02, CH/KF-01, CH/LSHAN), F → S at aa 20 (CH/FH), A → V (CH/YF-01, CH/QX-02, CH/KF-01, CH/LSHAN) and A → S (CH/LY-09, CH/FCH-01, CH/LH, CH/XIP-03, CH/ZHZ-04, CH/HEB) at aa 28, H → R at aa 32 (CH/YF-01, CH/QX-02, CH/KF-01, CH/LY-09), and K → N at aa 74 (CH/YF-01,CH/XY-02, CH/QX-02, CH/KF-01, CH/XIP-01, CH/FH, CH/LSHAN). More details are given in Table 3. Only one deletion was observed at aa 85 in the epitope region aa 7-146 of CH/XIP-03. In addition, B-cell epitope in the partial S protein (aa 7-146) was predicted using the BepiPred 1.0b Server. The results showed that there were three B-cell epitopes with higher scores between aa 7 and 146, located at positions 22-30, 32-40, and 64-75. In the current study, the amino acid mutations at aa 28, 32, and 74 were within the epitopes predicted above (aa 22-30, 32-40, 64-75, respectively; Fig. 3; Table 3), indicating that these changes might result in antigenicity differences.
Table 3.
Amino acid mutations in partial S proteins of 14 PEDV isolates
| Isolate | Amino acid position and change | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 11 | 14 | 20 | 28 | 32 | 74 | 77 | 85 | 147 | 159 | 230 | 274 | 285 | |
| CH/YF-01 | S → A | A → V | H → R | K → N | S → N | P → L | |||||||
| CH/XY-02 | K → N | ||||||||||||
| CH/QX-02 | S → A | A → V | H → R | K → N | S → N | P → L | |||||||
| CH/KF-01 | S → A | A → V | H → R | K → N | S → N | P → L | |||||||
| CH/XIP-01 | K → N | K → E | |||||||||||
| CHLY-09 | A → S | H → R | D → Y | T → K | |||||||||
| CH/FCH-01 | A → S | T → K | |||||||||||
| CH/LH | T → I | A → S | T → K | ||||||||||
| CH/FH | F → S | K → N | |||||||||||
| CH/LSHAN | T → A | S → A | A → V | K → N | T → M | ||||||||
| CH/XIP-03 | A → S | D → Y | – | K → E | |||||||||
| CH/JCH | T → K | ||||||||||||
| CH/ZHZ-04 | A → S | D → Y | T → K | ||||||||||
| CH/HEB | A → S | D → Y | T → K | ||||||||||
“–” represents one deletion at position aa 85 in the epitope region of the partial S protein of CH/XIP-03
Fig. 3.

Multiple alignment of partial S amino acid sequences of PEDV sample strains and reference strains. Shaded areas represent regions of greatest conservation. The red, yellow, and blue rectangles represent the locations of three neutralizing epitope regions reported previously, corresponding to aa 7-146, 245-252 and 271-278, respectively. The three green rectangles represent B-cell epitopes predicted in the region of aa 7-146, corresponding to aa 22-30, 32-40 and 64-75, respectively (color figure online)
Discussion
PEDV has been detected frequently in many provinces in China, including central China, since it was first identified in 1984, and has become one of the most important viral enteric diseases [7]. Despite the current vaccination strategy, the losses caused by PEDV infection are continuous and serious. It is necessary to understand the epidemiology of PEDV and to explore the antigenic variation of the virus. During the epidemiologic investigation, we confirmed that PEDV was the predominant causative pathogen contributing to outbreaks of clinical diarrhea in central China and determined that the positive rate of PEDV in all diarrhea samples tested was 78.6 %, followed by rotavirus (11.3 %) and transmissible gastroenteritis virus (TGEV) (6.4 %). Co-infections with multiple pathogens in diarrheal disease were very frequent, especially mixed infections with PEDV and porcine group A rotavirus (GARV). These findings are consistent with those reported by Zhang et al. [22]. The 14 samples used in this study were mostly from neonatal piglets (91.3 %), and they were obtained mainly in winter. To further investigate the molecular epidemiology of this virus, we chose the ORF3 gene and a partial S gene encoding a region including three neutralization epitopes for studying the evolutionary characteristic and antigenic variation of PEDV.
