

Abstract
Bats in Myanmar, Gabon, and Panama have been found to harbor diverse hepadnaviruses. Here, we report a novel hepadnavirus in 4 of 20 pomona roundleaf bats from Yunnan province, China. This virus contains 3,278 nucleotides (nt) in the full circularized genome, with four predicted open frames (ORFs) reading in the same direction. Full genomic sequence comparisons and evolutionary analysis indicate that this virus is a member of a new species within the genus Orthohepadnavirus
Keywords: Bat, Orthohepadnavirus, Genetic diversity
Introduction
Bats are important reservoir animals, harboring pathogenic zoonotic viruses including henipaviruses, lyssaviruses, and SARS-like coronaviruses [1]. The number of novel viruses identified in bats has been significantly increasing due to extensive studies by next-generation sequencing over the last few years [7]. Since the identification of a novel bat orthohepadnavirus (BtHV) from Miniopterus spp. in Myanmar [3], further similar viruses have been found in Uroderma spp. in Panama and Rhinolophus spp. and Hipposideros spp. in Gabon [2]. We report here another BtHV identified in bats in Yunnan province of China.
In 2011, 42 bats were collected in Pu’er city in Yunnan province from bats of three species: Hipposideros pomona (n = 20), Rhinolophus affinis (n = 20), and Rhinolophus sinicus (n = 2). The sampling of bats was approved by the Administrative Committee on Animal Welfare of the Military Veterinary Institute, Academy of Military Medical Sciences, China (Laboratory Animal Care and Use Committee Authorization, permit number JSY-DW-2010-02). All organs were pooled and subjected to viral metagenomic analysis as per our published method [4]. Results revealed 1,436 reads relating to orthohepadnaviruses, with an average length of 147 nucleotides (nt) and showing 68-74 % nt sequence identity to a Hipposideros BtHV from Gabon in its partial S and C genes. All liver samples were screened by PCR for orthohepadnavirus as described previously [3]. The results showed that 20 % (4/20) of pomona roundleaf bats (Hipposideros pomona) were positive, with all amplicons sharing >99 % nt sequence identity. All remaining bats were negative.
Three of the positives were randomly selected for full genomic amplification and sequencing. the results showed that their full genomic sequences (BtHV PEPR6, PEPR7 and PEPR13, GenBank accession numbers: KF939648-KF939650) were >99 % identical at the nucleotide level, with the same size of 3,278 nt; i.e., slightly larger than the genome of BtHV found in Myanmar (3230 nt) [3]. The genomic organizations were identical to those of other orthohepadnaviruses, containing four open reading frames (ORFs) encoding Pol, preS1/preS2/S, preC/C and X proteins in a circular and compact configuration. Orthohepadnaviral genomic characteristics, such as a highly conserved YMDD motif [6] and encapsidation signal ε [5], were also found in the PEPR genomes and were essentially identical to those of other orthohepadnaviruses. Predicted genomic structures and sequence comparisons are summarized in Table 1. The results showed that PEPR shared the highest nt and amino acid (aa) sequence identities in the Pol-, preS1/preS2/S-, and X genes with strain HBHBV identified in Hipposideros spp. from Gabon [2], while the preC/C gene of PEPR alone showed highest nt and aa sequence identity with isolate 776, a representative of Miniopterus BtHV in Myanmar [3].
Table 1.
Gene lengths and percent identity between BtHV and other hepadnaviruses
| Virus | Full genome | Pol gene | preS1/preS2/S gene | preC/C gene | X gene | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| nt | %ID | nt | %ID | aa | %ID | nt | %ID | aa | %ID | nt | %ID | aa | %ID | nt | %ID | aa | %ID | |
| PEPR6 | 3,278 | 2,607 | 868 | 1,248 | 415 | 654 | 217 | 429 | 142 | |||||||||
| 776 | 3,230 | 70.3 | 2,562 | 71.7 | 853 | 64.1 | 1,200 | 74.3 | 399 | 62.9 | 654 | 81.0 | 217 | 80.1 | 435 | 73.7 | 144 | 55.8 |
| TBHBV | 3,149 | 59.8 | 2,484 | 59.6 | 827 | 55.2 | 1,149 | 67.6 | 382 | 57.3 | 663 | 65.1 | 220 | 58.0 | 408 | 57.0 | 135 | 43.3 |
| HBHBV | 3,377 | 73.0 | 2,709 | 73.8 | 902 | 70.0 | 1,347 | 76.9 | 448 | 69.2 | 654 | 74.7 | 217 | 75.6 | 426 | 78.6 | 141 | 64.8 |