ORF3 is an important virulence gene that can be used to differentiate highly cell-adapted viruses and field isolates of PEDV and is a potential tool for studying the molecular epidemiology of PEDV [9, 23]. In the present study, 14 ORF3 genes of PEDV field strains collected in central China between 2012 and 2013 were amplified by RT-PCR, cloned, and sequenced to determine the genetic characteristics of viruses causing PED outbreaks in central China. The results confirmed that none of the 14 strains had the 51-nt deletion in ORF3. Sequence comparison with other PEDV reference strains selected from the GenBank database indicated that the ORF3 genes from the sample strains had a high degree of homology to most Chinese strains. For example, CH/LY-09, CH/FCH-01, CH/HEB, and CH/LH were distributed in the same subgroup with CH/ZMDZY/11; CH/YF-01 and CH/KF-01 were in the same subgroup with AH2012; and both CH/ZMDZY/11 and AH2012 were collected previously in central China. These results revealed that the PEDV strains prevailing in central China might originate from local regions in central China.
In addition, although relatively conserved when compared with the reference strains, variable sites were found in the ORF3 nucleotide sequences of these 14 isolates (alignment data not shown), leading to the following single amino acid changes: L → F at aa 3 (CH/FCH-01), Q → R at aa 40 (CH/XIP-03), S → A at aa 42 (CH/FH), T → A at aa 96 (CH/LY-09), Y → C at aa 140 (CH/SHANXI-02), L → S at aa 169 (CH/FCH-01), and E → K at aa 188 (CH/KF-01). We speculate that these mutations may cause differences in the virulence of PEDV in the field and consequently intensify the epidemic. Moreover, the results in this study did not show a 51-bp deletion in the complete ORF3 nucleotide sequence, indicating that the PEDV prevailing on swine farms of central China were wild-type strains.
As a glycoprotein on the viral surface, the S protein of PEDV is closely associated with its pathogenicity and immunogenicity [10]. In this study, 14 partial S genes from field PEDV strains were amplified by RT-PCR, cloned, sequenced, and analyzed to study their antigenic variation. Alignment of the deduced amino acid sequences of the partial S regions showed relatively high sequence identity among the PEDV strains. In particular, the 14 samples in this study were highly conserved in the neutralizing epitope region aa 245-252, but most of the changes occurred in the core epitope regions of aa 7-146 and aa 271-278. Of these, there were 10 samples with amino acid sequences changes at position 28: from A to V (CH/YF-01, CH/QX-02, CH/KF-01, CH/LSHAN) and A to S (CH/LY-09, CH/FCH-01, CH/LH, CH/XIP-03, CH/ZHZ-04, CH/HEB). The amino acid sequences in four samples (CH/YF-01, CH/QX-02, CH/KF-01, CH/LY-09) changed from H to R at aa 32, and seven samples (CH/YF-01, CH/XY-02, CH/QX-02, CH/KF-01, CH/XIP-01, CH/FH, CH/LSHAN) had a change from K to N at position 74. Furthermore, B-cell epitopes were predicted in the region of aa 7-146 using the BepiPred 1.0b Server. The results showed that there were three epitopes with the highest scores in the aa 7-146, located at positions 22-30, 32-40, 64-75. In this study, the amino acid mutation at positions 28, 32, and 74 were distributed within the epitope regions aa 22-30, 32-40, and 64-75, respectively (Fig. 3). These changes might alter the antigenicity of PEDV and consequently result in vaccination failure. Further study is needed to confirm the relationship between amino acid mutations in epitope regions and antigenicity, which would help to improve our understanding of the prevalence of PEDV in China.
Phylogenetic analysis based on the partial S gene showed that the 14 isolates were separated into two groups (Fig. 2), with CH/FCH-01, CH/LH, CH/LY-09, CH/JCH, CH/ZHZ-04, and CH/HEB forming a unique subgroup. Due to amino acid changes occurring mostly in epitope regions, these results indicated that the antigenicity of these strains differed from that of other strains, and these might be new PEDV variants prevailing in central China. Interestingly, although isolated from different geographical areas, two American strains (USA/Colorado/2013 and 13-019349), three Thai strains (VN12258, KU04RB08, and TH/NP-65/12) and one Korean strain (MK) were clustered within the same subgroup with some recent Chinese strains, indicating that these strains from different geographic regions might have similar antigenicity. In addition, one European strain (Br1/7), two Japanese strains (KH and NK), two Korean strains (DR13 and SM98), two early domestic strains (LZC and CH/S), and the vaccine strain (CV777) differed genetically from the other strains and clustered in the group G3 in the phylogenetic tree based on the partial S gene (Fig. 2). These results are consistent with those reported previously [24–26].