| RBHBV | 3,368 | 72.5 | 2,700 | 73.4 | 899 | 69.7 | 1,338 | 76.3 | 445 | 66.4 | 654 | 76.5 | 217 | 76.9 | 426 | 77.7 | 141 | 62.0 |
| HBV | 3,215 | 63.1 | 2,532 | 63.0 | 843 | 57.0 | 1,203 | 64.1 | 400 | 57.0 | 639 | 76.3 | 212 | 75.9 | 465 | 56.5 | 154 | 43.4 |
| WMHBV | 3,179 | 63.2 | 2,508 | 63.3 | 835 | 55.8 | 1,176 | 64.7 | 391 | 59.0 | 636 | 76.2 | 211 | 73.7 | 459 | 60.4 | 152 | 43.8 |
| WHV | 3,308 | 62.8 | 2,640 | 60.6 | 879 | 55.0 | 1,281 | 64.8 | 426 | 47.5 | 678 | 78.9 | 225 | 78.0 | 426 | 67.8 | 141 | 51.0 |
| ASHV | 3,302 | 63.4 | 2,634 | 60.5 | 877 | 54.5 | 1,284 | 64.5 | 427 | 48.5 | 654 | 79.3 | 217 | 77.6 | 417 | 68.0 | 138 | 55.0 |
| DHBV | 3,006 | 37.5 | 2,376 | 36.3 | 791 | 31.9 | 1,104 | 37.7 | 367 | 18.1 | 888 | 30.9 | 295 | 10.8 | NA | / | NA | / |
nt, the length of the gene in nucleotides; %ID, the percent identity of nucleotide sequence and amino acid sequence between BtHV PEPR6 and the others; aa, the length of the gene in amino acids; NA, X gene not available; the accession numbers of 776 (BtHV), TBHBV, HBHBV, RBHBV, HBV, WMHBV, WHV, ASHV and DHBV are JX941466, KC790381, KC790377, KC790376, D00329, AF046996, AY344076, U29144 and EU429324, respectively. The highest identities are in bold italic
To determine the phylogenetic relationship of PEPR to other orthohepadnaviruses, Pol genes from all known members of the family Hepadnaviridae [8] and including previously reported BtHVs [2, 3] were aligned using ClustalW, available in MEGA5.1 [9]. The phylogenetic tree showed that all sequences clustered into seven distinct groups sharing <74 % nt identity. Three consisted of avian, primate, and rodent hepatitis viruses and corresponded to the approved species listed in the Ninth Report of the International Committee on Taxonomy of Viruses (ICTV) from 2011 [8] (Fig. 1). The bat-associated orthohepadnaviruses identified in Myanmar, Panama, and Gabon [2, 3] and in this study were genetically diverse and classified into four distinct groups (Fig. 1). Comparisons of the full genome sequence of PEPR with those of BtHVs showed that sequence identity ranged from 60 % (with TBHBV) to 73 % (with HBHBV), and around 63 % with primate and rodent hepatitis viruses. Notably, PEPR was only 70 % identical to BtHVs in Myanmar, showing wide genetic heterogeneity. Although PEPR in Yunnan and HBHBV from Gabon both originated from Hipposideros spp., they shared only 73 % nt sequence identity; i.e., less than within both the primate hepatitis virus group (e.g., HBV and WMHBV; ~80 %) and the rodent hepatitis virus group (e.g., WHV and GSHV; ~85 %) [8]. These results show that BtHVs of wide genetic diversity exist within the genus Orthohepadnavirus, differing according to the bat genus or species and therefore requiring classification into different species.
Fig. 1.
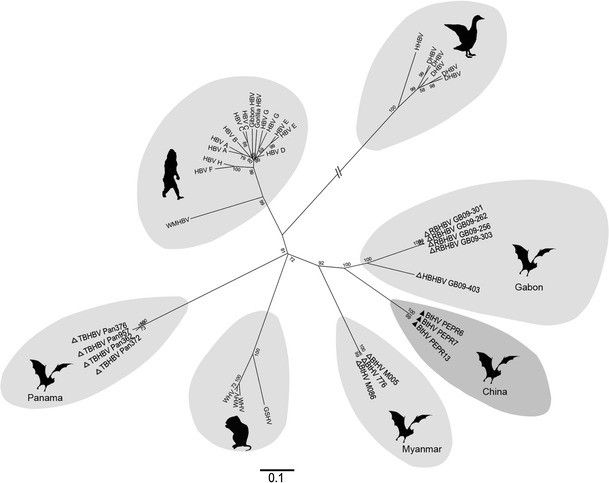
Evolutionary analysis of BtHV and representatives of other orthohepadnaviruses based on their complete Pol nucleotide sequences, with avian hepatitis viruses used as an outgroup. The phylogenetic tree was constructed by the maximum-likelihood method using MEGA 5.1. Viruses identified in this study are indicted by filled triangles; previous BtHVs, with open triangles. The hosts of RBHBV and HBHBV in Gabon, TBHBV in Panama, BtHV in Myanmar and BtHV in China are Rhinolophus alcyone, Hipposideros cf. ruber, Uroderma bilobatum, Miniopterus fuliginosus and Hipposideros pomona. The scale bar indicates nucleotide substitutions per site
Pu’er is about 430 km southeast of Sedon and Wutao in Myanmar, where BtHV 776 was found in Miniopterus spp. but not in Hipposideros and other bat species [3]. BtHVs were also absent from Rhinolophus spp. in Pu’er. This is in contrast to HBHBV and RBHBV, both detected in Gabon in Hipposideros and Rhinolophus spp., respectively, and sharing <82 % identity [2]. These results suggest the existence of diverse orthohepadnaviruses naturally circulating in various bat species worldwide, but further investigation with expanded bat sampling will be needed to determine if BtHVs have restricted host ranges and geographical distributions.