Based on the sequence analysis of ORF3 and partial S genes of PEDV, molecular epidemiology was conducted using 14 field strains from central China. Our results revealed that the PEDVs prevailing in the central China are still wild-type strains rather than the vaccine strain, and they mainly originated from the earlier strains in local regions. The amino acid mutations that occurred in the S epitope region might be associated with vaccination failure and the emergence of new PEDV variant strains. Further study should be done to examine the pathogenicity, antigenicity, and epidemiology of PEDV.
Acknowledgments
We gratefully acknowledge the invaluable comments and suggestions on the manuscript given by Yinbiao Wang and Qianyue Jin from Key Laboratory of Animal Immunology of the Ministry of Agriculture, China. This work was supported by grants from China Agriculture Research System (no. CARS-36), and the Pig Industry Technology System Innovation Team Project of Henan Province (S2012-06).
Footnotes
GenBank accession numbers of all sequences described in this study are listed in Table 2.
References
- 1.Bridgen A, Duarte M, Tobler K, Laude H, Ackermann M. Sequence determination of the nucleocapsid protein gene of the porcine epidemic diarrhoea virus confirms that this virus is a coronavirus related to human coronavirus 229E and porcine transmissible gastroenteritis virus. J Gen Virol. 1993;74:1795–1804. doi: 10.1099/0022-1317-74-9-1795. [DOI] [PubMed] [Google Scholar]
- 2.Pensaert MB, de Bouck P. A new coronavirus-like particles associated with diarrhea in swine. Arch Virol. 1978;58:243–247. doi: 10.1007/BF01317606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Duy DT, Toan NT, Puranaveja S, Thanawongnuwech R. Genetic characterization of porcine epidemic diarrhea virus PEDV isolates from southern Vietnam during 2009–2010 outbreaks. Thai J Vet Med. 2011;41:55–64. [Google Scholar]
- 4.Park SJ, Kim HK, Moon HJ, Song DS, Rho SM, Han JY, Nguyen VG, Park BK. Molecular detection of porcine kobuviruses in pigs in Korea and their association with diarrhea. Arch Virol. 2010;155:1803–1811. doi: 10.1007/s00705-010-0774-1. [DOI] [PubMed] [Google Scholar]
- 5.Puranaveja S, Poolperm P, Lertwatcharasarakul P, Kesdaengsakonwut S, Boonsoongnern A, Urairong K, Kitikoon P, Choojai P, Kedkovid R, Teankum K, Thanawongnuwech R. Chinese-like 19 strain of porcine epidemic diarrhea virus, Thailand. Emerg Infect Dis. 2009;15:1112–1115. doi: 10.3201/eid1507.081256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Xuan H, Xing D, Wang D, Zhu W, Zhao F, Gong H. Study on the culture of porcine epidemic diarrhea virus adapted to fetal porcine intestine primary cell monolayer. Chin J Vet Sci. 1984;4:202–208. [Google Scholar]
- 7.Gao Y, Kou Q, Ge X, Zhou L, Guo X, Yang H. Phylogenetic analysis of porcine epidemic diarrhea virus field strains prevailing recently in China. Arch Virol. 2013;158:711–715. doi: 10.1007/s00705-012-1541-2. [DOI] [PubMed] [Google Scholar]
- 8.Song DS, Park BK. Porcine epidemic diarrhea virus: a comprehensive review of molecular epidemiology, diagnosis, and vaccines. Virus Genes. 2012;44:167–175. doi: 10.1007/s11262-012-0713-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Song DS, Yang JS, Oh JS, Han JH, Park BK. Differentiation of a Vero cell adapted porcine epidemic diarrhea virus from Korean field strains by restriction fragment length polymorphism analysis of ORF 3. Vaccine. 2003;21:1833–1842. doi: 10.1016/S0264-410X(03)00027-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pospischil A, Stuedli A, Kiupel M. Diagnostic notes: update on porcine epidemic diarrhea. J Swine Health Prod. 2002;10:81–85. [Google Scholar]
- 11.Chang SH, Bae JL, Kang TJ, Kim J, Chung GH, Lim CW, Laude H, Yang MS, Jang YS. Identification of the epitope region capable of inducing neutralizing antibodies against the porcine epidemic diarrhea virus. Mol Cells. 2002;14:295–299. [PubMed] [Google Scholar]
- 12.Sun D, Feng L, Shi H, Chen J, Cui X, Chen H, Liu S, Tong Y, Wang Y, Tong G. Identification of two novel B cell epitopes on porcine epidemic diarrhea virus spike protein. Vet Microbiol. 2008;131:73–81. doi: 10.1016/j.vetmic.2008.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Godet M, Grosclaude J, Delmas B, Laude H. Major receptor-binding and neutralization epitope are located within the same domain of the transmissible gastroenteritis virus (coronavirus) spike protein. J Virol. 1994;68:8008–8016. doi: 10.1128/jvi.68.12.8008-8016.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sun DB, Feng L, Shi HY, Chen JF, Liu SW, Chen HY, Wang YF. Spike protein region (aa 636–789) of porcine epidemic diarrhea virus is essential for induction of neutralizing antibodies. Acta Virol. 2007;51:149–156. [PubMed] [Google Scholar]
- 15.Park SJ, Moon HJ, Yang JS, Lee CS, Song DS, Kang BK, Park BK. Sequence analysis of the partial spike glycoprotein gene of porcine epidemic diarrhea viruses isolated in Korea. Virus Genes. 2007;35:321–332. doi: 10.1007/s11262-007-0096-x. [DOI] [PubMed] [Google Scholar]
- 16.Chen J, Wang C, Shi H, Qiu H, Liu S, Chen X, Zhang Z, Feng L. Molecular epidemiology of porcine epidemic diarrhea virus in China. Arch Virol. 2010;155:1471–1476. doi: 10.1007/s00705-010-0720-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen X, Yang J, Yu F, Ge J, Lin T, Song T. Molecular characterization and phylogenetic analysis of porcine epidemic diarrhea virus (PEDV) samples from field cases in Fujian, China. Virus Genes. 2012;45:499–507. doi: 10.1007/s11262-012-0794-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li W, Li H, Liu Y, Pan Y, Deng F, Song Y, Tang X, He Q. New variants of porcine epidemic diarrhea virus, China. Emerg Infect Dis. 2012;18:1350–1353. doi: 10.3201/eid1803.120002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sun RQ, Cai RJ, Chen YQ, Liang PS, Chen DK, Song CX. Outbreak of porcine epidemic diarrhea in suckling piglets, China. Emerg Infect Dis. 2012;18:161–163. doi: 10.3201/eid1801.111259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol. 2011;28:2731–2739. doi: 10.1093/molbev/msr121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Larsen JE, Lund O, Nielsen M. Improved method for predicting linear B-cell epitopes. Immunome Res. 2006;2:2. doi: 10.1186/1745-7580-2-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang Q, Hu R, Tang X, Wu C, He Q, Zhao Z, Chen H, Wu B. Occurrence and investigation of enteric viral infections in pigs with diarrhea in China. Arch Virol. 2013;158:1631–1636. doi: 10.1007/s00705-013-1659-x. [DOI] [PubMed] [Google Scholar]
- 23.Shirato K, Matsuyama S, Ujike M, Taguchi F. Role of proteases in the release of porcine epidemic diarrhea virus from infected cells. J Virol. 2011;85:7872–7880. doi: 10.1128/JVI.00464-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Park SJ, Kim HK, Song DS, Moon HJ, Park BK. Molecular characterization and phylogenetic analysis of porcine epidemic diarrhea virus (PEDV) field isolates in Korea. Arch Virol. 2011;156:577–585. doi: 10.1007/s00705-010-0892-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yang X, Huo JY, Chen L, Zheng FM, Chang HT, Zhao J, Wang XW, Wang C-Q. Genetic variation analysis of reemerging porcine epidemic diarrhea virus prevailing in central China from 2010 to 2011. Virus Genes. 2013;46:337–344. doi: 10.1007/s11262-012-0867-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li Z, Chen F, Yuan Y, Zeng X, Wei Z, Zhu L, Sun B, Xie Q, Cao Y, Xue C, Ma J, Bee Y. Sequence and phylogenetic analysis of nucleocapsid genes of porcine epidemic diarrhea virus (PEDV) strains in China. Arch Virol. 2013;158:1267–1273. doi: 10.1007/s00705-012-1592-4. [DOI] [PMC free article] [PubMed] [Google Scholar]