HBV causes hepatitis B, cirrhosis and hepatocellular carcinoma in humans, resulting in >240 million chronic liver infections worldwide and about 600,000 deaths every year [10]. A previous study has shown that a BtHV cross-reacts with sera against HBV core protein, and reverse genetics has indicated that TBHBV from Panama has the ability to infect primary human hepatocytes [2], suggesting a potential zoonotic threat of BtHVs to humans.
In conclusion, BtHVs are a novel group of orthohepadnaviruses of wide genetic diversity that are widely distributed among different bat species. They may provide new insights into HBV transmission, infection, and prevention, and their impact on public health merits further investigation.
Acknowledgments
This study was supported by NSFC-Yunnan Province Joint Fund (U1036601), National “973’’ Program (Grant No. 2012CB722501) and National “863’’ Program (Grant No. 2012AA022006) to C. Tu.
Conflict of interest
The authors declare that they have no conflict of interest.
Footnotes
B. He, F. Zhang and L. Xia contributed equally to this work.
References
- 1.Calisher CH, Childs JE, Field HE, Holmes KV, Schountz T. Bats: important reservoir hosts of emerging viruses. Clin Microbiol Rev. 2006;19:531–545. doi: 10.1128/CMR.00017-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Drexler JF, Geipel A, Konig A, Corman VM, van Riel D, Leijten LM, Bremer CM, Rasche A, Cottontail VM, Maganga GD, Schlegel M, Muller MA, Adam A, Klose SM, Borges Carneiro AJ, Stocker A, Franke CR, Gloza-Rausch F, Geyer J, Annan A, Adu-Sarkodie Y, Oppong S, Binger T, Vallo P, Tschapka M, Ulrich RG, Gerlich WH, Leroy E, Kuiken T, Glebe D, Drosten C. Bats carry pathogenic hepadnaviruses antigenically related to hepatitis B virus and capable of infecting human hepatocytes. Proc Natl Acad Sci USA. 2013;110:16151–16156. doi: 10.1073/pnas.1308049110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.He B, Fan Q, Yang F, Hu T, Qiu W, Feng Y, Li Z, Li Y, Zhang F, Guo H, Zou X, Tu C. Hepatitis virus in long-fingered bats, Myanmar. Emerg Infect Dis. 2013;19:638–640. doi: 10.3201/eid1904.121655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.He B, Li Z, Yang F, Zheng J, Feng Y, Guo H, Li Y, Wang Y, Su N, Zhang F, Fan Q, Tu C. Virome profiling of bats from Myanmar by metagenomic analysis of tissue samples reveals more novel Mammalian viruses. PLOS One. 2013;8:e61950. doi: 10.1371/journal.pone.0061950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Knaus T, Nassal M. The encapsidation signal on the hepatitis B virus RNA pregenome forms a stem-loop structure that is critical for its function. Nucleic Acids Res. 1993;21:3967–3975. doi: 10.1093/nar/21.17.3967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ling R, Mutimer D, Ahmed M, Boxall EH, Elias E, Dusheiko GM, Harrison TJ. Selection of mutations in the hepatitis B virus polymerase during therapy of transplant recipients with lamivudine. Hepatology. 1996;24:711–713. doi: 10.1002/hep.510240339. [DOI] [PubMed] [Google Scholar]
- 7.Luis AD, Hayman DT, O’Shea TJ, Cryan PM, Gilbert AT, Pulliam JR, Mills JN, Timonin ME, Willis CK, Cunningham AA, Fooks AR, Rupprecht CE, Wood JL, Webb CT. A comparison of bats and rodents as reservoirs of zoonotic viruses: are bats special? Proc Biol Sci. 2013;280:20122753. doi: 10.1098/rspb.2012.2753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mason WS, Gerlich WH, Taylor JM, Kann M, Mizokami T, Loeb D, Sureau C, Magnius L, Norder H. Family Hepadnaviridae. In: King AMQ, Adams MJ, Carstens EB, Lefkowitz EJ, editors. Virus taxonomy: classification and nomenclature of viruses: ninth report of the International Committee on Taxonomy of Viruses. London: Academic Press Ltd.; 2011. pp. 445–455. [Google Scholar]
- 9.Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol. 2011;28:2731–2739. doi: 10.1093/molbev/msr121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. World Health Organization (2014) Hepatitis B, fact sheet no. 204. http://www.who.int/mediacentre/factsheets/fs204/en/index.html. Accessed 10 Mar 2014